Embed Size (px)
Citation preview

MOLECULAR AND CELLULAR BIOLOGY, May 2005, p. 4034–4045 Vol. 25, No. 100270-7306/05/$08.00�0 doi:10.1128/MCB.25.10.4034–4045.2005Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Restoration of Functional Gap Junctions through Internal RibosomeEntry Site-Dependent Synthesis of Endogenous Connexins in
Density-Inhibited Cancer CellsHicham Lahlou, Marjorie Fanjul, Lucien Pradayrol, Christiane Susini, and Stephane Pyronnet*
INSERM U531, Institut Louis Bugnard (IFR31), CHU Rangueil, TSA 50032, 31059 Toulouse Cedex 9, France
Received 8 October 2004/Returned for modification 29 October 2004/Accepted 3 January 2005
Gap junctions are composed of connexins and are critical for the maintenance of the differentiated state.Consistently, connexin expression is impaired in most cancer cells, and forced expression of connexinsfollowing cDNA transfection reverses the tumor phenotype. We have found that the restoration of densityinhibition of human pancreatic cancer cells by the antiproliferative somatostatin receptor 2 (sst2) is due tooverexpression of endogenous connexins Cx26 and Cx43 and consequent formation of functional gap junctions.Immunoblotting along with protein metabolic labeling and mRNA monitoring revealed that connexin expres-sion is enhanced at the level of translation but is not sensitive to the inhibition of cap-dependent translationinitiation. Furthermore, we identified a new internal ribosome entry site (IRES) in the Cx26 mRNA. Theactivity of Cx26 IRES and that of the previously described Cx43 IRES are enhanced in density-inhibited cells.These data indicate that the restoration of functional gap junctions is likely a critical event in the antiprolif-erative action of the sst2 receptor. We further suggest that the existence of IRESes in connexin mRNAs permitsconnexin expression in density-inhibited or differentiated cells, where cap-dependent translation is generallyreduced.
Gap junction intercellular communication plays an impor-tant function in tissue integrity. To form a gap junction chan-nel, each cell of an adjacent pair synthesizes a hemichannelcomposed of six connexin subunits called a connexon. A com-plete channel is then formed when a connexon of one celldocks with that of the adjacent cell. Connexins belong to alarge family of at least 20 protein isoforms in humans whoseexpression is tissue specific (48, 60). Normal differentiated andcontact-inhibited cells express at least one connexin isoformthat assembles, alone or in combination, into gap junctions.Consistently, the lack of growth control and the inability todifferentiate or apoptose in most cancer cells are related toaltered connexin expression and consequent dysfunctional gapjunction intercellular communication.
The alteration of connexin expression is of importance, astheir forced expression, following cDNA transfection, can pro-mote cell density inhibition and reverse the tumor phenotype(34). The loss of connexin function in cancer cells or in cellstreated with tumor promoters results from various defects,including alteration of transcription (51, 62), protein trafficking(16, 47), stability (26, 45), or phosphorylation (36, 52). Anotherproposed mechanism that could be altered is the translation ofconnexin mRNAs (40).
All nucleus-encoded eukaryotic mRNAs are modified attheir 5� end with a structure termed the cap (m7GpppN, whereN is any nucleotide). Cap-dependent translation is mediated bythe eukaryotic translation initiation factor (eIF) 4F, a proteincomplex composed of three subunits: eIF4E, which recognizes
the cap; eIF4A, an RNA helicase; and eIF4G, a scaffoldingprotein that simultaneously binds eIF4E and eIF4A (13).Through eIF3, eIF4G also binds the 40S ribosomal subunit(19) and therefore can bridge the ribosome to the mRNA 5�end by its simultaneous interaction with eIF4E. eIF4E activityis modulated by its reversible association with the bindingprotein 4E-BP1 (30, 41). Hypophosphorylated 4E-BP1 com-petes with eIF4G for a common binding site on eIF4E, whilehyperphosphorylated 4E-BP1 does not (18, 31). The binding ofhypophosphorylated 4E-BP1 to eIF4E disrupts the eIF4F com-plex and results in the inhibition of cap-dependent translation(41).
Although cap-dependent translation initiation is thought tobe the prevalent mode of ribosome binding to mRNAs ineukaryotes, a growing list of cellular mRNAs exhibit an inher-ent ability to bypass the requirement for the cap structure.Translation of these mRNAs is independent of eIF4E and thusinsensitive to the hypophosphorylated forms of 4E-BP1. Ribo-somes access these mRNAs by binding directly to an internalribosome entry site (IRES). IRESes were first identified inpicornavirus RNAs, which do not possess a 5� cap structure(21, 42). A restricted but increasing number of cellular IRESeshave since been discovered (57). It is thought that one functionof cellular IRESes is to permit the control of translation evenwhen cap-dependent translation is impaired. That the c-sisIRES is activated during differentiation supports this presump-tion (50).
The mRNAs coding for the connexin 32 (Cx32) and Cx43proteins can be translated by a cap-independent, IRES-medi-ated mechanism (20, 49). Since gap junctions play a vital func-tion in the maintenance of the differentiated state, it has beenhypothesized that the existence of IRESes in connexin mRNAs
* Corresponding author. Mailing address: INSERM U531, InstitutLouis Bugnard (IFR31), CHU Rangueil, TSA 50032, 31059 Toulousecedex 9, France. Phone: 33 (0)5 61 32 24 07. Fax: 33 (0)5 61 32 24 03.E-mail: [email protected].
4034
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from

would ensure connexin expression even in contact-inhibitedcells, where cap-mediated translation is impaired (59).
We have addressed this hypothesis in pancreatic cancer cellswhose tumor phenotype has been reversed by stable transfec-tion of somatostatin receptor 2 (sst2). Among the five soma-tostatin receptor subtypes, sst2 plays a critical role in tumorinhibition. However, since it is no longer expressed in mosthuman pancreatic adenocarcinomas (4), sst2 cannot transmitthe antitumoral signal of somatostatin. Conversely, reintroduc-ing sst2 is sufficient to inhibit cell proliferation and to attenuatemalignancy even in the absence of exogenous ligand. No ex-ogenous ligand is required because stable expression of sst2induces the synthesis and secretion of somatostatin, which per-manently activates the receptor (8, 46).
Given the important role of gap junction intercellular com-munication in the inhibition of cell proliferation (34), we pro-pose that the antiproliferative action of the sst2 receptor couldbe mediated, at least in part, by connexins. Here we show thatin sst2-expressing cells, the restoration of density inhibition isdependent on the formation of functional gap junctions com-posed of Cx26 and Cx43. We further show that, in addition toCx32 and Cx43, Cx26 mRNA also contains an IRES in its 5�untranslated region (UTR). The expression of both Cx26 andCx43 is ensured mainly by a posttranscriptional mechanismthrough cap-independent, IRES-mediated mRNA translation.
MATERIALS AND METHODS
Materials. Dulbecco’s modified Eagle’s medium with 1 g/liter glucose(DMEM) and other culture reagents were supplied by GIBCO except fetal calfserum (FCS, Sigma) and Plasmocin (InvivoGen). ExGen 500 transfection re-agent was from Euromedex. 18�-Glycyrrhetinic acid (�-GA) was purchased fromAldrich. Rhodamine-dextran (10,000 Da), biocytin, and Alexa Fluor 488-conju-gated avidin were obtained from Molecular Probes. The rabbit anti-sst2, whichwas generated against a peptide corresponding to a sequence of the carboxyl-terminal tail of the human sst2, was generously provided by S. Schulz (Depart-ment of Pharmacology and Toxicology, Otto-von-Guericke University, Magde-burg, Germany) (56). The anti-Cx43 rabbit polyclonal antibody was purchasedfrom Santa Cruz Biotechnology. Monoclonal anti-Cx43 (clone CX-1B1) (7, 39)and anti-Cx32 (clone CX-2C2) antibodies were from Zymed. The anti-Cx26rabbit polyclonal antibody used for immunofluorescence, immunoblotting, andimmunoprecipitation experiments and the anti-Cx26 monoclonal antibody (cloneCX-12H10) used for immunofluorescence experiments were purchased fromZymed. Monoclonal anti-cyclin D1 was purchased from Cell Signaling Technol-ogy. Polyclonal antibody against 4E-BP1 was kindly provided by N. Sonenberg(Department of Biochemistry and McGill Cancer Center, McGill University)(43). Monoclonal anti-�-tubulin was from Sigma. Rapamycin was purchasedfrom Calbiochem. L-[35S]methionine (1,000 Ci/mmol; 10 mCi/ml) was purchasedfrom Amersham Biosciences. All other materials were obtained from Sigmaunless otherwise stated.
Cell culture and growth assay. Human pancreatic tumor BxPC-3 cells stablyexpressing human sst2 or the mock vector (8, 46) were routinely grown inDMEM supplemented with 10% FCS, 2 mM L-glutamine, 400 �g/ml geneticin,2.5 �g/ml Fungizone, 5 U/ml streptomycin/penicillin, and 2.5 �g/ml Plasmocin.Cells were maintained in a water-saturated atmosphere of 5% CO2 at 37°C. Forthe growth assay, cells were plated in six-well dishes (105 cells/dish) and culturedfor 6 days. Cell proliferation was measured every 2 days by counting with aCoulter counter model Z1 (Coulter Electronics) as described previously (3). Toinhibit gap junction intercellular communication, �-GA was added to the culturemedium at the time of plating. Cells were fed every day with fresh mediumcontaining either 25 �M �-GA or vehicle (dimethyl sulfoxide). To analyze theeffect of �-GA on BxPC-3 cell proliferation, cells were plated at 105 cells/dish toreach confluence after 6 days of growth.
Detection of sst2. Cells grown on glass coverslips for 48 h were fixed with 3%paraformaldehyde for 20 min at �4°C and permeabilized for 1 min with 0.1%Triton X-100. After blocking for nonspecific antibody binding sites with 1%bovine serum albumin (BSA) in phosphate-buffered saline (PBS), cells were
incubated for 1 h at room temperature with anti-sst2 antibody (1:500) diluted inPBS-BSA. After washing, cells were incubated for 1 h with fluorescein isothio-cyanate-conjugated anti-rabbit immunoglobulin G (1:300) (Nordic Laboratories)diluted in PBS-BSA. Coverslips were mounted with fluorescent mounting me-dium (DAKO Corporation). The preparations were examined with a confocallaser scanning microscope (LSM 410, Carl Zeiss) using an argon laser adjustedto 488 nm.
Detection of connexins. Cells grown on glass coverslips for 48 h were fixed with�20°C absolute methanol for 20 min. Unspecific protein binding was preventedby 1 h incubation with PBS containing 1% FCS. Cells were then incubated for 1 hat room temperature with primary antibodies (1:100) diluted in PBS containing0.1% FCS (PBS-FCS). After washing, cells were incubated for 1 h at roomtemperature with Cy3-conjugated anti-mouse immunoglobulin G (1:250) or withAlexa Fluor 488-conjugated anti-rabbit immunoglobulin G (1:250) (MolecularProbes) diluted in PBS-FCS. Cells were then rinsed and stained with 0.4 �g/ml4�,6�-diamidino-2-phenylindole. The coverslips were mounted onto glass slideswith fluorescent mounting medium (DAKO Corporation). Coverslips wererinsed three times for 5 min in PBS-FCS between the steps. Immunofluorescenceimages were captured using a Nikon Eclipse E400 microscope and BiocomVisioLab 2000 software. Fluorescent controls were incubated without primary orsecondary antibodies. No significant cross-labeling or unspecific labeling wasobserved. For double immunofluorescence, cells were incubated simultaneouslywith mouse monoclonal anti-Cx26 and rabbit polyclonal anti-Cx43 antibodies (orvice versa), followed by incubation with Alexa Fluor 488- and Cy3-conjugatedsecondary antibodies.
Immunoblotting. Cells were plated in 100-mm-diameter culture dishes (106
cells/dish) and grown for 4 days unless otherwise noted. After a rapid wash withice-cold PBS, cells were lysed at 4°C in lysis buffer (50 mM HEPES, pH 7.5, 150mM NaCl, 100 mM NaF, 10 mM EDTA, 10 mM Na4P2O7, containing 1% TritonX-100, 1% sodium deoxycholate, 2 mM sodium orthovanadate, 1 mM phenyl-methylsulfonyl fluoride, 2 �g/ml aprotinin, and 20 �M leupeptin), sonicated for30 seconds on ice, then clarified by centrifugation at 13,000 rpm for 15 min at4°C. The protein content was determined in the supernatant using the Bradfordmethod (Bio-Rad). Equal amounts of protein were dissolved in 4� sample buffer(250 mM Tris-HCl, pH 6.8, 8% sodium dodecyl sulfate [SDS], 20% 2-mercap-toethanol, 50% glycerol, bromophenol blue) and left at room temperature for 30min. Total cell lysates were separated on SDS-polyacrylamide gel electrophoresis(SDS-PAGE) followed by electrophoretic transfer onto nitrocellulose mem-branes (Pall Life Sciences). Membranes were then subjected to immunoblottingusing horseradish peroxidase-conjugated secondary antibodies (Pierce) as de-scribed previously (25). Peroxidase activity was revealed using the enhancedchemiluminescence (ECL) system (Pierce). Quantitative analyses were carriedout by using a Biocom apparatus.
Scrape-loading and dye transfer assay for gap junction intercellular commu-nication. BxPC-3 cells were assessed for gap junction-mediated intercellularcommunication using a protocol adapted from that of Le and Musil (27, 28).Cells were plated on glass coverslips in 35-mm-diameter dishes (6 � 105 cells/dish) and grown for 4 days to confluence. The cells were rinsed three times withPBS containing 1% BSA, and the glass coverslip was removed from the dish; 3�l of a solution of PBS containing 0.5% rhodamine-dextran and 1% biocytin wasdirectly applied to the center of the coverslip, after which a scalpel was used tocreate a longitudinal scratch through the cell monolayer. Cells were incubated inthe dye mix for exactly 3 min, and then the coverslip was put back in the dish.Cells were quickly rinsed twice with PBS and immediately fixed for 15 min with2% paraformaldehyde (pH 7.0) in PBS. Fixed cells were then permeabilized for15 min with PBS containing 0.1% Triton X-100 and 0.2% BSA, followed byincubation with Alexa Fluor 488-conjugated avidin (1:200) diluted in 0.2% BSA–PBS for 30 min at room temperature. The coverslips were mounted onto glassslides with fluorescent mounting medium. Coverslips were rinsed twice in 1%BSA–PBS between the different steps. Rhodamine-dextran and (after reactionwith Alexa Fluor 488-conjugated avidin) biocytin were visualized by fluorescencemicroscopy using, respectively, G-2A and fluorescein isothiocyanate filter sets.Fluorescent controls were incubated without biocytin or without Alexa Fluor488-conjugated avidin. Only a small amount of unspecific labeling was obtainedwith Alexa Fluor 488-conjugated avidin.
In vivo labeling and measurement of protein synthesis. Cells were plated in60-mm-diameter culture dishes (4 � 105 cells/dish) and grown for 2 days. Cellswere starved for methionine for 30 min at 37°C in methionine-free DMEM andsupplemented with 10% FCS and 2 mM glutamine. The medium was thenreplaced with fresh methionine-free medium containing [35S]methionine (10�Ci/ml). The radioactive medium was removed after a 30-min pulse, and cellswere rinsed twice with ice-cold PBS. Cells were then lysed at 4°C in lysis bufferas described above, and protein content was quantified. Equal amounts were
VOL. 25, 2005 IRES-DRIVEN REGULATION OF CONNEXIN EXPRESSION 4035
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from

either immunoprecipitated using polyclonal antibodies directed against Cx26(Zymed) or Cx43 (Santa Cruz) or directly dissolved in 4� sample buffer asdescribed (25). Immunocomplexes or total cell lysates were submitted to SDS-PAGE, transferred onto nitrocellulose membranes, and analyzed by autoradiog-raphy using a PhosphorImager (Molecular Dynamics).
Real-time quantitative PCR. Cells were plated in 100-mm-diameter culturedishes (106 cells/dish) and grown for 4 days. Total RNA was extracted withTRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Aftera DNase I treatment (BD Biosciences-Clontech), total RNA was reverse tran-scribed by using random hexamers and Superscript II reverse transcriptase (In-vitrogen). Resulting cDNAs were used to perform quantitative analysis of Cx26and Cx43 mRNA expression by real-time PCR using a Gene Amp 5700 SequenceDetection System and SYBR green as a dye (Applied Biosystems). The se-quences of primers (MWG Biotech) were as follows: Cx26-forward primer,5�-CTGTGTTGTGTGCATTCGTCTTT-3�; reverse primer, 5�-CAGCGTGCCCCAATCC-3�; Cx43-forward primer, 5�-GACAAGGTTCAAGCCTACTCAACTG-3�; reverse primer, 5�-TGTCCCCAGCAGCAGGAT-3�; and glyceralde-hyde-3-phosphate dehydrogenase (GAPDH)-forward primer, 5�-GTCGGAGTCAACGGATTTGG-3�; reverse primer, 5�-AAAAGCAGCCCTGGTGACC-3�.PCR efficiencies were measured by preparing a standard curve for each primerpair (efficiency �90%). Target gene expression was normalized by usingGAPDH housekeeping gene expression as an internal control. Samples incu-bated without reverse transcriptase were used as negative template controls.
Plasmid constructs. The plasmid encoding human Cx32 (pORF/hCx32) andthe corresponding plasmid backbone were from InvivoGen. pRL-CMV was fromPromega. The Renilla luciferase- and firefly luciferase-based dicistronic vectorspSL2-Cx43 (containing the Cx43 mRNA 5� UTR) and pSL2-EMCV (containingthe encephalomyocarditis virus IRES) were kindly provided by R. Werner (De-partment of Biochemistry and Molecular Biology, University of Miami School ofMedicine, Miami, FL) (49). pSL2-Cx26 was obtained by substituting the Cx43 5�UTR for the PCR-amplified Cx26 5� UTR (forward primer: 5�-AAGAATTCAGGTGTTGCGGCCCCGCAGCG-3�, and reverse primer: 5�-TTTTCTCGAGCTTCTACTCTGGGCGGTTG-3�) in the EcoRI/XhoI-linearized pSL2-Cx43 vec-tor. The second stem loop (SL2, see the Results section for details), which waslost during such substitution, was reintroduced in the EcoRI restriction site. Thepromoterless pGL3-Cx43 dicistronic plasmid, which permits the detection ofcryptic promoter activities that could exist in the Cx43 5� UTR, was generated bysubcloning the NheI/NarI fragment of the pSL2-Cx43 vector (which contains theentire Renilla luciferase and Cx43 5� UTR entire sequences and part of fireflyluciferase) into a Nhe1/NarI-linearized pGL3basic vector (Promega). The pGL3-Cx26 vector was generated by substituting the Cx43 5� UTR and firefly luciferaseentire sequences for the Cx26 5� UTR and firefly luciferase entire sequences intothe pGL3-Cx43 vector using EcoRI and XbaI restriction sites.
Transient transfection and dual luciferase assays. For transient transfections,BxPC-3 cells were grown up to 50% confluence in 100-mm-diameter dishes inDMEM containing 10% FCS and transfected with 6 �g of the desired cDNAsusing polyethyleneimine (ExGen 500 transfection reagent; Euromedex) accord-ing to the manufacturer’s protocol. Cells transfected with the appropriate emptyvectors were used as a control. Transfected cells were then incubated for anadditional 48 h before lysis with 1� passive lysis buffer (Promega). After mea-suring the protein content in the supernatant, 20 �g of soluble cell lysate (in atotal volume of 50 �l) was assayed for Renilla and firefly luciferase activity withthe dual luciferase reporter assay system (Promega) following the manufacturer’sinstructions. A Luminoskan Ascent microplate luminometer with dual injectors(Thermo Labsystems) was used.
Statistical analysis. Statistical analysis was performed by using Student’s t test.P values � 0.05 indicate a statistically significant difference.
RESULTS
Description of cell lines. To evaluate the contribution oftranslational mechanisms in the expression of connexins andconsequent formation of gap junctions, we have utilized thepreviously described mock- (BxC) and stably sst2-transfected(Bx2.0) pools of human ductal pancreatic cancer cells BxPC3(8, 46). Pools were preferred to clonal populations becausethey are less likely to have additional alterations due to clonalexpansion. To further strengthen our observations, most of theexperiments were conducted in two additional sst2-expressingpools (Bx2.2 and Bx2.4). An immunostaining of cells visualized
by confocal microscopy was first performed to verify that thesetwo additional pools also express sst2. While sst2 was notdetected in BxC cells, both Bx2.2 and Bx2.4 pools expressedsst2 to a level similar to that of the already characterized Bx2.0cells (Fig. 1A). For clarity, the term Bx2 is used when a com-ment applies indifferently to any sst2-expressing pool (Bx2.0,Bx2.2, or Bx2.4).
Implication of gap junctions in sst2-mediated density inhi-bition. The proliferation rate of Bx2 cells was lower than thatof BxC cells (Fig. 1B). Depending on the pool analyzed, how-ever, proliferation was not uniformly inhibited. Bx2.4 cellsgrew faster than Bx2.0 or Bx2.2 cells. A more striking obser-vation was that all Bx2 cells were contact inhibited after 6 daysof culture, while BxC cells continued to proliferate. Theseobservations suggest that sst2 elicited antiproliferative mecha-nisms that could target both the rate of proliferation during thelog phase and density inhibition once cells attained confluence.
One cellular function that is related to density inhibition isgap junction intercellular communication (14). To test for theinvolvement of gap junction intercellular communication indensity inhibition of sst2-expressing cells, proliferation wasmonitored in the presence of 25 �M of 18�-glycyrrhetinic acid(�-GA). The dose of 25 �M was chosen because higher doses(50 or 100 �M) exhibited toxic effects (data not shown). �-GAis a chemical that reversibly precludes connexon packing in gapjunction plaques and thus blocks gap junction intercellularcommunication (15). Interestingly, while the log-phase growthwas not perturbed in any cell line, density inhibition of Bx2.0cells was abrogated by �-GA (Fig. 1C). Similar results wereobtained with Bx2.2 and Bx2.4 cells (data not shown). A sta-tistical analysis showed that the action of �-GA was significantat 6 days of cell culture (Fig. 1D). These results indicate thatsst2 provokes density inhibition through a mechanism depen-dent on gap junction intercellular communication.
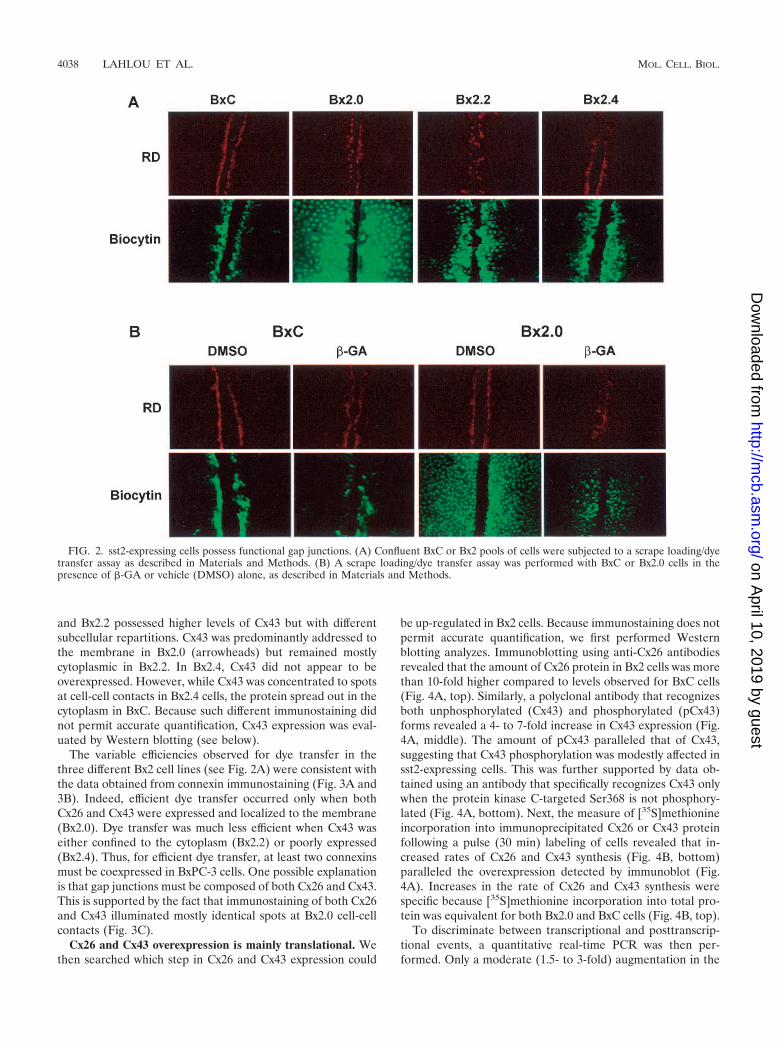
Functional gap junction intercellular communications arecomposed of Cx26 and Cx43 in sst2-expressing cells. Since�-GA precludes density inhibition exclusively in Bx2 cell lines,we hypothesized that sst2-expressing cells possess functionalgap junctions while BxC cells do not. To verify this hypothesis,a gap junction-dependent scrape-loading/dye transfer assaywas performed. The technique employs scraping to woundcells. Wounded cells are then loaded with a low-molecular-weight fluorescent dye, biocytin (molecular weight 372.48),that allows the monitoring of its transfer into contiguous intactcells (10). The involvement of gap junctions in biocytin transferis verified by the concurrent loading of rhodamine-dextran(molecular weight 10,000), a marker dye conjugate which can-not cross the narrow gap junction channel and therefore staysin wounded cells.
Figure 2A shows that biocytin stained mainly loaded(wounded) BxC cells, but largely diffused (albeit to a differentextent) from loaded to contiguous cells in the three sst2-ex-pressing cell lines. In contrast, rhodamine-dextran, used as anegative control, remained confined in wounded cells. To en-sure that the reversion of cell proliferation inhibition observedin the presence of �-GA (Fig. 1C) actually correlated withaltered gap junction intercellular communication, the scrape-loading/dye transfer assay was also performed using cells pre-treated with �-GA (Fig. 2B). �-GA severely impaired thetransfer of biocytin in Bx2.0 cells. These data demonstrate that
4036 LAHLOU ET AL. MOL. CELL. BIOL.
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from

reintroducing sst2 in the human pancreatic cancer cell lineBxPC-3 restores the formation of functional gap junctionswhich block cell proliferation.
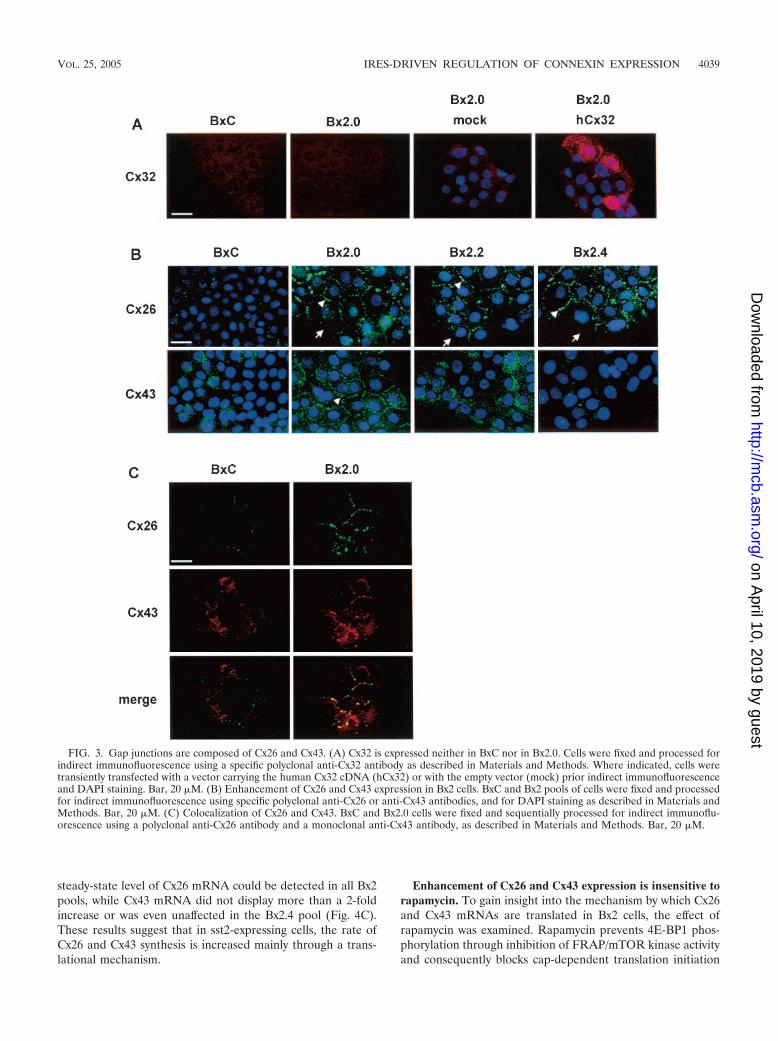
To identify which connexin(s) could compose gap junctionsin Bx2 cells, we performed indirect immunofluorescence anal-yses using antibodies that specifically recognize each of thethree connexins normally expressed in the pancreas (Cx26,Cx32, and Cx43). Cx26 and Cx32 have been shown to be spe-cifically expressed in the exocrine pancreas (32, 33). However,Cx32 was detected neither in BxC nor in Bx2 cells (Fig. 3A).This was not due to a defect in antibody efficacy, as Cx32 waseasily detectable following transient transfection of the humanCx32 cDNA into Bx2.0 cells (Fig. 3A). In contrast, Cx26 was
barely detectable in BxC but strongly overexpressed in all Bx2cells (Fig. 3B top).
Strikingly, immunostaining in all Bx2 cells was concentratedto bright spots at cell-cell contacts (arrowheads), but absentwhere cell membranes did not contact each other (arrows),indicating that Cx26 is correctly addressed to the membrane.We also found that BxC cells express Cx43 (Fig. 3B, bottom).This was surprising since Cx43 has been considered specific tothe endocrine pancreas. However, other mono- and polyclonalCx43 antibodies gave similar results in both immunostaining(not shown) and Western blotting (see below) and thus con-firmed that Cx43 is actually expressed in the exocrine pancre-atic cancer BxPC-3 cells. Compared to control cells, both Bx2.0
FIG. 1. sst2-dependent inhibition of BxPC-3 cell proliferation is reversed by an inhibitor of gap junction formation. (A) sst2 expression inBxPC-3 pools. BxC and Bx2 pools of cells were fixed and processed for indirect immunofluorescence coupled to confocal microscopy using aspecific polyclonal anti-sst2 antibody as described in Materials and Methods. Pictures show optical sections of cells median areas. Bar, 10 �m.(B) Proliferation rates of BxC and sst2-expressing Bx2 cells. Pools of cells were seeded at equivalent concentrations and replicates were countedevery two days as described in Materials and Methods. Results represent the means standard error of the mean of three independentexperiments performed in triplicates. (C) sst2-mediated inhibition of proliferation is prevented by �-GA. BxC or Bx2.0 cells were seeded atequivalent concentrations in the presence of �-GA or vehicle (DMSO) alone, and replicates were counted every two days. Results represent oneexperiment performed in triplicates. (D) Results obtained following 6 days of culture and shown in C were repeated and quantified. Valuesrepresent the means standard error of the mean of three separate experiments performed in triplicates. *, P � 0.05.
VOL. 25, 2005 IRES-DRIVEN REGULATION OF CONNEXIN EXPRESSION 4037
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from
and Bx2.2 possessed higher levels of Cx43 but with differentsubcellular repartitions. Cx43 was predominantly addressed tothe membrane in Bx2.0 (arrowheads) but remained mostlycytoplasmic in Bx2.2. In Bx2.4, Cx43 did not appear to beoverexpressed. However, while Cx43 was concentrated to spotsat cell-cell contacts in Bx2.4 cells, the protein spread out in thecytoplasm in BxC. Because such different immunostaining didnot permit accurate quantification, Cx43 expression was eval-uated by Western blotting (see below).
The variable efficiencies observed for dye transfer in thethree different Bx2 cell lines (see Fig. 2A) were consistent withthe data obtained from connexin immunostaining (Fig. 3A and3B). Indeed, efficient dye transfer occurred only when bothCx26 and Cx43 were expressed and localized to the membrane(Bx2.0). Dye transfer was much less efficient when Cx43 waseither confined to the cytoplasm (Bx2.2) or poorly expressed(Bx2.4). Thus, for efficient dye transfer, at least two connexinsmust be coexpressed in BxPC-3 cells. One possible explanationis that gap junctions must be composed of both Cx26 and Cx43.This is supported by the fact that immunostaining of both Cx26and Cx43 illuminated mostly identical spots at Bx2.0 cell-cellcontacts (Fig. 3C).
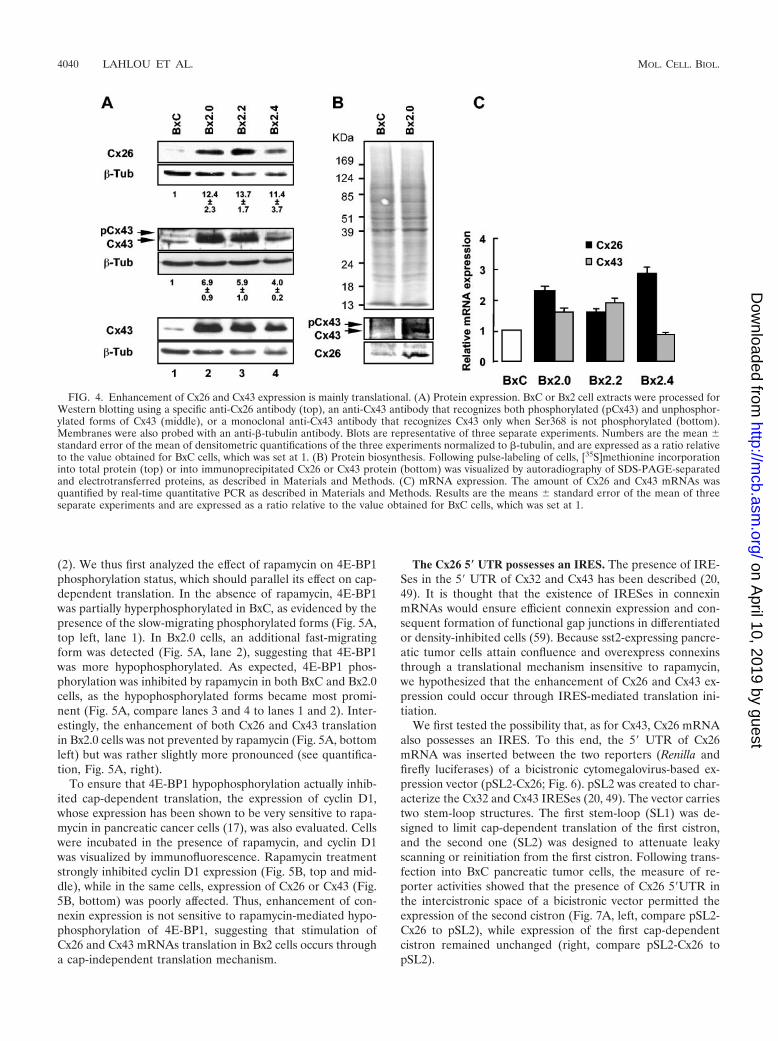
Cx26 and Cx43 overexpression is mainly translational. Wethen searched which step in Cx26 and Cx43 expression could
be up-regulated in Bx2 cells. Because immunostaining does notpermit accurate quantification, we first performed Westernblotting analyzes. Immunoblotting using anti-Cx26 antibodiesrevealed that the amount of Cx26 protein in Bx2 cells was morethan 10-fold higher compared to levels observed for BxC cells(Fig. 4A, top). Similarly, a polyclonal antibody that recognizesboth unphosphorylated (Cx43) and phosphorylated (pCx43)forms revealed a 4- to 7-fold increase in Cx43 expression (Fig.4A, middle). The amount of pCx43 paralleled that of Cx43,suggesting that Cx43 phosphorylation was modestly affected insst2-expressing cells. This was further supported by data ob-tained using an antibody that specifically recognizes Cx43 onlywhen the protein kinase C-targeted Ser368 is not phosphory-lated (Fig. 4A, bottom). Next, the measure of [35S]methionineincorporation into immunoprecipitated Cx26 or Cx43 proteinfollowing a pulse (30 min) labeling of cells revealed that in-creased rates of Cx26 and Cx43 synthesis (Fig. 4B, bottom)paralleled the overexpression detected by immunoblot (Fig.4A). Increases in the rate of Cx26 and Cx43 synthesis werespecific because [35S]methionine incorporation into total pro-tein was equivalent for both Bx2.0 and BxC cells (Fig. 4B, top).
To discriminate between transcriptional and posttranscrip-tional events, a quantitative real-time PCR was then per-formed. Only a moderate (1.5- to 3-fold) augmentation in the
FIG. 2. sst2-expressing cells possess functional gap junctions. (A) Confluent BxC or Bx2 pools of cells were subjected to a scrape loading/dyetransfer assay as described in Materials and Methods. (B) A scrape loading/dye transfer assay was performed with BxC or Bx2.0 cells in thepresence of �-GA or vehicle (DMSO) alone, as described in Materials and Methods.
4038 LAHLOU ET AL. MOL. CELL. BIOL.
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from
steady-state level of Cx26 mRNA could be detected in all Bx2pools, while Cx43 mRNA did not display more than a 2-foldincrease or was even unaffected in the Bx2.4 pool (Fig. 4C).These results suggest that in sst2-expressing cells, the rate ofCx26 and Cx43 synthesis is increased mainly through a trans-lational mechanism.
Enhancement of Cx26 and Cx43 expression is insensitive torapamycin. To gain insight into the mechanism by which Cx26and Cx43 mRNAs are translated in Bx2 cells, the effect ofrapamycin was examined. Rapamycin prevents 4E-BP1 phos-phorylation through inhibition of FRAP/mTOR kinase activityand consequently blocks cap-dependent translation initiation
FIG. 3. Gap junctions are composed of Cx26 and Cx43. (A) Cx32 is expressed neither in BxC nor in Bx2.0. Cells were fixed and processed forindirect immunofluorescence using a specific polyclonal anti-Cx32 antibody as described in Materials and Methods. Where indicated, cells weretransiently transfected with a vector carrying the human Cx32 cDNA (hCx32) or with the empty vector (mock) prior indirect immunofluorescenceand DAPI staining. Bar, 20 �M. (B) Enhancement of Cx26 and Cx43 expression in Bx2 cells. BxC and Bx2 pools of cells were fixed and processedfor indirect immunofluorescence using specific polyclonal anti-Cx26 or anti-Cx43 antibodies, and for DAPI staining as described in Materials andMethods. Bar, 20 �M. (C) Colocalization of Cx26 and Cx43. BxC and Bx2.0 cells were fixed and sequentially processed for indirect immunoflu-orescence using a polyclonal anti-Cx26 antibody and a monoclonal anti-Cx43 antibody, as described in Materials and Methods. Bar, 20 �M.
VOL. 25, 2005 IRES-DRIVEN REGULATION OF CONNEXIN EXPRESSION 4039
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from
(2). We thus first analyzed the effect of rapamycin on 4E-BP1phosphorylation status, which should parallel its effect on cap-dependent translation. In the absence of rapamycin, 4E-BP1was partially hyperphosphorylated in BxC, as evidenced by thepresence of the slow-migrating phosphorylated forms (Fig. 5A,top left, lane 1). In Bx2.0 cells, an additional fast-migratingform was detected (Fig. 5A, lane 2), suggesting that 4E-BP1was more hypophosphorylated. As expected, 4E-BP1 phos-phorylation was inhibited by rapamycin in both BxC and Bx2.0cells, as the hypophosphorylated forms became most promi-nent (Fig. 5A, compare lanes 3 and 4 to lanes 1 and 2). Inter-estingly, the enhancement of both Cx26 and Cx43 translationin Bx2.0 cells was not prevented by rapamycin (Fig. 5A, bottomleft) but was rather slightly more pronounced (see quantifica-tion, Fig. 5A, right).
To ensure that 4E-BP1 hypophosphorylation actually inhib-ited cap-dependent translation, the expression of cyclin D1,whose expression has been shown to be very sensitive to rapa-mycin in pancreatic cancer cells (17), was also evaluated. Cellswere incubated in the presence of rapamycin, and cyclin D1was visualized by immunofluorescence. Rapamycin treatmentstrongly inhibited cyclin D1 expression (Fig. 5B, top and mid-dle), while in the same cells, expression of Cx26 or Cx43 (Fig.5B, bottom) was poorly affected. Thus, enhancement of con-nexin expression is not sensitive to rapamycin-mediated hypo-phosphorylation of 4E-BP1, suggesting that stimulation ofCx26 and Cx43 mRNAs translation in Bx2 cells occurs througha cap-independent translation mechanism.
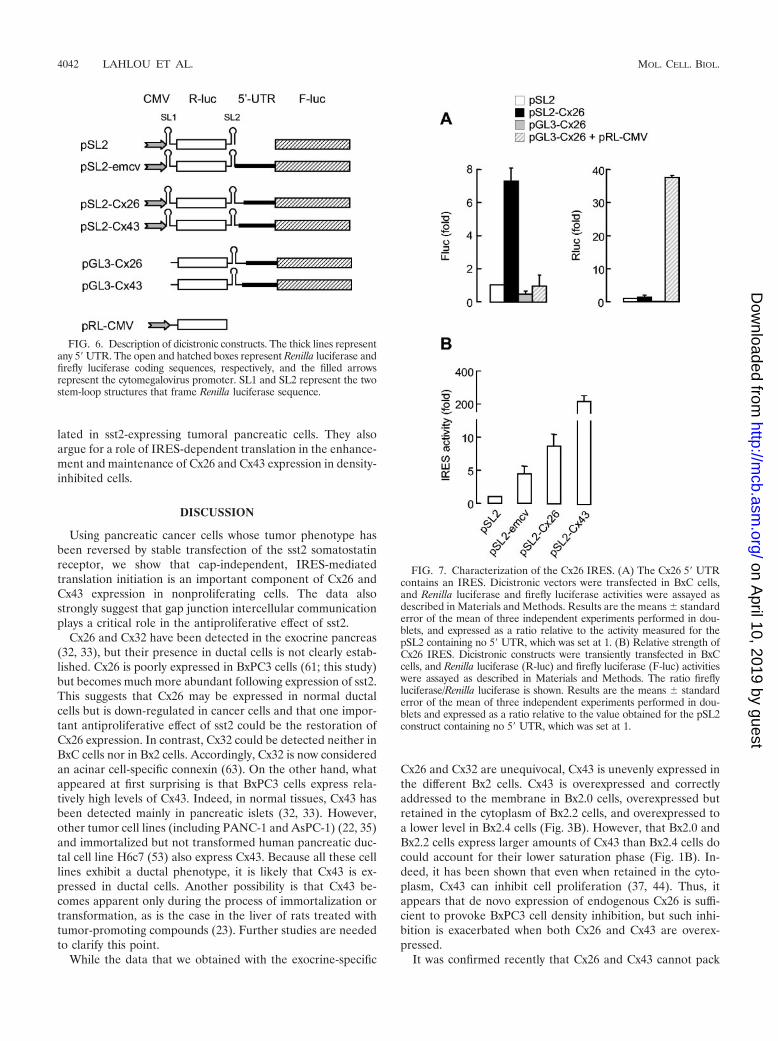
The Cx26 5� UTR possesses an IRES. The presence of IRE-Ses in the 5� UTR of Cx32 and Cx43 has been described (20,49). It is thought that the existence of IRESes in connexinmRNAs would ensure efficient connexin expression and con-sequent formation of functional gap junctions in differentiatedor density-inhibited cells (59). Because sst2-expressing pancre-atic tumor cells attain confluence and overexpress connexinsthrough a translational mechanism insensitive to rapamycin,we hypothesized that the enhancement of Cx26 and Cx43 ex-pression could occur through IRES-mediated translation ini-tiation.
We first tested the possibility that, as for Cx43, Cx26 mRNAalso possesses an IRES. To this end, the 5� UTR of Cx26mRNA was inserted between the two reporters (Renilla andfirefly luciferases) of a bicistronic cytomegalovirus-based ex-pression vector (pSL2-Cx26; Fig. 6). pSL2 was created to char-acterize the Cx32 and Cx43 IRESes (20, 49). The vector carriestwo stem-loop structures. The first stem-loop (SL1) was de-signed to limit cap-dependent translation of the first cistron,and the second one (SL2) was designed to attenuate leakyscanning or reinitiation from the first cistron. Following trans-fection into BxC pancreatic tumor cells, the measure of re-porter activities showed that the presence of Cx26 5�UTR inthe intercistronic space of a bicistronic vector permitted theexpression of the second cistron (Fig. 7A, left, compare pSL2-Cx26 to pSL2), while expression of the first cap-dependentcistron remained unchanged (right, compare pSL2-Cx26 topSL2).
FIG. 4. Enhancement of Cx26 and Cx43 expression is mainly translational. (A) Protein expression. BxC or Bx2 cell extracts were processed forWestern blotting using a specific anti-Cx26 antibody (top), an anti-Cx43 antibody that recognizes both phosphorylated (pCx43) and unphosphor-ylated forms of Cx43 (middle), or a monoclonal anti-Cx43 antibody that recognizes Cx43 only when Ser368 is not phosphorylated (bottom).Membranes were also probed with an anti-�-tubulin antibody. Blots are representative of three separate experiments. Numbers are the mean standard error of the mean of densitometric quantifications of the three experiments normalized to �-tubulin, and are expressed as a ratio relativeto the value obtained for BxC cells, which was set at 1. (B) Protein biosynthesis. Following pulse-labeling of cells, [35S]methionine incorporationinto total protein (top) or into immunoprecipitated Cx26 or Cx43 protein (bottom) was visualized by autoradiography of SDS-PAGE-separatedand electrotransferred proteins, as described in Materials and Methods. (C) mRNA expression. The amount of Cx26 and Cx43 mRNAs wasquantified by real-time quantitative PCR as described in Materials and Methods. Results are the means standard error of the mean of threeseparate experiments and are expressed as a ratio relative to the value obtained for BxC cells, which was set at 1.
4040 LAHLOU ET AL. MOL. CELL. BIOL.
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from

To exclude the possibility that the Cx26 5� UTR possesses anintrinsic promoter activity that could bias data interpretation, abicistronic vector carrying the Cx26 5� UTR but with no cyto-megalovirus promoter (Fig. 6, pGL3-Cx26) was also tested.Expression of the downstream cistron was not higher than thebackground obtained in the absence of Cx26 5� UTR (Fig. 7A,compare pGL3-Cx26 to pSL2), indicating that the Cx26 5�UTR did not contain a cryptic promoter. The lack of down-stream cistron expression observed following pGL3-Cx26 vec-tor transfection was not due to a defect in transfection effi-ciency as expression from a cotransfected vector carrying theRenilla luciferase reporter in a monocistronic context (pRL-CMV) was very high (compare pGL3-Cx26 plus pRL-CMV topGL3-Cx26). These results indicate that the Cx26 5� UTRpossesses an IRES.
We then evaluated the relative strength of Cx26 IRES inpancreatic cancer cells. Following transfection into BxC cells,the data show that the Cx26 IRES is much less active than thatof Cx43 but is around 2-fold stronger than the encephalo-myocarditis virus IRES which was used as a positive control(Fig. 7B).
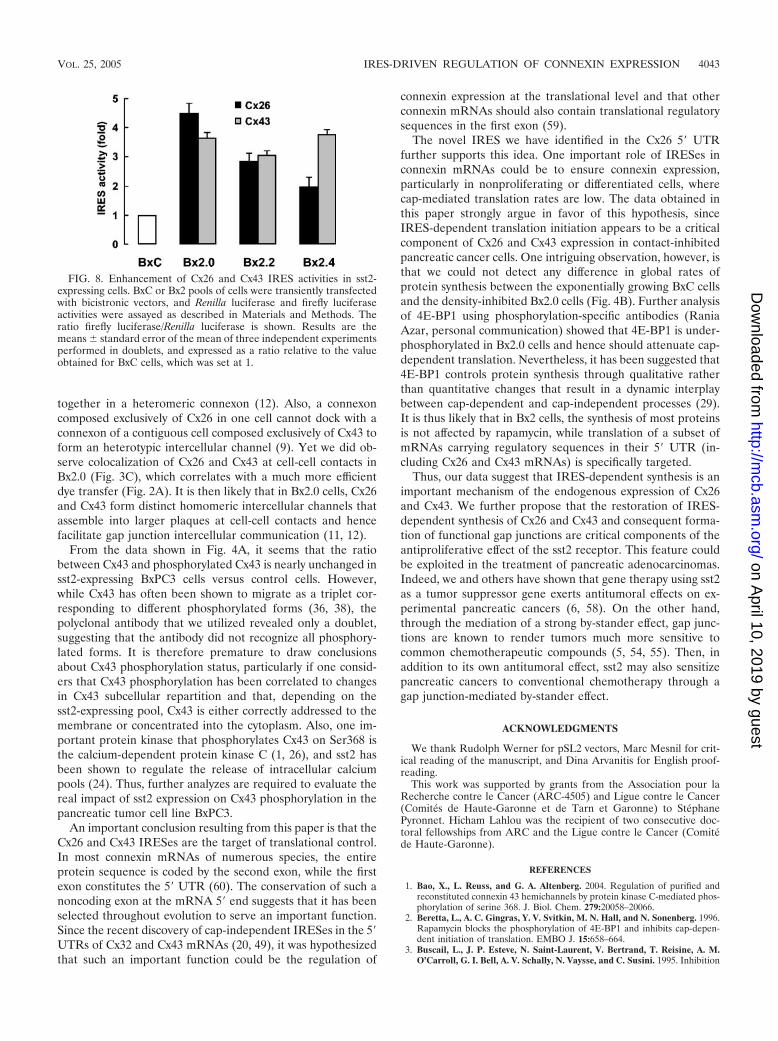
Cx26 and Cx43 IRESes are up-regulated in Bx2 cells. Theenhancement of connexin expression in Bx2 cells even in thepresence of the inhibitor of cap-dependent translation rapa-mycin could be explained by the existence of IRESes in Cx26and Cx43 mRNAs. To test this, IRES activity was measured inthe three Bx2 pools and in BxC cells following transfection ofbicistronic vectors. The activity of both Cx26 and Cx43 IRESeswas enhanced in all Bx2 pools compared to BxC cells (Fig. 8).These data indicate that Cx26 and Cx43 IRESes are up-regu-
FIG. 5. Enhancement of Cx26 and Cx43 expression is not sensitive to rapamycin. (A) Left. Rapamycin has no effect on enhanced connexinexpression. Extracts obtained from BxC or Bx2.0 cells treated or not with rapamycin were processed for Western blotting using a polyclonalanti-4E-BP1 or a monoclonal anti-�-tubulin antibody (top), and with anti-Cx43 or anti-Cx26 antibodies (bottom), as described in Materials andMethods. Right. The data obtained for Cx26 and Cx43 were subjected to a densitometric quantification normalized to �-tubulin. Histogramsrepresent the ratios between the values obtained for Bx2.0 and BxC. The ratios calculated for rapamycin-untreated cells were set at 100%.(B) Rapamycin blocks cyclin D1 expression. Cells treated with rapamycin or vehicle (DMSO) alone were fixed and sequentially processed forindirect immunofluorescence using a monoclonal anti-cyclin D1 antibody (top) and either a polyclonal anti-Cx26 or a polyclonal anti-Cx43 antibody(bottom), as described in Materials and Methods. Cells were also stained with DAPI and the pictures obtained were superposed with that obtainedfor cyclin D1 (middle). Bar, 20 �M.
VOL. 25, 2005 IRES-DRIVEN REGULATION OF CONNEXIN EXPRESSION 4041
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from
lated in sst2-expressing tumoral pancreatic cells. They alsoargue for a role of IRES-dependent translation in the enhance-ment and maintenance of Cx26 and Cx43 expression in density-inhibited cells.
DISCUSSION
Using pancreatic cancer cells whose tumor phenotype hasbeen reversed by stable transfection of the sst2 somatostatinreceptor, we show that cap-independent, IRES-mediatedtranslation initiation is an important component of Cx26 andCx43 expression in nonproliferating cells. The data alsostrongly suggest that gap junction intercellular communicationplays a critical role in the antiproliferative effect of sst2.
Cx26 and Cx32 have been detected in the exocrine pancreas(32, 33), but their presence in ductal cells is not clearly estab-lished. Cx26 is poorly expressed in BxPC3 cells (61; this study)but becomes much more abundant following expression of sst2.This suggests that Cx26 may be expressed in normal ductalcells but is down-regulated in cancer cells and that one impor-tant antiproliferative effect of sst2 could be the restoration ofCx26 expression. In contrast, Cx32 could be detected neither inBxC cells nor in Bx2 cells. Accordingly, Cx32 is now consideredan acinar cell-specific connexin (63). On the other hand, whatappeared at first surprising is that BxPC3 cells express rela-tively high levels of Cx43. Indeed, in normal tissues, Cx43 hasbeen detected mainly in pancreatic islets (32, 33). However,other tumor cell lines (including PANC-1 and AsPC-1) (22, 35)and immortalized but not transformed human pancreatic duc-tal cell line H6c7 (53) also express Cx43. Because all these celllines exhibit a ductal phenotype, it is likely that Cx43 is ex-pressed in ductal cells. Another possibility is that Cx43 be-comes apparent only during the process of immortalization ortransformation, as is the case in the liver of rats treated withtumor-promoting compounds (23). Further studies are neededto clarify this point.
While the data that we obtained with the exocrine-specific
Cx26 and Cx32 are unequivocal, Cx43 is unevenly expressed inthe different Bx2 cells. Cx43 is overexpressed and correctlyaddressed to the membrane in Bx2.0 cells, overexpressed butretained in the cytoplasm of Bx2.2 cells, and overexpressed toa lower level in Bx2.4 cells (Fig. 3B). However, that Bx2.0 andBx2.2 cells express larger amounts of Cx43 than Bx2.4 cells docould account for their lower saturation phase (Fig. 1B). In-deed, it has been shown that even when retained in the cyto-plasm, Cx43 can inhibit cell proliferation (37, 44). Thus, itappears that de novo expression of endogenous Cx26 is suffi-cient to provoke BxPC3 cell density inhibition, but such inhi-bition is exacerbated when both Cx26 and Cx43 are overex-pressed.
It was confirmed recently that Cx26 and Cx43 cannot pack
FIG. 6. Description of dicistronic constructs. The thick lines representany 5� UTR. The open and hatched boxes represent Renilla luciferase andfirefly luciferase coding sequences, respectively, and the filled arrowsrepresent the cytomegalovirus promoter. SL1 and SL2 represent the twostem-loop structures that frame Renilla luciferase sequence.
FIG. 7. Characterization of the Cx26 IRES. (A) The Cx26 5� UTRcontains an IRES. Dicistronic vectors were transfected in BxC cells,and Renilla luciferase and firefly luciferase activities were assayed asdescribed in Materials and Methods. Results are the means standarderror of the mean of three independent experiments performed in dou-blets, and expressed as a ratio relative to the activity measured for thepSL2 containing no 5� UTR, which was set at 1. (B) Relative strength ofCx26 IRES. Dicistronic constructs were transiently transfected in BxCcells, and Renilla luciferase (R-luc) and firefly luciferase (F-luc) activitieswere assayed as described in Materials and Methods. The ratio fireflyluciferase/Renilla luciferase is shown. Results are the means standarderror of the mean of three independent experiments performed in dou-blets and expressed as a ratio relative to the value obtained for the pSL2construct containing no 5� UTR, which was set at 1.
4042 LAHLOU ET AL. MOL. CELL. BIOL.
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from
together in a heteromeric connexon (12). Also, a connexoncomposed exclusively of Cx26 in one cell cannot dock with aconnexon of a contiguous cell composed exclusively of Cx43 toform an heterotypic intercellular channel (9). Yet we did ob-serve colocalization of Cx26 and Cx43 at cell-cell contacts inBx2.0 (Fig. 3C), which correlates with a much more efficientdye transfer (Fig. 2A). It is then likely that in Bx2.0 cells, Cx26and Cx43 form distinct homomeric intercellular channels thatassemble into larger plaques at cell-cell contacts and hencefacilitate gap junction intercellular communication (11, 12).
From the data shown in Fig. 4A, it seems that the ratiobetween Cx43 and phosphorylated Cx43 is nearly unchanged insst2-expressing BxPC3 cells versus control cells. However,while Cx43 has often been shown to migrate as a triplet cor-responding to different phosphorylated forms (36, 38), thepolyclonal antibody that we utilized revealed only a doublet,suggesting that the antibody did not recognize all phosphory-lated forms. It is therefore premature to draw conclusionsabout Cx43 phosphorylation status, particularly if one consid-ers that Cx43 phosphorylation has been correlated to changesin Cx43 subcellular repartition and that, depending on thesst2-expressing pool, Cx43 is either correctly addressed to themembrane or concentrated into the cytoplasm. Also, one im-portant protein kinase that phosphorylates Cx43 on Ser368 isthe calcium-dependent protein kinase C (1, 26), and sst2 hasbeen shown to regulate the release of intracellular calciumpools (24). Thus, further analyzes are required to evaluate thereal impact of sst2 expression on Cx43 phosphorylation in thepancreatic tumor cell line BxPC3.
An important conclusion resulting from this paper is that theCx26 and Cx43 IRESes are the target of translational control.In most connexin mRNAs of numerous species, the entireprotein sequence is coded by the second exon, while the firstexon constitutes the 5� UTR (60). The conservation of such anoncoding exon at the mRNA 5� end suggests that it has beenselected throughout evolution to serve an important function.Since the recent discovery of cap-independent IRESes in the 5�UTRs of Cx32 and Cx43 mRNAs (20, 49), it was hypothesizedthat such an important function could be the regulation of
connexin expression at the translational level and that otherconnexin mRNAs should also contain translational regulatorysequences in the first exon (59).
The novel IRES we have identified in the Cx26 5� UTRfurther supports this idea. One important role of IRESes inconnexin mRNAs could be to ensure connexin expression,particularly in nonproliferating or differentiated cells, wherecap-mediated translation rates are low. The data obtained inthis paper strongly argue in favor of this hypothesis, sinceIRES-dependent translation initiation appears to be a criticalcomponent of Cx26 and Cx43 expression in contact-inhibitedpancreatic cancer cells. One intriguing observation, however, isthat we could not detect any difference in global rates ofprotein synthesis between the exponentially growing BxC cellsand the density-inhibited Bx2.0 cells (Fig. 4B). Further analysisof 4E-BP1 using phosphorylation-specific antibodies (RaniaAzar, personal communication) showed that 4E-BP1 is under-phosphorylated in Bx2.0 cells and hence should attenuate cap-dependent translation. Nevertheless, it has been suggested that4E-BP1 controls protein synthesis through qualitative ratherthan quantitative changes that result in a dynamic interplaybetween cap-dependent and cap-independent processes (29).It is thus likely that in Bx2 cells, the synthesis of most proteinsis not affected by rapamycin, while translation of a subset ofmRNAs carrying regulatory sequences in their 5� UTR (in-cluding Cx26 and Cx43 mRNAs) is specifically targeted.
Thus, our data suggest that IRES-dependent synthesis is animportant mechanism of the endogenous expression of Cx26and Cx43. We further propose that the restoration of IRES-dependent synthesis of Cx26 and Cx43 and consequent forma-tion of functional gap junctions are critical components of theantiproliferative effect of the sst2 receptor. This feature couldbe exploited in the treatment of pancreatic adenocarcinomas.Indeed, we and others have shown that gene therapy using sst2as a tumor suppressor gene exerts antitumoral effects on ex-perimental pancreatic cancers (6, 58). On the other hand,through the mediation of a strong by-stander effect, gap junc-tions are known to render tumors much more sensitive tocommon chemotherapeutic compounds (5, 54, 55). Then, inaddition to its own antitumoral effect, sst2 may also sensitizepancreatic cancers to conventional chemotherapy through agap junction-mediated by-stander effect.
ACKNOWLEDGMENTS
We thank Rudolph Werner for pSL2 vectors, Marc Mesnil for crit-ical reading of the manuscript, and Dina Arvanitis for English proof-reading.
This work was supported by grants from the Association pour laRecherche contre le Cancer (ARC-4505) and Ligue contre le Cancer(Comites de Haute-Garonne et de Tarn et Garonne) to StephanePyronnet. Hicham Lahlou was the recipient of two consecutive doc-toral fellowships from ARC and the Ligue contre le Cancer (Comitede Haute-Garonne).
REFERENCES
1. Bao, X., L. Reuss, and G. A. Altenberg. 2004. Regulation of purified andreconstituted connexin 43 hemichannels by protein kinase C-mediated phos-phorylation of serine 368. J. Biol. Chem. 279:20058–20066.
2. Beretta, L., A. C. Gingras, Y. V. Svitkin, M. N. Hall, and N. Sonenberg. 1996.Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-depen-dent initiation of translation. EMBO J. 15:658–664.
3. Buscail, L., J. P. Esteve, N. Saint-Laurent, V. Bertrand, T. Reisine, A. M.O’Carroll, G. I. Bell, A. V. Schally, N. Vaysse, and C. Susini. 1995. Inhibition
FIG. 8. Enhancement of Cx26 and Cx43 IRES activities in sst2-expressing cells. BxC or Bx2 pools of cells were transiently transfectedwith bicistronic vectors, and Renilla luciferase and firefly luciferaseactivities were assayed as described in Materials and Methods. Theratio firefly luciferase/Renilla luciferase is shown. Results are themeans standard error of the mean of three independent experimentsperformed in doublets, and expressed as a ratio relative to the valueobtained for BxC cells, which was set at 1.
VOL. 25, 2005 IRES-DRIVEN REGULATION OF CONNEXIN EXPRESSION 4043
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from

of cell proliferation by the somatostatin analogue RC-160 is mediated bysomatostatin receptor subtypes SSTR2 and SSTR5 through different mech-anisms. Proc. Natl. Acad. Sci. USA 92:1580–1584.
4. Buscail, L., N. Saint-Laurent, E. Chastre, J. C. Vaillant, C. Gespach, G.Capella, H. Kalthoff, F. Lluis, N. Vaysse, and C. Susini. 1996. Loss of sst2somatostatin receptor gene expression in human pancreatic and colorectalcancer. Cancer Res. 56:1823–1827.
5. Carystinos, G. D., M. A. Alaoui-Jamali, J. Phipps, L. Yen, and G. Batist.2001. Upregulation of gap junctional intercellular communication and con-nexin 43 expression by cyclic-AMP and all-trans-retinoic acid is associatedwith glutathione depletion and chemosensitivity in neuroblastoma cells. Can-cer Chemother. Pharmacol. 47:126–132.
6. Celinski, S. A., W. E. Fisher, F. Amaya, Y. Q. Wu, Q. Yao, K. A. Youker, andM. Li. 2003. Somatostatin receptor gene transfer inhibits established pan-creatic cancer xenografts. J. Surg. Res. 115:41–47.
7. Cruciani, V., and S. O. Mikalsen. 1999. Stimulated phosphorylation of in-tracellular connexin43. Exp. Cell Res. 251:285–298.
8. Delesque, N., L. Buscail, J. P. Esteve, N. Saint-Laurent, C. Muller, G.Weckbecker, C. Bruns, N. Vaysse, C. Susini, and I. Rauly. 1997. sst2 soma-tostatin receptor expression reverses tumorigenicity of human pancreaticcancer cells. Cancer Res. 57:956–962.
9. Elfgang, C., R. Eckert, H. Lichtenberg-Frate, A. Butterweck, O. Traub, R. A.Klein, D. F. Hulser, and K. Willecke. 1995. Specific permeability and selec-tive formation of gap junction channels in connexin-transfected HeLa cells.J. Cell Biol. 129:805–817.
10. el-Fouly, M. H., J. E. Trosko, and C. C. Chang. 1987. Scrape-loading and dyetransfer. A rapid and simple technique to study gap junctional intercellularcommunication. Exp. Cell Res. 168:422–430.
11. Falk, M. M. 2000. Connexin-specific distribution within gap junctions re-vealed in living cells. J. Cell Sci. 113:4109–4120.
12. Gemel, J., V. Valiunas, P. R. Brink, and E. C. Beyer. 2004. Connexin43 andconnexin26 form gap junctions, but not heteromeric channels in co-express-ing cells. J. Cell Sci. 117:2469–2480.
13. Gingras, A. C., B. Raught, and N. Sonenberg. 1999. eIF4 initiation factors:effectors of mRNA recruitment to ribosomes and regulators of translation.Annu. Rev. Biochem. 68:913–963.
14. Goldberg, G. S., J. F. Bechberger, Y. Tajima, M. Merritt, Y. Omori, M. A.Gawinowicz, R. Narayanan, Y. Tan, Y. Sanai, H. Yamasaki, C. C. Naus, H.Tsuda, and B. J. Nicholson. 2000. Connexin43 suppresses MFG-E8 whileinducing contact growth inhibition of glioma cells. Cancer Res. 60:6018–6026.
15. Goldberg, G. S., A. P. Moreno, J. F. Bechberger, S. S. Hearn, R. R. Shivers,D. J. MacPhee, Y. C. Zhang, and C. C. Naus. 1996. Evidence that disruptionof connexon particle arrangements in gap junction plaques is associated withinhibition of gap junctional communication by a glycyrrhetinic acid deriva-tive. Exp. Cell Res. 222:48–53.
16. Govindarajan, R., S. Zhao, X. H. Song, R. J. Guo, M. Wheelock, K. R.Johnson, and P. P. Mehta. 2002. Impaired trafficking of connexins in andro-gen-independent human prostate cancer cell lines and its mitigation byalpha-catenin. J. Biol. Chem. 277:50087–50097.
17. Grewe, M., F. Gansauge, R. M. Schmid, G. Adler, and T. Seufferlein. 1999.Regulation of cell growth and cyclin D1 expression by the constitutivelyactive FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res.59:3581–3587.
18. Haghighat, A., S. Mader, A. Pause, and N. Sonenberg. 1995. Repression ofcap-dependent translation by 4E-binding protein 1: competition with p220for binding to eukaryotic initiation factor-4E. EMBO J. 14:5701–5709.
19. Hershey, J. W. 1991. Translational control in mammalian cells. Annu. Rev.Biochem. 60:717–755.
20. Hudder, A., and R. Werner. 2000. Analysis of a Charcot-Marie-Tooth diseasemutation reveals an essential internal ribosome entry site element in theconnexin-32 gene. J. Biol. Chem. 275:34586–34591.
21. Jang, S. K., H. G. Krausslich, M. J. Nicklin, G. M. Duke, A. C. Palmenberg,and E. Wimmer. 1988. A segment of the 5� nontranslated region of encepha-lomyocarditis virus RNA directs internal entry of ribosomes during in vitrotranslation. J. Virol. 62:2636–2643.
22. Kawasaki, Y., A. Tsuchida, T. Sasaki, S. Yamasaki, Y. Kuwada, M. Mu-rakami, and K. Chayama. 2002. Irsogladine malate up-regulates gap junc-tional intercellular communication between pancreatic cancer cells via PKApathway. Pancreas 25:373–377.
23. Krutovskikh, V. A., M. Mesnil, G. Mazzoleni, and H. Yamasaki. 1995. In-hibition of rat liver gap junction intercellular communication by tumor-promoting agents in vivo. Association with aberrant localization of connexinproteins. Lab. Investig. 72:571–577.
24. Lahlou, H., J. Guillermet, M. Hortala, F. Vernejoul, S. Pyronnet, C. Bous-quet, and C. Susini. 2004. Molecular signaling of somatostatin receptors.Ann. N. Y. Acad. Sci. 1014:121–131.
25. Lahlou, H., N. Saint-Laurent, J. P. Esteve, A. Eychene, L. Pradayrol, S.Pyronnet, and C. Susini. 2003. sst2 somatostatin receptor inhibits cell pro-liferation through Ras-, Rap1-, and B-Raf-dependent ERK2 activation.J. Biol. Chem. 278:39356–39371.
26. Lampe, P. D., E. M. TenBroek, J. M. Burt, W. E. Kurata, R. G. Johnson, and
A. F. Lau. 2000. Phosphorylation of connexin43 on serine368 by proteinkinase C regulates gap junctional communication. J. Cell Biol. 149:1503–1512.
27. Le, A. C., and L. S. Musil. 1998. Normal differentiation of cultured lens cellsafter inhibition of gap junction-mediated intercellular communication. Dev.Biol. 204:80–96.
28. Le, A. C., and L. S. Musil. 2001. A novel role for FGF and extracellularsignal-regulated kinase in gap junction-mediated intercellular communica-tion in the lens. J. Cell Biol. 154:197–216.
29. Li, S., N. Sonenberg, A. C. Gingras, M. Peterson, S. Avdulov, V. A. Pol-unovsky, and P. B. Bitterman. 2002. Translational control of cell fate: avail-ability of phosphorylation sites on translational repressor 4E-BP1 governs itsproapoptotic potency. Mol. Cell. Biol. 22:2853–2861.
30. Lin, T. A., X. Kong, T. A. Haystead, A. Pause, G. Belsham, N. Sonenberg,and J. C. Lawrence, Jr. 1994. PHAS-I as a link between mitogen-activatedprotein kinase and translation initiation. Science. 266:653–656.
31. Mader, S., H. Lee, A. Pause, and N. Sonenberg. 1995. The translationinitiation factor eIF-4E binds to a common motif shared by the translationfactor eIF-4 gamma and the translational repressors 4E-binding proteins.Mol. Cell. Biol. 15:4990–4997.
32. Meda, P. 1996. Gap junction involvement in secretion: the pancreas experi-ence. Clin. Exp. Pharmacol. Physiol. 23:1053–1057.
33. Meda, P., M. S. Pepper, O. Traub, K. Willecke, D. Gros, E. Beyer, B.Nicholson, D. Paul, and L. Orci. 1993. Differential expression of gap junctionconnexins in endocrine and exocrine glands. Endocrinology. 133:2371–2378.
34. Mesnil, M. 2002. Connexins and cancer. Biol. Cell 94:493–500.35. Mesnil, M., D. Rideout, N. M. Kumar, and N. B. Gilula. 1994. Non-com-
municating human and murine carcinoma cells produce alpha 1 gap junctionmRNA. Carcinogenesis. 15:1541–1547.
36. Mograbi, B., E. Corcelle, N. Defamie, M. Samson, M. Nebout, D. Segretain,P. Fenichel, and G. Pointis. 2003. Aberrant connexin 43 endocytosis by thecarcinogen lindane involves activation of the ERK/mitogen-activated proteinkinase pathway. Carcinogenesis 24:1415–1423.
37. Moorby, C., and M. Patel. 2001. Dual functions for connexins: Cx43 regu-lates growth independently of gap junction formation. Exp. Cell Res. 271:238–248.
38. Musil, L. S., and D. A. Goodenough. 1991. Biochemical analysis of con-nexin43 intracellular transport, phosphorylation, and assembly into gap junc-tional plaques. J. Cell Biol. 115:1357–1374.
39. Nagy, J. I., W. E. Li, C. Roy, B. W. Doble, J. S. Gilchrist, E. Kardami, andE. L. Hertzberg. 1997. Selective monoclonal antibody recognition and cel-lular localization of an unphosphorylated form of connexin43. Exp. Cell Res.236:127–136.
40. Neveu, M. J., J. R. Hully, K. L. Babcock, E. L. Hertzberg, B. J. Nicholson,D. L. Paul, and H. C. Pitot. 1994. Multiple mechanisms are responsible foraltered expression of gap junction genes during oncogenesis in rat liver.J. Cell Sci. 107:83–95.
41. Pause, A., G. J. Belsham, A. C. Gingras, O. Donze, T. A. Lin, J. C. Lawrence,Jr., and N. Sonenberg. 1994. Insulin-dependent stimulation of protein syn-thesis by phosphorylation of a regulator of 5�-cap function. Nature 371:762–767.
42. Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation ofeukaryotic mRNA directed by a sequence derived from poliovirus RNA.Nature 334:320–325.
43. Pyronnet, S., J. Dostie, and N. Sonenberg. 2001. Suppression of cap-depen-dent translation in mitosis. Genes Dev. 15:2083–2093.
44. Qin, H., Q. Shao, H. Curtis, J. Galipeau, D. J. Belliveau, T. Wang, M. A.Alaoui-Jamali, and D. W. Laird. 2002. Retroviral delivery of connexin genesto human breast tumor cells inhibits in vivo tumor growth by a mechanismthat is independent of significant gap junctional intercellular communication.J. Biol. Chem. 277:29132–29138.
45. Qin, H., Q. Shao, S. A. Igdoura, M. A. Alaoui-Jamali, and D. W. Laird. 2003.Lysosomal and proteasomal degradation play distinct roles in the life cycle ofCx43 in gap junctional intercellular communication-deficient and -compe-tent breast tumor cells. J. Biol. Chem. 278:30005–30014.
46. Rauly, I., N. Saint-Laurent, N. Delesque, L. Buscail, J. P. Esteve, N. Vaysse,and C. Susini. 1996. Induction of a negative autocrine loop by expression ofsst2 somatostatin receptor in NIH 3T3 cells. J. Clin. Investig. 97:1874–1883.
47. Roger, C., B. Mograbi, D. Chevallier, J. F. Michiels, H. Tanaka, D. Segre-tain, G. Pointis, and P. Fenichel. 2004. Disrupted traffic of connexin 43 inhuman testicular seminoma cells: overexpression of Cx43 induces membranelocation and cell proliferation decrease. J. Pathol. 202:241–246.
48. Saez, J. C., V. M. Berthoud, M. C. Branes, A. D. Martinez, and E. C. Beyer.2003. Plasma membrane channels formed by connexins: their regulation andfunctions. Physiol. Rev. 83:1359–1400.
49. Schiavi, A., A. Hudder, and R. Werner. 1999. Connexin43 mRNA contains afunctional internal ribosome entry site. FEBS Lett. 464:118–122.
50. Sella, O., G. Gerlitz, S. Y. Le, and O. Elroy-Stein. 1999. Differentiation-induced internal translation of c-sis mRNA: analysis of the cis elements andtheir differentiation-linked binding to the hnRNP C protein. Mol. Cell. Biol.19:5429–5440.
51. Singal, R., Z. J. Tu, J. M. Vanwert, G. D. Ginder, and D. T. Kiang. 2000.
4044 LAHLOU ET AL. MOL. CELL. BIOL.
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from

Modulation of the connexin26 tumor suppressor gene expression throughmethylation in human mammary epithelial cell lines. Anticancer Res. 20:59–64.
52. Spinella, F., L. Rosano, V. Di Castro, M. R. Nicotra, P. G. Natali, and A.Bagnato. 2003. Endothelin-1 decreases gap junctional intercellular commu-nication by inducing phosphorylation of connexin 43 in human ovarian car-cinoma cells. J. Biol. Chem. 278:41294–41301.
53. Tai, M. H., L. K. Olson, B. V. Madhukar, K. D. Linning, L. Van Camp, M. S.Tsao, and J. E. Trosko. 2003. Characterization of gap junctional intercellularcommunication in immortalized human pancreatic ductal epithelial cellswith stem cell characteristics. Pancreas. 26:e18–26.
54. Tanaka, M., and H. B. Grossman. 2001. Connexin 26 gene therapy of humanbladder cancer: induction of growth suppression, apoptosis, and synergy withcisplatin. Hum. Gene Ther. 12:2225–2236.
55. Tanaka, M., and H. B. Grossman. 2004. Connexin 26 induces growth sup-pression, apoptosis and increased efficacy of doxorubicin in prostate cancercells. Oncol. Rep. 11:537–541.
56. Tulipano, G., R. Stumm, M. Pfeiffer, H. J. Kreienkamp, V. Hollt, and S.Schulz. 2004. Differential beta-arrestin trafficking and endosomal sorting ofsomatostatin receptor subtypes. J. Biol. Chem. 279:21374–21382.
57. Vagner, S., B. Galy, and S. Pyronnet. 2001. Irresistible IRES. Attracting thetranslation machinery to internal ribosome entry sites. EMBO Rep. 2:893–898.
58. Vernejoul, F., P. Faure, N. Benali, D. Calise, G. Tiraby, L. Pradayrol, C.Susini, and L. Buscail. 2002. Antitumor effect of in vivo somatostatin recep-tor subtype 2 gene transfer in primary and metastatic pancreatic cancermodels. Cancer Res. 62:6124–6131.
59. Werner, R. 2000. IRES elements in connexin genes: a hypothesis explainingthe need for connexins to be regulated at the translational level. IUBMB Life50:173–176.
60. Willecke, K., J. Eiberger, J. Degen, D. Eckardt, A. Romualdi, M. Guldenagel,U. Deutsch, and G. Sohl. 2002. Structural and functional diversity of con-nexin genes in the mouse and human genome. Biol. Chem. 383:725–737.
61. Yang, L., Y. Chiang, H. J. Lenz, K. D. Danenberg, C. P. Spears, E. M.Gordon, W. F. Anderson, and D. Parekh. 1998. Intercellular communicationmediates the bystander effect during herpes simplex thymidine kinase/gan-ciclovir-based gene therapy of human gastrointestinal tumor cells. Hum.Gene Ther. 9:719–728.
62. Yano, T., F. Ito, K. Kobayashi, Y. Yonezawa, K. Suzuki, R. Asano, K.Hagiwara, H. Nakazawa, H. Toma, and H. Yamasaki. 2004. Hypermethyl-ation of the CpG island of connexin 32, a candiate tumor suppressor gene inrenal cell carcinomas from hemodialysis patients. Cancer Lett. 208:137–142.
63. Zhu, L., T. Tran, J. M. Rukstalis, P. Sun, B. Damsz, and S. F. Konieczny.2004. Inhibition of Mist1 homodimer formation induces pancreatic acinar-to-ductal metaplasia. Mol. Cell. Biol. 24:2673–2681.
VOL. 25, 2005 IRES-DRIVEN REGULATION OF CONNEXIN EXPRESSION 4045
on April 10, 2019 by guest
http://mcb.asm
.org/D
ownloaded from





























