Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
Biochimica et Biophysica Acta 1768 (2007) 3145–3161www.elsevier.com/locate/bbamem
Review
NMR studies on fully hydrated membrane proteins, with emphasis onbacteriorhodopsin as a typical and prototype membrane protein
Hazime Saitô a,b,⁎, Akira Naito c
a Center for Quantum Life Sciences, Hiroshima University, Higashi-Hiroshima 739-8526, Japanb Department of Life Science, Himeji Institute of Technology, Kamigori 678-1297, Japan
c Graduate School of Engineering, Yokohama National University, Hodogaya-ku, Yokohama 240-8501, Japan
Received 28 June 2007; received in revised form 24 August 2007; accepted 29 August 2007Available online 8 September 2007
Abstract
The 3D structures or dynamic feature of fully hydrated membrane proteins are very important at ambient temperature, in relation tounderstanding their biological activities, although their data, especially from the flexible portions such as surface regions, are unavailable from X-ray diffraction or cryoelectron microscope at low temperature. In contrast, high-resolution solid-state NMR spectroscopy has proved to be a veryconvenient alternative means to be able to reveal their dynamic structures. To clarify this problem, we describe here how we are able to reveal suchstructures and dynamic features, based on intrinsic probes from high-resolution solid-state NMR studies on bacteriorhodopsin (bR) as a typicalmembrane protein in 2D crystal, regenerated preparation in lipid bilayer and detergents. It turned out that their dynamic features are substantiallyaltered upon their environments where bR is present. We further review NMR applications to study structure and dynamics of a variety ofmembrane proteins, including sensory rhodopsin, rhodopsin, photoreaction centers, diacylglycerol kinases, etc.© 2007 Elsevier B.V. All rights reserved.
NMR; Membrane proteins; Fully hydrated; 13C NMR; Surface structure; Dynamics; Conformation
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31462. Intrinsic probes for conformation and dynamics analyses . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3146
2.1. Site-directed assignment of peaks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31462.2. Conformation-dependent 13C chemical shifts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31482.3. Dynamics-dependent 13C chemical shifts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31492.4. Dynamics-dependent suppressed peaks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3150
Abbreviations: bR, bacteriorhodopsin; bO, bacterioopsin; Bchl, bacteriochlorophylls; CP-MAS, cross polarization-magic angle spinning; 2D, two dimensional;3D, three dimensional; DARR, dipolar-assisted rotational resonance; DD-MAS, a single-pulse 13C dipolar decoupled-magic angle spinning; DGK, diacylglycerolkinase; DM, N-dodecylmaltoside; DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DSS, 2,2-dimethylsilapentane-5-sulfonic acid; EC, extracellular; GPCRs, G-protein-coupled receptors; HSQC, heteronuclear singlequantum coherence; HETCOR, heteronuclear correlation; HFIP, hexafluoroisopropanol; IP3, D-myo-inositol 1,4,5-trisphosphate; LH2, light-harvesting complex;MAS, magic angle spinning; MOVS, magnetically oriented vesicle system; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser effect; OTG, octyl ß-glucoside; pHtrII, pharaonis cognate transducer; PM, purple membrane; PISA, polarity index slant angle; PISEMA, polarization inversion spin exchange at the magicangle; PH, pleckstrin homology; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; POPC, 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine; pR,phoborhodopsin; ppR, pharaonis phoborhodopsin; RC, reaction center; REDOR, rotational echo double resonance; RFDR, radiofrequency-driven recoupling; Rho,rhodopsin; SDS, sodium dodecyl sulfate; SH3, Src homology 3; sR I, sensory rhodopsin I; sR II, sensory rhodopsin II; TET, trifluoroethylthio; TM, transmembrane;TMS, tetramethylsilane; TX-100, Triton X-100; TN-101, Triton N-101; TROSY, transverse relaxation-optimized NMR spectroscopy⁎ Corresponding author. Center for Quantum Life Science, Hiroshima University, Higashi-Hiroshima 739-8526, Japan. Tel./fax: +81 78 856 2876.E-mail address: [email protected] (H. Saitô).
0005-2736/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.bbamem.2007.08.026

3146 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
3. Fully hydrated membrane proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31503.1. bR in 2D crystals, distorted 2D crystal, lipid bilayers or detergents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31503.2. Surface structure of bR in 2D crystal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31533.3. Sensory rhodopsin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31533.4. Rhodopsin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31553.5. Photosynthetic antennae and photoreaction centers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31563.6. Other membrane proteins. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31563.7. Membrane proteins in magnetically aligned system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3157
4. Concluding remarks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3157References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3158
1. Introduction
Structural studies of membrane proteins, constituting anestimated 30% of all proteins, have beenmostly left aside in spiteof their crucial biological importance, because revealing 3Dstructures of membrane proteins by X-ray crystallography ormultidimensional NMR spectroscopy has been hampered byextreme difficulty in crystallization and too large molecularmass, respectively. Naturally, solid-state NMR approach mainlyutilizing 13C- or 15N-labeled proteins has been successfullyutilized as an alternative means, in order to increase sensitivityand selectivity of signals from the site of interest [1–6], as far asfrequencies of any possible molecular motions do not seriouslyinterfere with those of proton decoupling or magic anglespinning essential for averaging dipolar interactions or chemicalshift anisotropies, respectively [7,8]. Indeed, fully hydratedmembrane proteins are far from rigid body at ambient temper-ature, in spite of pictures obtained by X-ray diffraction studies,as demonstrated by extensive studies on bacteriorhodopsin (bR)as a typical membrane protein from the purple membrane (PM)ofHalobacterium salinarum [9–11]: bR in 2D crystal undergoesseveral types of motions depending upon the site of interest, fast(N108 Hz), intermediate (104 to 105 Hz) or slow (102 Hz)motions as manifested from selectively suppressed 13C NMRsignals from amino acid selective 13C-labeld preparations. Thefast motions are ascribed to the N- and C-terminal portionsundergoing isotropic motions which are suppressed by unsuc-cessful cross polarization due to time-averaged dipolar interac-tions. The second intermediate motions for the loops andtransmembrane α-helices result in failure of attempted peaknarrowing by interference of motional frequency with frequencyof proton decoupling or magic angle spinning, leading to com-pletely or partially suppressed peaks from areas underconsideration [5,6,12–14]. The last slow motions arise fromthe transmembrane α-helices which undergo exchange processwith a rate constant of 102 s−1 among several conformationsslightly different from each other [15].
In the presence of such low-frequency motions, spectralresolution of 13C NMR spectra of membrane proteins could besubstantially deteriorated, especially when they are densely13C-labeled as in [1,2,3-13C]Ala-bR [16] or uniformly [13C-,15N]-labeled as in LH2-light-harvesting complex [17]: verybroad featureless signals were dominant in the former due to theaccelerated transverse relaxation rate caused by the increasednumber of relaxation pathways through a number of 13C–13C
homo-nuclear dipolar interactions and scalar J couplings [16],while only about 40% of 13C NMR signals from the trans-membrane segments were detected in the latter [17]. Theassignment of peaks to globular proteins, on the other hand, wasnearly completed for residues 7–61 of a uniformly labeledglobular protein such as α-spectrin SH3 domain in the solid,although the signals of the N- and C-terminal residues 1–6 and62 were not detected [18]. This sort of inconvenience due tosuppressed peaks, however, could be effectively removed whenpathways of direct 13C–13C interactions are absent by use of asingle source of 13C labeled amino acid residue or partial 13Clabeling to prepare 13C-labeled proteins [12,19]. It is also veryimportant to clarify under what condition well-resolved solid-state 13C NMR spectra are available without suppressed peaksdue to such motions, even if they are essential for membraneproteins in maintaining various activities of cells [20].
It is demonstrated here that bR (see Fig. 1 for the primarystructure, by taking into account of the secondary structure asrevealed by X-ray diffraction) can be utilized as an excellentmodel to gain insight into real image of membrane proteinswhich may vary with a manner of preparations, 2D crystal,distorted 2D crystal, regenerated preparation in lipid bilayer, orin detergent, as revealed by 13C NMR [5,6,12–14]. Studies onsuch systems were made possible, because bR is readily avail-able in bulk from bacterial sources such as Halobacteria orEscherichia coli and considered as a prototype for a variety of G-protein-coupled receptors (GPCRs) involved in signal transduc-tion. Therefore, we are concerned with fully hydrated membraneproteins (with excess water) such as centrifuged pellet(40,000×g for 1 h) or fully hydrated lipid bilayer in the presenceof excess water, both of which corresponds with intactbiomembrane systems.
2. Intrinsic probes for conformation and dynamics analyses
2.1. Site-directed assignment of peaks
Well-resolved eleven 13C NMR signals of [3-13C]Ala-labeledbR were observed for fully hydrated pellet samples of purplemembrane (PM), although their spectral features differ substan-tially between the 13C cross polarization-magic angle (CP-MAS)and single-pulse, dipolar decoupled-magic angle spinning (DD-MAS) NMR spectra [21]. It is notable that the 13C CP-MASNMR signals of [3-13C]Ala-bR from the flexible regions such asN- or C-terminal residues anchored at the membrane surface

Fig. 1. Schematic representation of the primary structure of bR by taking into account of the secondary structure [transmembrane α-helices (A–G) in the boxedcolumn] revealed by X-ray diffraction studies. Cytoplasmic α-helix identified by the solid-state NMR, protruding from the membrane surface, is indicated by thecolumn G′. Ala (circled) or Val (boxed) residues labeled by either [3-13C]alanine, [1-13C]valine, or both are indicated.
3147H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
(see Fig. 1) were selectively suppressed, owing to the absence ofresidual dipolar interactions due to isotropic rapid motions [22].In contrast, the 13C NMR signals are fully visible by the DD-MAS experiment from the whole area of the membrane proteinbecause of shorter spin–lattice relaxation times (∼0.5 s) ascompared with the repetition time (4 s). Therefore, the graypeaks in the DD-MAS NMR (Fig. 2A) can be unambiguouslyascribed to the peaks from such flexible portions and the restpeaks are identical to those of the CP-MAS NMR. This assign-ment of peaks is consistent with the spectral change caused bycleavage of the C-terminal residues by proteolytic enzymes suchas papain and carboxylpeptidase A [23]. Such 13C NMR peaksfrom flexible portion, however, were still missing even iflyophilized samples were used instead of intact preparations,even if they are rehydrated up to 100% later because ofinsufficient manner of hydration [24].
Site-directed assignment of peaks can be straightforward bycomparing the 13C NMR signals of wild-type protein with thoseof a mutant in which a labeled amino acid residue of interest isreplaced by other type of amino acid residue [21]. For instance,the 13C NMR peaks for Ala 196 (F-G loop) and 126 (at thecorner of helix D) of [3-13C]Ala-labeled bR were unambigu-ously assigned to the peaks whose peak intensities were signifi-cantly reduced in the site-directed mutants, A196G and A126G,respectively [21]. This approach is still useful, even if an addi-tional chemical shift dispersion due to an accompanied con-formational change by a site-directed mutation is located near
the membrane surfaces. Indeed, spectral editing using Mn2+-induced spectral broadening turned out to be very effective toremove the effect of such additional spectral modifications atthe surface areas, by taking into account that these signals werebroadened beyond recognition to the extent more than 100 Hz inthe area within 8.7 Å of Mn2+ ion bound to the surface residues[25], based on accelerated paramagnetic relaxation rate [26,27].This approach is not useful, however, if wild-type or mutantproteins are subject to interference of fluctuation frequency withfrequencies of proton decoupling or magic angle spinning [7,8].
It turned out to be not easy, however, to extend this approachto a variety of membrane proteins in which global conforma-tional change occurs as in [3-13C]Ala-labeled D85N mutant ofbR in which the M-like state is achieved without photo-illumi-nation at alkaline condition (pH∼10) [28,29], because almostall 13C NMR peaks were displaced by taking the M-like photo-intermediate state at alkaline pH without photo-illumination(the abovementioned condition (1)). Very complicated spectralpatterns were also noted for A39V, A84G, A103C [30], A51G,A53G, A81G, A84G, A184G, A215G.[25], A106G, A160G,A160V, A228G [31].
Rotational echo double resonance (REDOR) filtered exper-iment in solid-state NMR is a powerful method to observeselectively a signal of a specific 13C–15N nuclear pair withstrong dipolar interaction by using 13C, 15N-doubly isotopiclabeling of a unique consecutive amino acid sequence in amembrane protein [32]. This method is particularly useful to

Table 113C chemical shifts of polypeptides characteristic of α-helix and β-sheet forms(ppm from TMS) a
Cα Cβ C_O
α-Helix
β-Sheet
Δ b α-Helix
β-Sheet
Δ b α-Helix
β-Sheet
Δ b
Ala 52.4 48.2 4.2 14.9 19.9 −5.0 176.4 171.8 4.6Leu 55.7 50.5 5.2 39.5 43.3 −3.8 175.7 170.5 5.2Glu (OBzl) 56.4 51.2 5.2 25.6 29.0 −3.4 175.6 171.0 4.6Asp (OBzl) 53.4 49.2 4.2 33.8 38.1 −4.3 174.9 169.8 5.1Val 65.5 58.4 7.1 28.7 32.4 −3.7 174.9 171.8 3.1Ile 63.9 57.8 6.1 34.8 39.4 −4.6 174.9 172.7 2.2Lys c 57.4 29.9 176.5Lys (Z) 57.6 51.4 6.2 29.3 28.5 −0.8 175.7 170.4 5.3Arg 57.1 28.9 175.2 169.0 6.2Phe 61.3 53.2 8.1 35.0 39.3 −4.3 175.1 170.6 4.5Met 57.2 52.2 5.0 30.2 34.8 −4.6 175.1 170.6 4.5Gly 43.2 168.4
171.6 168.5 3.1a Chemical shifts are referenced to neat TMS, initially referenced to the
carboxyl carbon of glycine at 176.03 ppm; taken from [6,34,35].b Difference of the 13C chemical shift of the α-helix with respect to that of the
β-sheet form.c Taken from the data obtained in aqueous solution.
Fig. 2. 13C DD-MAS (A) and CP-MAS NMR (B) spectra of [3-13C]Ala-labeledbR from purple membrane. Peaks in gray from the C-terminus are preferentiallysuppressed by the CP-MAS NMR, caused by a time-averaged 13C–1H dipolarinteraction in the presence of isotropic motions in the C-terminus [21]. Naturally,the rest peaks in the DD-MAS are identical to those of the CP-MAS NMR,except for the reduced peak intensities in the former.
3148 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
assign signal of a specific amino acid sequence, if there is noperturbation to induce any conformation in the membraneprotein among the heavily overlapped resonances. Actually,REDOR filtered experiments were performed to selectivelyobserve 13C NMR signals of [1-13C]Tyr-, [15N]Pro-bR [33]. Thetwo Tyr185 signals at 177.7 and 173.4 ppm which can beselected by a unique Tyr185-Pro186 pair were assigned to thepeaks due to all-trans and 13-cis and 15 syn configurations,respectively.
2.2. Conformation-dependent 13C chemical shifts
13C chemical shifts of polypeptides with known secondarystructure recorded in the solid turned out to be a very usefulmeans to determine secondary structure of a given peptide unitfor a variety of proteins or peptides, free from any possibleambiguity arising from their time averaging of chemical shifts,in solution, arising from conformational fluctuations [34–37].Indeed, the 13C chemical shifts of the backbone Cα and C=Ocarbons, as well as those of the side-chain Cβ, of respectiveamino acid residues in particular polypeptides are significantlydisplaced up to 8 ppm, depending on their local conformations,irrespective of there being neighboring residues [5,6,34–39]: theCα and C=O 13C shifts of theα-helix are displaced downfield by3.5–8.0 ppm with respect to those of the β-sheet form, while the
Cβ shifts taking the α-helix are displaced upfield by 3.4–5.2 ppm with respect to those of the β-sheet, as summarized inTable 1. The transferability of these conformation-dependent13C chemical shifts of particular residues from the model sys-tems to a more complicate protein has been proven to be ex-cellent and can be utilized as a diagnostic means to distinguishtheir local conformations, as far as they are rigid enough as insilk fibroin [40,41], collagen [42–44] and transmembranepeptides in the solid [45]. Naturally, it should be taken intoaccount that the chemical shift data with respect to a commonreference peak must be compared for this purpose. The 13Cchemical shifts summarized in Table 1 were referenced to thepeak position of neat TMS in a sealed capillary, by replacementmethod initially referenced to the carboxyl carbon of glycine(176.03 ppm from neat TMS) or CH2 carbon of adamantane(29.50 ppm for the high-field doublet from neat TMS). Thechemical shifts obtained by other reference systems [46,47],including DSS as additional reference, should be compared withthe data in Table 1 after subtracting or adding the correctionfactor shown in the right column in Table 2.
Chemical shifts from solution NMR are also utilized as refe-rence to the prediction of secondary structure for solid proteinsand peptides [48,49]. It should be cautioned, however, that use ofsuch data is not free from errors due to difference in the chemicalshift references between the solid and solution, includingcorrection of bulk susceptibility. As a result, there remains achemical shift difference between the solid and solution up to1–2 ppm depending on which reference is used. 13C chemicalshifts of respective amino acid residues from globular proteins insolution were further related with local conformations definedby a set of the torsion angles of the peptide unit, ϕ and φ, asrevealed by X-ray diffraction [50]. It is, therefore, tempted toextend this approach toward further refinement of the estimatedsecondary structure of membrane proteins initially obtained by

Fig. 3. Schematic representation of the distinction of 13C chemical shiftsbetween the α-helix and loops for Ala Cβ (A) and Val C=O (B) carbons of bR.The random coil peaks for both are located at the boundary between the 13CNMR peaks of the α-helix and loops.
Table 2Correction of 13C chemical shifts primarily referenced to glycine or adamatane(italic) and then secondarily referenced to TMS or DSS (ppm)
Standardreference
Primary reference (with referenced to therespective standard reference)
Chemical-shiftcorrection a
GlycineC_O
Adamantanelow field
Adamantanehigh field
TMS neat 176.03 38.04 29.00 0TMS neat 38.5 29.5 −0.5TMS 1%CDCl3 37.8 28.8 +0.2DSS solid 38.1 29.1 −0.15% D2O 40.4 31. 4 −2.41% D2O 40.6 31.5 −2.5a Chemical shifts were calibrated by the peak position expressed by italic as
the primary reference. Chemical shift correction should be made to compare thedata based on different reference system each other, after adding or subtracting“the chemical shift correction”.
3149H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
the solid-state 13C chemical shifts to determine the torsionangles based on such databases from solution NMR. It iscautioned, however, that the 13C chemical shifts of C=O andAla Cβ are not only varied with the abovementioned torsionangles but also with the effect of slow fluctuation motion[5,6,12–14,39], if any, and the manner of hydrogen bondinteractions [51,52], respectively.
Indeed, it should be noted that the 13C chemical shiftdispersions both in globular and membrane proteins are muchpronounced as compared with the reference data available fromthe simple polypeptides [5,6,48,49]. Displacement of the AlaCβ
13C signals of the α-helix conformation to lower field due toslow motions is recognized as the dynamics-dependentdisplacement of 13C chemical shift, as will be discussed later.In addition, we found that the theoretically calculated C=O 13Cchemical shifts as a function of the torsion angles were sub-stantially altered when a hydrogen bond interaction was takeninto account for C=O 13C shielding constant of N-acetyl-N-methyl-L-alanine amide as a model for (Ala)n [51]. For instance,the C=O 13C chemical shift of [1-13C]Val213-bR was resonatedat the lowermost peak position, 177.0 ppm [52], as comparedwith that of (Val)n taking the α-helix conformation in the solid(174.9 ppm) [5,6]. It is notable that this residue is indeedinvolved in formation of a very short hydrogen bond lengthtogether with that of the bifurcated hydrogen bond network, asrevealed by X-ray diffraction.
Instead of the turned structures for globular proteins, the 13CNMR signals of rather flexible loop regions can be distin-guished from those of the α-helical regions by the characteristicboundary peak to separate the loop and α-helical forms: they arelocated either at the lower or higher field of the random coilpeak at the boundary of the α-helical form for Ala Cβ or Val,Gly, Pro and Ala C=O peaks, respectively, as shown in Fig. 3.The random coil peak of Ala Cβ peak at 16.88 ppm (referencedto glycine COOH at 176.03 ppm from TMS) is located at thelowermost boundary of the α-helix peak [5,6], while the C=O13C peaks of Gly, Val, Ala, etc. are resonated at the highermostboundary of the α-helix form close to random coil peak (at171.7, 173.6, 174.4, and 175.2 ppm for Gly, Val, Pro, and Alaresidue, respectively) [5,6,16,52].
2.3. Dynamics-dependent 13C chemical shifts
The 13C NMR peaks for the α-helices are widely spreadfor membrane proteins, as compared with the data shown inTable 1. A possibility of the αII-helix form in bR was initiallyproposed by Krimm and Dwivedi [53] who claimed that themajority of the transmembrane α-helices takes a tilted αII-typeconformation rather than the usual α-helix conformation (αI-helix), based on the infrared spectral data of (Ala)n in HFIPsolution. However, no such tilted structure was available in thelater 3D structure revealed by cryoelectron microscope or X-raydiffraction studies at lower temperatures [9–11]. An intrachainhydrogen bond distance between residues i and i+4 in thetransmembrane peptide of bR turned out to be normal (4.5±0.1Å for αI-helix), as viewed from the 13C–15N interatomicdistance of 15N–H (Ala18) … O=13C (Ala14) in A (6–42)peptide measured by REDOR [54,55]. These findings areconsistent in that the α-helices involved in the transmembranemoieties are of the normal αI-helix, as viewed from theconformation-dependent 13C C=O chemical shift, in spite ofthe predicted αII-helix form by 13C chemical shifts of [3-13C]Ala residues [12–14].
Kimura et al. [45] showed that the Ala Cβ13C chemical
shifts of a variety of [3-13C]Ala-labeled transmembranepeptides of bR were displaced downfield by 0.3–1.2 ppm inDMPC bilayer, depending upon their respective sites, to thepeak positions of αII-helices as compared with those taking αI-helix in HFIP solution and solid, as summarized in Table 3. Thelowermost 13C NMR peak for Ala 160 of E (128–166) peptidein DMPC bilayer (as large as 1.5 ppm at 16.9 ppm), however,may be due to its location outside the bilayer by taking randomcoil form. This kind of the downfield displacement of peaksfrom the transmembrane helices is also in parallel with thatobserved in purple membrane, as shown in Table 1. It is notedthat such downfield displacement of peaks is present only in anenvironment of very flexible lipid bilayer and the extent differsamong the peptide sequence of the transmembrane peptides and

Table 3Ala Cβ
13C chemical shifts of transmembrane peptides of bR in HFIP solution,solid and DMPC bilayer (ppm from TMS)
Transmembranepeptides
In HFIPsolution(αI-helix)
Solid(αI-helix)
In DMPCbilayer(αII-helix)
13C shift differencebetween solid andDMPC bilayer
Ala 14 A (6–42) 15.2 15.8 0.618 A (6–35) 14.7 15.0 15.7 0.739 B (36–71) 15.7 16.9 1.251 B (36–71) 15.0 15.4 16.2 0.853 B (36–71) 15.2 15.3 15.7 0.498 C (72–107) 14.7 15.3 15.6 0.3114 D (102–131) 14.9 15.2 16.3 1.1160 E (128–166) 14.6 15.4 16.9 a 1.5a This peak is ascribed to random coil form, as a result of residue Ala 160 is
located outside the bilayer.
3150 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
also the manner of helix–helix and retinal–helix interactions forbR in a more pronounced manner [45].
It seems, therefore, to be more reasonable to interpretsuch downfield displacement of Ala Cβ peaks of α-helix form(αII-helix) in terms of the dynamics-dependent displacement ofpeaks [5,6,12–14,39], instead of the static picture initiallyproposed by Krimm and Dwivedi [53]. This means that the AlaCβ signals as defined by the αII-helices could be ascribed to atime-averaged conformation in which its torsion angles can bedeviated at ambient temperatures in membranes, caused by thepresence of a low-frequency motion associated with a localanisotropic fluctuation [38]. Further, it was demonstrated thatall the αII-helical
13C chemical shifts of [3-13C]Ala-bR andA160G mutant were displaced downfield when temperaturewas raised from −10 to 40 °C, whereas the corresponding 13Cchemical shifts of the αI-helices were not [31]. It is noted thatsuch downfield shift was most prominent for the C-terminal α-helix, even though this portion is not embedded in lipid bilayerbut is protruding from the membrane surface and is able toundergo fluctuation motions with correlation times in the orderof 10−6 s [16]. It is interesting to note that this kind of the αII-helix form is not present for α-helical polypeptides in HFIP oraqueous solution in which fast overall fluctuation motions arepossible, instead of allowed anisotropic slow motions.
2.4. Dynamics-dependent suppressed peaks
It should be realized that 13C NMR signals are not alwaysfully visible for fully hydrated membrane proteins of centri-fuged pellet of 2D crystal, depending upon the sites of interest[28,30,31,56–58], a variety of labeled amino acid residues suchas [1-13C]Ala, Leu, Phe, Pro, etc. [16,39,58–61], and themanner of sample preparations such as regenerated preparationin lipid bilayer or disrupted 2D crystals [21,59,60], recorded byboth CP-MAS and DD-MAS NMR when fluctuation frequencycould be interfered with frequencies of proton decoupling ormagic angle spinning [7,8]: incoherent frequency of molecularmotions with correlation time in the order of 10−5 s, if any,can interfere with coherent proton decoupling frequency (ca.50 kHz), which results in failure of attempted peak narrowingby high-power proton decoupling, leading to simultaneously
suppressed 13C NMR signals recorded by both CP-MAS andDD-MAS experiments. In a similar manner, incoherent fre-quency of molecular motions with correlation times of 10−4 s, ifany, can interfere with coherent frequency of magic anglespinning (ca. 4 kHz). Accordingly, one should always take intoaccount of a possibility whether or not several 13C NMR signalsfrom concerned 13C-labeled membrane proteins are missingunder the spectral conditions one used. One way to avoid suchinterference from fluctuation motions, therefore, will berecording spectra under modified fluctuation frequency recordedat higher or lower temperature. Alternatively, use of much higherproton decoupling or ultrafast magic angle spinning might behelpful. In the latter, however, extreme care will be necessary toprevent unavoidable heating of samples and also the effect ofpossible dehydration caused by centrifugation effect.
Nevertheless, the observation of such suppressed peaks byfluctuation motions for a variety of membrane proteins providesone an invaluable means to evaluate the presence of slow orintermediate millisecond or microsecond motions which playessential role in their biological activities, provided that 13CNMR spectral data of fully visible condition are available as areference. This is because it is very difficult to evaluate suchmotions by other means, together with the measurements of avariety of spin–lattice and spin–spin relaxation times. There-fore, it is emphasized that 13C NMR measurement using 50-kHz proton decoupling and 4 kHz MAS is very convenientmeans to reveal motional frequency in the order of 105 or104 Hz, respectively.
3. Fully hydrated membrane proteins
3.1. bR in 2D crystals, distorted 2D crystal, lipid bilayersor detergents
bR from PM is packed to form the trimeric unit in thepresence of certain endogenous lipids, which is further assem-bled into hexagonal lattice as a native 2D crystal [62], althoughfunctional unit of photocycle in bR is monomer itself [63]. It isdemonstrated, however, that bR dynamics as viewed from the13C spectral feature could be substantially altered by a choice ofa variety of 13C-labeled amino acid residues as probes[16,57,64]. In particular, 13C NMR spectra were fully visiblefrom [3-13C]Ala-bR in 2D crystal [22], although several 13CNMR signals could be suppressed from the surface areas when[1-13C]Gly-, Ala-, Leu-, Phe- and Trp-labeled bR wereexamined, owing to the presence of conformational fluctuationswith correlation time in the order of 10−4 s interfered withfrequency of magic angle spinning (4 kHz) (Fig. 4) [16,52,60].This is related to a possible conformational fluctuation, in 2Dcrystal, around the Cα–Cβ bond in the side chains of amino acidresidues as expressed by Cα–CβH2X system. Such conforma-tional space allowed for fluctuation could be limited to a verynarrow area for Val or Ile residues with bulky side-chain at Cβ,leading to limited χ1 rotation angle around the Cα–Cβ bond asdemonstrated by Cα–CβHYZ where Y and Z are CH3 and CH3
or CH2CH3, respectively [46]. This is the reason why the 13CNMR signals are fully visible from [1-13C]Val- or Ile-labeled bR

Fig. 5. 13C CP-MAS NMR spectra of [3-13C]Ala-labeled (left traces) and [1-13C]Ala-The 13C NMR peaks of the [3-13C]Ala-labeled transmembrane α-helices forW12L andexcept for the peaks from the C-terminal α-helices protruding from the membrane smutants gave rise to appreciably reduced, featureless peaks, due to modified backbonethe order of 10−4 s, in the absence of specific lipid–protein interactions (right). It is ahelices at the surface are also preferentially suppressed due to the presence of backb
Fig. 4. Comparison of the dynamic state of bR between PM taking 2D crystallinestate andmonomeric form in lipid bilayer. Dynamic states of the transmembraneα-helices, loops and C- or N-terminus can be expressed by their characteristiccorrelation times as derived by the spin–lattice relaxation times, spin–spinrelaxation times under CP-MAS condition, dynamics-dependent suppressedpeaks, and chemical exchange among helices taking slightly different con-formations (see text for the detail) [5]. The cytoplasmic surface structure (complex)between the C-terminal α-helix G′ and C–D/E–F loops are schematicallyindicated by the dotted lines. They are stabilized by formation of salt bridges andmetal-mediated linkages utilizingmono- or divalent cations (see text for the detail).
3151H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
preparations in 2D crystal, but partially suppressed for othertypes of [1-13C]amino acid labeled preparations.
Surprisingly, several Ala Cβ13C NMR signals from the
surface areas (8.7 Å depth), including the lowermost peaks in theloops (Ala 196,160 and 103) and the low-field region of thetransmembrane αII-helices (16–17 ppm) of intact bR from 2Dcrystal (Fig. 2B), were almost completely or partially sup-pressed, respectively, when 2D crystals were disrupted ordisorganized as in W80L and W12L mutants (Fig. 5 (left),caused by the absence of specific lipid–helix interaction, leadingto modified lipid–helix and helix–helix interactions) [60],bacterioopsin (in the absence of retinal) [30], or monomer inregenerated lipid bilayers [59]. At the same time, the carbonyl13C NMR signals from the transmembrane α-helices and loopsof monomeric bR were also almost completely suppressedamong preparations including [1-13C] Gly-, Ala-, Val-labeledbR due to acquisition of fluctuation motions with correlationtimes in the order of 10−4 s [52,59] (Fig. 5, right) (see alsoFig. 4). It is noted that the presence of such disrupted or dis-organized 2D lattice was readily visualized by the increasedrelative proportions of surrounding lipids [asterisked peak at19.7 ppm in Fig. 5 (left)] per protein, together with the remainingbroadened Ala Cβ
13C NMR signals [60]. This is in contrast tothe protein dynamics of ordinary 2D crystalline lattice with thecorrelation times in the order of 10−2 s: fluctuation motions (inthe order of 10−4 s) occur in the transmembrane α-helicesin the monomeric species in regenerated preparation in lipidbilayer in the absence of hexagonal packing [22]. Undoubtedly,
labeled (right traces) W12L (A) and (B) W80L mutants of (C) wild-type bR [60].W80Lmutants are substantially broadened as compared with those of wild type,
urface (left). Further, 13C NMR signals of [1-13C]Ala-labeled W80L and W12Ldynamics as a result of modified helix–helix interaction with correlation times inlso noted that the 13C NMR signals of the [1-13C]Ala-labeled transmembrane α-one dynamics with correlation times in the order of 10−4 s.
3152 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
formation of 2D lattice for bR is prerequisite to be able to recordfully visible 13C NMR signals. Indeed, bR from PM retainsstrongly bound specific polar lipids from H. salinarum which isessential for lattice formation, even if it is reconstituted in lipidbilayers.As a result, it tends to form 2D lattice at low temperature[52,59,65], although the 2D lattice thus formed is again disruptedwhen temperatures were raised to ambient temperature. There-fore, a search for suitable choice of lipid molecules which is ableto stabilize 2D crystal, together with use of appropriate aminoacid residues for 13C labeling, is a very important considerationprior to initiating 13C NMR studies on membrane proteins.
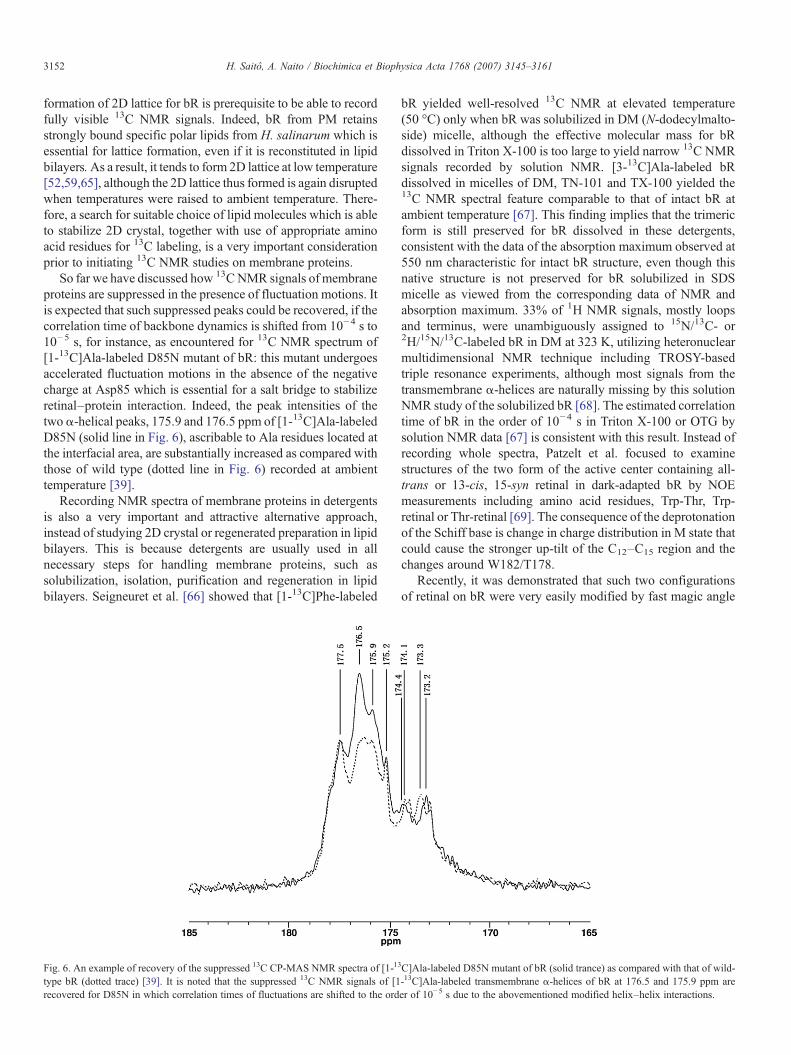
So far we have discussed how 13CNMR signals of membraneproteins are suppressed in the presence of fluctuation motions. Itis expected that such suppressed peaks could be recovered, if thecorrelation time of backbone dynamics is shifted from 10−4 s to10−5 s, for instance, as encountered for 13C NMR spectrum of[1-13C]Ala-labeled D85N mutant of bR: this mutant undergoesaccelerated fluctuation motions in the absence of the negativecharge at Asp85 which is essential for a salt bridge to stabilizeretinal–protein interaction. Indeed, the peak intensities of thetwo α-helical peaks, 175.9 and 176.5 ppm of [1-13C]Ala-labeledD85N (solid line in Fig. 6), ascribable to Ala residues located atthe interfacial area, are substantially increased as compared withthose of wild type (dotted line in Fig. 6) recorded at ambienttemperature [39].
Recording NMR spectra of membrane proteins in detergentsis also a very important and attractive alternative approach,instead of studying 2D crystal or regenerated preparation in lipidbilayers. This is because detergents are usually used in allnecessary steps for handling membrane proteins, such assolubilization, isolation, purification and regeneration in lipidbilayers. Seigneuret et al. [66] showed that [1-13C]Phe-labeled
Fig. 6. An example of recovery of the suppressed 13C CP-MAS NMR spectra of [1-13
type bR (dotted trace) [39]. It is noted that the suppressed 13C NMR signals of [1recovered for D85N in which correlation times of fluctuations are shifted to the ord
bR yielded well-resolved 13C NMR at elevated temperature(50 °C) only when bR was solubilized in DM (N-dodecylmalto-side) micelle, although the effective molecular mass for bRdissolved in Triton X-100 is too large to yield narrow 13C NMRsignals recorded by solution NMR. [3-13C]Ala-labeled bRdissolved in micelles of DM, TN-101 and TX-100 yielded the13C NMR spectral feature comparable to that of intact bR atambient temperature [67]. This finding implies that the trimericform is still preserved for bR dissolved in these detergents,consistent with the data of the absorption maximum observed at550 nm characteristic for intact bR structure, even though thisnative structure is not preserved for bR solubilized in SDSmicelle as viewed from the corresponding data of NMR andabsorption maximum. 33% of 1H NMR signals, mostly loopsand terminus, were unambiguously assigned to 15N/13C- or2H/15N/13C-labeled bR in DM at 323 K, utilizing heteronuclearmultidimensional NMR technique including TROSY-basedtriple resonance experiments, although most signals from thetransmembrane α-helices are naturally missing by this solutionNMR study of the solubilized bR [68]. The estimated correlationtime of bR in the order of 10−4 s in Triton X-100 or OTG bysolution NMR data [67] is consistent with this result. Instead ofrecording whole spectra, Patzelt et al. focused to examinestructures of the two form of the active center containing all-trans or 13-cis, 15-syn retinal in dark-adapted bR by NOEmeasurements including amino acid residues, Trp-Thr, Trp-retinal or Thr-retinal [69]. The consequence of the deprotonationof the Schiff base is change in charge distribution in M state thatcould cause the stronger up-tilt of the C12–C15 region and thechanges around W182/T178.
Recently, it was demonstrated that such two configurationsof retinal on bR were very easily modified by fast magic angle
C]Ala-labeled D85N mutant of bR (solid trance) as compared with that of wild--13C]Ala-labeled transmembrane α-helices of bR at 176.5 and 175.9 ppm areer of 10−5 s due to the abovementioned modified helix–helix interactions.

3153H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
spinning up to 12-kHz rotation corresponding to the samplepressure of 63 bar, as studied by 15N, 13C-labeled bR in 2Dcrystal [33,70]. In fact, backbone conformations of Tyr185located near retinal in bR were strongly perturbed by the retinalconfiguration as disclosed from REDOR-filtered experiment:the populations of the two conformations of bR, correspondingto all-trans and 13-cis configuration of retinal, change to the all-trans populated state for the light adapted state, while the 13-cispopulated state is dominant for the pressure-adapted state [33].Clearly, the two protein conformations are actually present inthe dark-adapted state of bR, because the two Tyr185 signals arenoticed due to the two retinal configurations in the REDOR-filtered experiment [33]. It is of interest to point out that thechange of retinal configuration can be performed by lightillumination to all-trans-rich state and pressure application to13-cis, 15-syn-rich state. On the other hand, only a single peakwas observed for Tyr 26 and 64, because these Tyr residues arelocated far from the retinal [70]. These experimental resultsindicate that retinal isomerization will perturb locally theprotein conformations with retinal through hydrogen bondnetworks. Microscopically, hydrogen bond network aroundretinal would be disrupted or distorted by a constantly increasedpressure induced by fast MAS frequencies, which generated theisomerization of retinal from all-trans to 13-cis state in bR [70].
3.2. Surface structure of bR in 2D crystal
Understanding the surface structure and dynamics ofmembrane proteins at ambient temperature is very importantin view of their biological responses initiated at the cytoplasmicor extracellular surfaces, although they cannot be clarified by X-ray diffraction data due to lack of their interaction. The presenceof the C-terminal α-helix for bR (as helix G′ in Fig. 1) in thecytoplasmic surface was already pointed out in 2.3 which wascharacterized by the specific peak position at 15.91 ppm for[3-13C]Ala-bR and also for the peak positions of Ala Cα andC=O carbons [16]. It is noted that the tilted C-terminal α-helixtoward the direction of the B and F helices is held together bythe loops at the cytoplasmic surface to form the cytoplasmicsurface complex or surface structure, which are particularlystabilized by formation of salt bridges and metal-mediatedlinkages utilizing mono- or divalent cations, as schematicallyillustrated by the dotted lines in Fig. 4. Such surface structures,however, could be naturally altered either by site-directedmutations at the C-terminal α-helix (R227Q) or at the loop(A160G, E166G, and A168G), and by the manner of alteredcation binding, as viewed from the 13C chemical shifts of Ala228 and 233 (C-terminal α-helix), Ala 103 (C–D loop), and Ala160 (E–F loop) [56].
The formed surface structure facilitates efficient protonuptake by preventing unnecessary fluctuations of the helices atthe site for proton uptake, because the cytoplasmic ends of the Band F helices are found to undergo fluctuation motions in theorder of 10−5 s, when such a surface structure is disrupted atlow temperature, low pH or mutants [28,56]. Indeed, the C-terminal α-helix in this case protrudes from the surface withoutinteractions with the cytoplasmic loops, at low temperatures or
in an M-like state of D85N mutant at pH 10 [56]. This view isconsistent with the previously published data for the “protonbinding cluster” consisting of Asp 104, Glu 166 and Glu 224[71–73]. Brown et al. [74] also showed that Arg 227 located atthe end of the helix G plays an important role through a clusterof interacting residues to facilitate the proton transfer from thecytoplasmic domain.
Several negatively charged amino acid residues, Glu or Aspresidues located at the cytoplasmic (CP) surfaces turns out to bealso involved as additional sites for the loosely bound divalentcations, essential for stabilization of extracellular (EC) structureand proton pump activity [75,76]. To clarify the contribution ofthe EC Glu residues to conformation and dynamics of bR, 13CNMR spectra of [3-13C]Ala- or [1-13C]Val-labeled bR mutantsin which extracellular Glu residues were replaced with Gln,E9Q/E194Q/E204Q (3Glu) and E9Q/E74Q/E194Q/E204Q(4Glu) were examined: the spectral lines of [3-13C]Ala-labeledthese mutants were significantly broadened owing to partiallyfailed proton decoupling interfering with the fluctuation fre-quency in the order of 105 Hz [7] and the presence of disruptedor disorganized trimeric structure [57].
The dynamics of bR and the lipid headgroups in oriented PMswere examined at various temperatures and relative humidity(rh) using solid-state NMR spectroscopy [77]. The 15NCP-MASpeak intensities of [15N]Gly-bR from fully hydrated PMs weresuppressed at 293 K, as compared with those of dry membranebut gradually recovered at low temperature or at lower hydration(75%) levels. The suppressed NMR signals due to interferencewith proton decoupling frequency (∼45 kHz) and short spin–spin relaxation time (T2) indicates that the loops of bR havemotional components around this frequency. The motion of thetransmembrane α-helices in bR was largely affected by thefreezing of excess water at low temperature. While between 253and 233 K, where a dynamic phase transition-like change wasobserved in the 31P NMR spectra for the phosphate lipid headgroups, the molecular motion of the loops and the C- and N-termini was slowed, suggesting lipid–loop interaction, althoughprotein–protein interaction between stacks cannot be excluded.
3.3. Sensory rhodopsin
Sensory rhodopsin I (sR I) and phoborhodopsin (pR orsensory rhodopsin II, sR II) are photoreceptors of retinal proteinsfrom Halobacteria active as positive and negative phototaxis,respectively [78]. These two photoreceptors are activated toyield signaling for phototaxis, through their plausible confor-mational changes, after receiving incoming light by tightlycomplexed to their respective cognate transducers consisting oftwo transmembrane helices (TM1 and TM2) of HtrI and HtrII,respectively [79].
[3-13C]Ala-labeled pharaonis ppR, corresponding to pRfrom Halobacteria with 50% homology in egg PC bilayer gaverise to 13C NMR spectra characteristic of the monomeric state:they are very similar to those of [3-13C]Ala-bR in lipid bilayer, inwhich signals from the loops and part of the transmembrane αII-helices are missing in spite of the three Ala residues present, asviewed from the primary structure taking into account of the

Fig. 7. Schematic representation of the manner of interaction of the C-terminal α-helical tip region in ppR (A) and D75N (B) with the transmembraneand cytoplasmic α-helices of pHtrII (1–159) [82]. In the former, the 2:2complex between ppR and pHtrII (1–159) is stabilized by the both types ofhelix–helix interactions between the transmembrane helix G ( ppR) and TM1( pHtrII) and the C-terminal α-helix ( ppR) and cytoplasmic α-helix ( pHtrII), asshown by the elliptical gray [Fig. 6A]. In the case of a signaling state for D75Nmutant, mutual interaction between the two cytoplasmic α-helices could beenhanced for D75N/pHtrII complex (as shown by elliptical gray) instead of theabovementioned interactions, involving the C-terminal tip of the cytoplasmicα-helix and the transmembrane α-helices (elliptical white), as schematicallydemonstrated in Fig. 6B.
3154 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
secondary structure, the essential portion of which beingpreserved between ppR and bR [80]. In particular, the 13CNMR peak of [3-13C]Ala-labeled ppR resonated at 15.9 ppm,ascribable to Ala residues located at the cytoplasmic α-helix asfound previously for bR, is readily distinguished in the DD-MAS NMR but is suppressed in the CP-MAS NMR. Distinctdynamics change, however, is notable for ppR as a result ofcomplex formation with the truncated cognate transducer pHtrII(1–159), as seen from the increased peak intensity of theabovementioned C-terminal α-helix at 15.9 ppm by 13C CP-MASNMR, together with the enhanced spectral resolution fromthe signals of whole area of the transmembrane α-helices as aresult of conformational rearrangement [80]. This observationwas interpreted in terms of the resultant spectral change causedby the increased “effective molecular mass” from the system of 7transmembrane α-helices (ppR alone) to 18 transmembrane α-helices of the complex [2×(7+2)] transmembrane α-helices as2:2 complex results in a change of fluctuation frequency from104–105 to104 Hz [13,80]. Therefore, mutual helix–helixinteractions among the extended TM1 and TM2 helices ofpHtrII (1–159) beyond the surface and the cytoplasmic α-helixof ppR plays an important role for stabilization of the complex,as schematically illustrated as elliptical gray portions wheremutual interactions were enhanced as shown in Fig. 7A [82].Here, both types of the helix–helix interactions arising from thehelix G (ppR) and TM1 (pHtrII) and also the C-terminal α-helix(ppR) and cytoplasmic α-helix (pHtrII) are illustrated forstabilization of the complex as shown by the elliptical gray. Itis also noteworthy that 13C CP-MAS NMR spectrum of thetransmembrane α-helices from [1-13C]Val-labeled ppR in eggPC bilayer is clearly visible, although the corresponding peaksfrom the loops are also partly suppressed.
As discussed already in 3.2, a single mutation at the E–F loopof [3-13C]Ala-bR (A160G and A160V) results in significantlyaltered spectral pattern arising from the modified surface struc-ture (complex) and dynamics of bR including the C-terminalα-helix and the E–F loop. Taking into account of the well-known structural homology between bR and ppR, Kawamuraet al. [81] examined a surface structure of wild-type ppRincluding the C-terminalα-helix and the E–F loop, together withits [3-13C]Ala-labeled A149S and A149V mutants, correspond-ing to A160G and A160V mutants for bR. They showed that analtered surface complex by mutation resulted in destabilizedcomplex as viewed from the 13C NMR spectrum of the surfaceareas, probably because of modified conformation at the cornerof the helix E, in addition to the change of hydropathy. It istherefore concluded that the surface complex of ppR, consistingof the C-terminal α-helix and the E–F loops, is directly involvedin the stabilization of the complex through conformationalstability of the helix E. Further, they studied a possible localdynamics changes of ppR and its D75N mutant as a prerequisiteactivated signaling state as complexed with pHtrII, using 13CNMR of [3-13C]Ala- and [1-13C]Val-labeled preparations [82].They divided the C-terminal α-helical portion of ppR or D75Ninto the two regions: more stable C-terminal stem and moreflexible tip portions. The local fluctuation frequency at the tipportion (108 Hz) of the C-terminal α-helix, however, was appre-
ciably decreased when ppR was bound to pHtrII (104–105 Hz),while it was increased when D75N was bound (106 Hz). D75Nmimics the signaling state because of disrupted salt-bridgeprerequisite for the signal transfer, when it was bound to pHtrII.At the same time, the fluctuation frequency of the cytoplasmicportion of pHtrII is lowered when ppR is replaced by D75N inthe complex with pHtrII. This means that the C-terminal tip isparticipates in binding with the linker region of pHtrII in thedark, but this portion might be released at the signaling statebecause of mutual association of the two transducers in thecytoplasmic regions within the ppR/pHtrII complex, as sche-matically demonstrated in Fig. 7B.
Etzkorn et al. [83] recorded a water-edited 13C, 13C spindiffusion spectrum of uniformly labeled ppR regenerated inlipid bilayer, with the exception of the four dominant residuetype valine, leucine, phenylalanine, and tyrosine, which occurin natural abundance (U[13C, 15N| (V, L, F, Y)]ppR). Theyrecorded the spectra at high field (18.8T or 14.1T) with MAS

3155H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
rates of 8–12.5 kHz and probe temperatures of −13–5 °C. Itwas shown that the reverse labeling of four dominant residuetypes (not labeled), which account for 34% of the entire aminoacid sequence, significantly improved spectral resolution.Further, they selected signals of the mobile water protons byutilizing a polarization transfer characteristics in the spectrawhich are sensitive to the distance between a given nuclear spinin the interior of the molecular complex and the surroundingwater environment. The assigned signals of 98 amino acidresidues were classified as mobile, rigid segments, not labeled,and also water-edited residues. The chemical shift values werefirst referenced to the upfield resonance of adamantane at31.47 ppm (supporting information to [83] ) and chemical shiftcorrection of −2.5 ppm is needed to compare the data with thoseof Table 1, if they are referenced to 1% DSS.
Yamaguchi et al. [84] examined 13C DD-MAS and CP-MASNMR spectra of [3-13C]Ala-labeled pHtrII (1–159) complexedwith ppR in egg PC bilayers. The intense, low-field αII-helical13C DD-MAS NMR peaks of pHtrII (1–159) resonated at 16.6and 16.3 ppm were ascribed to [3-13C]Ala residues located atthe coiled-coil portion protruding from the membrane surface,in view of the conformation-dependent displacement of peaks[5,6,34–36], together with their unique backbone dynamics bywhich they are suppressed in the CP-MAS NMR. It is alsointeresting to note that the low-field α-helical peaks of pHtrII(1–159) are not always fully visible in the absence of ppR,because the fluctuation frequency is close to the frequency ofthe proton decoupling to result in suppressed peaks. The high-field envelope peaks at 15.5 ppm were ascribed to [3-13C]Ala-labeled residues in the transmembrane α-helices of pHtrII (1–159). In contrast, the 13C NMR signals of [1-13C]Val-labeledpHtrII (1–159) were visible in the absence of ppR but werealmost suppressed in the presence of ppR. This means that suchspectral change was interpreted in terms of induced dynamicschange caused by the presence of fluctuation motion withfrequency in the order of 104 Hz which could be interfered withfrequency of magic angle spinning, as encountered formonomeric [1-13C]Val-labeled bR and ppR.
3.4. Rhodopsin
Bovine rhodopsin (Rho) is a typical GPCR active as amammalian photoreceptor protein of 40 kDa covalently linkedto 11-cis retinal through Lys 296. Absorption of a photon by the11-cis retinal causes its isomerization to all-trans retinal, leadingto a conformational change of the protein moiety, including thecytoplasmic surface. Its 3D structure of 2.8 Å has been resolvedby X-ray diffraction [85] and served as an important molecularbasis of understanding of vision as well as signal transductionsfor a variety of GPCRs. 13C and 15N MAS NMR spectra wererecorded on regenerated bovine Rho by the addition of 11-cisretinal to the apoprotein expressed by using suspension culturesof HEK293S cells in defined media containing 6-15N-lysine and2-13C glycine with yield of the protein 1.5–1.8mg/l, followed byreconstitution into DOPC vesicles [86]. The 15N shift of Schiffbase resonated at 156.8 ppm corresponds to an effective Schiffbase counterion distance of greater than 4 Å, consistent with
structural water in the binding site hydrogen bonded with theSchiff base nitrogen and the Glu 113 counterion. The recom-binant baculovirus Sf9 cell line (ATTC CRL-1711) was used toprepare 10 mg of [α,ε-15N]Lys-labeled Rho from two 5-l culturebatches and the protein was reconstituted into bovine retinalipids [87]. The resulting effective Schiff base counteriondistance of greater than 4Å is consistent with structural waterin the binding site hydrogen bonded with Schiff base nitrogenand Glu 113 counter ion.
13C DARR spectra of uniformly 13C labeled Rho wererecorded in order to establish its 13C–13C correlation in theground state [88]. In Rho containing [4′-13C]Tyr and [8,9-13C]retinal, two distinct tyrosine-to-retinal correlations were ob-served. The most intense cross peak arises from a correlationbetween Tyr 268 and the retinal [19-13CH3]retinal, which are4.8 Å apart in the Rho crystal structure. A second cross peakarises from a correlation between Tyr 191 and the [19-13CH3]retinal, which are 5.5 Å apart in the crystal structure. 13C DARRspectra of uniformly 13C labeled Rho in metarhodopsin II (metaII) intermediate, the active intermediate of Rho, were recordedin order to determine how retinal isomerization is coupled toreceptor activation of GPCRs [89]. In view of the retinalbinding pocket in Rho from the crystalline structure, 13C labelswere incorporated into tyrosine, serine, glycine, and threonineof the apoprotein opsin and Rho was regenerated with thepigment 11-cis retinal that have been 13C-labeled at the C12,C14, C15, C19 and C20 carbons. NMR measurements on bothRho and the metarhodopsin II intermediate show how retinalisomerization disrupts helix interactions that lock the receptoroff in the dark. The essential aspects of the isomerizationtrajectory are large rotation of the C20 methyl group towardextracellular loops 2 and a 4- to 5-Å translation of the retinalchromophore toward transmembrane helix 5. 15N NMR spectraof [u-13C, u-15N]Trp-labeled Rho were examined in order toprobe the changes in hydrogen bonding upon Rho activation[90]: the NMR chemical shifts of 15N-labeled histidine showthat His211 is neutral; the unprotonated imidazole nitrogen isnot coordinated to zinc in Rho and becomes more stronglyhydrogen bonded in metarhodopsin II. Mapping of binding sitecontacts in Rho and retinal was also examined by HETCORspectra, to resolve 1HGPCR (Rho)-13Clig (uniformly 13C-labeledligand, 11-cis retinal) signals: Thr 118-C19, Phe 261-C4 andThr 265-C8 [91].
Chemical shifts to all five tryptophan backbone 15N nucleiand partially to their bound protons of α,ε-15N labeled bovineRho were assigned, based on 13C, 15N-REDOR and HETCORexperiments of all possible 13C′i−1 carbonyl/15Ni-tryptophanlabeled amide pairs, and H/D exchange 1H,15N–HSQC ex-periments conducted in solution [92]. It was claimed that theintegrated solution and solid-state NMR approach provideshighly complementary information. Light-induced structuralchanges in Rho in detergent micelles were examined by meansof 19F signals of trifluoroethylthio (TET) [CF3-CH2-S]-labeledcysteine mutants in the cytoplasmic face at amino acid positions67, 140, 245, 248, 311 and 316 [93]. On illumination to formMeta II, upfield changes in chemical shifts were observed for19F labels at positions 67 and 140 and downfield changes for

3156 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
positions 248 and 316 whereas little or no change was observedat positions 245 and 311. Further, 19F nuclear Overhausereffects (NOEs) were measured to assess proximities betweenfluorine labels specifically incorporated into different regions ofthe cytoplasmic face in TET-labeled Rho in dodecylmaltoside.In particular, moderate and strong negative 19F–19F NOE en-hancements were observed for Cys-65–Cys-39 and Cys-139–Cys-251 pairs, respectively, indicating proximity between thesesites.
Only a single sharp peak from the C-terminal sequence wasobserved among 11 lysines of [α-15N]-labeled Rho in dodecylmaltoside as recorded by conventional and TROSY-type HSQCexperiments [94]. This failure to record 15N signals from 10lysines, including 8 in cyotoplasmic loops, is ascribed to thepresence of microsecond to millisecond motions in the back-bones [95]. In a similar manner, differential dynamic propertiesof side-chain versus backbone atoms were examined for[α,ε-15N] tryptophan-labeled Rho: large backbone motionswere found in the inactive dark state, while indole side-chain15N groups of tryptophan exhibit restriction to a single specificconformation [96].
3.5. Photosynthetic antennae and photoreaction centers
The photosynthetic reaction centers (RCs) from Rhodobac-ter sphaeroides is a transmembrane protein complex consistingof three polypeptide chains (L, M and H) and nine cofactors(two bacteriochlorophylls (BChl)2 forming the so-called specialpair (P), two accessory bacteriochlorophylls, two bacteriopheo-phytins (PA and PB), two quinones (QA and QB), and one Fe2+
ion arranged in an almost C2 symmetry [20]). Solid-state NMRstudies for RC from R. sphaeroides have been performed tolocate 13C NMR signals of [4′-13C]Tyr-enriched preparations[97]. The LH2 complex is a peripheral photosynthetic antennapigment proteins utilized to absorb light and to transfer theexcited-state energy to the light-harvesting LH1 complexsurrounding the RC [20]. The crystal structure of the LH2 ofRhodopseudomonas acidophilia [98] shows a ring structure ofnine identical units, each containing an α and a β polypeptide of53 and 41, respectively, which both span the membrane once asα-helices. The two polypeptides bind a total of three chloro-phyll molecules and two carotenoids. The nine heterodimericunits form a hollow cylinder with the α chains forming the innerwall and the β chains the outer walls. Instead of whole complex,Alia et al. [99] recorded 15N and 13C NMR spectra of oneprotomer of [13C6,
15N3]-, [π-15N]-, and [τ-15N]-histidine-labeled LH2 complex solubilized in detergent at 225 K, to gaininsight into charged state of histidines and hydrogen bondingstatus in this complex. Specific 15N labeling confirmed that it isthe τ-nitrogen of histidines which is ligated to Mg2+ ion of B50BChl molecules. Heteronuclear 2D correlation spectra ofuniformly [13C, 15N] labeled LH2 complex were recorded atfrozen state of detergent solubilized preparation [17]. Instead,narrowed peaks of the intrinsic transmembrane LH2 complexwere achieved by extensive and selective biosynthesis preparedfrom [1,4-13C]succinic acid or [2,3-13C]succinic labeled mediaand sequence-specific assignment of peak based on 2D dipolar
correlation experiments [100]. Further, selective chemical shiftassignment of B800 and B850 bacteriochlorophylls (Bchl) inuniformly [13C,15N]-labeled LH2 complexes was performed by2D RFDR correlation spectroscopy [101]. Brief account of 13CNMR studies on photosynthetic antennae and reaction centerswas recently summarized [102].
3.6. Other membrane proteins
E. coli diacylglycerol kinase (DGK) is a membrane-boundenzyme to catalyze conversion of diacylglycerol and MgATP tophosphatic acid and MgADP. It is believed to be assembled intoa trimer to be active as a catalytic unit, each consisting of threetransmembrane α-helices, together with the additional two am-phiphilic α-helices located at the membrane surface [103,104].Surprisingly, very broad featureless 13C CP-MAS NMR spectraof [3-13C]Ala-labeled DGK in POPC bilayers (protein/lipidratio of 1:20) were observed at temperatures of liquid crystallinephase, under which this enzyme exhibits activity, whereas theirspectral resolution was substantially improved at −10 °C takingthe gel phase lipid, just below the phase transition temperatureof POPC bilayers (−5 °C) [105]. 13C NMR signals of [1-13C]Val-labeled DGK were almost completely suppressed at tem-peratures adopting liquid crystalline phase either in POPC orDPPC bilayers. Such a suppression of peaks is obviously causedby interference of fluctuation frequency with frequency ofmagic angle spinning and proton decoupling (104–105 Hz)under physiological condition [105]. The fact that the enzymaticactivity is low under conditions where motion is restricted andhigh when conformational fluctuation can occur suggests thatacquisition of low-frequency backbone motions, on the micro-second to millisecond time scale, may facilitate the efficientenzymatic activity of DGK.
Pleckstrin homology (PH) domain of phospholipase C-δ1(PLC-δ1) as peripheral membrane proteins is not integratedbut bound to membrane surface and regulates the membranelocalization of PLC-δ1 [106] through its high affinity specificinteraction with phosphatidylinositol 4,5-bisphosphate (PIP2)and D-myo-inositol 1,4,5-trisphosphate (IP3). Tuzi et al.[107,108] examined 13C DD-MAS and CP-MAS NMR spectraof [3-13C]Ala-labeled PH domain complexed with PIP2 in PC/PIP2 bilayer with reference to the peak positions available fromthe complex with IP3. They proposed that conformationalchanges originate from a hydrophobic interaction between theamphipathic α-helix located in the β5/β6 loop and thehydrophilic layer of the membrane and contribute to themembrane binding affinity, interdomain interactions andintermolecular interactions of PLC-δ1. Uekama et al. [109]showed that characteristic local conformations in the vicinity ofAla88 and Ala112 induced by the hydrophobic interaction werelost in the surface of the PC/PS/PIP2 vesicle, however. Thus,they proposed that the structure and functional relationshipsamong PLCs and other peripheral membrane proteins that havesimilar PH domain would be affected by the local lipidcomposition of membranes.
So far, we have discussed high-resolution NMR studies onmembrane proteins in 2D crystals, lipid bilayer or detergents. It

3157H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
is worthwhile, however, to discuss NMR features of such mem-brane proteins in 3D crystal, even though 3D crystallization isstill extremely difficult. Bovine heart cytochrome c oxidase is atransmembrane protein which catalyzes the reduction of O2 toH2O, utilizing electrons obtained from reduced cytochrome c[110] and its 3D structure at 2.8 Å resolution is currently avail-able [111]. Tuzi et al. recorded the 13C CP-MASNMR spectra of3D crystalline cytochrome c oxidase which is a membrane pro-tein of 400 kDa containing 70 detergent molecules per protein[112]. The observed 13C NMR signals gave rise to a spectralresolution comparable to that of crystalline lysozyme. It is dif-ficult, however, to assign those signals to individual residues,unless otherwise specifically 13C-labeled proteins were used. Itis emphasized that the 13C NMR signals were not seriouslyoverlapped with the detergent signals, because the observedpeak intensity of the polar heads in detergent BL8SY, is onlyabout 10% of the anticipated values at 1-ms contact time, owingto the presence of rapid tumbling motions in the crystal asdetected by the spin–lattice relaxation times.
Cytochrome bo3 oxidase is also a member of heme-copperoxidase superfamily which includes the abovementionedmitochondrial cytochrome c oxidase [113]. Its X-ray structureis now available at 3.5 Å [114]. Frericks et al. [115] recordedmultidimensional NMR spectra at high field (17.6T) of dialyzedpellet (approximately 25% protein, 5% lipids and 60% buffer)of uniformly 13C,15N-labeled E. coli cytochrome bo3 oxidasesolubilized in n-dodecyl β-D-maltoside. They tried partialassignment of peaks based on multidimensional spectraincluding 2D 13C–13C, 15N–13C, 3D N[i]-C′[i−1]-CX[i−1],13C–13C–13C correlation spectra. They claimed, for instance,that resultant Ala Cα and Cβ chemical shifts in the range of54.1–57.4 and 16.7–20.3 ppm were ascribed to α-helicalportions, although they are substantially deviated from the dataof (Ala)n summarized in Table 1. Again, the same precaution asgiven in [83] is necessary to compare the chemical shiftdatabase, although no explicit description as to the reference tothe chemical shifts was made. The problem arising from use ofsolution NMR database, however, was already discussed in 2.2.
3.7. Membrane proteins in magnetically aligned system
We did not include so far many important solid-state NMRworks of mechanically oriented membrane proteins [3,116,117],analyzed by PISEMA (polarization inversion spin exchange atthe magic angle) experiments [118–120] and PISA (polar indexslant angles) wheel [121,122] which generates patterns ofuniform helical structures and reflects their tilt and polarity in thebilayers. It has been questioned whether PISAwheels would beobserved in membrane proteins, because it is very difficult topredict such wheels from relatively low resolution of the mem-brane protein structures and the high sensitivity of the wheels tolocal structural variations [123]. Li et al. [124] observed 15NPISEMA spectra of two (a 30-residue protein, KdpF, from My-cobacterium tuberculosis and an 82-kDa protein of octamericstate Pv1861 from Mtb) of three proteins in mechanicallyaligned lipid bilayers. They revealed that DAGK (the sameprotein as DGK in 3.5) as the third protein has TM helices
displaying small tilt angles. For KdpF, the tilt of this helixis characterized as 34±3°. Rv1861 has three putative TMhelices and a characteristic PISAwheel is observed indicating atilt of 37±3°. Therefore, PISEMA spectra show that helicalstructure in membrane protein can have a very regular structureresulting in resonance pattern known as PISAwheels.
Park et al. extended 15N PISEMA NMR approach to studyuniformly 15N-labeled and selectively [15N] Ile-labeled che-mokine receptors as GPCRs, CXCR1, in magnetically alignedbicelles [125]. They showed that nearly all residues are struc-tured and immobilized along with the rest of the protein in thephospholipid bilayers. In particular, more than half of the Ileresidues are in transmembrane helices with the others in inter-helical loop or terminal regions. It was not possible to determinehelix tilt or polarity by inspection because signals from PISAwheel are from residues in six different helices. Resonancesfrom residues in loop and terminal regions that are likely to haveirregular structure have chemical shift in the 100–140 ppmrange.
It is worthwhile to point out that use of magnetically orientedvesicle system (MOVS) such as DMPC–melittin or –dynorphinbilayers provides an excellent means to able to study orientedmembrane proteins under fully hydrated condition [126–128].It is also possible to examine NMR behavior of surface portionsprotruding from the membrane surface in more detail.
4. Concluding remarks
We have demonstrated here how conformation and dynamicsof fully hydrated membrane proteins, with emphasis on bR as atypical membrane protein as well as a prototype of GPCRs, canbe revealed by site-directed NMR based on selective isotopelabeling (conducted at moderate field strength of 9.4 T withspinning rate of ca. 4 kHz), with reference to the conformation-dependent 13C chemical shifts and dynamics-dependentsuppressed peaks, respectively. It should be taken into accountthat fully hydrated membrane proteins as centrifuged pellets arenaturally not always rigid in spite of 2D or 3D crystallinesamples. Indeed, they consist of a variety of flexible portions,undergoing fluctuation motions with the correlation times from10−4 to 10−9 s. Therefore, it is essential to record both CP-MASand DD-MAS NMR spectra to cover 13C signals from the wholeareas of membrane proteins, including the surface areas of thetransmembrane α-helices, loops and N- or C-termini.
It turned out that 13C NMR signals are almost fully visible asfar as one records 13C NMR spectra of bR from PM, labeledwith [3-13C]Ala, [1-13C]Val, or Ile. Otherwise, it should bealways borne in mind that 13C NMR spectra are not always fullyvisible at ambient temperature: considerable proportions ofsignals could be suppressed by failure of attempted peak nar-rowing by interference of incoherent fluctuation frequency withcoherent frequencies of either proton decoupling or magic anglespinning (104–105 Hz), depending upon the manner of samplepreparations (2D crystalline sample, disotorted or partiallydisrupted 2D crystal, regenerated preparation in lipid bilayer),types of amino acid residues for 13C labeling, site of interest,etc. It is advised, therefore, to search the best condition to be

3158 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
able to promote formation of the 2D crystals together with asearch for special lipids for this purpose.
Nevertheless, it is demonstrated that this sort of peaksuppression provides one excellent means to detect the presenceof intermediate motions of millisecond or microsecond atcertain portions, which are not easy to determine by othermeans. The use of such low spinning rate for MAS (∼4 kHz)for this purpose is also essential to avoid unnecessary heating ofprobe and prevent dehydration due to centrifugal effect. Further,it is pointed out that care should be exercised as to use ofsolution NMR data which could inherently contain varioussources of errors as discussed in 2.2 when they are applied tosolid-state NMR without any further refinement by a moleculardynamics simulation.
Conformation and dynamics are obviously in conflict witheach other especially when membrane proteins are concerned,even though both characteristics are essential aspects to describethem at ambient temperature. It seems, therefore, to be veryimportant to compromise the dynamic aspect so far accumu-lated by site-directed NMR approach with the forthcomingmultidimensional NMR approach to emphasize the conforma-tional aspect. For this purpose, it is also very important to clarifythe dynamic aspect of bR at higher magnetic field, decouplingand spinning frequency, etc.
References
[1] R. Fu, T.A. Cross, Solid-state nuclear magnetic resonance investigation ofprotein and polypeptide structure, Annu. Rev. Biophys. Biomol. Struct.28 (1999) 235–268.
[2] H.J.M. de Groot, Solid-state NMR spectroscopy applied to membraneproteins, Curr. Opin. Struct. Biol. 10 (2000) 598–600.
[3] S.J. Opella, C. Ma, F.M. Marassi, Nuclear magnetic resonance ofmembrane-associated peptides and proteins, Methods Enzymol. 339 (2001)285–313.
[4] A. Watts, S.K. Straus, S.L. Grage, M. Kamihira, Y.H. Lam, X. Zhao,Membrane protein structure determination using solid state NMR,Methods Mol. Biol. 278 (2004) 403–473.
[5] H. Saitô, Site-directed solid-state NMR on membrane proteins, Annu.Rep. NMR Spectrosc. 57 (2006) 99–175.
[6] H. Saitô, I. Ando, A. Naito, Solid-state NMR for Biopolymers: Principlesand Application, Springer, 2006.
[7] W.P. Rothwell, J.S. Waugh, Transverse relaxation of dipolar coupled spinsystems under RF irradiation: detecting motions in solids, J. Chem. Phys.74 (1981) 2721–2732.
[8] D. Suwelack, W.P. Rothwell, J.S. Waugh, Slow molecular motiondetected in the NMR spectra of rotating solids, J. Chem. Phys. 73 (1980)2559–2569.
[9] N. Grigorieff, T.A. Ceska, K.H. Downing, J.M. Baldwin, R. Henderson,Electron-crystallographic refinement of the structure of bacteriorhodop-sin, J. Mol. Biol. 259 (1996) 393–421.
[10] E. Pebay-Peyroula, G. Rummel, J.P. Rosenbusch, E.M. Landau, X-raystructure of bacteriorhodopsin at 2.5 angstroms from microcrystals grownin lipidic cubic phases, Science 277 (1997) 1676–1681.
[11] H. Luecke, H.T. Richter, J.K. Lanyi, Proton transfer pathways inbacteriorhodopsin at 2.3 angstrom resolution, Science 280 (1998)1934–1937.
[12] H. Saitô, S. Tuzi, S. Yamaguchi, M. Tanio, A. Naito, Conformation andbackbone dynamics of bacteriorhodopsin revealed by 13C NMR,Biochim. Biophys. Acta 1460 (2000) 39–18.
[13] H. Saitô, Dynamic pictures of membrane proteins in two-dimensionalcrystal, lipid bilayer and detergent as revealed by site-directed solid-state13C NMR, Chem. Phys. Lipids 132 (2004) 101–112.
[14] H. Saitô, Y. Kawase, A. Kira, K. Yamamoto, M. Tanio, S. Yamaguchi, A.Naito, S. Tuzi, Surface and dynamic structures of bacteriorhodopsin in2D crystal, distorted or disrupted lattice, revealed by site-directed solid-state 13C NMR, Photochem. Photobiol. 83 (2007) 253–262.
[15] S. Tuzi, A. Naito, H. Saitô, Temperature-dependent conformationalchange of bacteriorhodopsin as studied by solid-state 13C NMR, Eur. J.Biochem. 239 (1996) 294–301.
[16] S. Yamaguchi, S. Tuzi, K. Yonebayashi, A. Naito, R. Needleman, J.K.Lanyi, H. Saitô, Surface dynamics of bacteriorhodopsin as revealed by13C NMR studies on [13C]Ala-labeled proteins: detection of millisecondor microsecond motions in interhelical loops and C-terminal α-helix,J. Biochem. (Tokyo) 129 (2001) 373–382.
[17] T.A. Egorova-Zachernyuk, J. Hollander, N. Fraser, P. Gast, A.J. Hoff, R.Cogdell, H.J.M. de Groot, M. Baldus, Heteronuclear 2D-correlations in auniformly [13C, 15N]labeled membrane-protein complex at ultra-highmagnetic fields, J. Biomol. NMR 19 (2001) 243–253.
[18] J. Pauli, M. Baldus, B. van Rossum, H. de Groot, H. Oschkinat, Backboneand side-chain 13C and 15N signal assignments of the α-spectrin SH3domain by magic angle spinning solid-state NMR at 17.6 tesla,ChemBioChem 2 (2001) 272–281.
[19] A.J. van Gammeren, F.B. Hulsbergen, J.G. Hollander, H.J.M. de Groot,Biosynthetic site-specific 13C labeling of the light-harvesting 2 proteincomplex: a model for solid state NMR structure determination of trans-membrane proteins, J. Biomol. NMR 30 (2004) 267–274.
[20] C. Branden, J. Tooze, Introduction to Protein Structure, Second edition,Garland publishing, New York, N.Y., 1991 1999.
[21] S. Tuzi, S. Yamaguchi, M. Tanio, H. Konishi, S. Inoue, A. Naito, R.Needleman, J.K. Lanyi, H. Saitô, Location of a cation-binding site in theloop between helices F and G of bacteriorhodopsin as studied by 13CNMR, Biophys. J. 76 (1999) 1523–1531.
[22] S. Tuzi, S. Yamaguchi, A. Naito, R. Needleman, J.K. Lanyi, H. Saitô,Conformation and dynamics of [3-13C]Ala-labeled bacteriorhodopsin andbacterioopsin, induced by interactionwith retinal and its analogs, as studiedby 13C nuclear magnetic resonance, Biochemistry 35 (1996) 7520–7527.
[23] S. Tuzi, A.Naito, H. Saitô, 13CNMRstudy on conformation and dynamicsof the transmembrane α-helices, loops, and C-terminus of [3-13C]Ala-labeled bacteriorhodopsin, Biochemistry 33 (1994) 15046–15052.
[24] S. Tuzi, A. Naito, H. Saitô, A high-resolution solid-state 13C-NMR studyon [1-13C]Ala and [3-13C]Ala and [1-13C]Leu and Val-labelledbacteriorhodopsin. Conformation and dynamics of transmembranehelices, loops and termini, Eur. J. Biochem. 218 (1993) 837–844.
[25] S. Tuzi, J. Hasegawa, R. Kawaminami, A. Naito, H. Saitô, Regio-selective detection of dynamic structure of transmembrane α-helices asrevealed from 13C NMR spectra of [3-13C]Ala-labeled bacteriorhodopsinin the presence of Mn2+ ion, Biophys. J. 81 (2001) 425–434.
[26] I. Solomon, Relaxation processes in a system of two spins, Phys. Rev.99 (1955) 559–565.
[27] N. Bloembergen, Proton relaxation times in paramagnetic solutions,J. Chem. Phys. 27 (1957) 572–573.
[28] Y. Kawase, M. Tanio, A. Kira, S. Yamaguchi, S. Tuzi, A. Naito, M.Kataoka, J.K. Lanyi, R. Needleman, H. Saitô, Alteration of conformationand dynamics of bacteriorhodopsin induced by protonation of Asp 85and deprotonation of Schiff base as studied by 13C NMR, Biochemistry39 (2000) 14472–14480.
[29] A. Kira, M. Tanio, S. Tuzi, H. Saitô, Significance of low-frequency localfluctuation motions in the transmembrane B and C α-helices ofbacteriorhodopsin, to facilitate efficient proton uptake frm the cytoplas-mic surface, as revealed by site-directed solid-state 13C NMR, Eur.Biophys. J. 33 (2004) 580–588.
[30] S. Yamaguchi, S. Tuzi, M. Tanio, A. Naito, J.K. Lanyi, R. Needleman, H.Saitô, Irreversible conformational change of bacterio-opsin induced bybinding of retinal during its reconstitution to bacteriorhodopsin, asstudied by 13C NMR, J. Biochem. (Tokyo) 127 (2000) 861–869.
[31] S. Yamaguchi, K. Yonebayashi, H. Konishi, S. Tuzi, A. Naito, J.K. Lanyi,R. Needleman, H. Saitô, Cytoplasmic surface structure of bacteriorho-dopsin consisting of interhelical loops and C-terminal a-helix, modifiedby a variety of environmental factors as studied by 13C NMR, Eur. J.Biochem. 268 (2001) 2218–2228.

3159H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
[32] J.C. Lansing, J.G. Hu, M. Belenky, R.G. Griffin, J. Herzfeld, Solid-stateNMR investigation of the buried X-proline peptide bonds of bacter-iorhodopsin, Biochemistry 42 (2003) 3586–3593.
[33] I. Kawamura, N. Kihara, M. Ohmine, K. Nishimura, S. Tuzi, H. Saitô, A.Naito, Solid-state NMR studies of two backbone conformations atTyr185 as a function of retinal configurations in the dark, light, andpressure adapted bacteriorhodopsins, J. Am. Chem. Soc. 129 (2007)1016–1017.
[34] T. Taki, S. Yamashita, M. Satoh, A. Shibata, T. Yamashita, R. Tabeta, H.Saitô, 13C chemical shifts of solid polypeptides by cross polarization/magic angle spinning (CP/MAS) NMR spectroscopy: conformation-dependent 13C chemical shifts characteristic of α-helix and β-sheetforms, Chem. Lett. (1981) 1803–1806.
[35] H. Saitô, R. Tabeta, A. Shoji, T. Ozaki, I. Ando, Conformationalcharacterization of polypeptides in the solid state as viewed from theconformation-dependent 13C chemical shifts determined by the 13Ccross polarization/magic angle spinning method: oligo(L-alanine), poly(L-alanine), copolymers of L- and D-alanine, and copolymers of L-alaninewith N-methyl- or N-benzyl-L-alanine, Macromolecules 16 (1983)1050–1057.
[36] H. Saitô, Conformation-dependent 13C chemical shifts: a new means ofconformational characterization as obtained by high-resolution solid-stateNMR, Magn. Reson. Chem. 24 (1986) 835–852.
[37] D.-K. Lee, A. Ramamoorthy, Determination of the solid-state conforma-tions of polyalanine using magic-angle spinning NMR spectroscopy,J. Phys. Chem., B 103 (1999) 271–275.
[38] H. Saitô, I. Ando, High-resolution solid-state NMR studies on syntheticand biological macromolecules, Annu. Rep. NMR Spectrosc. 21 (1989)209–290.
[39] H. Saitô, S. Tuzi, M. Tanio, A. Naito, Dynamic aspects of membraneproteins and membrane-associated peptides as revealed by 13C NMR:lessons from bacteriorhodopsin as a intact protein, Annu. Rep. NMRSpectrosc. 47 (2002) 39–108.
[40] H. Saitô, R. Tabeta, T. Asakura, Y. Iwanaga, A. Shoji, T. Ozaki, I. Ando,A high resolution 13C NMR study of silk fibroin in the solid state by thecross polarization-magic angle spinning method. Conformational char-acterization of silk I and II type forms of Bombyx mori fibroin by theconformation-dependent 13C chemical shifts, Macromolecules 17 (1984)1405–1412.
[41] M. Ishida, T. Asakura, M. Yokoi, H. Saitô, Solvent- and mechanical-induced conformational transition of silk fibroins studied by high-resolution solid-state 13C NMR spectroscopy, Macromolecules 23 (1990)88–94.
[42] H. Saitô, R. Tabeta, A. Shoji, T. Ozaki, I. Ando, T. Miyata, A highresolution 13C NMR study of collagen fibrils in solid state studied by thecross polarization-magic angle spinning method. Manifestation ofconformation-dependent 13C chemical shifts and application to confor-mational characterization, Biopolymers 23 (1984) 2279–2297.
[43] H. Saitô, M. Yokoi, A 13C NMR study on collagens in the solid state:hydratio/dehydration-induced conformational change of collagen anddetection of internal motions, J. Biochem. (Tokyo) 111 (1992) 376–382.
[44] D. Huster, J. Schiller, K. Arnold, Comparison of collagen dynamics inarticular cartilage and isolated fibrils by solid-state NMR spectroscopy,Magn. Reson. Med. 48 (2002) 624–632.
[45] S. Kimura, A. Naito, S. Tuzi, H. Saitô, A 13C NMR study on [3-13C]-,[1-13C]Ala- or [1-13C]Val-labeled transmembrane peptides of bacterior-hodopsin in lipid bilayers: insertion, rigid-body motions and local con-formational fluctuations at ambient temperature, Biopolymers 58 (2001)78–88.
[46] C.R. Morcombe, K.W. Zilm, Chemical shift referencing in MAS solidstate NMR, J. Magn. Reson. 162 (2003) 479–486.
[47] R.K. Harris, E.D. Becker, S.M. Cabral de Menezes, P. Granger, R.E.Hoffman, K.W. Zilm, Further conventions for NMR shielding andchemical shifts (IUPAC Recommendations in press), Pure Appl. Chem.
[48] S. Spera, A. Bax, Empirical correlation between protein backboneconformation and Cα and Cβ
13C nuclear magnetic resonance chemicalshifts, J. Am. Chem. Soc. 113 (1991) 5490–5492.
[49] D.S. Wishart, B.D. Sykes, F.M. Richards, Relationship between nuclear
magnetic resonance chemical shift and protein secondary structure,J. Mol. Biol. 222 (1991) 311–333.
[50] G. Cornilescu, F. Delaglio, A. Bax, Protein backbone angle restraintsfrom searching a database for chemical shift and sequence homology,J. Biomol. NMR 13 (1999) 289–302.
[51] I. Ando, H. Saitô, R. Tabeta, A. Shoji, T. Ozaki, Conformation-dependent13C chemical shifts of N-acetyl-N-methyl-L-alanine amide as a modelcompound of poly(L-alanine), Macromolecules 17 (1984) 457–461.
[52] H. Saitô, J. Mikami, S. Yamaguchi, M. Tanio, A. Kira, T. Arakawa, K.Yamamoto, S. Tuzi, Site-directed 13C solid-state NMR studies onmembrane proteins: strategy and goals toward revealing conformationand dynamics as illustrated for 13C-labeled bacteriorhodopsin, Magn.Reson. Chem. 42 (2004) 218–230.
[53] S. Krimm, A.M. Dwivedi, Infrared spectra of the purple membrane: clueto a proton conduction mechanism? Science 216 (1982) 407–408.
[54] S. Kimura, A. Naito, S. Tuzi, H. Saitô, Dynamic structure of trans-membrane α-helical fragments of bacteriorhodopsin in lipid bilayercharacterized by 13C chemical shift tensor and hydrogen bond distance byREDOR NMR, J. Mol. Struct. 602–603 (2002) 125–131.
[55] S. Kimura, A. Naito, H. Saitô, K. Ogawa, A. Shoji, Characterization ofa-helix structures in polypeptides, revealed by 13C=O .. H–15N hydrogenbond lengths determined by 13C REDORNMR, J.Mol. Struct. 562 (2001)197–203.
[56] K. Yonebayashi, S. Yamaguchi, S. Tuzi, H. Saitô, Cytoplasmic surfacestructures of bacteriorhodopsin modifed by site-directed mutations andcation-bindings as revealed by 13C NMR, Eur. Biophys. J. 32 (2003)1–11.
[57] H. Saitô, S. Yamaguchi, K. Ogawa, M. Marquez, C. Sanz, E. Padros,Glutamic acid residue(s) of bacteriorhodopsin at the extracellular surfaceas determinants for conformation and dynamics as revealed by site-directed solid-state 13C NMR, Biophys. J. 86 (2004) 1673–1681.
[58] A. Kira, M. Tanio, S. Tuzi, H. Saitô, Significance of low-frequency localfluctuation motions in the transmembrane B and C α-helices of bac-teriorhodopsin, to facilitate efficient proton uptake from the cytoplasmicsurface, as revealed by site-directed solid-state 13C NMR, Eur. Biophys. J.33 (2004) 580–588.
[59] H. Saitô, K. Yamamoto, S. Tuzi, S. Yamaguchi, Backbone dynamics ofmembrane proteins in lipid bilayers: the effect of two-dimensional arrayformation as revealed by site-directed solid-state 13C NMR studies on[3-13C]Ala- and [1-13C]Val-labeled bacteriorhodopsin, Biochim. Bio-phys. Acta 1616 (2003) 127–136.
[60] H. Saitô, T. Tsuchida, K. Ogawa, T. Arakawa, S. Yamaguchi, S. Tuzi,Residue-specific millisecond to microsecond fluctuations in bacterior-hodopsin induced by disrupted or disorganized two-dimensionalcrystalline lattice, through modified lipid–helix and helix–helix interac-tions, as revealed by 13C NMR, Biochim. Biophys. Acta 1565 (2002)97–106.
[61] S. Tuzi, A. Naito, H. Saitô, Local protein structure and dynamics atkinked transmembrane α-helices of [1-13C]Pro-labeled bacteriorhodopsinas revealed by site-directed solid-state 13C NMR, J. Mol. Struct. 654(2003) 205–214.
[62] C. Sternberg, C. L'Hostis, C.A. Whiteway, A. Watts, The essential role ofspecific Halobacterium halobium polar lipids in 2D-array formation ofbacteriorhodopsin, Biochim. Biophys. Acta. 1108 (1992) 21–30.
[63] N.A. Dencher, K.-D. Kohl, M.P. Heyn, Photochemical cycle and light-dark adaptation of monomeric and aggregated bacteriorhodopsin invarious lipid environments, Biochemistry 22 (1983) 1323–1334.
[64] H. Saitô, S. Yamaguchi, H. Okuda, A. Shiraishi, S. Tuzi, Dynamic aspectof bacteriorhodopsin as a typical membrane protein as revealed by site-directed solid-state 13C NMR, Solid State Nucl. Magn. Reson. 25 (2004)5–14.
[65] K. Yamamoto, S. Tuzi, H. Saitô, I. Kawamura, A. Naito, Conformationand dynamics changes of bacteriorhodopsin and its D85N mutant in theabsence of 2D crystalline lattice as revealed by site-directed 13C NMR,Biochim. Biophys. Acta 1758 (2006) 181–189.
[66] M. Seigneuret, J.M. Neumann, J.L. Rigaud, Detergent delipidation andsolubilization strategies for high-resolution NMR of the membraneprotein bacteriorhodopsin, J. Biol. Chem. 266 (1991) 1066–10069.

3160 H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
[67] M. Tanio, S. Tuzi, S. Yamaguchi, H. Konishi, A. Naito, R. Needleman,J.K. Lanyi, H. Saitô, Evidence of local conformational fluctuations andchanges in bacteriorhodopsin, dependent on lipids, detergents andtrimeric structure, as studied by 13C NMR, Biochim. Biophys. Acta 1375(1998) 84–92.
[68] M. Schubert, M. Kolbe, B. Kessler, D. Oesterhelt, P. Schmieder,Heteronuclear multidimensional spectroscopy of solubilized membraneproteins: resonance assignment of native bacteriorhodopsin, ChemBio-Chem 3 (2002) 1019–1023.
[69] H. Patzelt, B. Simon, A. terLaak, B. Kessler, R. Kuhne, P. Schmieder, D.Oesterhelt, G. Oschkinat, The structures of the active center in dark-adapted bacteriorhodopsin by solution-state NMR spectroscopy, Proc.Natl. Acad. Sci. U. S. A. 99 (2002) 9765–9770.
[70] I. Kawamura, Y. Degawa, S. Yamaguchi, K. Nishimura, S. Tuzi, H. Saitô,A. Naito, Pressure-induced isomerization of retinal on bacteriorhodopsinas disclosed by fast magic angle spinning NMR, Photochem. Photobiol.83 (2007) 346–350.
[71] J. Riesle, D. Oesterhelt, N.A. Dencher, J. Heberle, D38 is an essential partof the proton translocation pathway in bacteriorhodopsin, Biochemistry35 (1996) 6635–6643.
[72] S. Checover, E. Nachliel, N.A. Dencher, M. Gutman, Mechanism ofproton entry into the cytoplasmic section of the proton-conductingchannel of bacteriorhodpsin, Biochemistry 36 (1997) 13919–13928.
[73] S. Checover, Y. Marantz, M. El Nachliel, M. Gutman, J. Pfeiffer, D.Tittor, N.A. Oesterhelt, Dynamics of the proton transfer reaction on thecytoplasmic surface of bacteriorhodopsin, Biochemistry 40 (2001)4281–4292.
[74] L.S. Brown, Y. Yamazaki, A. Maeda, L. Sun, R. Needleman, J.K. Lanyi,The proton transfer in the cytoplasmic domain of bacteriorhodopsin of bRby a cluster of interacting residues, J. Mol. Biol. 239 (1994) 401–414.
[75] C. Sanz, T. Lazarova, F. Seplucre, R. González-Moreno, J.-L.Bourdelande, E. Querol, E. Padros, Opening the Schiff base moiety ofbacteriorhodopsin by mutation of the four extracellular Glu side chains,FEBS Lett. 4556 (1999) 191–195.
[76] C. Sanz, M. Márquez, A. Peralvarez, S. Elouatik, F. Seplucre, E. Querol,T. Lazarova, E. Padros, Contribution of extracellular Glu residues to thestructure and function of bacteriorhodopsin. Presence of specific cation-binding sites, J. Biol. Chem. 276 (2001) 40788–40794.
[77] M. Kamihira, A. Watts, Functionary relevant coupled dynamic profileof bacteriorhodopsin and lipids in purple membranes, Biochemistry 45(2006) 4304–4313.
[78] W.D. Hoff, K.-H. Jung, J.L. Spudich, Molecular mechanism ofphotosignaling by archael sensory rhodopsin, Annu. Rev. Biophys.Biomol. Struct. 26 (1997) 223–258.
[79] X.-N. Zhang, J. Zhu, J.L. Spudich, The specificity of interaction ofarchael transducers with their cognate sensory rhodopsins is determinedby their transmembrane helices, Proc. Natl. Acad. Sci. U. S. A. 96 (1999)857–862.
[80] T. Arakawa, K. Shimono, Y. S.Tuzi, N. Sudo, H. Kamo, Dynamicstructure of pharaonis phoborhodopsin (sensory rhodopsin II) andcomplex with a cognate truncated transducer as revealed by site-directed13C solid-state NMR, FEBS Lett. 536 (2003) 237–240.
[81] I. Kawamura, Y. Ikeda, Y. Sudo, M. Iwamoto, K. Shimono, S.Yamaguchi, S. Tuzi, H. Saitô, N. Kamo, A. Naito, Participation of thesurface structure of Pharaonis phoborhodopsin, ppR and its A149S andA149V mutants, consisting of the C-terminal α-helix and E–F loop, inthe complex-formation with the cognate transducer pHtrII, as revealed bysite-directed 13C solid-state NMR, Photochem. Photobiol. 83 (2007)339–345.
[82] I. Kawamura, H. Yoshida, Y. Ikeda, S. Yamaguchi, S. Tuzi, H. Saitô, N.Kamo, A. Naito, Dynamics change of phoborhodopsin and transducer byactivation: study using D75N mutant of the receptor by site-directedsolid-state 13C NMR manuscript in preparation.
[83] M. Etzkorn, S. Martell, O.C. Andronesi, K. Seidel, M. Engelhard, M.Baldus, Secondary structure, dynamics, and topology of a seven-helixreceptor in native membranes, studied by solid-state NMR spectroscopy,Angew. Chem., Int. Ed. Engl. 45 (2006) 1–5.
[84] K. Yamaguchi, S. Shimono, Y. Sudo, S. Tuzi, A. Naito, N. Kamo, H. Saitô,
Conformation and dynamics of the [3-13C]Ala,-[1-13C]Val-labeledtruncated pharaonis transducer, pHtrII(1–159) as revealed by site-directed13C solid-state NMR: changes due to association with phoborhodopsin(sensory rhodopsin II), Biophys. J. 86 (2004) 3131–3140.
[85] K. Palczewski, T. Kumasaka, T. Hori, C.A. Behnke, H. Motoshima, B.A.Fox, I. Le Trong, D.C. Teller, T. Okada, R.E. Stenkamp, M. Yamamoto,M. Miyano, Crystal structure of rhodopsin: a G protein-coupled receptor,Science 289 (2000) 739–745.
[86] M. Eilers, P.J. Reeves, W. Ying, H.G. Khorana, S.O. Smith, Magic anglespinning NMR of the protonated retinylidene Schiff base nitrogen inrhodopsin: expression of 15N-lysine- and 13C-glycine-labeled opsin in astable cell line, Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 487–492.
[87] A.F.L. Creemers, C.H.W. Klaassen, P.H.M. Bovee-Geurts, R. Kelle, U.Kragl, J. Raap, W.J. de Grip, J. Lugtneburg, H.J.M. de Groot, Solidstate 15N NMR evidence for a complex Schiff base counterion in thevisual G-protein-coupled receptor rhodopsin, Biochemistry 38 (1999)7195–7199.
[88] E. Crocker, A.B. Patel, M. Eilers, S. Jayaraman, E. Getmanova, P.J.Reeves, M. Ziliox, H.G. Khorana, M. Sheves, Dipolar assisted rotationalresonance NMR of tryptophan and tyrosine in rhodopsin, J. Biomol.NMR 29 (2004) 11–20.
[89] A.B. Patel, E. Crocker, M. Eilers, A. Hirshfeld, M. Sheves, S.O. Smith,Coupling of retinal isomerization to the activation of rhodopsin, Proc.Natl. Acad. Sci. U. S. A. 101 (2004) 10048–10053.
[90] A.B. Patel, E. Crocker, P.J. Reeves, E.V. Getmanova, M. Eilers, H.G.Khorana, S.O. Smith, Changes in interhelical hydrogen bonding uponrhodopsin activation, J. Mol. Biol. 347 (2005) 803–812.
[91] S.R. Kiihne, A.F.L. Creemers, W.J. de Grip, P.H.M. Bovee-Geurts, J.Lugtenburg, H.J.M. de Groot, Selective interface detection: mappingbinding site contacts in membrane proteins by NMR spectroscopy, J. Am.Chem. Soc. 127 (2005) 5734–5735.
[92] K. Werner, I. Lehner, K. Dhiman, C. Richter, C. Glaubitz, H. Schwalbe, J.Klein-Seetharman, H.G. Khorana, Combined solid state and solutionNMR studies of α,ε-15N labeled bovine rhodopsin, J. Biomol. NMR 37(2007) 303–312.
[93] J. Klein-Seetharman, E.V. Getmanova, M.C. Loewen, P.J. Reeves, H.G.Khorama, NMR spectroscopy in studies of light-induced structuralchanges in mammalian rhodopsin: applicability of solution 19F NMR,Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 13744–13749.
[94] M.C. Loewen, J. Klein-Seetharman, E.V. Getmanova, P.J. Reeves, H.Schwalbe, H.G. Khorana, Solution 19F nuclear Overhauser effects instructural studies of the cytoplasmic domain of mammalian rhodopsin,Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 4888–4892.
[95] J. Klein-Seetharman, P.J. Reeves, M.C. Loewen, E.V. Getmanova, J.Chung, H. Schwalbe, P.J. Wright, H.G. Khorama, Solution NMRspectroscopy of [α-15N]lysine-labeled rhodopsin: the single peakobserved in both conventional and TROSY-type HSQC spectra is ascribedto Lys-339 in the carboxyl-terminal peptide sequence, Proc. Natl. Acad.Sci. U. S. A. 99 (2002) 3452–3457.
[96] J. Klein-Seetharman, N.V.K. Yanamala, F. Javeed, P.J. Reeves, E.V.Getmanova, M.C. Loewen, H. Schwalbe, H.G. Khorana, Differentialdynamics in the G protein-coupled receptor rhodopsin revealed bysolution NMR, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 3409–3413.
[97] M.R. Fischer, H.J.M. de Groot, J. Raap, C. Winkel, A.J. Hoff, J.Lugtenburg, Carbon-13 magic angle spinning NMR study of the light-induced and temperature-dependent changes in Rhodobacter sphaeroidesR26 reaction centers enriched in [4′-13C]tyrosine, Biochemistry 31 (1992)11038–11049.
[98] G. McDermott, S.M. Prince, A.A. Freer, A.M. Hawthornthwaite-Lawless, M.Z. Papiz, R.J. Cogdell, N.W. Isaacs, Crystal structure of anintegral membrane light-harvesting complex from photosyntheticbacteria, Nature 374 (1995) 517–521.
[99] J. Alia, C. Matysik, M. Soede-Huijbregts, J. Baldus, J. Raap, P.Lugtenburg, H.J. Gast, A.J. van Gorkom, H.J.M. Hoff, Ultrahigh fieldMAS NMR dipolar correlation spectroscopy of the histidine residues inlight-harvesting complex II from photosynthetic bacteria reveals par-tial internal charge transfer in the B850/His complex, J. Am. Chem. Soc.123 (2001) 4803–4809.

3161H. Saitô, A. Naito / Biochimica et Biophysica Acta 1768 (2007) 3145–3161
[100] A.J. van Gammeren, F.B. Hulsbergen, J.G. Hollander, H.J.M de Groot,Biosynthetic site-specific 13C labeling of the light-harvesting 2 proteincomplex: a model for solid state NMR structure determination oftransmembrane proteins, J. Biomol. NMR 30 (2004) 267–274.
[101] A.J. van Gammeren, F. Buda, F.B. Hulsbergen, S. Kiihne, J.G. Hollander,T.A. Egorova-Zachernyuk, N.J. Fraser, R.J. Cogdell, H.J.M. de Groot,Selective chemical shift assignment of B800 and B850 bacteriochlor-ophylls in uniformly [13C,15N]-labeled light-harvesting complexes bysolid-state NMR spectroscopy at ultra-high magnetic field, J. Am. Chem.Soc. 127 (2005) 3213–3219.
[102] H.J.M. de Groot, Photosynthetic antennae and reaction centers, in: G.A.Webb (Ed.), Modern Magnetic Resonance, Springer, 2006, pp. 323–329.
[103] C.R. Sanders II, L. Czerski, O. Vinogradova, P. Badola, D. Song, S.O.Smith, Escherichia coli diacylglycerol kinase is an α-helical polytopicmembrane protein and can spontaneously insert into preformed lipidvesicles, Biochemistry 35 (1996) 8610–8618.
[104] F.W. Lau, X. Chen, J.U. Bowie, Active sites of diacylglycerol kinase fromEscherichia coli are shared between subunits, Biochemistry 38 (1999)5521–5527.
[105] S. Yamaguchi, S. Tuzi, J.U. Bowie, H. Saitô, Secondary structure andbackbone dynamics of Escherichia coli diacylglycerol kinase, as revealedby site-directed solid-state 13C NMR, Biochim. Biophys. Acta 1698(2004) 97–105.
[106] M.E. Cifuentes, L. Honkanen, M. Rebecchi, Proteolytic fragments ofphosphoinositide-specific phospholipase C-δ1. Catalytic and membranebinding properties, J. Biol. Chem. 268 (1993) 11586–11593.
[107] S. Tuzi, N. Uekama, M. Okada, S. Yamaguchi, H. Saitô, H. Yagisawa,Structure and dynamics of the phospholipase C-δ1 pleckstrin homologydomain located at the lipid bilayer surface, J. Biol. Chem. 278 (2003)28019–28025.
[108] S. Tuzi, N. Uekama, M. Okada, H. Yagisawa, Structure of membrane-binding proteins revealed by solid-state NMR, in: G.A. Webb (Ed.),Modern Magnetic Resonance, Springer, 2006, pp. 295–299.
[109] N. Uekama, T. Sugita, M. Okada, H. Yagisawa, S. Tuzi, Phosphatidyl-serine induces functional and structural alterations of the membrane-associated pleckstrin homology domain of phospholipase C-δ1, FEBS J.274 (2007) 177–187.
[110] W.S. Caughey, W.J. Wallace, J.A. Volpe, S. Yoshikawa, in The enzyme,3rd edn. Vol. 13C, Academic Press, New York, 1976, pp. 299–344.
[111] T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K.Shinzawa-Itoh, R. Nakashima, R. Yaono, S. Yoshikawa, Structures ofmetal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A,Science 269 (1995) 1069–1074.
[112] S. Tuzi, K. Shinzawa-Itoh, T. Erata, A. Naito, S. Yoshikawa, H. Saitô, Ahigh-resolution solid-state 13C-NMR study on crystalline bovine heartcytochrome-c oxidase and lysozyme. Dynamic behavior of protein anddetergent in the complex, Eur. J. Biochem. 208 (1992) 713–720.
[113] J.A. Garcia-Horsman, B. Barquera, J. Rumbley, J. Ma, R.B. Gennis, The
superfamily of heme-copper respiratory oxidases, J. Bacteriol. 176 (1994)5587–5600.
[114] J. Abramson, J. Riistama, G. Larsson, A. Jasaitis, M. Svensson-Ek, L.Laakkonen, A. Puustinen, S. Iwata, M. Wikstrom, The structure of theubiquinol oxidase from Escherichia coli and its ubiquinone binding site,Nat. Struct. Biol. 7 (2000) 910–917.
[115] H.L. Frericks, D.H. Zhou, L.L. Yap, R.B. Gennis, C.M. Rienstra, Magic-angle spinning solid-state NMR of a 144 kDa membrane protein complex:E. coli cytochrome bo3 oxidase, J. Biomol. NMR. 36 (2006) 55–71.
[116] T.A. Cross, Solid-state nuclear magnetic resonance characterization ofgramicidin channel structure, Methods Enzymol. 289 (1997) 672–696.
[117] S.J. Opella, F. Marassi, Structure determination of membrane proteins byNMR spectroscopy, Chem. Rev. 104 (2004) 3587–3606.
[118] C.H. Wu, A. Ramamoorthy, S.J. Opella, High-resolution heteronucleardipolar solid-state NMR spectroscopy, J.Magn. Reson. Sec. A. 109 (1994)270–272.
[119] A. Ramamoorthy, C.H. Wu, S.J. Opella, Experimental aspects ofmultidimensional solid-state NMR correlation spectroscopy, J. Magn.Reson. 140 (1999) 131–140.
[120] A. Ramamoorthy, Y. Wei, D.-K. Lee, PISEMA solid-state NMRspectroscopy, Annu. Rep. NMR Spectrsc. 52 (2004) 1–52.
[121] F.M. Marassi, S.J. Opella, Solid-state NMR index of helical mem-brane protein structure and topology, J. Magn. Reson. 140 (1999)150–155.
[122] J. Wang, J. Denny, C. Tian, S. Kim, Y. Mo, F. Kovacs, Z. Song, K.Nishimura, Z. Gan, R. Fu, J.R. Quine, T.A. Cross, Imaging membraneprotein helical wheels, J. Magn. Reson. 144 (2000) 162–167.
[123] S.K. Straus, W.R. Scott, A. Watts, Assessing the effects of time andspatial averaging in 15N chemical shift/15N–1H dipolar correlation solidstate NMR experiments, J. Biomol. NMR. 26 (2003) 283–295.
[124] C. Li, P. Gao, H. Qin, R. Chase, P.L. Gor'kov, W.W. Brey, T.A. Cross,Uniformly aligned full-length membrane proteins in liquid crystallinebilayers for structural characterization, J. Am. Chem. Soc. 129 (2007)5304–5305.
[125] S.H. Park, S. Prytulla, A.A. De Angelis, J.M. Brown, H. Kiefer, S.J.Opella, High-resolution NMR spectroscopy of GPCR in aligned bicelles,J. Am. Chem. Soc. 128 (2006) 7402–7403.
[126] A. Naito, T. Nagao, K. Norisada, T. Mizuno, S. Tuzi, H. Saitô,Conformation and dynamics of melittin bound to magnetically orientedlipid bilayers by solid-state 31P and 13C NMR spectroscopy, Biophys J.78 (2000) 2405–2417.
[127] A. Naito, T. Nagao, M. Obata, Y. Shindo, M. Okamoto, S. Yokoyama, S.Tuzi, H. Saitô, Dynorphin induced magnetic ordering in lipid bilayers asstudied by 31P NMR spectroscopy, Biochim. Biophys. Acta 1558 (2002)34–44.
[128] A. Naito, S. Toraya, K. Nishimura, Nuclear magnetic resonance oforiented bilayer systems, in: G.A. Webb (Ed.), Modern MagneticResonance, Springer, 2006, pp. 237–243.





























