Embed Size (px)
Citation preview

MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedSample Type: EDTA BloodDate of Collection: 02/06/2014Date Received: 02/16/2014Date Tested: 02/16/2014Barcode: 11200000000334Indication: Screening for geneticdisease carrier status
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedSample Type: EDTA BloodDate of Collection: 02/06/2014Date Received: 02/16/2014Date Tested: 02/16/2014Barcode: 11200000000223Indication: Screening for geneticdisease carrier status
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul Smith123 Main StreetCity, CA 10231Phone: (800) 555-1212Fax: (800) 555-1212NPI: 3652760645Report Date: 02/18/2014
Family Prep Screen POSITIVE: CARRIER AT RISK FOR SYMPTOMSABOUT THIS TEST
The Counsyl Family Prep Screen (version 2.0) utilizes sequencing,maximizing coverage across all DNA regions tested, to help you learnabout your chance to have a child with a genetic disease.
PANEL DETAILS
Universal Plus + Walker-Warburg Syndrome with Fragile X Syndrome(108 conditions tested)
VERSION
JANE MILLER (Family Prep Screen 2.0)JOHN MILLER (Family Prep Screen 2.0)
RESULTS SUMMARY
Risk Details JANE MILLER JOHN MILLER
POSITIVE: CARRIER APOSITIVE: CARRIER AT RISK FOR ST RISK FOR SYYMPTMPTOOMSMS
Factor XI DeficiencyReproductive Risk: < 1 in 2,100Inheritance: Autosomal Recessive
NEGATIVENo disease-causing mutationsdetected.
CARRIER AT RISK FORSYMPTOMSNM_000128.3(F11):c.901T>C(F301L,aka F283L) heterozygote †
POSITIVE: CARRIERPOSITIVE: CARRIER
Wilson DiseaseReproductive Risk: < 1 in 570Inheritance: Autosomal Recessive
CARRIER*
NM_000053.3(ATP7B):c.3207C>A(H1069Q) heterozygote
NEGATIVENo disease-causing mutationsdetected.
POSITIVE: CARRIERPOSITIVE: CARRIER
Smith-Lemli-Opitz SyndromeReproductive Risk: < 1 in 640Inheritance: Autosomal Recessive
NEGATIVENo disease-causing mutationsdetected.
CARRIER*
DHCR7{NM_001360.2}:c.111G>Aheterozygote †
POSITIVE: MILD CPOSITIVE: MILD CONDITIONONDITION
MTHFR DeficiencyReproductive Risk: Not CalculatedInheritance: Autosomal Recessive
MILD CONDITIONNM_005957.4(MTHFR):c.665C>T(A222V) heterozygoteNM_005957.4(MTHFR):c.1286A>C(E429A) heterozygote
MILD CONDITIONNM_005957.4(MTHFR):c.1286A>C(E429A) heterozygote
POSITIVE: MILD CPOSITIVE: MILD CONDITIONONDITION
HFE-associatedHereditary HemochromatosisReproductive Risk: Not CalculatedInheritance: Autosomal Recessive
NEGATIVENo disease-causing mutationsdetected.
MILD CONDITIONNM_000410.3(HFE):c.845G>A(C282Y) heterozygote
†Likely to have a negative impact on gene function.*Carriers generally do not experience symptoms.
No disease-causing mutations were detected in any other gene tested. A complete list of all conditions tested can be found on page 16.
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 1 of 21Version: 3.2.5

CLINICAL NOTES
• Ethnicity unknown or not reported for JANE and JOHN. Riskcalculation is based on the assumption of Northern Europeanancestry.
NEXT STEPS
• Patients may wish to discuss any positive results with blood relatives,as there is an increased chance that they are also carriers.
• To schedule a complimentary appointment with a genetic counselor,visit counsyl.com/my/consults/.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 2 of 21Version: 3.2.5

POSITIVE: CARRIER AT RISK FOR SYMPTOMS
Factor XI DeficiencyGene: F11 | Inheritance Pattern: Autosomal Recessive
Reproductive risk: < 1 in 2,100Risk before testing: < 1 in 1,000,000
Patient JANE MILLER JOHN MILLER
Result Negative Carrier At Risk for Symptoms
Variant(s) No disease-causing mutations detected. NM_000128.3(F11):c.901T>C(F301L, aka F283L)heterozygote †
Methodology Targeted genotyping and sequencing Targeted genotyping and sequencing
Interpretation This does not rule out the possibility of being acarrier. The post‑test risk of being a carrier,assuming a negative family history, is < 1 in 500.
This individual is a carrier of factor XI deficiency.Carriers may be at risk for bleeding problems.
JANE MILLER's detection rate: > 10%. JOHN MILLER's detection rate: > 10%.Gene: F11. Variants Genotyped (4): E135*, F301L, c.1716+1G>A, c.1714_1716+11del14. Exons Sequenced: NM_000128:1-15.
†Likely to have a negative impact on gene function.
What is Factor XI Deficiency?Factor XI deficiency, also called factor 11 deficiency or hemophilia C, is an inherited disorder which is usually mild but can cause uncontrolledbleeding. This bleeding tends to be more severe after surgery, injury, or childbirth. Bleeding can be a particular problem after tooth extraction,dental surgery, tonsil surgery, or urinary tract surgery.
People with factor XI deficiency may also be prone to bruising, nosebleeds, or blood in their urine. Rarely, male children with the disease willbleed heavily following circumcision. More than half of women with the disease have abnormally heavy and prolonged menstrual periods.
It is uncommon for people with factor XI deficiency to bleed spontaneously for no obvious reason. There may be a delay in bleeding following aninjury.
Factor XI is a protein produced by the liver and found in the blood. It helps platelets in the blood to clot following an injury to a blood vessel. Inpeople with factor XI deficiency, levels of factor XI are lower than normal. Bleeding problems tend to occur when factor XI levels are lower than15% of the normal level. Bleeding problems can occur, however, when levels are as high as 70% of the normal level. The severity of the bleedingvaries widely from person to person, even among members of the same family.
Carriers of factor XI deficiency are at elevated risk for bleeding problems. Studies have suggested that 20 to 50% of carriers of the disease show"excessive bleeding," although the definition of this phrase varies. Rarely, carriers have shown major bleeding problems.
How Common is Factor XI Deficiency?Factor XI deficiency is fairly common among Ashkenazi Jews. One in eight Ashkenazi Jews is thought to be a carrier of factor XI deficiency. In onestudy, 1 in 190 Ashkenazi Jews has a severe bleeding problem related to factor XI deficiency, while a different study found a much lower rate ofsevere symptoms, 1 in 450. Among non-Jews in the United States, only 1 in 1,000,000 have factor XI deficiency, making it very rare.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 3 of 21Version: 3.2.5

The disease is also common among families in northwest England, where 1 in 10,000 people has the disease. Other groups at greater risk forcarrying mutations that cause factor XI deficiency are Iraqi Jews, Sephardic Jews, and people of Arab background living in Israel.
How is Factor XI Deficiency Treated?In the United States, factor XI deficiency is treated with infusions of fresh frozen blood plasma. This blood plasma contains normal quantities offactor XI, thus temporarily enhancing the body’s ability to clot. However, significant amounts of plasma may be required to achieve the desiredclotting effect due to the low concentration of factor XI in plasma.
In Europe, there are several commercially available concentrated doses of factor XI. One is manufactured in the United Kingdom, the other inFrance.
In the case of bleeding in the mouth, nose, intestines, or uterus, there are several medications which may be helpful, though they are noteffective for major internal bleeding and can cause clotting throughout the body.
What is the Prognosis for Someone With Factor XI Deficiency?Factor XI deficiency is not known to affect lifespan. In people who do not realize they have the disease, life-threatening bleeding is possiblefollowing surgery or injury.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 4 of 21Version: 3.2.5

POSITIVE: CARRIER
Wilson DiseaseGene: ATP7B | Inheritance Pattern: Autosomal Recessive
Reproductive risk: < 1 in 570Risk before testing: 1 in 30,000
Patient JANE MILLER JOHN MILLER
Result Carrier Negative
Variant(s) NM_000053.3(ATP7B):c.3207C>A(H1069Q)heterozygote
No disease-causing mutations detected.
Methodology Targeted genotyping and sequencing Targeted genotyping and sequencing
Interpretation This individual is a carrier of Wilson disease.Carriers generally do not experience symptoms.
This does not rule out the possibility of being acarrier. The post‑test risk of being a carrier,assuming a negative family history, is < 1 in 140.
JANE MILLER's detection rate: > 40%. JOHN MILLER's detection rate: > 40%.Gene: ATP7B. Variants Genotyped (2): H1069Q, R778L. Exons Sequenced: NM_000053:1-3,5-21.
What is Wilson Disease?Wilson disease is an inherited disease that causes the body to retain too much copper. Copper deposits in the liver, brain, kidneys, and eyeseventually cause tissue damage and scarring that makes the affected organs stop working properly. If not diagnosed and treated early, thecondition causes organ failure and death.
Symptoms typically first appear in childhood or early adolescence, but they can appear as early as age 3 and as late as age 70. The most commonsymptom is liver disease, which first appears as fatigue, abdominal pain, or jaundice. In some cases, it progresses quickly to liver or kidneyfailure, and will require a liver transplant.
Symptoms can also include neurological problems such as tremors, poor coordination, loss of fine motor skills, problems walking, muscle rigidityin the body or face, or difficulty swallowing. Some people with the condition also develop psychiatric problems including depression, poorimpulse control, phobias, aggression, and compulsive behavior. Wilson disease may also interfere with memory and attention span.
Copper deposits also accumulate in the eyes, creating characteristic brown circles around the colored part of the eye. These circles do notinterfere with vision.
Even with ongoing treatment to remove excess copper from the body, people with Wilson disease sometimes develop arthritis, heart problems,and endocrine disorders caused by copper accumulation.
How Common is Wilson Disease?Worldwide, approximately 1 in 30,000 people have Wilson disease. It is most common in China, Japan, and Sardinia, where it may affect as manyas 1 in 10,000 people.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 5 of 21Version: 3.2.5

How is Wilson Disease Treated?Wilson disease requires lifelong, regular treatment to remove copper from the body. Most people with the condition take a medication calledpenicillamine (brand name: Cuprimine or Depen) several times a day by mouth, combined with vitamin B6. This traps and removes copper fromthe body through the urine. However, some people react to penicillamine with fever, rash, and other serious complications. These people may betreated with other oral medications such as trientine or high-dose zinc. For individuals who do not respond to medication or have severe sideeffects, liver transplant is a final treatment option.
With careful treatment prior to the first symptom's appearance, most symptoms can be prevented. If treatment begins after symptoms appear,these symptoms can often show marked improvement. Stopping treatment, however, will cause health problems to return.
People with Wilson disease should not use copper cooking utensils. They should avoid foods high in copper, such as liver, chocolate, mushrooms,nuts, and shellfish. If they live in an area with copper water pipes, they should drink distilled water.
What is the Prognosis for Someone With Wilson Disease?With proper treatment, Wilson disease can be managed for many years after diagnosis. Its effect on lifespan is unclear.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 6 of 21Version: 3.2.5
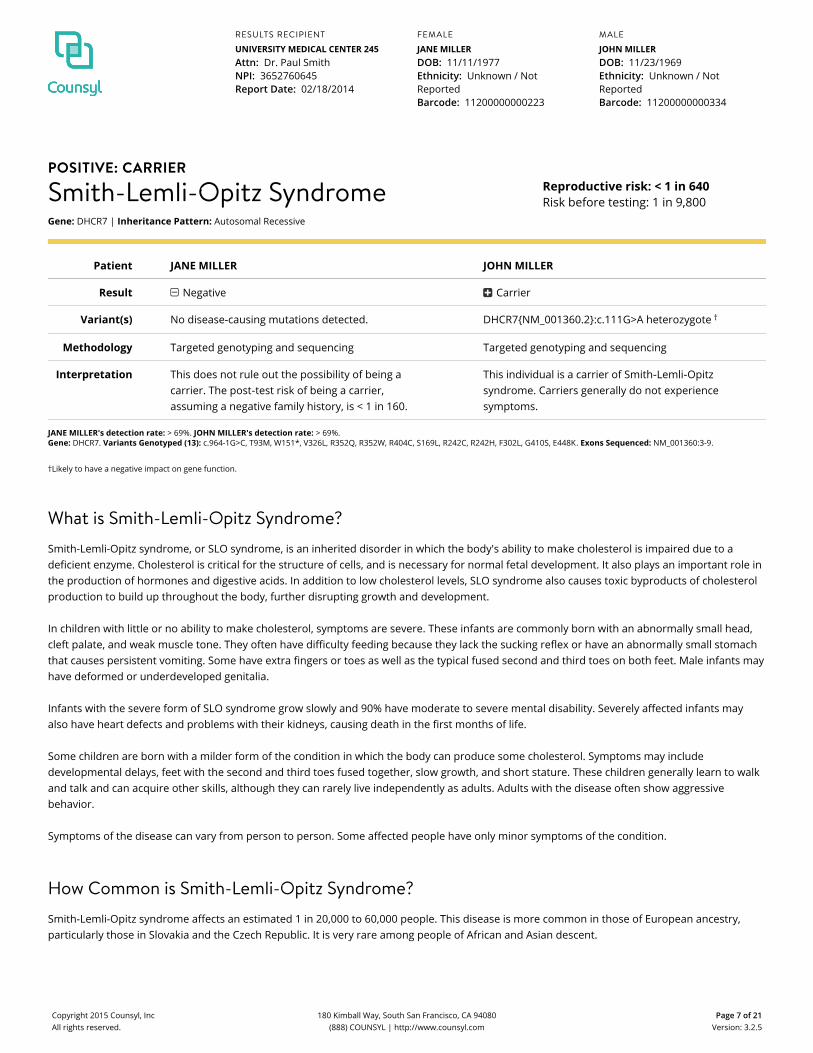
POSITIVE: CARRIER
Smith-Lemli-Opitz SyndromeGene: DHCR7 | Inheritance Pattern: Autosomal Recessive
Reproductive risk: < 1 in 640Risk before testing: 1 in 9,800
Patient JANE MILLER JOHN MILLER
Result Negative Carrier
Variant(s) No disease-causing mutations detected. DHCR7{NM_001360.2}:c.111G>A heterozygote †
Methodology Targeted genotyping and sequencing Targeted genotyping and sequencing
Interpretation This does not rule out the possibility of being acarrier. The post‑test risk of being a carrier,assuming a negative family history, is < 1 in 160.
This individual is a carrier of Smith‑Lemli‑Opitzsyndrome. Carriers generally do not experiencesymptoms.
JANE MILLER's detection rate: > 69%. JOHN MILLER's detection rate: > 69%.Gene: DHCR7. Variants Genotyped (13): c.964-1G>C, T93M, W151*, V326L, R352Q, R352W, R404C, S169L, R242C, R242H, F302L, G410S, E448K. Exons Sequenced: NM_001360:3-9.
†Likely to have a negative impact on gene function.
What is Smith-Lemli-Opitz Syndrome?Smith-Lemli-Opitz syndrome, or SLO syndrome, is an inherited disorder in which the body's ability to make cholesterol is impaired due to adeficient enzyme. Cholesterol is critical for the structure of cells, and is necessary for normal fetal development. It also plays an important role inthe production of hormones and digestive acids. In addition to low cholesterol levels, SLO syndrome also causes toxic byproducts of cholesterolproduction to build up throughout the body, further disrupting growth and development.
In children with little or no ability to make cholesterol, symptoms are severe. These infants are commonly born with an abnormally small head,cleft palate, and weak muscle tone. They often have difficulty feeding because they lack the sucking reflex or have an abnormally small stomachthat causes persistent vomiting. Some have extra fingers or toes as well as the typical fused second and third toes on both feet. Male infants mayhave deformed or underdeveloped genitalia.
Infants with the severe form of SLO syndrome grow slowly and 90% have moderate to severe mental disability. Severely affected infants mayalso have heart defects and problems with their kidneys, causing death in the first months of life.
Some children are born with a milder form of the condition in which the body can produce some cholesterol. Symptoms may includedevelopmental delays, feet with the second and third toes fused together, slow growth, and short stature. These children generally learn to walkand talk and can acquire other skills, although they can rarely live independently as adults. Adults with the disease often show aggressivebehavior.
Symptoms of the disease can vary from person to person. Some affected people have only minor symptoms of the condition.
How Common is Smith-Lemli-Opitz Syndrome?Smith-Lemli-Opitz syndrome affects an estimated 1 in 20,000 to 60,000 people. This disease is more common in those of European ancestry,particularly those in Slovakia and the Czech Republic. It is very rare among people of African and Asian descent.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 7 of 21Version: 3.2.5

How is Smith-Lemli-Opitz Syndrome Treated?There is no cure for SLO syndrome, but its symptoms can be addressed. The primary treatment is to supplement the person's diet with largeamounts of dietary cholesterol, either in the form of purified cholesterol or in foods such as egg yolks and cream. This has been shown toimprove symptoms. Early intervention and therapy helps with speech and physical disabilities. Medication may treat symptoms such as vomiting,constipation, and gastroesophageal reflux. Surgery and orthotics can help muscle spasms and improve mobility.
Because the condition can cause extreme sun sensitivity, people with SLO syndrome should always wear sunblock, sunglasses, and appropriateclothing when they go outdoors.
What is the Prognosis for Someone With Smith-Lemli-Opitz Syndrome?Although serious internal malformations can lead to early death, with good nutrition and medical care many people with SLO syndrome can havea normal lifespan. Mental disability typically prevents people with this disease from living independently.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 8 of 21Version: 3.2.5

POSITIVE: MILD CONDITION
MTHFR DeficiencyGene: MTHFR | Inheritance Pattern: Autosomal Recessive
Patient JANE MILLER JOHN MILLER
Result Mild Condition Mild Condition
Variant(s) NM_005957.4(MTHFR):c.665C>T(A222V)heterozygoteNM_005957.4(MTHFR):c.1286A>C(E429A)heterozygote
NM_005957.4(MTHFR):c.1286A>C(E429A)heterozygote
Methodology Targeted genotyping Targeted genotyping
Interpretation This result is consistent with a diagnosis of MTHFRdeficiency.
This individual is a carrier of MTHFR deficiency.Carriers generally do not experience symptoms.
Gene: MTHFR. Variants Genotyped (2): A222V, E429A.
What is MTHFR Deficiency?MTHFR deficiency is a mild condition associated with a slightly higher risk of neural tube defects and pregnancy loss. Roughly 40% of Americansare carriers of MTHFR deficiency, while 10% have the condition. For the vast majority, it causes no problems with their health or the health oftheir children. The mild MTHFR deficiency for which Counsyl tests should not be confused with severe MTHFR deficiency.
The MTHFR enzyme is involved in the conversion of the amino acid homocysteine to another amino acid, methionine. People with mild MTHFRdeficiency have a decreased ability to perform this conversion, and as a result they have higher levels of homocysteine in their body and lowerlevels of the vitamin folate.
There are two common mutations found in the MTHFR gene, A222V and E429A (also known as C677T and A1298C respectively). Having one copyof either mutation (being a carrier) is not thought to have any impact on one’s health or that of one’s children.
The following gene combinations may be significant. Note that they are only significant if they raise the level of homocysteine in the blood, andthis does not happen in everyone:
A222V/A222VFor women, two copies of the A222V mutation has been associated with a 2 to 3-fold higher risk of having a child with severe neural tube defectssuch as spina bifida. This type of birth defect normally affects 1 in 1,000 births, so women with two A222V mutations would face a 2 to 3 in 1,000risk, which is still low.
Taking folate supplements has been shown to reduce neural tube defects by 75% and may be doing so by normalizing levels of homocysteine inthe body.
Some studies have shown that mild MTHFR deficiency may be beneficial in lowering the risks for colorectal cancer or boosting immunity tocertain pathogens. These findings are tentative, however.
A222V/E429AThis mutation pairing is thought to share the same risks as described above for the A222V/A222V mutation pairing.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 9 of 21Version: 3.2.5

E429A/E429AThis mutation pairing is NOT associated with any elevated risks for health problems.
How Common is MTHFR Deficiency?Mild MTHFR deficiency is very common. The table below shows the results of numerous studies conducted worldwide looking at the A222Vmutation. The first number represents the percentage of the population who is a carrier and the second number represents the percentage ofthe population thought to be affected.
Ethnic Group Carrier Rate Affected Rate
Hispanic American 48% 15%
Caucasian American 45% 12%
Japanese 45% 12%
German 37% 6%
Asian 29% 3%
African American 24% 2%
Sub-Saharan African 12% 1%
The E429A mutation is less well-studied, but is also thought to be quite common. In three studies, the A222V/E429A mutation pairing was foundin 17% of Americans, 15% of Canadians, and 20% of Dutch people.
How is MTHFR Deficiency Treated?Most people with mild MTHFR deficiency require no treatment. Because food in the United States is often fortified with vitamins, most people eatsufficient amounts of folate to compensate for higher levels of homocysteine.
Pregnant women with the condition—and all pregnant women—are advised to take folate supplements (folic acid) before and during pregnancyto reduce the risk of birth defects by as much as 75 to 85%. These vitamins are particularly important for women with MTHFR deficiency.
What is the Prognosis for Someone With MTHFR Deficiency?Based on current scientific knowledge, most people with MTHFR deficiency will be totally unaffected by it. Women face a slightly elevated risk ofhaving a child with neural tube defects, however the risk is still low.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 10 of 21Version: 3.2.5

POSITIVE: MILD CONDITION
HFE-associated Hereditary HemochromatosisGene: HFE | Inheritance Pattern: Autosomal Recessive
Patient JANE MILLER JOHN MILLER
Result Negative Mild Condition
Variant(s) No disease-causing mutations detected. NM_000410.3(HFE):c.845G>A(C282Y) heterozygote
Methodology Targeted genotyping Targeted genotyping
Interpretation This does not rule out the possibility of being acarrier.
This individual is a carrier of HFE‑associatedhereditary hemochromatosis. Carriers generally donot experience symptoms. Clinical symptoms arealso uncommon in C282Y homozygotes.
Gene: HFE. Variants Genotyped (2): C282Y, H63D.
What is HFE-associated Hereditary Hemochromatosis?HFE-associated hereditary hemochromatosis (HFE-HHC) is a common and treatable inherited disease in which the body absorbs and stores toomuch iron, potentially damaging organs such as the liver, heart, and pancreas. If the disease is diagnosed and treated before symptoms develop,people with HFE-HHC typically have a normal lifespan. If the disease is untreated, however, it can lead to fatal liver and heart failure.
For reasons not well understood, the majority of people with the genetic mutations that cause HFE-HHC do not develop symptoms of the diseaseat any point in their lives. For these people, simple blood tests can determine whether or not the body is storing too much iron. If it is, beginningtreatment early can leave a person virtually symptom-free for life.
Depending on the specific mutation(s) a person has, he or she can be more or less likely to develop the iron overload symptoms of HFE-HHC.
People who have two copies of the C282Y mutation are most likely to have dangerously elevated levels of iron in their blood. Studies have foundthat among those with the C282Y/C282Y combination, men are more likely to develop symptoms of iron overload than women, perhaps becausewomen's menstrual cycles lower their iron levels on a regular basis. Do keep in mind, however, that the majority of people with two copies of theC282Y mutation do not develop any symptoms of HFE-HHC.
Those who have C282Y in combination with another HFE-HHC mutation are much less likely to develop symptoms of the disease. Only 0.5% to2% of people with C282Y in combination with another mutation are thought to have clinical signs of the disease. People with this geneticcombination who have another disease of the liver may be more likely to develop HFE-HHC symptoms.
Among people who have two copies of any other HFE-HHC mutation, including a very common mutation known as H63D, the likelihood ofdeveloping symptoms is extremely low. In the absence of another liver disease, two copies of any HFE-HHC mutation other than C282Y is unlikelyto cause any health problems.
In men who have not been treated for HFE-HHC, the first symptoms of the disease typically begin between the ages of 30 to 50; for untreatedwomen, symptoms usually begin later, after menopause.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 11 of 21Version: 3.2.5

Early symptoms often include weakness, abdominal pain, joint pain, weight loss, loss of interest in sex, chest pain, and a progressive gray orbronze pigmentation to the skin. Liver disease (either fibrosis or the more serious cirrhosis) is a common problem associated with HFE-HHC.Cirrhosis can lead to fatal liver failure and/or an increased likelihood of developing cancer of the liver.
The heart can also be affected by HFE-HHC, seen as an irregular heartbeat and/or congestive heart failure. Other problems caused by HFE-HHCcan include diabetes, arthritis, impotence (in men), early menopause (in women), thyroid problems, and adrenal gland problems.
How Common is HFE-associated Hereditary Hemochromatosis?HFE-HHC mutations are extremely common, particularly among Caucasians. Roughly one-third (33%) of Caucasians are carriers of the condition,most commonly the H63D mutation. The H63D mutation is almost always associated with asymptomatic cases unless paired with the C282Ymutation. In the general population, 1 in 200 to 400 has two copies of the C282Y genetic mutation, the combination of mutations which is mostlikely to cause symptoms of HFE-HHC.
Please bear in mind that most people who have these genetic mutations do not develop the disease.
The disease is less common among Hispanics, African Americans, Asians, and Native Americans. Roughly 13% of Hispanics, 8.5% of Asians, and6% of African Americans is a carrier for the mild mutation, H63D. An additional 3% of Hispanics, 2.3% of African Americans are carriers of thepotentially disease-causing C282Y mutation.
How is HFE-associated Hereditary Hemochromatosis Treated?Ideally HFE-HHC is treated before the organs of the body are damaged. However, not everyone who has the mutations that cause HFE-HHCdevelops symptoms or requires treatment. A simple blood test of iron levels in the blood—physicians specifically look at serum ferritinconcentration and transferrin-iron saturation levels—can determine whether the body is absorbing too much. When iron reaches a certainthreshold, treatment is recommended. If iron levels have not reached that threshold, no treatment is necessary. Blood tests must be repeatedperiodically to check these iron levels.
If a person has a high level of iron, treatment involves removing a certain quantity of blood at regular intervals. This is known as phlebotomy.Typically phlebotomy is performed frequently—perhaps weekly or twice weekly—until certain iron levels are reached, and then performed lessfrequently—often 2 to 4 times a year—on an indefinite basis. This treatment is simple, inexpensive, and safe.
If a person is already suffering from symptoms of HFE-HHC, treatment can lessen or relieve some of the symptoms. Cirrhosis is unlikely toimprove with treatment, although treatment may slow its progression. If liver disease has reached severe levels, liver transplantation may be anoption. Those who have any amount of liver damage are advised to avoid alcohol.
All people with symptoms of HFE-HHC are advised to eat only moderate amounts of iron-rich foods, avoid taking iron supplements or excessvitamin C, and refrain from eating uncooked seafood, as they are highly susceptible to a particular kind of bacterial infection.
What is the Prognosis for Someone With HFE-associated Hereditary Hemochromatosis?The prognosis for a person with the genetic mutations that cause HFE-HHC is generally good, as the majority of people in that situation do notdevelop symptoms of the disease. Most will not have dangerously elevated levels of iron in their blood, and therefore will not have any iron-overload problems.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 12 of 21Version: 3.2.5

For those that do have dangerously high iron levels in their blood, beginning treatment before symptoms appear is a critical part of ensuring along, healthy life. Nearly all symptoms of the disease can be prevented with early and ongoing treatment. If a person with HFE-HHC is treatedbefore he or she develops cirrhosis of the liver, he or she can expect a normal lifespan. Among people who already have cirrhosis associatedwith HFE-HHC, 72% will survive at least 5 more years and 62% will survive at least 10 more years. People who already have cirrhosis are at anincreased risk for developing a type of liver cancer.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 13 of 21Version: 3.2.5

Methods and Limitations
JANE MILLER [Family Prep Screen 2.0]: sequencing, targeted genotyping, triplet repeat detection, and copy number analysis.JOHN MILLER [Family Prep Screen 2.0]: sequencing, targeted genotyping, and copy number analysis.
Targeted genotyping: Targeted DNA mutation analysis is used to simultaneously determine the genotype of 412 variants associated with 106 diseases. The test isnot validated for detection of homozygous mutations, and although rare, asymptomatic individuals affected by the disease may not be genotyped accurately.
Sequencing: High-throughput sequencing is used to analyze 1435 exons in 99 genes, as well as selected intergenic and intronic regions. These regions are sequencedto high coverage and the sequences are compared to standards and references of normal variation. Mutations may not be detected in areas of lower sequencecoverage. On average, more than 99% of all bases in the exons listed for each gene are sequenced at the minimum read depth. Variants discovered in other exons ofthese genes will also be reported if they meet quality control criteria. Triplet repeats and large deletions and duplications may not be detected. Small insertions anddeletions may not be as accurately determined as single nucleotide variants. Genes that have closely related pseudogenes are not well analyzed by this method.
High-throughput sequencing detects, on average, 94% of known clinically significant variants. Disease-specific detection rates and residual risks are reported as"greater than (>)" and "less than (<)" the values for targeted genotyping, respectively. More precise values are not currently available, but may become available in thefuture.
All variants that are a recognized cause of the disease will be reported. In addition, variants that have not previously been established as a recognized cause ofdisease may be identified. In these cases, only variants classified as "predicted" or "likely" pathogenic are reported. Predicted/likely pathogenic variants are describedelsewhere in the report as "predicted/likely to have a negative impact on gene function". In general, predicted pathogenic variants are those which are predicted to bepathogenic based on the nature of the sequence change, while likely pathogenic variants are evaluated by reviewing reports of allele frequencies in cases andcontrols, functional studies, variant annotation and effect prediction, and segregation studies. Benign variants, variants of uncertain significance, and variants notdirectly associated with the intended disease phenotype are not reported. Literature citations validating reported variants are available upon request.
Triplet repeat detection: PCR is used to size the CGG repeat in the 5' UTR of FMR1 (NM_002024.4: c.1-131CGG[1_n]). PCR products generated from fluorescentlylabeled primers are detected by capillary electrophoresis. Reported sizes are accurate to +/- 1 repeat for up to 200 repeats. Alleles above 200 CGG repeats (fullmutations), while identified, are not sized. Nearby mutations may interfere with detection of CGG expansions. Deletion of the CGG repeat and other FMR1 mutationsmay not be detectable. Methylation will not be detected. Small degrees of size mosaicism, including gonadal mosaicism, will not be detected as the test has beencalibrated to yield results that are equivalent to the results from Southern blot.
Copy number analysis: Targeted copy number analysis is used to determine the copy number of exon 7 of the SMN1 gene relative to other genes. Other mutationsmay interfere with this analysis. Some individuals with two copies of SMN1 are carriers with two SMN1 genes on one chromosome and a SMN1 deletion on the otherchromosome. In addition, a small percentage of SMA cases are caused by nondeletion mutations in the SMN1 gene. Thus, a test result of two SMN1 copiessignificantly reduces the risk of being a carrier; however, there is still a residual risk of being a carrier and subsequently a small risk of future affected offspring forindividuals with two or more SMN1 gene copies. Some SMA cases arise as the result of de novo mutation events which will not be detected by carrier testing.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 14 of 21Version: 3.2.5

Limitations: In an unknown number of cases, nearby genetic variants may interfere with mutation detection. Other possible sources of diagnostic error includesample mix-up, trace contamination, bone marrow transplantation, blood transfusions and technical errors. If more than one variant is detected in a gene, additionalstudies may be necessary to determine if those variants lie on the same chromosome or different chromosomes. The Counsyl test does not fully address all inheritedforms of intellectual disability, birth defects and genetic disease. A family history of any of these conditions may warrant additional evaluation. Furthermore, not allmutations will be identified in the genes analyzed and additional testing may be beneficial for some patients. For example, individuals of African, Southeast Asian, andMediterranean ancestry are at increased risk for being carriers for hemoglobinopathies, which can be identified by CBC and hemoglobin electrophoresis or HPLC(ACOG Practice Bulletin No. 78. Obstet. Gynecol. 2007;109:229-37) and additional Tay-Sachs disease testing can be performed using a biochemical assay (Gross et al.Genet. Med. 2008:10(1):54-56).
This test was developed and its performance characteristics determined by Counsyl, Inc. It has not been cleared or approved by the US Food and Drug Administration(FDA). The FDA does not require this test to go through premarket review. This test is used for clinical purposes. It should not be regarded as investigational or forresearch. This laboratory is certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) as qualified to perform high-complexity clinical testing.These results are adjunctive to the ordering physician's evaluation. CLIA Number: #05D1102604.
H. Peter Kang, MD, MS, FCAP Rebecca Mar-Heyming, PhD, DABMG
LAB DIRECTORS
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 15 of 21Version: 3.2.5

Conditions TestedAutosomal Recessive Disorders
SEQUENCING AND TARGETED GENOTYPING
ABCC8-related Hyperinsulinism - Gene: ABCC8. Variants (3): c.4160_4162delTCT,V187D, 3992-9G>A. Exons: NM_000352:2-39. Detection rate: Unknown due to rarityof disease.Achromatopsia - Gene: CNGB3. Variants (3): R403Q, c.819_826del8, c.1148delC.Exons: NM_019098:1-18. Detection rate: Ethnicity Unknown > 62%.Alkaptonuria - Gene: HGD. Variants (11): G161R, G270R, P230S, S47L, V300G,M368V, c.16-1G>A, c.342+1G>A, c.457dupG, c.175delA, c.1111dupC. Exons:NM_000187:1-14. Detection rate: Ethnicity Unknown > 80%.Alpha-1 Antitrypsin Deficiency - Gene: SERPINA1. Variant (1): E366K. Exons:NM_000295:2-5. Detection rate: Ethnicity Unknown > 95%.Alpha-Mannosidosis - Gene: MAN2B1. Variant (1): R750W. Exons:NM_000528:1-15,17-24. Detection rate: Ethnicity Unknown > 32%.Andermann Syndrome - Gene: SLC12A6. Variants (2): c.2436+1delG, R1011*.Exons: NM_133647:1-25. Detection rate: Unknown due to rarity of disease.ARSACS - Gene: SACS. Variants (2): c.8844delT, R2502*. Exons: NM_014363:2,4-10.Detection rate: Unknown due to rarity of disease.Aspartylglycosaminuria - Gene: AGA. Variant (1): C163S. Exons: NM_000027:1-9.Detection rate: Unknown due to rarity of disease.Ataxia With Vitamin E Deficiency - Gene: TTPA. Variant (1): c.744delA. Exons:NM_000370:1-5. Detection rate: Ethnicity Unknown > 10%.Ataxia-Telangiectasia - Gene: ATM. Variants (8): R35*, Q1970*,c.7517_7520delGAGA, 5762ins137, c.7638_7646del9, c.3245_3247delATCinsTGAT,K1192, E1978*. Exons: NM_000051:1-63. Detection rate: Ethnicity Unknown > 65%.Autosomal Recessive Polycystic Kidney Disease - Gene: PKHD1. Variants (4):c.5895dupA, T36M, R496*, V3471G. Exons: NM_138694:2-67. Detection rate:Ethnicity Unknown > 18%.Bardet-Biedl Syndrome, BBS1-related - Gene: BBS1. Variant (1): M390R. Exons:NM_024649:1-17. Detection rate: Ethnicity Unknown > 79%.Bardet-Biedl Syndrome, BBS10-related - Gene: BBS10. Variant (1): c.271dupT.Exons: NM_024685:1-2. Detection rate: Ethnicity Unknown > 46%.Biotinidase Deficiency - Gene: BTD. Variants (4): c.98_104del7ins3, D252G, Q456H,R538C. Exons: NM_000060:1-4. Detection rate: Ethnicity Unknown > 45%.Bloom Syndrome - Gene: BLM. Variant (1): c.2207_2212del6ins7. Exons:NM_000057:2-22. Detection rate: Ethnicity Unknown > 10%.Canavan Disease - Gene: ASPA. Variants (4): E285A, Y231*, A305E, c.433-2A>G.Exons: NM_000049:1-6. Detection rate: Ethnicity Unknown > 53%.Carnitine Palmitoyltransferase IA Deficiency - Gene: CPT1A. Variant (1): G710E.Exons: NM_001876:2-19. Detection rate: Ethnicity Unknown > 10%.Carnitine Palmitoyltransferase II Deficiency - Gene: CPT2. Variants (3):c.1239_1240delGA, S113L, R124*. Exons: NM_000098:1-5. Detection rate: EthnicityUnknown > 80%.Cartilage-Hair Hypoplasia - Gene: RMRP. Variant (1): g.70A>G. Exon:NR_003051:1. Detection rate: Ethnicity Unknown > 48%.Citrullinemia Type 1 - Gene: ASS1. Variants (2): c.421-2A>G, G390R. Exons:NM_000050:3-16. Detection rate: Ethnicity Unknown > 20%.CLN3-related Neuronal Ceroid Lipofuscinosis - Gene: CLN3. Variant (1):461_677del. Exons: NM_001042432:2-16. Detection rate: Ethnicity Unknown > 96%.CLN5-related Neuronal Ceroid Lipofuscinosis - Gene: CLN5. Variant (1):c.1175_1176delAT. Exons: NM_006493:1-4. Detection rate: Unknown due to rarityof disease.Cohen Syndrome - Gene: VPS13B. Variant (1): c.3348_3349delCT. Exons:NM_017890:2-62. Detection rate: Unknown due to rarity of disease.Congenital Disorder of Glycosylation Type Ia - Gene: PMM2. Variants (4): V231M,F119L, R141H, P113L. Exons: NM_000303:1-8. Detection rate: Ethnicity Unknown >72%.Congenital Disorder of Glycosylation Type Ib - Gene: MPI. Variant (1): R295H.Exons: NM_002435:1-8. Detection rate: Unknown due to rarity of disease.Congenital Finnish Nephrosis - Gene: NPHS1. Variants (2): c.121_122delCT,R1109*. Exons: NM_004646:2-23,26-27,29. Detection rate: Unknown due to rarityof disease.
Costeff Optic Atrophy Syndrome - Gene: OPA3. Variant (1): c.143-1G>C. Exons:NM_025136:1-2. Detection rate: Unknown due to rarity of disease.Cystic Fibrosis - Gene: CFTR. Variants (99): G85E, R117H, R334W, R347P, A455E,G542*, G551D, R553*, R560T, R1162*, W1282*, N1303K, c.1521_1523delCTT,c.1519_1521delATC, c.2052delA, c.3528delC, c.489+1G>T, c.579+1G>T, c.1585-1G>A,c.1766+1G>A, 2789+5G>A, c.2988+1G>A, 3849+10kbC>T, E60*, R75*, E92*, Y122*,G178R, R347H, Q493*, V520F, S549N, P574H, M1101K, D1152H, c.2012delT,c.262_263delTT, c.313delA, c.948delT, c.3744delA, c.3773dupT, c.1680-1G>A,3272-26A>G, c.2051_2052delAAinsG, S549R(c.1645A>C), R117C, L206W, G330*,T338I, R352Q, S364P, G480C, C524*, S549R(c.1647T>G), Q552*, A559T, G622D,R709*, K710*, R764*, Q890*, R1066C, W1089*, Y1092X, R1158*, S1196*, W1204*,Q1238*, S1251N, S1255*, c.3067_3072del6, c.442delA, c.531delT, c.803delA,c.805_806delAT, c.1545_1546delTA, 1949del84, c.1911delG, c.1923_1931del9ins1,c.1976delA, c.3039delC, c.3536_3539delCCAA, c.3659delC, c.1155_1156dupTA,c.2052dupA, c.2175dupA, c.2738insG, 296+12T>C, c.273+1G>A, 405+3A>C,c.274-1G>A, 711+5G>A, c.580-1G>T, c.1766+1G>T, 1898+5G>T, Q996,c.325_327delTATinsG, 3849+4A>G, c.1075_1079del5ins5. Exons: NM_000492:1-27.IVS8-5T allele analysis is only reported in the presence of the R117H mutation.Detection rate: Ethnicity Unknown > 91%.Cystinosis - Gene: CTNS. Variants (4): 57 kb deletion, 537del21, W138*, L158P.Exons: NM_004937:1,3-12. Detection rate: Ethnicity Unknown > 67%.D-Bifunctional Protein Deficiency - Gene: HSD17B4. Variants (2): G16S, N457Y.Exons: NM_000414:1-24. Detection rate: Ethnicity Unknown > 35%.Factor XI Deficiency - Gene: F11. Variants (4): E135*, F301L, c.1716+1G>A,c.1714_1716+11del14. Exons: NM_000128:1-15. Detection rate: Ethnicity Unknown> 10%.Familial Dysautonomia - Gene: IKBKAP. Variants (2): IVS20+6T>C, R696P. Exons:NM_003640:19-20,26. Detection rate: Unknown due to rarity of disease.Familial Mediterranean Fever - Gene: MEFV. Variants (4): M694V, V726A, M680I,M694I. Exons: NM_000243:1-10. Detection rate: Unknown due to rarity of disease.Fanconi Anemia Type C - Gene: FANCC. Variants (3): IVS4+4A>T, c.67delG, R548*.Exons: NM_000136:2-15. Detection rate: Ethnicity Unknown > 54%.Galactosemia - Gene: GALT. Variants (8): S135L, Q188R, F171S, L195P, K285N,c.253-2A>G, T138M, Y209C. Exons: NM_000155:1-11. Detection rate: EthnicityUnknown > 80%.GJB2-related DFNB1 Nonsyndromic Hearing Loss and Deafness - Gene: GJB2.Variants (7): c.35delG, c.167delT, c.235delC, c.358_360delGAG, W24*, W77R, L90P.Exons: NM_004004:1-2. Detection rate: Ethnicity Unknown > 79%.Glutaric Acidemia Type 1 - Gene: GCDH. Variant (1): R402W. Exons:NM_000159:2-12. Detection rate: Ethnicity Unknown > 40%.Glycogen Storage Disease Type Ia - Gene: G6PC. Variants (7): R83C, Q347*,c.79delC, c.379_380dupTA, R83H, G188R, Q242*. Exons: NM_000151:1-5. Detectionrate: Ethnicity Unknown > 61%.Glycogen Storage Disease Type Ib - Gene: SLC37A4. Variants (2):c.1042_1043delCT, G339C. Exons: NM_001164277:3-11. Detection rate: EthnicityUnknown > 46%.Glycogen Storage Disease Type III - Gene: AGL. Variants (3): c.4456delT, Q6*,c.18_19delGA. Exons: NM_000642:2-34. Detection rate: Ethnicity Unknown > 45%.Glycogen Storage Disease Type V - Gene: PYGM. Variants (4): R50*, G205S,c.715_717delGTC, W798R. Exons: NM_005609:1-20. Detection rate: EthnicityUnknown > 80%.GRACILE Syndrome - Gene: BCS1L. Variant (1): S78G. Exons: NM_004328:3-9.Detection rate: Unknown due to rarity of disease.Hb Beta Chain-Related Hemoglobinopathy (Including Beta Thalassemia andSickle Cell Disease) - Gene: HBB. Variants (28): E7V, K18*, Q40*, c.126_129delCTTT,c.27dupG, IVS-II-654, IVS-II-745, c.315+1G>A, IVS-I-6, IVS-I-110, IVS-I-5, c.92+1G>A,-88C>T, -28A>G, -29A>G, c.25_26delAA, c.217dupA, c.316-2A>C, c.316-2A>G, G25,-87C>G, E7K, W16*, c.51delC, c.20delA, E27K, E122Q, E122K. Exons: NM_000518:1-3.Detection rate: Ethnicity Unknown > 83%.Hereditary Fructose Intolerance - Gene: ALDOB. Variants (3): A150P, N335K,A175D. Exons: NM_000035:2-9. Detection rate: Ethnicity Unknown > 75%.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 16 of 21Version: 3.2.5

Hereditary Thymine-Uraciluria - Gene: DPYD. Variant (1): c.1905+1G>A. Exons:NM_000110:1-23. Detection rate: Ethnicity Unknown > 52%.Herlitz Junctional Epidermolysis Bullosa, LAMA3-related - Gene: LAMA3. Variant(1): R661*. Exons: NM_000227:1-16,18-38. Detection rate: Ethnicity Unknown >10%.Herlitz Junctional Epidermolysis Bullosa, LAMB3-related - Gene: LAMB3.Variants (3): R42*, Q243*, R635*. Exons: NM_000228:2-23. Detection rate:Ethnicity Unknown > 48%.Herlitz Junctional Epidermolysis Bullosa, LAMC2-related - Gene: LAMC2. Variant(1): R95*. Exons: NM_005562:1-23. Detection rate: Unknown due to rarity ofdisease.Hexosaminidase A Deficiency (Including Tay-Sachs Disease) - Gene: HEXA.Variants (9): c.1274_1277dupTATC, c.1421+1G>C, G269S, c.1073+1G>A, R178H,c.805+1G>A, 7.6kb del, G250D, R170W. Exons: NM_000520:1-14. Detection rate:Ethnicity Unknown > 23%.Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency - Gene:CBS. Variant (1): G307S. Exons: NM_000071:3-17. Detection rate: EthnicityUnknown > 14%.Hypophosphatasia, Autosomal Recessive - Gene: ALPL. Variants (4): c.1559delT,F327L, D378V, E191K. Exons: NM_000478:1-12. Detection rate: Ethnicity Unknown >30%.Inclusion Body Myopathy 2 - Gene: GNE. Variants (2): M712T, V572L. Exons:NM_005476:2-12. Detection rate: Unknown due to rarity of disease.Isovaleric Acidemia - Gene: IVD. Variant (1): A314V. Exons: NM_002225:1-12.Detection rate: Ethnicity Unknown > 47%.Joubert Syndrome 2 - Gene: TMEM216. Variant (1): R73L. Exons:NM_001173990:1-5. Detection rate: Unknown due to rarity of disease.Krabbe Disease - Gene: GALC. Variants (2): Ex11-17del, T529M. Exons:NM_000153:2-17. Detection rate: Ethnicity Unknown > 58%.Limb-Girdle Muscular Dystrophy Type 2D - Gene: SGCA. Variant (1): R77C. Exons:NM_000023:1-9. Detection rate: Ethnicity Unknown > 32%.Limb-Girdle Muscular Dystrophy Type 2E - Gene: SGCB. Variant (1): S114F.Exons: NM_000232:2-6. Detection rate: Ethnicity Unknown > 12%.Lipoamide Dehydrogenase Deficiency - Gene: DLD. Variants (2): c.104dupA,G229C. Exons: NM_000108:1-14. Detection rate: Unknown due to rarity of disease.Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency - Gene: HADHA.Variant (1): E510Q. Exons: NM_000182:1-20. Detection rate: Ethnicity Unknown >87%.Maple Syrup Urine Disease Type 1B - Gene: BCKDHB. Variants (3): R183P, G278S,E372*. Exons: NM_183050:1-10. Detection rate: Unknown due to rarity of disease.Medium Chain Acyl-CoA Dehydrogenase Deficiency - Gene: ACADM. Variants (2):K329E, Y67H. Exons: NM_000016:1-12. Detection rate: Ethnicity Unknown > 78%.Megalencephalic Leukoencephalopathy With Subcortical Cysts - Gene: MLC1.Variants (4): c.135dupC, G59E, S93L, IVS2-10T>A. Exons: NM_015166:2-12.Detection rate: Ethnicity Unknown > 13%.Metachromatic Leukodystrophy - Gene: ARSA. Variants (5): P428L, c.465+1G>A,c.1210+1G>A, I181S, T411I. Exons: NM_000487:1-7. Detection rate: EthnicityUnknown > 53%.Mucolipidosis IV - Gene: MCOLN1. Variants (2): 511_6944del, c.406-2A>G. Exons:NM_020533:2-14. Detection rate: Ethnicity Unknown > 10%.Muscle-Eye-Brain Disease - Gene: POMGNT1. Variant (1): c.1539+1G>A. Exons:NM_017739:2-22. Detection rate: Ethnicity Unknown > 75%.NEB-related Nemaline Myopathy - Gene: NEB. Variant (1): R2478_D2512del.Exons: NM_004543:7-8,18,25,28,33,36,45,48,54-55,58,61,71,73-74,91,94,101,111-112,114,118-119,122-123,127,129,132-135,138,140,143,146-147. Detection rate:Unknown due to rarity of disease.Niemann-Pick Disease Type C - Gene: NPC1. Variant (1): I1061T. Exons:NM_000271:2-25. Detection rate: Ethnicity Unknown > 17%.Niemann-Pick Disease, SMPD1-associated - Gene: SMPD1. Variants (4):c.996delC, L304P, R498L, c.1829_1831delGCC. Exons: NM_000543:1-6. Detectionrate: Ethnicity Unknown > 38%.Nijmegen Breakage Syndrome - Gene: NBN. Variant (1): c.657_661del5. Exons:NM_002485:1-16. Detection rate: Ethnicity Unknown > 78%.Northern Epilepsy - Gene: CLN8. Variant (1): R24G. Exons: NM_018941:2-3.Detection rate: Unknown due to rarity of disease.
Pendred Syndrome - Gene: SLC26A4. Variants (5): c.1001+1G>A, L236P, E384G,T416P, H723R. Exons: NM_000441:1-21. Detection rate: Ethnicity Unknown > 69%.PEX1-related Zellweger Syndrome Spectrum - Gene: PEX1. Variants (2):c.2097dupT, G843D. Exons: NM_000466:1-24. Detection rate: Ethnicity Unknown >68%.Phenylalanine Hydroxylase Deficiency - Gene: PAH. Variants (13): IVS-10int-546,I65T, R261Q, R408W, IVS12+1G>A, R408Q, Y414C, L48S, R158Q, G272*, P281L, E280K,S349P. Exons: NM_000277:1-13. Detection rate: Ethnicity Unknown > 43%.Polyglandular Autoimmune Syndrome Type 1 - Gene: AIRE. Variants (2): Y85C,R257*. Exons: NM_000383:1-14. Detection rate: Ethnicity Unknown > 65%.Pompe Disease - Gene: GAA. Variants (4): D645E, R854*, IVS1-13T>G, c.525delT.Exons: NM_000152:2-20. Detection rate: Ethnicity Unknown > 67%.PPT1-related Neuronal Ceroid Lipofuscinosis - Gene: PPT1. Variants (3): T75P,R122W, R151*. Exons: NM_000310:1-9. Detection rate: Ethnicity Unknown > 53%.Primary Carnitine Deficiency - Gene: SLC22A5. Variant (1): R254*. Exons:NM_003060:2-10. Detection rate: Unknown due to rarity of disease.Primary Hyperoxaluria Type 1 - Gene: AGXT. Variants (2): G170R, I244T. Exons:NM_000030:1-11. Detection rate: Ethnicity Unknown > 42%.Primary Hyperoxaluria Type 2 - Gene: GRHPR. Variants (2): c.103delG,c.404+3_404+6delAAGT. Exons: NM_012203:1-9. Detection rate: Ethnicity Unknown> 37%.PROP1-related Combined Pituitary Hormone Deficiency - Gene: PROP1. Variant(1): c.301_302delAG. Exons: NM_006261:1-3. Detection rate: Ethnicity Unknown >55%.Pseudocholinesterase Deficiency - Gene: BCHE. Variant (1): D98G. Exons:NM_000055:2-4. Detection rate: Ethnicity Unknown > 83%.Pycnodysostosis - Gene: CTSK. Variant (1): c.990A>G. Exons: NM_000396:2-8.Detection rate: Ethnicity Unknown > 10%.Rhizomelic Chondrodysplasia Punctata Type 1 - Gene: PEX7. Variants (4): G217R,A218V, L292*, c.903+1G>C. Exons: NM_000288:2-10. Detection rate: EthnicityUnknown > 70%.Salla Disease - Gene: SLC17A5. Variant (1): R39C. Exons: NM_012434:2-11.Detection rate: Unknown due to rarity of disease.Segawa Syndrome - Gene: TH. Variant (1): R202H. Exons: NM_000360:1-8,10-13.Detection rate: Ethnicity Unknown > 10%.Short Chain Acyl-CoA Dehydrogenase Deficiency - Gene: ACADS. Variant (1):R107C. Exons: NM_000017:2-10. Detection rate: Unknown due to rarity of disease.Sjogren-Larsson Syndrome - Gene: ALDH3A2. Variant (1): P315S. Exons:NM_000382:1-10. Detection rate: Ethnicity Unknown > 24%.Smith-Lemli-Opitz Syndrome - Gene: DHCR7. Variants (13): c.964-1G>C, T93M,W151*, V326L, R352Q, R352W, R404C, S169L, R242C, R242H, F302L, G410S, E448K.Exons: NM_001360:3-9. Detection rate: Ethnicity Unknown > 69%.Steroid-Resistant Nephrotic Syndrome - Gene: NPHS2. Variants (2): R138Q,R138*. Exons: NM_014625:1-8. Detection rate: Ethnicity Unknown > 33%.Sulfate Transporter-Related Osteochondrodysplasia - Gene: SLC26A2. Variants(4): C653S, R178*, R279W, c.699+2T>C. Exons: NM_000112:2-3. Detection rate:Ethnicity Unknown > 75%.TPP1-related Neuronal Ceroid Lipofuscinosis - Gene: TPP1. Variants (3): G284V,R208*, c.509-1G>C. Exons: NM_000391:1-13. Detection rate: Ethnicity Unknown >60%.Tyrosinemia Type I - Gene: FAH. Variants (6): IVS12+5G>A, Q64H, P261L, W262*,E357*, c.554-1G>T. Exons: NM_000137:1-14. Detection rate: Ethnicity Unknown >50%.Usher Syndrome Type 1F - Gene: PCDH15. Variant (1): R245*. Exons:NM_033056:2-33. Detection rate: Unknown due to rarity of disease.Usher Syndrome Type 3 - Gene: CLRN1. Variant (1): N48K. Exons: NM_174878:1-3.Detection rate: Unknown due to rarity of disease.Very Long Chain Acyl-CoA Dehydrogenase Deficiency - Gene: ACADVL. Variant(1): V283A. Exons: NM_000018:1-20. Detection rate: Ethnicity Unknown > 20%.Walker-Warburg Syndrome - Gene: FKTN. Variant (1): c.1167dupA. Exons:NM_001079802:3-11. Detection rate: Unknown due to rarity of disease.Wilson Disease - Gene: ATP7B. Variants (2): H1069Q, R778L. Exons:NM_000053:1-3,5-21. Detection rate: Ethnicity Unknown > 40%.
TARGETED GENOTYPING
Factor V Leiden Thrombophilia - Gene: F5. Variant (1): R506Q.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 17 of 21Version: 3.2.5

Gaucher Disease - Gene: GBA. Variants (10): N409S, L483P, c.84dupG, c.115+1G>A,V433L, R535H, D448H, D448V, R502C, R502H. Detection rate: Ethnicity Unknown60%.HFE-associated Hereditary Hemochromatosis - Gene: HFE. Variants (2): C282Y,H63D.
Hurler Syndrome - Gene: IDUA. Variants (2): W402*, Q70*. Detection rate:Ethnicity Unknown 67%.MTHFR Deficiency - Gene: MTHFR. Variants (2): A222V, E429A.Prothrombin Thrombophilia - Gene: F2. Variant (1): G20210A.
COPY NUMBER ANALYSIS
Spinal Muscular Atrophy - Gene: SMN1. Variant (1): SMN1 copy number.Detection rate: Ethnicity Unknown 95%.
X-linked Dominant Disorders
TRIPLET REPEAT DETECTION
Fragile X Syndrome - Gene: FMR1. Variant (1): NM_002024.4:c.1-131CGG[1_n].
X-linked Recessive Disorders
SEQUENCING AND TARGETED GENOTYPING
Choroideremia - Gene: CHM. Variant (1): c.1609+2dupT. Exons: NM_000390:1-15.Detection rate: Unknown due to rarity of disease.
X-Linked Juvenile Retinoschisis - Gene: RS1. Variants (3): E72K, G74V, G109R.Exons: NM_000330:1-6. Detection rate: Ethnicity Unknown > 20%.
TARGETED GENOTYPING
Glucose-6-Phosphate Dehydrogenase Deficiency - Gene: G6PD. Variants (7):S188F, V68M, R459P, R459L, A335T, G163S, V291M.
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 18 of 21Version: 3.2.5
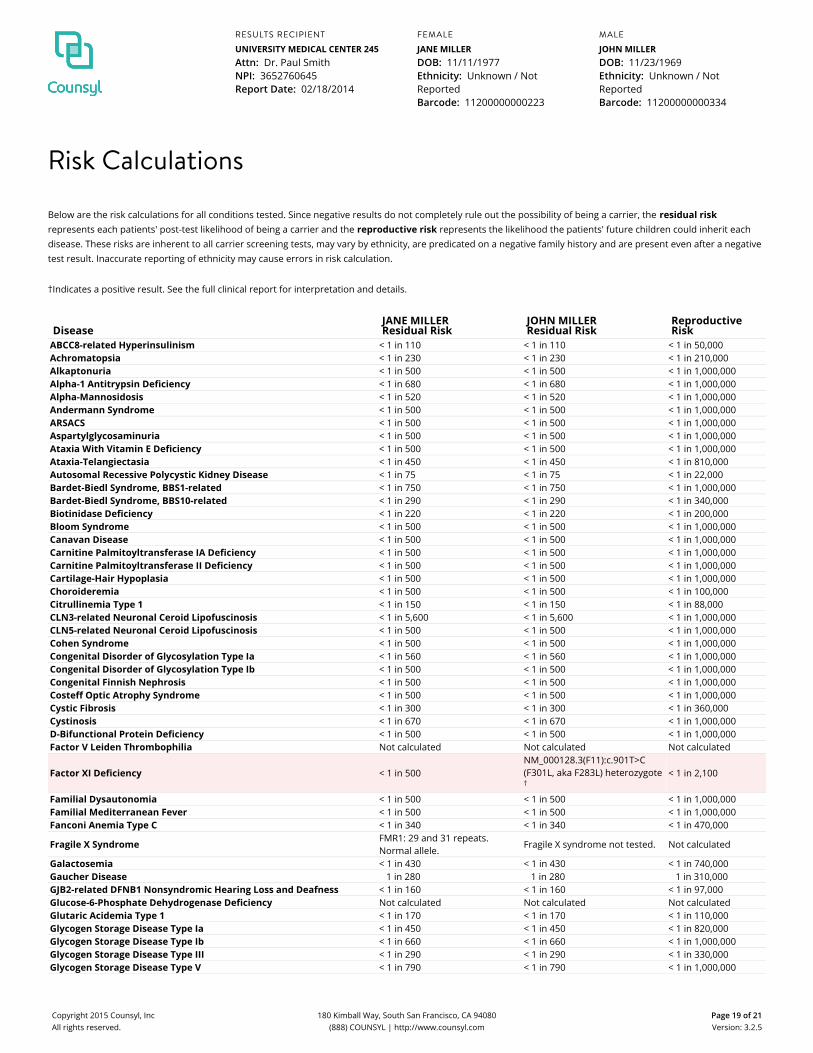
Risk Calculations
Below are the risk calculations for all conditions tested. Since negative results do not completely rule out the possibility of being a carrier, the residual riskrepresents each patients' post-test likelihood of being a carrier and the reproductive risk represents the likelihood the patients' future children could inherit eachdisease. These risks are inherent to all carrier screening tests, may vary by ethnicity, are predicated on a negative family history and are present even after a negativetest result. Inaccurate reporting of ethnicity may cause errors in risk calculation.
†Indicates a positive result. See the full clinical report for interpretation and details.
DiseaseJANE MILLERResidual Risk
JOHN MILLERResidual Risk
ReproductiveRisk
ABCC8-related Hyperinsulinism < 1 in 110 < 1 in 110 < 1 in 50,000Achromatopsia < 1 in 230 < 1 in 230 < 1 in 210,000Alkaptonuria < 1 in 500 < 1 in 500 < 1 in 1,000,000Alpha-1 Antitrypsin Deficiency < 1 in 680 < 1 in 680 < 1 in 1,000,000Alpha-Mannosidosis < 1 in 520 < 1 in 520 < 1 in 1,000,000Andermann Syndrome < 1 in 500 < 1 in 500 < 1 in 1,000,000ARSACS < 1 in 500 < 1 in 500 < 1 in 1,000,000Aspartylglycosaminuria < 1 in 500 < 1 in 500 < 1 in 1,000,000Ataxia With Vitamin E Deficiency < 1 in 500 < 1 in 500 < 1 in 1,000,000Ataxia-Telangiectasia < 1 in 450 < 1 in 450 < 1 in 810,000Autosomal Recessive Polycystic Kidney Disease < 1 in 75 < 1 in 75 < 1 in 22,000Bardet-Biedl Syndrome, BBS1-related < 1 in 750 < 1 in 750 < 1 in 1,000,000Bardet-Biedl Syndrome, BBS10-related < 1 in 290 < 1 in 290 < 1 in 340,000Biotinidase Deficiency < 1 in 220 < 1 in 220 < 1 in 200,000Bloom Syndrome < 1 in 500 < 1 in 500 < 1 in 1,000,000Canavan Disease < 1 in 500 < 1 in 500 < 1 in 1,000,000Carnitine Palmitoyltransferase IA Deficiency < 1 in 500 < 1 in 500 < 1 in 1,000,000Carnitine Palmitoyltransferase II Deficiency < 1 in 500 < 1 in 500 < 1 in 1,000,000Cartilage-Hair Hypoplasia < 1 in 500 < 1 in 500 < 1 in 1,000,000Choroideremia < 1 in 500 < 1 in 500 < 1 in 100,000Citrullinemia Type 1 < 1 in 150 < 1 in 150 < 1 in 88,000CLN3-related Neuronal Ceroid Lipofuscinosis < 1 in 5,600 < 1 in 5,600 < 1 in 1,000,000CLN5-related Neuronal Ceroid Lipofuscinosis < 1 in 500 < 1 in 500 < 1 in 1,000,000Cohen Syndrome < 1 in 500 < 1 in 500 < 1 in 1,000,000Congenital Disorder of Glycosylation Type Ia < 1 in 560 < 1 in 560 < 1 in 1,000,000Congenital Disorder of Glycosylation Type Ib < 1 in 500 < 1 in 500 < 1 in 1,000,000Congenital Finnish Nephrosis < 1 in 500 < 1 in 500 < 1 in 1,000,000Costeff Optic Atrophy Syndrome < 1 in 500 < 1 in 500 < 1 in 1,000,000Cystic Fibrosis < 1 in 300 < 1 in 300 < 1 in 360,000Cystinosis < 1 in 670 < 1 in 670 < 1 in 1,000,000D-Bifunctional Protein Deficiency < 1 in 500 < 1 in 500 < 1 in 1,000,000Factor V Leiden Thrombophilia Not calculated Not calculated Not calculated
Factor XI Deficiency < 1 in 500NM_000128.3(F11):c.901T>C(F301L, aka F283L) heterozygote†
< 1 in 2,100
Familial Dysautonomia < 1 in 500 < 1 in 500 < 1 in 1,000,000Familial Mediterranean Fever < 1 in 500 < 1 in 500 < 1 in 1,000,000Fanconi Anemia Type C < 1 in 340 < 1 in 340 < 1 in 470,000
Fragile X Syndrome FMR1: 29 and 31 repeats.Normal allele.
Fragile X syndrome not tested. Not calculated
Galactosemia < 1 in 430 < 1 in 430 < 1 in 740,000Gaucher Disease 1 in 280 1 in 280 1 in 310,000GJB2-related DFNB1 Nonsyndromic Hearing Loss and Deafness < 1 in 160 < 1 in 160 < 1 in 97,000Glucose-6-Phosphate Dehydrogenase Deficiency Not calculated Not calculated Not calculatedGlutaric Acidemia Type 1 < 1 in 170 < 1 in 170 < 1 in 110,000Glycogen Storage Disease Type Ia < 1 in 450 < 1 in 450 < 1 in 820,000Glycogen Storage Disease Type Ib < 1 in 660 < 1 in 660 < 1 in 1,000,000Glycogen Storage Disease Type III < 1 in 290 < 1 in 290 < 1 in 330,000Glycogen Storage Disease Type V < 1 in 790 < 1 in 790 < 1 in 1,000,000
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 19 of 21Version: 3.2.5
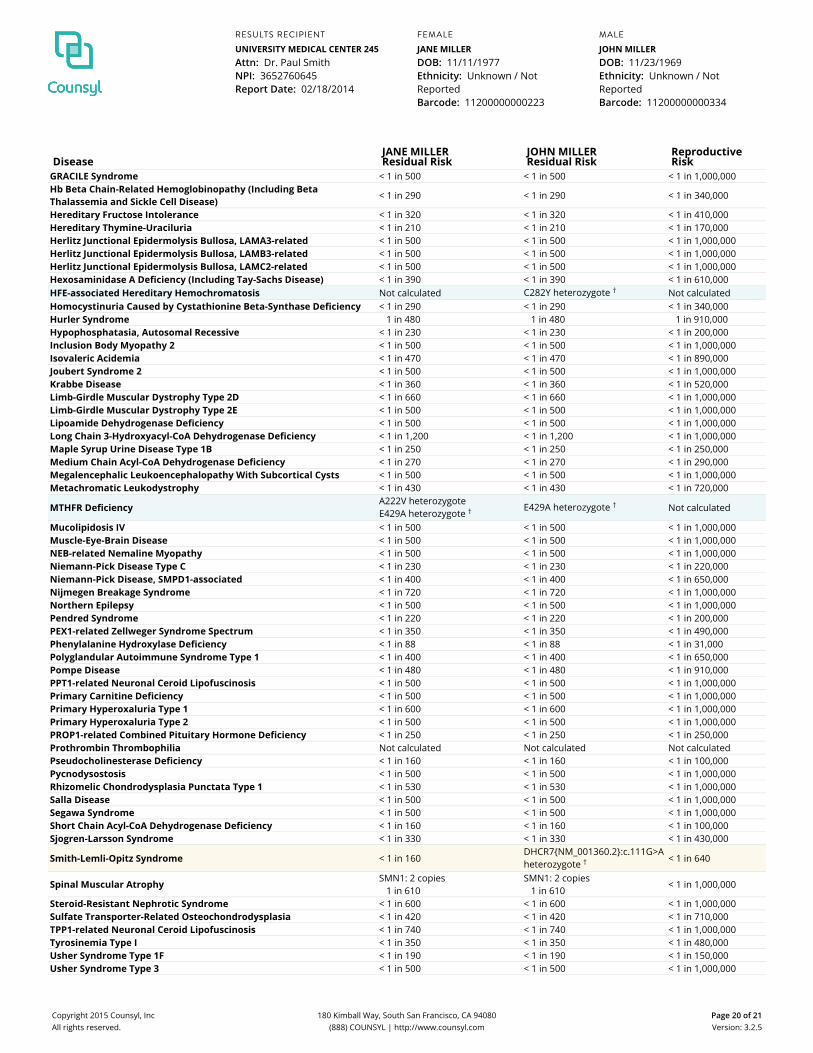
DiseaseJANE MILLERResidual Risk
JOHN MILLERResidual Risk
ReproductiveRisk
GRACILE Syndrome < 1 in 500 < 1 in 500 < 1 in 1,000,000Hb Beta Chain-Related Hemoglobinopathy (Including BetaThalassemia and Sickle Cell Disease) < 1 in 290 < 1 in 290 < 1 in 340,000
Hereditary Fructose Intolerance < 1 in 320 < 1 in 320 < 1 in 410,000Hereditary Thymine-Uraciluria < 1 in 210 < 1 in 210 < 1 in 170,000Herlitz Junctional Epidermolysis Bullosa, LAMA3-related < 1 in 500 < 1 in 500 < 1 in 1,000,000Herlitz Junctional Epidermolysis Bullosa, LAMB3-related < 1 in 500 < 1 in 500 < 1 in 1,000,000Herlitz Junctional Epidermolysis Bullosa, LAMC2-related < 1 in 500 < 1 in 500 < 1 in 1,000,000Hexosaminidase A Deficiency (Including Tay-Sachs Disease) < 1 in 390 < 1 in 390 < 1 in 610,000HFE-associated Hereditary Hemochromatosis Not calculated C282Y heterozygote † Not calculatedHomocystinuria Caused by Cystathionine Beta-Synthase Deficiency < 1 in 290 < 1 in 290 < 1 in 340,000Hurler Syndrome 1 in 480 1 in 480 1 in 910,000Hypophosphatasia, Autosomal Recessive < 1 in 230 < 1 in 230 < 1 in 200,000Inclusion Body Myopathy 2 < 1 in 500 < 1 in 500 < 1 in 1,000,000Isovaleric Acidemia < 1 in 470 < 1 in 470 < 1 in 890,000Joubert Syndrome 2 < 1 in 500 < 1 in 500 < 1 in 1,000,000Krabbe Disease < 1 in 360 < 1 in 360 < 1 in 520,000Limb-Girdle Muscular Dystrophy Type 2D < 1 in 660 < 1 in 660 < 1 in 1,000,000Limb-Girdle Muscular Dystrophy Type 2E < 1 in 500 < 1 in 500 < 1 in 1,000,000Lipoamide Dehydrogenase Deficiency < 1 in 500 < 1 in 500 < 1 in 1,000,000Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency < 1 in 1,200 < 1 in 1,200 < 1 in 1,000,000Maple Syrup Urine Disease Type 1B < 1 in 250 < 1 in 250 < 1 in 250,000Medium Chain Acyl-CoA Dehydrogenase Deficiency < 1 in 270 < 1 in 270 < 1 in 290,000Megalencephalic Leukoencephalopathy With Subcortical Cysts < 1 in 500 < 1 in 500 < 1 in 1,000,000Metachromatic Leukodystrophy < 1 in 430 < 1 in 430 < 1 in 720,000
MTHFR DeficiencyA222V heterozygoteE429A heterozygote † E429A heterozygote † Not calculated
Mucolipidosis IV < 1 in 500 < 1 in 500 < 1 in 1,000,000Muscle-Eye-Brain Disease < 1 in 500 < 1 in 500 < 1 in 1,000,000NEB-related Nemaline Myopathy < 1 in 500 < 1 in 500 < 1 in 1,000,000Niemann-Pick Disease Type C < 1 in 230 < 1 in 230 < 1 in 220,000Niemann-Pick Disease, SMPD1-associated < 1 in 400 < 1 in 400 < 1 in 650,000Nijmegen Breakage Syndrome < 1 in 720 < 1 in 720 < 1 in 1,000,000Northern Epilepsy < 1 in 500 < 1 in 500 < 1 in 1,000,000Pendred Syndrome < 1 in 220 < 1 in 220 < 1 in 200,000PEX1-related Zellweger Syndrome Spectrum < 1 in 350 < 1 in 350 < 1 in 490,000Phenylalanine Hydroxylase Deficiency < 1 in 88 < 1 in 88 < 1 in 31,000Polyglandular Autoimmune Syndrome Type 1 < 1 in 400 < 1 in 400 < 1 in 650,000Pompe Disease < 1 in 480 < 1 in 480 < 1 in 910,000PPT1-related Neuronal Ceroid Lipofuscinosis < 1 in 500 < 1 in 500 < 1 in 1,000,000Primary Carnitine Deficiency < 1 in 500 < 1 in 500 < 1 in 1,000,000Primary Hyperoxaluria Type 1 < 1 in 600 < 1 in 600 < 1 in 1,000,000Primary Hyperoxaluria Type 2 < 1 in 500 < 1 in 500 < 1 in 1,000,000PROP1-related Combined Pituitary Hormone Deficiency < 1 in 250 < 1 in 250 < 1 in 250,000Prothrombin Thrombophilia Not calculated Not calculated Not calculatedPseudocholinesterase Deficiency < 1 in 160 < 1 in 160 < 1 in 100,000Pycnodysostosis < 1 in 500 < 1 in 500 < 1 in 1,000,000Rhizomelic Chondrodysplasia Punctata Type 1 < 1 in 530 < 1 in 530 < 1 in 1,000,000Salla Disease < 1 in 500 < 1 in 500 < 1 in 1,000,000Segawa Syndrome < 1 in 500 < 1 in 500 < 1 in 1,000,000Short Chain Acyl-CoA Dehydrogenase Deficiency < 1 in 160 < 1 in 160 < 1 in 100,000Sjogren-Larsson Syndrome < 1 in 330 < 1 in 330 < 1 in 430,000
Smith-Lemli-Opitz Syndrome < 1 in 160DHCR7{NM_001360.2}:c.111G>Aheterozygote † < 1 in 640
Spinal Muscular Atrophy SMN1: 2 copies1 in 610
SMN1: 2 copies1 in 610
< 1 in 1,000,000
Steroid-Resistant Nephrotic Syndrome < 1 in 600 < 1 in 600 < 1 in 1,000,000Sulfate Transporter-Related Osteochondrodysplasia < 1 in 420 < 1 in 420 < 1 in 710,000TPP1-related Neuronal Ceroid Lipofuscinosis < 1 in 740 < 1 in 740 < 1 in 1,000,000Tyrosinemia Type I < 1 in 350 < 1 in 350 < 1 in 480,000Usher Syndrome Type 1F < 1 in 190 < 1 in 190 < 1 in 150,000Usher Syndrome Type 3 < 1 in 500 < 1 in 500 < 1 in 1,000,000
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 20 of 21Version: 3.2.5

DiseaseJANE MILLERResidual Risk
JOHN MILLERResidual Risk
ReproductiveRisk
Very Long Chain Acyl-CoA Dehydrogenase Deficiency < 1 in 110 < 1 in 110 < 1 in 49,000Walker-Warburg Syndrome < 1 in 500 < 1 in 500 < 1 in 1,000,000
Wilson DiseaseNM_000053.3(ATP7B):c.3207C>A(H1069Q) heterozygote † < 1 in 140 < 1 in 570
X-Linked Juvenile Retinoschisis < 1 in 500 < 1 in 500 < 1 in 62,000
MALE
JOHN MILLERDOB: 11/23/1969Ethnicity: Unknown / NotReportedBarcode: 11200000000334
FEMALE
JANE MILLERDOB: 11/11/1977Ethnicity: Unknown / NotReportedBarcode: 11200000000223
RESULTS RECIPIENT
UNIVERSITY MEDICAL CENTER 245Attn: Dr. Paul SmithNPI: 3652760645Report Date: 02/18/2014
Copyright 2015 Counsyl, IncAll rights reserved.
180 Kimball Way, South San Francisco, CA 94080(888) COUNSYL | http://www.counsyl.com
Page 21 of 21Version: 3.2.5





























