Embed Size (px)
Citation preview

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Open AccessR E S E A R C H A R T I C L E
Research articleProgesterone reverses the mesenchymal phenotypes of basal phenotype breast cancer cells via a membrane progesterone receptor mediated pathwayLian Zuo, Wei Li and Shaojin You*
AbstractIntroduction: Basal phenotype breast cancers (BPBC) are often associated with apparent epithelial to mesenchymal transition (EMT). The role of progesterone (P4) in regulating EMT of BPBC has not been reported.
Methods: The EMT relevant biology was investigated in vitro using human BPBC cell models (MDA-MB468 and MDA-MB231) with P4, PR agonist (RU486), and PR antagonist (R5020) treatments. The essential role of membrane progesterone receptor α (mPRα) in the P4-regulated EMT was demonstrated by knocking down the endogenous gene and/or stably transfecting exogenous mPRα gene in the BPBC cell models.
Results: The expression of snail and down-stream EMT proteins such as occludin, fibronectin, and E-cadherin was significantly regulated by P4 incubation, which was accompanied by cell morphological reversion from mesenchymal to epithelial phenotypes. In searching for the cell mediator of P4' action in the MDA-MB468 (MB468) cells, it was found that mPRα but not the nuclear PR has an essential role in the P4 mediated EMT inhibition. Knocking down the expression of mPRα with specific siRNA blocked the P4's effects on expression of the EMT proteins. In another BPBC cell line - MDA-MB231 (MB231), which is mPRα negative by Western blotting, P4 treatment did not alter cell proliferation and EMT protein expressions. Introduction of the exogenous mPRα cDNA into these cells caused cell proliferation, but not EMT, to become responsive to P4 treatment. In further studies, it was found that activation of the PI3K/Akt pathway is necessary for the P4-induced EMT reversion. To define the potential inter-mediate steps between mPRα and PI3K, we demonstrated that mPRα, caveolin-1 (Cav-1), and epidermal growth factor receptor (EGFR) are colocalized in the membrane of caveolar vesicle and the P4-repressed EMT in MB468 cells can be blocked by EGFR inhibitor (AG1478) and PI3K inhibitor (wortmannin).
Conclusions: Our data suggest that the signaling cascade of P4 induced mesenchymal repression is mediated through mPRα and other caveolae bound signaling molecules namely Cav-1, EGFR, and PI3K. This novel finding may have great impact on fully understanding the pathogenesis of BPBC and provide an essential clue for developing a targeted therapeutic strategy for treatment of BPBC.
IntroductionBasal phenotype breast cancer (BPBC) is a subtype ofcancer with apparent mesenchymal phenotypes. Boyerand colleagues first described a morphologic changefrom epithelial-like sheets of cultured cancer cells to scat-tered, fibroblast-like cells capable of invading the base-
ment membrane, so called epithelial to mesenchymaltransition (EMT) [1]. The morphologic criteria of EMTin vitro involve changes in cell polarity, separation intoindividual cells and acquisition of cell motility [2]. Thesechanges can be either stable or reversible. The essentialchanges in gene expression that disrupt cell polarity andcause mesenchymal transition have been identified. Snail,twist, and slug have been shown as key regulators of EMTin both animal and human cancers [3]. Among these
* Correspondence: [email protected] Research & Educational Foundation (151F), Atlanta VA Medical Center, 1670 Clairmont Road, Decatur, GA 30033, USAFull list of author information is available at the end of the article
© 2010 Zuo et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons At-tribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in anymedium, provided the original work is properly cited.

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 2 of 12
genes, snail acts as a transcriptional factor to repressgenes that encode the cell-cell junctional apparatus, suchas E-cadherin and occludin; and to enhance genes thatencode mesenchymal or tumor interstitial components,such as fibronectin and vimentin, resulting in a dediffer-entiated mesenchymal transition characterized byincreased cell motility [4,5].
The roles of female sex hormones such as progesterone(P4) in the pathogenesis of BPBC remain unclear. Classi-cally, the actions of P4 on cancer cells are attributed tothe binding of nuclear progesterone receptor (PR), trans-location of P4/PR complex into the nucleus and subse-quent activation of target genes over the course of severalhours. These mechanisms, however, are not applicable toBPBC due to a lack or very low level of PR expression inthese cancers. The mechanisms for P4's actions in modu-lating the cancer biology of BPBC remain largelyunknown. Recently, the cell membrane hormonal recep-tors, such as membrane progesterone receptor (mPR)family and progestin membrane receptor component 1(PGMRC1), were identified and demonstrated functionalin BPBC [6,7]. It is believed that the rapid responses of P4are initiated at the cell surface by binding to the mem-brane receptors [8-10]. For examples, progestin, a syn-thetic P4, has been shown to activate a variety of signalingpathways through mPRα [6]. The binding of progestin tomPRα alters the secondary messenger pathways throughactivation of the pertussis toxin-sensitive inhibitory G-proteins and then activates the mitogen activated proteinkinases (MAPK)/Erk 1/2 pathway [6,7,11,12]. However,this theory has been debated because others failed todemonstrate mPRs on the cell surface or mediate P4-dependent signaling events, such as coupling to G pro-teins [13]. Moreover, mPRs were shown to be primarilysituated in the endoplasmic reticulum [13,14]. In thisstudy, we co-localized mPRα, caveolin-1 (Cav-1), and epi-dermal growth factor receptor (EGFR) at a specifiedmembrane structure, so called caveolar vesicle, and dem-onstrated that P4 reverses the mesenchymal phenotypesof human BPBC cells (MB468 and MB231) via a caveolaebound signaling complex namely mPRα, Cav-1, EGFR,and PI3K/Akt. Further study on this unique molecularpathway may afford great potential to discover novelmolecular targets for treatment of BPBC.
Materials and methodsChemicals and antibodiesRU486 (mifepristone), AG1498, wortmannin, PP1 andPD98052 were purchased from EMD Chemicals (Gibb-stown, NJ, USA); R5020 (promegestone) and bpV (phen)were from PerkinElmer (Waltham, MA, USA) andThermo Fisher Scientific (Pittsburgh, PA, USA), respec-tively. Anti-snail antibody was from Abcam (1:1000,
Cambridge, MA, USA); anti-E-cadherin and anti-fibronectin antibodies were obtained from EPITMICS(1:1000, Burlingame, CA, USA); anti-mPRα, anti-GAPDH (1:500) and secondary antibodies (1:2000) werepurchased from Santa Cruz Biotechnology (Santa Cruz,CA, USA); anti-occludin antibody was from BD trans-duction (1:500, San Jose, CA, USA); and anti-α-tubulinantibody was from Sigma (1:2000, St. Louis, MO, USA).
Cell cultureThe human breast cancer cell lines MDA-MB468(MB468), MDA-MB231 (MB231) and human embryonickidney 293 (HEK 293) cells were obtained from theAmerican Type Culture Collection (Rockville, MD, USA).Both human breast cancer cell lines were negative forestrogen receptor (ER) and human epidermal growth fac-tor receptor (Her)-2 and classified as 'basal phenotype A'cells [15]. The cultured MB468 cells at early passages typ-ically appear like epithelial cells with oval and/or polygo-nal shapes [16]; and after multiple passages (50+), thesecells exhibit apparent mesenchymal phenotypes withspindle and elongated shapes [see Additional file 1],which are ideal for the proposed studies. Long-term cellculture in vitro may generate genetic instabilities and thederived cell lines with altered cell biological features havebeen utilized as cell models for in vitro studies [17,18].The late passage MB468 cells and early passage MB231cells with apparent mesenchymal phenotypes [16] werecultured and maintained at 37°C with 21% oxygen and 5%carbon dioxide (without 5% carbon dioxide for MB231and HEK293 culture) in DMEM (Sigma, St. Louis, MO,USA) containing 10% FBS (Gibco, Carlsbad, CA, USA), 2mM L-glutamine, 100 U/ml penicillin, and 100 μg/mlstreptomycin (Gibco, Carlsbad, CA, USA) and main-tained in a humidified incubator. The XTT cell prolifera-tion assay kit was from Cayman Chemicals (Ann Arbor,MI, USA).
ImmunoblottingWestern blot assays were performed as described previ-ously [19]. After treatment with or without P4 and/ordiverse pathway inhibitors, the growth-arrested cellswere lysed with 500 μl ice-cold lysis buffer, pH 7.4 (50mM HEPES, 5 mM EDTA, 50 mM NaCl), 1% Triton X-100, protease inhibitors (10 μg/ml aprotinin, 1 mM phe-nylmethylsulfonyl fluoride, 10 μg/ml leupeptin) andphosphatase inhibitors (50 mM sodium fluoride, 1 mMsodium orthovanadate, 10 mM sodium pyrophosphate).Cell lysates (25 μg) were separated using SDS-PAGE andtransferred to nitrocellulose membranes, blocked over-night in PBS containing 6% nonfat dry milk and 0.1%Tween 20, and incubated for one hour with primary anti-bodies at proper dilutions. After incubation with second-ary antibodies, proteins were detected by ECL

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 3 of 12
chemiluminescence. Image J was used for quantitativeanalysis.
Cell morphological changes of MB468 treated with P4MB468 cells (105 cells/dish) were seeded and grown in 35mm cell culture dishes (Bioptechs Inc., Butler, PA, USA)for 24 hours. The medium was changed to complete cul-ture medium with or without 30 ng/ml of P4 for 48 hoursand then cultured as indicated. Nomarski differentialinterference contrast (DIC) images were taken using aconfocal microscopy (Olympus FV1000, Tokyo, Japan)with a transmitted light at 400× magnification.
Cell proliferation assayThe XTT cell proliferation assay was performed accord-ing to the manufacturer's protocol. Briefly, cells wereseeded in a 96-well plate in 100 μl of culture medium withor without the compounds to be tested and incubated for24 to 48 hours at 37°C. The reconstituted XTT mixture(10 μl/well) was added and the cells were incubated fortwo hours. The absorbance of each sample was subse-quently measured using a microplate reader at a wave-length of 450 nm.
Knocking down mPRα expression with small interference RNACells were transfected with mPRα small interferenceRNA (siRNA) or an equal amount of nonspecific controlsiRNA (Dharmacon, Lafayette, CO, USA) using the Olig-ofectamine reagents according to the manufacturer's pro-tocol (Invitrogen, Carlsbad, CA, USA). Two days aftertransfection with siRNA, the cells were incubated withdiverse experimental reagents.
Transfection of mPRα DNA plasmidThe MB231 cells were cultured and split when the cellconfluence reached about 90%. The human mPRα cDNAconstructed in a pUC-based plasmid with CMV pro-moter (pBK-CMV) vector [20] was purified and thentransfected into the cells using Lipofectamine 2000reagent following the manufacturer's instructions (Invit-rogen, Carlsbad, CA, USA). Two days after transfection,the mPRα expressing cells were selected with 1000 μg/mlG418 (Gibco, Carlsbad, CA, USA). The resistant colonieswere then isolated and propagated with 500 μg/ml G418in order to produce the stably transfected cell lines.
Isolation of caveolar fractionsCaveolae membranes were isolated as described previ-ously [21]. Briefly, MB468 cells was homogenized in 1 mlof 2-(N-morpholino) ethanesulfonic acid (MES)-bufferedsaline (24 mM MES, pH 6.5, and 0.15 NaCl) plus 1% Tri-ton X-100 and spun down at 3,000 g for five minutes at4°C. The supernatant (4 ml) was used to dissolve sucroseand compose 40% of sucrose solution. This solution was
placed in a 12.5-ml Beckman centrifuge tube (BeckmanCoulter, Fullerton, CA, USA) with a 5 to 30% sucrose gra-dient layered on top and then centrafuged at 39,000 rpmfor 24 hours at 4°C in a Beckman SW-41 rotor. After thecentrafuge, 600 μl fractions of the solution were collectedand subjected to further analysis.
Immunohistochemical analysisIn brief, two tissue microarray slides consisting of humanbreast cancer (140 and 70) cores and adjacent benignbreast tissue (10 and 24) cores were purchased from theBiomax US (Rockville, MD, USA). These tissue microar-ray slides were constructed with complete different setsof tissue blocks. There were total 105 breast cancer and17 benign breast tissues in these two tissue arrays. Two1.5 mm-cores from each breast tissue block were con-structed in the tissue microarray slides. After deparaffini-zation, rehydration, antigen retrieval, and endogenousperoxidase blocking, the slides were blocked with 5% nor-mal horse serum for one hour and sequentially incubatedwith anti-mPRα antibody (1:200 dilution) at 4°C over-night and then incubated with a secondary antibody atroom temperature (see manual of the ImmPRESSREAGENT kit, VECTOR Lab, CA). The color was devel-oped with the ImmPACT DAB kit (VECTOR Lab, Burl-ingame, CA, USA). Between the incubations, the slideswere washed twice with 1× PBS buffer (5 minutes each)[19,22]. Two negative controls were included: (1) controlslides were stained without the primary antibody; (2)control slides were incubated with a specific blockingpeptide (cat# sc-50111p, Santa Cruz, CA, USA) prior tothe primary antibody incubations. The immunostainedslides were counterstained with hematoxylin and evalu-ated using a Nikon microscope with an Olympus digitalcamera (Tokyo, Japan). The immunohistochemical resultwas evaluated using a semi-quantitative scoring systemby a trained research pathologist, who was blinded topatients' clinical data provided by the company. Theintensity of the immunostaining was defined into fourcategories (strong positive, moderate, weak, and nega-tive) [see Additional file 2].
Statistical analysisThe quantitative data was expressed as mean ± standarderror and statistical significance was assessed by Stu-dent's paired two-tailed t-test. The positive rates of mPRαimmunostains in different groups of human breast can-cers and benign diseases was compared and analyzed byFisher's exact test. P < 0.05 was considered significant.
ResultsP4 regulates expression of snail and other EMT-relevant proteins in MB468 but not in MB231 cellsIn this study, we focused on the effects of P4 on expres-sion of snail and other EMT marker proteins. As shown
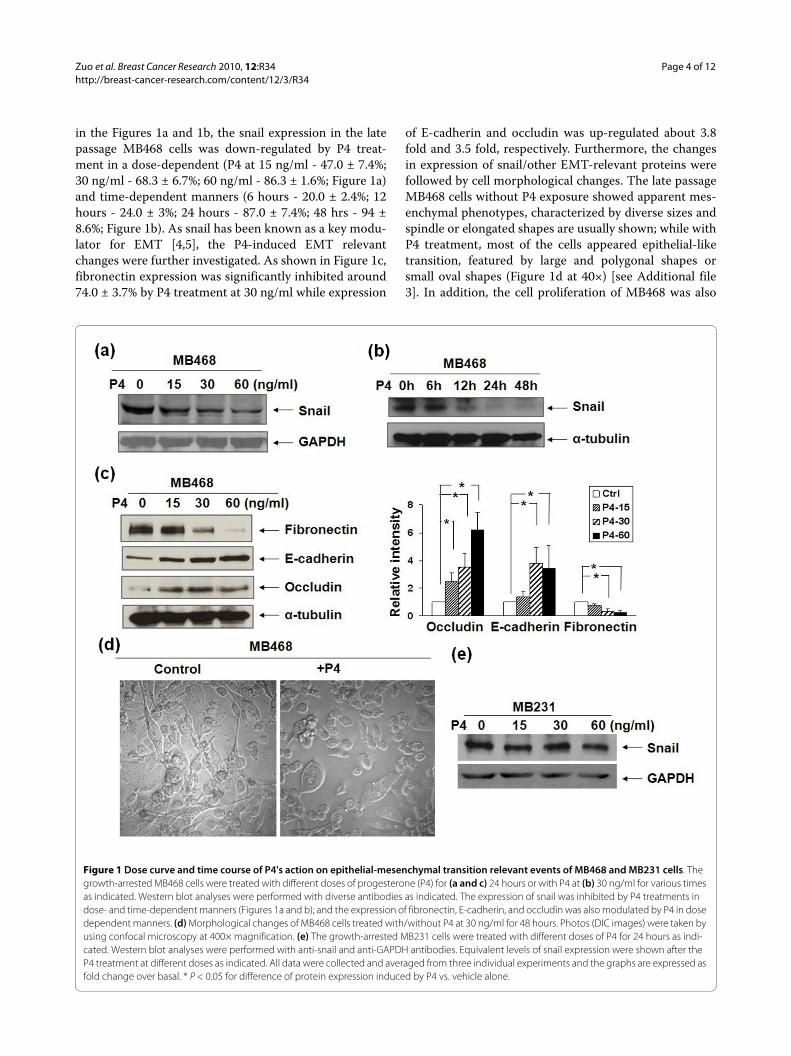
Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 4 of 12
in the Figures 1a and 1b, the snail expression in the latepassage MB468 cells was down-regulated by P4 treat-ment in a dose-dependent (P4 at 15 ng/ml - 47.0 ± 7.4%;30 ng/ml - 68.3 ± 6.7%; 60 ng/ml - 86.3 ± 1.6%; Figure 1a)and time-dependent manners (6 hours - 20.0 ± 2.4%; 12hours - 24.0 ± 3%; 24 hours - 87.0 ± 7.4%; 48 hrs - 94 ±8.6%; Figure 1b). As snail has been known as a key modu-lator for EMT [4,5], the P4-induced EMT relevantchanges were further investigated. As shown in Figure 1c,fibronectin expression was significantly inhibited around74.0 ± 3.7% by P4 treatment at 30 ng/ml while expression
of E-cadherin and occludin was up-regulated about 3.8fold and 3.5 fold, respectively. Furthermore, the changesin expression of snail/other EMT-relevant proteins werefollowed by cell morphological changes. The late passageMB468 cells without P4 exposure showed apparent mes-enchymal phenotypes, characterized by diverse sizes andspindle or elongated shapes are usually shown; while withP4 treatment, most of the cells appeared epithelial-liketransition, featured by large and polygonal shapes orsmall oval shapes (Figure 1d at 40×) [see Additional file3]. In addition, the cell proliferation of MB468 was also
Figure 1 Dose curve and time course of P4's action on epithelial-mesenchymal transition relevant events of MB468 and MB231 cells. The growth-arrested MB468 cells were treated with different doses of progesterone (P4) for (a and c) 24 hours or with P4 at (b) 30 ng/ml for various times as indicated. Western blot analyses were performed with diverse antibodies as indicated. The expression of snail was inhibited by P4 treatments in dose- and time-dependent manners (Figures 1a and b); and the expression of fibronectin, E-cadherin, and occludin was also modulated by P4 in dose dependent manners. (d) Morphological changes of MB468 cells treated with/without P4 at 30 ng/ml for 48 hours. Photos (DIC images) were taken by using confocal microscopy at 400× magnification. (e) The growth-arrested MB231 cells were treated with different doses of P4 for 24 hours as indi-cated. Western blot analyses were performed with anti-snail and anti-GAPDH antibodies. Equivalent levels of snail expression were shown after the P4 treatment at different doses as indicated. All data were collected and averaged from three individual experiments and the graphs are expressed as fold change over basal. * P < 0.05 for difference of protein expression induced by P4 vs. vehicle alone.
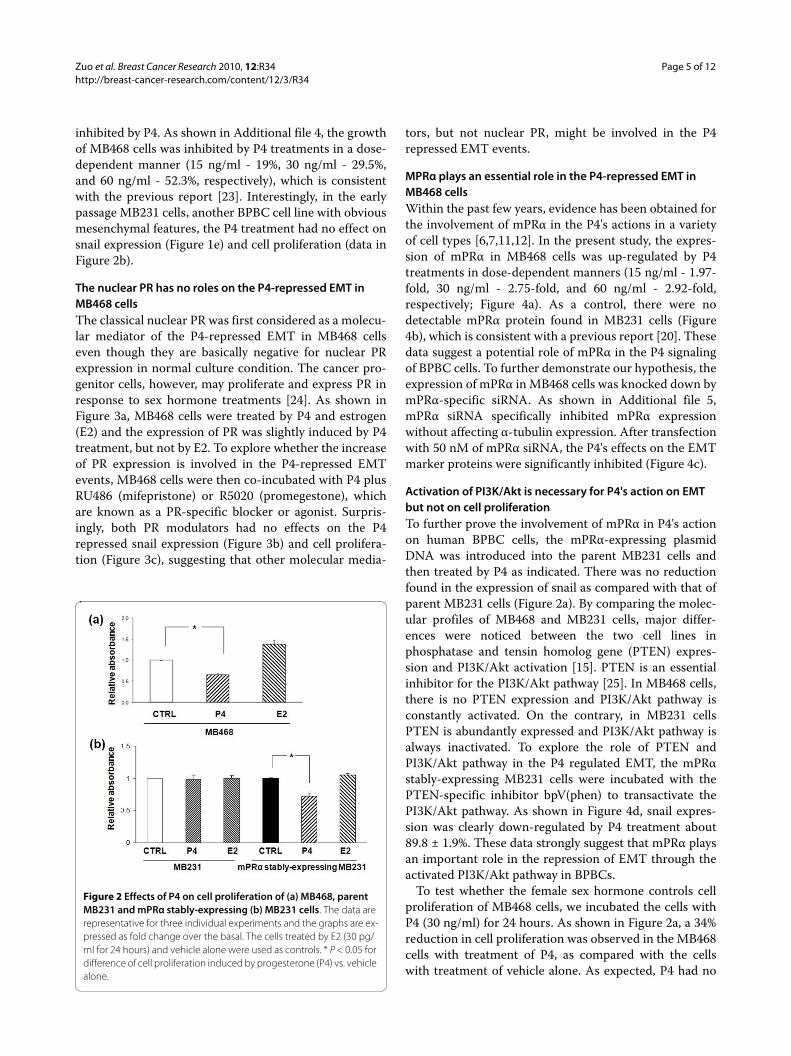
Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 5 of 12
inhibited by P4. As shown in Additional file 4, the growthof MB468 cells was inhibited by P4 treatments in a dose-dependent manner (15 ng/ml - 19%, 30 ng/ml - 29.5%,and 60 ng/ml - 52.3%, respectively), which is consistentwith the previous report [23]. Interestingly, in the earlypassage MB231 cells, another BPBC cell line with obviousmesenchymal features, the P4 treatment had no effect onsnail expression (Figure 1e) and cell proliferation (data inFigure 2b).
The nuclear PR has no roles on the P4-repressed EMT in MB468 cellsThe classical nuclear PR was first considered as a molecu-lar mediator of the P4-repressed EMT in MB468 cellseven though they are basically negative for nuclear PRexpression in normal culture condition. The cancer pro-genitor cells, however, may proliferate and express PR inresponse to sex hormone treatments [24]. As shown inFigure 3a, MB468 cells were treated by P4 and estrogen(E2) and the expression of PR was slightly induced by P4treatment, but not by E2. To explore whether the increaseof PR expression is involved in the P4-repressed EMTevents, MB468 cells were then co-incubated with P4 plusRU486 (mifepristone) or R5020 (promegestone), whichare known as a PR-specific blocker or agonist. Surpris-ingly, both PR modulators had no effects on the P4repressed snail expression (Figure 3b) and cell prolifera-tion (Figure 3c), suggesting that other molecular media-
tors, but not nuclear PR, might be involved in the P4repressed EMT events.
MPRα plays an essential role in the P4-repressed EMT in MB468 cellsWithin the past few years, evidence has been obtained forthe involvement of mPRα in the P4's actions in a varietyof cell types [6,7,11,12]. In the present study, the expres-sion of mPRα in MB468 cells was up-regulated by P4treatments in dose-dependent manners (15 ng/ml - 1.97-fold, 30 ng/ml - 2.75-fold, and 60 ng/ml - 2.92-fold,respectively; Figure 4a). As a control, there were nodetectable mPRα protein found in MB231 cells (Figure4b), which is consistent with a previous report [20]. Thesedata suggest a potential role of mPRα in the P4 signalingof BPBC cells. To further demonstrate our hypothesis, theexpression of mPRα in MB468 cells was knocked down bymPRα-specific siRNA. As shown in Additional file 5,mPRα siRNA specifically inhibited mPRα expressionwithout affecting α-tubulin expression. After transfectionwith 50 nM of mPRα siRNA, the P4's effects on the EMTmarker proteins were significantly inhibited (Figure 4c).
Activation of PI3K/Akt is necessary for P4's action on EMT but not on cell proliferationTo further prove the involvement of mPRα in P4's actionon human BPBC cells, the mPRα-expressing plasmidDNA was introduced into the parent MB231 cells andthen treated by P4 as indicated. There was no reductionfound in the expression of snail as compared with that ofparent MB231 cells (Figure 2a). By comparing the molec-ular profiles of MB468 and MB231 cells, major differ-ences were noticed between the two cell lines inphosphatase and tensin homolog gene (PTEN) expres-sion and PI3K/Akt activation [15]. PTEN is an essentialinhibitor for the PI3K/Akt pathway [25]. In MB468 cells,there is no PTEN expression and PI3K/Akt pathway isconstantly activated. On the contrary, in MB231 cellsPTEN is abundantly expressed and PI3K/Akt pathway isalways inactivated. To explore the role of PTEN andPI3K/Akt pathway in the P4 regulated EMT, the mPRαstably-expressing MB231 cells were incubated with thePTEN-specific inhibitor bpV(phen) to transactivate thePI3K/Akt pathway. As shown in Figure 4d, snail expres-sion was clearly down-regulated by P4 treatment about89.8 ± 1.9%. These data strongly suggest that mPRα playsan important role in the repression of EMT through theactivated PI3K/Akt pathway in BPBCs.
To test whether the female sex hormone controls cellproliferation of MB468 cells, we incubated the cells withP4 (30 ng/ml) for 24 hours. As shown in Figure 2a, a 34%reduction in cell proliferation was observed in the MB468cells with treatment of P4, as compared with the cellswith treatment of vehicle alone. As expected, P4 had no
Figure 2 Effects of P4 on cell proliferation of (a) MB468, parent MB231 and mPRα stably-expressing (b) MB231 cells. The data are representative for three individual experiments and the graphs are ex-pressed as fold change over the basal. The cells treated by E2 (30 pg/ml for 24 hours) and vehicle alone were used as controls. * P < 0.05 for difference of cell proliferation induced by progesterone (P4) vs. vehicle alone.
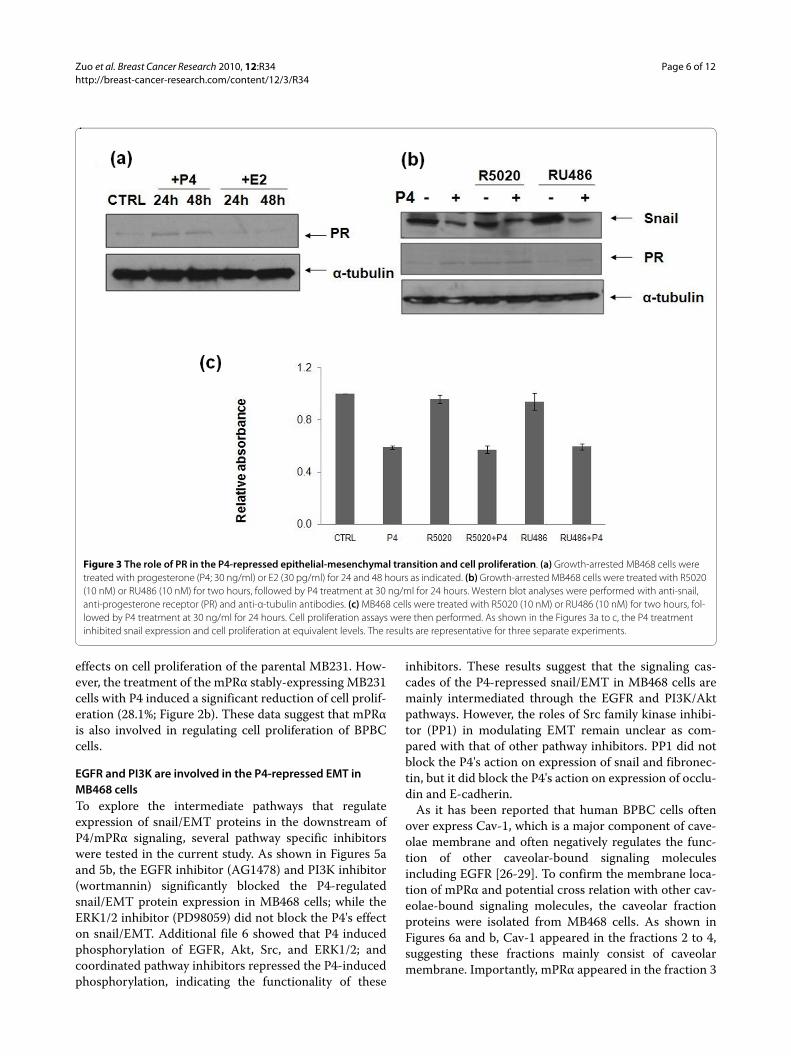
Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 6 of 12
effects on cell proliferation of the parental MB231. How-ever, the treatment of the mPRα stably-expressing MB231cells with P4 induced a significant reduction of cell prolif-eration (28.1%; Figure 2b). These data suggest that mPRαis also involved in regulating cell proliferation of BPBCcells.
EGFR and PI3K are involved in the P4-repressed EMT in MB468 cellsTo explore the intermediate pathways that regulateexpression of snail/EMT proteins in the downstream ofP4/mPRα signaling, several pathway specific inhibitorswere tested in the current study. As shown in Figures 5aand 5b, the EGFR inhibitor (AG1478) and PI3K inhibitor(wortmannin) significantly blocked the P4-regulatedsnail/EMT protein expression in MB468 cells; while theERK1/2 inhibitor (PD98059) did not block the P4's effecton snail/EMT. Additional file 6 showed that P4 inducedphosphorylation of EGFR, Akt, Src, and ERK1/2; andcoordinated pathway inhibitors repressed the P4-inducedphosphorylation, indicating the functionality of these
inhibitors. These results suggest that the signaling cas-cades of the P4-repressed snail/EMT in MB468 cells aremainly intermediated through the EGFR and PI3K/Aktpathways. However, the roles of Src family kinase inhibi-tor (PP1) in modulating EMT remain unclear as com-pared with that of other pathway inhibitors. PP1 did notblock the P4's action on expression of snail and fibronec-tin, but it did block the P4's action on expression of occlu-din and E-cadherin.
As it has been reported that human BPBC cells oftenover express Cav-1, which is a major component of cave-olae membrane and often negatively regulates the func-tion of other caveolar-bound signaling moleculesincluding EGFR [26-29]. To confirm the membrane loca-tion of mPRα and potential cross relation with other cav-eolae-bound signaling molecules, the caveolar fractionproteins were isolated from MB468 cells. As shown inFigures 6a and b, Cav-1 appeared in the fractions 2 to 4,suggesting these fractions mainly consist of caveolarmembrane. Importantly, mPRα appeared in the fraction 3
Figure 3 The role of PR in the P4-repressed epithelial-mesenchymal transition and cell proliferation. (a) Growth-arrested MB468 cells were treated with progesterone (P4; 30 ng/ml) or E2 (30 pg/ml) for 24 and 48 hours as indicated. (b) Growth-arrested MB468 cells were treated with R5020 (10 nM) or RU486 (10 nM) for two hours, followed by P4 treatment at 30 ng/ml for 24 hours. Western blot analyses were performed with anti-snail, anti-progesterone receptor (PR) and anti-α-tubulin antibodies. (c) MB468 cells were treated with R5020 (10 nM) or RU486 (10 nM) for two hours, fol-lowed by P4 treatment at 30 ng/ml for 24 hours. Cell proliferation assays were then performed. As shown in the Figures 3a to c, the P4 treatment inhibited snail expression and cell proliferation at equivalent levels. The results are representative for three separate experiments.

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 7 of 12
where EGFR was also located, indicating the potentialcrosstalk between mPRα and EGFR.
MPRα expression in human benign and malignant breastsTo evaluate expression of mPRα in human breasts, tissuemicroarray slides were studied by immunohistochemis-try. As shown in Table 1, 94 of 105 breast cancer tissueswere stained positive for anti-mPRα. The positive signalsare mainly observed in cytoplasm (Figure 7a) and/or cellmembrane of cancer cells (Figure 7b). There were 14 tri-ple-negative breast cancers (TNBC) among these breastcancer tissues. Most of these TNBC (13 of 14) were mod-erate to strong positive for mPRα stain. In addition,mPRα was also detected in all normal and/or benignbreast tissues. The ductal and alveolar epithelial cells ofbreast were shown to be negative or weak positive whilethe myoepithelial cells were shown to be strong positivefor mPRα (Figure 7c).
DiscussionClassically, the actions of P4 on breast cancer cells areattributed to the binding of nuclear PR and subsequent
activation of the downstream target genes. Lange and col-leagues proposed that P4 acts as a priming agent in breastcancer and, in his scenario, breast cells can be directedtoward one path or another by crosstalking between theP4/PR complex and other signaling pathways [30]. In thePR-negative MB231 cells, P4 showed no effect on cellproliferation and invasion. However, after introducingexogenous PR cDNA into MB231 cells, the PR-expressingMB231 cells exhibited less proliferative activities after P4treatment than the parental MB231 cells [31]. With thisPR-expressing cell model, Sumida and colleagues demon-strated that P4 induced remarkable EMT-like changes incell morphology and surface adhesion structures [31]. Inthe current study, we showed that P4 treatment in vitroinhibited EMT relevant proteins in the late passageMB468 cells. A negative association between P4 and snailexpression was observed (Figure 1b). Consistent withdown-regulation of snail expression, other EMT-relevantproteins, such as E-cadherin, occludin, and fibronectin,were subsequently modulated by P4 (Figure 1c); andthese molecular changes were accompanied with cell
Figure 4 The role of mPRα in the P4-repressed EMT of MB468 and MB231 cells. (a) The growth-arrested MB468 cells were treated with diverse concentrations of progesterone (P4) for 24 hours. (PC: HEK293 cell lysate as a positive control for membrane progesterone receptor (mPR) α expres-sion.) (b) MB231 cells and MB468 cells were treated with/without P4 at 30 ng/ml for 24 hours. Western blot analyses were performed with anti-mPRα, anti-snail, and anti-α-tubulin antibodies. (c) MB468 cells were transfected with 50 nM of mPRα siRNA and 50 nM of scramble siRNA; and then treated with P4 at 30 ng/ml for 24 hours. The Western blot analyses showed similar patterns of snail/epithelial-mesenchymal transition (EMT) protein expres-sions in the cells transfected with scramble siRNA and equivalent levels of snail/EMT proteins in the cells transfected by mPRα siRNA, indicating the roles of specifically knocking down mPRα in MB231 cells. (d) The parent MB231 and mPRα stably-expressing MB231 cells were treated with/without bpV(phen) at 1 μM for one hour, followed by P4 treatment at 30 ng/ml. Western blot analyses were performed with anti-snail, anti-mPRα, and anti-α-tubulin antibodies. The data are representative for three individual experiments.

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 8 of 12
morphological reversion from mesenchymal- to epithe-lial-like phenotypes (Figure 1d). Our results indicate thatP4 functions as an anti-EMT hormone in MB468 cells invitro. It is still unclear how P4 regulates these EMT eventsand what the cell mediators of P4 are.
The membrane progestin receptor, mPRα, has recentlybeen identified as an intermediary factor of the proges-tin-induced intracellular signaling cascades in the PR-negative breast cancer cell lines in vitro [6,12]. Theexpression of mPRα in human breast cancer tissues, how-ever, has not been well evaluated. With PCR assay, Dress-ing and Thomas reported expression of mPRα mRNA inboth normal and malignant breast tissues [7]. Using an invitro hormone binding technique and a FITC-conjugatedBSA-progesterone, Pelekanou and colleagues detectedthe 'membrane-associate receptor for progesterone' in 57of 61 breast cancers (94%) [32]. In this report, the proteinexpression of mPRα was detected in both human benignand malignant breasts (Table 1), which is quite consistentwith Pelekanou's result. The receptor was also demon-
strated in all but one triple-negative breast cancer - a typeof cancer that shares many common features with BPBC[33,34]. Moreover, in the benign breasts, strong positivestain for mPRα was detected in the basal myoepithelialcells. Recently we showed that the mammary ducts ofnormal mice were positive for both PR and mPRα. ThePR was predominantly seen in the ductal epithelium,while mPRα was mostly observed in the basal myoepithe-lial cells [22]. The synergistic roles of mPRα and PR innormal mammary glands remain to be explored.
The mPRα receptor has been associated with manyphysiologic functions in vertebrates. It induces oocytematuration, stimulates sperm hypermotility, down-regu-lates GnRH secretion, modulates T cell functions, andadjusts human myometrial cell contractility [6,7,11-13,35]. In agreement with the earlier studies performed inhuman myometrial cells and fish oocytes [12,13], wefound that P4 up-regulated the expression of mPRα inMB468 cells (Figure 4a). Importantly, P4's actions onexpression of snail/EMT-relevant proteins were signifi-
Figure 5 The molecular pathways involved in the P4-repressed EMT of MB468 cells. Growth-arrested MB468 cells were treated with (a) AG1478 (1 μM) or PP1 (10 μM) and wortmannin (0.1 μM) or (b) PD98059 (50 μM) for one hour, followed by progesterone (P4) treatment at 30 ng/ml for 24 hours. Western blot analyses were performed with diverse anti-snail and/or epithelial-mesenchymal transition (EMT) relevant antibodies as indicated. The data are representative for three individual experiments.

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 9 of 12
cantly blocked by the mPRα specific siRNA (Figure 4c).In contrast, P4 treatment alone had no effect on snailexpression in the parent MB231 cells, in which mPRαprotein is undetectable by western blot assay (Figure 4b).We thought that the exogenous mPRα cDNA stabletransfection would cause the cell EMT responding to theP4 treatment. Unexpectedly, the expression of snail/EMTrelevant markers remained unchanged after P4 treat-ments, indicating other factors in the P4/mPRα signalingpathway were still blocked.
The mesenchymal phenotype of MB231 cells undernormoxic culture conditions has been associated withhigh levels of urokinase-type plasminogen activator(uPA) and uPA receptor (uPAR) expression and silencinguPA expression decreased expression of vimentin andsnail and induced epithelial-like transition in the cells
[16]. In the current study, we showed that the P4repressed EMT in MB231 cells is correlated to themutant pten and activation of PI3K/Akt signaling path-way. PTEN is a major inhibitor of the PI3K/Akt signalingpathway. Loss of PTEN protein expression occurs com-monly in breast cancer, which has been associated withloss of ER [36] and resistance to cancer therapies [37].The PTEN-deficient cell lines displayed greater sensitiv-ity to the growth inhibitory effects of the PI3K inhibitor,LY294002, as compared with the PTEN-positive cell lines[38]. Recently major differences have been reported inthe status of PI3K/Akt pathway and function of PTENbetween MB468 and MB231 cells [15,39]. It was assumedthat the activation of PI3K/Akt pathway, resulting from adysfunctional PTEN, is essential for the P4-repressedEMT. In further study, we demonstrated that the expres-sion of snail/EMT-relevant proteins in the mPRα express-ing MB231 cells was significantly modulated afterincubating the cells with P4 plus PTEN inhibitor -bpV(phen) (Figure 4d). However, activation of PI3K/Aktseems not to be essential for the P4-repressed cell prolif-eration because the growth reduction of the mPRα-expressing MB231 cells could be induced by P4 treatmentalone. It is assumed that the P4 inhibited cell proliferationmay go through other pathways, such as the secondarymessenger pathway through activation of pertussis toxin-sensitive inhibitory G proteins and MAPK/Erk1/2[12,20].
When exploring the intermediate pathways that regu-late snail/EMT in P4 signaling, we showed that P4's
Figure 6 Co-localization of mPRα and EGFR in caveolar mem-brane of MB468 cells. Caveolar (Cav) fractions were isolated and im-munoblotted for detecting Cav-1, membrane progesterone receptor (mPR) α, and epidermal growth factor receptor (EGFR) after the MB468 cells were treated (a) without and (b) with progesterone (P4) at 30 ng/ml for 24 hours. The Figures 6a and b show that Cav-1, mPRα, and EGFR are co-localized in the caveolar fraction #3 component regardless of P4 treatments. The results are representative for two separate experi-ments. PC, mPRα positive cells (HEK293).
Table 1: Positivity of mPRα expression in human breast cancers
mPRα reactivity TNBC Non TNBC Total
Positive (%) 13 (92.9) 81 (89) 94(89.5)
Negative 1 10 11
Total 14 91 105
No significant difference was found between the positive rates of mPRα (TNBC vs. non TNBC groups, P = 0.312, Fisher's exact test). MPRα, membrane progesterone receptor α; TNBC, triple-negative cancers.
Figure 7 Expression of mPRα in human breast cancer tissues. (a and b) The low to intermediate intensities of membrane progesterone receptor (mPR) α stains in the cytoplasm of most human breast cancer cells. Apparent cytoplasm-membrane stains were observed in some of the cancer cells (black arrows). (c) The immunostain of mPRα in normal human breast. EP,ductal epithelium; MEP, myoepithelium.

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 10 of 12
actions on EMT were significantly blocked in the latepassage MB468 cells by AG1478 (an EGFR inhibitor) andwortmannin (PI3K inhibitor), suggesting EGFR andPI3K/Akt pathways are involved in the P4 repressed EMTevents. Studies have shown that along with other signal-ing molecules such as PDGFR, Ha-ras, and c-Src, bothEGFR and PI3K are distributed in the caveolar vesicles inwhich Cav-1 serves as a main structure component [40].Cav-1 usually functions as a negative regulator of othercaveolar-bound signaling molecules [26-29]. Existingdata has shown that BPBC is associated with high expres-sion of Cav-1 [41,42] and EMT of cancer cells is depen-dent upon the presence of Cav-1 [40]. Okamoto andcolleagues showed that long-term EGF treatmentreduced expression of Cav-1 in cancer cells; and subse-quently up-regulated snail and down-regulated E-cad-herin expression [40]. Lu and colleagues demonstratedthat EGF treatment of human tumor cells that overexpress EGFR caused a dramatic alteration in cell-cellcontacts and internalization of E-cadherin [43]. It wasassumed that upon binding to EGF, EGFR forms homodi-mers or heterodimers which result in the activation oftheir intrinsic kinases and autophosphorylation of spe-cific tyrosine residues within their cytoplasmic domains[44]. The activated EGFR may recruit other molecularsignaling complexes such as PI3K, via several potentialpaths. For example, EGFR may bind to and recruit PI3Kdirectly because the canonical binding sites for the regu-latory subunit of PI3K are not found on EGFR [45]; it mayalso employ the docking protein Gab1 to recruit PI3K[46]. In addition, the EGFR adapter (Shc) may recruitPI3K by assembly of a Shc-Grb2-Gab2-PI3K complex[47]. The role of PI3K/Akt pathway in cancer EMT hasbeen well documented in various human malignancies[48-50]. The proposed mPRα dependent molecular path-ways that inhibit EMT of BPBC are schematically illus-trated in Figure 8.
The essential roles of c-Src pathway in the P4/PR sig-naling pathways have been demonstrated in humanbreast cancer cells that is T47 D cells. The cell anchorage-independent growth was stimulated by progestin andblocked by inhibition of Erk1/2, c-Src, EGFR, or RNAinterference of Wnt-1 [51]. Recently Lester and col-leagues reported that when MB468 breast cancer cellswere cultured in a hypoxia condition expression of uPARwas increased, cell-cell junctions were disrupted, vimen-tin expression was increased, and E-cadherin was lostfrom cell surfaces, indicating enhancement of EMT[16,52]. Lester and colleagues proposed a model in whichSrc family kinases may concert with other cell signalingfactors, including PI3K and ERK1/2 and play an essentialrole in the regulation of uPA and uPAR and EMT [16]. Inthis report, we found that in the late passage MB468 cells,
the Src family kinases inhibitor (PP1) did not block theP4's action on snail and fibronectin (one of the mesenchy-mal phenotypes), but it blocked the P4's action on expres-sion of occludin and E-cadherin (epithelial phenotypes).The roles of Src family kinases on the P4-repressed EMTremain to be explored.
ConclusionsIn summary, using two human BPBC cell lines as models,we identified a PR-independent pathway that involves thesignaling cascade of EMT through a caveolae-bound sig-naling complex namely mPRα, Cav-1, EGFR, and PI3K/Akt. It is assumed that mPRα receptor is the key modula-tor of EMT located on the caveolar membrane of BPBCcells. Through the receptor-mediated mechanisms, P4directly inactivates the PI3K-snail-EMT pathway or inter-acts with Cav-1 and modulates the activities of the EGFRpathway, which then cross inhibit PI3K pathway, andeventually suppresses the cell EMT. The proposed path-way is attractive for further understanding the molecularmechanisms of EMT and for developing novel therapeu-tic strategies against BPBC.
Figure 8 Schematic illustration of molecular pathway initiated by P4 signaling in BPBC cells. MB468 cells usually maintain the activities of PI3K/Akt at high levels because these cells contain a mutant pten gene and over expressed epidermal growth factor receptor (EGFR). Ac-tivation of PI3K/Akt pathway is a vital signal for cell survival and epithe-lial to mesenchymal transition (EMT). It is assumed that the binding of progesterone (P4) to membrane progesterone receptor (mPR) α in the caveolar membrane of the cells inhibits EMT-relevant events either di-rectly through Caveolar (Cav) 1 activation or through a cross interac-tion with EGFR and trans-activation of Cav-1 which subsequently inactivate PI3K/Akt pathway. Inactivation of PI3K/Akt pathway subse-quently inhibits the nuclear translocation of snail and then modulates expression of other EMT-relevant proteins.

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 11 of 12
Additional material
AbbreviationsBPBC: basal phenotype breast cancers; Cav-1: caveolar-1; DMEM: Dulbecco'smodified Eagle's medium; E2: estrogen; EGF: epidermal growth factor; EGFR:epidermal growth factor receptor; EMT: epithelial-mesenchymal transition; ER:estrogen receptor; FBS: fetal bovine serum; HEK: human embryonic kidney;HER: human epidermal growth factor receptor; MB231: MDA-MB231 cells;MB468: MDA-MB468 cells; MES: 2-(N-morpholino) ethanesulfonic acid; mPRα:membrane progesterone receptor α; P4: progesterone; PBS: phosphate-buff-ered saline; PCR: polymerase chain reaction; PR: progesterone receptor; PTEN:phosphatase and tensin homolog gene; siRNA: small interferring RNA; TNBC:triple-negative breast cancers; uPA: urokinase-type plasminogen activator;uPAR: uPA receptor.
Competing interestsThe authors declare that they have no competing interests.
Authors' contributionsLZ functioned as a main researcher in this study and her work covered most ofthe assays. WL functioned as a researcher in this study whose contribution wasmainly focused on some of the protein expression. SY proposed the hypothe-sis and designed the study, functioned as the supervisor and PI, and composedthe manuscript.
AcknowledgementsWe thank Dr. Peter Thomas (Marine Science Institute, University of Texas at Aus-tin) for providing mPRα plasmid and kind consultant; Dr. Hima Bindu Chunduri and Dr. Prem Sharma (Atlanta Research & Educational Foundation, Atlanta VA Medical Center, GA) for their assistance in plasmid DNA isolation. This study was supported by the DoD BCRP Synergy Idea Award (S. You) and Atlanta Research and Education Foundation Bridge Fund (S. You).
Author DetailsAtlanta Research & Educational Foundation (151F), Atlanta VA Medical Center, 1670 Clairmont Road, Decatur, GA 30033, USA
References1. Boyer B, Tucker GC, Valles AM, Gavrilovic J, Thiery JP: Reversible transition
towards a fibroblastic phenotype in a rat carcinoma cell line. Int J Cancer Suppl 1989, 4:69-75.
2. Thiery JP: Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002, 2:442-454.
3. Lee JM, Dedhar S, Kalluri R, Thompson EW: The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006, 172:973-981.
4. Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA: The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000, 2:76-83.
5. Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A: The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000, 2:84-89.
6. Thomas P: Characteristics of membrane progestin receptor alpha (mPRalpha) and progesterone membrane receptor component 1 (PGMRC1) and their roles in mediating rapid progestin actions. Front Neuroendocrinol 2008, 29:292-312.
7. Dressing GE, Thomas P: Identification of membrane progestin receptors in human breast cancer cell lines and biopsies and their potential involvement in breast cancer. Steroids 2007, 72:111-116.
8. Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M: Multiple actions of steroid hormones--a focus on rapid, nongenomic effects. Pharmacol Rev 2000, 52:513-556.
9. Losel R, Wehling M: Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol 2003, 4:46-56.
10. Losel R, Breiter S, Seyfert M, Wehling M, Falkenstein E: Classic and non-classic progesterone receptors are both expressed in human spermatozoa. Horm Metab Res 2005, 37:10-14.
11. Sleiter N, Pang Y, Park C, Horton TH, Dong J, Thomas P, Levine JE: Progesterone receptor A (PRA) and PRB-independent effects of progesterone on gonadotropin-releasing hormone release. Endocrinology 2009, 150:3833-3844.
12. Zhu Y, Rice CD, Pang Y, Pace M, Thomas P: Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci USA 2003, 100:2231-2236.
13. Karteris E, Zervou S, Pang Y, Dong J, Hillhouse EW, Randeva HS, Thomas P: Progesterone signaling in human myometrium through two novel membrane G protein-coupled receptors: potential role in functional progesterone withdrawal at term. Mol Endocrinol 2006, 20:1519-1534.
14. Ashley RL, Clay CM, Farmerie TA, Niswender GD, Nett TM: Cloning and characterization of an ovine intracellular seven transmembrane receptor for progesterone that mediates calcium mobilization. Endocrinology 2006, 147:4151-4159.
15. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW: A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10:515-527.
16. Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL: Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J Biol Chem 2009, 284:22825-22833.
17. Fernandez P, Burghardt R, Smith R, Nodland K, Safe S: High passage T47 D human breast cancer cells: altered endocrine and 2,3,7,8-tetrachlorodibenzo-p-dioxin responsiveness. Eur J Pharmacol 1994, 270:53-65.
18. Koli KM, Ramsey TT, Ko Y, Dugger TC, Brattain MG, Arteaga CL: Blockade of transforming growth factor-beta signaling does not abrogate antiestrogen-induced growth inhibition of human breast carcinoma cells. J Biol Chem 1997, 272:8296-8302.
Additional file 1 Morphology of early and late passage MB468 cells. The cultured MB468 cells at early passages (6 passages) appeared as oval and/or polygonal shapes; and after multiple passages (50+ passages), these cells exhibit apparent mesenchymal phenotypes with spindle and elon-gated shapes as indicated. Photos (DIC images) were taken by confocal microscopy at 200× magnification.Additional file 2 Diverse intensities of mPRα immunostains in human breast cancers. (a) Strong positive stain - most of the cancer cells are stained dark brown. (b) Modulate positive - most of the cancer cells are stained modulate brown. (c) Weak positive - light brown. (d) Negative stain - very light brown or no stain. mPRα, membrane progesterone receptor α.
Additional file 3 Cell morphology of late passage MB468 cells at low magnification with/without P4 treatment. Photos (DIC images) were taken by confocal microscopy at 200× magnification. This is an enlarged view of the Figure 1d. P4, progesterone.Additional file 4 Dose curve of the P4-repressed cell proliferation of MB 468 cells. The growth-arrested MB468 cells were treated with different doses of progesterone (P4) as indicated. The cell proliferation was inhibited in a dose-dependent manner (15 ng/ml - 20%, 30 ng/ml - 25%, 60 ng/ml - 48%). The data were averaged from three experiments and the graph repre-sents an averaged data expressed as fold change over basal. * P < 0.05 for difference of cell proliferation induced by P4 vs. vehicle alone.
Additional file 5 Knocking down expression of mPRα by siRNA in MB468 cells. MB468 cells were transfected with indicated amount of mem-brane progesterone receptor α (mPRα) siRNA. Western blot analysis was performed with anti-mPRα and anti-α-tubulin antibodies. As shown in the figure, more than 90% of mPRα expression was inhibited by transfection of mPRα siRNA at 50 nM. The data are representative for three experiments.
Additional file 6 Treatment of MB468 cells with P4 alone. A figure showing that the treatment of MB468 cells with progesterone (P4) alone significantly promotes phosphorylation of (a) epidermal growth factor receptor (EGFR), (b) Akt, (c) Src and (d) ERK1/2; and co-treatment of the cells with P4 and the specific pathway inhibitors abolishes the P4-induced phosphorylation on diverse pathway components (i.e. EGFR, Akt, Src and ERK1/2), indicating the effectiveness of P4 treatments in the activation of diverse molecular pathways.
Received: 18 November 2009 Revised: 15 April 2010 Accepted: 11 June 2010 Published: 11 June 2010This article is available from: http://breast-cancer-research.com/content/12/3/R34© 2010 Zuo et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.Breast Cancer Research 2010, 12:R34

Zuo et al. Breast Cancer Research 2010, 12:R34http://breast-cancer-research.com/content/12/3/R34
Page 12 of 12
19. Zuo L, Li L, Wang Q, Fleming TP, You S: Mammaglobin as a potential molecular target for breast cancer drug delivery. Cancer Cell Int 2009, 9:8.
20. Thomas P, Pang Y, Dong J, Groenen P, Kelder J, de Vlieg J, Zhu Y, Tubbs C: Steroid and G protein binding characteristics of the seatrout and human progestin membrane receptor alpha subtypes and their evolutionary origins. Endocrinology 2007, 148:705-718.
21. Brainard AM, Miller AJ, Martens JR, England SK: Maxi-K channels localize to caveolae in human myometrium: a role for an actin-channel-caveolin complex in the regulation of myometrial smooth muscle K+ current. Am J Physiol Cell Physiol 2005, 289:C49-57.
22. You S, Zuo L, Varma V: Broad tissue expression of membrane progesterone receptor alpha in normal mice. J Mol Histol 2010 in press.
23. Purmonen S, Manninen T, Pennanen P, Ylikomi T: Progestins regulate genes that can elicit both proliferative and antiproliferative effects in breast cancer cells. Oncol Rep 2008, 19:1627-1634.
24. Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA: Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA 2008, 105:5774-5779.
25. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275:1943-1947.
26. Zhang B, Peng F, Wu D, Ingram AJ, Gao B, Krepinsky JC: Caveolin-1 phosphorylation is required for stretch-induced EGFR and Akt activation in mesangial cells. Cell Signal 2007, 19:1690-1700.
27. Galbiati F, Volonte D, Brown AM, Weinstein DE, Ben-Ze'ev A, Pestell RG, Lisanti MP: Caveolin-1 expression inhibits Wnt/beta-catenin/Lef-1 signaling by recruiting beta-catenin to caveolae membrane domains. J Biol Chem 2000, 275:23368-23377.
28. Razani B, Zhang XL, Bitzer M, von Gersdorff G, Bottinger EP, Lisanti MP: Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem 2001, 276:6727-6738.
29. Felley-Bosco E, Bender FC, Courjault-Gautier F, Bron C, Quest AF: Caveolin-1 down-regulates inducible nitric oxide synthase via the proteasome pathway in human colon carcinoma cells. Proc Natl Acad Sci U S A 2000, 97:14334-14339.
30. Lange CA, Richer JK, Horwitz KB: Hypothesis: Progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol 1999, 13:829-836.
31. Sumida T, Itahana Y, Hamakawa H, Desprez PY: Reduction of human metastatic breast cancer cell aggressiveness on introduction of either form a or B of the progesterone receptor and then treatment with progestins. Cancer Res 2004, 64:7886-7892.
32. Pelekanou V, Kampa M, Kafousi M, Dambaki K, Darivianaki K, Vrekoussis T, Sanidas E, Tsiftsis DD, Stathopoulos EN, Castanas E: Erythropoietin and its receptor in breast cancer: correlation with steroid receptors and outcome. Cancer Epidemiol Biomarkers Prev 2007, 16:2016-2023.
33. Kuroda N, Ohara M, Inoue K, Mizuno K, Fujishima N, Hamaguchi N, Lee GH: The majority of triple-negative breast cancer may correspond to basal-like carcinoma, but triple-negative breast cancer is not identical to basal-like carcinoma. Med Mol Morphol 2009, 42:128-131.
34. Rakha EA, Ellis IO: Triple-negative/basal-like breast cancer: review. Pathology 2009, 41:40-47.
35. Tubbs C, Thomas P: Progestin signaling through an olfactory G protein and membrane progestin receptor-alpha in Atlantic croaker sperm: potential role in induction of sperm hypermotility. Endocrinology 2009, 150:473-484.
36. Depowski PL, Rosenthal SI, Ross JS: Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol 2001, 14:672-676.
37. Pandolfi PP: Breast cancer--loss of PTEN predicts resistance to treatment. N Engl J Med 2004, 351:2337-2338.
38. DeGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB, Hidalgo M: Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann Oncol 2004, 15:1510-1516.
39. Pfeiler G, Horn F, Lattrich C, Klappenberger S, Ortmann O, Treeck O: Apoptotic effects of signal transduction inhibitors on human tumor cells with different PTEN expression. Oncol Rep 2007, 18:1305-1309.
40. Okamoto T, Schlegel A, Scherer PE, Lisanti MP: Caveolins, a family of scaffolding proteins for organizing "preassembled signaling complexes" at the plasma membrane. J Biol Chem 1998, 273:5419-5422.
41. Elsheikh SE, Green AR, Rakha EA, Samaka RM, Ammar AA, Powe D, Reis-Filho JS, Ellis IO: Caveolin 1 and Caveolin 2 are associated with breast cancer basal-like and triple-negative immunophenotype. Br J Cancer 2008, 99:327-334.
42. Pinilla SM, Honrado E, Hardisson D, Benitez J, Palacios J: Caveolin-1 expression is associated with a basal-like phenotype in sporadic and hereditary breast cancer. Breast Cancer Res Treat 2006, 99:85-90.
43. Lu Z, Ghosh S, Wang Z, Hunter T: Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4:499-515.
44. Schlessinger J: Cell signaling by receptor tyrosine kinases. Cell 2000, 103:211-225.
45. Bjorge JD, Chan TO, Antczak M, Kung HJ, Fujita DJ: Activated type I phosphatidylinositol kinase is associated with the epidermal growth factor (EGF) receptor following EGF stimulation. Proc Natl Acad Sci USA 1990, 87:3816-3820.
46. Rodrigues GA, Falasca M, Zhang Z, Ong SH, Schlessinger J: A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol Cell Biol 2000, 20:1448-1459.
47. Gu H, Maeda H, Moon JJ, Lord JD, Yoakim M, Nelson BH, Neel BG: New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. Mol Cell Biol 2000, 20:7109-7120.
48. Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, Testa JR: Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene 2005, 24:3574-3582.
49. Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q: PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res 2007, 67:2922-2926.
50. Micalizzi DS, Ford HL: Epithelial-mesenchymal transition in development and cancer. Future Oncol 2009, 5:1129-1143.
51. Faivre EJ, Lange CA: Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol 2007, 27:466-480.
52. Lester RD, Jo M, Montel V, Takimoto S, Gonias SL: uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol 2007, 178:425-436.
doi: 10.1186/bcr2588Cite this article as: Zuo et al., Progesterone reverses the mesenchymal phe-notypes of basal phenotype breast cancer cells via a membrane progester-one receptor mediated pathway Breast Cancer Research 2010, 12:R34





























