Embed Size (px)
Citation preview

Response Rates and Survival in Primary Systemic Amyloidosis
By Morie A. Gertz, Robert A. Kyle, and Philip R. Greipp
Patients (153) with biopsy-proven primary systemic amyloi- dosis (AL) were evaluated for their response rate to alkylat- ing agent-based chemotherapy. Twenty-seven of the pa- tients (18%) responded. The serum creatinine concentration had an adverse effect on response rate (P = .05). In patients with nephrotic syndrome, a normal serum creatinine value, and no echocardiographic evidence of cardiac amyloidosis, the response rate was 39% (12 of 31). Five of 34 patients with amyloid cardiomyopathy responded. Two of these five are alive 10 years after diagnosis. None of the 18 patients with amyloid peripheral neuropathy showed regression of their disease. The median time to achieve response was 11.7 months. The median survival of the 27 patients was 89.4
URING THE PAST 17 years there has been over a D score of reports describing responses of patients with primary systemic amyloidosis (AL) treated with cytotoxic chemotherapy.’-25 Unfortunately, the bulk of the papers are single case reports, and there is no literature to guide the clinician to expected response rates. Moreover, it is unclear which patients with AL will derive the greatest benefit from therapy. Although responses have been reported, no large series has reported a sufficient number of responders to determine whether responding patients truly derive mean- ingful survival benefit. There are no guidelines for the clinician as to what duration of therapy is required before concluding that a trial has failed. We undertook this investigation to address these three questions.
MATERIALS AND METHODS
The subjects in this study were 153 patients with AL diagnosed and subsequently evaluated at the Mayo Clinic, Rochester, MN as part of two prospective sequential studies on therapy.’,’ All 153 patients had biopsy-proven amyloidosis. Patients with overt multi- ple myeloma were excluded. The separation of patients with myeloma-associated amyloidosis from those whose immunocyte dyscrasia is not malignant is difficult. There are no absolute established criteria to distinguish the two syndromes because these manifestations represent different parts of the spectrum of the same fundamental process. One-fourth of patients with amyloido- sis in the absence of myeloma will have greater than 10% plasma cells in the bone marrow.26 Patients whose bone marrow contained greater than 20% plasma cells were excluded. Patients whose bone marrow showed sheets or clusters of plasma cells were excluded. Patients with evidence suggestive of myeloma bone diseases such as osteoporotic compression fractures, any lytic lesions, or unex- plained fractures of the ribs or long bones were excluded. Patients whose serum M protein value exceeded the range generally established for monoclonal gammopathy of undetermined signifi- cance were excluded. Patients with renal insufficiency on the basis of myeloma kidney rather than renal amyloidosis were excluded. No patient with hypercalcemia was included.
These criteria are more stringent than those of the Chronic Leukemia Myeloma Task Force.” Only 12 of the 153 patients (8%) had a serum monoclonal protein spike that exceeded 2 g/dL. Only three in the responding group had an M protein value greater than 2 g/dL. The highest M protein value in the responding group was 2.25 g/dL, in the range associated with monoclonal gammopathy of undetermined significance. No patient with senile, secondary, familial, or localized (including dialysis-related) amyloidosis was included in this analysis. The diagnosis of immunocyte-derived
months and 21 of 27 survived 5 years (78%). Eight patients remain alive with a minimum follow-up of 90 months. Seven died of acute leukemia or dysmyelopoietic syndrome, a presumed complication of melphalan therapy. In the group of 126 patients who showed no response to alkylating agent- based therapy, the median survival was 14.7 months and 9 (7%) survived over 5 years. All 126 patients have died. Alkylating agent-based chemotherapy for AL is beneficial in a subset of patients and a trial of chemotherapy is strongly recommended. Those patients who do respond demonstrate survival benefit. o 1991 by The American Society of Hematology.
amyloid was verified by the presence of a free monoclonal light chain in the serum or urine in 132 (86%) of the patients at presentation. In the 21 patients (14%) who did not have a detectable monoclonal protein in the serum or urine, the diagnosis of amyloidosis was made clinically by the absence of any family history of amyloidosis, the absence of any underlying inflammatory process that could produce secondary amyloidosis, and typical organ distribution for primary amyloidosis. Only three in the responding group had no detectable M protein in the serum or urine and were classified clinically as having AL. Routine immuno- histochemical confirmation of the composition of the amyloid deposits and amino acid sequence data on the amyloid fibril protein were not gathered.
Before entry to the study, all patients gave informed, written consent, and the study was approved by the Mayo Clinic Institu- tional Review Board. The date of diagnosis of amyloidosis ranged from November 1969 to August 1982. All patients were previously untreated before their Mayo Clinic evaluation and were willing to return to the Mayo Clinic for ongoing follow-up; this arrangement allowed accurate monitoring of their therapy and the duration of therapy required to produce benefit. All 153 patients were followed up to within 3 months of this analysis or until death. At this time 145 patients have died, and none have been lost to follow-up. The shortest follow-up of a surviving patient is 8 years. Survival data were analyzed by both log-rank and Gehan-Wilcoxon tests.
Patients were treated with melphalan and prednisone. Melphalan therapy was initiated at a dosage of 0.15 mg/kg of body weight per day in two divided doses. Prednisone therapy was administered at a dosage of 0.8 m a g of body weight per day in four divided doses. Each cycle of treatment was 7 days long. Cycles were repeated every 6 weeks, and the leukocyte and platelet counts were monitored every 3 weeks. The dose of melphalan was subsequently adjusted with each successive cycle to induce a moderate degree of leukopenia (leukocytes, 2,500 to 3,500lpL). There was no difference in the amount of melphalan
Treatment schema.
From the Dysproteinemia Clinic, Mayo Clinic and Mayo Founda-
Submitted April 16,1990; accepted September 24, 1990. Supported in part by the Quade Amyloidosis Research Fund. Address reprint requests to Morie A. Gem, MD, Dysproteinemia
Clinic, Mayo Clinic, 200 First St SWj Rochester, MN 55905. The publication costs of this article were defrayed in part by page
charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section 1734 solely to indicate this fact.
tion, Rochester, MN,
0 1991 by The American Society of Hematology. 0006-4971 /91/7702-OO03$3.OOlO
Blood, Vol77, No 2 (January 15). 1991: pp 257-262 257
For personal use only.on April 15, 2019. by guest www.bloodjournal.orgFrom

258 GERTZ, KYLE, AND GREIPP
administered during the first 6 months of therapy to responders and nonresponders. The total planned duration of melphalan and prednisone treatment ranged between 24 and 36 months. Therapy was continued by the treating clinicians at their discretion. Al- though an attempt was made to treat patients for 24 months, progressive cytopenias may have required earlier cessation of drug treatment. All patients had their treatment discontinued by 36 months. Patients with significant renal insufficiency generally had treatment initiated at a one-third dose reduction for melphalan. No other adjunctive therapy was administered during the study.
To avoid classification of a response based on reduction in the production of M protein, all responding patients were required to have evidence of regression of their organ manifestations of amyloidosis. A 50% reduction in the serum M protein value without clear-cut evidence of improved function of organs involved with amyloidosis was not considered a response. Patients with nephrotic range proteinuria required a 50% reduc- tion in 24-hour protein loss without an increase in serum creatinine level during follow-up and a complete return to normal creatinine level if it had been increased before treatment to be considered a response. Patients with hepatic involvement had to have complete return to normal such that the liver was no longer palpable below the right costal margin and a complete return to normal of the alkaline phosphatase level. Patients with cardiomyopathy had to have total resolution of congestive heart failure. In addition, a complete disappearance of any monoclonal protein present in the serum or urine had to be shown. It was required that an echocardio- gram be performed on all patients in the study to monitor cardiac status for evidence of a response. No attempt was made to show histologic regression of amyloid deposits in patients who had evidence of resolution of their organ dysfunction. It cannot be assumed that the response criteria designed to recognize clinical regression of manifestations of amyloid indicate histologic regres- sion of amyloid.4
Definition of response.
RESULTS
A discriminant analysis was performed between the group of responders and nonresponders to ensure that they were homogeneous at the outset of therapy. Discriminant analysis was performed using the variables known to affect survival in amyloidosis: age, presence or absence of he- patomegaly, congestive heart failure, creatinine level, uri- nary light chain, sex, serum &-microglobulin, and hemoglo- bin. None of these factors showed any statistically significant differences between the two groups (stepwise logistic regres- sion,P > .1).
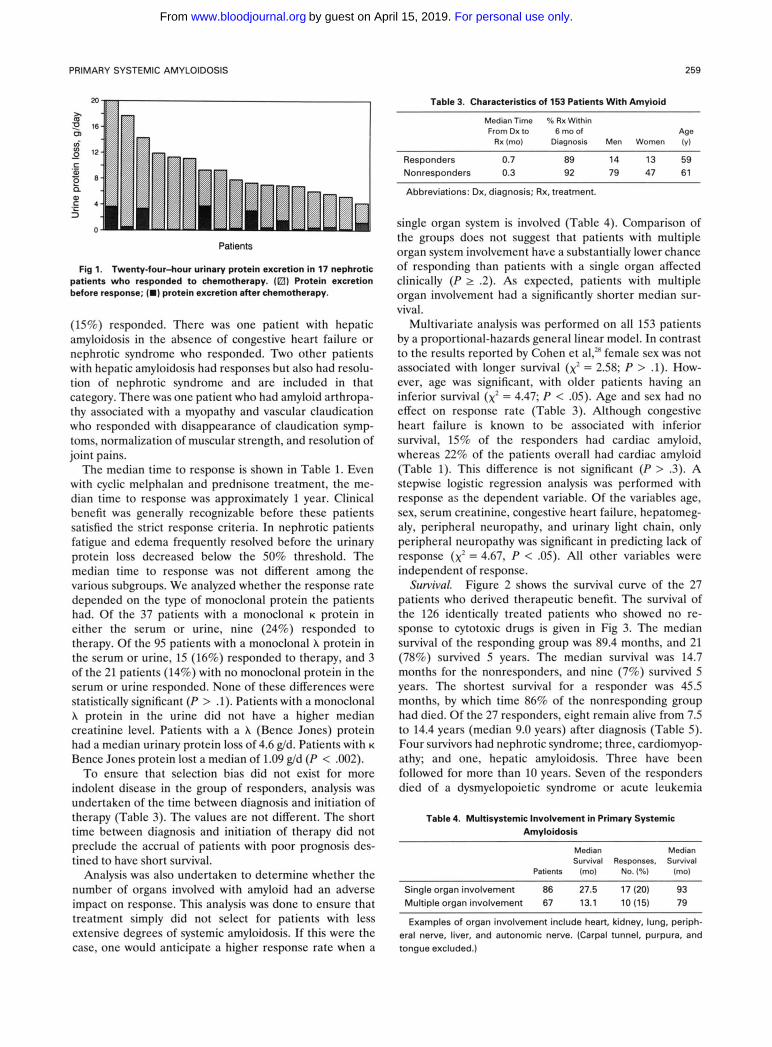
Table 1 lists the responses observed in this cohort of patients. Of the 153 patients treated, 27 (18%) responded to melphalan and prednisone therapy. The best response rates were shown in patients with renal amyloidosis- overall response rate of 25%. Of the 17 responders with renal amyloid, 11 had complete resolution of the nephrotic syndrome and 6 others had a 50% reduction in their urinary protein loss (Table 2, Fig 1). Eleven had reduction of their nephrotic range proteinuria to less than or equal to 0.5 g/d. Only 3 of the 17 patients had greater than 3 g of protein excretion per day after therapy; their reduction in urinary protein loss was 5.5, 10.9, and 16.3 g/d. Thirteen of the 17 had a greater than 5 g/d reduction in 24-hour urine protein excretion. Two of the 17 also had hepatic amyloidosis. In both, hepatomegaly and alkaline phosphatase levels normal- ized.
Table 1. Responses in AL
Responders Median Time (mol
Predominant Syndrome Patients No. Percentage to Response (range)
All 153 27 18 11.7 (3.4-49.1) Nephrotic 68 17 25 11.7 (3.4-43.9)
prnol/L 42 13 31 11.7 (5.5-43.9)
associated 31 12 39 12.2 (5.5-43.9)
pmol/L 26 4 15 9.3 (3.4-13.7)
Creatinine I 115
No cardiac amyloid
Creatinine > 115
Cardiac 34 5 15 10.6 (4.9-18.7) Renal (proteinuria not
nephrotic range) 13 3 23 9.3 (5.8-23.4) Liver 8 1 13 -
Miscellaneous 12 1 8
- Neuropathy 18 0 0 -
Four of the 17 nephrotic patients had an elevated pretreatment serum creatinine level (133,159,177, and 248 pmol/L) and the serum creatinine level returned to normal in all. No patient with a creatinine level greater than 265 pmoVL responded to treatment. Of all patients with a creatinine level greater than 115 kmol/L, the response rate was 9.4% (5 of 53). Renal insufficiency decreases the likelihood of a treatment response (x’ = 3.76; P = .052). When patients were subdivided into those with and without renal insufficiency, the response rate in patients with nephrotic-range proteinuria and a normal creatinine level was 31% (Table 1). Of the 42 patients with nephrotic-range proteinuria and normal creatinine levels, 11 had echocardio- graphic evidence of amyloid cardiomyopathy. When these patients were excluded from analysis, the remaining 31 with amyloid nephrotic syndrome, normal creatinine level, and normal echocardiogram at the time treatment was initiated had an overall response rate of 39%. In the patients with nephrotic syndrome, urinary light chains (Bence Jones proteins) were present in 13 (76%). Following therapy, the light chains disappeared from the urine in 12. The disappear- ance of the light chains occurred at the same time as the total urinary protein loss was decreased. One patient had a recognizable free light chain by immunoelectrophoresis only. No discrete spike was recognizable. In the 108 nonresponders with a detectable M protein, a 50% reduc- tion was documented in 11.
Three additional patients with renal amyloidosis re- sponded to therapy but had insufficient proteinuria at diagnosis to qualify as nephrotic (1.4, 1.9, and 1.9 g/d). All three had a normal pretreatment serum creatinine value. There were 34 patients with amyloid cardiomyopathy who did not have nephrotic syndrome. Of these patients, five
Table 2. Characteristics of Response in Patients With Nephrotic Syndrome (n = 17)
Protein Loss Decrease in Median Protein at Best Urinary Reduction in
Loss Median Response Median Protein Urine Protein Ig/d) (g/d) (gld) bid) (gld)
4.1-19.9 7.7 0.1-3.7 0.4 2.9-17.1 6.8
For personal use only.on April 15, 2019. by guest www.bloodjournal.orgFrom
PRiMARY SYSTEMIC AMYLOIDOSIS 259
Table 3. Characteristics of 153 Patients With Amyloid
m ?? 16 or vi g 12 - c al
Q al
.- -
._ c 4
5 0
Patients
Fig 1. Twenty-four-hour urinary protein excretion in 17 nephrotic patients who responded to chemotherapy. (El) Protein excretion before response; (m) protein excretion after Chemotherapy.
(15%) responded. There was one patient with hepatic amyloidosis in the absence of congestive heart failure or nephrotic syndrome who responded. Two other patients with hepatic amyloidosis had responses but also had resolu- tion of nephrotic syndrome and are included in that category. There was one patient who had amyloid arthropa- thy associated with a myopathy and vascular claudication who responded with disappearance of claudication symp- toms, normalization of muscular strength, and resolution of joint pains.
The median time to response is shown in Table 1. Even with cyclic melphalan and prednisone treatment, the me- dian time to response was approximately 1 year. Clinical benefit was generally recognizable before these patients satisfied the strict response criteria. In nephrotic patients fatigue and edema frequently resolved before the urinary protein loss decreased below the 50% threshold. The median time to response was not different among the various subgroups. We analyzed whether the response rate depended on the type of monoclonal protein the patients had. Of the 37 patients with a monoclonal K protein in either the serum or urine, nine (24%) responded to therapy. Of the 95 patients with a monoclonal A protein in the serum or urine, 15 (16%) responded to therapy, and 3 of the 21 patients (14%) with no monoclonal protein in the serum or urine responded. None of these differences were statistically significant (P > .1). Patients with a monoclonal A protein in the urine did not have a higher median creatinine level. Patients with a A (Bence Jones) protein had a median urinary protein loss of 4.6 g/d. Patients with K
Bence Jones protein lost a median of 1.09 g/d (P < .002). To ensure that selection bias did not exist for more
indolent disease in the group of responders, analysis was undertaken of the time between diagnosis and initiation of therapy (Table 3). The values are not different. The short time between diagnosis and initiation of therapy did not preclude the accrual of patients with poor prognosis des- tined to have short survival.
Analysis was also undertaken to determine whether the number of organs involved with amyloid had an adverse impact on response. This analysis was done to ensure that treatment simply did not select for patients with less extensive degrees of systemic amyloidosis. If this were the case, one would anticipate a higher response rate when a
MedianTime % Rx Within From Dx to 6 mo of Age
Rx(mo) Diagnosis Men Women (y)
Responders 0.7 89 14 13 59 Nonresponders 0.3 92 79 47 61
Abbreviations: Dx, diagnosis; Rx, treatment.
single organ system is involved (Table 4). Comparison of the groups does not suggest that patients with multiple organ system involvement have a substantially lower chance of responding than patients with a single organ affected clinically (P 2 .2). As expected, patients with multiple organ involvement had a significantly shorter median sur- vival.
Multivariate analysis was performed on all 153 patients by a proportional-hazards general linear model. In contrast to the results reported by Cohen et al,’” female sex was not associated with longer survival (xz = 2.58; P > .1). How- ever, age was significant, with older patients having an inferior survival (xz = 4.47; P < .05). Age and sex had no effect on response rate (Table 3). Although congestive heart failure is known to be associated with inferior survival, 15% of the responders had cardiac amyloid, whereas 22% of the patients overall had cardiac amyloid (Table 1). This difference is not significant (P > 3). A stepwise logistic regression analysis was performed with response as the dependent variable. Of the variables age, sex, serum creatinine, congestive heart failure, hepatomcg- aly, peripheral neuropathy, and urinary light chain, only peripheral neuropathy was significant in predicting lack of response (x’ = 4.67, P < .OS). All other variables were independent of response.
Figure 2 shows the survival curve of the 27 patients who derived therapeutic benefit. The survival of the 126 identically treated patients who showed no re- sponse to cytotoxic drugs is given in Fig 3. The median survival of the responding group was 89.4 months, and 21 (78%) survived 5 years. The median survival was 14.7 months for the nonresponders, and nine (7%) survived 5 years. The shortest survival for a responder was 45.5 months, by which time 86% of the nonresponding group had died. Of the 27 responders, eight remain alive from 7.5 to 14.4 years (median 9.0 years) after diagnosis (Table 5). Four survivors had nephrotic syndrome; three, cardiomyop- athy; and one, hepatic amyloidosis. Three have been followed for more than 10 years. Seven of the responders died of a dysmyelopoietic syndrome or acute leukemia
Survival.
Table 4. Multisystemic Involvement in Primary Systemic Amyloidosis
Median Median Survival Responses. Survival
Patients (mo) No. (%I (mol ~~
Single organ involvement 86 27.5 17 (20) 93 Multiple organ involvement 67 13.1 10 (15) 79
Examples of organ involvement include heart, kidney, lung, periph- eral nerve, liver, and autonomic nerve. (Carpal tunnel, purpura, and tongue excluded.)
For personal use only.on April 15, 2019. by guest www.bloodjournal.orgFrom

260 GERTZ, KYLE, AND GREIPP
Table 5. Status of Amyloidosis Patients Responding to Chemotherapy
Cause of Death Patients
o ~ l l l " l ' l " " " l ' l l l l ' 0 1 2 3 4 5 6 7 6 9 10
Years
Fig 2. Survival from time of diagnosis of 27 responding patients with AL.
induced by prolonged melphalan therapy (4.0 to 7.7 years; median, 5.2 years). All of the 126 nonresponders have died; acute leukemia developed in one. The amyloid syndromes in the nonresponding 5-year survivors were: neuropathy in five patients, nephrotic syndrome and slow progression of disease in two, hepatic amyloidosis in one, and amyloid limited to the soft tissues in one. All nine nonresponding survivors had a creatinine level less than 133 p,mol/L at diagnosis, and none had congestive heart failure. Six of the nine had a monoclonal protein at diagnosis and one had disappearance of the M protein after chemotherapy. It has been reportedz9 that amyloid neuropathy patients have a better prognosis, and we have reported five patients with hepatic amyloidosis who survived 5 years despite progres- sive disease.%
DISCUSSION
It is generally considered that no effective therapy exists for AL.31 Kyle et a1 previously reported that melphalan and prednisone therapy did not produce a significantly better survival compared with either placebo or c~lchicine.'.~ The results reported here are not inconsistent with those re- ports. Although we strongly believe that a subset of patients will derive benefit, the percentage of patients who evidence this degree of response is small and insufficient to shift the
0 1 2 3 4 5 6 7 6 9 10
Years
Fig 3. Survival from time of diagnosis of 126 similarly treated patients who failed to respond to melphalan.
Leukemia/dysmyelopoietic syndrome Amyloid renal failure Amyloid heart failure Amyloid inanition Suicide Solid tumor
7 3 5 1 1 2 \
Alive, 8; dead, 19.
median survival. Improved survival for certain subsets may be overlooked.
One may question whether the small response rate and risk of treatment-related leukemia justify a trial of alkylat- ing agent chemotherapy. This question cannot be answered on absolute terms, but the short median survival and the small number of long-term survivors in the nonresponding group (7% at 5 years) makes a trial of therapy worthwhile. The prolonged survival of the responding group was specif- ically related to their response to chemotherapy and not because of ancillary factors such as a younger age, absence of congestive heart failure, hepatomegaly, or anemia. Histo- logic demonstration of regression of amyloid deposits was not attempted to corroborate the clinical evidence of resolution of organ dysfunction. Although amyloid deposits may persist on repeat tissue biopsy, it is ultimately the resolution of all evidence of organ impairment and return to a normal life-style that is of the utmost importance to the patient. Because repeat biopsy is often impractical, the standard of clinical regression should be the criterion for continued treatment with alkylating agents.
Although the median survival for patients with conges- tive heart failure is only 6 m0nths,2~'~' we are currently following up on two patients with endomyocardial biopsy- proven primary cardiac amyloidosis who had congestive heart failure at diagnosis and now are alive and well 10 years after the initiation of therapy.
We believe it reasonable that chemotherapy be contin- ued for a minimum of 1 year. If there is no evidence of progressive disease, treatment may be continued beyond 1 year even if the patient does not have evidence of objective response to therapy. Alternatively, one must keep in mind that the response criteria used in these studies might overestimate the duration of therapy required to achieve a response. For example, although a 50% decrease in urinary protein is required to be defined as a response in this study, reduction of proteinuria may be seen well before the 50% level is achieved. Improvement may actually have been taking place for many months before achievement of the criteria set in this study.
Treatment does carry risks; 7 of the 27 responding patients died as a result of dysmyelopoietic syndrome or acute leukemia related to prolonged exposure to mel~halan.3~ The optimal duration of treatment for patients with amyloidosis is unknown and, in view of the high incidence of acute leukemia, one wonders whether a shorter period of melphalan exposure in responders might
For personal use only.on April 15, 2019. by guest www.bloodjournal.orgFrom

PRIMARY SYSTEMIC AMYLOIDOSIS 261
decrease the risk of alkylator-induced damage to the bone marrow. The fact that treatment is effective in only a minority of patients has previously been emphasi~ed,’~ and there is no evidence to suggest that if single-agent alkylating- agent treatment fails, patients would derive benefit from more aggressive multi-agent ~hemotherapy.’~-’~
The mechanism whereby treatment of AL with melpha- Ian and prednisone produces a response is unknown. Chemotherapy is used to decrease or eradicate the synthe- sis of monoclonal light chains by decreasing the number of plasma cells producing the light chain in the bone marrow. This treatment should retard further deposition of AL in tissues. Whether there is also an effect on fibril formation or rate of mobilization of AL, deposits from tissues remains to be shown?’
Colchicine has been used in the treatment of AL, and survival analysis based on the use of retrospective controls suggests prolongation of s u r ~ i v a l . ~ ~ ~ ~ ~ ~ ~ However, there is no evidence that colchicine is capable of producing disease regressions of the type reported here.
We are unaware of any reports in which spontaneous remissions of AL have occurred. There are also no pub- lished reports of corticosteroid-induced regressions of amy-
loidosis, and animal studies have suggested that steroids can accelerate amyloid dep~sition.~’ It has never been shown that treatment with melphalan and prednisone is superior to melphalan alone. The previously reported cases of response in AL have included prednisone in the regi-
In multiple myeloma, a prospective study showed the superiority of melphalan and prednisone therapy to melphalan alone. Because the rationale for treating AL with chemotherapy is based on the experience with my- eloma, steroids have consistently been used, although their ultimate value remains unknown. Multiple case reports have been published on the use of dimethyl sulfoxide (DMSO) in amyloido~is.~~”~ Clear-cut regressions in organ involvement in AL have not been shown in systemic cases.
In conclusion, alkylating agent-based chemotherapy for AL can produce regressions of disease in a small percent- age of patients. Patients who show regression of disease benefit as measured by long-term survival. The optimal duration of treatment is unknown. It is not clear that this type of regression has been shown in AL with DMSO or colchicine. Whether combinations of cytotoxic therapy with DMSO or colchicine will be of greater benefit remains to be shown with prospective randomized studies.
men.1-4,14-18
REFERENCES
1. Kyle RA, Greipp PR: Primary systemic amyloidosis: Compar- ison of melphalan and prednisone versus placebo. Blood 52:818, 1978
2. Buxbaum JN, Hurley ME, Chuba J, Spiro T: Amyloidosis of the AL type: Clinical, morphologic, and biochemical aspects of the response to therapy with alkylating agents and prednisone. Am J Med 67:867,1979
3. Kyle RA, Greipp PR, Garton JP, Gertz MA: Primary sys- temic amyloidosis: Comparison of melphalan/prednisone versus colchicine. Am J Med 79708,1985
4. Kyle RA, Wagoner RD, Holley KE: Primary systemic amyloid- osis: Resolution of the nephrotic syndrome with melphalan and prednisone. Arch Intern Med 1421445,1982
5. Jones NF, Hilton PJ, Tighe JR, Hobbs JR: Treatment of “primary” renal amyloidosis with melphalan. Lancet 2616, 1972
6. Emmerich B, Kircher A, Fink U, Schmid L, Rastetter J: Primare amyloidose mit nephrotiscbem syndrom: 5 jahriger verlauf unter kombinierter chemotherapie. Klin Wochenschr 58:1207, 1980
7. Jones NF: Renal amyloidosis: Pathogenesis and therapy. Clin Nephrol6459,1976
8. Manoharan A: Successful treatment of primary amyloidosis (letter to the editor). Mayo Clin Proc 61:835,1986
9. Corkery J, Bern MM, Tullis JL: Resolution of amyloidosis and plasma-cell dyscrasia with combination chemotherapy (letter to the editor). Lancet 2425,1978
10. Mehta AD: Regression of amyloidosis in multiple myeloma. Br J Clin Pract 32:358,1978
11. Cohen HJ, Lessin LS, Hallal J, Burkholder P: Resolution of primary amyloidosis during chemotherapy: Studies in a patient with nephrotic syndrome. Ann Intern Med 82:466,1975
12. Horne MK 111: Improvement in amyloidosis (letter to the editor). Ann Intern Med 83:281,1975
13. Bradstock K, Clancy R, Uther J, Basten A, Richards J: The successful treatment of primary amyloidosis with intermittent Chemotherapy. Aust N Z J Med 8:176,1978
14. Schwartz RS, Cohen JR, Schrier S L Therapy of primary amyloidosis with melphalan and prednisone. Arch Intern Med 139:1144,1979
15. Farhangi M, Thakur VM, Durham JB: Objective response in amyloidosis treated with intermittent chemotherapy. South Med J 77:775, 1984
16. Gertz MA, Kyle R A Response of primary hepatic amyloid- osis to melphalan and prednisone: A case report and review of the literature. Mayo Clin Proc 61:218,1986
17. Benson MD: Treatment of AL amyloidosis with melphalan, prednisone, and colchicine. Arthritis Rheum 29:683, 1986
18. Fielder K, Durie BGM: Primary amyloidosis associated with multiple myeloma: Predictors of successful therapy. Am J Med 80413,1986
19. Camoriano JK, Greipp PR, Bayer GK, Bowie EJW: Resolu- tion of acquired factor X deficiency and amyloidosis with melpha- Ian and prednisone therapy. N Engl J Med 316:1133,1987
20. Sheehan-Dare RA, Simmons A V Amyloid myopathy and myeloma: Response to treatment. Postgrad Med J 63:141,1987
21. Plamieniak Z, Zareba-Bogdal E, Bednorz W Dlugotrwala remisja skrobiawicy pienvotnej PO leczeniu azatiopryna. Pol Tyg Lek 35:1387,1980
22. ElezoviC I, DjukanoviC R, RoloviC Z: Successful treatment of hemorrhagic syndrome due to an acquired, combined deficiency of factors VI1 and X in a patient with multiple myeloma and amyloidosis (letter to the editor). Eur J Haematol42105, 1989
23. Fritz DA, Luggen ME, Hess EV: Unusual longevity in primary systemic amyloidosis: A 19-year survivor. Am J Med 86245,1989
24. Schattner A, Varon D, Green L, Hurwitz N, Bentwich Z Primary amyloidosis with unusual bone involvement: Reversibility with melphalan, prednisone, and colchicine. Am J Med 86:347, 1989
25. Brown MP, Walls RS: Amyloidosis of immunoglobulin origin: Useful treatment? Med J Aust 152:95, 1990
For personal use only.on April 15, 2019. by guest www.bloodjournal.orgFrom

262 GERTZ, KYLE, AND GREIPP
26. Kyle RA, Greipp PR: Amyloidosis (AL): Clinical and laboratory features in 229 cases. Mayo Clin Proc 58:665,1983
27. Committee of the Chronic Leukemia-Myeloma Task Force: Proposed guidelines for protocol studies: 11. Plasma cell myeloma. Cancer Chemother Rep 4145,1973
28. Cohen AS, Rubinow A, Anderson JJ, Skinner M, Mason JH, Libbey C, Kayne H: Survival of patients with primary (a) amyloidosis: Colchicine-treated cases from 1976 to 1983 compared with cases seen in previous years (1961 to 1973). Am J Med 821182,1987
29. Duston MA, Skinner M, Anderson J, Cohen AS: Peripheral neuropathy as an early marker of AL amyloidosis. Arch Intern Med 149:358,1989
30. Gertz MA, Kyle RA: Hepatic amyloidosis (primary [AL], immunoglobulin light chain): The natural history in 80 patients. Am J Med 8573,1988
31. Editorial Treatment of primary amyloidosis. Lancet 2:1187, 1978
32. Kyle RA, Greipp PR, OFallon WM: Primary systemic amyloidosis: Multivariate analysis for prognostic factors in 168 cases. Blood 68:220,1986
33. Kyle RA, Pierre RV, Bayrd ED: Primary amyloidosis and acute leukemia associated with melphalan therapy. Blood 44:333, 1974
34. Cohen HJ: Combination chemotherapy for primary amy- loidosis reconsidered (letter to the editor). Ann Intern Med 89572, 1978
35. Case DC Jr, Lee BJ 111: Combination chemotherapy (M-2) for amyloidosis-Failure to effect synthesis of paraprotein and deposition of amyloid (abstract). Blood 50186,1977
36. E v y Y, Belghiti-Deprez D, Sobel A Traitement de l'amylose AL sans mydome. Ann Med Interne (Paris) 139:190,1988
37. Frustaci A, Gentiloni N, Feoli F: Amiloidosi a pattern misto: Sensibiliti a colchicina + melphalan. Minerva Med 72:957,1981
38. Stone MJ: Amyloidosis: A final common pathway for protein deposition in tissues. Blood 75531, 1990
39. Rubinow A, Cohen AS, Kayne H, Libbey CA. Colchicine
therapy in primary amyloidosis. A preliminary report (abstract). Arthritis Rheum 24:124,1981
40. Ravid M, Robson M, Kedar I: Prolonged colchicine treat- ment in four patients with amyloidosis. Ann Intern Med 87568, 1977
41. Akoglu E, Akoglu T, Erken E: Colchicine in systemic amyloidosis (letter to the editor). Ann Rheum Dis 43:857, 1984
42. Hardt F: Acceleration of casein induced amyloidosis in mice by immunosuppressive agents. Acta Pathol Microbiol Scand [A] 7961,1971
43. Wang W-J, Lin C-S, Wong C-K Response of systemic amyloidosis to dimethyl sulfoxide. J Am Acad Dermatol 15402, 1986
44. Hsieh SD, Yamamoto R, Saito K, Iwamoto Y, Kuzuya T, Ohba S, Kobori S, Saito K Amyloidosis presented with whitening and loss of hair which improved after dimethylsulfoxide (DMSO) treatment. Jpn J Med 26:393,1987
45. Tokunaka S, Osanai H, Morikawa M, Yachiku S: Experience with dimethyl sulfoxide treatment for primary localized amyloid- osis of the bladder. J Urol135:580,1986
46. Ravid M, Shapira J, Lang R, Kedar I: Prolonged dimethylsul- phoxide treatment in 13 patients with systemic amyloidosis. Ann Rheum Dis 41587,1982
47. Takahashi A, Matsumoto J, Nishimura S, Tanida N, Imura S, Isobe T, Shimoyama T: Improvement of endoscopic and histo- logic findings of AA-type gastrointestinal amyloidosis by treatment with dimethyl sulfoxide and prednisolone. Gastroenterol Jpn 20:143,1985
48. Scheinberg MA, Pernambuco JC, Benson MD: DMSO and colchicine therapy in amyloid disease. Ann Rheum Dis 43:421, 1984
49. Ossennan EF, Sherman WH, Kyle RA: Further studies of therapy of amyloidosis with dimethylsulfoxide (DMSO). Excerpta Medica International Congress Series No. 497,1979, p 563
50. Crovato F, De Marchi R, Nazzari G, Desire110 G, Fusco F: Dimethyl sulphoxide in primary AL amyloidosis (letter to the editor). Haematologica (Pavia) 69:234,1984
For personal use only.on April 15, 2019. by guest www.bloodjournal.orgFrom
1991 77: 257-262
MA Gertz, RA Kyle and PR Greipp Response rates and survival in primary systemic amyloidosis
http://www.bloodjournal.org/content/77/2/257.full.htmlUpdated information and services can be found at:
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American
For personal use only.on April 15, 2019. by guest www.bloodjournal.orgFrom





























