Embed Size (px)
Citation preview

JPET #237578
1
Title Page
Idiosyncratic, Drug-induced Liver Injury: Is Drug-Cytokine Interaction the
Linchpin?
Robert A. Roth, PhD, Ashley R. Maiuri, PhD, Patricia E. Ganey, PhD
Department of Pharmacology and Toxicology, Michigan State University, East Lansing,
MI, USA
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
2
Running Title Page
Drug/Cytokine-Synergy in Idiosyncratic Liver Injury
Corresponding Author:
Robert Roth
Food Safety and Toxicology Bldg., Room 221
1129 Farm Lane, East Lansing, MI, 48824
517-353-9841
Number of text pages: 23
Number of figures: 3
Number of references: 117
Number of words in the abstract: 196
Number of words in the body of the manuscript: 6,718
Recommended section assignment: Toxicology
Non-standard abbreviations:
Apaf: apoptosis protease-associated factor
APAP: acetaminophen
Bid: Bcl interacting protein
CHOP: CCAAT-enhancer-binding protein homologous protein
ConA: concanavalin A
DOX: doxorubicin
ER: endoplasmic reticulum
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
3
ERK: extracellular signal-regulated kinase
GM-CSF: granulocyte macrophage-colony stimulating factor
HLA: human leukocyte antigen
IDILI: idiosyncratic drug-induced liver injury
iNOS: inducible nitric oxide synthase
IFNγ: interferon-gamma
IRF: interferon regulatory factor
JAK: janus kinase
JNK: c-Jun N-terminal kinase
LPS: lipopolysaccharide
LVX: levofloxacin
MAPK: mitogen-activated protein kinase
MHC: major histocompatibility complex
MPT: mitochondrial permeability transition
NFκB: nuclear factor kappa B
NK: natural killer
NSAID: non-steroidal anti-inflammatory drug
PERK: protein kinase RNA-like ER kinase
ROS: reactive oxygen species
SNP: single nucleotide polymorphism
STAT: signal transducer and activator of transcription
TCDD: 2,3,7,8-tetrachoro-dibenzo-p-dioxin
TLR: toll-like receptor
TNFα: tumor necrosis factor-alpha
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
4
TNFR: TNF receptor
TVX: trovafloxacin
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
5
Abstract
Idiosyncratic, drug-induced liver injury continues to be a human health problem in part because
drugs that cause these reactions are not identified in current preclinical testing and because
progress in prevention is hampered by incomplete knowledge of mechanisms that underlie
these adverse responses. Several hypotheses involving adaptive immune responses,
inflammatory stress, inability to adapt to stress, and multiple, concurrent factors have been
proposed. Still, much remains unknown about how drugs interact with the liver to effect death of
hepatocytes. Evidence supporting hypotheses implicating adaptive or innate immune responses
in afflicted patients has begun to emerge and is bolstered by results obtained in experimental
animal models and in vitro systems. A commonality in adaptive and innate immunity is the
production of cytokines, including interferon-γ (IFNγ). IFNγ initiates cell signaling pathways that
culminate in cell death or inhibition of proliferative repair. Tumor necrosis factor-α (TNFα),
another cytokine prominent in immune responses, can also promote cell death. Furthermore,
TNFα interacts with IFNγ, leading to enhanced cellular responses to each cytokine. In this short
review we propose that the interaction of drugs with these cytokines contributes to idiosyncratic,
drug-induced liver injury, and mechanisms by which this could occur are discussed.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
6
Introduction
Idiosyncratic adverse drug responses occur in a minority of patients during drug therapy. The
liver is a frequent target of such reactions (Gunawan and Kaplowitz, 2007). For example,
trovafloxacin (TVX) is a broad-spectrum antibiotic that was introduced to the US market in 1998.
About a year later, several patients who consumed the drug suffered serious liver injury, leading
to curtailing of its use (Ball et al., 1999). Similarly, the nonsteroidal, anti-inflammatory drug
(NSAID) diclofenac has been associated with rare occurrence of liver injury in patients
(Boelsterli, 2003). Halothane was once a widely used, volatile anesthetic that caused severe
liver injury (“halothane hepatitis”) in approximately 1 in 30,000 patients who were anesthetized
with the drug; it has largely been replaced by other halogenated anesthetics that do not share
its IDILI liability (Ray and Drummond, 1991). These are but a few examples of drugs that cause
idiosyncratic, drug-induced liver injury (IDILI) reactions that remain a public health concern,
pose major challenges in drug development, and have led to the market withdrawal of otherwise
therapeutically effective drugs. Unlike typical (“intrinsic”) toxic responses to xenobiotic agents,
IDILI reactions happen at therapeutic dosing regimens and often occur with inconsistent
temporal patterns in relation to drug exposure (Zimmerman, 2000).
The infrequency with which IDILI reactions occur in humans and animals has rendered them
difficult to study. Importantly, these reactions are not predicted from tests used currently in
preclinical safety evaluation and often are not discovered in clinical trials, since the numbers of
volunteers in clinical trials are too few to reveal rare adverse reactions. The basis for the
reactions is incompletely understood, and consequently several hypotheses to explain them
have emerged. The most longstanding is that drugs with IDILI liability precipitate damaging
adaptive immune responses. Within the last several years, other hypotheses have been
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
7
proposed, among which are the multiple determinant hypothesis, the inflammatory stress
hypothesis and the failure-to-adapt hypothesis. What is known is that IDILI responses are
driven by both sensitivity of the individual patient and characteristics of the drug. Determinants
of individual sensitivity include genetic differences and environmental stressors. Much effort
has been devoted in recent years to identifying factors that contribute to individual sensitivity.
Less is known about the specific events that drive hepatocellular injury during IDILI reactions.
This article offers a short review of the supporting evidence and a perspective about how
immune mediators interact with drug exposure to effect killing of hepatocytes, with a particular
emphasis on the role of the cytokines, tumor necrosis factor-alpha (TNFα) and interferon-
gamma (IFNγ).
Interferon-gamma and its interaction with tumor necrosis factor-alpha cause diverse
cellular effects, including cell death.
Interferon-gamma (IFNγ). IFNγ is a cytokine that exists as a soluble dimer encoded by the
IFNG gene. It is not produced in substantial amounts by hepatic parenchymal cells but is
expressed and secreted by several immune cell types, including CD4+ T-helper cells (ie, Th1
cells), CD8+ cytotoxic T-cells, natural killer (NK) T-cells, NK cells and eosinophils. These cells
can be activated to express and release IFNγ by cytokines such as interleukin (IL)-12, IL-18 and
IL-27 (Trinchieri and Scott, 1995; Okamura et al., 1998; Batten and Ghilardi, 2007). Moreover,
interaction among different types of leukocytes occurs; for example, using CD1 to present
glycolipid stimuli, NKT-cells can activate NK cells to produce IFNγ (Carnaud et al., 1999;
Hayakawa et al., 2001). This stimulation involves IFNγ itself.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
8
The biologic activities of IFNγ result mostly from binding to two transmembrane receptors that
reside on hepatocytes and on certain nonparenchymal cells, including Kupffer cells. Ligation of
IFNγ receptors activates an intracellular signaling pathway involving janus kinase (JAK) and
signal transducer and activator of transcription (STAT). This activation results in the
transcription of dozens of genes, the protein products of which produce a variety of cellular
responses (Schroder et al., 2004). Many of these responses involve immune cells. For
example, IFNγ can increase the activity of antigen-presenting macrophages, promote leukocyte
adhesion required for migration into tissue and enhance NK cell activation. Kupffer cells
activated by IFNγ produce cytokines, including TNFα, which can modulate hepatocellular
function, promote an inflammatory response and participate in cell death (see below). Both
CD8+ and CD4+ T-cells have receptors for IFNγ (Whitmire et al., 2005). IFNγ promotes
differentiation of naïve CD4+ (Th0) cells into Th1 cells and inhibits their differentiation into Th2
cells. Since Th1 cells produce IFNγ, this differentiation can enhance IFNγ secretion and provide
positive feedback to increase Th1 differentiation.
In addition to these activities, IFNγ can inhibit proliferation of many types of cells. Proliferation
of hepatocytes occurs slowly in normal liver but can increase markedly when the liver is
stressed by partial hepatectomy or by exposure to pathogens or toxicants. Expression of IFNγ
receptors increases on hepatocytes when liver injury occurs, and receptor ligation by IFNγ
inhibits hepatocyte proliferation (Volpes et al., 1991; Dong et al., 2007). This occurs through
inhibition of cell cycle progression involving STAT-1 activation of IFNγ-responsive genes. One
result is the expression of IFN regulatory factor-1 (IRF-1), which in turn promotes expression of
p53 (Kano et al., 1999; Sun et al., 2006). STAT-1 and p53 activate the promoter for p21, the
expression of which leads to inhibition of S-phase progression and, consequently, to the
inhibition of cell proliferation (reviewed in Tura et al., 2001 and Horras et al., 2011).
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
9
IFNγ can also induce cell death, including apoptosis of hepatocytes, by mechanisms
independent of p53 (Kano et al., 1997). Mechanisms by which this occurs are not well
understood (reviewed in Horras et al., 2011). According to one proposed mechanism, IFNγ acts
through STAT-1 activation and IRF-1 induction to express inducible nitric oxide synthase
(iNOS); this results in enhanced production of NO, which under conditions of redox stress can
initiate intracellular signaling that culminates in apoptosis. However, other cell death pathways,
some not involving STAT-1, can also be activated by IFNγ, depending on cell type. Finally,
IFNγ can cause expression of Fas ligand (FasL) on cells and thereby contribute to Fas-FasL-
induced cell killing (Boselli et al., 2007). Thus, IFNγ-mediated cell killing may occur by several
different mechanisms (Fig. 1).
Tumor Necrosis Factor-alpha (TNFα). IFNγ often acts through interaction with TNFα. TNFα is
a cytokine that can induce a variety of cellular responses and plays a critical role in liver
physiology. It is produced and released by a variety of immune cell types including, but not
limited to NK cells, macrophages, and T-cells (both CD4+ and CD8+). TNFα signaling can
initiate either hepatocyte proliferation or hepatocyte apoptosis, and an appropriate balance
between these two conditions is critical to preserving homeostasis in the liver (Wullaert et al.,
2007). It binds to and activates two distinct plasma membrane receptors, TNFα receptor 1
(TNFR1, p55) and TNFα receptor 2 (TNFR2, p75). TNFR1 is expressed constitutively in most
cell types, whereas the expression of TNFR2 is restricted mainly to immune cells. TNFR1 is
responsible for initiating most of the biological activities of TNFα (Chen and Goeddel, 2002).
Whether TNFα initiates intracellular signaling for cell survival and proliferation or for apoptosis
depends on the state of the cell (Wajant et al. 2003). To initiate cell survival, TNFα binding to
the extracellular domain of TNFR1 drives receptor trimerization followed by recruitment of a
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
10
complex of adapter proteins, which can result in activation of NFκB. NFkB then translocates to
the nucleus, where it promotes transcription of many genes involved in cell survival and
proliferation and in inhibition of signaling for apoptosis (Wullaert et al. 2007).
When hepatocytes become stressed or damaged, TNFα can lead to activation of cell death
signaling pathways. Activation of TNFR1 can recruit and activate procaspase-8, resulting in
apoptosis via two possible routes. The first involves direct cleavage and activation of the
executioner caspases-3 and 7, which cleave a number of proteins leading to apoptosis. The
second, mitochondrial pathway can also be initiated by caspase-8 and entails cleavage and
activation of the proapoptotic protein, Bcl-interacting protein (Bid). The truncated form of Bid
(tBid) translocates to the outer mitochondrial membrane where it facilitates formation of the
mitochondrial permeability transition (MPT) pore. Formation of the MPT pore allows for release
of cytochrome c into the cytosol where it interacts with apoptosis protease-associated factor
(Apaf1) and procaspase-9, resulting in cleavage and activation of the latter. Activated caspase-9
subsequently cleaves and activates the executioner caspases 3 and 7, which effect apoptosis
(Green 1998, Wullaert et al. 2007).
In addition to the pathways discussed above, TNFα signaling via TNFR1 can result in activation
of the mitogen-activated protein kinases (MAPKs), c-jun N-terminal kinase (JNK) and p38.
Importantly, activation of these MAPKs can promote signaling for either cell survival or
apoptosis depending on their subcellular localization, duration of activation, health state of the
cell and other factors (Cowan and Storey, 2003). For instance, when it is activated transiently,
JNK, in particular, activates transcription factors that promote cell survival, including AP-1 and
NFκB (Hasselblatt et al., 2007). However, when it is activated for a prolonged period of time,
JNK can lead to activation of substrates that promote cell death, including p53. Specifically,
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
11
phosphorylation of p53 by JNK promotes its stabilization and resistance to proteasomal
degradation (Fuchs et al., 1998). Moreover, JNK can lead to activation of the transcription factor
c-MYC, which can give rise to apoptosis under certain conditions (Hoffman and Liebermann,
2008). Finally, persistent activation of JNK can result in a decrease in mitochondrial membrane
potential (by an unknown mechanism) leading to MPT, apoptosome formation and activation of
caspase-3 and thereby bring about apoptosis (Gross et al., 1999).
IFNγ -TNFα Interaction. Importantly, IFNγ and TNFα are sometimes incapable of causing the
responses described above on their own or are required in very large concentrations to do so;
however, a pronounced synergy between IFNγ and TNFα can lead to various responses at
relevant cytokine concentrations. For example, IFNγ and TNFα can synergize with each other
in causing DNA fragmentation and apoptosis in vitro in primary mouse hepatocytes (Morita et
al., 1995). Additionally, it has been suggested that IFNγ can synergize with TNFα and other
inflammatory mediators to induce expression of the iNOS gene; as noted above, in the
presence of redox stress, iNOS induction can lead to production of oxidizing species that
promote hepatocyte apoptosis (Vodovotz et al., 2004; Fig. 1).
Although interaction between IFNγ and TNFα seems to be important in some IDILI models
(addressed below), this interaction is incompletely understood at the molecular level. The IFNγ
-mediated binding of STAT-1 to gamma activation sequences (GASs) in DNA and the binding of
IRF-1 to IFN-activated response elements in DNA were enhanced in a human epithelial cell line
after cotreatment of the cells with IFNγ and TNFα, and this may be due, in part, to increased
expression of IFNγ receptor (IFNγ R) (Robinson et al., 2003). TNFα enhanced IFNγ-stimulated
JAK2 phosphorylation and activation in human sarcoma cells, and tyrosine phosphorylation of
IFNγ R chain 1 was elevated in cells cotreated with IFNγ and TNFα (Han et al., 1999). These
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
12
results suggest that both the expression and activation of IFNγ R can be enhanced by TNFα
coexposure. Conversely, expression of TNFRs can be enhanced by IFNγ (Wang et al., 2006).
Moreover, in human monoblastic Mono-Mac-6 cells stimulated with lipopolysaccharide (LPS),
IFNγ prolonged TNFα expression (Lee and Sullivan, 2001). In microglial cells in vitro, IFNγ and
TNFα cooperated in enhancing expression of iNOS and other prooxidant enzymes (Mir et al.,
2009); the enhanced expression depended on MEK (MAPK kinase) and on extracellular signal-
regulated kinase (ERK) signaling that resulted in the release of TNFα (Mir et al., 2008). Also,
IFNγ activation of the JAK/STAT pathway potentiated TNFα-induced NFkB binding to DNA and
activated IRF-1 needed for iNOS expression. In the AML-12 hepatocyte cell line, cotreatment
with IFNγ and TNFα caused cell cycle arrest that was independent of apoptosis and mediated
by p53 and NO (Brooling et al., 2005). Together, these results suggest that the IFNγ -TNFα
interaction can involve (1) enhanced IFNγ R expression and activation by TNFα exposure; (2)
enhanced TNFR expression and prolonged TNFα expression by IFNγ exposure (Fig. 1); (3)
potentiation by IFNγ of TNFα-induced NFkB binding to DNA and (4) synergistic cell cycle arrest
that is mediated by NO. However, most of these studies were conducted in extrahepatic,
transformed cells so additional study is needed to understand the molecular mechanisms that
underlie IFNγ-TNFα interactions in liver parenchymal and nonparenchymal cells and whether
these interactions become significant only in stressed cells.
IFNγ and TNFα: uniting hypotheses regarding the etiology of IDILI
The Adaptive Immunity Hypothesis of IDILI. The adaptive immunity hypothesis has
remained for decades the most popular of the theories to explain IDILI. The classical thinking
has been that a reactive metabolite of a drug binds to a protein, and the resulting adducted
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
13
protein acts as a hapten that is recognized by and sensitizes the adaptive immune system.
Drug rechallenge or continued drug exposure then precipitates an adaptive immune response
that injures the liver (Fig. 2, upper right). The “pharmacological interaction” hypothesis is a
more recent modification, proposing that a drug might bind directly and reversibly with antigen-
presenting molecules, stimulating a damaging immune response that does not require prior
sensitization to the drug (Wuillemin et al., 2013).
The observation that fever, skin rash and eosinophilia accompany some IDILI reactions has
been taken as evidence for an adaptive immune etiology. However, the most compelling
evidence for adaptive immune system involvement in IDILI has arisen from recent studies in
humans that revealed associations between HLA polymorphisms and cases of IDILI for several
drugs. For example, patients who suffered IDILI from amoxicillin/clavulanate had one or more
HLA single nucleotide polymorphisms (SNPs), suggesting involvement of the adaptive immune
system in the pathogenesis (Lucena et al., 2011). Most of the SNPs occurred in HLA Class II
genes. Importantly, although various HLA SNPs were associated with increased IDILI risk, the
predictive value of the SNPs was very small; this suggests that some other genetic or
environmental factor(s) is needed to precipitate IDILI, even in individuals with the associated
HLA SNPs. Interestingly, a SNP in the gene encoding TNFα was also found in the study by
Lucena et al. (2011) to be significantly associated with amoxicillin/clavulanate IDILI, suggesting
the possibility of a role for inflammatory cytokines. Indeed, the linkage of the TNFα gene to the
HLA-B locus raises the possibility that some IDILI reactions associated with various HLA-B
alleles could be due to linkage with variations in the TNFα gene (Inoko et al., 1987).
The adaptive immune response is complex and interacts with the innate immune system to
effect killing of pathogens and damage to host tissue. Haptens comprising drug metabolite-
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
14
protein adducts are endocytosed and degraded into peptides by antigen-presenting cells, which
can then present specific peptides bound to HLA II molecules to CD4+ T-helper cells (Th cells)
or to CD8+ T-cells that express specific receptors for the peptide-HLA complex. This binding
activates the T-cells, and upon a second, costimulatory signal (provided by CD28, CD80, CD86)
and subsequent autocrine signals, the cells proliferate and differentiate. One antigen-
presenting pathway involving CD8+ T-cells results in differentiation into cytotoxic T-cells, which
can kill pathogen-infected cells by expressing cytotoxic proteins (see below). Another pathway
results in differentiation of Th0 cells into either Th1 cells or Th2 cells. Differentiated Th1 cells
produce several cytokines, a major one of which is IFNγ. Th2 cell differentiation results in a
population of B-cells that produce antibodies. Th2 cells also produce factors such as IL-3, IL-5
and GM-CSF (granulocyte macrophage-colony stimulating factor) that stimulate differentiation of
myeloid precursor cells into eosinophils; these cells produce large amounts of IFNγ in response
to TNFα, IL-12 or IL-4 (Spencer et al., 2009).
Killing of host cells by activated T-cells can occur through several mechanisms. Perforin
expressed by cytotoxic CD8+ T-cells can incorporate into the plasma membranes of stressed
host cells, forming a pore and allowing the passage of granzyme B into the cells, which initiates
caspase-dependent cell death signaling (Fig. 1). Cytotoxic CD8+ T-cells can also express FasL
on their surfaces, which can bind to Fas on target cells to initiate killing through caspase-
dependent apoptotic pathways. Cytotoxic CD8+ T-cells also express IFNγ receptors, the
activation of which promotes CD8+ T-cell expansion as well as cell killing by these mechanisms
(Whitmire et al., 2005). Alternatively, IFNγ produced by CD4+ Th1 or other cells can initiate cell
death signaling by binding to an IFNγ R on the surface of host cells (Fig. 1). Such binding
initiates intracellular signaling involving STAT-1 activation of IFNγ -responsive genes, including
the gene expressing IRF-1, which in turn induces iNOS and other genes that could be involved
in initiation of cell death. IFNγ can also act indirectly by stimulating macrophages to release
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
15
cytotoxic factors such as NO, reactive oxygen species (ROS) and TNFα (Fig. 1); as noted
above, TNFα acts synergistically with IFNγ in hepatocellular killing.
In addition to its ability to stimulate T-cell-mediated killing, IFNγ might influence liver
regeneration by inhibiting hepatocyte proliferation. In a study in partially hepatectomized rats,
IFNγ stimulated MHC II antigen expression on Kupffer cells; the authors speculated that these
cells then present hepatocytes as antigen to Th cells and cytotoxic T-cells, which suppresses
reparative hepatocyte proliferation (Sato et al., 1993).
Despite the longstanding popularity of the adaptive immunity hypothesis, no animal models
have emerged in which substantial liver injury occurs from an adaptive immune response after
sensitization with a drug that has caused IDILI in people (Ng et al., 2012; Metushi et al., 2015b).
Recent animal models have been developed which implicate a role for adaptive immunity in
IDILI responses to the drugs amodiaquine and halothane; however, it is important to note that
the liver injury produced in these models is mild and does not reflect the severity of liver injury
that occurs in human IDILI responses to these drugs. Nevertheless, these models provide some
insight concerning the mechanisms underlying adaptive immune-mediated IDILI responses.
Metushi et al. (2015a) found in mice that depletion of NK cells attenuated the mild liver injury
induced by amodiaquine exposure. When activated, NK cells release IFNγ which is known to
activate signaling pathways that lead to cell death. Additionally, Chakraborty et al. (2015) found
that depletion of CD4+ T cells, which also release IFNγ, protected mice from the delayed onset
of halothane hepatitis. Accordingly, it is possible that IFNγ by itself or in the presence of other
cytokines promotes hepatocellular killing in cases of human IDILI induced by amodiaquine or
halothane.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
16
Insight into which pathways predominate in T-cell-mediated killing of hepatocytes has been
provided by studies by Dr. G. Tiegs and colleagues in an animal model employing concanavalin
A (con A). This plant lectin is a T-cell mitogen that causes T-cell-dependent liver injury in mice.
Antibody-mediated depletion of CD4+ T-cells protected completely against liver injury from con
A, whereas depletion of CD8+ T-cells did not prevent liver injury, suggesting the importance of
Th1 cells (Tiegs et al., 1992; Tiegs, 1997; Cao et al., 1998). NKT cells also appear to contribute
(Erhardt and Tiegs, 2010; Zhang et al., 2013). Con A treatment was associated with
appearance of IFNγ and TNFα in plasma, and inactivation of macrophages or neutralizing either
one of these cytokines prevented con A-induced liver injury. Moreover, liver injury development
correlated with IFNγ activation of STAT-1 and IRF-1 and with TNFα activation of the JNK
pathway (Streetz et al., 2001; Hong et al., 2002). Findings in IL-27 conditional knockout mice
supported the importance of dysregulated production of IFNγ by CD4+ T-cells in con A-induced
liver injury in mice (Zhang et al., 2012). Interestingly, IL-5 derived from NKT cells led to
maturation of eosinophils, which contributed to the liver injury in this model (Louis et al., 2002).
These results suggest that IFNγ is critically important in mediating liver injury from Th1 cell
activation and that it acts synergistically with TNFα produced by Kupffer cells.
Recent studies with 2,3,7,8-tetrachoro-dibenzo-p-dioxin (TCDD) showed that this environmental
contaminant enhances con A-induced liver injury in mice and that both IFNγ and NK cells, which
produce IFNγ, are involved (Fullerton et al., 2013). This result indicates that xenobiotic agents
can interact with Th1 cell-dependent pathways to cause hepatocellular killing by mechanisms
involving IFNγ and IFNγ-producing cells and suggests the possibility that some drugs
associated with IDILI might evoke similar responses.
The paucity of animal models employing IDILI-associated drugs has limited the progress into
understanding the contribution of the adaptive immune system to IDILI and the factors that
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
17
might govern such reactions. Nevirapine used in the treatment of HIV infections has caused
skin and liver reactions in patients. Uetrecht and colleagues developed a model of nevirapine-
induced skin injury in Brown Norway rats that is clearly adaptive immune-mediated (Popovic et
al., 2010). The skin rash that developed depended on CD4+ T-cells, and cells isolated from
rechallenged rats released IFNγ and other cytokines. No liver injury developed in this animal
model; however, if nevirapine-induced liver injury in humans arises from the same mechanism
as the skin rash in rats, then IFNγ could be a player in the IDILI pathogenesis from this drug.
Clearly, more animal models in which liver injury occurs from IDILI-associated drugs by an
adaptive immune-mediated mechanism are needed to understand the importance of IFNγ,
TNFα and other mediators in such reactions.
The Multiple Determinant Hypothesis of IDILI. It has been theorized that some IDILI
reactions result from the intersection of several factors or events (ie, “determinants”) that
together precipitate hepatocellular necrosis (Li, 2002; Ulrich, 2007). Inasmuch as the probability
of a reaction would be the product of the probabilities of each factor/event, and since the
product would usually be very small, this hypothesis could explain why IDILI responses are
typically rare. Important factors are proposed to relate to chemical properties of the drug, drug
exposure and metabolism/bioactivation, and genetic and/or environmental factors that
determine individual susceptibility to injury. Examples of likely genetic factors could include
polymorphisms in drug metabolizing enzymes or transporters that could lead to enhanced
production of a toxic metabolite or altered hepatic accumulation of a drug or its metabolites,
respectively. Other factors could include race/ethnicity, nutrition, pre-existing chronic liver
diseases and differences in intestinal microbiome. For example, recent studies indicate that
alterations in the intestinal microbiome determine sensitivity of animals to liver injury from
numerous hepatotoxiciants (Lv et al., 2014; Chiu et al, 2015; Shen et al., 2015; Dubey et al.,
2015; Tian et al., 2015).
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
18
A murine model of halothane-induced liver injury has been developed based on this hypothesis.
Halothane is metabolized to a reactive metabolite that binds covalently to cellular
macromolecules, and the formation of halothane-protein adducts is thought to be required for
liver injury. Among the known human risk factors for halothane hepatitis are female sex, middle
age and genetic predisposition (Inman and Mushin, 1974; Cousins et al., 1989). Fasting may be
an additional risk factor, since all patients are fasted prior to surgery that requires general
anesthesia. Halothane given to fasted, mature, female Balb/c mice caused pronounced liver
injury (Dugan et al., 2010). Fed mice were less sensitive to the liver injury, male Balb/c mice
and female C57Bl/6 mice were markedly resistant, and immature female Balb/c mice were less
sensitive too. Moreover, isoflurane, which is not extensively metabolized to a reactive
intermediate and does not share the human IDILI liability of halothane, failed to cause liver
injury in the mouse model, indicating that chemical structure of the anesthetic is a determinant
of the hepatotoxic response. These results are consistent with the multiple determinant
hypothesis, inasmuch as the confluence of factors known to increase risk in humans
(femaleness, mature age, halothane structure, genetics) was required to produce halothane
hepatitis in mice.
In this murine model of halothane hepatitis, plasma IFNγ concentration was elevated 10-fold in
halothane-treated females compared with similarly treated male mice or ovariectomized female
mice, which were insensitive to injury (Dugan et al., 2011). IFNγ knockout mice were resistant to
halothane-induced liver injury, indicating the importance of IFNγ in the pathogenesis. Halothane
treatment increased the activation (CD69 expression) of NK cells, and inactivation of NK cells
attenuated both the rise in plasma IFNγ and the liver injury. A recent study also implicated
eosinophils in this model (Proctor et al., 2013); these cells are another potential source of IFNγ.
Interestingly, TNFα concentration in plasma also rose as a result of halothane exposure. These
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
19
results suggested that IFNγ released from NK cells and perhaps eosinophils plays an essential
role in the development of severe halothane-induced hepatotoxicity in mice and raised the
possibility of synergistic interaction between IFNγ and TNFα in the pathogenesis.
The Inflammatory Stress Hypothesis of IDILI. The erratic temporal and dose-response
relationships that characterize idiosyncratic reactions suggest that some event occurring during
the course of therapy precipitates IDILI. If this is true, then the precipitating event must happen
occasionally and irregularly to account for the infrequent and erratic occurrence of these
reactions. Inflammatory cell infiltrates often characterize liver lesions in patients who suffer
IDILI (e.g., see Khouri et al., 1987; Fukano et al., 2000; Murphy et al., 2000). This raised the
possibility that some IDILI reactions might be explained by an episode of modest inflammation
occurring during the course of drug therapy that interacts with some action of the drug to initiate
liver injury. Such inflammatory episodes occur commonly in people and are associated with
various diseases, infections and intestinal translocation of inflammagenic bacterial products
such as endotoxin (ie, lipopolysaccharide, LPS). Indeed, episodes of mild, subclinical
endotoxemia appear to be a normal occurrence in people (reviewed in Roth et al., 1997; Ganey
and Roth, 2001; Ganey et al., 2004).
LPS and other inflammagens bind to “pattern recognition receptors” such as toll-like receptors
(TLRs) on cells of the innate immune system (Fig. 2, upper left). This initiates intracellular
signaling that leads to activation of transcription factors and expression of inflammatory
mediators such as TNFα and IFNγ (Arbour et al., 2000; Beutler, 2000). These mediators are
essential in defense against pathogens, but as noted above they are also capable of altering
homeostasis of host cells. It is well known that modest activation of the innate immune system
by inflammagens such as small doses of LPS can markedly augment hepatotoxic responses to
numerous chemicals, including some drugs (Ganey et al., 2004; Roth and Ganey, 2010).
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
20
Accordingly, when an inflammatory episode of sufficient magnitude occurs during drug therapy,
it could interact with a drug to render an individual susceptible to a hepatotoxic reaction that
would not otherwise occur (i.e., an IDILI response). The episodic and variable nature of
exposure to LPS and other inflammagens and genetic variations (e.g., in TLRs or genes
encoding cytokines) that influence individual responses to inflammagens could explain
individual susceptibility, the infrequency of idiosyncratic reactions and their erratic temporal
relationship to drug exposure.
The inflammatory stress hypothesis led to attempts to determine if liver injury could be produced
in animals by concurrent exposure to an IDILI-associated drug and an otherwise noninjurious
inflammatory episode. Indeed, several drugs that cause human IDILI also caused liver injury in
animals upon cotreatment with a small, nontoxic dose of LPS, whereas drugs without human
IDILI liability failed to synergize with LPS to cause hepatotoxicity (summarized in Deng et al.,
2009 and Shaw et al., 2010). It is of interest that drugs frequently associated with human IDILI
are nonsteroidal anti-inflammatory drugs and antibiotics, ie, drugs used in conditions associated
with inflammation. Drug-LPS interaction models in rodents have now been developed with
chlorpromazine, halothane, ranitidine, diclofenac, sulindac, amiodarone, doxorubicin (DOX) and
TVX (see Deng et al., 2009 and Shaw et al., 2010). In each of these models, liver injury occurs
from drug-LPS cotreatment but not from exposure to either agent alone. Thus, the inflammatory
stress hypothesis has provided the first animal models in which pronounced liver injury occurs
from numerous drugs associated with human IDILI.
An example is the TVX-LPS model of drug-inflammation interaction in mice. A robust
hepatotoxic response occurred when a small (ie, nonhepatotoxic but modestly inflammatory)
dose of LPS was given to mice 3 hr after doses of TVX that were nontoxic by themselves (Shaw
et al., 2007). Interestingly, levofloxacin (LVX), a drug in the same pharmacological class that
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
21
does not share the same propensity for causing IDILI, showed no synergistic interaction with
LPS. Of interest, both IFNγ and TNFα were elevated in the plasma of TVX-LPS cotreated
mice, and either (1) neutralization of TNFα with etanercept or knockout of either TNFR1 or
TNFR2 or (2) knockout of the gene encoding IFNγ provided protection from hepatocellular
necrosis (Shaw et al., 2008; Shaw et al., 2009a; Shaw et al., 2009b). Interestingly, TVX
enhanced LPS-stimulated TNFα production in the RAW 264.7 murine macrophage cell line in
vitro (Poulsen et al., 2014a and Poulsen et al., 2014b), suggesting that TVX can directly
enhance macrophage activation and TNFα release.
Together, these results indicated that both IFNγ and TNFα are critical for the
hepatopathogenesis of LPS-TVX interaction in vivo. Moreover, the liver injury in TVX-LPS
cotreated mice depended on a modest prolongation of TNFα appearance in the plasma by TVX
above that caused by LPS alone (Shaw et al., 2009a). IFNγ knockout reduced plasma TNFα,
and conversely neutralization of TNFα markedly reduced the appearance of IFNγ in plasma,
suggesting that each cytokine amplified the production of the other in a dysregulated cycle
(Shaw et al., 2008).
Replacing LPS administration with TNFα administration in this murine model also led to
hepatotoxic interaction with TVX (Shaw et al., 2009a). Interestingly, TNFα administration alone
caused appearance of IFNγ in plasma, and coadministration of TVX enhanced this response.
These findings emphasize the importance of TNFα in initiating IFNγ production and are
consistent with studies in vitro, since TNFα, by acting through macrophages or other cell types,
can stimulate IFNγ production by cultured NK cells and conversely IFNγ can enhance TNFα
production by macrophages (Berner et al., 2005; Makarenkova et al., 2005; Vila-del Sol et al.,
2008).
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
22
Transcriptomic analysis of the livers of TVX-LPS-cotreated mice revealed selective
enhancement of expression of several genes involved in IFNγ signaling (Shaw et al., 2008).
These genes included IRF-1, which is involved in IFNγ -mediated apoptosis and inhibition of cell
proliferation. In the human HepG2 hepatocyte cell line, TVX enhanced cell killing from exposure
to a combination of IFNγ and TNFα, indicating that the IDILI-associated antibiotic increased the
sensitivity of hepatocytes to killing from this cytokine combination (Maiuri et al., 2016b).
Studies in other inflammatory stress models also point to importance of IFNγ. One study
revealed that DOX-LPS cotreatment synergistically enhanced liver injury in rodents, and this
enhancement depended on IFNγ (Hassan et al., 2008). Ju and colleagues showed that the viral
RNA mimetic and TLR3 agonist, polyinosinic-polycytidylic acid (polyI:C), markedly enhanced
halothane-induced liver injury in mice. This was accompanied by activation of Kupffer cells and
NK cells and upregulation of TNFα expression (Cheng et al., 2009). Although IFNγ was not
evaluated in that study, it is produced by NK cells activated by polyI:C (Zhang et al., 2009), so
that it seems likely that IFNγ interaction with TNFα contributes to injury in that model. Recently,
we have shown that IFNγ synergizes with TNFα in sensitizing hepatocytes in vitro to killing by a
number of drugs that cause IDILI in people (Maiuri et al., 2015 and Maiuri et al., 2016b).
Together, these results suggest that IFNγ -TNFα interactions could be critical to IDILI
pathogenesis from a number of IDILI-associated drugs.
The Failure-to-adapt Hypothesis of IDILI. A majority of human volunteers given a therapeutic
dose of acetaminophen (APAP) daily for two weeks experienced early, modest increases in
serum ALT activity which subsequently subsided toward normal despite continued drug
treatment (Watkins et al., 2006). An interpretation of this result was that APAP causes minor
hepatotoxicity, to which the liver adapts over time, thereby returning to normal. It is possible
that this injury-adaptation phenomenon occurs commonly with many drugs and that people who
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
23
develop IDILI responses to a drug are those whose livers fail to adapt, permitting progression to
fulminant injury (Watkins, 2005). Such adaptation to minor injury has been demonstrated in
mice treated with amodiaquine, a drug that has caused IDILI in humans (Metushi et al., 2015b).
Stimulation of immune cells by TNFα or IFNγ leads to IL-10 expression, which in turn
downregulates TNFα and IFNγ production, thereby preventing their organ-damaging actions.
Failure of this regulation by IL-10 in susceptible patients could result in unrestricted and
damaging cytotoxic actions of cytokines such as TNFα and IFNγ. In this regard, it is of interest
that one study found worse outcomes from IDILI in patients who had IL-10 polymorphisms that
resulted in lower plasma IL-10 concentrations (Pachkoria et al., 2008) and another found an
association between polymorphisms resulting in low plasma IL-10 and diclofenac-induced liver
injury (Aithal et al., 2004). Accordingly, it is possible that in susceptible patients an inability of
IL-10 to control production of cytokines such as TNFα and IFNγ could lead to hepatocellular
death from these cytokines, which would present clinically as “failure to adapt” to their damaging
effects.
Recovery from liver injury typically entails proliferation of hepatocytes. For example, after loss
of liver tissue from partial hepatectomy the organ responds with cell proliferation that restores
liver mass and function. In a phenomenon that has been termed “autoprotection,” repeated,
small doses of APAP given to rodents reduced liver injury when a larger, hepatotoxic dose was
subsequently administered, and cell proliferation stimulated by the APAP pretreatments was
thought to play a major role in the reduced hepatic sensitivity (Dalhoff et al., 2001). Indeed,
tissue repair via cell proliferation has been shown to be critical to halting the progression of
injury for numerous hepatotoxicants, including APAP, and inhibiting cell proliferation results in
injury progression (Chanda et al., 1995; Mehendale, 2005). These observations suggest the
possibility that failure to adapt to modest injury caused by drugs might be due at least in part to
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
24
reduced hepatocellular proliferative ability in susceptible individuals, leading to injury
progression and IDILI rather than adaptation (Fig. 2, bottom right).
As noted above, IFNγ is known to cause cell cycle arrest in hepatocytes in vitro and in vivo and
might therefore play a role in inhibiting cell proliferation during treatment with certain drugs
(Kano et al., 1999; Tura et al., 2001; Brooling et al., 2005; Sun et al., 2006; Dong et al., 2007).
Moreover, numerous drugs that cause human IDILI have been shown to inhibit cell proliferation
in vitro. Examples include diclofenac (Rajabalian et al., 2009), sulindac (Chennamaneni et al.,
2012), TVX (Beggs et al., 2015), halothane (Waxler et al., 1994), DOX (Supino et al., 1997) and
chlorpromazine (Basta-Kaim et al., 2006). Additionally, a series of NSAIDs associated with IDILI
inhibited HepG2 cell proliferation in vitro, whereas an NSAID not associated with IDILI, aspirin,
did not have this effect (Maiuri et al. 2014). This raises the possibility that a drug’s ability to
inhibit proliferative repair could contribute to IDILI. Although speculative, of interest is the
possibility that IFNγ produced during drug exposure might interact synergistically with direct,
antiproliferative effects of a drug to inhibit hepatocellular regeneration, thereby prompting failure
to adapt.
Although these cytokines might prompt adaptation failure by inhibiting hepatocyte proliferation,
an effect on immune cells is also possible. There exists some evidence that hepatic failure to
adapt might be due to a failure of immune tolerance in the liver (reviewed in Dara et al., 2016).
Accordingly, cytokine-induced inhibition of proliferation of lymphocytes that are needed for
immune tolerance could contribute to the initiation of severe IDILI.
Drug Interaction with Cytokine-mediated Cell Death Signaling
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
25
Drugs associated with IDILI synergize with cytokines including IFNγ and TNFα in vitro to kill
hepatocytes (Cosgrove et al., 2009; Zou et al. 2009; Gandhi et al. 2010; Fredriksson et al. 2011;
Lu et al. 2013; Beggs et al. 2014; Maiuri et al. 2015). TNFα augmented the cytotoxicity of
sulindac sulfide in primary rat hepatocytes and in HepG2 cells (Zou et al., 2009). The interaction
between sulindac sulfide and TNFα depended on caspase 3/7 activity and also involved
sulindac sulfide-induced oxidative stress (Zou et al., 2010). TNFα potentiated cytotoxicity of
chlorpromazine in primary mouse hepatocytes via activation of JNK (Gandhi et al., 2010). TVX
synergized with TNFα to cause cell death in HepG2 cells and primary mouse hepatocytes
(Beggs et al., 2015). The TVX/TNFα-induced cytotoxic interaction depended on the MAPKs JNK
and ERK and also on ataxia telangiectasia Rad3-related (ATR), which is activated in response
to replication stress and DNA damage (Beggs et al., 2014, Beggs et al., 2015). Several drugs
associated with IDILI synergized with an inflammagen mixture containing TNFα, IFNγ, IL-1α,
and LPS, causing cytotoxicity in HepG2 cells and primary human hepatocytes (Cosgrove et al.,
2009). Another study demonstrated that diclofenac synergized with TNFα to kill HepG2 cells,
and this depended on caspase activation and JNK activation (Fredriksson et al., 2011).
Additionally, diclofenac dysregulated NFkB signaling, which likely promoted apoptosis by
interfering with its ability to dampen the apoptotic pathway. In a subsequent study, endoplasmic
reticulum (ER) stress sensors protein kinase RNA-like ER kinase (PERK) and CCAAT-
enhancer-binding protein homologous protein (CHOP) were also involved in the cytotoxic
interaction mediated by TNFα in combination with either diclofenac or carbamazepine
(Fredriksson et al., 2014). Maiuri et al. (2015) found that NSAIDs associated with IDILI
synergize with TNFα to kill HepG2 cells, and IFNγ enhanced this interaction. Interestingly, an
NSAID not associated with IDILI, aspirin, did not synergize with cytokines to kill cells. The
cytotoxic interaction between NSAIDs associated with IDILI and the cytokines TNFα and IFNγ
depended on activation of caspases and MAPKs. An interesting observation was that certain
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
26
NSAIDs were more likely to synergize with IFNγ than others, and this propensity was associated
with degree of IDILI liability and the chemical structure of the NSAID (Maiuri et al., 2015).
As noted above, many of the actions of IFNγ are mediated by intracellular activation of STAT-1.
IFNγ phosphorylates STAT-1 at the tyrosine701 site, but maximal activation of STAT-1 as a
transcription factor necessitates additional phosphorylation at serine727. Studies in HepG2
cells revealed that diclofenac stimulates phosphorylation of IFNγ at serine727. This
phosphorylation is mediated in part via JNK and ERK and explains how IFNγ initiates
enhancement of cytotoxicity from diclofenac-TNFα interaction (Maiuri et al., 2015). In contrast,
the propionic acid derivative ibuprofen, which causes IDILI that is of less clinical concern,
ablated STAT-1 phosphorylation in response to IFNγ. This finding could explain the differential
propensities of certain NSAIDs to synergize with IFNγ to enhance cell death. As mentioned
earlier, diclofenac causes ER stress (Fredriksson et al., 2014). The cytotoxic interaction
between diclofenac and TNFα/IFNγ required availability of intracellular Ca++ which is known to
be released from the ER during ER stress (Maiuri et al., 2016).
These in vitro studies demonstrate that a number of drugs associated with IDILI activate various
stress responses in the cell which ultimately lead to a cytotoxic interaction with TNFα and IFNγ.
For instance, TVX caused DNA damage and induced replication stress leading to cytotoxic
synergy with TNFα (Beggs et al., 2015). Sulindac sulfide caused oxidative stress which led to
synergy with TNFα, and diclofenac caused ER stress resulting in synergy with both TNFα and
IFNγ. Moreover, many of these drugs, irrespective of pharmacological class, led to activation of
the MAPKs JNK and ERK, and this MAPK activation was required for synergy with these two
cytokines. Importantly, although drugs associated with IDILI induce cellular stress via different
mechanisms, in the presence of cytokines these diverse stress responses ultimately culminate
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
27
in persistent MAPK activation which is crucial to the drug/cytokine cytotoxic interaction in
hepatocytes (Fig. 3).
Conclusion
IFNγ and TNFα are produced during innate and adaptive immune responses (Fig. 2); each can
enhance production of the other. Enhanced IFNγ production and interaction with TNFα occurs
and plays a critical role in liver injury that develops in animal models based on different
hypotheses about IDILI etiology. In addition, recent studies in vitro are revealing interaction
between IDILI-associated drugs and TNFα/IFNγ that lead to hepatocyte killing (Fig. 3). These
observations suggest that TNFα and IFNγ, perhaps acting synergistically with each other, could
be critical to the pathogenesis of IDILI, irrespective of the mechanism by which a drug increases
their production. IFNγ probably contributes to IDILI pathogenesis by (1) enhancing the
production of cytokines such as TNFα, (2) promoting death of hepatocytes and/or (3) inhibiting
proliferative repair of liver. It seems likely that one or more biological activities and TNFα act in
concert with direct actions of a drug to precipitate IDILI, but more study is needed to understand
how cytokines interplay with drug exposure in producing adverse outcomes.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
28
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Roth R.A., Maiuri A.R., Ganey P.E.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
29
References
Aithal GP, Ramsay L, Daly AK, Sonchit N, Leathart JB, Alexander G, Kenna JG, Caldwell J,
Day CP (2004) Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients
with diclofenac hepatotoxicity. Hepatology. 39:1430-40.
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA
(2000) TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat
Genet. 25:187-91.
Ball P, Mandell L, Niki Y, Tillotson G (1999) Comparative tolerability of the newer
fluoroquinolone antibacterials. Drug Saf. 21:407-421.
Basta-Kaim A, Budziszewska B, Jagla G, Nowak W, Kubera M, Lason W (2006) Inhibitory effect
of antipsychotic drugs on the Con A- and LPS-induced proliferative activity of mouse
splenocytes: a possible mechanism of action. J Physiol Pharmacol. 57: 247-264.
Batten M, Ghilardi N (2007) The biology and therapeutic potential of interleukin 27. J Mol Med
(Berl). 85: 661-672.
Beggs KM, Fullerton AM, Miyakawa K, Ganey PE, Roth RA (2014) Molecular mechanisms of
hepatocellular apoptosis induced by trovafloxacin-tumor necrosis factor-alpha interaction.
Toxicol Sci. 137:91-101.
Beggs KM, Maiuri AR, Fullerton AM, Poulsen KL, Breier AB, Ganey PE, Roth RA (2015)
Trovafloxacin-induced replication stress sensitizes HepG2 cells to tumor necrosis factor-alpha-
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
30
induced cytotoxicity mediated by extracellular signal-regulated kinase and ataxia telangiectasia
and Rad3-related. Toxicology. 331:35-46.
Berner MD, Sura ME, Alves BN, Hunter KW (2005) IFN-gamma primes macrophages for
enhanced TNF-alpha expression in response to stimulatory and non-stimulatory amounts of
microparticulate beta-glucan. Immunol Lett. 98: 115-122.
Beutler B (2000) Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr
Opin Microbiol. 3:23-8.
Boelsterli UA (2003) Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity.
Toxicol Appl Pharmacol. 192:307-22.
Boselli D, Losana G, Bernabei P, Bosisio D, Drysdale P, Kiessling R, Gaston JS, Lammas D,
Casanova JL, Kumararatne DS, Novelli F (2007) IFN-gamma regulates Fas ligand expression in
human CD4+ T lymphocytes and controls their anti-mycobacterial cytotoxic functions. Eur J
Immunol. 37:2196-204.
Brooling JT, Campbell JS, Mitchell C, Yeoh GC, Fausto N (2005) Differential regulation of
rodent hepatocyte and oval cell proliferation by interferon gamma. Hepatology. 41: 906-915.
Cao Q, Batev R, Pang G, Russell A, Clancy R (1998) IL-6, IFN-gamma and TNF-alpha
production by liver-associated T cells and acute liver injury in rats administered concanavalin A.
Immunol Cell Biol. 76: 542-549.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
31
Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, Bendelac A (1999) Cutting
edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK
cells. J Immunol. 163: 4647-4650.
Chakraborty M, Fullerton AM, Semple K, Chea LS, Proctor WR, Bourdi M, Kleiner DE, Zeng X,
Ryan PM, Dagur PK, Berkson JD, Reilly TP, Pohl LR (2015) Drug-induced allergic hepatitis
develops in mice when myeloid-derived suppressor cells are depleted prior to halothane
treatment. Hepatology. 62:546-57.
Chanda S, Mangipudy RS, Warbritton A, Bucci TJ, Mehendale HM (1995) Stimulated hepatic
tissue repair underlies heteroprotection by thioacetamide against acetaminophen-induced
lethality. Hepatology. 21: 477-486.
Chen G, Goeddel DV (2002) TNF-R1 signaling: a beautiful pathway. Science. 296:1634-5.
Cheng L, You Q, Yin H, Holt M, Franklin C, Ju C (2009) Effect of polyI:C cotreatment on
halothane-induced liver injury in mice. Hepatology. 49: 215-226.
Chennamaneni S, Zhong B, Lama R, Su B (2012) COX inhibitors indomethacin and sulindac
derivatives as antiproliferative agents: synthesis, biological evaluation, and mechanism
investigation. Eur J Med Chem. 56: 17-29.
Chiu WC, Huang YL, Chen YL, Peng HC, Liao WH, Chuang HL, Chen JR, Yang SC. (2015)
Synbiotics reduce ethanol-induced hepatic steatosis and inflammation by improving intestinal
permeability and microbiota in rats. Food Funct. 6: 1692-700.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
32
Cosgrove BD, King BM, Hasan MA, Alexopoulos LG, Farazi PA, Hendriks BS, Griffith LG,
Sorger PK, Tidor B, Xu JJ, Lauffenburger DA (2009) Synergistic drug-cytokine induction of
hepatocellular death as an in vitro approach for the study of inflammation-associated
idiosyncratic drug hepatotoxicity. Toxicol Appl Pharmacol. 237:317-30.
Cousins MJ, Plummer JL, Hall PD (1989) Risk factors for halothane hepatitis. Aust N Z J. 59: 5-
14.
Cowan KJ, Storey KB (2003) Mitogen-activated protein kinases: new signaling pathways
functioning in cellular responses to environmental stress. J Exp Biol. 206:1107-15.
Dalhoff K, Laursen H, Bangert K, Poulsen HE, Anderson M, Grunnet N, Tygstrup N (2001)
Autoprotection in acetaminophen intoxication in rats: the role of liver regeneration. Pharmacol
Toxicol. 88: 135-141.
Dara L, Liu ZX, Kaplowitz N (2016) Mechanisms of adaptation and progression in idiosyncratic
drug induced liver injury, clinical implications. Liver Int. 36:158-65.
Deng X, Luyendyk JP, Ganey PE, Roth RA (2009) Inflammatory stress and idiosyncratic
hepatotoxicity: hints from animal models. Pharmacol Rev. 61:262-82.
Dong Z, Zhang J, Sun R, Wei H, Tian Z (2007) Impairment of liver regeneration correlates with
activated hepatic NKT cells in HBV transgenic mice. Hepatology. 45: 1400-1412.
Dubey V, Ghosh AR, Bishayee K, Khuda-Bukhsh AR. (2015) Probiotic Pediococcus
pentosaceus strain GS4 alleviates azoxymethane-induced toxicity in mice. Nutr
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
33
Res. 35: 921-9.
Dugan CM, MacDonald AE, Roth RA, Ganey PE (2010) A mouse model of severe halothane
hepatitis based on human risk factors. J Pharmacol Exp Ther. 333:364-72.
Dugan CM, Fullerton AM, Roth RA, Ganey PE. Natural killer cells mediate severe
liver injury in a murine model of halothane hepatitis. (2011) Toxicol Sci. 120: 507-18.
Erhardt A, Tiegs G (2010) Tolerance induction in response to liver inflammation. Dig Dis. 28: 86-
92.
Fredriksson L, Herpers B, Benedetti G, Matadin Q, Puigvert JC, de Bont H, Dragovic S,
Vermeulen NP, Commandeur JN, Danen E, de Graauw M, van de Water B (2011) Diclofenac
inhibits tumor necrosis factor-α-induced nuclear factor-κB activation causing synergistic
hepatocyte apoptosis. Hepatology. 53:2027-41.
Fredriksson L, Wink S, Herpers B, Benedetti G, Hadi M, de Bont H, Groothuis G, Luijten M,
Danen E, de Graauw M, Meerman J, van de Water B (2014) Drug-induced endoplasmic
reticulum and oxidative stress responses independently sensitize toward TNFα-mediated
hepatotoxicity. Toxicol Sci.140:144-59.
Fuchs SY, Adler V, Pincus MR, Ronai Z (1998) MEKK1/JNK signaling stabilizes and activates
p53. Proc Natl Acad Sci USA. 95:10541-6.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
34
Fukano M, Amano S, Sato J, Yamamoto K, Adachi H, Okabe H, Fujiyama Y, Bamba T (2000)
Subacute hepatic failure associated with a new antidiabetic agent, troglitazone: a case report
with autopsy examination. Hum Pathol. 31:250-3.
Fullerton AM, Roth RA, Ganey PE (2013) Pretreatment with TCDD exacerbates liver injury from
Concanavalin A: critical role for NK cells. Toxicol Sci. 136:72-85.
Gandhi A, Guo T, Ghose R (2010) Role of c-Jun N-terminal kinase (JNK) in regulating tumor
necrosis factor-alpha (TNF-alpha) mediated increase of acetaminophen (APAP) and
chlorpromazine (CPZ) toxicity in murine hepatocytes. J Toxicol Sci. 35:163-73.
Ganey PE, Roth RA (2001) Concurrent inflammation as a determinant of susceptibility to toxicity
from xenobiotic agents. Toxicology. 169:195-208.
Ganey PE, Luyendyk JP, Maddox JF, Roth RA (2004) Adverse hepatic drug reactions:
inflammatory episodes as consequence and contributor. Chem Biol Interact. 150:35-51.
Green DR (1998) Apoptotic pathways: the roads to ruin. Cell. 94: 695-8.
Gross A, McDonnell J, Korsmeyer S (1999) BCL-2 family members and the mitochondria in
apoptosis. Genes Dev. 13, 1899-1911.
Gunawan BK, Kaplowitz N (2007) Mechanisms of drug-induced liver disease. Clin Liver Dis. 11:
459-75.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
35
Han Y, Rogers N, Ransohoff RM (1999) Tumor necrosis factor-alpha signals to the IFN-gamma
receptor complex to increase Stat1alpha activation. J Interferon Cytokine Res. 19: 731-740.
Hassan F, Morikawa A, Islam S, Tumurkhuu G, Dagvadori J, Koide N, Naiki Y, Mori I, Yoshida
T, Yokochi T (2008) Lipopolysaccharide augments the in vivo lethal action of doxorubicin
against mice via hepatice damage. Clin Exp Immunol. 151: 334-340.
Hasselblatt P, Rath M, Komnenovic V, Zatloukal K, Wagner EF (2007) Hepatocyte survival in
acute hepatitis is due to c-Jun/AP-1-dependent expression of inducible nitric oxide synthase.
Proc Natl Acad Sci USA. 104: 17105-10.
Hayakawa Y, Takeda K, Yagita H, Kakuta S, Iwakura Y, Van Kaer L, Saiki I, Okumura K (2001)
Critical contribution of IFN-gamma and NK cells, but not perforin-mediated cytotoxicity, to anti-
metastatic effect of alpha-galactosylceramide. Eur J Immunol. 31: 1720-1727.
Hoffman B, Liebermann DA (2008) Apoptotic signaling by c-MYC. Oncogene 27
Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, Nguyen VA, Gao B (2002)
Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin
Invest. 110: 1503-1513.
Horras CJ, Lamb CL, Mitchell K (2011) Regulation of hepatocyte fate by interferon-γ. Cytokine
Growth Factor Rev. 22: 35-43.
Inman WH, Mushin WW (1974) Jaundice after repeated exposure: an analysis of Reports to the
Committee on Safety of Medicines. Br Med J. 1: 5-10.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
36
Inoko H, Trowsdale J. (1987) Linkage of TNF genes to the HLA-B locus. Nucleic Acids
Res. 15: 8957-62.
Kano A, Haruyama T, Akaike T, Watanabe Y (1999) IRF-1 is an essential mediator in IFN-
gamma-induced cell cycle arrest and apoptosis of primary cultured hepatocytes. Biochem
Biophys Res Commun. 257: 672-677.
Kano A, Watanabe Y, Takeda N, Aizawa S, Akaike T (1997) Analysis of IFN-gamma-induced
cell cycle arrest and cell death in hepatocytes. J Biochem. 121: 677-683.
Khouri MR, Saul SH, Dlugosz AA, Soloway RD (1987) Hepatocanalicular injury associated with
vitamin A derivative etretinate. An idiosyncratic hypersensitivity reaction. Dig Dis Sci. 32: 1207-
11.
Lee JY, Sullivan KE (2001) Gamma interferon and lipopolysaccharide interact at the level of
transcription to induce tumor necrosis factor alpha expression. Infect Immun. 69: 2847-2852.
Li AP (2002) A review of the common properties of drugs with idiosyncratic hepatotoxicity and
the “multiple determinant hypothesis” for the manifestation of idiosyncratic drug toxicity. Chem
Biol Interact. 142: 7-23.
Louis H, Le Moine A, Flamand V, Nagy N, Quertinmont E, Paulart F, Abramowicz D, Le Moine
O, Goldman M, Deviere J (2002) Critical role of interleukin 5 and eosinophils in concanavalin A
induced hepatitis in mice. Gastroenterology. 122: 2001-2010.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
37
Lu J, Miyakawa K, Roth RA, Ganey PE (2013) Tumor necrosis factor-alpha potentiates the
cytotoxicity of amiodarone in Hepa1c1c7 cells: roles of caspase activation and oxidative stress.
Toxicol Sci. 131: 164-78.
Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, Day CP, Ruiz-Cabello F,
Donaldson PT, Stephens C, Pirmohamed M, Romero-Gomez M, Navarro JM, Fontana RJ,
Miller M, Groome M, Bondon-Guitton E, Conforti A, Stricker BH, Carvajal A, Ibanez L, Yue QY,
Eichelbaum M, Floratos A, Pe'er I, Daly MJ, Goldstein DB, Dillon JF, Nelson MR, Watkins PB,
Daly AK (2011) Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by
multiple HLA class I and II alleles. Gastroenterology. 141: 338-47.
Lv LX, Hu XJ, Qian GR, Zhang H, Lu HF, Zheng BW, Jiang L, Li LJ. (2014) Administration of
Lactobacillus salivarius LI01 or Pediococcus pentosaceus LI05 improves acute liver injury
induced by D-galactosamine in rats. Appl Microbiol Biotechnol. 98: 5619-32.
Maiuri AR, Breier AB, Gora LF, Parkins RV, Ganey PE, Roth RA (2015) Cytotoxic Synergy
Between Cytokines and NSAIDs Associated With Idiosyncratic Hepatotoxicity Is Driven by
Mitogen-Activated Protein Kinases. Toxicol Sci. 146: 265-80.
Maiuri AR, Breier AB, Turkus JD, Ganey PE, Roth RA (2016a) Calcium Contributes to the
Cytotoxic Interaction Between Diclofenac and Cytokines. Toxicol Sci. 149: 372-84.
Maiuri AR, Gora L, Parkins R, Ganey PE, Roth RA (2014) NSAIDs synergize with inflammatory
cytokines to kill hepatocytes: Implications in idiosyncratic reactions. The Toxicologist. 138: 316.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
38
Maiuri AR, Wassink B, Turkus JD, Breier A, Lansdell T, Kaur G, Ganey PE, Roth RA (2016b) A
promising, in vitro approach to classify drugs according to their potential to cause idiosyncratic,
drug-induced liver injury. The Toxicologist. 150: 237.
Makarenkova V, Chakrabarti AK, Liberatore JA, Popovic P, Lu G, Watkins S, Vujanovic NL
(2004) Dendritic cells and natural killer cells interact via multiple TNF family molecules. J
Leukoc Biol. 77: 408-413.
Metushi IG, Hayes MA, Uetrecht J (2015a) Treatment of PD-1(-/-) mice with amodiaquine and
anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology. 61:
1332-42.
Metushi IG, Cai P, Dervovic D, Liu F, Lobach A, Nakagawa T, Uetrecht J (2015b) Development
of a novel mouse model of amodiaquine-induced liver injury with a delayed onset. J
Immunotoxicol. 12: 247-60.
Mehendale HM (2005) Tissue repair: an important determinant of final outcome of toxicant-
induced injury. Toxicol Pathol. 33: 41-51.
Mir M, Asensio VJ, Tolosa L, Gou-Fabregas M, Soler RM, Llado J, Olmos G (2009) Tumor
necrosis factor alpha and interferon gamma cooperatively induce oxidative stress and
motoneuron death in rat spinal cord embryonic explants. Neuroscience. 162: 959-971.
Mir M, Tolosa L, Asensio VJ, Llado J, Olmos G (2008) Complementary roles of tumor necrosis
factor alpha and interferon gamma in inducible microglial nitric oxide generation. J
Neuroimmunol. 204: 101-109.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
39
Morita M., Watanabe Y., Akaike T. 1995. Protective Effect of Hepatocyte Growth Factor on
Interferon-gamma-Induced Cytotoxicity in Mouse Hepatocytes. Hepatology. 21, 1585-1593.
Murphy EJ, Davern TJ, Shakil AO, Shick L, Masharani U, Chow H, Freise C, Lee WM, Bass NM
(2000) Troglitazone-induced fulminant hepatic failure. Acute Liver Failure Study Group. Dig Dis
Sci. 45: 549-53.
Ng W, Lobach AR, Zhu X, Chen X, Liu F, Metushi IG, Sharma A, Li J, Cai P, Ip J, Novalen M,
Popovic M, Zhang X, Tanino T, Nakagawa T, Li Y, Uetrecht J (2012) Animal models of
idiosyncratic drug reactions. Adv Pharmacol. 63: 81-135
Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K (1998) Regulation of
interferon-gamma production by IL-12 and IL-18. Curr Opin Immunol. 10: 259-264.
Pachkoria K, Lucena MI, Crespo E, Ruiz-Cabello F, Lopez-Ortega S, Fernandez MA, Romero-
Gomez M, Madrazo A, Durán JA, de Dios AM, Borraz Y, Navarro JM, Andrade RJ (2008)
Analysis of IL-10, IL-4 and TNF-alpha polymorphisms in drug-induced liver injury (DILI) and its
outcome. J Hepatol. 49: 107-14.
Popovic M, Shenton JM, Chen J, Baban A, Tharmanathan T, Mannargudi B, Abdulla D,
Uetrecht JP (2010) Nevirapine hypersensitivity. Handb Exp Pharmacol. 196: 437-451.
Poulsen KL, Olivero-Verbel J, Beggs KM, Ganey PE, Roth RA (2014a) Trovafloxacin enhances
lipopolysaccharide-stimulated production of tumor necrosis factor-α by macrophages: role of the
DNA damage response. J Pharmacol Exp Ther. 350: 164-70.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
40
Poulsen KL, Albee RP, Ganey PE, Roth RA (2014b) Trovafloxacin potentiation of
lipopolysaccharide-induced tumor necrosis factor release from RAW 264.7 cells requires
extracellular signal-regulated kinase and c-Jun N-Terminal Kinase. J Pharmacol Exp Ther. 349:
185-91.
Proctor WR, Chakraborty M, Chea LS, Morrison JC, Berkson JD, Semple K, Bourdi M, Pohl LR
(2013) Eosinophils mediate the pathogenesis of halothane-induced liver injury in mice.
Hepatology. 57: 2026-2036.
Rajabalian S, Meimandi MS, Badinloo M (2009) Diclofenac inhibits proliferation but NGF-
induced differentiation of PC12 cells. Pak J Pharm Sci. 22: 259-262.
Ray DC, Drummond GB (1991) Halothane hepatitis. Br J Anaesth. 67: 84-99.
Robinson CM, Shirey KA, Carlin JM (2003) Synergistic transcriptional activaton of indoleamine
dioxygenase by IFN-gamma and tumor necrosis factor-alpha. J Interferon Cytokine Res. 23:
413-421.
Roth RA, Harkema JR, Pestka JP, Ganey PE (1997) Is exposure to bacterial endotoxin a
determinant of susceptibility to intoxication from xenobiotic agents? Toxicol Appl Pharmacol.
147: 300-11.
Roth RA, Ganey PE (2010) Intrinsic versus idiosyncratic drug-induced hepatotoxicity--two
villains or one? J Pharmacol Exp Ther. 332: 692-7.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
41
Sato Y, Tsukada K, Matsumoto Y, Abo T (1993) Interferon-gamma inhibits liver regeneration by
stimulating major histocompatibility complex class II antigen expression by regeneration liver.
Hepatology. 18: 340-346.
Schroder K, Hertzog PJ, Ravasi T, Hume DA (February 2004). "Interferon-gamma: an overview
of signals, mechanisms and functions". J. Leukoc. Biol. 75 (2): 163–89.
Shaw PJ, Hopfensperger MJ, Ganey PE, Roth RA. Lipopolysaccharide and
trovafloxacin coexposure in mice causes idiosyncrasy-like liver injury dependent
on tumor necrosis factor-alpha. (2007) Toxicol Sci. 100: 259-66.
Shaw PJ, Ditewig AC, Waring JF, Liguori MJ, Blomme EA, Ganey PE, Roth RA (2008)
Coexposure of mice to trovafloxacin and lipopolysaccharide, a model of idiosyncratic
hepatotoxicity, results in a unique gene expression profile and interferon gamma-dependent
liver injury. Toxicol Sci. 107: 270-280.
Shaw PJ, Beggs KM, Sparkenbaugh EM, Dugan CM, Ganey PE, Roth RA. Trovafloxacin
enhances TNF-induced inflammatory stress and cell death signaling and reduces TNF
clearance in a murine model of idiosyncratic hepatotoxicity. (2009a) Toxicol Sci. 111: 288-301.
Shaw PJ, Ganey PE, Roth RA. Tumor necrosis factor alpha is a proximal mediator
of synergistic hepatotoxicity from trovafloxacin/lipopolysaccharide coexposure. (2009b) J
Pharmacol Exp Ther. 328: 62-8.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
42
Shaw PJ, Ganey PE, Roth RA (2010) Idiosyncratic drug-induced liver injury and the role of
inflammatory stress with an emphasis on an animal model of trovafloxacin hepatotoxicity.
Toxicol Sci. 118: 7-18.
Shen TC, Albenberg L, Bittinger K, Chehoud C, Chen YY, Judge CA, Chau L, Ni J, Sheng M,
Lin A, Wilkins BJ, Buza EL, Lewis JD, Daikhin Y, Nissim I, Yudkoff M, Bushman FD, Wu GD.
(2015) Engineering the gut microbiota to treat hyperammonemia. J Clin Invest. 125: 2841-50.
Spencer LA, Szela CT, Perez SA, Kirchhoffer CL, Neves JS, Radke AL, Weller PF (2009)
Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines
that are secreted rapidly and differentially. J Leukoc Biol. 85: 117-123.
Streetz K, Fregien B, Plumpe J, Kubicka S, Sass G, Bischoff SC, Manns MP, Tiegs G,
Trautwein C (2001) Dissection of the intracellular pathways in hepatocytes suggests a role for
Jun kinase and IFN regulatory factor-1 in Con A-induced liver failure. J Immunol. 167: 514-523.
Sun R, Park O, Horiguchi N, Kulkarni S, Jeong WI, Sun HY, Radaeva S, Gao B (2006) STAT1
contributes to dsRNA inhibition of liver regeneration after partial hepatectomy in mice.
Hepatology. 44: 955-966.
Supino R, Casazza AM, Di Marco A (1977) Effect of daunorubicin and Adriamycin on nucleic
acid synthesis of serum stimulated mouse embryo fibroblasts. Tumori. 63: 31-42.
Tian F, Chi F, Wang G, Liu X, Zhang Q, Chen Y, Zhang H, Chen W. (2015) Lactobacillus
rhamnosus CCFM1107 treatment ameliorates alcohol-induced liver injury in a mouse
model of chronic alcohol feeding. J Microbiol. 53: 856-63.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
43
Tiegs G, Hentschel J, Wendel A (1992) A T cell-dependent experimental liver injury in mice
inducible by concanavalin A. J Clin Invest. 90: 196-203.
Tiegs G (1997) Experimental hepatitis and role of cytokines. Acta Gastroenterol Belg. 60: 176-
179.
Trinchieri G, Scott P (1995) Interleukin-12: a proinflammatory cytokine with immunoregulatory
functions. Res Immunol. 146: 423.
Tura BJ, Bunyan KE, Harrison DJ (2001) The effect of IFNgamma on the hepatocyte: cell cycle
and apoptosis. Int J Exp Pathol. 82: 317-326.
Ulrich RG (2007) Idiosyncratic toxicity: a convergence of risk factors. Annu Rev Med. 58: 17-34.
Vila-del Sol V, Punzon C, Fresno M (2008) IFN-gamma-induced TNF-alpha expression is
regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J Immunol. 181:
4461-4470.
Vodovotz Y, Kim PK, Bagci EZ, Ermentrout GB, Chow CC, Bahar I, Billiar TR (2004)
Inflammatory modulation of hepatocyte apoptosis by nitric oxide: in vivo, in vitro, and in silico
studies. Curr Mol Med. 4: 753-762.
Volpes R, Van den Oord JJ, De Vos R, Depla E, De Ley M, Desmet VJ (1991) Expression of
interferon-gamma receptor in normal pathological human liver tissue. J Hepatol. 12: 195-202.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
44
Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ.
10: 45-65.
Wang F, Schwarz B, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR (2006)
IFN-gamma-induced TNFR2 upregulation is required for TNF-dependent intestinal epithelial
barrier dysfunction. Gastroenterology. 131: 1153-1163.
Watkins PB (2005) Idiosyncratic liver injury: challenges and approaches. Toxicol Pathol. 33: 1-5.
Watkins PB, Kaplowitz N, Slattery JT, Colonese CR, Colucci SV, Stewart PW, Harris SC (2006)
Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a
randomized controlled trial. JAMA. 296: 87-93.
Waxler B, Zhang X, Wezeman FH (1994) Anesthetic agents modify tissue proteinase inhibitor
content and tumor behavior. J Lab Clin Med. 123: 53-58.
Wen Z, Zhong Z, Darnell JE (1995) Maximal activation of transcription by Stat1 and Stat3
requires both tyrosine and serine phosphorylation. Cell. 82: 241-250.
Whitmire JK, Tan JT, Whitton JL (2005) Interferon-gamma acts directly on CD8+ T cells to
increase their abundance during virus infection. J Exp Med. 201: 1053-1059.
Wuillemin N, Adam J, Fontana S, Krähenbühl S, Pichler WJ, Yerly D (2013) HLA haplotype
determines hapten or p-i T cell reactivity to flucloxacillin. J Immunol. 190: 4956-64.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
45
Wullaert A, van Loo G, Heyninck K, Beyaert R (2007) Hepatic tumor necrosis factor signaling
and nuclear factor-kappaB: effects on liver homeostasis and beyond. Endocr Rev. 28: 365-86.
Zhang M, Han Y, Han C, Xu S, Bao Y, Chen Z, Gu Y, Xia D, Cao X (2009) The beta2 integrin
CD11b attenuates polyinosinic:polycytidylic acid-induced hepatitis by negatively regulating
natural killer cell functions. Hepatology. 50: 1606-1616.
Zhang S, Liang R, Luo W, Liu C, Wu X, Gao Y, Hao J, Cao G, Chen X, Wei J, Xia S, Li Z, Wen
T, Wu Y, Zhou X, Wang P, Zhao L, Wu Z, Xiong S, Gao X, Gao X, Chen Y, Ge Q, Tian Z, Yin Z
(2013) High susceptibility to liver injury in IL-27 p28 conditional knockout mice involves intrinsic
interferon-gamma dysregulation of CD4(+) T cells. Hepatology. 57: 1620-1631.
Zimmerman HJ (2000) Drug-induced liver disease. Clin Liver Dis. 4: 73-96
Zou W, Beggs KM, Sparkenbaugh EM, Jones AD, Younis HS, Roth RA, Ganey PE (2009)
Sulindac metabolism and synergy with tumor necrosis factor-alpha in a drug-inflammation
interaction model of idiosyncratic liver injury. J Pharmacol. Exp Ther. 331: 114-21.
Zou W, Roth RA, Younis HS, Burgoon LD, Ganey PE. Oxidative stress is important
in the pathogenesis of liver injury induced by sulindac and lipopolysaccharide
cotreatment. (2010) Toxicology. 272:2-8.
Legends for Figures
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
46
Figure 1. IFNγ-mediated hepatocyte killing by T cells. Both CD8+ T-cells and CD4+ T-cells
as well as NK and other cells produce and release interferon-gamma (IFNγ), which can kill cells
through activation of cell death pathways involving STAT1 and IRF-1 activation (right). This
killing can be enhanced by TNFα and/or by ROS/NO released from inflammatory cells, including
macrophages such as Kupffer cells (Mphage). IFNγ can also kill hepatocytes via autocrine or
paracrine activation of CD8+ T-cells involving pathways initiated by Fas-Fas ligand (FasL)
interaction or by formation of perforin pores that allow granzyme B translocation (left). During
adaptive immune responses, CD8+ T-cells recognize altered hepatocytes via receptors (TCRs),
prompting release of IFNγ. See text for references.
Figure 2. Hypotheses regarding drug-cytokine interactions in IDILI pathogenesis.
Evidence in humans and animals suggests that drugs can be bioactivated to reactive
metabolites that can initiate an adaptive immune response (top right). Alternatively, initiation of
an innate immune response through Toll-like receptor activation (top left) can prompt an
hepatotoxic interaction with drugs. Both of these immune responses result in the generation of
mediators such TNFα and IFNγ. Some drugs also directly increase production of these
cytokines by activated immune cells. At the level of the hepatocyte, IDILI-associated drugs can
enhance hepatocellular sensitivity to the cytotoxic actions of TNFα and IFNγ (bottom center). In
addition, IFNγ and at least some IDILI-associated drugs inhibit hepatocyte proliferation, thereby
impairing the ability of the liver to adapt to drug-induced stress (bottom right).
DAMPs, damage associated molecular pattern molecules. Other abbreviations are
defined in the text.
Figure 3. Cell death signaling in IDILI drug-cytokine interaction. Hypothesized pathway of
IDILI drug/cytokine synergy based on studies in vitro (see text for references). TNFα exposure
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from

JPET #237578
47
causes early and transient activation of JNK that is innocuous by itself, probably in part due to
activation of the NFkB survival pathway. MAPKs, in particular JNK and ERK, play key roles in
IDILI drug/cytokine-mediated cell death. Some drugs including diclofenac and carbamazepine
induce increased intracellular calcium and ER stress that leads to activation of MAPKs and
synergy with cytokines to cause cell death whereas others such as TVX induce the DNA
damage response pathway which also leads to synergy with TNFα. In the context of early
activation of JNK by TNFα, JNK activation is prolonged by drug exposure, leading to activation
of cell death signaling. Simultaneous disruption by diclofenac of the NFkB-mediated cell
survival pathway amplifies diclofenac/TNFα-mediated cell death signaling. Activation of JNK
also leads to ERK activation, which results in phosphorylation of cytosolic STAT1 at serine727.
Full activation of STAT1 requires additional phosphorylation at tyrosine701, which is effected by
IFNγ exposure. By as yet unknown mechanisms, this amplifies cell killing initiated by
drug/TNFα interaction. Not shown is ER stress-mediated activation of an additional apoptotic
pathway that involves expression of CHOP.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on November 15, 2016 as DOI: 10.1124/jpet.116.237578
at ASPE
T Journals on M
ay 4, 2018jpet.aspetjournals.org
Dow
nloaded from
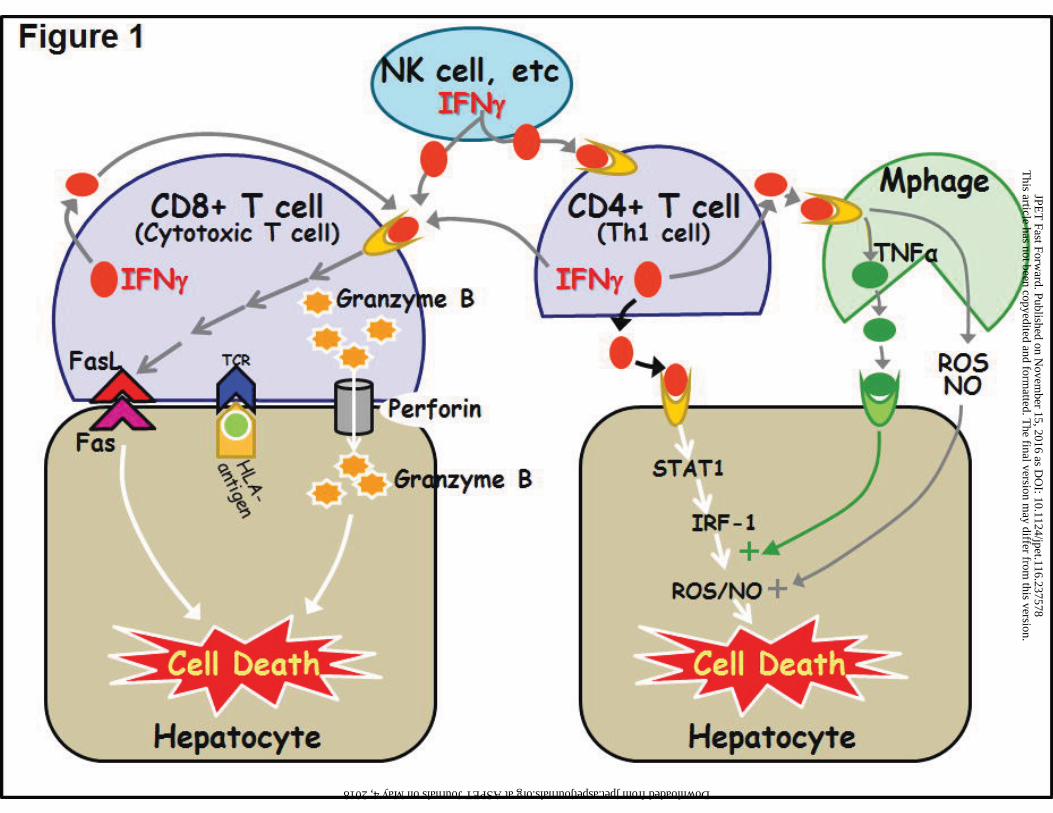
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
JPET
Fast Forward. Published on N
ovember 15, 2016 as D
OI: 10.1124/jpet.116.237578
at ASPET Journals on May 4, 2018 jpet.aspetjournals.org Downloaded from
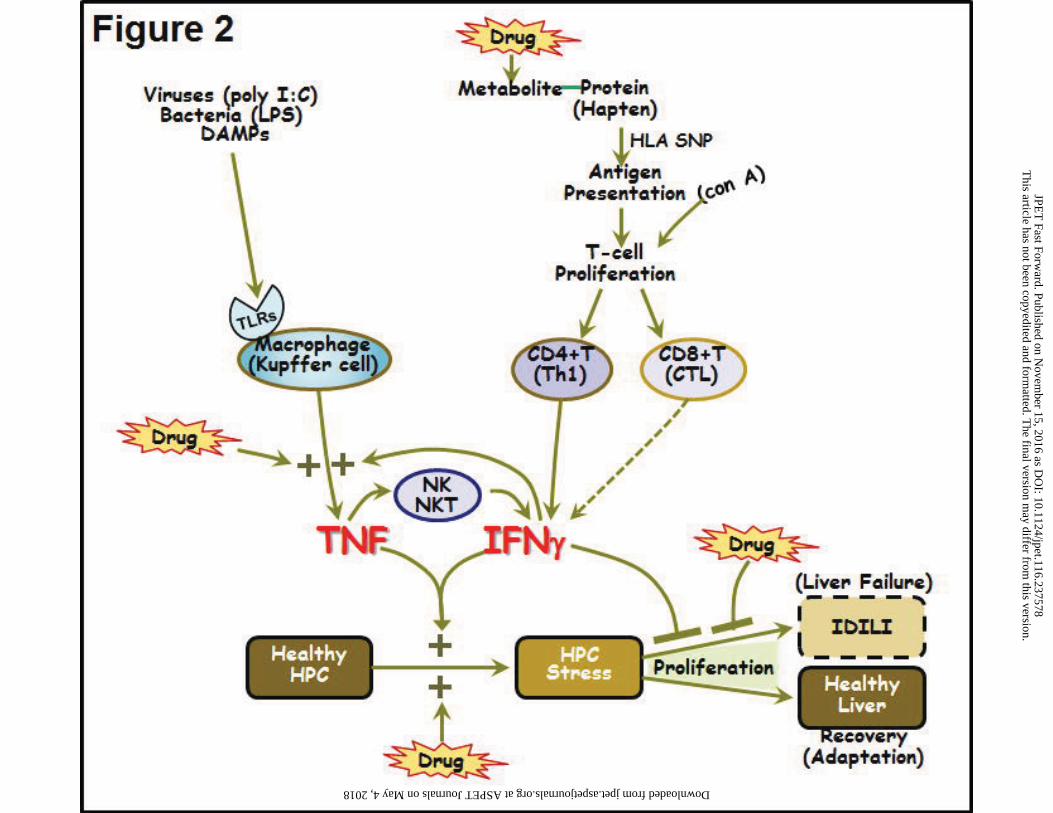
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
JPET
Fast Forward. Published on N
ovember 15, 2016 as D
OI: 10.1124/jpet.116.237578
at ASPET Journals on May 4, 2018 jpet.aspetjournals.org Downloaded from
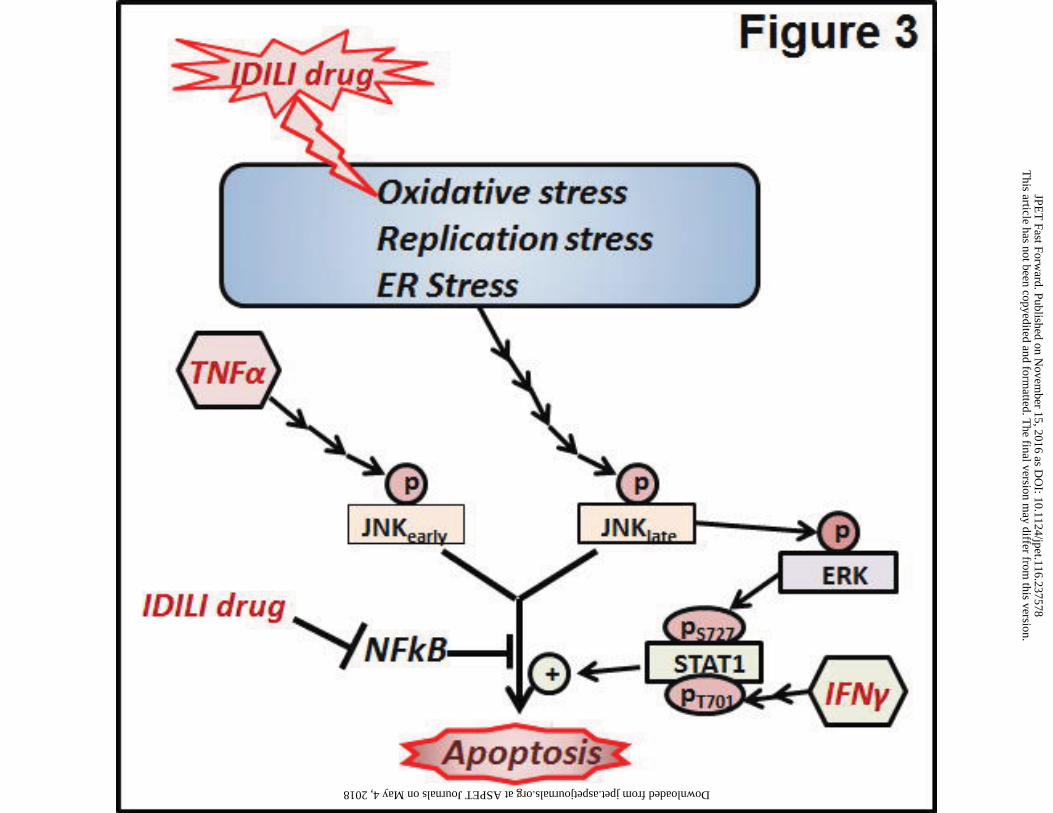
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
JPET
Fast Forward. Published on N
ovember 15, 2016 as D
OI: 10.1124/jpet.116.237578
at ASPET Journals on May 4, 2018 jpet.aspetjournals.org Downloaded from





























