Embed Size (px)
Citation preview

Safety WorkshopSafety WorkshopLing Chin, MD, MPH
Safety Pharmacovigilance Team,
OPCRO, DAIDS, NIAID
Ling Chin, MD, MPH
Safety Pharmacovigilance Team,
OPCRO, DAIDS, NIAID

2
At the conclusion of this workshop, participants will be able to demonstrate an understanding of:−Current context regarding safety in clinical trials
−The concept of safety and safety monitoring and how it relates to clinical trials research
−Protocol requirements pertaining to areas relevant to safety
−Key roles and responsibilities related to safety
−Safety and adverse event terminology
−Expedited reporting of adverse events
ObjectivesObjectives

3
ObjectivesObjectives
At the conclusion of this workshop, participants will be able to demonstrate an understanding of:
−Ensuring safety in clinical trials
−The adverse event life cycle
−What makes a well-documented adverse event, including a comprehensive narrative
−How to assess an adverse event case, including causality (attribution)

4
Food and Drug Administration Amendments Act of 2007 (FDAAA)
Provides FDA with additional requirements, authorities, and resources with regard to both pre- and postmarket drug safety
• New authorities to require postmarket studies and clinical trials, safety labeling changes, and Risk Evaluation and Mitigation Strategies (REMS)
• Increased activities for active post market risk identification and analysis
• New reporting of adverse events related to food and new regulations for pet food labeling, ingredients, and processing standards
Current FDAAA EnvironmentCurrent FDAAA Environment

5
FDAAA Implementation – Examples Required postmarketing studies, or clinical trials to address safety issues
(21 letters); postmarketing commitments now required (previously voluntary) and established timeframes are enforceable
Required safety label changes (4 times)
Can require REMS to ensure that the benefits of a drug outweigh its risks (13 REMS)
Sentinel Initiative (launched May 08):
• Create and implement a nation-wide active, electronic surveillance system for monitoring medical product safety
• Develop methods to obtain access to disparate data sources and to establish a postmarket risk identification and analysis system
• Requires FDA to work closely with partners from public, academic, and private entities
Current FDAAA EnvironmentCurrent FDAAA Environment

6
Impact relevant to DAIDS:
• When DAIDS holds the Investigational New Drug (IND): increased requirements for safety assessment, pharmacovigilance, risk evaluation
• Clinical Trial Agreements (CTAs ) with pharmaceutical companies:
– Increasing demands for safety data and safety reports, increasingly shortened timelines
– Postmarketing requirements on Marketing Authorizing Holders (MAHs) irrespective of DAIDS obligations for IND studies vs. non-IND studies: expect final safety reports in 15 days or less
Framing the contextFraming the context

7
Impact relevant to pharmaceutical companies:• Increased requirements for safety assessment,
pharmacovigilance, risk evaluation, and risk mitigation/ minimization
– Parallel and additional requirements may be required by other authorities globally, including European Medicine Evaluation Agency (EMEA)
• FDA can penalize pharmaceutical companies for a variety of noncompliance issues
• Civil monetary penalties (CMPs): can be assessed for an assortment of violations such as violations of new REMS provisions, postmarket study requirements, or labeling violation
• CMPs can reach substantial amounts, e.g. maximum of $10 million in a single proceeding
Framing the contextFraming the context

8
Increasing demands for safety data:• All Serious Adverse Events (SAEs)
• Follow all AEs/SAEs till resolved or stable
• Additional adverse events of interest:– Cancers– Myocardial Infarctions (MIs)– Hepatic events
• Pregnancy outcomes
• Global reporting to EMEA and regulatory agencies of European Union (EU) member states
– Use of CIOMS form– Country of origin of AE
Framing the contextFraming the context

9
The times they are a changin’The times they are a changin’
Then (1990’s) Now (2009)
HIV/AIDS Fatal disease Chronic illness
Focus Any Treatment Efficacy (mortality)
Trade-offs between Treatment & Safety (morbidity)
Focus HIV and Opportunistic Infections (OIs)
HIV and OIs, Co-morbidities and Other Chronic Illnesses, ART toxicities
Perspective Largely Clinical Clinical and Regulatory

10
Clinical Trial Continuum: From Drug Development to Optimal Regimensto Treatment Strategies
Clinical Trial Continuum: From Drug Development to Optimal Regimensto Treatment Strategies

11
Safety MonitoringSafety Monitoring
Why is safety monitoring required in all clinical trials?
To ensure Subject Safety and Study Integrity
To Ensure Subject Safety and
Study Integrity

12
Purpose of Safety MonitoringPurpose of Safety Monitoring
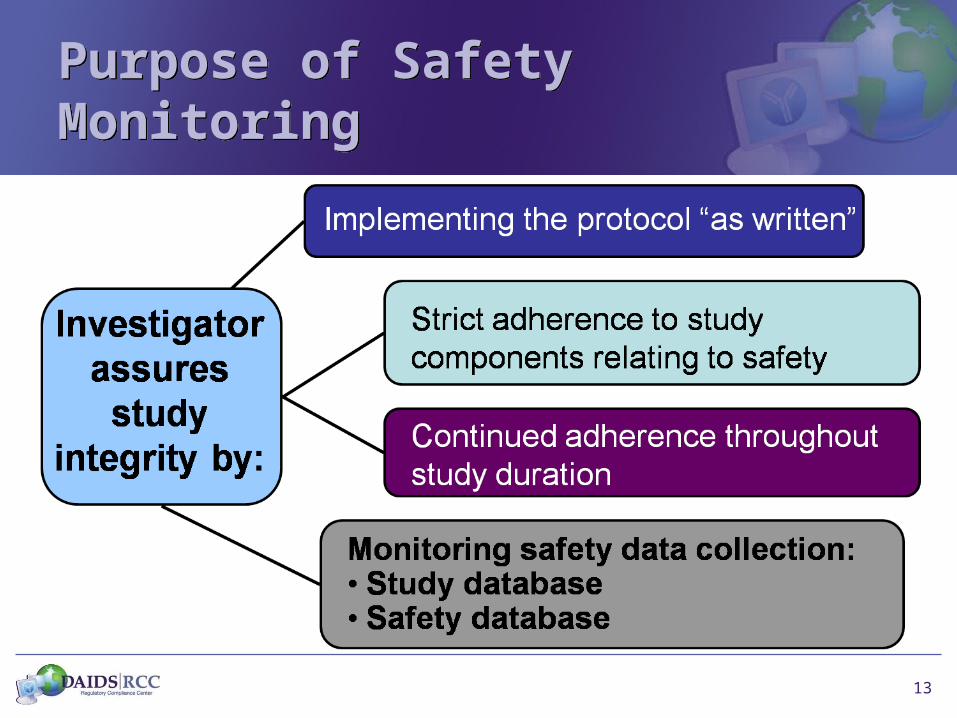
13
Purpose of Safety MonitoringPurpose of Safety Monitoring

Clinical Role vs. Research RoleBalancing Both Roles
Clinical Role vs. Research RoleBalancing Both Roles
Clinical Role: Subject OK• Is subject in imminent jeopardy?
• Provide appropriate management commensurate with clinical situation, e.g. toxicity management
• Provide appropriate referral: emergent care or back to regular care
• Follow up with subject status
Not Subject’s Primary Clinician
Research Role: Study/Data OK• Identification of adverse event
• Immediate notification necessary? To whom? [per protocol and safety monitoring plans]
• Complete documentation of adverse event. Follow until resolution/stability including updating records
• Determine if AE meets criteria for SAE/EAE
• Adhere to reporting requirements
• Adhere to toxicity management as specified
• Adhere to stopping rules as specified
14

15
Roles and Responsibilities – Research StaffRoles and Responsibilities – Research Staff
• Record AE and/or SAE/EAE per protocol specifications
• Follow protocol toxicity management section
• Follow site SOP for emergencies
• Follow site SOP to notify study clinician/physician
• Record the AE/SAE
Is immediate/emergency intervention needed?
Yes No

16
Roles and Responsibilities – Study Clinician/PhysicianRoles and Responsibilities – Study Clinician/Physician
Subject reports AE
Documentation Follow until AE resolution
or condition stabilizes
Study clinician/physician will assess and manage the
AE. Decide if SAE/EAE
Emergency intervention vs.
Non-emergency care
Research provisions vs. Clinical care

17
Assurance of Safety and Well-Being:Research vs. Medical RolesAssurance of Safety and Well-Being:Research vs. Medical Roles
Emergency intervention vs. Non-emergency care• Acute on-site management, as necessary, and per site SOP• Referral to care when stable
Research provisions vs. Clinical care• Provide interventions permitted by the protocol• Follow protocol specifications for toxicity management • Beyond protocol specifications, refer out for clinical care
Therapeutic misconception• Subjects think they are receiving proven interventions, per their usual
clinical care, despite being in a research study• Physicians think they can provide interventions, per usual practice

18
Roles and Responsibilities – Study Clinician/PhysicianRoles and Responsibilities – Study Clinician/Physician
Study product: Per site, per study?
Study status: Safety pause, clinical hold, early termination?
Study product: Dose held, changed, or discontinued?
Study participation:
Continue, withdraw?
Action taken with Study product
Subject Study

19
Roles and Responsibilities – Study TeamRoles and Responsibilities – Study Team
Safety: Ensure safety and well being of subjects at all times
Data: Ensure data integrity to assess the risks/safety profile of the study intervention
Data capture; in particular, the collection of safety data
Overall monitoring of adverse events occurring in study
Take timely action regarding subject participation status in study
Be cognizant of expedited reporting requirements for safety data

20
Roles and Responsibilities – Study Team vs. Sponsor/RCCRoles and Responsibilities – Study Team vs. Sponsor/RCC
Safety monitoring by study team
• Acute on-site management and discussion with study team
• Periodic review by study team and monitoring committees
– Data generated by Data Management Centers (DMC)
Expedited reporting to sponsor/RCC
• SAE/EAE sent to RCC
• RCC is not part of discussions that occur within study/safety monitoring teams regarding the event
• The RCC only has information about the event from the EAE Form; site should include relevant information from study team discussions
• RCC processes event and sends queries to site to obtain additional information
• All follow-up information should be provided to RCC

Land’s End at Cabo San LucasThe majestic stone arch at the southern tip of Baja, where the
Sea of Cortez meets the Pacific Ocean
MentalBreakMentalBreak

22
Safety Monitoring EnvironmentSafety Monitoring Environment
IND Trials
Pre-marketPostmarket
OHRP 45 CFR 46 OHRP 45 CFR 46
FDA
21 CFR Part 312 - IND
21 CFR 312.32 (IND Safety Reports)
21 CFR 312.33 (Annual Reports)
21 CFR 812.150 (IDE Reports)
21 CFR Part 314 - NDA
21 CFR 314.80 (Postmarketing)
21 CFR 314.98 (Generics)
21 CFR 600.80 (Biologics)
21 CFR 803 (Medical Devices)
ICH E2A (Oct 1994) ICH E2D (Nov 2003)
NIH Policy NIH Policy
Country/State Regulations Country/State Regulations
IRBs/ECs IRBs/ECs
Sponsor Sponsor 22

23
ICH: E Documents on SafetyICH: E Documents on Safety
Clinical Safety
ICH E1 – The Extent of Population Exposure to Assess Clinical Safety for Drugs Intended for Long-Term Treatment of Non-Life Threatening Conditions
ICH E2A – Clinical Safety Data Management: Definitions and Standards for Expedited Reporting
ICH E2B – Clinical Safety Data Management: Data Elements for Transmission of Individual Case Safety Reports
ICH E2C – Clinical Safety Data Management: Periodic Safety Update Reports for Marketed Drugs
ICH E2D – Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting
ICH E2E – Pharmacovigilance Planning
ICH E2F – Development Safety Update Report
Good Clinical Practice
ICH E6 – Good Clinical Practice
http://www.ich.org/cache/compo/276-254-1.html

24
Drug Development Model:Safety Data Flow in Clinical TrialsDrug Development Model:Safety Data Flow in Clinical Trials

25
Safety Data from Clinical TrialsSafety Data from Clinical Trials
Obligations to report data from DAIDS clinical trials irrespective of IND status. For both IND or Non-IND studies:
Data for non-expedited reporting: • recorded on AE CRF, goes to clinical trial database
Data for expedited reporting:
• recorded on AE CRF and linked to an SAE type form• goes to safety database
• IND timelines: 24o to sponsor, 7/15 days to FDA, EMEA
• Post-marketing timelines: depends on sponsor, 15 days to FDA, EMEA
Annual/Periodic Reports :• Need safety data from clinical and safety database• Must be reconciled
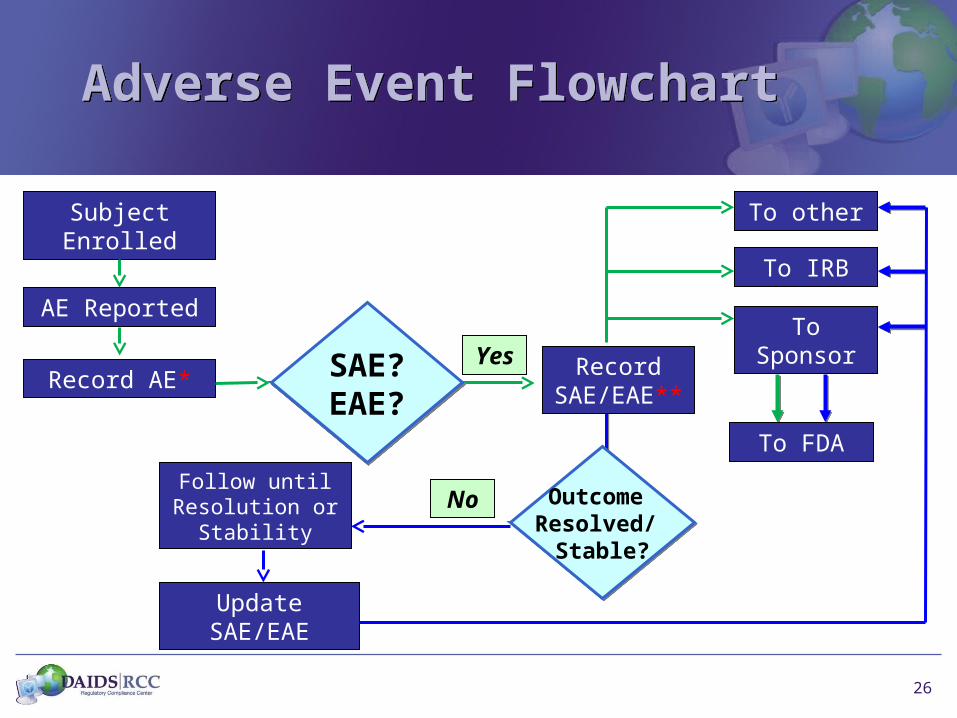
26
Adverse Event FlowchartAdverse Event Flowchart
Subject Enrolled
AE Reported
Record AE*YesSAE?
EAE?SAE?EAE?
To Sponsor
Record SAE/EAE**
To IRB
To FDA
To other
Follow until Resolution or
Stability
Outcome Resolved/
Stable?
Outcome Resolved/
Stable?
Update SAE/EAE
No

27
Adverse EventAdverse Event
* Protocol specifications for AE• When to collect e.g., study visit• Method of collection e.g., in person, telephone call• What to collect e.g., all AEs, only certain AEs by body
system, only certain AEs by severity• What forms to use e.g. AE CRF, study CRFs
** Protocol specifications for SAE/EAE• Criteria• Expedited time frames• Form e.g. SAE, EAE form
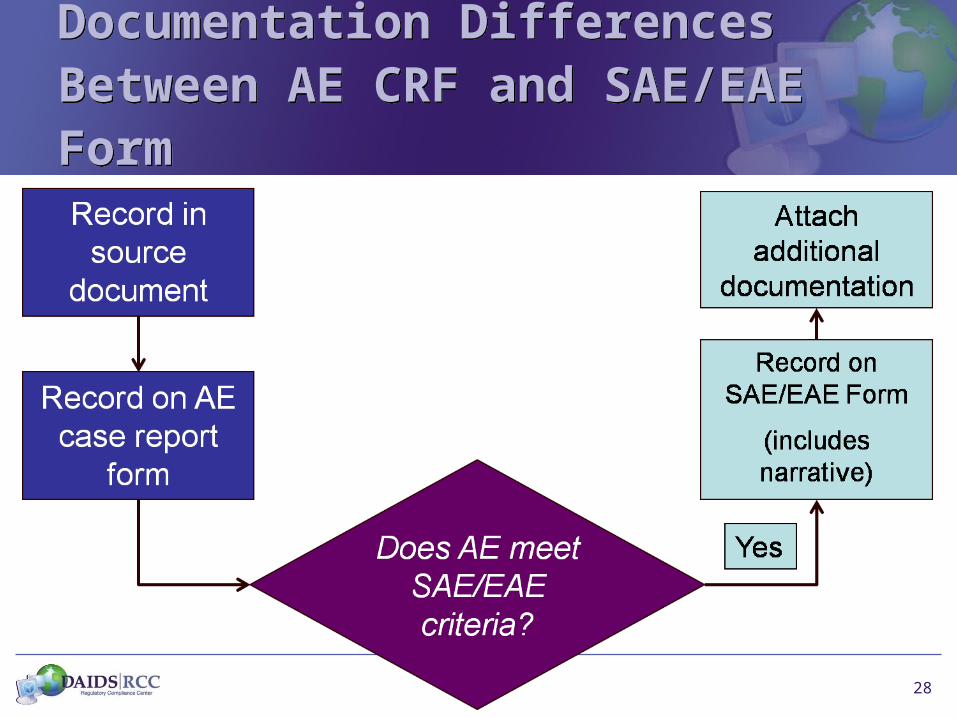
28
Documentation Differences Between AE CRF and SAE/EAE FormDocumentation Differences Between AE CRF and SAE/EAE Form

Documentation Differences Between AE CRF and SAE/EAE Form: Data Elements
Documentation Differences Between AE CRF and SAE/EAE Form: Data Elements
AE CRF Data Elements
• AE
• Start Date
• Start Date / Continuing
• Is it SAE?
• Severity
• Relatedness
• Action taken with Study Agent
• Outcome (study participation)
SAE/EAE Form Data Elements
• Participant Identifiers
• Study Agent details
• Narrative
• Past medical history
• Relevant labs, tests, procedures
• Concomitant meds
• Outcome of SAE/EAE
• Other supporting information
29

StrechBreakStrechBreak

31
Adverse EventAdverse Event
Any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have to have a causal relationship with this treatment.
(ICH E2A)

32
Adverse Event TermAdverse Event Term
The AE should best describe what the subject says (i.e. verbatim description)
Can be extracted from medical records
Can incorporate medical assessment (including a diagnosis if available)
The more accurate the AE term, the more accurate the safety database. The AE terms will be coded using a standard dictionary e.g. Medical Dictionary for Regulatory Activities (MedDRA); this is NOT site responsibility

33
AE Term - ExamplesAE Term - Examples
If “anaphylactic reaction” is reported with “rash, dyspnea, hypotension, and laryngospasm,” selecting “anaphylactic reaction” alone is considered appropriate.
If “myocardial infarction” is reported with “chest pain, dyspnea, diaphoresis, ECG changes and jaundice,” it would be considered appropriate to select terms for both “myocardial infarction” and “jaundice.”
EAE Form: List only one primary AE. Choose the one term that best describes the nature of the adverse event being reported.

34
Serious Adverse Event (SAE)Serious Adverse Event (SAE)
A serious adverse event (experience) or reaction is any untoward medical occurrence that at any dose:
Results in death,
Is life-threatening,
Requires inpatient hospitalization or prolongation of existing hospitalization,
Results in persistent or significant disability/incapacity, or
Is a congenital anomaly/birth defect.
In addition, “…important medical events that may not be immediately life-threatening or result in death or hospitalization but may jeopardize the patient or may require intervention to prevent one of the other outcomes listed in the definition above…should also usually be considered serious.”
(ICH E2A)

35
Adverse Event vs. Event OutcomeAdverse Event vs. Event Outcome
Death
Death is an outcome and is not usually considered to be an AE.
• Example: If “death due to myocardial infarction” is reported, “myocardial infarction” can be selected and death should be captured as the outcome.
If the only information reported is death, then the most specific death term available should be selected.
• Example: If a reporter states only that “a study subject was found dead,” “found dead” can be selected.

36
Adverse Event vs. Event OutcomeAdverse Event vs. Event Outcome
Hospitalization
Hospitalization is a consequence and is not usually considered an AE.
• Example: If “hospitalization due to congestive heart failure” is reported, “congestive heart failure” can be selected and hospitalization should be captured as the consequence of the event.
If the only information reported is the outcome term, then the most specific term available should be selected.
• Example: If a reporter states only that “a study subject was hospitalized,” “hospitalization” can be selected.

37
HospitalizationHospitalization
Hospitalization in the absence of a medical AE is not in itself an AE and does not need to be reported in an expedited time frame.
Admission for treatment of a pre-existing condition (can include target disease) not associated with the development of a new AE or with a worsening of the pre-existing condition
Diagnostic admission (e.g. for work-up of existing condition persistent such as pre-treatment lab abnormality)
Protocol-specified admission (e.g. procedure required by study protocol)
Administrative admission (e.g. for yearly physical exam)
Social admission (e.g. study subject has no place to sleep)
Elective admission (e.g. elective surgery)

38
SeveritySeverity
Describes the intensity of the event
Events are graded on a severity scale
Example:
• Mild, Moderate, Severe
• Numeric Scale e.g. 1 to 5

39
Severity vs. SeriousSeverity vs. Serious
Severity is not the same as Serious
Severity = Intensity
• e.g., chest pain, moderate severity
Serious (SAE) = Based on patient/event outcome or action criteria
• Used to define regulatory reporting obligations
AE classification: must divorce usage of serious in a clinical sense from serious in a regulatory sense
For clinical connotations, use severity descriptors

40
Action Taken with DrugAction Taken with Drug
Action Taken with Drug:
• Withdrawn
• Dose reduced
• Dose increased
• Dose not changed
• Unknown
• Not applicable
Refer to protocol
Refer to DAIDS EAE Form
> ICH E2B (R3)

41
OutcomeOutcome
Outcome of reaction/event at the time of last observation • Recovered/resolved• Recovering/resolving• Not recovered/not resolved• Recovered/resolved with sequelae• Fatal• Unknown
Outcome of subject in study• Remains in Study• Withdrawn• Lost to follow-up• Death

42
ExpectednessExpectedness
Pertains to whether an event is expected or unexpected (on the basis of previous observation, not what might be anticipated from the pharmacological properties of the product)
Unexpected: the nature or severity of the adverse event is not consistent with the applicable product information (e.g. Investigator’s Brochure for unapproved product, Package Insert for approved product)

43
Relatedness (Causality)Relatedness (Causality)
No standard international nomenclature
Conveys that a “causal relationship” between the study product and the adverse event is “at least a reasonable possibility” [ICH E2A]
• Facts (evidence) exist to suggest the relationship
• Information on SAEs/EAEs generally incomplete when first received
• Follow-up information actively pursued
Judged by:
• Reporting health professional
• Sponsor

44
Standard determinations include:
• Is there [Drug Exposure] and [Temporal Association]?
• Is there [Dechallenge/Rechallenge] or [Dose Adjustments]?
• Any known association per [Investigator’s Brochure] or [Package Insert]?
• Is there [Biological Plausibility]?
• Any other possible [Etiology]?
Determination of RelatednessDetermination of Relatedness

45
Determination of RelatednessDetermination of Relatedness
‘May be more art than science’• 1st level of review: NR• 2nd level of review: any degree R• 3rd level of review: any evidence to compel moving in one
direction or the other; i.e. from NR to any reasonable possibility of R, or from any R to NR
Clear-cut case; easy to make a determination
Not so clear-cut: use your best judgment based on available information; assure adequacy of information; obtain more whenever
Unless clear-cut case, there’s no absolute right or wrong; give your best judgment; substantiate and follow-up
Err on conservative side; contribute to knowledge base

46
Comprehensive, stand-alone “medical story”• Written in logical time sequence• Include key information from supplementary records• Include relevant autopsy or post-mortem findings
Summarize all relevant clinical and related information, including:• Study subject characteristics• Therapy details• Medical history• Clinical course of the event(s)• Diagnosis (workup, relevant tests/procedures, lab results)• Other information that supports or refutes an AE
> ICH E2D
NarrativeNarrative

47
Narrative TemplateNarrative Template
This is a [Age] year old [Race] [Male/Female] in [Study] who reported [Primary AE] on [Date of AE]. Enrolled into study on [Date Enrolled], Study medication was started on [Date], which is [Study Day _/Week _], taken for [Duration]. The event occurred during the [Treatment/Follow-up Phase].
If fetus: provide [Gestational Age], (or mother’s LMP), at time of event. Also, [Gestational Age/Trimester] at first drug exposure and duration of exposure. If birth, provide details of [Infant Status] at birth. If hospital stay is complicated, provide details of hospital stay.
Provide details of the [AE] in chronological order, along with other [Signs/Symptoms]. Provide details of [Physical Exam], along with all relevant [Procedures] and [Lab Results].

48
Narrative TemplateNarrative Template
Provide details of [Treatment] and [Treatment Rationale] on basis of [Findings/Test Result(s)]. Describe [Treatment Response].
If hospitalization, provide [Dates Hospitalization], describe relevant [Hospital Course], [Diagnostic Work-up], [Procedures/Tests and Results], [Treatment], [Treatment Response].
Provide [Discharge Diagnosis], and any [Follow-up Information]. List [Discharge Meds].
Provide pertinent [Past Medical Hx], [Family Hx], [Concomitant Meds], [Alcohol/Tobacco/Substance Use] and any previous similar [AEs].

49
Review and Assessment of SAE/EAEReview and Assessment of SAE/EAE
Assemble all information available and use medical judgment
Standard for each AE:
• Select [Seriousness Criteria]
• Grade [Severity] per DAIDS Toxicity Table
• Specify [Actions Taken on Study Product]
• Specify [Outcome of SAE/EAE]. If Outcome is not resolved at time of evaluation, follow until resolution or stability at each study visit
• Is it [Expected]?
• Is it [Related]?

50
Sponsor role: (ICH E2D)
• Information about the case should be collected from the healthcare professionals who are directly involved in the study subject’s care
• Clearly identified evaluations by the sponsor are considered appropriate and are required by some regulatory authorities
• Opportunity to render another opinion; may be in disagreement with; and/or provide another alternative to the diagnosis/assessment given by initial reporter
• Sponsor makes an assessment of causality (attribution) just as the PI makes an assessment of causality (attribution)
• If causality (attribution) is different between the sponsor and the investigator, both assessments are reported
Clinical Case EvaluationClinical Case Evaluation

51
Site vs. Sponsor AssessmentSite vs. Sponsor Assessment
Balancing Both RolesSite Assessment Sponsor Assessment
• Site advantage: has access to subject; may elicit further info, perform PE, obtain tests, labs, records
• Information from self-report (may lack validation)
• Know subject best
• Judgment stands
• Open to dialog with sponsor
• Information limited to what was submitted from site
• May initiate queries to site: incur time and delay
• Constraint: Must adhere to reporting timelines to FDA
• MO level: Serious? Unexpected? Related?
• SPT level: sign-off, agree with Site PI, agree with DAIDS MO. Any critical flaw in reasoning?
• Open to dialog with Site PI, DAIDS MO

52
AcknowledgementsAcknowledgements
Safety Teams at DAIDS/SPT and RCC, especially:
• Larry Allan
• Adeola Adeyeye, and
• Daniel Dondero
DAIDS Training Group, especially
• Jane Reynolds
Members present at this Workshop:
• DAIDS/SPT: Larry Allan, Ling Chin
• RCC: Oluwadamilola Ogunyankin, Albert Yoyin, Anu Jasti, Sandy Butler, Nadine Pakker





























