Embed Size (px)
Citation preview

Ruthenium Catalysed Oxidations of Organic Compounds By Ernest S. Gore Johnson Matthey Inc., West Chester, Pennsylvania
Ruthenium and i ts complexes can be used to catalyse the oxidation, both homogeneous and heterogeneous, of a wide range of organic subs- trates. These include olefins, alkynes, arenes, alcohols, aldehydes, ketones, ethers, sulphides, amines, and phosphines. A wide variety of oxidants can be used under mild conditions; conversions and selectivities are usually high, and the catalyst can be easily recovered. Ruthenium can also be used to catalyse the oxidative destruction of pollutants in both gas and liquid phases.
The synthetic use of ruthenium tetroxide as an oxidant for organic compounds was first reported in I 95 3 by Djerassi and Engle (I). The scope of ruthenium oxidations was greatly expanded by Berkowitz and Rylander in 1958 (2). All of these early workers used ruthenium tetroxide as a stoichiometric oxidant; however, ruthenium tetroxide is rather inconvenient to use in this way. It is troublesome to prepare, expensive, and its strong oxidising power tends to make it less selective than other oxidants. Caution-ruthenium tetroxide is an extremely powerful and volatile oxidant; it should only be handled in a well ventilated area and when wearing appropriate protective clothing.
Thus it is not surprising that work was soon initiated on using ruthenium in catalytic quantities in oxidation reactions. The first such use of ruthenium seems to have been in an obscure publication in 1956 (3). A more readily available report appeared in 1959 (4). The advantages of catalytic ruthenium oxidations over stoichiometric ruthenium tetroxide have proved to be so convincing that today virtually all ruthenium mediated oxidations are perfor- med catalytically.
While several reviews (5-9) have been written on ruthenium mediated oxidations, the last one available in the West appeared ten years ago and dealt equally with ruthenium catalysed reactions and stoichiometric
ruthenium tetroxide reactions (9). With the emphasis shifting to ruthenium catalysed reac- tions, new reactions have been discovered and conditions have been found which have improved the selectivity and yields of these reactions. Thus it is appropriate to survey the subject again to summarise the state of the art.
Experimental Conditions Many different ruthenium catalysts and
oxidants have been used. Of these, the most common catalysts are RuCldPPhJ, RuCl,.xH,O, and RuO,.xH,O, and the most common oxidants are HOOAc, NaIO,, O,, and NaOCl. Some catalysdoxidant systems are very selective indeed. For example both RuCl,(PPh,)JPh(IOAc), and RuCl,(PPh),)@- methylmorpholine-N-oxide specifically convert primary alcohols to aldehydes in high yields (10, I I ) and RuCl#Ph,)JPhCH=CHCOCH, converts vicinal diols to vicinal diketones (I 2).
Conditions for ruthenium catalysed oxida- tions are very mild; usually a few hours or less at room temperature is sufficient. A variety of solvent systems can be used, and depending on the oxidant a wide range of pH’s can be tolerated. Oxidations with oxygen can be carried out at atmospheric pressure.
Many ruthenium catalysed reactions have been performed in the H,O-CCl, solvent system. But slow or incomplete reactions are
platinum M e l d s Rev., 1983,27, (3), 111-125 111

occasionally encountered in this system, especially in the presence of carboxylic acids. Recently it has been found that adding CH3CN to the system greatly improves yields and reac- tion times (13, 14). When (Eh~decene was oxidised in HzO-CC1, only 20 per cent conver- sion occurred in 2 hours; but on adding C H F N the reaction was complete in the same period.
Oxidation of Olefins Cleavage of the Double Bond
When the oxidation of an olefin is catalysed by ruthenium in the oxidation state +3 or higher, the usual result is cleavage of the double bond. Ketones are produced if the carbons are fully substituted; otherwise acids, or occasionally, aldehydes are obtained, see Table I. On the other hand, osmium tetroxide when used catalytically converts olefins to aldehydes rather than to acids (21).
Non-Cleavage of the Double Bond Only a few non-cleavage reactions are
known, and these are given in Table 11. The catalyst is usually a +2 ruthenium complex and the products are unpredictable.
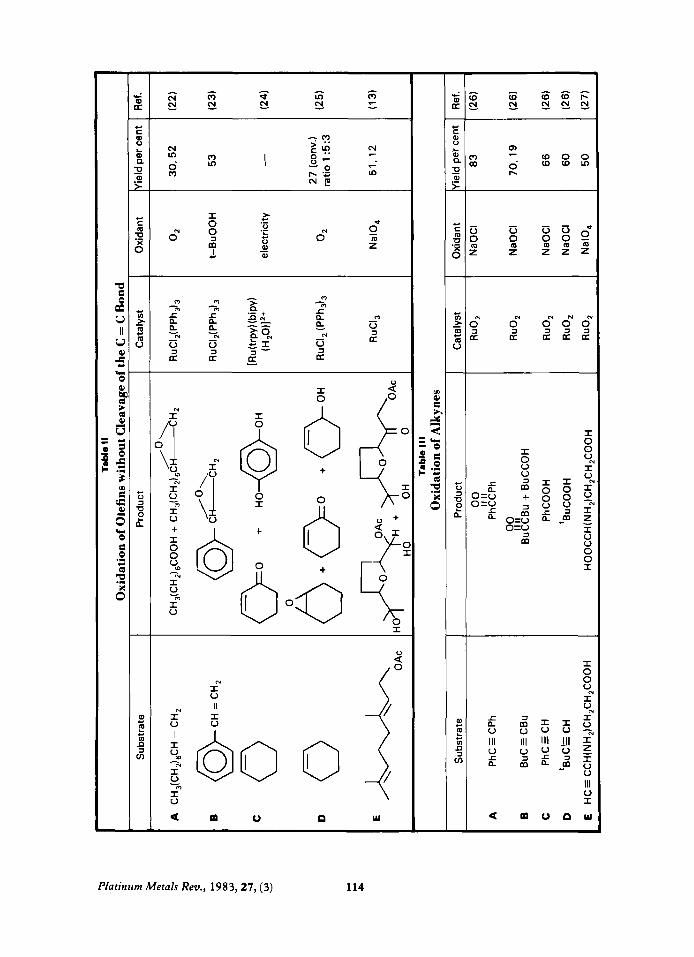
Oxidation of Alkynes Terminal alkynes are cleaved to give acids
while internal alkynes yield diketones with no cleavage, see Table 111.
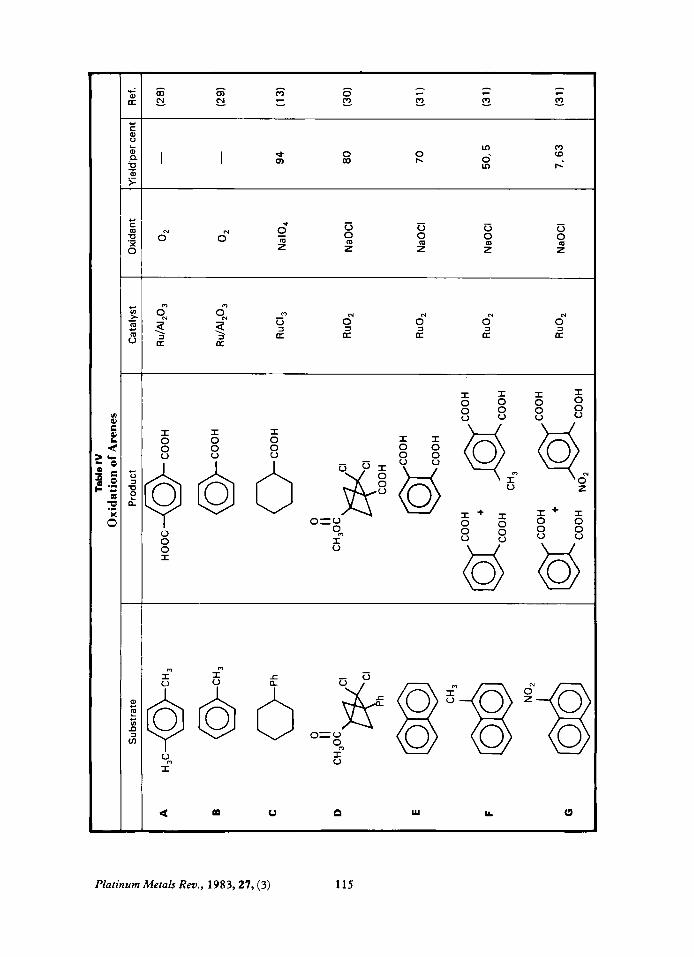
Oxidation of Arenes Ruthenium catalysed oxidations of arenes
can proceed in three ways, see Table IV: Alkyl side chains on the phenyl ring can be con- verted to -COOH (IV-A, B). The phenyl ring can be cleaved from R-Ph to
The phenyl ring can be degraded to form a dicarboxylic acid (IV-E, F , G). In almost all cases where an alkyl side chain is replaced by a carboxyl group, a heterogeneous catalyst was used, for example IV-A, B. This is one of the few cases in which a heterogeneous catalyst is used in ruthenium oxidations. Oxygen is used as the oxidant since at temperatures below 4ooOC it can only oxidise
form R-COOH (IV-C, D).
ruthenium as far as RuO,, which is insoluble. Stronger oxidants such as NaOCl or NaIO, will oxidise ruthenium to the + 7 or +8 oxidation state and these are soluble.
The effect of the electronegativity of the sub- stituent on the products of the oxidation of naphthalenes can be seen in reactions IV-F and IV-G. An electrondonating substituent favours cleavage of the substituted ring, while an electron-withdrawing substituent favours cleavage of the unsubstituted ring.
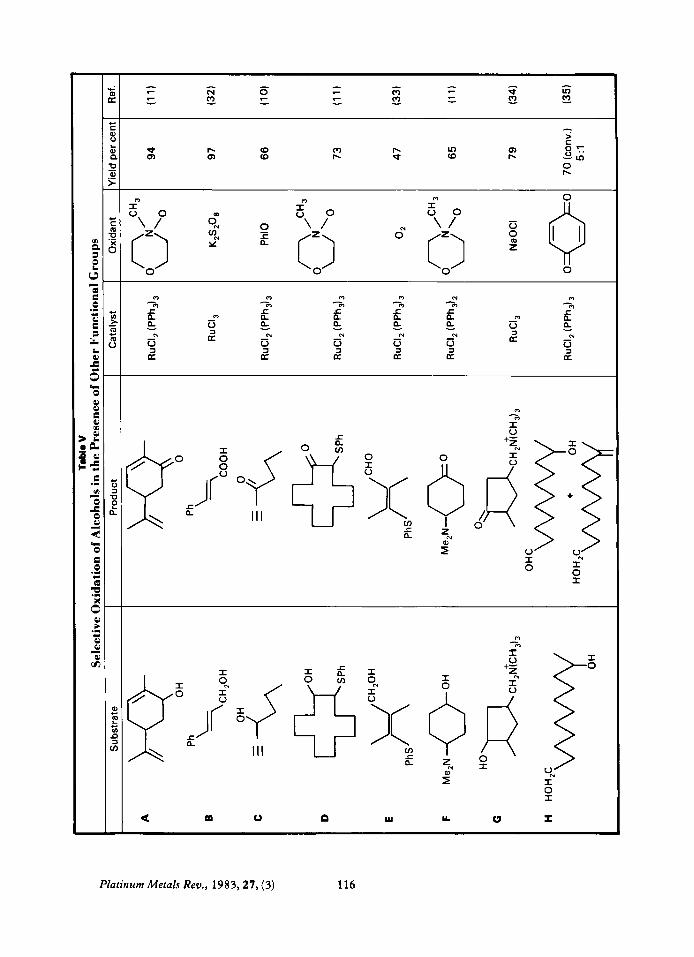
Oxidation of Alcohols This is the most common synthetic use of
ruthenium catalysed oxidations. Highly selec- tive conditions are readily available; alcohols can be converted to aldehydes rather than acids and vicinal diols can be readily oxidised to either cleaved or non-cleaved products depend- ing on conditions. In alcohols containing another oxidisable group such as a C=C double bond, a CGC triple bond, an arene, nitrogen or sulphur, the hydroxyl group is oxidised pre- ferentially (Table V). In substrates containing both primary and secondary alcohols the primary alcohol is oxidised preferentially, (see V-H).
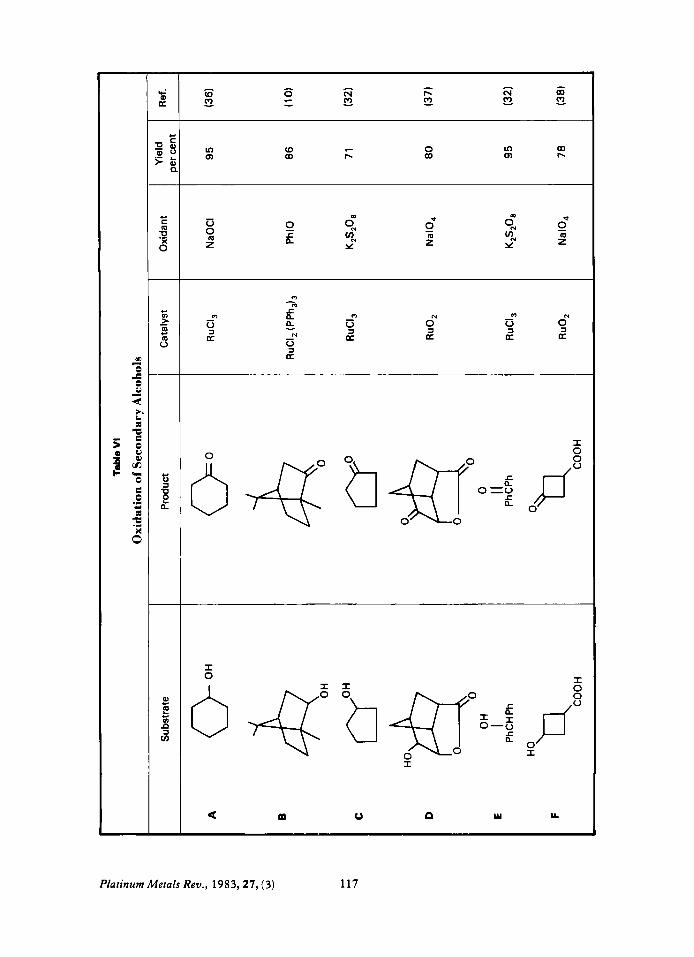
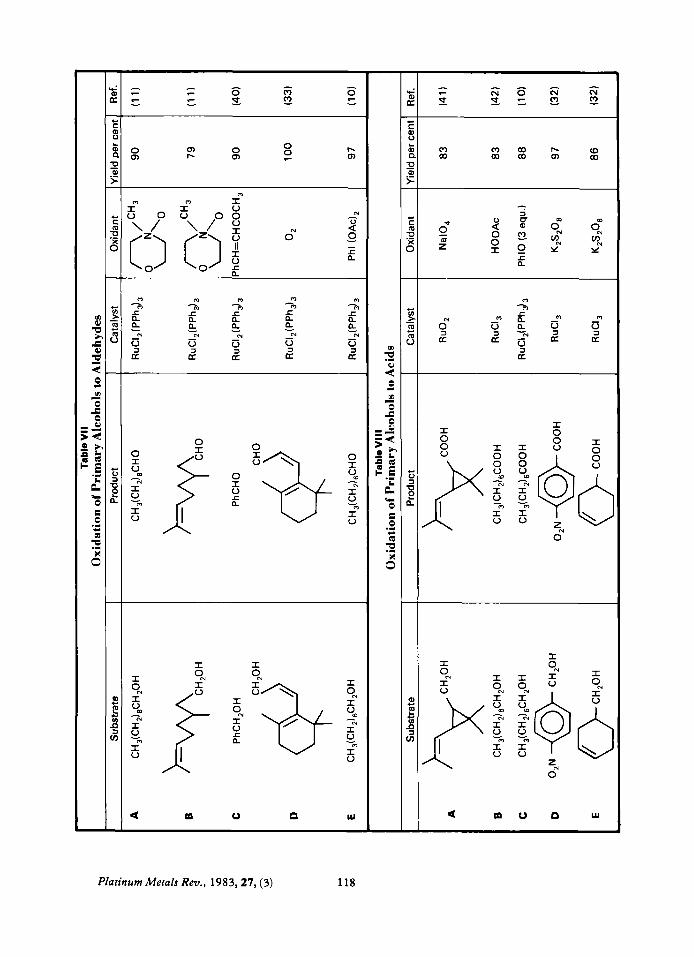
Secondary Alcohols These are oxidised cleanly and in good yields
to ketones, see Table VI. Cyclobutanols can be oxidised to cyclobutanones (38) (VI-F) in yields higher than with CrOJoxalic acid (39).
Primary Alcohols Primary alcohols are oxidised either to
aldehydes (Table VII) or to acids (Table VIII). The outcome of the reaction can be highly selective depending on the conditions used. For example: RuClXPPh,), with N- methylmorpholine-N-xide ( I I), O, (33), PhCH=CHCOCH, (40) or Ph(IOAc), (10)
always gives aldehydes. RuCIXPPhJ, with excess PhIO (to), or RuC1, with HOOAc (42) or K&OR (32) always gives acids.
Diols Oxidation of diols can give either cleaved or
non-cleaved products, see Table IX. Stronger
Platinum Metals Rev., 1983,27, (3) 112

73 P. i?
f t 5 2 c
F c
W
m
W
h)
21
W
h
v
c
c
W
Tab
le I
Oxi
datio
n of
Ole
fins w
ith C
leav
age o
f the
C =
C B
ond
Subs
trat
e
CH
3(C
H,),
CH
=
CH
(CH
,l,C
OO
H
CH
,(CH
,),C
H
= C
H,
Ph' ph
3Frh
PhC
H =
CH
,
PhC
H =
CH
,
Br + Br
Prod
uct
CH
,(CH
,),C
OO
H+
HO
OC
(CH
,l,C
OO
H
CH
3(C
H,),
CO
OH
HO
OC
(CH
,),,C
OO
H
PhC
OO
H
PhC
HO
f CHO
Br
Br
Cat
alys
t
RuO
,
RuC
I,
RuO
,
RuC
I,
RuO
,
RuO
,"
RuO
,
RuO
,
Oxi
dant
NaO
Cl
Nal
O,
02
NaO
Cl
Nal
O,
0,
Nal
O,
Nal
O,
Yiel
d pe
r cen
t
94
89
94
83
62
92
82
86
Ref
.
"3 p
pm c
obal
t nap
hthe
nate
add
ed
5 F
E
HC
E C
CH
(NH
,)CH
,CH
,CO
OH
c.
W
m
W
h)
21
HO
OC
CH
(NH
,)CH
,CH
,CO
OH
R
uO,
Nal
O,
50
(2
7)
h
v
W
Tab
le II
Oxi
dati
on of
Ole
fins
wit
hout
Cle
avag
e o
Sub
stra
te
A
CH,(C
H,),C
H =
CH,
B
QcH
=
w,
0
C
D
E
OA
c
Sub
stra
te
A
PhC
C
Ph
B
BuC
C
Bu
C
PhC
E C
H
D
‘Bu
ck
CH
Pro
duct
n
40 py+
+0AC
Tab
le I I I
O
xida
tion
of A
lkvn
es
Pro
duct
00
I1
II
PhC
CPh
88
BuC
CB
u +
BuC
CO
H
PhC
OO
H
‘BU
CO
OH
the
C =
C B
ond
Cat
alys
t
Ru C
I ,( PP
h 3) ,
RuCI
,(PPh
,) ,
[Ru(
trpv
) (bip
v)
(H ,011
’+
RuC
I, (P
Ph,),
RuC
I,
Oxi
dant
0,
t-BuO
OH
elec
tric
ity
0,
Nal
O,
ield
per
cen
t
30
,52
53
-
27 (
conv
.) at
io 1
:5:3
51,1
2
Cat
alys
t O
xida
nt
NaO
Cl
NaO
Cl
I 70, 1
9 1 (26)
Ruo
z I
RuO
, N
aOC
l 6
6
(26)
I NaO
Cl
I 60
I (26)
Platinum Meto
2 - 5 Q
P
Q
* cn 0 W
0 l-
m sr: m (9
r--
u P
u % z
I 0
V 0 0 I
I 0
& u
I + I 0 0 8 8
' 6 z
I + I 0 0 8 8
0 I
0
I
b 1
b J- 0
U m 0 n
Yatinum Metals Rev., 1983, 27, (3) 115
Tab
le I
V
Oxi
datio
n of
Are
nes
d 0)
I- Q)
(D (D
I- d
In (0
Q) Ic
I
> C 8' -In 0 I-
n
r n n -
N s K
n s K
n n 1
I V +- I V
2,
0 / Q 0 0 I 5 .'t
ON I 0 I
I
I 0,
>c v) L
n
I 0
+- I =N
0 I d I 0
I 0 % I i U m u 0 w LL (1 I
Platinum Metals Rev., 1983,27, (3) 116
Tabi
e V
S
elec
tive
Oxi
dati
on o
f A
lcoh
ols
in t
he P
rese
nce
of O
ther
Fun
ctio
nal G
roup
s
W m - hl m - zi
s 1 m s -
I- hl
Lo OD 0) I- Lo W 7 0
m OD b OD
9 4 Y c n
P
0 m
4 Y z
Q a n Q a 2
K
I 0
0 d'
a m u n w LL
Platinum Metals Rev., 1983,27, (3) 117
Tab
le V
I
Oxi
dati
on o
f Se
cond
ary
Alc
ohol
s
5 F
I P
hl (O
Ac),
c.
W
m
W
h)
21
W
h
v
Oxi
dant
Nal
O,
HO
OA
c
Phl
O (3
equ
.)
K,S,
O,
KZ
SZ
O8
c
L
oo
Yie
ld p
er c
ent
Ref
.
83
(4
1 I
83
(4
2)
88
(1 0)
97
(32)
86
(32)
Sub
stra
te
A
CH,(C
H,),C
H,OH
0
C
PhCH
,OH
D
E
CH,(C
H,l,C
H,OH
Sub
stra
te CH
,OH
A
B CH
,(CH
,)&H
,OH
C
CH,(C
H,),C
H ,O
H
Tab
le V
II
Oxi
dati
on o
f P
rim
arv
Alc
ohol
s to
Ald
ehyd
es
Pro
duct
CH,(C
H,),C
HO
%CHO
PhC
HO
CH,(C
H,),C
HO
Cat
alys
t
RuCI
,( PP
h,),
RuCI
,(PPh
,),
RuCI
,(PPh
,),
Ru C
I ( P
Ph,) ,
RuCI
,(PPh
,),
Tab
le V
lll
Oxi
dati
on o
f P
rim
ary
Alc
ohol
s to
Aci
ds
Pro
duct
0
CO
OH
Oxi
dant
n
PH
3
3 wN
\o
WN
Lo
3 hC
H=C
HC
OC
H,
Cat
alys
t
RuO
,
RuCI
,
RuCI
,(PPh
3),
RuCI
,
RuCI
,
field
per
cen
t
90
79
90
100
97
Platinum Metals Platinum Metals Platinum Metals
Platinum Metals
Y
c
L
\o
I
t S
ubst
rate
PhC
(CH
,)HC
H(O
H)C
H,O
H
6 OH
OH
O
H
I I
CH
3CH
C
HC
H,
do"
dO
H
OH
OH
I
I
CH
3CH
CH
CH
,
I S
ubst
rate
a CH
,(CH
,),CHO
PhC
HO
CI
C
D
Tab
le I
X
Oxi
dati
on of
Dio
ls
Pro
duct
PhC
(CH
,) H
CO
OH
HO
OC
(CH,
),CO
OH
CH,C
HO
00
00
1111
CH,C
CCH,
Cat
alvs
t
RuCI
,
RuCI
,
RuCI
,
RuCI
,
RuCI
, (P
Ph,),
RuCI
, (P
Ph,),
Tab
le X
O
xida
tion
of A
ldeh
ydes
and
Ket
ones
P
rodu
ct
CH
,(CH
,),COO
H
PhC
OO
H
CI
@-C
OO
H
HO
OC
(CH
,),C
OO
H
+ HO
OC(
CH,),
COO
H
+ HO
OC(
CH,),
COO
H
Oxi
dant
Nal
O,
NaO
Cl
H,Oz
H2O
z
'hC
H=C
HC
OC
H3
'hC
H=C
HC
OC
H,
Cat
alys
t O
xida
nt
RuCI
, (P
Ph,),
RuCI
, (P
Ph,),
RuCI
,
RuCI
, N
aOC
l
field
Der
cen
i
92
90
88
-
85
70
fiel
d pe
r cen
t 88
96
99
3,5
6,2
8
-
Ref
.
Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals Platinum Metals
Platinum Metals
Platinum Metals
Platinum Metals
c C m n 6
c
- r J m c
- I
2 2 1 -
N O L D N
P f f P P 0 f
0 0 0 0 0 0 0 i i z m m m m m z z z z
t
0 i
o s a
o I u 0
3= u, A
N I
I V
+
0
c)
I
I u
a m u
0 I u 0 t
+
11 I V 0
$ n
0
YI LL
Platinum Metals Rev., 1983,27, (3) 120
Tab
le X
I O
xida
tion
of
Acv
clic
Eth
ers
ri al a
P 9 P 4 P
0 0 0 0 0 i z z z z m m
Q a Q a Q a Q a Q LT
I 0 0
I N 0
u, I 0 0 I u N
I V
0
u
I
0 - 70 0 0
4 m u D W
Platinum Metals Rev., 1983,27, (3) 121
Tabl
a X
I1
Oxi
dati
on o
f C
yclic
Eth
ers
0 m
B a
B a
N
0 % m
0
m
+
cnN
I
I V
O= V,
LAooQoac I I I I I 0=3 o=v
Om o=v
I I V V
I& 0 v1, V
Q D I o=v
I V
LJ I 0" WCI I V
0 I o=v,
LL V
II z I
Om
o=v o=v I V
a m o n u U (1 I -
Platinum Metals Rev., 1983,27, (3) 122
Tab
le X
lll
Oxi
dati
on o
f Su
lphi
des a
nd A
min
es

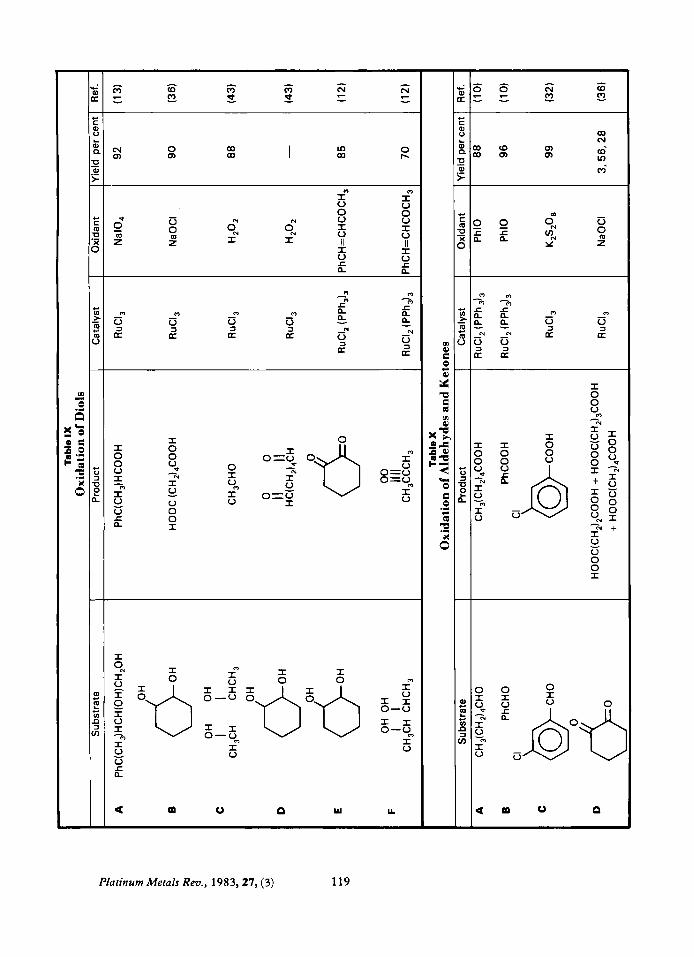
oxidants cleave the diol, generally giving acids (IX-A, B), although under carefully controlled conditions aldehydes can be obtained as the major product (IX-C, D). The use of RuClXPPhJ, and PhCH=CHCOCH, selectively produces non-cleaved diketones from diols (IX-E, F).
Carbohydrates Ruthenium catalyses the oxidation of
hydroxyl groups in carbohydrates, secondary hydroxyl groups being converted into carbonyls and primary groups to acids. For example L- sorbose is converted to a mixture of erythrose and glycolic acid (44) and sugar, I, is converted to its keto sugar, 11, in 100 per cent yield (45).
I I1
The yields are usually better when ruthenium is used than when CrO, is used (9).
Oxidation of Aldehydes and Ketones
Aldehydes are readily oxidised to carboxylic acids, see Table X. There has been little published work on the oxidation of ketones. In one paper dealing with kinetics the authors reported that ketones were converted to diketones (46), for example:
CH,COCH&H, + CH,COCOCH,
However in this case a ten fold excess of subs- trate was used. Diketones are cleaved to give a mixture of acids (X-D).
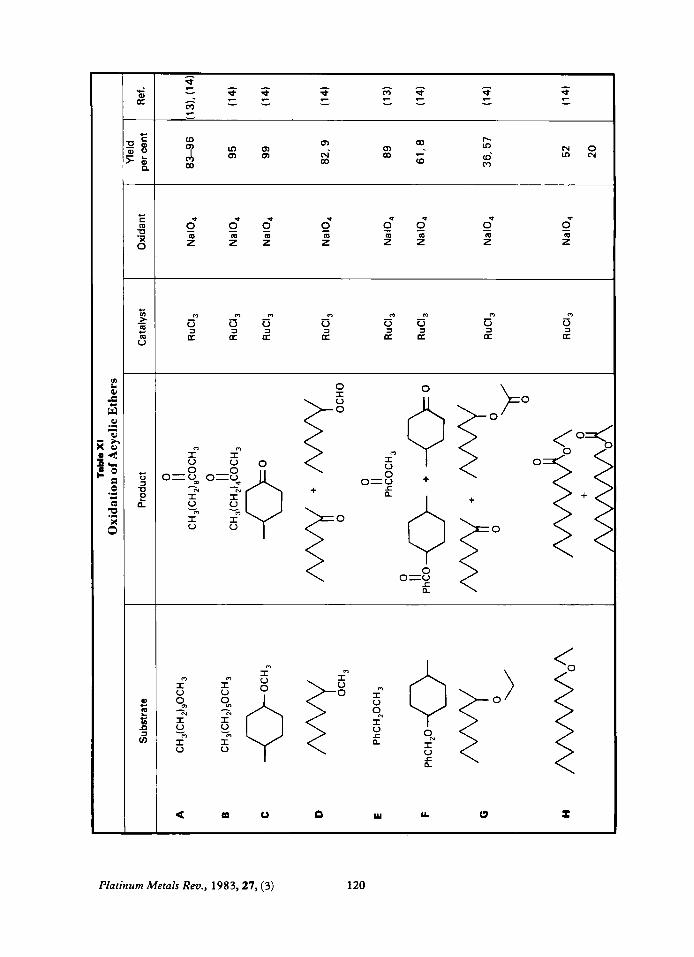
Oxidation of Ethers Acyclic Ethers
Primary methyl ethers, RCH,OCH,, are oxidised to methyl esters, RCOOCH,, in excellent yields, (XI-A, B). Secondary methyl ethers, RR'CHOCH,, on the other hand undergo cleavage to give ketones, RCORI (XI- C, D). Benzyl ethers, PhCH,OR, undergo oxidation of the benzyl group to give esters,
PhCOOR, in fair to good yields (XI-E, F). Oxidation of unsymmetric ethers, RORI where one of the substituents is not aromatic, gives unpredictable results with either R or R1 being oxidised in roughly equal proportions.
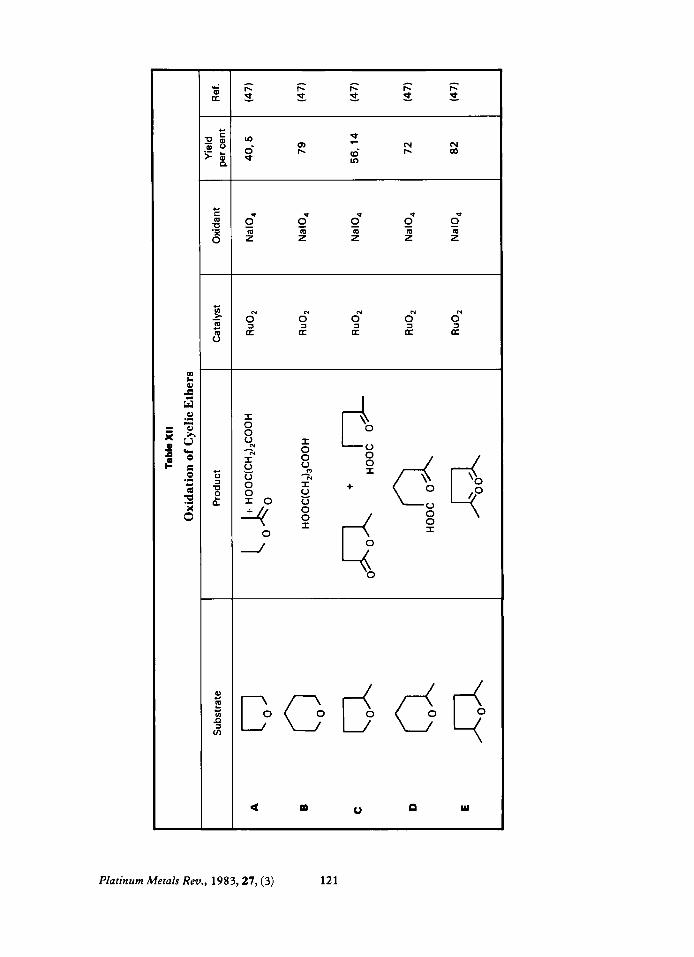
Cyclic Ethers Only carbons next to the ether linkage are
oxidised. If both carbons are secondary, the products are mainly lactones with some car- boxylic acids depending on the sensitivity of the lactone to hydrolysis (XII-A, B). If one carbon is secondary and the other tertiary the secondary carbon is oxidised preferentially giving a lactone (XII-C, D). Some hydrolysis to keto acids can occur. If both carbons are tertiary, cleavage to diketones occurs (XII-E).
Ethers are a class of compounds for which the yields and selectivities differ significantly when they are oxidised catalytically or stoichiometrically with ruthenium tetroxide. For example, tetrahydrofuran oxidised stoichiometrically with ruthenium tetroxide gives only y-butyrolactone in 65 to IOO per cent yield (2,47) but when oxidised catalytically with RuO, and NaIO, (47) the products are y- butyrolactone, 40 per cent, and succinic acid, 5 per cent.
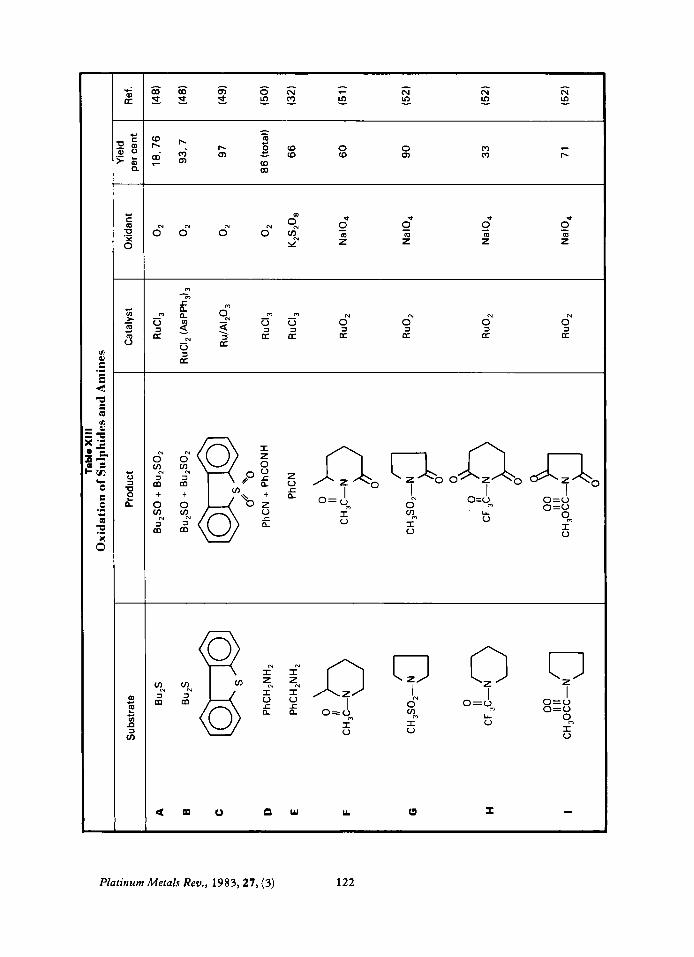
Oxidation of Sulphides and Amines
Sulphides are usually oxidised to a mixture of sulphoxides and sulphones (XIII-A, B), but in at least one case a sulphone was obtained exclusively (XIII-C).
Linear primary amines are oxidised to nitriles with some hydrolysis to the amide (XIII- D, E). Cyclic amines are oxidised to either lactams (XIII-F, G) or imides (XIII-H, I). The yields range from poor to good. As with ethers, only the carbon adjacent to the heteroatom is oxidised and secondary carbons are oxidised preferentially to tertiary carbons.
Oxidation of Steroids Steroids generally undergo the same reac-
tions that have already been discussed- oxidative cleavage of C=C double bonds to
Platinum Metals Rev., 1983,27, (3) 123

acids (4, 53), degradation of aromatic rings (54), suggested (56) for removing sulphur containing and oxidation of secondary alcohols to ketones impurities from various petroleum fractions. ( 1 0 , I I ) . Thus sulphur (500 ppm) in an n-paraffin frac-
It is interesting that cholesterol, which has tion was reduced to less than 50 ppm in 4 hours both a secondary hydroxyl group and a C=C by treatment with RdNaOCl at 2oOC. double bond, does not undergo any reaction Ruthenium has also been suggested (57,58) (10, XI). for the removal of ammonia from waste water
An atypical reaction which some steroids by treating the waste at elevated temperatures undergo is simultaneous oxidation of a tertiary with oxygen and a supported ruthenium CH group to a tertiary alcohol, and a secondary catalyst. Chlorophenols and highly toxic CH, ~ to a ketone (54): ~ polychlorodibenzodioxins effectively destroyed were shown by ruthen- to be
ium catalysed oxidations (59). Finally ruthenium has been
demonstrated to remove pollu- tants in the gas phase. Thus 5500 ppm vinyl chloride in air Ac 0 AcO
- - 0
Pollution Control via Ruthenium Catalysed Oxidations
Wet scrubbing with KMnO, is used com- mercially to control air pollution. However with some pollutants, notably thiophenes, the reac- tions are too slow to be useful. It has been demonstrated (5 5 ) that oxidation of thiophenes with RdNaOCl is more than IOO times faster than with KMnO,. This means that residence times are within the range that wet scrubbing of airborne thiophenes and other sulphur con- taining pollutants is practicable.
Ruthenium catalysed oxidations have been
was reduced to 2 ppm by pass- ing the gas over a 0.5 per cent ruthenium on alumina catalyst at a temperature of 376OC (60).
Conclusion Ruthenium and its complexes are extremely
versatile oxidation catalysts. They will catalyse the oxidation of virtually any oxidisable organic functional group and, by choosing the appropriate conditions, the oxidations can be made to proceed in high yield and selectivity even in the presence of other oxidisable groups. Thus they offer a useful alternative to the more classical oxidation reagents.
I
2
3
4 5 6
7
8
9
10
References C. Djerassi and R. Engle, 3. Am. Chem. Soc., 11 K. B. Sharpless, K. Akashi and K. Oshima, 1953, 757 3838 L. M. Berkowitz and P. N. Rylander, 3. Am. 12 S. L. Regen and G. M. Whitesides,J. Org. Chem.,
R. Pappo and A. Becker, Isr. Res. Counc. Bull., 13 P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. 19569 5A, 300 B. Sharpless,3. Org. Chem., I 981,46, ( 1 9 ) ~ 3936 S.SarrlandY.Yan~3.7.0rg.Chern.,1959,~4,2018 14 P. H. J. Carlsen and K. B. Sharpless, personal
communication PN;Rylander, EngeIhard Tech. Bull., 1969,9,135 1 5 T. A. Foglia, P. A. Barr, A. J. Malloy and M. J.
“Oxidation” Vol. I , ed. R. L. Augustine, Marcel Costanzo, 3. Am. Oil Chem. Soc., 1977, 54, (I I), Dekker, New York, I 969 870A
Tetrahedron Lett., 1976, (29), 2503
Chem. SOC., 1958,80,6682. 1972,377 (1 I), 1832
T. Yamanaka, Kagaku Kogvo, 1979, 30, (6), 619; Chem. Abstr., 1979,91, I 12953 T. Yamanaka, Kagaku Kogyo, 1979,30, (7), 747; Chem. Abstr., 1979,91, 199460 D. G. Lee and M. van den Engh in “Oxidation in Organic Chemistry”, Part B, ed. W. S. Trahanovsky, Academic Press, New York, 1973 P. Miiller and J. Godoy, Tetrahedron Lett., I 98 I , 22, (251,2361
16 K. Kawamoto and T. Yoshioka, European Parent
17 K. A. Keblys and M. Dubeck, U.S. Patent
I 8 W.-D. SchrGer and W. Friedrichsen; Liebigs Ann.
19 Mitsubishi Petrochemical Co., LtG., ’ fapawe Purenr, 80,087,739; 1980; C‘lrem. Abstr., 1981,
Appl. 21, 118; 1981
134099649; I 968
C h . , 197891978, (91, 1648
94, 103012
Platinum Metals Rev. , 1983,27, (3) 124
effectively destroyed by ruthen-

20
21
22
23
24
25
26
27
28
29 30
3’
32
33
34
35
36
37
K. H. Holm, D. G. Lee and L. Skattebgjl, Acta Chem. Scand., 1978, Bjz, (9), 693 M. Schroder, Chem. Rev., 1980,80, (2), 187 M. E. Pudel, L. G. Privalova, Z. K. Maizus, L. V. Revenko, M. L. Khidekel and I. V. Kalechits, Neftekhimiya, 1973, 13,64 J. 0. Turner and J. E. Lyons, German Patent
B. A. Moyer, M. S. Thompson and T. J. Meyer, J . Am. Chem. SOC., 1980,1oz, (7), 2310 S. Cenini, A. Fusi and F. Porta, Gazz. Chim. Ital., 1978, 108, (3-4), 109 H. Gopal and A. J. Gordon, Tetrahedron Lett.,
M. J. Jung, B. W. Metcalf, B. Lippert and P. Casara, Biochem., (Washington), 1978, 17, (I 3), 2628 D. C. Cronauer and L. G. Baumgard, US. Patent
S. N. Massie, U.S. Patent 3,775,472; 1973 D. E. Applequist and J. W. Wheeler, Tetrahedron
U. A. Spitzer and D. G. Lee, J. Org. Chem., I 974,
M. Schrijder and W. P. Griffith, 3. Chem. SOC., Chem. Commun., 1979, (I), 58 M. Matsumoto and S. Ito, 3. Chem. SOC., Chem. Commun., I 98 I, (I 7), 907 R. S. Givens and D. R. Rademacher, 3. Med.
H. Tomioka, K. Takai, K. Oshima and H. Nozaki, Tetrahedron Lett., I 98 I, zz, (I 7), I 605 S. Wolfe, S. K. Hasan and J. R. Campbell, Chem. Commun., 1970, (21), 1420 H. Gopal, T. Adams and R. M. Moriarty,
2,231,678; I973
‘971,(3’)1294’
3,865370; 197
Lett., 1977, (39)s 34’1
39, (161,2468
Chem.9 I9747 17, (41, 457
40 Y. Sasson and J. Blum, Tetrahedron Lett., 1971,
41 A. I. Dalton, Jr., H. J. Doran and R. D. H. Murray, U.S. Parent 4,225,694; 1980
42 M. N. Sheng, U.S. Patent 3,997,578; I 976 43 Mitsui Petrochemical Industries, Ltd., Japaness
Patent, 80, 102, 528; 1980; Chem. Abstr., 1981,
44 K. N. Reddy, V. Devi and P. K. Saiprakash, Nail. Acad. Sci. Lett., (India), 1981, 4, (7), 297; Chem. Abst., 1982,96, 85864
45 G. Descotes, J. P. Praly and D. Sinou, J. Mol. Catal., I 979,6, (6),42 I
46 P. S. Radhakrishnamurti and D. K. Mahapatro, Ind.J. Chem., 1979, I ~ A , (I), 53
47 A. B. Smith, I11 and R. M. Scarborough, Jr., Synth. Comm., 1980, 10, (3), 205
48 I. V. Howell, M. A. Ledlie and R. C. Pitkethly, British Patent 1,404,s I 3; I 975
49 M. A. Ledlie and I. V. Howell, Tetrahedron Lett., 1976, (101,785
50 R. Tang, S. E. Diamond, N. Neary and F. Mares, J . Chem. SOC., Chem. Commun., 1978, (13), 562
51 N. Tangari and V. Tortorella, J. Chem. SOC., Chem. Commun., 1975, (3), 71
52 J. C. Sheehan and R. W. Tulis, 3. Org. Chem., 19743 3% (1 51,2264
53 D. M. Piatak, H. B. Bhat and E. Caspi, J. Org.
54 D. M. Piatak and 0. Ekundayo, Steroids, 1973,
55 D. C. Ayres and C. M. Scott, Enuiron. Sci.
56 V. Cariati and V. Viani, British Patent 1,549,363;
57 N. Okada, Y. Nakanishi and Y. Harada, German
(241,2167
94, 46774
Chem., 1969~34, (11, I 12
219 475
Technol., 1979, 13, (I I), 1 3 8 3
‘979
Patent 2,735,892; 1978 Tetrahedron, 1972, 28, (16), 4259
1967, (47h 4729
60,20
58 Osaka Gas Co., Ltd.,Japanese Patent 80,086,854;
59 D.C.Ayres, Plarinum Metals Rev., I 98 I ,25,(4), I 60 60 K. Yang, J. F. Scamehorn, J. D. Reedy and R. C.
38 J. A. Caputo and R. Fuchs, Tetrahedron Lett.,
39 M. Krumpolc and J. Rocek, Org. Synth., 1981,
1980, Chem- Abstr.3 19809 939 244898
Lindberg, German Patent 2,640,906; I 977
Platinum Group Metals in Organic Synthesis Modern Synthetic Methods, Volume 3,1983-Transition Metals in Organic Synthesis EDITED BY R. SCHEFFOLD, Salle and Sauerlander, and John Wiley & Sons, 440 pages.
To provide chemists with an easy access to important and rapidly developing areas of syn- thetic organic chemistry triennial seminars on modern synthetic methods are held at Interlaken, sponsored and organised by The Association of Swiss Chemists. The May 1983 conference was devoted to the transition metals, and the above named volume of contributions is being co-produced by Salle and Sauerlander, with distribution rights in Austria, Germany and Switzerland (sfr./DM 48), and by John Wiley & Sons for the rest of the world.
The platinum group metals are featured in two of the five sections, these being “Principles of Transition Metals Chemistry” by Professor J. K. Stille and “Group VIII Metals in Organic Synthesis” by Professor L. S. Hegedus, both of Colorado State University. The contributions are well supported by references and this most useful book will undoubtedly fulfil its main purpose of serving as a guide for chemists interested in the application of transition metal chemistry to organic synthesis, in addition to aiding participants at the May conference.
Platinum Metals Rev., 1983,27, ( 3 ) 125





























