Embed Size (px)
Citation preview

SANFILIPPO SYNDROME CLINICAL GUIDELINES

Sanfilippo Syndrome Clínical Guidelines I 3
SANFILIPPO SYNDROMECLINICAL GUIDELINES

4 I Sanfilippo Syndrome Clínical Guidelines
01. Pathophysiology.Diagnosis.Correlación genotipo-fenotipo ............................................................................. 07
1.1 Pathophysiology .......................................................................................................... 091.2 Biochemical and genetic diagnosis .......................................................................... 14 – Detecting GAGs in urine .............................................................................................. 14 – Determining enzyme activity ........................................................................................ 16 – Molecular-genetic analysis ........................................................................................... 18 – Prenatal diagnosis ....................................................................................................... 19 – Carrier diagnosis ......................................................................................................... 19 – Future prospects: neonatal screening and whole exome sequencing ........................... 191.3 Genotype-phenotype correlation .............................................................................. 21 – MPS III-A ..................................................................................................................... 22 – MPS III-B ..................................................................................................................... 23 – MPS III-C .................................................................................................................... 25 – MPS III-D ..................................................................................................................... 26Web resources ................................................................................................................... 27
02. MPS III. Clinic. Clinical forms.Differential diagnosis ............................................................................................ 29
2.1 Early symptoms ........................................................................................................... 302.2 Behavioral disorder ..................................................................................................... 31 – Sleep disorders ........................................................................................................... 322.3 Neurodegeneration. Neurodevelopmental regression ............................................ 322.4 Extraneuroligical symptoms ...................................................................................... 33
– Coarse facies. Facial appearance ................................................................................ 33 – Height and musculoskeletal disorders ......................................................................... 34 – Precocious puberty ..................................................................................................... 35 – Hepatomegaly ............................................................................................................. 35 – Hernias ....................................................................................................................... 35 – Gastrointestinal involvement. Diarrhea ......................................................................... 35 – Dental disorders .......................................................................................................... 35 – Cardiac involvement .................................................................................................... 35 – Afectación ORL ........................................................................................................... 36 – Hearing and vision ....................................................................................................... 362.5 Neuroimaging .............................................................................................................. 37
CONTENTS

Sanfilippo Syndrome Clínical Guidelines I 5
2.6 Differences between types ........................................................................................ 382.7 Attenuated forms of MPS III ....................................................................................... 402.8 Intrafamilial variability ................................................................................................ 412.9 Clinical diagnosis and differential diagnosis ............................................................ 42
03. Neuropsychology ofSanfilippo syndrome .............................................................................................. 49
3.1 Dementia in children: Sanfilippo disease ................................................................. 503.1.1 Progression of cognitive and behavioral disorders in MPS III ....................................... 503.1.2 Cognitive and behavioral disorders in subtypes of MPS III ............................................ 52 – Sanfilippo syndrome types A ....................................................................................... 52 – Sanfilippo syndrome types B ....................................................................................... 55 – Sanfilippo syndrome types C & D ................................................................................ 553.2 Neuropsychological assessment in MPS III ............................................................. 563.2.1 Evaluation Methods ..................................................................................................... 563.2.2 Cognitive assessment .................................................................................................. 56 – Instruments ................................................................................................................. 573.2.3 Behavior assessment ................................................................................................... 593.2.4 Specific instruments in MPS III ..................................................................................... 60Conclusions ........................................................................................................................ 61
04. Monitoring mucopolysaccharidosistype III or Sanfilippo syndrome ............................................................................ 65
4.1 Medical history ............................................................................................................ 664.2 Physical examination .................................................................................................. 664.3 Neurological and functional assessments ............................................................... 66
05. Treatment forSanfilippo syndrome .............................................................................................. 75
5.1 Supportive treatment ................................................................................................. 765.2 Enzyme replacement therapy ................................................................................... 785.3 Hematopoietic stem cell transplantation ................................................................. 795.4 Substrate reduction therapy ...................................................................................... 815.5 Gene therapy ............................................................................................................... 82 Conclusions ........................................................................................................................ 84

Sanfilippo Syndrome Clínical Guidelines I 7
01.Pathophysiology.Diagnosis. Genotype-phenotype correlation.Luis Aldámiz-Echevarría, Marta Llarena, Fernando Andrade.Unit for Metabolic Diseases. Cruces University Hospital - BioCruces Health Research Institute. Reference Center for Metabolic Diseases (CSUR Ebro region). Clinical group related to ClBER of Rare Diseases (CIBERER).

8 I Sanfilippo Syndrome Clínical Guidelines
Type III (MPS III) mucopolysaccharidosis or Sanfilippo disease is one of the 40 currently described lysosomal diseases. It is considered to be the most common of the MPS11, with an estimated prevalence of 0.28 - 4.1 cases per 100,000 births2. The disease is inherited in an autosomal recessive manner.
1. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249-54.2. Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 2008; 31: 240-252.
FATHER UNAFFECTED “CARRIER”
MOTHER UNAFFECTED “CARRIER”
UNAFFECTED
1 of 4AFFECTED
1 of 4UNAFFECTED “CARRIER”
2 of 4
Figure 1. Autosomal recessive gene.

Sanfilippo Syndrome Clínical Guidelines I 9
The four subtypes of MPS III (A, B, C and D) are categorized into four different enzyme deficiencies in the path of heparan sulfate degradation, which in turn are caused by different mutations (Table 1). It was first described by pediatrician Dr Silvestre Sanfilippo3, although it was discovered in 1961 when the clinical manifestations of a girl with hepatosplenomegaly, normal skeletal evaluation and excretion of large amounts of heparan sulfate in urine were published4. In 1963, Sanfilippo et al. described eight children with mental retardation and a high excretion of a single glycosaminoglycan (GAG), the heparan sulfate3. Most children excreting heparan sulfate only had a less coarse facial appearance and milder somatic and radiographic manifestations compared to Hunter and Hurler patients (MPS II and MPS I, respectively), which showed greater severity. The MPS III - Sanfilippo disease is mainly characterized by a severe degeneration of the central nervous system.
Table 1. MPS III Classification - Sanfilippo disease
Subtype MPS III (MIM #)
Gen (MIM #)
Deficient enzyme GAG accumulated
MPS III-A (#252900) SGSH (#605270) heparán N-sulfatase HS
MPS III-B (#252920) NAGLU (#609701) α-N-acetylglucosaminidase HS
MPS III-C (#252930) HGSNAT (#610453)
acetyil-CoA:α-glucosaminide-N-acetyltransferase
HS
MPS III-D (#252940) GNS (#607664) N-acetylglucosamine 6-sulfatase HS
MIM #: Online Mendelian Inheritance in Man® (https:/www.omim.org); HS: Heparan sulfate
1.1 PATHOPHYSIOLOGYThe MPS III - Sanfilippo disease is caused by the lysosomal accumulation of heparan sulfate, due to a congenital deficiency in one of the four enzymes involved in its sequential degradation: heparan N-sulfatase (SGSH, MPS III-A; MIM #252900), α-N- acetylglucosaminidase (NAGLU, MPS III-B; MIM #252920), acetyl-CoA:α-glucosaminide-N- acetyltransferase (HGSNAT, MPS III-C; MIM #252930) and N-acetylglucosamine 6-sulfatase (GNS, MPS III-D; MIM #252940)2,5. Heparan sulfate belongs to the family of glycosaminoglycans (GAGs), which are components of proteoglycans present on the cell’s surface and on the extracellular matrix. Heparan sulfate degradation takes place in cellular
3. Sanfilippo S, Podosin R, Langer LO, Good RA. Mental retardation associated with acid mucopolysacchariduria (hepari-tin sulfate type). J Pediat 1963; 63: 837-838.4. Harris RC. Mucopolysaccharides disorder: A possible new genotype of Hurler´s syndrome. Am J Dis Child 1961;102:714.5. Neufeld EF, Muenzer J. The mucopolysaccharidoses. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease. 8th Ed. New York: McGraw-Hill; 2001. p. 3421-3452.

10 I Sanfilippo Syndrome Clínical Guidelines
lysosomes. In MPS III, the progressive accumulation of heparan sulfate in the lysosomes and the excretion in urine, eventually leads to the clinical manifestation of the disease. In addition, it is suggested that the excess of heparan sulfate could alter the synthesis and/or traffic of gangliosides, causing a secondary accumulation of gangliosides GM2 and GM3 which can also contribute to the pathology of MPS III6. Recently, Yubero et al. have observed a combined deficit of Coenzyme Q10 and pyridoxal phosphate (PLP) (coenzyme and active form of vitamin B6) in patients with MPS III7.
This disease of lysosomal storage has severe neurological manifestations. It has been observed that in the MPS III, the primary target organ affected is the central nervous system, whereas somatic manifestations are relatively mild in contrast to other types of mucopolysaccharidosis2. Although the pathological mechanism is not known in the MPS III, several mechanisms which potentially contribute to neurodegeneration have been described.
First, it is suggested that there is an interaction between neuroinflammation, microglia activation and adaptive immunity in the MPS8. In this line of research, several studies in animal model of MPS III-A and III-B have shown the role of the immune system in the pathology of MPS III. It has been observed that the heparan sulfate can trigger an immune response in the MPS III and it has been shown that it is involved in the microglial activation in the central nervous system in mouse animal model of MPS III-B9. Microglia cells, like the astrocytes and the entire immune system in the brain, are activated by the interaction of non-degraded fragments of heparan sulphate with their respective TLR4 (Toll-like receptor 4)9,10,11,12. Thus, resident mononuclear phagocytes in the brain (microglia) appear to be key in the neuroinflammatory processes associated with MPS13; because once activated, they induce the synthesis of inflammatory cytokines, including TNF-α (tumor necrosis factor α) and IL-1β (interleukin 1β). At the
6. McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides, and glycosamino-glycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol. 2004;480(4):415-26. 7. Yubero D, Montero R, O’Callaghan M, Pineda M, Meavilla S, Delgadillo V, et al. Coenzyme Q10 and Pyridoxal Phospha-te Deficiency is a common feature in Mucopolysaccharidosis Type III. JIMD Rep. 2015 Jul 24.8. Archer LD, Langford-Smith KJ, Bigger BW, Fildes JE. Mucopolysaccharide diseases: a complex interplay between neuroinflammation, microglial activation and adaptive immunity. J Inherit Metab Dis. 2014;37(1):1-12.9. Ausseil J, Desmaris N, Bigou S, Attali R, Corbineau S, Vitry S, et al. Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice. PLoS One. 2008;3(5):e2296.10. Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA. 2003;100(4):1902-7. 11. Villani GR, Gargiulo N, Faraonio R, Castaldo S, Gonzalez Y Reyero E, et al. Cytokines, neurotrophins, and oxidative stress in brain disease from mucopolysaccharidosis IIIB. J Neurosci Res. 2007;85(3):612-22.12. DiRosario J, Divers E, Wang C, Etter J, Charrier A, Jukkola P, et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J Neurosci Res. 2009;87(4):978-90.13. Wilkinson FL, Holley RJ, Langford-Smith KJ, Badrinath S, Liao A, Langford-Smith A, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7(4):e35787.

Sanfilippo Syndrome Clínical Guidelines I 11
same time, although the inhibition of the signaling path TLR4 in mouse MPS III-B, causes a delay in the swelling in the brain, it does not stop the progression of the neurodegenerative process9. Other studies have demonstrated the capacity of the heparan sulfate in initiating an adaptive immune response by activating T lymphocytes and B lymphocytes in mice MPS III-B12,14 which leads to the synthesis of inflammatory cytokines. All these complex processes of neuroinflammation and systemic immune inflammatory response seem to be responsible for the severe and progressive neurodegeneration seen in the MPS III A and B subtypes.
Second, gangliosides storage GM2 and GM3 taking place in the neurons of the cerebral cortex and cerebellum9,15 has been previously described as the cause of neuronal apoptosis in the Tay-Sachs and Sandhoff diseases, where the accumulation of GM2 is caused by genetic defects in hexosaminidases A and B, respectively16,17. Furthermore, considering that gangliosides, cholesterol and GPI-linked proteins (glycosylphosphatidylinositol) create clusters in lipid rafts; a possible involvement of rafts in the brains of mice with MPS III-A and III-B is suggested6 In fact, the increase of GM3 observed in a mouse model for MPS III-B would reflect an enrichment of these substances in rafts in the plasma membranes of neurons in the medial entorhinal cortex region in the mouse’s brain with MPS III-B18. On the other hand, it has been suggested that the absence of gangliosides can have harmful effects in MPS III19. Thus, a significant reduction in life expectancy as well as an increase neurodegeneration in double mutant mouse models MPS III-A or III-B with knock-out of the GalNAc transferase (Galnt3), a crucial enzyme for ganglioside synthesis, when compared with mutant mouse models of MPS III-A or III-B have been observed19.
Third, the neurodegeneration could be caused by the presence of protein aggregates18,20,21,22. In a post-mortem analysis of brains from patients with MPS III-B, it was observed a reduction in the density of GABAergic interneurons that could be related to mental illness;
14. Killedar S, Dirosario J, Divers E, Popovich PG, McCarty DM, Fu H. Mucopolysaccharidosis IIIB, a lysosomal storage disease, triggers a pathogenic CNS autoimmune response. J Neuroinflammation. 2010;7:39.15. Crawley AC, Gliddon BL, Auclair D, Brodie SL, Hirte C, King BM, et al. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006;1104(1):1-17.16. Huang JQ, Trasler JM, Igdoura S, Michaud J, Hanal N, Gravel RA. Apoptotic cell death in mouse models of GM2 gangliosidosis and observations on human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1997;6:1879-85.17. Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is su-ppressed by bone marrow transplantation. Proc Natl Acad Sci USA. 2000;97(20):10954-9.18. Ohmi K, Zhao HZ, Neufeld EF. Defects in the medial entorhinal cortex and dentate gyrus in the mouse model of Sanfili-ppo syndrome type B. PLoS One 2011;6(11):e27461.19. Mohammed EE, Snella EM, Rutz-Mendicino MM, Echevarria FD, Awedikian R, Whitley EM, et al. Accelerated clinical disease and pathology in mucopolysaccharidosis type IIIB and GalNAc transferase double knockout mice. Mol Genet Metab. 2012;107(1-2):129-35. 20. Ginsberg SD, Galvin JE, Lee VM, Rorke LB, Dickson DW, Wolfe JH, et al. Accumulation of intracellular amyloid-beta peptide (A beta 1-40) in mucopolysaccharidosis brains. J Neuropathol Exp Neurol. 1999;58(8):815-24.21. Hamano K, Hayashi M, Shioda K, Fukatsu R, Mizutani S. Mechanisms of neurodegeneration in mucopolysaccharido-ses II and IIIB: analysis of human brain tissue. Acta Neuropathol. 2008;115(5):547-59.22. Ohmi K, Kudo LC, Ryazantsev S, Zhao HZ, Karsten SL, Neufeld EF. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc Natl Acad Sci USA. 2009;106(20):8332-7.

12 I Sanfilippo Syndrome Clínical Guidelines
and it was also detected an accumulation of α-synuclein which would be related to the neurodegeneration of swollen-inflamed neurons21. In studies with mouse model of MPS III-B, hyperphosphorylated tau protein (p-tau) and amyloid beta protein were detected in neurons of the medial entorhinal cortex regions and the dentate gyrus18,22. In addition, in these two brain regions, high levels of protein associated with autophagy dysfunction were observed; proteins modified with nitrotyrosine, which is a marker of oxidative stress observed in the brains of MPS III-B11; and modified proteins linked to O-GlcNAc (N-acetylglucosamine)), which is a marker of metabolic stress; and an increased level of heparan sulfate proteoglycans, called glypicans outside the lysosome compartment18. It is suggested that this accumulation of glypicans outside the lysosome could be harmful for neurons, because they are potentially involved in the formation of p-tau and beta amyloid aggregates, which are toxic and difficult to degrade18. The accumulation of these glypicans and other markers identified in MPS III-B has also been observed in mice with MPS III-A. However, its levels were lower in the case of mice with MPS I and MPS II; and none was detected in the animal model of mouse MPS VI, which shows no blockage in the path of heparan degradation18. Although there are differences between the MPS III and the Alzheimer disease (for example, there is no formation of extracellular plaques in MPS III); their pathology is similar, such as the presence of p-tau and amyloid beta. Probably, tau hyperphosphorylation is one of the late events in the pathogenesis leading to neurodegeneration.
Fourth, the accumulation of heparan sulfate fragments, which in MPS III-A and III-B are excessively and abnormally sulfated13, can cause backlash signaling in brain neurons. In particular, they induce an overexpression of the GM130 protein and therefore alterations in the Golgi apparatus23; an increase in the proliferation and a resulting neuritis24, as well as an alteration in cell polarization and the migration of neural cells25; they all potentially contribute to the neuropathology. Most of the knowledge on the physiological mechanism of the MPS III comes from studies in animal models as the knockout mouse model of MPS III-B26 and MPS III-A27.
23. Roy E, Bruyère J, Flamant P, Bigou S, Ausseil J, Vitry S, et al. GM130 gain-of-function induces cell pathology in a model of lysosomal storage disease. Hum Mol Genet. 2012;21(7):1481-95.24. Hocquemiller M, Vitry S, Bigou S, Bruyère J, Ausseil J, Heard JM. GAP43 overexpression and enhanced neurite out-growth in mucopolysaccharidosis type IIIB cortical neuron cultures. J Neurosci Res. 2010;88(1):202-13.25. Bruyère J, Roy E, Ausseil J, Lemonnier T, Teyre G, Bohl D, et al. Heparan sulfate saccharides modify focal adhesions: implication in mucopolysaccharidosis neuropathophysiology. J Mol Biol. 2015;427(4):775-91.26. Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, et al. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc Natl Acad Sci U S A. 1999;96(25):14505-10.27. Bhaumik M, Muller VJ, Rozaklis T, Johnson L, Dobrenis K, Bhattacharyya R, et al. A mouse model for mucopolysac-charidosis type III A (Sanfilippo syndrome). Glycobiology. 1999;9(12):1389-96.

Sanfilippo Syndrome Clínical Guidelines I 13
Figure 2. Diagnostic algorithm for MPS III - Sanfilippo disease (modified from references30,31).
+++ : high levels of GAGs in urine / - : normal levels of GAGs in urine / HS: heparan sulfate / DS: dermatan sulfate / CS: chondroitin sulphate / KS: keratan sulfate / SGSH: heparan N-sulfatase / NAGLU: α-N-acetylglucosaminidase / HGSNAT: acetyl-CoAlpha-glucosaminide acetyltransferase / GNS: N-acetylglucosamine 6-sulfatase.
Progressive cognitive loss, behavioral problems, speech and sleep disorder
Coarse facial features, recurrent infections, epilepsy, thick hair, orthopedic malformations
X-rays and suspected MPS
Quantification of GAGs in urine (spectrophotometric methods: dimethylmethylene blue test)
Typification of GAGs in urine (electrophoresis, thin-layer chromatography
or mass spectrometry in tandem)
Determination of enzymatic activity (HS enzyme degradation: SGSH,
NAGLU, HGSNAT, GNS)
Molecular-genetic analysis (genes SGSH, NAGLU, HGSNAT, GNS)MPS III diagnosis confirmation
Observing the evolution of symptoms.Refer to specialist
in metabolic diseases
Diagnosis for other MPS
Clinical suspicion of genetic disorder
Refer to specialistin metabolic disorders
DSHSCSKS
HS

14 I Sanfilippo Syndrome Clínical Guidelines
Future studies may show whether the pathophysiological mechanism is the same in the four MPS III subtypes, despite the genetic and biochemical differences and the severity of clinical manifestations. Studies in mouse model for MPS III-C, have recently confirmed general signs of brain inflammation, including the activation of astrocytes, microglia and cytokines production, similar to those previously described for other MPS28. In addition, Martins et al. have observed a mitochondrial dysfunction in neurons and a neuronal loss that would explain why the MPS III-C manifests itself primarily as a neurodegenerative disease. However, the precise sequence of events starting with the accumulation of heparan sulfate leading to a widespread brain pathology and malfunction, loss and neuronal death is still to be determined.
1.2 BIOCHEMICAL AND GENETIC DIAGNOSISBecause MPS III is mainly manifested as a neurological disease and it presents few somatic clinical manifestations, early diagnosis is sometimes difficult29. The following is a summary of the current practices of laboratory in the diagnosis of MPS III, with the aim of facilitating the patient’s early identification and diagnosis. The diagnostic approach is based on clinical suspicion, radiological examinations and detecting GAGs in urine (in particular, the increased excretion of heparan sulfate). The definitive diagnosis is obtained by evaluating the corresponding enzyme activity (according to each subtype) in leukocytes or fibroblasts and using molecular genetic studies. Figure 2 shows an algorithm for the diagnosis of MPS III30,31.
Detecting GAGs in urine When there is a clinical suspicion of MPS III, the first step is the request of a quantitative test to detect the presence of GAGs in urine through spectrophotometric methods using dimethylmethylene blue (DMB)32,33. The DMB test is based on the union of GAGs to the dimethylmetilene blue and the quantification of the GAG-DMB complex with a spectrophotometer34. The sensitivity of this test is 100%, with a specificity of 75-100%. In
28. Martins C, Hůlková H, Dridi L, Dormoy-Raclet V, Grigoryeva L, Choi Y, et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain. 2015;138(Pt 2):336-55.29. Wijburg FA, Węgrzyn G, Burton BK, Tylki-Szymańska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013;102(5):462-70.30. Andrade F, Llarena M, Aldámiz-Echevarría L, de las Heras J, Sanjurjo P. Síndrome de Sanfilippo. En: Sanjurjo P, Balde-llou A, eds. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. 4ª Ed. Madrid: Ergon; 2014. Capítulo 62; p. 889-894. 31. Andrade F, Aldámiz-Echevarría L, Llarena M, Couce ML. Sanfilippo syndrome: Overall review. Pediatr Int. 2015;57(3):331-8.32. Andrade F, Prieto JA, Elorz J, Martín S, Sanjurjo P, Aldámiz-Echevarría L. Stability of urinary glycosaminoglycans in patients with mucopolysaccharidoses. Clin Chim Acta. 2008;388(1-2):73-7.33. Lage S, Prieto JA, Andrade F, Sojo A, Sanjurjo P, Aldámiz-Echevarría LJ. Reliability of a visual test for the rapid detec-tion of mucopolysaccharidoses: GAG-test(®). J Clin Lab Anal. 2011;25(3):179-84.34. De Jong JG, Wevers RA, Liebrand-van Sambeek R. Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidoses based on dimethylmethylene blue. Clin Chem. 1992;38(6):803-7.

Sanfilippo Syndrome Clínical Guidelines I 15
addition, a recent study has shown that the DMB test can be performed with urine samples dried on filter paper without losing of precision, facilitating its delivery to laboratories35. This quantitative detection of GAGs in urine is a simple screening test, non-invasive, inexpensive and useful before the suspicion of MPS; therefore it is recommended to test all patients with developmental delay and/or abnormal behavior. Although there are other quantitative tests for determining GAGs in urine (Alcian blue test, carbazole reaction and turbidity test with cetylpiridinium), these tests are less sensitive or more laborious than the DMB test and therefore it is not advisable to use them in the diagnosis of MPS III. On the other hand, the semi-quantitative tests in urine using cationic stains in filter paper (for example, Berry spot and Ames spot test) give relatively high percentages of false-positive and false-negative and are obsolete36,37,38.
It should be borne in mind that a negative result when detecting GAGs in urine does not rule out the existence of MPS III due to the fact that in some patients with attenuated forms of the disease, the levels of GAGs excretion with healthy controls can overlap and the increased excretion of heparan sulfate in the MPS III can be ignored39,40. In addition, the excretion of GAGs in urine tends to decrease with age in patients with more attenuated forms; so it is less likely that older patients and/or more attenuated phenotypes show such high levels as the ones in younger and more affected children41, 42. Another factor to take into account is the dilution of the urine sample, for determining GAGs it is advisable to collect the first morning urine or a combination of several samples of urine. Finally, given that the levels of GAGs in urine decrease with age, leveling off at approximately the 15 years of age; the interpretation of urinary GAG levels requires comparison with a normal range for each age. Therefore, clinicians should be careful when comparing GAGs tests conducted in different laboratories; since normal values and measurement units may vary between laboratories.
35. Civallero G, Bender F, Gomes A, Marasca G, Guidobono R, De Mari J, et al. Reliable detection of mucopolysacchari-duria in dried-urine filter paper samples. Clin Chim Acta. 2013;415:334-6.36. De Jong JG, Hasselman JJ, van Landeghem AA, Vader HL, Wevers RA. The spot test is not a reliable screening procedure for mucopolysaccharidoses. Clin Chem. 1991;37(4):572-5.37. Chih-Kuang C, Shuan-Pei L, Shyue-Jye L, Tuen-Jen W. MPS screening methods, the Berry spot and acid tur-bidity tests, cause a high incidence of false-negative results in sanfilippo and morquio syndromes. J Clin Lab Anal. 2002;16(5):253-8.38. Mabe P, Valiente A, Soto V, Cornejo V, Raimann E. Evaluation of reliability for urine mucopolysaccharidosis screening by dimethylmethylene blue and Berry spot tests. Clin Chim Acta. 2004;345(1-2):135-40.39. Gray G, Claridge P, Jenkinson L, Green A. Quantitation of urinary glycosaminoglycans using dimethylene blue as a screening technique for the diagnosis of mucopolysaccharidoses: an evaluation. Ann Clin Biochem. 2007;44(Pt 4):360-3.40. Neufeld, E., Muenze, J. Lysosomal Disorders. En: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, eds. The Online Metabolic and Molecular Bases of Inherited Diseases. 2011. Chapter 136, Part 16, p. 1-73. http://ommbid.mhmedical.com/book.aspx?bookid=971.41. De Ruijter J, Ijlst L, Kulik W, van Lenthe H, Wagemans T, van Vlies N, et al. Heparan sulfate derived disaccharides in plasma and total urinary excretion of glycosaminoglycans correlate with disease severity in Sanfilippo disease. J Inherit Metab Dis. 2013;36(2):271-9.42. Gallegos-Arreola MP, Machorro-Lazo MV, Flores-Martínez SE, Zúñiga-González GM, Figuera LE, González-Noriega A, et al. Urinary glycosaminoglycan excretion in healthy subjects and in patients with mucopolysaccharidoses. Arch Med Res. 2000;31(5):505-10.

16 I Sanfilippo Syndrome Clínical Guidelines
The quantitative determination of urinary GAGs is a sensitive but nonspecific test. Therefore, all urine samples testing positive in the quantitative test for GAGs must be confirmed by electrophoresis or thin layer chromatography, as both techniques improve sensitivity and specificity. These techniques may identify abnormal GAGs pattern, which in the case of patients with MPS III corresponds to a band marker for an excess of heparan sulfate only, without the presence of dermatan or keratan sulfate43. In addition, there are other methods for detecting and quantifying specific types of abnormal species of GAGs in urine by tandem mass spectrometry44,45,46. The analysis of GAGs using this technique would be useful for screening, but not for giving a definitive diagnosis. In addition, one of the disadvantages is the need for high cost equipment, so they are only available in some laboratories. Lawrence et al. are developing a method of mass spectrometry for the analysis of nonreducing ends carbohydrates (non-reducing end (NRE) analysis) as biomarkers for MPS47,48.
Determining enzyme activityThe gold standard technique for diagnosis is the determination of enzyme activity in cultured skin fibroblasts, leukocytes, plasma or serum. The specific diagnosis of MPS III-A, -B, -C or -D is confirmed by showing a decrease or absence of one of thefour enzymatic activities involved in the degradation of heparan sulfate in the patient’s leukocytes or fibroblasts; the reduction should be less than 10% when compared to the activity in healthy individuals, with normalcy in other sulfatases. Because the disease due to deficiency in multiple sulfatases also shows a reduction in the activity ofthe heparan N-sulfatase , N-acetylglucosamine 6-sulfatase and other sulfatases, biochemical analysis of at least other sulfatase is required to confirm the diagnosis of MPS III and thus rule out multiple sulfatases deficiency49.
43. Lukacs Z. Mucopolysaccharides. En: Blau N, Duran M, Gibson KM, eds. Laboratory Guide to the Methods in Bioche-mical Genetics. Springer Berlin Heidelberg; 2008. Chapter 4, p. 287-324.44. Fuller M, Rozaklis T, Ramsay SL, Hopwood JJ, Meikle PJ. Disease-specific markers for the mucopolysaccharidoses. Pediatr Res. 2004;56(5):733-8.45. Auray-Blais C, Lavoie P, Zhang H, Gagnon R, Clarke JT, Maranda B, et al. An improved method for glycosaminoglycan analysis by LC-MS/MS of urine samples collected on filter paper. Clin Chim Acta. 2012;413(7-8):771-8.46. Zhang H, Young SP, Millington DS. Quantification of glycosaminoglycans in urine by isotope-dilution liquid chromato-graphy-electrospray ionization tandem mass spectrometry. Curr Protoc Hum Genet. 2013;Chapter 17:1-14.47. Lawrence R, Brown JR, Al-Mafraji K, Lamanna WC, Beitel JR, Boons GJ, et al. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat Chem Biol. 2012;8:197-204.48. Lawrence R, Brown JR, Lorey F, Dickson PI, Crawford BE, Esko JD. Glycan-based biomarkers for mucopolysacchari-doses. Mol Genet Metab. 2014;111(2):73-83.49. Hopwood JJ, Ballabio A. Multiple sulfatase deficiency and the nature of the sulfatase family. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease. 8th Ed. New York: McGraw-Hill; 2001. P. 3725-32.

Sanfilippo Syndrome Clínical Guidelines I 17
There are different methods for enzymatic determination in MPS III: radioactively labeled oligosaccharides trials50,51,52,53,54, spectrophotometric assays55,56 and fluorescence - based assays57,58,59,60,61. Currently existing tests are based on fluorescence, where the enzyme activities are determined using the corresponding fluorogenic substrates (4-methylumbelliferil-), which are commercially available. Enzyme assays have a higher sensitivity and specificity than methods based on urine samples. It should be noted that although the enzymatic determination for MPS III-B in dried blood drop is available, it is only valid for screening and positive results should be confirmed in plasma samples62; future studies will confirm the use of this technique in the regular clinical practice63. Furthermore, the fluorimetric enzyme tests for MPS III-A, -C and -D does not leave room to fluorescent products, and therefore, they require the addition of a hydrolytic enzyme coupled to the enzyme reaction to generate the fluorescent substrate 4-methylumbelliferone.
50. Hopwood JJ, Elliott H. The diagnosis of the Sanfilippo C syndrome, using monosaccharide and oligosaccharide subs-trates to assay acetyl-CoA: 2-amino-2-deoxy-α-glucoside N-acetyltransferase activity. Clin Chim Acta. 1981;112(1):67-75.51. Hopwood JJ, Elliott H. Selective depolymerisation of heparin to produce radio-labelled substrates for sulfamidase, 2-acetamido-2-deoxy-alpha-D-glucosidase, acetyl-CoA:2-amino-2-deoxy-alpha-D-glucoside N-acetyltransferase, and 2-acetamido-2-deoxy-D-glucose 6-sulfate sulfatase. Carbohydr Res. 1981;91(2):165-90.52. Hopwood JJ, Elliott H. Detection of the Sanfilippo type B syndrome using radiolabelled oligosaccharides as substrates for the estimation of α-N-acetylglucosaminidase. Clin Chim Acta.1982;120(1):77-86.53. Reynertson R, Campbell P, Ford JD, Jacobsson I, Rodén L, Thompson JN. New oligosaccharides from heparin and heparan sulfate and their use as substrates for heparin-degrading enzymes. J Biol Chem. 1983;258(12):7449-59.54. Thompson JN, Rodén L, Reynertson R. Oligosaccharide substrates for heparin sulfamidase. Anal Biochem. 1986;152(2):412-22.55. Nowakowski RW, Thompson JN, Taylor KB. Sanfilippo syndrome, type D: a spectrophotometric assay with prenatal diagnostic potential. Pediatr Res. 1989;26(5):462-6.56. Thompson JN, Huffman PS, McConkie-Rosell A, Hessling J. Prenatal diagnosis of Sanfilippo syndrome type A by early amniocentesis. Biochem. Mol Biol Int. 1993;29(5):793-7.57. He W, Voznyi YaV, Boer AM, Kleijer WJ, van Diggelen OP. A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease type D (MPS IIID). J Inherit Metab Dis. 1993;16(6):935-41.58. He W, Voznyi Ya V, Huijmans JG, Geilen GC, Karpova EA, Dudukina TV, et al. Prenatal diagnosis of Sanfilippo disease type C using a simple fluorometric enzyme assay. Prenat Diagn. 1994;14:17-22.59. Karpova EA, Voznyi YaV, Keulemans JL, Hoogeveen AT, Winchester B, Tsvetkova IV, et al. A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease type A (MPS IIIA). J Inherit Metab Dis. 1996;19(3):278-85.60. Marsh J, Fensom AH. 4-Methylumbelliferyl alpha-N-acetylglucosaminidase activity for diagnosis of Sanfilippo B disea-se. Clin Genet. 1985;27(3):258-62.61. Voznyi YaV, Karpova EA, Dudukina TV, Tsvetkova IV, Boer A, Janse H, et al. A fluorimetric enzyme assay for the diagno-sis of Sanfilippo disease C (MPS III C). J Inherit Metab Dis. 1993;16(2):465-72.62. Civallero G, Michelin K, de Mari J, Viapiana M, Burin M, Coelho JC, et al. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin Chim Acta. 2006;372(1-2):98-102.63. Civallero G, De Mari J, Viapiana Camelier M, Burin M, Giugliani R. Assay of heparan-Nsulfamidase in dried leukocytes impregnated in filter paper: a new tool for the identification of mucopolisaccharidosis IIIA and potentially other lysosomal disorders. Mol Genet Metab. 2013;108(4):267-8.

18 I Sanfilippo Syndrome Clínical Guidelines
Therefore, depending on the method used, patients with deficient beta-hexosaminidase activity may give false-positive for MPS III-C61; and in the case of the MPS III-A and -D (both caused by sulfatase deficiencies) the activity of a second sulfatase to rule out deficiency of multiple sulfatases should be measure64. Currently, new methods are being developed for the quantification of enzymatic activities in the MPS III through fluorimetry and tandem mass spectrometry65, but its use is not yet available.
Molecular genetic analysisThe diagnosis for identification of mutations originating each MPS III subtype is confirmed through molecular studies. In this technique, the coding regions (exons) and flanking intronic regions of each of the four genes responsible for MPS III are amplified and sequenced, and the results are compared to reference sequences and databases of known mutations and polymorphisms. The molecular genetic analysis should be offered to the families of all patients for providing genetic counseling. For a limited number of genetic variants the genotype-phenotype correlations which can help in prognosis (more details in the next section of this chapter) have been described.
Mutations described to date are available on The Human Gene Mutation Database (HGMD®) del Institute of Medical Genetics (Cardiff, Reino Unido)66. The SGSH gene encoding heparan sulfamidase or N-sulfatase, whose deficiency results in MPS III-A, is located in chromosome 17q25.367. To date, 142 mutations have been described, missense being the majority (64.1%). Nonsense mutations have also been found (with premature shutdown), small insertions and small deletions. In MPS III-B, the NAGLU gene encoding the α-N-acetylglucosamine minidase is located in chromosome 17q2168. 154 mutations have been described in this gene, including missense (94), nonsense, deletions, insertions and splicing. The HGSNAT gene encoding the acetyl-CoA:α-glucosaminide-N-acetyltransferase is located in a pericentromeric region of chromosome 8 (8p11.2-p11.1), and its mutations are involved in the MPS III-C69. HTo date 66 mutations have been described (51.5% are missense mutations). The deficit of the N-acetylglucosamine 6-sulfatase enzyme triggering the MPS
64. Dierks T, Schlotawa L, Frese MA, Radhakrishnan K, von Figura K, Schmidt B. Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease - Lysosomal storage disorders caused by defects of non-lyso-somal proteins. Biochim Biophys Acta. 2009;1793(4):710-25.65. Wolfe BJ, Ghomashchi F, Kim T, Abam CA, Sadilek M, Jack R, et al. New substrates and enzyme assays for the de-tection of mucopolysaccharidosis III (Sanfilippo Syndrome) types A, B, C, and D by tandem mass spectrometry. Bioconjug Chem. 2012;23(3):557-64.66. The Human Gene Mutation Database en Institute of Medical Genetics in Cardiff, Cardiff University 2003. http://www.hgmd.cf.ac.uk/ac/index.php (acceso 11 Noviembre 2015).67. Scott HS, Blanch L, Guo XH, Freeman C, Orsborn A, Baker E, et al. Cloning of the sulphamidase gene and identifica-tion of mutations in Sanfilippo A syndrome. Nat Genet. 1995;11(4):465-7.68. Zhao HG, Li HH, Bach G, Schmidtchen A, Neufeld EF. The molecular basis of Sanfilippo syndrome type B. Proc Natl Acad Sci U S A. 1996;93(12):6101-5.69. Fan X, Zhang H, Zhang S, Bagshaw RD, Tropak MB, Callahan JW, et al. Identification of the gene encoding the enzy-me deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C). Am J Hum Genet. 2006;79:738-44.

Sanfilippo Syndrome Clínical Guidelines I 19
III-D is due to mutations in the GNS gene, located on chromosome 12q1470,71. A total of 23 mutations, with a very high percentage of large deletions have been described, and only 7 are missense.
Prenatal DiagnosisThe prenatal test by testing enzymatic activity in cells from the amniotic fluid or chorionic villi is available in a very limited number of specialized centers at the global level; therefore for the prenatal diagnosis, when possible, we prefer the molecular genetic analysis72. In this method we must know a priori the mutated alleles for the specific family subjected to the test, so that the molecular diagnosis in chorionic villi can be easier requiring less amount of samples40. On the other hand, if we know the MPS III subtype but there is no information on the mutations, it is possible to make a direct enzymatic test (for example, the one described in reference58). In advanced stages of pregnancy, we can make a qualitative analysis of GAGs in amniotic fluid73. Before performing these tests it is important to provide appropriate genetic counseling74.
Carriers diagnosisCarriers diagnosis is a service frequently requested by families with MPSIII. Nowadays, as long as we know the mutation in the specific family requesting it, it is possible to provide final information of the status of carriers, being the molecular diagnosis based on the analysis of mutations in the DNA the only ultimate test for the determination. If the mutation is unknown, it would be necessary to use other biochemical and/or enzymatic methods, but those techniques have certain limitations2,5,75,76.
Future prospects: neonatal screening and whole exome sequencingDuring the last decade a potential neonatal screening test for MPS III is available based on automated determination of GAGs in dry urine samples on paper77. However, the sensitivity and specificity of this MPS test has not been evaluated in a pilot study. In addition, this test has never been included in the current neonatal screening programs, since those metabolic genetic disorders not counting with approved therapies, such as the MPS III, do not meet the criteria for inclusion in these screening programs. This
70. Beesley CE, Burke D, Jackson M, Vellodi A, Winchester BG, Young EP. Sanfilippo syndrome type D: identification of the first mutation in the N-acetylglucosamine-6-sulphatase gene. J Med Genet. 2003;40(3):192-4.71. Mok A, Cao H, Hegele RA. Genomic basis of mucopolysaccharidosis type IIID (MIM 252940) revealed by sequencing of GNS encoding N-acetylglucosamine-6-sulfatase. Genomics. 2003;81:1-5.72. Hopwood JJ. Prenatal diagnosis of Sanfilippo syndrome. Prenat Diagn. 2005;25(2):148-50.73. Whiteman P, Henderson H. A method for the determination of amniotic-fluid glycosaminoglycans and its application to the prenatal diagnosis of Hurler and Sanfilippo diseases. Clin Chim Acta. 1977;79(1):99-105.74. Fletcher J, Wilcken B. Neonatal screening for lysosomal storage disorders. Lancet. 2012;379(9813):294-5.75. Vance JM, Conneally PM, Wappner RS, Yu PL, Brandt IK, Pericak-Vance MA. Carrier detection in Sanfilippo syndrome type B: report of six families. Clin Genet. 1981;20(2):135-40.76. Hopwood JJ, Muller V, Harrison JR, Carey WF, Elliott H, Robertson EF, et al. Enzymatic diagnosis of the mucopolysac-charidoses: experience of 96 cases diagnosed in a five-year period. Med J Aust. 1982;1(6):257-60.77. Whitley CB, Spielmann RC, Herro G, Teragawa SS. Urinary glycosaminoglycan excretion quantified by an automated semimicro method in specimens conveniently transported from around the globe. Mol Genet Metab. 2002;75(1):56-64.

20 I Sanfilippo Syndrome Clínical Guidelines
could change with the revision of these criteria78, as well as the new therapies in the clinical trial stage for MPS III. In addition, the evolution of new technologies based on tandem mass spectrometry for determining enzymatic activity62,65 or the quantification of oligosaccharides derived from GAGs79, will offer new possibilities for considering neonatal screening for one or more MPS III subtypes. For example, pilot newborn screening programs for other MPS are currently on development phase80,81.
Finally, advances in the methodology for whole exome sequencing, which corresponds to the coding regions of proteins in the genome, offer future possibilities for screening and diagnosis. Although to date this technique has several limitations82, there are some cases of patients with MPS III that have been identified through this methodology83,84. In the case of MPS III, the mutational spectrum of the four subtypes consists primarily in point mutations, not being common mutations caused by large deletions or changes of reading frame. Therefore, the whole exome sequencing would completely change the diagnostic algorithms for MPS III, allowing the simultaneous screening of several neuropathic congenital disorders, identifying previously unknown or private mutations, and assisting in the understanding of the phenotypic variants by identifying genetic modifiers85.
Without a doubt, the future development of a neonatal screening in dried blood drop and high-throughput methods for the whole exome sequencing will mean significant changes for the diagnosis of MPS III.
78. Calonge N, Green NS, Rinaldo P, Lloyd-Puryear M, Dougherty D, Boyle C, et al. Committee report: Method for evalua-ting conditions nominated for population-based screening of newborns and children. Genet Med. 2010;12(3):153-9.79. De Ruijter J, de Ru MH, Wagemans T, Ijlst L, Lund AM, Orchard PJ, et al. Heparan sulfate and dermatan sulfate de-rived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol Genet Metab. 2012;107(4):705-10.80. Lin SP, Lin HY, Wang TJ, Chang CY, Lin CH, Huang SF, et al. A pilot newborn screening program for Mucopolysaccha-ridosis type I in Taiwan. Orphanet J Rare Dis. 2013;8:147.81. Ruijter GJ, Goudriaan DA, Boer AM, Van den Bosch J, Van der Ploeg AT, Elvers LH, et al. Newborn screening for hunter disease: a small-scale feasibility study. JIMD Rep. 2014;14:23-7.82. Ku CS, Naidoo N, Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum Genet. 2011;129(4):351-70.83. Brady J, Trehan A, Landis D, Toro C. Mucopolysaccharidosis type IIIB (MPS IIIB) masquerading as a behavioural disorder. BMJ Case Rep. 2013;2013.84. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neu-ro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78.85. Bodamer OA, Giugliani R, Wood T. The laboratory diagnosis of mucopolysaccharidosis III (Sanfilippo syndrome): A changing landscape. Mol Genet Metab. 2014;113(1-2):34-41.

Sanfilippo Syndrome Clínical Guidelines I 21
1.3 GENOTYPE-PHENOTYPE CORRELATIONMost mutations identified in MPS III are private - unique to individual families, making it difficult to establish genotype-phenotype correlations as well as the screening of the general population. Furthermore, the interpretation of the clinical phenotype resulting from the different mutations is very difficult for this type of disease in which the predominant feature is the degeneration of the central nervous system and most clinical evaluations are the result of independent observations not always comparable. In addition, the prediction of genotype-phenotype correlations is complicated due to the presence of polymorphisms, which could modify the residual enzyme activity and influence the clinical phenotype. In fact, the MPS III phenotype varies considerably from severe, intermediate and attenuated. In order to predict a personalized clinical severity for a particular patient, it would be crucial to have a detailed analysis of each patient indicating genotype record, a determination of the amount and activity of the enzyme in fibroblasts derived from patients, a detailed phenotype description, the natural history and accurate measurements of the amount and nature of accumulated compounds.
Mutations responsible for the four MPS III subtypes are countless (HGMD®)66, although in some cases one or a few mutations can be dominant in a particular geographical region or ethnic group. Several studies have shown that subtype B is more prevalent in southern Europe86,87,88,89 and subtype A in northern Europe90,91,92,93. ASome mutations described can clearly be classified as null, that is that as a result they lead to the complete absence of enzyme activity. They include deletions of large DNA fragments, rearrangements within the chromosome, frameshift mutation, mutations in consensus regions for splicing, and nonsense mutations. A priori it is expected that the presence of two null alleles cause increased severity in the disease; but if one allele has some residual enzyme activity, this could result in a milder form. In general, the clinical effect (phenotype) of missense mutations can only be predicted based on the previous experience with these mutations.
86. Michelakakis H, Dimitriou E, Tsagaraki S, Giouroukos S, Schulpis K, Bartsocas CS. Lysosomal storage diseases in Greece. Genet Couns. 1995;6(1):43-7.87. Weber B, Guo XH, Kleijer WJ, van de Kamp JJ, Poorthuis BJ, Hopwood JJ. Sanfilippo type B syndrome (mucopoly-saccharidosis III B): allelic heterogeneity corresponds to the wide spectrum of clinical phenotypes. Eur J Hum Genet. 1999;7(1):34-44.88. Beesley C, Moraitou M, Winchester B, Schulpis K, Dimitriou E, Michelakakis H. Sanfilippo B syndrome: molecular defects in Greek patients. Clin Genet. 2004;65(2):143-9.89. Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, et al. Prevalence of lysosomal storage diseases in Portu-gal. Eur J Hum Genet. 2004;12(2):87-92.90. Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet. 1997;101(3):355-8.91. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105(1-2):151-6.92. Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28(6):1011-7.93. Malm G, Månsson JE. Mucopolysaccharidosis type III (Sanfilippo disease) in Sweden: clinical presentation of 22 chil-dren diagnosed during a 30-year period. Acta Paediatr. 2010;99(8):1253-7.

22 I Sanfilippo Syndrome Clínical Guidelines
Even the knowledge of the structure and catalytic center of the enzyme is not always useful for predicting the effect of missense mutations, because many changes/replacements of amino acids can cause problems in the transport of the newly synthesized protein from the endoplasmic reticulum to the lysosome, without interfering with the enzyme’s catalytic activity40. Therefore, in order to establish correlations between the type of mutation and the severity of the biochemical and clinical phenotype, which is so useful for the clinical prognosis, it is necessary to combine studies of populations with studies on the expression of mutated enzymes in mammalian cell lines, which allow typifying the effects of each mutation in the enzymatic activity, the folding, stability, processing and intracellular traffic.
MPS III-AThe MPS III-A is the most common subtype in northern Europe90,91,92,93. Several revisions of the correlation between the genotype and clinical manifestations in patients with MPS III-A94,95,96, confirming the great phenotypic variability associated with the genotype have been published. In addition, these studies of molecular characterization of SGSH gene in patients with MPS III-A indicated a high incidence of certain mutations in different geographical origins, which could prove to be very useful for the molecular diagnosis of this subtype94. Thus, common mutations have been identified by their geographical distribution, in the SGSH gene: p.R245H mutation is more common in the Australian, Dutch and German people97; p.S66W in the Italian population, especially in Sardinia98; c.1079delC in the Spanish population99,100 and p.R74C in Poland101.
94. Yogalingam G, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: Diagnostic, clinical, and biological implications. Hum Mutat 2001;18:264-8195. Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJ, Wevers RA, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010;68(6):876-87.96. Héron B, Mikaeloff Y, Froissart R, Caridade G, Maire I, Caillaud C, et al. Incidence and natural history of mucopolysac-charidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011;155A(1):58-68.97. Weber B, Guo XH, Wraith JE, Cooper A, Kleijer WJ, Bunge S, et al. Novel mutations in Sanfilippo A syndrome: implica-tions for enzyme function. Hum Mol Genet. 1997;6(9):1573-9.98. Di Natale P, Balzano N, Esposito S, Villani GR. Identification of molecular defects in Italian Sanfilippo A patients inclu-ding 13 novel mutations. Hum Mutat. 1998;11(4):313-20.99. Montfort M, Vilageliu L, Garcia-Giralt N, Guidi S, Coll MJ, Chabás A, et al. Mutation 1091delC is highly prevalent in Spanish Sanfilippo syndrome type A patients. Hum Mutat. 1998;12(4):274-9.100. Chabás A, Montfort M, Martínez-Campos M, Díaz A, Coll MJ, Grinberg D, et al. Mutation and haplotype analyses in 26 Spanish Sanfilippo syndrome type A patients: possible single origin for 1091delC mutation. Am J Med Genet. 2001;100(3):223-8.101. Bunge S, Ince H, Steglich C, Kleijer WJ, Beck M, Zaremba J, et al. Identification of 16 sulfamidase gene mutations in-cluding the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo A). Hum Mutat. 1997;10(6):479-85.

Sanfilippo Syndrome Clínical Guidelines I 23
P.R245H, p.Q380R, p.S66W and c.1080delC mutations are associated with the severe classic phenotype95,97,101,102,103,104. On the other hand, the presence of p.G122R, p.R206P, p.S298P, p.I322S and p.E369K mutations is considered responsible for a more attenuated Sanfilippo phenotype94,105,106. In particular, patients with the p.S298P mutation on one or more alleles showed an attenuated phenotype, with a significantly longer preservation of psychomotor functions and higher survival rate, even in combination with the severe mutations p.R245H, p.Q380R, p.S66W and c.1080delC95. Furthermore, missense mutations p.L12Q, p.180L and p.T421R seem to result in a very gentle MPS III-A phenotype95.
The prediction of the phenotype based on genotype is hindered by the presence of polymorphisms in the SGSH gene, such as p.V226A, p.V361I and p.R456H97,98, which could modify the residual activity of the enzyme mutated94.
In Spain, the MPS III-A is the most common subtype107, in contrast with previous studies indicating that this MPS III subtype is more common in northern Europe90,91,92,93. This retrospective study sets the natural history of the MPS III in Spain, with the participation of 55 patients (62%, MPS III-A; 20%, MPS III-B and 18%, MPS III-C). As in other studies, diversity was observed in the clinical manifestations due to allelic heterogeneity. Most mutations for MPS III-A were previously described99,100; and 3 new mutations were identified107.
MPS III-BMost mutations found in the NAGLU gene are private, indicating the high molecular heterogeneity of subtype B, which is most common in Southern Europe. In Greece it was noted that subtype B is more prevalent than A88,108. In Portugal, where the MPS III-B is also the most common subtype, p.R234C mutation was identified as a founder mutation which is common in patients from Spain and Portugal, suggesting a single and relatively recent origin in the Iberian Peninsula109.
102. Blanch L, Weber B, Guo XH, Scott HS, Hopwood JJ. Molecular defects in Sanfilippo syndrome type A. Hum Mol Genet. 1997;6(5):787-91.103. Weber B, van de Kamp JJ, Kleijer WJ, Guo XH, Blanch L, van Diggelen OP, et al. Identification of a common mutation (R245H) in Sanfilippo A patients from The Netherlands. J Inherit Metab Dis. 1998;21(4):416-22.104. Beesley CE, Young EP, Vellodi A, Winchester BG. Mutational analysis of Sanfilippo syndrome type A (MPS IIIA): identi-fication of 13 novel mutations. J Med Genet. 2000;37(9):704-7.105. Gabrielli O, Coppa GV, Bruni S, Villani GR, Pontarelli G, Di Natale P. An adult Sanfilippo type A patient with homozy-gous mutation R206P in the sulfamidase gene. Am J Med Genet A. 2005;133A(1):85-9.106. Meyer A, Kossow K, Gal A, Steglich C, Mühlhausen C, Ullrich K, et al. The mutation p.Ser298Pro in the sulphamidase gene (SGSH) is associated with a slowly progressive clinical phenotype in mucopolysaccharidosis type IIIA (Sanfilippo A syndrome). Hum Mutat. 2008;29(5):770.107. Delgadillo V, O’Callaghan Mdel M, Gort L, Coll MJ, Pineda M. Natural history of Sanfilippo syndrome in Spain. Orpha-net J Rare Dis. 2013;8:189.108. Beratis NG, Sklower SL, Wilbur L, Matalon R. Sanfilippo disease in Greece. Clin Genet. 1986;29(2):129-32.109. Mangas M, Nogueira C, Prata MJ, Lacerda L, Coll MJ, Soares G, et al. Molecular analysis of mucopolysaccharido-sis type IIIB in Portugal: evidence of a single origin for a common mutation (R234C) in the Iberian Peninsula. Clin Genet. 2008;73(3):251-6.

24 I Sanfilippo Syndrome Clínical Guidelines
All described mutations occur once or at relatively low frequencies, reflecting the broad phenotypic spectrum observed in MPS III-B87,94. Nonsense mutations, including p.R297X and p.E336X, are associated with severe phenotypes87,94,110,111. Most missense mutations identified are unique, it is noted that among them mutations p.Y140C, p.R674H, p.R643C, p.R565W and p.P521L appeared with relative frequencies of 3.4 - 5.4% in patients with MPS III-B94. The p.R674H, p.R565W and p.P521L mutations have been identified in patients with severe phenotype87, 110,111,112. On the other hand, mutations p.F48L, p.G69S, p.S612G, p.R643C, p.E634K and p.L497V appear to reduce the clinical severity of the MPS III-B phenotype, so that enzymes with these mutations have some residual activity94,111. Finally, mutations with deletions and insertions identified in the NAGLU gene are associated with severe phenotypes111, probably due to increased instability and/or absence of residual enzyme activity.
As in the case of the MPS III-A, several polymorphisms have been identified in the MPSIII-B, including a missense change p.G737R87,110, which could potentially modify the severity of the disease94.
In Spanish population, several new mutations were identified in the NAGLU gene113. Among them, the p.W168X and p.R234C mutations were found in more than one patient. Subsequent studies suggest that the p.R234C mutation originated in the Iberian Peninsula109. Delgadillo et al., in addition to the mutations described above, have recently identified a new mutation causing the MPS III-B in the retrospective study of the natural history of MPS III in Spain (11 patients with MPS III-B)107. The high allelic heterogeneity makes it difficult to establish a clear genotype-phenotype correlation.
110. Zhao HG, Aronovich EL, Whitley CB. Genotype-phenotype correspondence in Sanfilippo syndrome type B. Am J Hum Genet. 1998;62(1):53-63.111. Valstar MJ, Bruggenwirth HT, Olmer R, Wevers RA, Verheijen FW, Poorthuis BJ, et al. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis. 2010;33(6):759-67.112. Beesley CE, Young EP, Vellodi A, Winchester BG. Identification of 12 novel mutations in the alpha-N-acetylgluco-saminidase gene in 14 patients with Sanfilippo syndrome type B (mucopolysaccharidosis type IIIB). J Med Genet. 1998; 35(11):910-4.113. Coll MJ, Antón C, Chabás A. Allelic heterogeneity in Spanish patients with Sanfilippo disease type B. Identification of eight new mutations. J Inherit Metab Dis. 2001;24(1):83-4.

Sanfilippo Syndrome Clínical Guidelines I 25
MPS III-CIn 2009, Feldhammer et al. conducted a review of all mutations described in the HGSNAT gene identifying 10 new mutations114114. Although the spectrum of mutations in patients with MPS III-C showed a great heterogeneity, and there was no obvious genotype-phenotype correlation, it was possible to identify some mutations with high frequency in certain populations, suggesting a founder effect of missense mutations p.R344C and P.S518F in the Netherlands (22% and 29.3%, respectively)115, mutation insertion c.525dupT in Portugal (83%)116 and mutation c.852-1G>A in the south of Italy117; which was very useful for the molecular studies of MPS III-C in these countries. There are other described mutations p.R384X, c.493+1G>A, p.R344H and p.S541L, which although having a relatively high frequency in families with MPS III-C, are characterized by a fairly wide geographical distribution114.
Most patients with MPS III-C show severe clinical phenotypes accompanied by a total or almost total loss of the HGSNAT’s enzymatic activity. It has been observed that both the severity and the clinical course are highly variable even among siblings, complicating the prediction of clinical phenotype for each patient. Still, two mutations probably associated with an attenuated phenotype p.G262R and p.S539C have been described115. For the MPS III-C, several polymorphisms p.P237Q, p.V481L, p.K523Q and p.A615T were identified, resulting in amino acid changes not affecting the enzyme activity, and therefore without clinical relevance114.
In Spain, Canals et al. identified 9 pathogenic mutations (7 of them new) in the HGSNAT gene in 11 patients with MPS III-C (7 of Spanish origin, 1 Argentine and 3 of Moroccan origin)118. The most frequent mutations were c.372-2A>G and c.234+1G>A; the haplotype analysis suggested a single source for both. Each one of the 7 new mutations identified appeared only in a patient, 4 of them are missense mutations without residual enzyme activity (0-1.19%)118.
114. Feldhammer M, Durand S, Mrázová L, Boucher RM, Laframboise R, Steinfeld R, et al. Sanfilippo syndrome type C: mutation spectrum in the heparan sulfate acetyl-CoA: alpha-glucosaminide N-acetyltransferase (HGSNAT) gene. Hum Mutat. 2009;30(6):918-25.115. Ruijter GJ, Valstar MJ, van de Kamp JM, van der Helm RM, Durand S, van Diggelen OP, et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol Genet Metab. 2008;93:104-11.116. Coutinho MF, Lacerda L, Prata MJ, Ribeiro H, Lopes L, Ferreira C, et al. Molecular characterization of Portuguese patients with mucopolysaccharidosis IIIC: two novel mutations in the HGSNAT gene. Clin Genet. 2008;74:194-5.117. Fedele AO, Filocamo M, Di Rocco M, Sersale G, Lübke T, di Natale P, et al. Mutational analysis of the HGSNAT gene in Italian patients with mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Hum Mutat. 2007;28(5):523.118. Canals I, Elalaoui SC, Pineda M, Delgadillo V, Szlago M, Jaouad IC, et al. Molecular analysis of Sanfilippo syndrome type C in Spain: seven novel HGSNAT mutations and characterization of the mutant alleles. Clin Genet. 2011;80(4):367-74.

26 I Sanfilippo Syndrome Clínical Guidelines
MPS III-DThe MPS III-D is the rarest subtype of MPS III. In 2003, for the first time two mutations in the GNS gene responsible for MPS III-D70,71, were identified, although the gene had been identified in 1988119. In 2010, Valstar et al. conducted a review of 22 mutations previously identified in only 31 patients from 26 families120. In 2015, a total of 23 mutations in the GNS gene (HGMD®data base) were identified66; a very high percentage of them are large deletions, a fact that has not been observed in genes involved in other lysosomal diseases. Although it is still difficult to establish genotype-phenotype correlations for the MPS III-D; it can be predicted that nonsense mutations and insertions or deletions causing reading frame changes would be considered pathogenic.
119. Robertson DA, Callen DF, Baker EG, Morris CP, Hopwood JJ. Chromosomal localization of the gene for human glucosamine-6-sulphatase to 12q14. Hum Genet. 1988;79(2):175-8.120. Valstar MJ, Bertoli-Avella AM, Wessels MW, Ruijter GJ, de Graaf B, Olmer R, et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010;31(5):E1348-60.

Sanfilippo Syndrome Clínical Guidelines I 27
RECURSOS WEB- Stop Sanfilippo Foundation (Spain).
http://www.stopsanfilippo.org
- Fondation Sanfilippo Suisse (Switzerland). http://www.fondation-sanfilippo.ch
- Team Sanfilippo (EEUU). http://teamsanfilippo.org/
- Sanfilippo Network (Mexico). http://www.redsanfilippo.org/
- Sanfilippo Children’s Foundation (Australia). https://www.sanfilippo.org.au/
- Sanfilippo Children’s Research Foundation (Canada). http://www.alifeforelisa.org/
- OMIM®: Online Mendelian Inheritance in Man®. An Online Catalog of Human Genes and Genetic Disorders. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine. http://omim.org/
- Genetics Home Reference. http://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-iii U.S. National Library of Medicine (NLM), Campus of the National Institutes of Health (NIH), Bethesda, Maryland.
- HGMD®: The Human Gene Mutation Database. Institute of Medical Genetics in Cardiff, Cardiff University 2003. http://www.hgmd.cf.ac.uk/ac/index.php
- Book “The Online Metabolic and Molecular Bases of Inherited Diseases”. Part 16:“Lysosomal Disorders”. http://ommbid.mhmedical.com/book.aspx?bookid=971
- The Spanish Forum for Lysosomal diseases. http://www.enfermedadeslisosomales.es
- MedlinePlus Medical Encyclopedia. Sanfilippo Syndrome: https://www.nlm.nih.gov/medlineplus/spanish/ency/article/001210.htm

Sanfilippo Syndrome Clínical Guidelines I 29
02.MPS III. Clinic. Clinicalforms. Differential diagnosis. Luis González Gutiérrez-Solana. Deputy of Neuropaediatrics. Unit for Neurodegenerative Diseases. Niño Jesús University Children’s Hospital Madrid.

30 I Sanfilippo Syndrome Clínical Guidelines
Mucopolysaccharidosis (MPS) type III or Sanfilippo syndrome is clinically characterized by a serious dysfunction of the central nervous system, with only slight somatic involvement1. The disparity between severe cerebral degeneration (with progressive cognitive and behavioral impairment) and the skeleton’s relatively slight involvement, the viscera and facial features is unique among the MPS2,3.
Most individuals with MPS III have a severe form of the disease, in which there are three stages: an initial phase of psychomotor retardation, increased in the speech areas, starting between 1 to 4 years of age; a phase of slowly progressive cognitive impairment and prominent behavioral disorders, which begins between the 3 and 6 years of age; and a final phase of severe dementia and neurodegeneration with motor impairment during adolescence, which often leads to death at the end of the second or the beginning of the third decade of life. Other symptoms include sleep disorders, recurring diarrhea, ENT repeated infections, hearing and visual impairment and epilepsy. However, a wide variability is observed in the onset and severity of the disease in all the MPS III types (A, B, C and D), and even within the same family4,5.
2.1 EARLY SYMPTOMSChildren are apparently normal at birth. They seem to develop normally during the first year of life6; although in the Meyer et al series4 67.6% of patients show symptoms in the first year, and on the Buhrman et al series7 40% of patients show symptoms in the first 12 months and 74% in the first 24 months.
The onset of the disease generally between the 2 and 6 years of age is variable, with psychomotor retardation, behavioral problems or a combination of both8. Other symptoms
1. Neufeld EF, Muenzer J. The mucopolysaccharidoses. En: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The metabolic and molecular basis of inherited disease. 8th ed. New York: McGraw-Hill; 2001. p. 3421-52.2. Sanfilippo disease / mucopolysaccharidosis type III. En: Nyhan WL, Barshop BA, Al-Aqeel AI, eds. Atlas of inherited metabolic diseases. 3th ed. London: Hodder Arnold; 2012. p. 580-7.3. Wraith JE. Mucopolysaccharidoses and oligosaccharidoses. En: Saudubray JM, van der Berghe G, Walter JH, eds. Inborn metabolic diseases: diagnosis and treatment. 5th ed. Berlin: Springer-Verlag; 2012. p. 580-90. 3. Wraith JE. Mucopolysaccharidoses and oligosaccharidoses. En: Saudubray JM, van der Berghe G, Walter JH, eds. Inborn metabolic diseases: diagnosis and treatment. 5th ed. Berlin: Springer-Verlag; 2012. p. 580-90. 4. Meyer A, Kossow K, Gal A, Mühlhausen C, Ullrich K, Braulke T, Muschol N. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics 2007;120:e1255-61.5. Ruijter GJ, Valstar MJ, van de Kamp JM, van der Helm RM, Durand S, van Diggelen OP, Wevers RA, Poorthuis BJ, Pshezhetsky AV, Wijburg FA. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol Genet Metab 2008;93:104-11.6. Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJ, Wevers RA, van Diggelen OP, Poorthuis BJ, Halley DJ, Wijburg FA. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol 2010;68:876-87. 7. Buhrman AD, Thakkar K, Poe M, Escolar ML. Natural history of Sanfilippo syndrome type. J Inherit Metab Dis 2014;37:431-7.8. Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 2008;31:240-52.

Sanfilippo Syndrome Clínical Guidelines I 31
can be coarse hair, hirsutism, sleeping disorders and mild hepatomegaly (common in young patients but not in adolescents and adults)1. At the time of diagnosis speech delay is much higher than motor delay5,9. Speech is delayed, with poor coordination and content, and some patients never learn to speak1. Cleary and Wraith10 describe 62 patients with MPS III (47 type A, 12 type B and 3 type C). The diagnosis was made at an average age of 4.9 years. The initial symptoms in most patients were a combination of psychomotor retardation, especially language, and recurrent ENT infections. More than half had behavioral disorders and diarrhea, and 16 had hearing loss. Meyer et al4 studied 71 patients with MPS IIIA. In the initial development they noted that 66.7% had speech delay (two patients never got to speak) and 33.3% motor retardation. Symptoms of more frequent onset were sleeping disorders (38%), behavioral disorders (38%), diarrhea (31%), recurrent infections (23%), speech delay (20%), and hernias (20%). In the only Spanish series published11 the most common symptoms were delayed speech (85%), coarse facies (78%), hyperactivity (65%), recurrent diarrhea (50%) and recurrent otitis (46%). In the series of Buhrman et al4 the most common initial symptoms were delayed speech (48%), dysmorphic facies (22%), hearing loss (20%), motor retardation (13%), developmental delay (11%), behavioral problems (9%), somatic symptoms (4%) and otitis (4%).
2.2 BEHAVIORAL DISORDERThe progressive behavioral disorder is characteristic of Sanfilippo syndrome. It usually starts around the 3-5 years of age and consists of hyperactive, chaotic, anxious, and sometimes aggressive behavior4. Children with MPS III may appear stubborn and isolated, and the interaction with other children may be difficult. Self-stimulatory behaviors12 and biting or licking objects are frequent9. They may have temper tantrums being destructive and dangerous to their siblings, so they require continuous monitoring. The presence of severe cognitive impairment along with important behavioral disorders in individuals with a normal physical strength, makes the management of these children particularly difficult1. Some patients are so difficult to control requiring placement in an institution13. Behavioral problems are decreased with age until disappearing due to the progression of neurodegeneration eventually leading to a complete loss of initiative8.
9. Van de Kamp JJ, Niermeijer MF, von Figura K, Giesberts MA. Genetic heterogeneity and clinical variability in the Sanfili-ppo syndrome (types A, B, and C). Clin Genet 1981;20:152-60.10. Cleary MA, Wraith JE. Management of mucopolysaccharidosis type III. Arch Dis Child 1993;69:403-6 .11. Delgadillo V, O’Callaghan M del M, Gort L, Coll MJ, Pineda M. Natural history of Sanfilippo syndrome in Spain. Orpha-net J Rare Dis 2013;8:189. 12. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: an epidemiological study. J Ment Defic Res 1983;27:185-203.13. Lindor NM, Hoffman A, O’Brien JF, Hanson NP, Thompson JN. Sanfilippo syndrome type A in two adult sibs. Am J Med Genet 1994;53:241-4.

32 I Sanfilippo Syndrome Clínical Guidelines
Behavioral disorders were present in 84/87 patients with MPS IIIA6, 74% before age 5 years and 94% before age 10 years. Some patients with milder form showed no behavioral problems until adulthood.
Rumsey et al14 studied a group of 21 children with severe MPS IIIA, of which 13 met the criteria for autism spectrum disorder in the ADOS scale (Autism Diagnostic Observation Schedule). A positive result was strongly associated with age: 11/13 were older than 46 months and only 2 patients under the age of 46 months met the diagnostic criteria. Social and affective were the most frequent disorders, and they did not show restricted interests or repetitive behaviors, except for biting objects. Between the 3 and 4 years, a decrease in the following areas was most frequently observed: social communication behaviors, including the use of nonverbal behaviors such as eye contact, facial expressions and gestures; directed vocalizations; response to name and shared enjoyment interaction.
Sleep disordersSleep disorders and insomnia have an incidence rate of around 80-90%4,15. Sleep disorders include difficulty to sleep, early wake up, frequent awakenings during sleep, and even a complete change of the day-night rhythm8. Some patients will spend all night up, at least occasionally. It is frequent that they cry suddenly, bite the bedding clothes; and less common, laugh or sing inappropriately16. In many patients sleep problems are episodic, with normal sleep between incidents4.
2.3 NEURODEGENERATION. NEURODEVELOPMENTAL REGRESSION
In patients with a severe phenotype, after an initial phase of slowed psychomotor development from the 2-4 years onwards, in a maximum age development of approximately 3.5-4 years17, a more or less rapid deterioration phase of cognitive and motor functions begins between the 4 and 6 years4,12,17. There is great variability in the rate of regression, even among siblings, with some showing rapid loss of function and others who exhibit a much slower progression of the disease8. A third of patients with MPS IIIA showed regression before the 4 years of age and 78.9% before 644. The speech and the child’s acquired understanding skills are lost. The regression of intellectual functions, especially of speech, precedes the regression of motor functions4,5,9.
14. Rumsey RK, Rudser K, Delaney K, Potegal M, Whitley CB, Shapiro E. Acquired autistic behaviors in children with mucopolysaccharidosis type IIIA. J Pediatr 2014;164:1147-1151.15. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child 1995;73:77-81.16. Colville GA, Watters JP, Yule W, Bax M. Sleep problems in children with Sanfilippo syndrome. Dev Med Child Neurol 1996;38:538-44.17. Valstar MJ, Marchal JP, Grootenhuis M, Colland V, Wijburg FA. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J Rare Dis 2011;6:43.

Sanfilippo Syndrome Clínical Guidelines I 33
Receptive speech remains unaltered more time than the expressive speech; the cognitive function is the most affected area; however, the adaptive behavior remains unchanged over time7.
Sanfilippo patients often develop a crisis in the second or third stage of the disease3, generally easily controlled2.16/62 patients had crisis in the Cleary and Wraith series10,most after the 8 years of age, widespread or mixed, 39% in the Van de Kamp et al series9, 52% in the Meyer et al series4, 66% in the Valstar et al series6 and 45% in Delgadillo et al series11. The frequency increases with age: 73.9% of those over 15 years of age had crisis4. Crises are earlier in patients with a severe phenotype6.
Neurological problems are progressive. The walk is clumsy and coordination is poor. Pyramidalism and involuntary athetoid movements appear. There may be constant drooling and problems with chewing and swallowing. In this last phase, patients are “isolated” losing contact with the environment as a result of the progressive dementia1. Finally the patient is bedridden, in a vegetative state2.
Death can occur from before the age of 10 until the third or fourth decades of life2. Individuals with the more severe form of the disease usually do not survive beyond the age of 20 years, while those with milder forms can live considerably longer. The average age of death in the MPS IIIA was 15,2 years (range, 8,5 to 25,5 years) in the series of Meyer et al4 and 15 years (range, 11,5 to 26 years) in Delgadillo et al11. There is variability in survival between types and within each one of them (see below).
2.4 EXTRANEUROLOGICAL SYMPTOMSAlthough in many patients with MPS III somatic impairment is absent or mild, a wide variety of systemic symptoms and some cases with prominent systemic manifestations have been described in literature. 93% of children in the Buhrman et al series7 showed significant somatic disease. Barone et al18 18 present a 18 months old child with Sanfilippo B, with severe somatic involvement, including macrocephaly, coarse facies, marked hepatosplenomegaly, umbilical and inguinal hernias, dysostosis multiplex and with normal psychomotor development at that time (although, with subsequent neurodegeneration).
Facies tosca. Aspecto facialThe coarse facies is not a prominent component of Sanfilippo syndrome, especially in early stages, and some patients have a normal appearance even in adulthood1. In the Meyer et al series4 only 7% (5/71) of patients with MPS IIIA had coarse facies at the beginning, 86% showed coarse facies in evolution and 14% (10/71) did not have it at ages between the 6,8 and 32,8 years. Thus, many children develop a coarse facial
18. Barone R, Fiumara A, Villani GRD, Di Natale P, Pavone L. Extraneurologic symptoms as presenting signs of Sanfilippo disease. Pediatr Neurol 2001;25:254-7.

34 I Sanfilippo Syndrome Clínical Guidelines
appearance with age7,9,10. However, in the Delgadillo et al series11 78% had coarse facies at the beginning. The nasal bridge may be slightly flattened and the lips slightly coarse. Many children with Sanfilippo are hirsute, with bushy eyebrows and a low frontotemporal hairline2. Some have macrocephaly: 48% in the van de Kamp et al series9 and 73.2% in the Meyer et al series9 y 73,2% en la serie de Meyer et al4. In Figs. 2-9 we can see the facial appearance of some children with MPS.
Musculoskeletal disorders and heightContractures, usually mild, will primarily affect elbows8. In a Valstar et al series6 53% of patients with MPS IIIA had contractures, especially in elbows, hands and feet. Knees and hips can also be rigid, but they generally do not limit mobility until later. In advanced stages, the distal spasticity of the lower limbs may worsen the walk.
The skeletal involvement of MPS III is minimal, with only a slight dysostosis multiplex, which predominantly affects the skull, vertebrae and hip9. There is thickening of the cranial vault in the 73% of cases. There may be spine abnormalities (kyphosis or scoliosis) mild or moderate. Over half of patients have abnormalities in the dorsolumbar vertebrae (ovoid, hooked or small)9.
The musculoskeletal disorders of the MPS III are less severe than those caused by other MPS, but some individuals with Sanfilippo show symptoms, especially through spine or hip disease. In a series of 18 children with MPS III, with an average age of 10,3 years, 3 had significant scoliosis (21-99º), 2 had L1 hypoplasia, 8 hip dysplasia, 4 had bilateral osteonecrosis of the femoral heads, 3 had equinus (one of them with fixed contractures), one was operated for carpal tunnel syndrome and another for trigger thumb19. The hip may be poorly developed, with x-ray similar to the Perthes disease, causing pain and difficulties in walking. The presence of radiographic abnormalities and hip pain was examined in 33 patients with MPS III. Eight patients (24%) had signs of osteonecrosis of the femoral head (ONFH) (Ficat degree ≥ I), 6 of them bilateral. None of the 14 patients with an attenuated form had ONFH. The 7 patients with Ficat degree ≥ II had hip pain20. Therefore, the ONFH is frequent in patients with severe Sanfilippo and evaluation should be considered in the follow-up of these patients20.
19. White KK, Karol LA, White DR, Hale S. Musculoskeletal manifestations of Sanfilippo Syndrome (mucopolysaccharidosis type III). J Pediatr Orthop 2011;31:594-8.20. De Ruijter J, Maas M, Janssen A, Wijburg FA. High prevalence of femoral head necrosis in Mucopolysaccharidosis type III (Sanfilippo disease): a national, observational, cross-sectional study. Mol Genet Metab 2013;109:49-53.

Sanfilippo Syndrome Clínical Guidelines I 35
Height is almost always normal before the age of 10, but it is affected in half of the children over 12 years9. Growth is retarded in children over 6 years; but with less intensity than in other MPS. The severity of MPS III correlates with the growth pattern and final height (however, growth seems accelerated in the first 6 years in patients with severe phenotype)21.
Precocious pubertySome children with Sanfilippo and central precocious puberty have been reported22,23. The 5 patients reported were male. Four were treated with GnRH agonists, which appear to improve final height and behavioral problems23.
HepatomegalyThere may be mild hepatomegaly, especially during childhood2. The frequency is variable depending on the series, from less than 50% up to 90%4,6,7,9. Splenomegaly is less frequent and prominent.
HerniasDepending on the series, recurrent hernias with variable frequencies ranging between 8% and 62%, are reported4 ,5,7,9,10.
Gastrointestinal involvement. DiarrheaDiarrhea episodes are common; even a few patients suffer persistent diarrhea9,10. In general, it improves with age1. Constipation is common in older patients6.
A 5 year old child with psychomotor retardation and elevated transaminases for 18 months eventually diagnosed with MPS IIIA was reported24.
Alteraciones dentales 39% in the Buhrman et al series presented abnormal dentition7.
Cardiac involvement Cardiac abnormalities in Sanfilippo syndrome are rare but sometimes severe. In the Cleary and Wraith series10, 5/62 patients had echocardiographic abnormalities, one of them with severe hypertrophic cardiomyopathy which was lethal. In the Meyer et al series 38% of patients with MPS IIIA in whom cardiac evaluation was done had valvular disease
21. De Ruijter J, Broere L, Mulder MF, van der Ploeg AT, Rubio-Gozalbo ME, Wortmann SB, Visser G, Wijburg FA. Growth in patients with mucopolysaccharidosis type III (Sanfilippo disease). J Inherit Metab Dis 2014;37:447-54.22. Tylki-Szymanska A, Metera M. Precocious puberty in three boys with Sanfilippo A (mucopolysaccharidosis III A). J Pediatr Endocrinol Metab 1995;8:291-3.23. Concolino D, Muzzi G, Pisaturo L, Piccirillo A, Di Natale P, Strisciuglio P. Precocious puberty in Sanfilippo IIIA disease: diagnosis and follow-up of two new cases. Eur J Med Genet 2008;51:466-71.24. Krawiec P, Pac-Kozuchowska E, Mełges B, Mroczkowska-Juchkiewicz A, Skomra S, Pawłowska-Kamieniak A, Kominek K. From hypertransaminasemia to mucopolysaccharidosis IIIA. Ital J Pediatr 2014;40:97.

36 I Sanfilippo Syndrome Clínical Guidelines
(especially mitral valve)4. In a Valstar et al series of patients with MPS IIIB25, 10/44 they had mild valvular disease and 2 had left ventricular hypertrophy with severe aortic stenosis. There have been some cases of MPS III with severe valvular dysfunction26,27 27 and conduction disturbances28,29. An exceptionally atypical case of a woman beginning with arterial hypertension at the age of 45 developing a severe biventricular cardiomyopathy later on, and dying at the age of 56 years after a heart transplant has been reported. She did no show neurological impairment, or other somatic symptoms, nor facial dysmorphia, or alterations in the CMR related to MPS. The sulfamidase enzymatic activity was deficient using various substrates, although with a significant residual activity, and there was a clear deposit of mucopolysaccharides in the myocardial cells30.
ENT involvementRespiratory infections and otitis are common in children with Sanfilippo2,7,10. In the Cleary and Wraith series10, 26/62 patients had undergone ENT surgery before diagnosis (Cleary 93), and in Delgadillo et al11 adenoidectomy was performed to 23/55 children and tonsillectomy to 15/55 before diagnosis. In the Buhrman et al series7 tympanocentesis was performed to 91% of children, at an average age of 24 months, and adenoidectomy and/ or tonsillectomy to 72% of patients.
Hearing and visionA reliable assessment of hearing and vision is sometimes impossible in patients with MPS III due to their behavioral and cognitive problems.
There may be progressive and severe hearing loss2. Hearing may be affected as a result of recurrent otitis. Hearing loss is common in patients with moderate to severe impairment1, and 45% in the Meyer et al series had impaired hearing4. . In the Buhrman et al series7 73% of children under 2 years and 91% of children between 2 and 7 years had hearing loss. Most were light to moderate sensorineural hearing loss, and 46% wore hearing aids. Interestingly, older children with a mild phenotype had no hearing loss. The most common ocular abnormality in the MPS III is retinitis pigmentosa, with associated changes in the
25. Valstar MJ, Bruggenwirth HT, Olmer R, Wevers RA, Verheijen FW, Poorthuis BJ, Halley DJ, Wijburg FA. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis 2010;33:759-67.26. Herd JK, Subramanian S, Robinson H. Type 3 mucopolysaccharidosis: report of a case with severe mitral valve involvement. J Pediatr 1973;82:101-4.27. Muenzer J, Beekman RH, Profera LM, Bove EL. Severe mitral insufficiency in mucopolysaccharidosis type III-B (Sanfilippo syndrome). Pediatr Cardiol 1993;14:130-2.28. Herranz Jordán B, Campo Sampedro F, Pedrola Guixé D, Cruz Hernández M, Cardesa García JJ. [Mucopolysaccharidosis type III A in a girl with atrial septal defect and paroxysmal supraventricular tachycardia]. An Esp Pediatr 1990;32:85-7.29. Misumi I, Chikazawa S, Ishitsu T, Higuchi S, Shimazu T, Ikeda C, Uchino M, Shibata Y, Ebihara K, Akahoshi R. Atrioventricular block and diastolic dysfunction in a patient with Sanfilippo C. Intern Med 2010;49:2313-6.30. Van Hove JL, Wevers RA, Van Cleemput J, Moerman P, Sciot R, Matthijs G, Schollen E, de Jong JG, Carey WF, Muller V, Nicholls C, Perkins K, Hopwood JJ. Late-Onset visceral presentation with cardiomyopathy and without neurological symptoms of adult Sanfilippo A syndrome. Am J Med Genet A 2003;118A:382-7.

Sanfilippo Syndrome Clínical Guidelines I 37
electroretinogram31. In a Valstar et al series6, retinitis pigmentosa was observed in 6 of 18 patients with Sanfilippo A, in which fundoscopy was possible, all of them older than 20 years; while the 12 patients with normal ocular fundus were under 20 years. In 3 of the 6 patients with retinitis, a preliminary examination was normal, therefore, retinitis pigmentosa could be interpreted as a sign of neurodegeneration6,32. Retinitis pigmentosa was also found in 6 patients with MPS IIIB25 and in 3 patients with Sanfilippo C5,32. Rare cases of corneal opacity, optic atrophy or papilledema have been reported31.
2.5 NEUROIMAGINGAt the beginning of the regression, computerized tomography (CT) scan shows mild to moderate cortical atrophy in most patients. The progression to severe cortical atrophy occurs in late stages of disease1.
The findings on brain magnetic resonance (BMRI) more frequently described in patients with Sanfilippo disease are: increased perivascular spaces in the corpus callosum, basal ganglia and white matter, hyperintensities in white matter and brain atrophy33,34,35, similar to those found in others MPS36.
In three children followed with BMRI for three years, Barone et al33, showed that brain atrophy and changes in the white matter may precede the onset of obvious neurological symptoms and the progression of neurological impairment is accompanied by varying degrees of progressive atrophy. Another serial assessment with BMRI in 3 children with MPS IIIB showed similar findings34. Neither one nor the other study correlated the intensity of changes in the BMRI with the clinical severity33,34. In a child with Sanfilippo A, enlarged perivascular spaces of moderate size in the corpus callosum and clivus abnormalities of the cervical vertebrae were observed35.
31. Fenzl CR, Teramoto K, Moshirfar M. Ocular manifestations and management recommendations of lysosomal storage disorders I: mucopolysaccharidoses. Clin Ophthalmol 2015;9:1633-44.32. Berger-Plantinga EG, Vanneste JA, Groener JE, van Schooneveld MJ. Adult-onset dementia and retinitis pigmentosa due to mucopolysaccharidosis III-C in two sisters. J Neurol 2004;251:479-81.33. Barone R, Nigro F, Triulzi F, Musumeci S, Fiumara A, Pavone L. Clinical and neuroradiological follow-up in mucopolysaccharidosis type III (Sanfilippo syndrome). Neuropediatrics 1999;30:270-4.34. Zafeiriou DI, Savvopoulou-Augoustidou PA, Sewell A, Papadopoulou F, Badouraki M, Vargiami E, Gombakis NP, Katzos GS. Serial magnetic resonance imaging findings in mucopolysaccharidosis IIIB (Sanfilippo’s syndrome B). Brain Dev 2001;23:385-9.35. Kara S, Sherr EH, Barkovich AJ. Dilated perivascular spaces: an informative radiologic finding in Sanfilippo syndrome type A. Pediatr Neurol 2008;38:363-6.36. Calleja Gero ML, González Gutiérrez-Solana L, López Marín L, López Pino MA, Fournier Del Castillo C, Duat Rodríguez. Neuroimaging findings in patient series with mucopolysaccharidosis. Neurologia 2012;27:407-13.

38 I Sanfilippo Syndrome Clínical Guidelines
2.6 DIFFERENCES BETWEEN TYPESRecognition of clinical variability in MPS III is important for clinical diagnosis, prognosis and genetic advice and, in the near future, for adequate therapeutic evaluation. It is difficult to distinguish individual patients with any of the four types of MPS III based on clinical criteria, given the great clinical heterogeneity within each of the types, and even within the same family.
Cleary and Wraith10 do not see differences between types A and B in their series, but most authors noted that type A is more serious than type B. However, there are adults with MPS IIIA, as well as seriously ill patients with type B. C and D types appear to be clinically heterogeneous1. Type A is most common in northern Europe and in our country; type B seems more prevalent in Southeast Europe8,11.
Van de Kamp et al9 studied 76 patients with Sanfilippo in the Netherlands (36 with type A, 23 with type B and 14 with type C) concluding that type A has an earlier onset, more severe manifestations and earlier death than types B and C.
From 55 Spanish patients with Sanfilippo studied by Delgadillo et al11, 34 were type A, 11 type B and 10 type C. Crises were generally earlier in type A than in type B and C. Language loss was around the 5 years of age in types A and B and later in type C. Walk loss also occurred later in type C than types in A and B.
Valstar et al reported 92 patients with MPS IIIA (32 alive and 60 deceased). In the overall cohort analysis, most have a normal development in the first year of life, with onset of symptoms at an average age of 2.5 years (range 0.5-7 years). A wide phenotypic variability is observed among patients; so, speech is lost on average at the age of 7.5 years (range, 2-51 years), and walk is lost at an average age of 13 years (range, 5-51 years). Death occurred at an average age of 18 years (range, 6-59), mostly due to pneumonia. Considering the clinical course, three phenotypic groups can be seen: severe, intermediate and attenuated. Patients with severe phenotype, the most common, are totally dependent on the second half of the first decade, usually dying in the first two decades of life. Individuals with MPS IIIA intermediate have a slower regression and can live into adulthood. Those with attenuated form can reach higher levels of development, with some degree of speech and walking, and survive well into adulthood6.
In the Buhrman et al review7 of 46 children with Sanfilippo A, the average age of onset of symptoms was 17 months (range, 0-36 months), and the average age of diagnosis 35 months (range, 0.5- 9.8 years), with an average delay between the age of onset of symptoms of 24 months (1-82 months) and diagnosis. Six cases were diagnosed by family history.
MPS IIIB also shows the broad spectrum of disease severity. In a Dutch series of 44 patients25, only 9 patients (21%) showed the classic form of the disease with psychomotor retardation especially in the speech at an average age of 3 years, lost the ability to speak to an average age of 7.5 years (range, 5-10 years), walk loss at an average age of 12 years (range, 8-18 years) and death in the first two decades of life.

Sanfilippo Syndrome Clínical Guidelines I 39
Type C is generally somewhat less severe than type A. A Dutch series examines 29 patients with MPS IIIC5. Psychomotor development was normal in the first year. The first symptoms occurred at an average age of 3.5 years (range 1-6 years) and consisted of psychomotor retardation and behavioral problems. Two patients had an attenuated form and intrafamily variability was observed. Symptoms in MPS IIIC were similar to those found in MPS IIIA, but with a slower global evolution. Three patients over 30 years had retinitis pigmentosa and no patients under 16 had epilepsy. Verbal communication is often lost before the age of 10 years in patients with MPS IIIA4 and around the15 years in patients with MPS IIIC5. Most patients with MPS IIIA lose the ability to walk by the age of 15, while the majority of patients with MPS IIIC lose it up between the 20 and 30 years of age4,5,10. Death occurred at an average age of 34 years (range, 25-48).
Type D is very rare and heterogeneous, with onset between 15 months and 11 years, and various clinical presentations, with patients dying within the 14-17 years37 and patients surviving in the fourth decade38. Kaplan et al39 describe two brothers with MPS IIID of 11 and 3 years with coarse facies, hirsutism, limited elbows extension and overall slight development delay, mostly in the speech, but without neurological impairment until that time. Tylki et al40 described a child with Sanfilippo D beginning in the second year of life with hyperactivity and language delay, light hirsutism,inguinal hernia, mild hepatomegaly and dysostosis multiplex, without neurological regression until at least the 11 years of age. Ozand et al41 described a young girl with MPS IIID with an acquired language disorder, suggestive of receptive verbal agnosia, without dysmorphia or skeletal disorders, with cognitive regression and white matter disorders and brain atrophy in the BMRI. Valstar et al42 described 12 patients with MPS IIID. The clinical signs and symptoms are similar to others MPS III. The initial psychomotor development was normal, with onset of behavioral problems at 4 years, followed by development stagnation, language impairment and subsequent motor impairment.
37. Jones MZ, Alroy J, Rutledge JC, et al. Human mucopolysaccharidosis IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J Neuropathol Exp Neurol 1997;56:1158-67.38. Jansen AC, Cao H, Kaplan P, Silver K, Leonard G, De Meirleir L, Lissens W, Liebaers I, Veilleux M, Andermann F, Hegele RA, Andermann E. Sanfilippo syndrome type D: natural history and identification of 3 novel mutations in the GNS Gene. Arch Neurol 2007;64:1629-34.39. Kaplan P, Wolfe LS. Sanfilippo syndrome type D. J Pediatr 1987;110:267-71.40. Tylki-Szymańska A, Czartoryska B, Górska D, Piesiewicz-Grzonkowska E. Type III D mucopolysaccharidosis (Sanfilippo D): clinical course and symptoms. Acta Paediatr Jpn 1998;40:492-4.41. Ozand PT, Thompson JN, Gascon GG, Sarvepalli SB, Rahbeeni Z, Nester MJ, Brismar J. Sanfilippo type D presenting with acquired language disorder but without features of mucopolysaccharidosis. J Child Neurol 1994;9:408-11.42. Valstar MJ, Bertoli-Avella AM, Wessels MW, Ruijter GJ, de Graaf B, Olmer R, Elfferich P, Neijs S, Kariminejad R, Suheyl Ezgü F, Tokatli A, Czartoryska B, Bosschaart AN, van den Bos-Terpstra F, Puissant H, Bürger F, Omran H, Eckert D, Filocamo M, Simeonov E, Willems PJ, Wevers RA, Niermeijer MF, Halley DJ, Poorthuis BJ, van Diggelen OP. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat 2010;31:E1348-60.

40 I Sanfilippo Syndrome Clínical Guidelines
2.7 MPS III ATTENUATED FORMSIn the attenuated phenotype of MPS III the onset is usually around the age of 4 years with speech and/or psychomotor retardation. Later the mild cognitive deficit may remain stable during adolescence and even adulthood before proceeding further. Behavioral problems are also present in patients with attenuated phenotype; however, they appear later and are easier to treat. The third phase and death can occur between the fourth and seventh decades of life2525,43. Diagnosis is particularly difficult and almost always late in these forms of MPS III.
Lindor et al13 studied 2 adult siblings with MPS IIIA and attenuated signs. The sister shows an oppositional and aggressive behavior between the1 and 4 years of age, and delayed speech development, although normalizing later. At the 7-8 years of age, cognitive impairment is observed requiring special schooling. By age 24 begins with physical and verbal aggression and is admitted. Mild coarse facies, slight elbows and hips contractures and slight signs of dysostosis multiplex (thickened skull and widened ribs) are observed. Her brother who is 30 years old with intellectual disabilities, a little bit impulsive and cruder than his sister is also analyzed. In the early years he showed speech and sociability delay, and hearing loss from 5 years (hearing aids from 7 years) with special educational needs. Gabrielli et al44 described a 20- year-old woman, with onset at 6 years, low height and moderate intellectual disabilities, without intellectual regression or behavioral disorders at the time of publication. In the Meyer et al series4, 7/71 patients with MPS IIIA over the 12.5 years of age, showed speech, cognitive and motor functions partly preserved up to 23.8 years. Valstar et al6 describe 8 patients of attenuated phenotype from 6 families with MPS IIIA. In one case, the onset of behavioral disturbances at the age of 33 led to a diagnosis in a man with slight intellectual disabilities. Two siblings aged 9 and 11 years, studied after the diagnosis of MPS III in a cousin, had an IQ of 70 and 92.
Many cases of attenuated MPS IIIB, especially in Holland have been described. Valstar et al25 described 33 attenuated patients of a total of 42 (79%) evaluated, with a much slower cognitive and motor regression and survival into adulthood. The first symptoms of the disease, mild psychomotor retardation, were observed at an average age of 4 years; but patients later showed stable development stagnation and intellectual disabilities for many years. Speech loss occurred at an average age of 35 years (range, 8-68 years) and the walk loss at 42.5 years (range, 18-68 years). Almost all survived into adulthood. The coexistence of behavioral disorders, present in almost all patients, was key to make
43. Moog U, van Mierlo I, van Schrojenstein Lantman-de Valk HM, Spaapen L, Maaskant MA, Curfs LM. Is Sanfilippo type B in your mind when you see adults with mental retardation and behavioral problems? Am J Med Genet C Semin Med Genet 2007;145C:293-301.44. Gabrielli O, Coppa GV, Bruni S, Villani GR, Pontarelli G, Di Natale P. An adult Sanfilippo type A patient with homozygous mutation R206P in the sulfamidase gene. Am J Med Genet A 2005;133A:85-9.

Sanfilippo Syndrome Clínical Guidelines I 41
the diagnosis. Twelve patients from two families have been previously published9,43,45,46. The van de Kamp et al series9, 14/23 Sanfilippo B is about three families with attenuated phenotype, dementia in the most of them in the third or fourth decade of life and prolonged survival. Van Schrojenstein-de Valk et al45 describe 7 patients between the 30 and 43 years of age with MPS IIIB attenuated, with dementia and late onset of behavioral disorders. Moog et al43 published 20 patients with the attenuated form of MPS IIIB. All patients had an intellectual disability beginning in the first decade (average age 5 years), with slowly progressive deterioration. Most patients have prominent behavioral problems and were difficult to treat, and developed physical problems such as heart disease, arthritis, swallowing difficulty, visual disturbances and crisis. They suffered loss of mobility between the 36 and 68 years. Six died between the 28 and 69 years. Except for 3 patients, the diagnosis was made on the 3rd-7th decade of life. Verhoeven et al46 described a 57 years old woman with MPS IIIB attenuated, psychomotor retardation and behavioral disorders from the 6 years of age and slowly progressive dementia in the fourth decade of life. A 66 year old brother, admitted since age 54 had a similar clinical picture.
Berger-Plantinga et al32 described two asymptomatic sisters until de age of 28 and 36, with attenuated Sanfilippo C, they start with a slowly progressive mental deterioration, behavioral disorders and visual impairment through retinitis pigmentosa. Due to their mental and behavioral impairment, they required admission to a nursing home at the age of 42 and 46 years.
2.8 INTRAFAMILY VARIABILITYIntrafamily variability has been described in all types of MPS III, which underlines the need for prudence in family council and the limitations of using siblings as references to assess the treatment’s outcome.
McDowell et al47 describe two siblings with MPS IIIA, with similar physical findings, but marked differences in mental status. The least affected sister had early bladder control and a “trainable mental retardation”. At the age of 12, although she had lost some academic skills and developed behavioral disorders, her social skills and speech were practically intact. However, her brother never acquired sphincters control, lost speech at the age of 2 and showed an evolution with cognitive regression. At the age of 11 he had severe behavioral disorders and total lack of communication.
45. Van Schrojenstein-de Valk HM, van de Kamp JJ. Follow-up on seven adult patients with mild Sanfilippo B-disease. Am J Med Genet 1987;28:125-9.46. Verhoeven WM, Csepán R, Marcelis CL, Lefeber DJ, Egger JI, Tuinier S. Sanfilippo B in an elderly female psychiatric patient: a rare but relevant diagnosis in presenile dementia. Acta Psychiatr Scand 2010;122:162-5.47. McDowell GA, Cowan TM, Blitzer MG, Greene CL. Intrafamilial variability in Hurler syndrome and Sanfilippo syndrome type A: implications for evaluation of new therapies. Am J Med Genet 1993;47:1092-5.

42 I Sanfilippo Syndrome Clínical Guidelines
In a family with MPS IIIB published by Di Natale et al48, the least affected 23 year old brother works in a shop and has an IQ of 44; and his sister, 26, has no contact with the environment due to progressive dementia and shows coarse facies, severe hearing loss, uncontrollable hyperactivity, destructive behavior and aggressiveness.
In 2 families interrelated, 6 final and 2 suspected patients with attenuated Sanfilippo B, with a wide phenotypic variability, are reported. In general, speech development was quite normal, and either lost late or it was never lost; however, one patient never spoke and another lost his speech at the age of 6. Mental decline came generally late, and most went to primary school; but one patient was delayed from the first year and another was admitted at the age of 11. Few mobility difficulties were reported, except for a patient who never walked well. Two patients had typical coarse facies, while the rest did not49.
In three families with two siblings affected with Sanfilippo C, the difference in age at which they completely lost speech ranged from 6 to16 years and they lost walking between the 4 and 10 years5.
2.9 CLINICAL DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS (see Table 2).
Like all lysosomal diseases, Sanfilippo syndrome can present a wide spectrum of severity, with symptoms starting from the first year until the third or fourth decade of life. There may be a significant delay in diagnosis from the beginning of the disease, because the somatic and radiographic involvements are lightweight and there is a high incidence of false negatives in the screening with urinary GAG1. Late diagnosis is very common in patients with a slowly progressive or attenuated phenotype, being from 1.5 to 9 years depending on the subtype50. Today, with the possibility of effective treatments, early diagnosis is more important than in the past.
In general, we should think about Sanfilippo syndrome before a child with delay or speech impairment, especially if it is associated with somatic characteristic findings or behavioral disorders. It is not uncommon for MPS III to start with an isolated speech delay and normal development in other areas, leading to the misdiagnose of idiopathic language delay. In other cases there is a more comprehensive development delay which can be understood as an autism spectrum disorder or idiopathic psychomotor retardation50.
48. Di Natale P. Sanfilippo B disease. A re-examination of a sibship after 12 years. J Inherit Metab Dis 1991;14:23-8.49. Van de Kamp JJ, van Pelt JF, Liem KO, Giesberts MA, Niepoth LT, Staalman CR. Clinical variability in Sanfilippo B disease: a report on six patients in two related sibships. Clin Genet 1976;10:279-84.50. Wijburg FA, Węgrzyn G, Burton BK, Tylki-Szymańska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr 2013;102:462-70.

Sanfilippo Syndrome Clínical Guidelines I 43
We should think of Sanfilippo syndrome before a child showing delayed language or cognitive regression with an acquired autistic social behavior14.
In a second phase of the MPS III, psychomotor retardation is associated with conduct disorder and sleep disorders. Hyperactivity is very strong and easily confused with attention deficit disorder and hyperactivity or autism spectrum disorder. Characteristically children with Sanfilippo respond poorly to stimulant medication and behavioral treatments50. Other diseases with prominent language delay and behavioral disorders in childhood are creatine disorders, 4-hydroxybutyric aciduria, adenylosuccinate lyase deficiency, and some mitochondrial diseases (especially when associating hearing loss)51.
The existence of attenuated cases, forces to suspect MPS III in patients with psychomotor retardation, even in the absence of regression. Sanfilippo syndrome must be included in the differential diagnosis of patients with static psychomotor retardation, especially in combination with behavioral problems25. MPS III should also be considered in adults with a history of mental retardation who have progressive dementia43,45,46, especially if it is accompanied by behavioral disorder and/or retinitis pigmentosa32.
The presence of coarse facies features in a child with psychomotor and/or speech retardation with or without behavioral disorders, suggests the diagnosis of MPS III. The presence of signs and symptoms of the “Hurler phenotype” (coarse facies, visceromegaly, hernias, valvular heart disease, hearing loss, ENT infections, dysostosis multiplex, limited joint range) requires thinking of Sanfilippo, in other MPS, and other lysosomal diseases52. The first approach is the MPS’ GAG and enzymatic activity quantitative assessment in urine and blood samples. Any of these symptoms and also the presence of hirsutism, episodic or permanent diarrhea, sleep disorders, epilepsy or the existence of other cases in the family, in a child or adult with intellectual disabilities and/or behavioral disorders should encourage the research for MPS III.
The MPS’ typical findings in the BMRI (increased perivascular spaces, patched white matter involvement or brain atrophy) may suggest the diagnosis in a child with consistent symptoms, but the absence of alterations does not exclude the diagnosis.
51. García-Cazorla A, Wolf NI, Hoffmann GF. Neurological disease. En: Hoffmann GF, Zschocke J, Nyhan WL, eds. Inherited metabolic diseases: a clinical approach. Berlin: Springer-Verlag; 2010. p. 127-59.52. López Marín L, Cantarín Extremera V, González Gutiérrez-Solana L. Mucopolisacaridosis. En: Pediatría y enfermedades raras. Enfermedades lisosomales. Madrid: Ergón; 2015. p. 79-104.

44 I Sanfilippo Syndrome Clínical Guidelines
Figures 3-9. Facial appearance of children with Sanfilippo syndrome (MPSIII A).
PABLO 4 YEARS
MANUELA 5 YEARS
JAVIER 9 YEARS
BORJA 6 YEARS EVA 4 YEARSDANIELA 4 YEARS IÑAKI 5 YEARS

Sanfilippo Syndrome Clínical Guidelines I 45
Figures 10-12. Facial appearance of children with Sanfilippo syndrome (MPSIII B).
DANIEL 9 YEARS
ANDREA 7 YEARS IRUNE 4 YEARS

46 I Sanfilippo Syndrome Clínical Guidelines
IKER 6 YEARS
MIKEL 34 YEARS
Figures 13-14. Facial appearance of children with Sanfilippo syndrome (MPSIII C).

Sanfilippo Syndrome Clínical Guidelines I 47
Table 2. Diagnostic algorithm for MPS III (adapted from49).
FA: family history. FO: fundus oculi.
Psycho-motor and/or speechretardation with/without regression and
Behavioral disorder (hyperactivity, impulsivity,
aggression, agitation, anxiety, autistic behavior), sleep
disorder, epilepsy, progressive dementia.
Quantify GAGs in urine, GAG pattern.
Enzyme activity. Genetic study.
Refer to referral center.
Coarse facies, hirsutism, ENT infections, hearing loss,
diarrhea, hepatomegaly, hernias, valvular heart disease,
limited joint range. FA
FO: retinitis pigmentosa. Dysostosis multiplex.
BMRI: atrophy, increased EPV, WM involvement.

Sanfilippo Syndrome Clínical Guidelines I 49
03.Neuropsychology forSanfilippo syndrome. Mª de la Concepción Fournier del Castillo, Javier Melero Llorente. Unit of Clinical Neuropsychology. Niño Jesús University Children’s Hospital Madrid.

50 I Sanfilippo Syndrome Clínical Guidelines
3.1 DEMENTIA IN CHILDREN: THE SANFILIPPO DISEASE
Sanfilippo disease, or mucopolysaccharidosis type III (MPS III) is a form of dementia in children. Dementia in children can be defined as an alteration in the normal course of development of a specific function or cognitive function characterized by the patient’s declined previous intellectual level and altered cognitive domains, usually irreversible. As in other neurodegenerative diseases, a slowdown in the child ‘s development which will be initially reported as a delay in procurement is at first observed, followed by stagnation in which new learning cannot occur, and subsequently, the loss of skills previously acquired1.
In such diseases, the lack of treatments to slow the progressive deterioration of a child’s cognitive status is the fundamental reason for the absence of neuropsychological studies. It is rare that patients are referred to child clinical neuropsychology consultations, so few clinical psychologists are familiar with these disorders. In recent years, advances in genetics and knowledge of causative conditions of some of these diseases, are opening the door to possible treatments, making a neuropsychological evaluation a useful tool to functionally monitor disease progression and the effects of various treatments2.
Sanfilippo syndrome is primarily a neurodegenerative disease with only mild somatic disease3, it is characterized by severe cognitive and behavioral disturbances, but neither the cognitive nor behavioral impairments have been evolutionarily carefully studied.
3.1.1 COGNITIVE AND BEHAVIORAL DISORDERS POGRESSION IN MPS IIIThe progression of the disease initially described in three stages4, has been confirmed
1. Shapiro E, Klein KA. Dementia in Chilhood: Issues in Neuropsychological Assessment with Application to the Natural History ad Treatment of Degenerative Storage Diseases. En: Tramontana MG, Hooper SR (Eds.), Advances in Child Neu-ropsychology, vol.2. New York: Springer-Verlag, 1994, 119-171.2. Fournier MC (2002). Neuropsicología de las demencias infantiles: Adrenoleucodistrofia ligada a X. Mafre Medicina 2002; 12: 337-358. 3. López L, Cantarín V, Gónzalez L. Mucopolisacaridosis. En: Pediatría y Enfermedades raras. Enfermedades Lisosomales. Madrid: Ergon; 2015, 79-107.4. Cleary MA, Wraith JE. Management of mucopolysaccharidosis type III. Arch Dis Child 1993; 69:403-406.

Sanfilippo Syndrome Clínical Guidelines I 51
later with different natural history studies5,6,7,8. The diagnosis is carried out usually after a consultation requested by the parents due to an increase in behavioral changes, speech skills loss or developmental delays observed in their children9. Table 1 shows the signs and symptoms reported in different stages of the disease.
During the first phase happening between the age of 1-3 years, usually before diagnosis, the clinical pattern is just a developmental delay, especially in speech. Different series6,7,10 show that at 18 months there is an obvious delay in speech, which has equivalent frequencies in the 4 subtypes of MPS III7. Hearing loss occurs at the same time that speech delay is observed which would explain why the language is affected before cognition11. CWith regard to language development there are no differences between the 4 subtypes of MPS III, only 64.5% of patients acquire the ability to associate two words, and only 43% will achieve it before the 3 years of age7, most patients never develop a fully expressive language6.
During the second stage which takes place from the 3 years of age, severe behavioral and sleep disturbances are observed6,7,10 an indicator of disease progression, and later the speech, cognitive, and ultimately motor skills regression occurs5. The schooling of patients, as an indirect measure of the decline rate in patients with MPS III, shows that the proportion of patients who are kept in standard education is lower in patients with form A, followed by B, D and C9. A recent study in our country indicates that patients begin special education around the age of 68.
In the third phase beginning around the age of 10, the usual course is the loss of speech functions (speech gradually becomes unintelligible), loss of mobility and progressive dementia in the form of isolation and decreased contact with the environment6,7,10. The slow deterioration and loss of skills leads into a vegetative state9. Only a small proportion of patients with MPS III over the 12 years of age (9.9%) preserve some motor and speech
5. Meyer A, Kossow K, Gal A, Mühlhausen C, Ullrich K, Braulke T, Muschol N. Scoring Evaluation of the Natural Course of Mucopolysaccharidosis Type IIIA (Sanfilippo Syndrome Type A). Pediatrics 2007; 120(5): 1255-1261. 6. Valstar MJ, Ruijter GJG, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: A mini-review. J Inherit Metab Dis 2008; 31:240-252.7. Héron B, Mikaeloff Y, Froissart R, Caridade G, Maire I, Caillaud C, Levade T, Chabrol B, Feillet F, Ogier H, Valayannopou-los V, Michelakakis H, Zafeiriou D, Lavery L, Wraith E, Danos O, Heard JM, Tardieu M. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A 2011; 155(1): 58-68.8. Delgadillo V, O’Callaghan MM, Gort L, Coll MJ, Pineda M. Natural history of Sanfilippo syndrome in Spain. Orphanet J Rare Dis 2013; 8: 189-200. 9. Hopwood JJ (2007). Sanfilippo Syndrome: Clinical Genetic Diagnosis and Therapies. En: Barranger JA, Cabrera-Salazar MA (Eds.). Lysosmal Storage Disorders. New York: Springer; 2007, 415-432.10. Wijburg FA, Wegrzyn G, Burton BK, Tylki- Szymańska A. Muchopolysaccharidosis type III (Sanfilippo Syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr 2013, 102(5):462-470.11. Buhrman D, Thakkar K, Poe M, Escolar ML. Natural history of Sanfilippo syndrome type A. J Inherit Metab Dis 2014; 37:431-437.

52 I Sanfilippo Syndrome Clínical Guidelines
skills (unintelligible speech and aided gait) to adulthood55. As in other degenerative diseases loss of speech precedes the loss of motor functions and occurs earlier in types A and B than in C6,7,10. The loss of social interaction occurs significantly earlier in form A than in C, there is no difference between form A and B6,7,10. From the 10 years of age, the intensity of problems decreases progressively, between the 10 and 15 years of age, 60% of patients remain unresponsive most of the time and only 10% have behavioral problems12.
As in the rest of MPS, there is a considerable heterogeneity in Sanfilippo disease and not all patients will follow this pattern of deterioration, severe and attenuated forms even in the same type of MPS will be observed13. The brain atrophy, with which it occurs, sometimes precedes the dementia, but there is no correlation between the severity of the findings in imaging tests and the severity of the phenotype14. While an onset of dementia has been reported in adult patients with MPS IIIB, in patients with MPS IIIA it is already evident at the age of 6 years15. It is believed that the progression of dementia is faster in type A than in the B and C types, but the clinical phenotype of patients with the same subtype of MPS III is highly variable, which has been attributed to the high number of mutations identified in patients7. With the exception of mutations associated with attenuated forms of clinical expression of MPS IIIA5 and MPS IIIB16, correlations between genotype and phenotype have not been established.
3.1.2 BEHAVIORAL AND COGNITIVE CHANGES IN MPSIII SUBTYPES
Sanfilippo syndrome type AThe most frequently studied form is also the most severe one, with earlier beginning and a more rapid progression3. It has been suggested recently that the behavioral phenotype of this this subtype17 has similar characteristics to those described in the Klüver-Bucy Syndrome18, consistently associated with amygdala dysfunction in many species19. During childhood the Klüver-Bucy syndrome is characterized by hyperactive behavior, irritability, aggression, disinhibition and sleep disorders; social interactions and eye contact are poor,
12. Cross EM, Grant S, Jones S, Bigger BW, Wraith JE, Mahon LV, et al. An investigation of the middle and late behaviou-ral phenotypes of Mucopolysaccharidosis Type-III. J Neurodev Disord 2014; 6(1): 46-55. 13. Wraith JE. Mucopolysaccharidoses and Oligosaccharidoses. En: SaudubrayJM, van den Berghe G y Walter JH (Eds). Inborn Metabolic Diseases Diagnosis and Treatment (5ª edición). Berlín: Springer- Verlag; 2012, 579-590. 14. Barone R, Nigro F, Triulzi F, Musumeci S, Fiumara A, Pavone L. Clinical and neuroradiological follow up in mucopoly-saccharidosis type III (Sanfilippo syndrome). Neuropediatrics 1999; 30: 270-274. 15. Nyhan WL, Barshop BA, Ozand PT (Eds). Atlas of Metabolic Diseases (2ª edición). Londres: Hodder Education; 2005: 524-531.16. Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJ, Wevers RA, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol 2010; 68(6): 876-87. 17. Potegal M, Yund B, Rudser K, Ahmed A, Delaney K, Nestrasil I, et al. Mucopolysaccharidosis Type IIIA presents as a variant of Klüver-Bucy syndrome. J Clin Exp Neuropsychol 2013; 35(6): 608-616. 18. Klüver H, Bucy PC. Preliminary analysis of functions of the temporal lobes in monkeys. Arch Neurol Psychiatry 1939;42: 979–1000.19. Neylan T. Temporal lobe and behavior: Klüver and Bucy’s classic. J Neuropsychiatry 1997; 9: 606-620.

Sanfilippo Syndrome Clínical Guidelines I 53
some children show visual agnosia, distractibility caused by visual stimuli, hyperorality and absent or decreased emotional attachment to parents20. Although there are very few reports of children and most are during pubertal and prepubertal period21, it has also been described in young children. The literature contains complete or incomplete clinical pictures for herpes simplex encephalitis22, hypoxic-ischemic encephalopathies23, congenital malformations and bi-temporal epilepsy24. The characteristic behavior of the Klüver-Bucy syndrome describes the behavioral problems in the second stage of MPS IIIA17. Parents report “orality” children tend to put objects in their mouth not swallowing, though25, but the biggest concern is reckleness, requiring constant attention on their side. Also, the ability of empathy and concern for others are reported to be handicapped compared to their age group. They often exhibit oppositional behavior and disobedience, not responding to punishment, they may also show aggressive behavior. These characteristics distinguish them from other children with MPS who are equally cognitively impaired26,27.
A recent study17 analyzes the spontaneous behavior of patients with MPS IIIA, compared with patients with MPS I, in order to monitor the effect of cognitive impairment, and correlates it with a volumetric analysis of the amygdala and hippocampus through high - resolution MRI (3 Tesla) . It is suggested that the MPS IIIA is the first pediatric disease with a behavioral phenotype identified as a variant of the Kluver-Bucy syndrome associated to a marked atrophy of the amygdala in children with MPS IIIA diagnosed before the age of 6, which may be responsible for the reduction of fear and reckleness usually shown by these children.
In the MPS IIIA, the course of decline in motor skills is more variable than the loss of cognitive skills; fine motor skills deteriorate faster than gross motor skills28. Parents report a decline in gross motor skills later, which suggest direct measures; this is explained by
20. Lippé S, Gonin-Flambois C, Jambaqué I. The neuropsychology of the Klüver-Bucy syndrome in children. Handb Clin Neurol. 2013; 112: 1285-1288. 21. Jha S, Ansari MK. Partial Kluver–Bucy syndrome in a patient with acute disseminated encephalomyelitis. J Clin Neu-rosci 2010; 17:1436-1438. 22. De Tiège X, De Laet C, Mazoin N, Christophe C, Mewasingh LD, Wetzburger C, Dan B. Postinfectious immune-edia-ted encephalitis after pediatric herpes simplex encephalitis. Brain Dev 2005, 27: 304-307.23. Jha S, Kumar R, Kumar M, Kumar R. Cerebral birth anoxia, seizures and Klüver- Bucy syndrome: some observations. J Ped Neurol 2005; 3: 227-232.24. Pestana EM, Gupta A. Fluctuating Klüver–Bucy syndrome in a child with epilepsy due to bilateral anterior temporal congenital malformations. Epilepsy Behav 2007; 10: 340-343.25. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis in Child 1995; 73: 77-81.26. Colville GA, Bax MA. Early presentation in the mucopolysaccharide disorders. Child Care Health Dev 1996; 22(1): 31-36.27. Robertson SP, Klug GL, Rogers JG. Cerebrospinal fluid shunts in the management of behavioural problems in Sanfili-ppo syndrome (MPS III). Eur J Pediatr 1998; 157(8): 653-655.28. Delaney K, Nestrasil I, Yund B, King K, Whitley C, Rudser K, Haslett P, Shapiro E. Motor function decline and motor apraxia in Sanfilippo syndrome type A. Mol Gen Metab 2012; 108(2): S34.

54 I Sanfilippo Syndrome Clínical Guidelines
the presence of a motor apraxia in patients, patients are able to spontaneously perform activities involving gross motor but not by imitation or on instructions28.
Evolutionarily in children with MPS IIIA29 the disease progresses with age, towards increasing social and behavioral problems, difficulties in regulating emotions and reckleness. Hyperactivity, orality, irritability and aggression slowed in older ages, hyperactivity particularly decreases from the age of 6. The progression of lack of fear is associated with a significantly reduced volume of the amygdala and is a sensitive tracer of disease progression29. Decreased volume in the hippocampus is also associated with worsening impairments in social and emotional interaction29; this finding is consistent with studies involving the hippocampus in processessetting bonding30,31 and social emotions32.
The progressive loss of language and the characteristics poor social interactions of Sanfilippo syndrome type A also resemble those occurring on autism spectrum disorders. In fact, around the age of 4, their social and emotional dysfunctions are severe enough to meet criteria for diagnosis; however restricted interests or repetitive behaviors are largely absent, autism spectrum traits are more prominent as the disease progresses33. Recent studies34 show reduced volume in the left amygdala when children with MPS IIIA with autism spectrum features (average age 6 years) are compared with younger patients (average age 3 years) not showing autistic symptoms. Other studies longitudinally evaluating autistic behaviors in types A and B, have also shown that autism spectrum traits manifest at a different chronological age in children with autism35. In MPS IIIA diagnosed early (before the age of 5), behavioral problems and autistic-like symptoms are reported less frequently than in patients diagnosed later7.
29. Shapiro EG, Nestrasil I, Ahmed A, Wey A, Rudser KR, Delaney KA, et al. Quantifying behaviors of children with Sanfilippo syndrome: the Sanfilippo Behavior Rating Scale. Mol Genet Metab 2015; 114(4): 594-598.30. Buchheim, S. Erk, C. George, H. Kachele, T. Kircheret, P. Martius, Pokorny D, Ruchsow M, Spitzer M, Walter H. Neural correlates of attachment trauma in borderline personality disorder: a functional magnetic resonance imaging study, Psychiatry Res. Neuroimaging 2008; 163(3): 223-235.31. Quirin M, Gillath O, Pruessner JC, Eggert LD. Adult attachment insecurity and hippocampal cell density. Soc. Cogn. Affect. Neurosci 2010; 5(1): 39-47.32. Britton JC, Phan KL, Taylor SF, Welsh RC, Berridge KC, Liberzon I. Neural correlates of social and nonsocial emotions: an fMRI study. NeuroImage 2006; 31(1): 397-409.33. Rumsey RK, Rudser K, Delaney K, Potegal M, Whitley CB, Shapiro E. Acquired autistic behaviors in children with mucopolysaccharidosis type IIIA. J Pediatr 2014; 164(5):1147-1151.34. Wakumoto A, Ahmed A, Rumsey R, Kovac V, Rudser K, Whitley C, Potegal M, Shapiro E, Nestrasil I. Amygdalar volumes and acquired autistic symptoms in MPS IIIA. Mol Genet Metab 2015; 114: S11-S130.35. Buhrman DZ, Escolar ML, Poe M, Dalsimer D. Longitudinal assessment of autistic behaviors in children with Sanfilippo syndromes: Types A and B. Mol Genet Metab 2015; 114: S25-S26.

Sanfilippo Syndrome Clínical Guidelines I 55
Sanfilippo syndrome type BKnowledge on the progression of MPS IIIB is scarce, some series36 report that the classic severe phenotype, similar course to type A, is only observed in a small proportion of patients (21%), the rest (79% ) had a much milder form, with a slow regression of both intellectual and motor skills, most surviving to adulthood. The first symptoms of the disease are mild developmental delays most frequently reported around the 4 years of age. Then, a slowdown and subsequent stagnation in development is later reported. Patients with an attenuated phenotype had stable intellectual disabilities for many years. Certain missense mutations were found only in patients with attenuated phenotype36. Some authors have suggested that the MPS IIIB should be considered in patients with developmental delay, even in the absence of a progressive decline in intellectual abilities; the coexistence of behavioral problems is a key feature to determine the need for metabolic assessments36. Cases of such a slow progressive deterioration have been reported that MPS IIIB diagnosis has been made in the sixth decade of life37. The loss of volume of substance with the thinning of the cortical layer is the MRI pattern to be associated to cognitive deterioration in MPS IIIB38.
Sanfilippo syndrome types C and DThe MPS IIIC and IIID are often attenuated forms. Very few studies analyze the progression of deterioration in MPS IIIC. A recent study39 shows slower motor and verbal functions deterioration rate than in previous studies40, although it varies even among affected siblings, i.e. the course is highly variable. Psychomotor development is reported as normal in all patients during the first year of life. Intellectual regression accompanied by speech loss precedes motor impairment in over 10 years39. There have been reports of dementia in adults due to MPS IIIC with intelligence kept into adulthood and slowly progressive deterioration41. Despite the diagnosis of patients with MPS IIIC at a higher age, autism spectrum disorders are less common than in types A and B7.
36. Valstar MJ, Bruggenwirth HT, Olmer R, Wevers RA, Verheijen FW, Poorthuis BJ, Halley DJ, Wijburg FA. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J Inherit Metab Dis 2010; 33:759-767.37. Moog U, van Mierlo I, van Schrojenstein Lantman-de Valk HM, Spaapen L, Maaskant MA, Curfs LM. Is Sanfilippo Type B in Your Mind When You See Adults With Mental Retardation and Behavioral Problems? Am J Med Genet C Semin Med Genet 2007; 145(3): 293-301.38. Nestrasil I, Ahmed A, Kovac V, Rudser K, Delaney K, Barbier A, Whitley CB, Shapiro E. Brain volumes and cognitive function in MPS IIIB (Sanfilippo syndrome type B): Cross-sectional study. Mol Gen Metab 2015; 114: S87. 39. Ruijter GTG, Valstar MJ, van de Kamp JM, van der Helm RM, Durand S, van Diggelen OP, Wevers RA, Poorthuis BJ, Pshezhetsky AV, Wijburg FA. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol Genet Metab 2008; 93: 104-111.40. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: an epidemiological study. J Ment Defic Res 1983; 27: 185-203.41. Berger-Plantinga EG, Vanneste JAL, Groener JEM, van Schooneveld MJ. Adult-onset dementia and retinitis pigmentosa due to mucopolysaccharidosis III-C in two sisters. J Neurol 2004; 251: 479-481.

56 I Sanfilippo Syndrome Clínical Guidelines
There are no conclusive studies on the MPSIII-D because only isolated cases that do not allow setting their natural history are recorded in literature, although their survival rate is higher42,43.
3.2 NEUROPSYCHOLOGICAL ASSESSMENT IN MPS IIINeuropsychological evaluation should be performed by a clinical neuropsychologist with experience in pediatric evaluation of degenerative diseases but also with enough clinical experience in handling children with severe behavioral disorders.Clinical Neuropsychology consultations may help guide families regarding the care needed by children in every moment of the disease to avoid inadequate liability on their behavior; they should help in the clinical management of patients, provide educational recommendations and identify the rehabilitation treatment required for specific deficits.
It is important to establish baselines at the time of diagnosis and continue to monitor cognitive and behavioral functions as long as the disease progresses or changes depending on the treatments received. Almost all children will require special education services. The neuropsychologist must provide recommendations based on the needs of each child, but also based on the knowledge of the disease’s natural history, this means that assessments should be carried out in centers having enough experience as it is a rare disease.
3.2.1 ASSESSMENT METHODSA detailed description reviewing different valuation methods in children with Sanfilippo syndrome can be seen in Delaney et al.44 Given the specific features detailed above, neuropsychological assessment in children with Sanfilippo syndrome should include different approaches to account for possible heterogeneity that we will find when evaluating the different phenotypes/genotypes with consequent cognitive and behavioral variability. At the time of diagnosis, it will be necessary to determine the degree of evolutionarily and cognitive development, as well as behavioral changes and their impact on adaptive functioning. Revisions are made once a year from diagnosis, or sooner if closer monitoring requires to be determined.
3.2.2 COGNITIVE ASSESSMENTOne of the most important aspects in the evaluation of patients with MPS III is that the methods employed must be flexible at all times, so that they can adapt to the needs and
42. Tylki-Szymanska A, Czartoryska B, Górska D, Piesiewicz-Grzonkowska E. Type III D MPS (Sanfilippo D): clinical course and symptoms. Acta Paediatr Jpn 1998; 40:492-494.43. Jansen AC, Cao H, Kaplan P, Silver K, Leonard G, De Meirleir L, Lissens W, Liebaers I, Veilleux M, Andermann F, Hegele RA, Andermann E. Sanfilippo syndrome type D: Natural history and identification of 3 novel mutations in the GNS gene. Arch Neurol 2007; 64:1629-1634.44. Delaney KA, Rudser KR, Yund BD, Whitley CB, Haslett PA, Shapiro EG. Methods of neurodevelopmental assessment in children with neurodegenerative disease: Sanfilippo syndrome. J Inherit Metab Dis 2014; 13: 129-137.

Sanfilippo Syndrome Clínical Guidelines I 57
possibilities of the patient, their evolutionary moment and the behavioral changes that may arise. On the one hand cognitive tests used should, as far as possible, be appropriate to the child’s age; it is desirable that the techniques used are standard for patient‘s age. On the other hand, they should be appropriate to their level of development at the moment of the evaluation, test selection with low floors is ideal to start evaluation with very easy items for the child and thus facilitate their collaboration, it is advisable to avoid demanding tests requiring multiple assessment sessions with the consequent fatigue for the child and family. In patients with progressive deterioration the lack of cooperation may be secondary to the inability to perform the task, it cannot always be interpreted as secondary to the child’s behavioral problems and lack of collaboration, for example, the apraxia that can observed in patients with MPS III can determine that the patient will not be able to perform tasks before instructions or by imitation, tasks which parents or teachers report they conduct in familiar surroundings. In patients with great difficulty to control behavior or with advanced deterioration, it can be virtually impossible to use typical standardized tasks of cognitive performance, being required in that case an approach based on scales of development and/or adaptive behavior, with indirect measures using parents and teachers as informants. Figure 2 shows the algorithm for test selection based on the patient’s particularities; we should not forget the importance of knowing the technical characteristics of assessment tools.
InstrumentsThe Scales of development will always be of choice for children under 30 months and in those with cognitive development equivalent to less than 3 years; they will also be useful for patients with a suspected higher development, but that because of their behavioral difficulties cannot collaborate on tasks of greater complexity:
a) Bayley Scales of Infant Development, third edition (BSID-III)45. Provide scaling for children up to the 42 months of age, for cognitive, motor and language scales (as well as reports for adaptive behavior and social-emotional development), and equivalent ages for each domain allowing the calculation of development quotients in older children . Its main advantage is that it is possibly the scale of development of wider dissemination, being a clear benchmark in clinical trials and studies of natural history. As a disadvantage, it should be noted that due to its extension, it is often a complex tool to use during routine clinical practice.
b) Merrill-Palmer Scales of development, revised version46. Scaled for children up to the 78 months of age. As the above, they allow the assessment of cognitive, motor and speech development (as well as adaptive behavior and socio-emotional development) and have equivalent ages in each scale. The main advantages of these scales of development are the use of some highly attractive valuation materials and its ease of application, since
45. Bayley N. Bayley-III, Escalas Bayley de desarrollo infantil-III. Madrid:Pearson 2015.46. Roid GH, Sampers JL. Merrill-Palmer-R, Escalas de desarrollo. Madrid:Tea; 2011.

58 I Sanfilippo Syndrome Clínical Guidelines
the construction of the scale allows relatively rapid assessments and do not involve fatigue for the child. It can be applied with a cognitive developmental age of less than 6 and a half years.
c) Battelle Developmental Inventory47. This test consists of the same domains as the ones mentioned above, but has a screening subtest that allows establishing equivalent developmental ages up to 95 months with reasonable reliability. Given the great speed with which this screening can performed, with parents as informants, it is an interesting addition to the above tools, so it can direct the overall level of development of the child and then proceed with a more targeted assessment. It allows setting an age of cognitive development that can be applied to patients with serious deterioration and/or unable to collaborate in direct tests.
The tests for general skills and intellectual level may be used in selected patients with an attenuated phenotype. Although these tests are usually scaled for children from 30 months of age in the general population, it is recommended to use them only with patients who have reached a development equivalent to at least 36 months of age, so that we avoid subjecting the child to an excessively demanding situation resulting in poor collaboration:
a) Wechsler Intelligence Scales (fourth and fifth revisions). It has a version for pre and primary school (WPPSI-IV)48 which covers ages between 36 months and 7 years and 7 months, a version (WISC-V)49 for children aged 6-16 years and another one for over 16 years and adults. (WAIS-IV)50. They can be a valid alternative for patients with mild/moderate intellectual disability and ability to collaborate, not recommended in cases of cognitive impairment or behavioral difficulties.
b) British Ability Scales (second revision, BAS-II)51. As above, it has a version for children from 36 months to 7 years and 11 months (with differentiated forms for children older and younger than 42 months), and a school version for children 6-18 years.Unlike the Wechsler scales they do not require assessment of working memory or processing speed. In principle this lower level of demand could enable the assessment of children with major difficulties; unfortunately the unattractiveness of test materials will hamper implementation. The American version of the test (Differential Ability Scales,
47. Newborg J, Stock JR, Wnek L. Inventario de desarrollo Battelle. Madrid:Tea; 1996.48. Wechsler, D. WPPSI-IV, Escala de Inteligencia de Wechsler para preescolar y primaria. Madrid: Pearson; 2015.49. Wechsler, D. WISC-V, Escala de inteligencia de Wechsler para niños-V. Madrid: Pearson; En prensa.50. Wechsler, D. WAIS-IV, Escala de Inteligencia de Wechsler para adultos. Madrid: Pearson; 2012.51. Elliot CD, Smith P, McCullogh K. BAS-II, Escalas de aptitudes intelectuales. Madrid:Tea; 2011.

Sanfilippo Syndrome Clínical Guidelines I 59
second revision, DAS-II)52, also based on Elliott’s original scale53 with the same factorial structure and the same subtests, features more attractive materials which facilitates collaboration. However, the children’s version, up to 8 years and 11 months, is the only one scaled for the Spanish-speaking population.
c) Kaufman Assessment Battery for Children (second revision, K-ABC 2)54. In its latest review, Kaufman scales are of choice in children with difficulties thanks to its simple elements and attractive materials. Its limitation is that a scaling in Spanish is not yet available, but it allows calculating a non-verbal index which is a good measure of overall capacity. In children with cognitive difficulties but capable of working, the K-ABC 2, and in particular its non-verbal index, is an advisable choice.
3.2.3 BEHAVIORAL ASSESSMENTFor assessing the behavior of children with MPS III it will be essential both direct observation of behavior by the clinician and information provided by the family, especially with regard to the evolution of the difficulties and the adaptive possibilities of children in their usual environment. When observing the behavior of a child with Sanfilippo in the consultation, it should be noted that the behavior may not be equivalent to that reported by parents at home; therefore the indispensable complementary information provided by parents is of huge value to determine the degree of behavioral difficulties. Thus, any valuation made to a child with Sanfilippo, assessing the degree of progress or deterioration in adaptive behavior (activities of daily living, socialization, play, etc.) and disruptive behaviors should be a prime axis, as it will often be one of the main determinants of the quality of life for the child and the family. The instruments that may be useful in the assessment are:
a) Vineland adaptive behavior scales, second edition55. For the assessment of adaptive behavior, Vineland scales are undeniably useful since they are standardized from birth to age 90, which means that the same instrument allows monitoring the patient throughout life. Through an interview to parents, it provides indexes for the domains of communication, daily living skills, socialization, motor skills and maladaptive behavior, as well as subscripts in each domain and developmental age equivalent to the subscripts. They correlate well with the level of valued cognitive development, with scales of development, and tests of overall capacity, so they are also a good instrument to select the most appropriate assessment strategies for each individual patient.
52. Elliot CD. Differential ability scales, second edition. San Antonio, TX: Pearson; 2007.53. Elliot CD. Differential ability scales. San Antonio, TX: The Psychological Corporation; 1990.54. Kaufman AS, Kaufman NL. Kaufman assessment battery for children, second edition. Circle Pines, MN: AGS Publishing; 2004.55. Sparrow SS, Cicchetti DV, Balla DA. Vinelan Adaptive Behavior Scales-II (2ªed). USA: Pearson; 2008.

60 I Sanfilippo Syndrome Clínical Guidelines
b) Achenbach Child Behavior Checklist (version for children under 6 years)56. The CBCL scale version for children from 18 months to 5 years is a very easy to use self-administered questionnaire for parents. It provides information on the amount of maladjusted behaviors in relation to age in internalizing and externalizing domains. Its usefulness is limited to the disease’s early stages to detect the moments of progression of behavioral difficulties.
c) Brief Rating Inventory for Executive Functions - Preschool Form (version for children under 6 years)57. Complementary to the above, this other parental self-applied questionnaire allows detecting emerging difficulties in executive functions in children from 2 to 5 years of age. It has scales for assessing behavioral and metacognitive executive functions, and although the latter may be less useful, the behavioral regulation rates may be sensitive to early behavioral problems. It is not useful in intermediate and advanced stages of the disease and, given the content of its elements, the form for over 6 years is not recommended for all children with cognitive impairment or developmental levels who are not above this age.
3.2.4 SPECIFIC INSTRUMENTS FOR MPS IIISanfilippo Behavior Rating Scale (SBRS)29 is a newly developed scale that allows the evaluation of specific behavioral difficulties more frequently appearing in children with MPS III. It is a self-administered questionnaire for parents comprising three sections: Communication, Emotional regulation and Behavior. Communication section allows the assessment of current and past communication skills, in case the child’s communication has been better in the past. Emotional regulation section quantifies the frequency, duration and predominant emotion in the child’s uncontrolled emotions episodes. Finally, Behavior section is built specifically to cover all the possible behavioral problems that may occur in children with MPS III (especially in type A); through 68 items it asks for about 15 Kluver-Bucy type behavioral domains, autistic-like behavior, and attention difficulties and oppositional/aggressive behaviors. Given the expected variability, greater internal consistency domains are those more visible and frequent.
The SBRS has a baseline form for a more detailed initial evaluation and a follow-up form to document changes in behavior throughout the follow-up. It allows some standardization of results through averages and standard deviations of three age groups and a group of slow progression. Used in a complementary manner to other assessments, it is a good characterization of behavior in children with MPS III, especially in the more severe forms. However, it should be noted that it is a self-administered questionnaire for parents with complex questions, also retrospective, so parents may need help from the clinician to answer some of the elements.
56. Achenbach TM. Child Behavior Checklist. Burlington, VT: ASE-BA, University of Vermont; 2007.57. Gioia GA, Espy KA y Isquith PK. BRIEF-P. USA: PAR Inc.; 2003.

Sanfilippo Syndrome Clínical Guidelines I 61
3.3 CONCLUSIONESThe progression of Sanfilippo syndrome involves a dysfunction of the central nervous system, neurocognitive decline and marked behavioral disturbances. Although thebehavioral abnormalities even today have not been fully typified, their pattern may be the key to understanding the neurobehavioral pathology of this disease29.
To date, there are no longitudinal studies with different types of MPS III and phenotypic severity (severe and attenuated forms), however, neuropsychological follow-up studies in neurodegenerative diseases such as Sanfilippo can provide prognostic information and help clarify the risk-benefit of future treatment options. The prospect of new therapeutic trials in this rare disease emphasizes the need for reliable and quantitative markers to evaluate potential benefits. Several studies17,58,59 show that despite the deterioration of patients and their behavioral problems, most cases are open to cognitive/behavioral assessment if performed by professionals experienced in degenerative diseases in childhood.
It is important to have quantitative and specific neuropsychological measures to clarify the progression of Sanfilippo syndrome in its different stages, knowing the pattern of behavioral change can help guide and support the families of these children. Such knowledge would also provide useful markers for assessing disease future therapeutic interventions.
58. Valstar MJ, Marchal JP, Grootenhuis M, Colland V, Wijburg FA. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J Rare Dis 2011; 6: 43-49. 59. Martin HR, Poe MD, Reinhartsen D, Pretzel RE, Roush J, Rosenberg A, Dusing SC, Escolar ML. Methods for assessing neurodevelopment in lysosomal storage diseases and related disorders: a multidisciplinary perspective. Acta Paediatr 2008; 97(457): 69-75.

62 I Sanfilippo Syndrome Clínical Guidelines
SIG
NS
/ S
YM
PTO
MS
PHASE 1 Developmental delay Speech delay
PHASE 2
Progressive cognitive difficulties Speech regressionRegression of motor skillsBehavioral difficulties
Hyperactivity/impulsivity Decreased attention capacity AggressivenessOralityLack of emotional control Reckleness Autistic-like behavior
PHASE 3
Profound intellectual disability Loss of speech and communicationLoss of motor skillsCessation of behavioral difficultiesProgressive isolationProgressive loss of responses to the environment
Table 3. Signs and symptoms associated with the three phases of MPS III. Not all symptoms and signs may occur in the same patient.

Sanfilippo Syndrome Clínical Guidelines I 63
Figure 15. Algorithm for test selection in the neuropsychological assessment of patients with MPS III.
Interview with parents (all patients)• VABS-II• Bayelle (Screening)
Family questionnairesDirect assessment with the child
Initial phases or non- serious
behavioral problems
Intermediate/ advanced
phases with serious behavioral problems
• CBCL• BRIEF• SBRS • SBRS
If Developmental Age is
< 36 months
If Developmental Age is
> 36 months
Scales of development MP-R
(in research contexts, preferable BSID-III)
Significant cognitive difficulties
Slight or absent cognitive difficulties
Mandatory direct speech assessment?
Yes No
BAS-II K-ABC 2
Wechsler scales according to
chronological age

Sanfilippo Syndrome Clínical Guidelines I 65
04.Monitoringmucopolysaccharidosis type III or Sanfilippo syndrome. Laura López Marín. Deputy of Neuropaediatrics. Niño Jesús University Children’s Hospital Madrid.

66 I Sanfilippo Syndrome Clínical Guidelines
The progressive nature of mucopolysaccharidosis (MPS) requires a continuous assessment of the clinical situation, mainly including vision, hearing, joint mobility, cardiopulmonary and neurological function, and intellectual level. Therefore, monitoring of these children involves a multidisciplinary team to assess the progression of symptoms and the most appropriate treatment in each case1,2,3. However, contrary to what happens in other MPS, Sanfilippo syndrome is characterized by the severe degeneration of the central nervous system generally with mild somatic disease, so, in most cases, frequent revisions as in other types of MPS will not be necessary. The follow-up protocol will be established for each patient individually depending on the organs affected and the severity4,5 (Table 4).
1. MEDICAL HISTORY. REGISTERING PROBLEMS SINCE THE LAST REVIEW (EVERY 6 MONTHS):
If the patient had an infection, has been subjected to any surgery, has been hospitalized and why and whether any drug has been administered, it must be recorded on the patient’s medical history.
2. PHYSICAL EXAMINATION (EVERY 6 MONTHS):
Will reflect the vital signs (blood pressure, heart rate, respiratory rate and oxygen saturation) and some anthropometric data (weight, height, head circumference). Perform a detailed general pediatric examination, including measurement of visceromegalies (in mid-axillary line) and assessment of joint mobility angles.
3. FUNCTIONAL AND NEUROLOGICAL ASSESSMENTS (EVERY 6 MONTHS):
The neurological evaluation should include an account of the milestones of psychomotor development and activities of daily living as well as possible changes in gait, strength, fine motor skills, potty training, behavior, hearing and vision, paying particular attention to sleep and behavior disorders in order to conduct the most appropriate therapeutic management. Some scales may be useful for evaluating these aspects (Tables 2 and 3).
1. Aguirre Rodríguez FJ, Aldámiz-Echevarría Azuara L, Dalmau Serra J, González Gutiérrez-Solana L, González-Meneses López A, Pérez López J, Ruiz Gómez MA, Torralba Cabeza MA, Vitoria Miñana I, Sanjurjo Crespo P. Guía clínica de la mucopolisacaridosis tipo I (MPS I). Acta Pediatr Esp 2014; 72(Supl.): S1-S20. 2. Guía clínica de la mucopolisacaridosis tipo I (MPS I). Acta Pediatr Esp 2014; 72: S1-S20. 3. Guillén-Navarro E, Blasco AJ, Gutiérrez-Solana LG, Couce ML, Cancho-Candela R, Lázaro P, López-Marín L; grupo de trabajo Hunter España. Clinical practice guideline for the management of Hunter syndrome. Med Clin (Barc) 2013;141:453.e1-13.4. González Gutiérrez-Solana L, López Marín L. Guía de la mucopolisacaridosis tipo VI o síndrome de Maroteaux-Lamy. En: Guía para el manejo de las MPS. Madrid: Ergón; 2015.5. Pérez J, Ceberio L. Mucopolisacaridosis tipo III o enfermedad de Sanfilippo. En: Guía para el manejo de las MPS. Madrid: Ergón; 2015.

Sanfilippo Syndrome Clínical Guidelines I 67
Seizures are frequent as the disease progresses; we must record their type and frequency to decide the most appropriate antiepileptic pattern6,7,8.
The neurological examination will be complete and adjusted to the patient’s age, including strength, pyramidal signs and examination of superficial and deep sensitivity.
Although spinal cord compression is rare in MPSIII, signs of upper motor neuron involvement, loss of proprioception, decreased resistance to walk or abnormal gait, bowel or bladder dysfunction, DTR abnormalities and clonus should be evaluated periodically by history and physical examination.
SUPPLEMENTARY TESTS:
1. Cranio-cervical MRI (from baseline and then, depending on the patient’s clinical condition), which should be interpreted by expert radiologists to assess white matter, Virchow-Robin spaces, ventricular size and signs of atrophy9.
2. Sensory and motor speed conduction of the median and ulnar nerves: (from baseline and then, depending on the patient’s clinical condition), which should be interpreted by expert radiologists to assess white matter, Virchow-Robin spaces, ventricular size and signs of atrophy.
3. EEG should be performed every 6-12 months, if patients have seizures, and only occasionally if they do not have them clinically.
1. ENT evaluation (every 6-12 months): Hearing loss is common in patients suffering from MPSIII, but it is often very difficult to assess due to their behavioral disorders and cognitive impairment. For this reason, ENT periodic monitoring with audiometry or brainstem auditory evoked potentials is very important to indicate, when necessary, the placement of transtympanic drainage and/or hearing aids.
Overnight polysomnography should be performed if there is suspicion of obstructive sleep apnea (OSA) to assess the need for adeno-tonsillectomy7,9,10.
6. Cleary MA, Wraith JE. Management of mucopolysaccharidosis type III. Arch Dis Child 1993; 69: 403-6. 7. Wijburg FA, Węgrzyn G, Burton BK, Tylki-Szymańska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr 2013; 102: 462-70. 8. Delgadillo V, O’Callaghan M del M, Gort L, Coll MJ, Pineda M. Natural history of Sanfilippo syndrome in Spain. Orphanet Journal of Rare Diseases 2013; 8:189.9. Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 2008; 31: 240-52. 10. Andrade F, Aldámiz-Echevarría L, Llarena M, Couce ML. Sanfilippo syndrome: Overall review. Pediatr Int 2015; 57:331-8.

68 I Sanfilippo Syndrome Clínical Guidelines
2. Cardiological evaluation (every 24 months):Unlike other MPS, valvular pathology is uncommon11, therefore, cardiological revisions may initially be periodical on those not showing abnormalities4,8. It should include an ECG and an echocardiogram. Additional cardiological scans should be performed before major surgical procedures.
3. Orthopedic evaluation (every 12 months):Although bone problems are less frequent than in other types of MPS, some patients with MPS III develop kyphoscoliosis, clubfoot deformities or equinovarus, carpal tunnel syndrome and trigger finger12. Hip osteonecrosis is a frequent complication (especially in children with a more severe phenotype) and may result in the placement of prosthesis if the diagnosis is delayed13. Periodic clinical examination is useful in monitoring the progression of bone deformities. Hip, spine and long bones x-rays will be performed at baseline and then the frequency will be decided on an individual basis depending on how severe the disturbance will be. The surgery indication should be established not only in terms of orthopedic findings, but also taking into account the patient’s clinical condition and the degenerative nature of the disease12.
Many children must receive physiotherapy. It is possible that throughout evolution, they will need orthotics, adapted chairs and the administration of botulinum toxin for spasticity.
4. Ophthalmological evaluation (every 12-24 months):These patients often develop corneal opacity but may have pigment retinopathy or optic nerve atrophy9,14.
Ophthalmological examination should include visual acuity, refraction, examination of the cornea with a slit lamp, fundus oculi and measurement of intraocular pressure, as well as electroretinography if there is data suggesting retinopathy.
5. Neuropsychological evaluation (every 12 months):The periodical neuropsychological examination (motor area, cognitive, behavioral) is essential to define the evolution of these patients and conduct the appropriate speech therapy, psycho-educational and schooling supports10. It should be performed by neuropsychologists experts in these diseases. The frequency of these evaluations will be established individually according to each patient and their current situation, as a general rule they will be conducted once a year.
11. Buhrman AD, Thakkar K, Poe M, Escolar ML. Natural history of Sanfilippo syndrome type. J Inherit Metab Dis 2014; 37: 431-7.12. Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, Ponder KP, Roberts WC, Rosenfeld HM, Giugliani R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis 2011; 34:1183-97.13. White KK, Karol LA, White DR, Hale S. Musculoskeletal manifestations of Sanfilippo Syndrome (mucopolysaccharido-sis type III). J Pediatr Orthop 2011; 31: 594-8.14. De Ruijter J, Maas M, Janssen A, Wijburg FA. High prevalence of femoral head necrosis in Mucopolysaccharidosis type III (Sanfilippo disease): a national, observational, cross-sectional study. Mol Genet Metab 2013; 109: 49-53.

Sanfilippo Syndrome Clínical Guidelines I 69
6. Psychiatric evaluation:The management of behavioral disorders is not easy and sometimes these children will require assessment and monitoring by a pediatric psychiatrist8.
7. Anesthetic evaluation: Anesthetic procedures pose a high risk in patients with MPS, due to the alteration in airway anatomy, the accumulation of GAGs and progressive lung disease (restrictive and obstructive). Postoperative recovery may be slow and complications such as airway obstruction can occur. For all this, patients with MPS should be anesthetized in well-endowed centers with expert anesthetists. In addition, cardiological, respiratory and airway assessment should be performed before any procedure requiring sedation or anesthesia.
8. Nutritional evaluation (if required):In the late stages of the disease an alteration in the swallowing is common (weight loss, risk of aspiration), therefore, nutritional supplements or placing a gastrostomy tube may be required7.
9. Dental examination (every 2 years):Although less frequent than in other types of MPS, these patients have a higher incidence of abnormal gums, tooth decay or dental abscesses than the general population, therefore, periodic evaluations are recommended and subject to unknown cause irritability10,15.
10. Other additional tests:• Blood tests with blood count, liver and kidney function, ions, nutritional markers (ferritin,
vitamin B12, vitamin D) every 12 months.• Abdominal ultrasound every 24 months.
15. Wilkin J, Kerr NC, Byrd KW, et al. Characterization of a Case of Pigmentary Retinopathy in Sanfilippo Syndrome Type IIIA Associated with Compound Heterozygous Mutations in the SGSH Gene. Ophthalmic Genet 2015; 2: 1-11. [Epub ahead of print].

70 I Sanfilippo Syndrome Clínical Guidelines
BASAL EVERY 6 MONTHS
EVERY YEAR
EACH2 YEARS
ACCORDING TO CLINICAL
Medical history X X
Physical examination X X
Weight, height, HC X X
BP, HR, RR, SO2 X X
Neurological examination X X
Neuropsychological assessment X X
ENT-audiometry evaluation X X
Cardiology-ECG and ultrasound scan
X X
Ophthalmology X X
Orthopedic evaluation:
X-rays (hips, skeletal system)
X X
X X
Electrophysiology -EEG
-NCV
X
X
Polysomnography X
MRI craniocervical X X
Abdominal ultrasound X X
Blood test X X
Nutritional assessment X
Dental exam X
Table 4. Multidisciplinary monitoring.
ECG: electrocardiogram / HR: heart rate / RR: Respiratory rate / HC: head circumference / MRI: magnetic resonance / SO2: oxygen saturation / BP: blood pressure / NCV: nerve conduction velocity.

Sanfilippo Syndrome Clínical Guidelines I 71
Table 5. Functional scale (adapted from16). It is a valid and reliable instrument that can measure and document the course of the disease; it is easy to use and provides important results regarding the clinical phenotype and disease progression.
(1) Some patients have motor and speech delay from the earliest stages. In these cases, start with a score of 2.
16. Mellara TS, Azevedo DT, Faria G, et al. Dental Findings and Management in a Mucopolysaccharidosis Type IIIB Patient. J Dent Child (Chic) 2012; 79: 176-80.
FUNCTION SCORE
Motor (1) Normal gait 3
Clumsy gait 2
Aided gait 1
Fails to walk 0
Speech (1) Normal 3
Mild Involvement 2
Difficult to understand or unintelligible 1
Absent 0
Cognitive Normal 3
Cognitive impairment 2
Loss of interest towards the environment 1
Unresponsive to stimuli 0
TOTAL SCORE
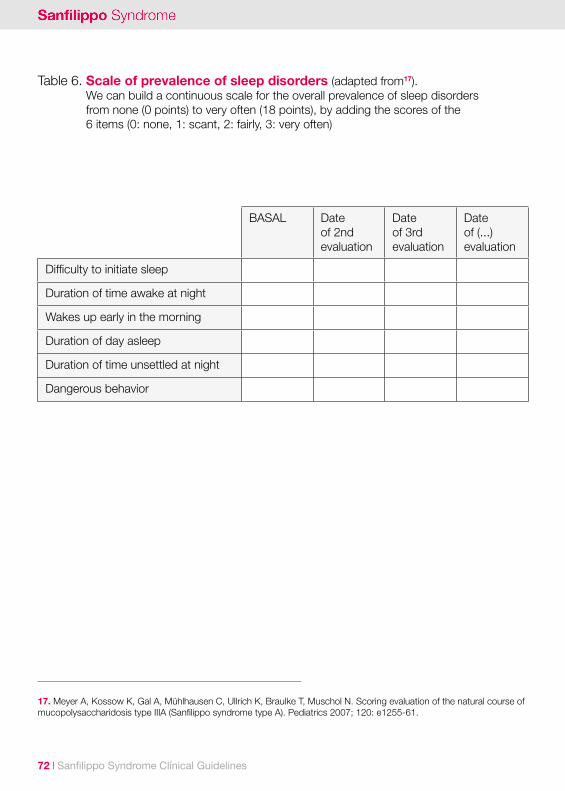
72 I Sanfilippo Syndrome Clínical Guidelines
Table 6. Scale of prevalence of sleep disorders (adapted from17). We can build a continuous scale for the overall prevalence of sleep disorders from none (0 points) to very often (18 points), by adding the scores of the 6 items (0: none, 1: scant, 2: fairly, 3: very often)
17. Meyer A, Kossow K, Gal A, Mühlhausen C, Ullrich K, Braulke T, Muschol N. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics 2007; 120: e1255-61.
BASAL Dateof 2ndevaluation
Dateof 3rdevaluation
Dateof (...)evaluation
Difficulty to initiate sleep
Duration of time awake at night
Wakes up early in the morning
Duration of day asleep
Duration of time unsettled at night
Dangerous behavior

Sanfilippo Syndrome Clínical Guidelines I 75
05.Treatment forSanfilippo syndrome.Mireia del Toro.Pediatric Neurology Service. Vall d’Hebron University Hospital, Barcelona.

76 I Sanfilippo Syndrome Clínical Guidelines
Treatment advances, in recent years, in some lysosomal diseases have been a major change in their prognosis and the patient’s quality of life.However, the neurodegenerative nature of mucopolysaccharidosis type III (MPS III) and the difficulty for drugs to cross the blood-brain barrier, has determined that it is proving difficult to have an effective therapy for these patients. However, at present, several therapies focused on the central nervous system are undergoing testing.
Classically treatments for mucopolysaccharidosis are based on different approaches:
• Multidisciplinary support treatment from the different specialists involved.
• Deficient enzyme replacement, either with external administration by hematopoietic stem cell or chaperones transplantation which stabilize the protein.
• Reduction of substrate accumulation by inhibiting the enzymes synthesizing it.
• Gene therapy.
En este capítulo se revisará la situación de las diferentes alternativas terapéuticas para MPS III.
5.1 SUPPORTIVE TREATMENTDespite the therapeutic advances that may arise in the coming years, supporting the multidisciplinary treatment will remain very important to ensure patient’s best possible quality of life. These measures should address both the various aspects of involvement as their general needs for integration into everyday life. Patients with MPS III essentially will require a neurological monitoring in the initial stages but later they will require the attention of other specialists.
Conduct disorder is one of the most prominent problems in patients with MPS, more usually precocious in the form MPS IIIA. The fact that these children have no motor difficulties and are usually strong is an added difficulty in handling and a source of major stress at the household level. Antipsychotic drugs have been used for years with acceptable results, but risperidone seems to have more positive effects with fewer side effects. Risperidone at doses between 0.2 and 2 mg at night or divided in two doses obtained good results by reducing hyperactivity and oppositional behavior, thereby reducing rates of maternal anxiety and depression1.
1. Kalkan Ucar S, Ozbaran B, Demiral N, Yuncu Z, Erermis S, Coker M. Clinical overview of children with mucopolysaccha-ridosis type III A and effect of Risperidone treatment on children and their mothers psychological status. Brain Dev 2010; 32:156-61.

Sanfilippo Syndrome Clínical Guidelines I 77
In the sleep disorder it seems that the dysregulation of melatonin synthesis plays an important role, therefore, it is advisable to supplement melatonin in doses up to 15 to 18 mg if necessary2. In advanced stages of the disease, the frequent occurrence of seizures will require specific treatment with anticonvulsant drugs. A movement disorder that can simulate crises but has no electroencephalogram translation and which is having good response to levomepromazine has also been described3.
Bone problems, although less prevalent and severe than in other MPS, require follow-up by orthopedics and physical therapists for prevention and correction. Special attention should be paid to the hip, as a high prevalence of avascular necrosis of the femoral head has been described, requiring a specific diagnosis and treatment, including the placement of prostheses in advanced cases. There is also a need to monitor the emergence of carpal tunnel syndrome. Both conditions can result in pain and discomfort that patients do not know how to explain and adversely affect behavior.
Proper nutritional support is essential for all patients at any stage of their disease. This includes planning a proper diet to meet the caloric needs, vitamin and minerals as well as the introduction of artificial feeding routes such as nasogastric tube or gastrostomy if swallowing disorders appear. Likewise, the tendency to diarrhea of unknown cause, probably caused by intestinal dysfunction secondary to GAGs deposits should be considered in follow-ups as it may worsen the quality of life significantly. A secondary coenzyme Q10 deficiency in fibroblasts in patients with MPS IIIA and IIIB has been described recently. The treatment of cell lines with coenzyme Q improved the residual enzyme activity in cells MPS IIIB but not in IIIA; the treatment with an antioxidant cocktail had no effect and the combination of both reduced the accumulation of glycosaminoglycans4. . Is pending check if the supplementation may have positive effects on patients.
The problems of the upper airway, often entailing obstructive apneas may require tonsillectomy and adenoidectomy in most patients as well as placement of ear tubes. We should monitor the auditory function because a hearing loss will impact the speech negatively. The possibility of ophthalmologic disorders (retinal changes and alterations of the optic nerve) also requires regular monitoring.
2. Mahon LV, Lomax M, Grant S, Cross E, Hare DJ, Wraith JE, et al. Assessment of sleep in children with mucopolysac-charidosis type III. PLoS One 2014; 9:e84128.3. Santra S, Simmons L, Wassmer E. Levomepromazine as a treatment for non-epileptic movement disorder in advanced Sanfilippo disease. J Inher Metab Dis 2014; 37 (Suppl 1):S42.4. Matalonga L, Arias A, Coll MJ, Garcia-Villoria J, Gort L, Ribes A. Treatment effect of coenzyme Q(10) and an antioxidant cocktail in fibroblasts of patients with Sanfilippo disease. J Inherit Metab Dis 2014; 37:439-46.

78 I Sanfilippo Syndrome Clínical Guidelines
Psychological support is necessary for both the patient and his family. Both must adapt to the different phases of the process and be prepared to face the strong health, family and social burden resulting from these diseases. Also, in the early stages, the early measures of care for psychomotor development are relevant for the child’s maximum developmental capacity.
The assistance should also involve all necessary specialists to cover the possible facets of involvement: pulmonologists, cardiologists, endocrinologists. This monitoring should try to be properly coordinated since the large number of visits may interfere relevantly in the organization of the everyday life .
5.2 ENZYME REPLACEMENT THERAPYEnzyme replacement therapy involves the periodic administration of the deficient enzyme labeled with a biochemical signal. Its basis lies in the finding that an enzyme administered exogenously to a cellular medium is able to enter cells via the mannose 6-phosphate receptors and act accordingly. It has also been observed that a 1-5% of metabolic activity is able to correct the metabolic defect in the cell line. As a result, in the last 20 years, using different techniques, appropriate enzymes for treating various lysosomal diseases have been developed.
The limitations of enzyme replacement therapy (ERT) in MPS III deals with the difficulty of drugs to cross the blood-brain barrier covering the nervous system. To overcome this difficulty different strategies are being designed: on the one hand the possibility of direct administration on the spinal or intraventricular space and on the other hand, the association of the modified enzymes to a molecule which will help them to cross the barrier once administered intravenously.
In MPS IIIA, the first trials of enzyme therapy with sulfamidase were carried out in 2007 with direct injections into the brain before moving on to the intrathecal injections in the cisternal space. The results of this latest study showed a decrease in the accumulation of GAGs and other secondary substances, an improvement in nervous system injuries and favorable changes in the behavior of animals5. This prompted the execution of a clinical trial of intrathecal administration through a lumbar catheter in patients with MPS IIIA and severe impairment. The first trial was conducted in patients with severe impairment, reporting the safety of the treatment. Currently a second clinical trial is being conducted in younger patients (2-4 years) with milder involvement at baseline (IQ above 60). In an attempt to cross the blood-brain barrier from the bloodstream, they conducted a study in monkeys with intravenous administration of the enzyme sulfamidase merged with the
5. Hemsley KM, King B, Hopwood JJ. Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol Genet Metab 2007; 90:313-28.

Sanfilippo Syndrome Clínical Guidelines I 79
antibody against the insulin receptor. This association appears to permeate the barrier in sufficient quantities to be effective6.
As for the MPS IIIB, the problem is that the NAGLU enzyme lacks the required mannose 6-phosphate receptor to be taken up by cells. To solve this problem, a research on the combination of NAGLU with a growth factor (insulin growth factor II or IGF II) in intraventricular administration to an animal model of MPS IIIB is being carried. The resultsas regards to increasing the enzyme activity both inside and outside of the nervous system and reducing accumulation of GAGs have been positive7. As for patients, a clinical trial is being conducted with intravenous administration of enzyme to assess safety.
A different way to increase enzyme activity is the use of chaperones. It is based on the fact that some mutations cause a qualitative but not quantitative defect of the enzyme, i.e., the configuration changes resulting in inactive and misfolded proteins. These proteins are degraded by the proteases and removed from the cell. The chaperones are small molecules responsible for ensuring the functionality of a protein able to act at different levels: as substrate analogues, agonists or antagonists receptors, modulators and as ligands to epitopes ensuring the configuration of the enzyme. In this context, it has been shown that glucosamine corrects the folding defect caused by some mutations in MPS IIIC and restores the enzyme activity8. As for the MPS IIIB, several molecules that at low doses could act as chaperones were proposed but they are in initial experimental phase9.
5.3 HEMATOPOIETIC STEM CELL TRANSPLANTATION
Hematopoietic cell transplantation has been an effective therapy in different inborn errors of metabolism since the 1980s, especially in lysosomal and peroxisomal diseases. Its efficiency is based on the provision of enzymes to the various tissues from donor cells migrating from the cardiocirculatory system. The enzyme can be transferred from one cell to another through a cross-correction phenomenon mediated by the mannose 6-phosphate receptors. Cells from the monocyte-macrophage cell lines can cross the blood brain barrier and be transformed into glial cells capable of synthesizing enzymes.
6. Boado RJ, Lu JZ, Hui EK, Pardridge WM. Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type A cells. Mol Pharm 2014; 11:2928-34.7. Kan SH, Aoyagi-Scharber M, Le SQ, Vincelette J, Ohmi K, Bullens S, et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc Natl Acad Sci U S A 2014; 111:14870-5.8. Matos L, Canals I, Dridi L, Choi Y, Prata MJ, Jordan P et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J Rare Dis 2014; 9:180.9. De Ruijter J, Valstar MJ, Wijburg FA. Mucopolysaccharidosis type III (Sanfilippo Syndrome): emerging treatment strate-gies. Curr Pharm Biotechnol 2011; 12:923-30.

80 I Sanfilippo Syndrome Clínical Guidelines
There are now two possibilities of donation: bone marrow hematopoietic cells from healthy donor (preferably related) or cord blood hematopoietic cells. The main advantage of this therapy is the definitive correction of the enzymatic defect in those tissues in which the donated cells are well planted.
However, it has some drawbacks preventing it from being used as a general treatment for all lysosomal diseases. On the one hand, the benefit depends on the severity of the disease and the time of transplantation. On the other, the effect varies in different tissues: it is very effective in organs with reticuloendothelial system, has little impact on bone, and the CNS cells need a period of 6-12 months for improvement to occur. Early transplantation is able to preserve cognitive function and improve some somatic manifestations in MPS I (Hurler syndrome), although with considerable morbidity, mortality and residual disease10. However, their lack of effectiveness in MPS III may be because the cells crossing the blood-brain barrier are not able to synthesize enough enzyme to stop the neurological progression11.
There has been a similar progression of neuropsychological impairment as the one in patients undergoing HSCT, despite achieving nearly normal blood enzyme levels12,13,14. Most referred cases have been transplanted after onset of symptoms, making the results difficult to interpret. The case of an asymptomatic child transplanted at the10 months of age, with biochemical correction without serious complications related to HSCT, who at the 8 years of age had a neurological impairment similar to his untreated brother is particularly conspicuous14. In the series of Prasad et al15, 19 children were transplanted umbilical cord stem cells, 12 survive and 9 show stable disease, although with little effect on cognitive function (higher in the 2 patients younger than 2 years), although there appear to be fewer behavioral and sleep problems. Some authors suggest that a transplant performed in the first month of life, could correct or prevent build up in the nerve cells of the host, before occurring irreversible damage16. In a recent publication, two patients with MPS III (A and B) underwent transplantation of umbilical cord cells before the onset of neurological symptoms. Both decreased urine GAGs levels and improved
10. Aldenhoven M, Wynn RF, Orchard PJ, O’Meara A, Veys P, Fischer A, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood 2015;125:2164-72.11. Welling L, Marchal JP, van Hasselt P, van der Ploeg AT, Wijburg FA, Boelens JJ. Early umbilical cord blood-deri-ved stem cell transplantation does not prevent neurological deterioration in mucopolysaccharidosis Type III. JIMD Rep 2015;18:63-8. 12. Vellodi A, Young E, New M, Pot-Mees C, Hugh-Jones K. Bone marrow transplantation for Sanfilippo disease type B. J Inherit Metab Dis 1992; 15: 911-8.13. Cleary M A, Wraith J E. Management of mucopolysaccharidosis type III. Arch Dis Child 1993; 69: 403-6. 14. Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: a comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis 1999; 22: 849-50. 15. Prasad VK, Mendizabal A, Parikh SH, Szabolcs P, Driscoll TA, Page K, et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: influence of cellular compo-sition of the graft on transplantation outcomes. Blood 2008; 112: 2979-89. 16. Hopwood JJ. Sanfilippo syndrome: clinical genetic diagnosis and therapies. En: Barranger JA, Cabrera-Salazar MA, eds. Lysosomal storage disorders. New York: Springer; 2007. p. 415-32.

Sanfilippo Syndrome Clínical Guidelines I 81
somatic impairment; however, they showed a neurological impairment similar to that of untreated children11.
Transplantation of genetically modified hematopoietic precursors has been assayed in MPS IIIB, to express the enzyme and injected via a viral vector. This technique achieves an increase in the NAGLU activity and a significant reduction of matter build up17. It has also proved a positive effect at both the cerebral anatomical and the behavioral levels of MPS IIIB mice with repeated injection of mononuclear cells from umbilical cord18.
5.4 SUBSTRATE REDUCTION THERAPYThe substrate reduction therapy is based on inhibiting the path of synthesis of GAGs that cannot be degraded to thus avoid their accumulation. The treatments are usuallyoral and with small molecules that can cross the blood-brain barrier. Different molecules were tested, of which genistein has been more tested.
Genistein is a soy isoflavone inhibiting a tyrosine kinase in the epidermal growth factor receptor. The activity of this enzyme in the receptor is required for the synthesis of GAGs. In early studies in vitro, the addition of genistein to fibroblast cultures from patients with MPS IIIA and IIIB induced a significant reduction of GAGs levels. Subsequently the genistein’s ability to decrease GAG deposits both in peripheral tissues and the central nervous system was tested in mouse models, resulting in positive changes in the behavior of these animals19. In 2007 and 2008 some trials were presented with Polish patients treated with daily doses of 5 mg/kg/day of genistein and in addition to a reduction in eliminating GAGs, improvement was observed in cognitive functions and behavior20. In a follow-up to 3 years these changes were only kept in a low percentage of children21. Subsequent studies of a larger number of patients at doses of 5 or 10 mg/kg/day have not been able to confirm the beneficial effects
17. Zheng Y, Ryazantsev S, Ohmi K, Zhao HZ, Rozengurt N, Kohn DB, et al. Retrovirally transduced bone marrow has a therapeutic effect on brain in the mouse model of mucopolysaccharidosis IIIB. Mol Genet Metab 2004; 82:286-95.18. Willing AE, Garbuzova-Davis SN, Zayko O, Derasari HM, Rawls AE, James CR, et al. Repeated administrations of human umbilical cord blood cells improve disease outcomes in a mouse model of Sanfilippo syndrome type III B. Cell Transplant 2014; 23:1613-30.19. Malinowska M, Wilkinson FL, Langford-Smith KJ, Langford-Smith A, Brown JR, Crawford BE, et al. Genistein improves neuropathology and corrects behaviour in a mouse model of neurodegenerative metabolic disease. PLoS One 2010; 5:e14192.20. Piotrowska E, Jakóbkiewicz-Banecka J, Tylki-Szymanska A, Liberek A, Maryniak A, Malinowska M, et al. ßGenistin-rich soy isoflavone extract in substrate reduction therapy for Sanfilippo syndrome: An open-label, pilot study in 10 pediatric patients. Curr Ther Res Clin Exp 2008; 69: 166–179.21. Piotrowska E, Jakobkiewicz-Banecka J, Maryniak A, Tylki-Szymanska A, Puk E, Liberek A, et al. Two-year follow-up of Sanfilippo Disease patients treated with a genistein-rich isoflavone extract: assessment of effects on cognitive functions and general status of patients. Med Sci Monit 2011; 17:196-202.

82 I Sanfilippo Syndrome Clínical Guidelines
of genistein22,23. At the moment several studies with high doses of pure genistein (150 mg/kg/day) are being conducted to evaluate the safety and efficacy of this treatment for MPS III. The first results from 22 patients with severe MPS II and MPS III, after at least 12 months of treatment have shown that the treatment is well tolerated and safe. The clinical and analytical controls every 3 months showed no major biochemical alterations except for a slight alteration of transaminases, alkaline phosphatase, amylase or lipase. There was no clear decrease on GAG levels or changes in the patients’ severity scale, although the main objective was to assess safety24. The fact of being a safe treatment and despite its dubious effectiveness at the clinical level, many patients suffering from MPS III are being treated with genistein at different doses.
Rhodamine B is a dye used in cosmetics which inhibits the synthesis of GAGs chains by blocking the formation of the glucidic precursors. In human fibroblasts it shows capability to reduce GAGs synthesis and in MPSIIIA animal models this results in decreased visceromegalies and cerebral accumulation, and an improvement of cognitive skills25. Studies in 4 successive generations of mice treated with rhodamine B have not yet demonstrated toxicity or teratogenicity. There are no studies in patients yet.
Other approaches through genetic therapies have been reported recently. RNAs, silencers inhibiting gene expression (EXTL2 and EXTL3) required for the synthesis of GAGs in fibroblasts of patients with MPS IIIC have been synthesized26. The in vitro results are encouraging and are under processing stage in animal model.
5.5 GENE THERAPYGene therapy consists of obtaining a functional gene which produces the appropriate enzyme to replace the deficient. This has been attempted through different methods: the addition of a foreign gene, inhibition of a specific area of the gene or complete inhibition of mutated genes to achieve a gain-of-function. These techniques can be applied in vivo, by introducing the appropriate gene into the cells or tissues, or ex vivo when the genetic change is made in hematopoietic cell lines that are then transplanted to the patient.
22. De Ruijter J, Valstar MJ, Narajczyk M, Wegrzyn G, Kulik W, Ijlst L, et al. Genistein in Sanfilippo disease: a randomized controlled crossover trial. Ann Neurol 2012; 71:110-20.23. Delgadillo V, O’Callaghan Mdel M, Artuch R, Montero R, Pineda M. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis 2011; 34:1039-44.24. Kim KH, Dodsworth C, Paras A, Burton BK. High dose genistein aglycone therapy is safe in patients with mucopoly-saccharidoses involving the central nervous system. Mol Genet Metab 2013; 109:382-5.25. Roberts AL, Rees MH, Klebe S, Fletcher JM, Byers S. Improvement in behavior after substrate deprivation therapy with rhodamine B in a mouse model of MPS IIIA. Mol Genet Metab 2007; 92:115-21.26. Canals I, Benetó N, Cozar M, Vilageliu L, Grinberg D. EXTL2 and EXTL3 inhibition with siRNAs as a promising substra-te reduction therapy for Sanfilippo C syndrome. Sci Rep 2015; 5: 13654.

Sanfilippo Syndrome Clínical Guidelines I 83
This is the most pathophysiological therapeutic approach and it has two factors adding benefits once applied: on the one hand, the observation that a small amount of enzyme (1-10%) is sufficient for proper cell function and secondly, the cross-correction phenomenon by which the enzyme may be transferred from one cell to another making it normofunctional.
There are, however, important limitations at the present time. One of them is the ability to transfer a sufficient amount of active genes or the appropriate cell population into the affected tissue. Numerous vectors have been tested, some of which apparently can cross the blood-brain barrier administered via bloodstream, and others are directly injected into the nervous system at the intracerebral or intracisternal level. A second problem is the level and duration of the expression of transgenic products administered in their site of action. It is known that these cells may be lost spontaneously, or as a result of an immunologic reaction and the potential toxicity of the administration via vectors is not yet established27.
In MPS IIIA early studies in mouse model with intraventricular sulfamidase injections with a modifying factor which increased the enzyme activity and carried by an adenovirus vector (AAV5) showed good uptake in all the nervous system, reducing build-up and improving the motor and cognitive functions28. This has been further confirmed and refined in various studies with major animal models (dog), different vectors (AAV8, AAV9, AAV10) and different ways to approach (intravenous, intracerebral, intracisternal and intraventricular) with equally encouraging results29,30. It has been possible to objectify that the effect is progressive for at least the first twelve months of administration.
In MPS IIIB, the positive effects of the simultaneous intravenously and intraventricular injection of NAGLU enzyme with adenovirus in animal model have been described31.
27. Aronovich EL, Hackett PB. Lysosomal storage disease: gene therapy on both sides of the blood-brain barrier. Mol Genet Metab 2015; 114:83-93.28. Fraldi A, Hemsley K, Crawley A, Lombardi A, Lau A, Sutherland L, et al. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet 2007; 16:2693-702.29. Haurigot V, Marcó S, Ribera A, Garcia M, Ruzo A, Villacampa P, et al. Whole body correction of mucopolysaccharido-sis IIIA by intracerebrospinal fluid gene therapy. J Clin Invest 2013; 8: 3254-71. 30. Ruzo A, Marcó S, García M, Villacampa P, Ribera A, Ayuso E, et al. Correction of pathological accumulation of glyco-saminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum Gene Ther 2012; 23:1237-46.31. Fu H, Kang L, Jennings JS, Moy SS, Perez A, Dirosario J, et al. Significantly increased lifespan and improved behavio-ral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther 2007; 14:1065-77.

84 I Sanfilippo Syndrome Clínical Guidelines
Subsequently the equally positive effect of a single intravenous administration of AAV9 vector in mouse model32 as well as in superior animal (monkey) was demonstrated33.
As for MPS IIIC there are several groups performing gene therapy research but no results are reported at the moment.
One approach being evaluated is the combination of gene therapy with stem cell transplantation as a way for administrating the vector17,34. The only clinical trial results published to date is made with four MPS IIIA patients who received intracerebral injection of sulfamidase enzyme performed in 12 points with its activator (SUMF1) carried by an adenovirus (AVV10). The first year of treatment was safe, well tolerated without significant adverse effects. Efficacy results are preliminary but appear to be positive for 3 patients although the brain atrophy progressed for 2 of them who had greater involvement at baseline35.
CONCLUSIONSIn the coming years, the treatment for patients with MPS III will have a major change with the coming of new therapies. At the moment, clinical trials with enzyme replacement therapy for MPS IIIA (intrathecal) and MPS IIIB (intravenous) as well as the extension study of patients with intracerebral gene therapy ( “clinical trials”) are being conducted. It is expected that soon, trials with intravenous and intraventricular gene therapy will be initiated in patients with MPS IIIA and MPS IIIB.
32. Fu H, Dirosario J, Killedar S, Zaraspe K, McCarty DM. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol Ther 2011; 19:1025-33.33. Murrey DA, Naughton BJ, Duncan FJ, Meadows AS, Ware TA, Campbell KJ, et al. Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: toxicology, biodistribution, and immunological assess-ments in primates. Hum Gene Ther Clin Dev 2014; 25:72-84.34. Langford-Smith A, Wilkinson FL, Langford-Smith KJ, Holley RJ, Sergijenko A, Howe SJ, et al. Hematopoietic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol Ther 2012; 20:1610-21.35. Tardieu M, Zérah M, Husson B, de Bournonville S, Deiva K, Adamsbaum C, et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccha-ridosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther 2014; 25:506-16.

610 893 873 / [email protected] / follow us on www.stopsanfilippo.org







































