Embed Size (px)
Citation preview

1SEMINARIO N°03: ENFERMEDADES
MUTACIONALES
Alumnos : Contreras Villegas, Rosa Cotrina Morán, Carlos
Cotrina Cerquera, Maria Curo Bancayán, Diego Dávila Burga, Jhonatan
Docente : Dr. Aníbal Monge Moyano
Asignatura : Biología celular
Ciclo : 2015 – I
ÍNDICE
Introducción……………………………………………………………………………………………………………………………………… 3Objetivos ………………………………………………………………………………………………………………………………………… 3
ContenidoGenética – generalidades………………………………………………………………………………………………………………… 4Mutaciones……………………………………………………………………………………………………………………………………… 6
A.-Mutaciones Génicas……………………………………………………………………………………………………………………… 8
UNIVERSIDAD NACIONAL “PEDRO RUIZ GALLO”
FACULTAD DE MEDICINA HUMANA

2TalasemiaRetinoblastomaEnfermedad de HuntingtonAcondroplasiaHemofilia ASíndrome de MarfanAlbinismoGalactosemiaFibrosis Quística
B.-Mutaciones Cromosómicas…………………………………………………………………………………………………………… 20B.1) DeleccionesSíndrome de Cri du ChatB.2)TranslocacionesLeucemia promielocítica agudaB.3) DuplicacionesB.4) Inversiones
C.-Mutaciones Genómicas………………………………………………………………………………………………………………… 24C.1) Anomalías autosómicas numéricas:Síndrome de DownSíndrome de EdwarsSíndrome de PatauC.2) Anomalías sexuales numéricas: Síndrome de KlinefelterSíndrome de TurnerSíndrome de doble YSíndrome de doble XC.3) OtrosDisomías (Prader-Willi, Angelman)Tetrasomías y pentasomías (Leucemia neutrofílica crónica)
Conclusiones ……………………………………………………………………………………………………………………………………… 43BIBLIOGRAFÍA…………………………………………………………………………………………………………………………………… 43
INTRODUCCIÓN

3Durante los últimos 50 años, especialmente desde el establecimiento del número correcto
de cromosomas del cariotipo humano en 46 para las células somáticas, la genética ha ido
introduciéndose en el campo de la medicina. Las ideas acerca de la individualidad química de los
seres vivos, y especialmente del ser humano, cuando se habla de los "errores congénitos del
metabolismo", se consolida la idea de que el defecto se debía a un gen defectuoso, es decir,
mutante. Esta genética, junto con el estudio de las anomalías cromosómicas, dio lugar a una
incipiente genética médica.
En 1953 Watson y Crick describieron la estructura replicativa de la doble hélice del ácido
desoxirribonucleico (DNA), la molécula portadora de la información genética y de la herencia. El
posterior desarrollo de la biología molecular y de la ingeniería genética nos ha ofrecido una nueva
compresión de qué es un gen. Desde una perspectiva molecular el gen es la unidad de DNA
formada por elementos controladores y reguladores. El conocimiento de la estructura de los genes,
cómo se regulan, se transcriben y se traducen en proteínas, ha sido fundamental para empezar a
comprender las causas y los mecanismos de producción de las enfermedades genéticas.
Este trabajo pretende mostrar un resumen de las principales enfermedades mutacionales
ocasionadas por diversas afecciones genéticas, incidiendo en aquellas que en el futuro veremos
como caos clínicos frecuentes. Esperamos que la información aquí presentada sea de provecho
para quienes accedan a la lectura de las páginas siguientes, para sentar las bases de la formación
como profesionales de la salud.
Atentamente, Los alumnos
Objetivos
Conceptualizar términos básicos y fundamentales de la genética
Clasificar a las enfermedades mutacionales para su mejor estudio

4 Establecer y aprender la definición, causas y síntomas de cada enfermedad que
estudiaremos.
I. GENÉTICA- GENERALIDADES
La diferencia en un gen, de las 20000 a 25000 que posee cada ser humano, puede cambiar la vida.
Si nuestro ADN es similar en un 99.98%; un 0.02% es lo que nos diferencia de Newton, A. Einstein,
S. Ramón y Cajal, la madre Teresa de Calcuta, etc. Si se puede llegar a ser tan grandes.
Todos poseemos genes que influyen en nuestra vida. Afectan nuestra altura, peso, el color de los
cabellos y la pigmentación de la piel. También nuestra susceptibilidad a muchas enfermedades y
trastornos y hasta contribuyen a nuestra inteligencia y personalidad. Los genes son fundamentales
para determinar lo que somos.
La genética provee uno de los principios unificadores de la biología: todos los organismos utilizan el
mismo sistema genético (código genético), desde las bacterias hasta los seres humanos.
El código genético tiene cuatro características básicas:
Esta formado por tripletes o codones del ARNm.
Es específico, porque identifica a un aminoácido.
Es universal, porque todos los seres vivos lo utilizan.
Es degenerado, porque existen 64 códigos para identificar solo a 20 aminoácidos.
El conjunto completo de instrucciones genéticas de cualquier organismo es su genoma y todos los
genomas están codificados en ácidos nucleicos, sea DNA o RNA. El sistema de codificación de la
información genética también es común a todas las formas de vida: las instrucciones genéticas se
encuentran en un mismo formato y, con raras excepciones, las palabras del código son idénticos.
En forma semejante, el proceso por el cual la información genética se copia y decodifica es
notablemente similar para todas las formas de vida.
La genética también desempeña un papel sustancial en la medicina. Los médicos reconocen que
muchas enfermedades y trastornos tienen un componente hereditario, incluidos algunos trastornos
genéticos bien conocidos, como la anemia falciforme y la enfermedad de Huntington y muchas
enfermedades comunes, como el asma, la diabetes y la hipertensión. Los avances en la genética
molecular han permitido esclarecer el origen del cáncer y la creación de numerosas pruebas
diagnósticas.
La terapia génica, la alteración directa de genes para el tratamiento de enfermedades humana, se
ha practicado hasta hoy en miles de pacientes.
El estudio de casi cualquier campo de la biología o la medicina está incompleto sin una
comprensión acabada de los genes y de los métodos genéticos.
CONCEPTOS BÁSICOS DE GENÉTICA
Existen células de dos tipos básicos: eucariontes y procariontes.
El gen es la unidad fundamental de la herencia.
Los genes se presentan en múltiples formas denominadas alelos.

5 Los genes codifican los fenotipos.
La información genética es transportada por el DNA y el RNA.
Los genes se localizan en los cromosomas.
Los cromosomas se separan a través de los procesos de mitosis y meiosis.
La información genética es transmitida desde el DNA al RNA y de este a la proteína.
Las mutaciones son cambios permanentes y heredables en la información genética.
Algunos rasgos se ven afectados por múltiples factores.
La evolución es el cambio genético.
CARACTERÍSTICAS DEL MATERIAL GENÉTICO
Si bien la vida se caracteriza por una enorme diversidad, las instrucciones de codificación de todos
los organismos vivientes están escritos en el mismo lenguaje genético: el lenguaje de los ácidos
nucleicos.
El material genético debe poseer tres características importantes:
1. El material genético del contener información compleja.
a) El material genético debe poder almacenar gran cantidad de información: instrucciones
para todos los rasgos y funciones de un organismo.
Esta información debe tener la capacidad de variar porque las diferentes especies, e
incluso cada miembro de una especie, poseen una dotación genética distinta.
El material genético debe ser estable porque la mayor parte de las alteraciones de las
instrucciones genéticas (mutaciones) suelen ser perjudiciales.
2. El material genético debe replicarse fielmente.
La vida de cualquier organismo comienza en una célula única, que debe sufrir millones de
divisiones celulares para producir una criatura multicelular compleja como cada uno de
nosotros.
Cuando los organismos se reproducen y pasan los genes a su progenie, las instrucciones de
codificación deben copiarse con fidelidad.
3. El material genético debe codificar fenotipos
El material genético (el genotipo) debe ser capaz de codificar para (determinar los rasgos (el
fenotipo). El producto de un gen suele ser una proteína; por ende, debe existir un mecanismo para
que las instrucciones genéticas puedan traducirse en la secuencia aminoacídica de una proteína
II. MUTACIONES
Una mutación es una alteración o cambio en la información genética (genotipo) de un ser vivo y
que, por lo tanto, va a producir un cambio de características, que se presenta súbita y
espontáneamente, y que se puede transmitir o heredar a la descendencia. La unidad genética
capaz de mutar es el gen que es la unidad de información hereditaria que forma parte del ADN. En
los seres multicelulares, las mutaciones sólo pueden ser heredadas cuando afectan a las células
reproductivas. Una consecuencia de las mutaciones puede ser una enfermedad genética, sin
embargo, aunque en el corto plazo puede parecer perjudicial, a largo plazo las mutaciones son

6esenciales para nuestra existencia. Sin mutación no habría cambio y sin cambio la vida no podría
evolucionar.[][]
- Históricamente se han Génicos afectan a un solo gen
dividido en Cromosómicas afectan el número a la estructura de los
cromosomas.
¿Por qué?, esta distinción surge a raíz de que:
Cromosómicas podían observarse directamente, al mirar a los cromosomas con un
microscopio
Génicas Solo podían detectarse mediante la observación de sus efectos fenotipos.
- Hoy en día ¿Cómo las distinguimos?
De manera arbitraria, sobre la base del tamaño de la lesión del DNA
¿Gracias a qué? Secuenciación del DNA
A.- MUTACIONES GÉNICAS
Encontramos varias formas de clasificar a las mutaciones génicas; el más apropiado dependerá del
motivo de estudio de la mutación, así tenemos:
Por la naturaleza del efecto fenotipo.
Por el agente causante de la mutación.
Por la naturaleza molecular del defecto.
Para su división nos basaremos en la naturaleza molecular del defecto:
1. SUSTITUCIONES DE BASES
Es el tipo más simple de mutación génica.
Consiste en la alteración de un solo nucleótido en el DNA, pero cuando se altera la base de
un nucleótido también se alterará la base del nucleótido correspondiente en la cadena
opuesta, es la siguiente ronda de replicación:
Transición: Se reemplaza una purina por otra
Tenemos 2 tipos diferente, lo mismo con la pirimidina
Transversión: una purina pirimidina o
Una pirimidina purina
2. INVERSIONES Y DELECCIONES
Consiste en la adición o la eliminación, respectivamente, de uno o más pares de
nucleótidos.
Estos, dentro de secuencias que codifican proteína, pueden conducir a:
Mutaciones de cambio de marco de lectura de un gen.
Suelen tener efectos drásticos sobre el fenotipo
Enfermedades monogénicas
Poliposis adenomatasa de colon

7 Poliquistosis real del adulto
Déficit de 1 – antitripsina
Fibrosis quística
Distrofia muscular de Duchenne
Hipocolesterolemia familiar
Síndrome del cromosoma x frágil
Hemocromatosis (hereditaria)
Hemofilia A
Carcinoma colon-rectal hereditario no poliposo
Enfermedad de Huntington
Síndrome de Marfan
Distrofia mictónica
Neurofibromatosis de tipo I
Ontogénesis Imperfecta
Fenilectonoria
Retinoblastoma
Drepanocitosis
Enfermedad de Tay–Sachs
Talasemia
Expansión por repetición de trinucleótidos
Mutación en la cual aumentar considerablemente el número de copias de un trinucleótido.
A continuación describimos algunas enfermedades:
1.-TALASEMIA
1.1.-NOMBRES ALTERNATIVOS: Anemia mediterránea; Anemia de Cooley
1.2.-DEFINICIÓN
Las talasemias son trastornos hereditarios, caracterizados por la producción anormal de
hemoglobina, que ocasionan disminución en su producción y destrucción excesiva de los glóbulos
rojos.

81.3.-CAUSAS, INCIDENCIA Y FACTORES DE RIESGO
La hemoglobina contiene dos cadenas, la globina alfa y beta. Las anomalías genéticas, que causan
un desequilibrio en la producción de cualquiera de las cadenas, pueden ser hereditarias.
Las talasemias beta son causadas por una mutación en la cadena de la globina beta. Para adquirir
la forma mayor de esta enfermedad, los genes mutados se deben heredar de ambos padres. Si se
hereda un solo gen mutado, la persona será portadora de la enfermedad, pero no experimenta los
síntomas, lo cual corresponde a la forma menor de la enfermedad.
En la forma mayor, los niños son normales al nacer, pero desarrollan anemia durante el primer año
de vida. Algunos problemas que se pueden presentar son: insuficiencia en el crecimiento,
deformidades de los huesos, y agrandamiento del hígado y del bazo. Las transfusiones de sangre
podrían modificar algunos de los signos de la enfermedad, pero la sobrecarga de hierro por las
transfusiones puede causar daño a los sistemas cardíaco, hepático y endocrino.
La forma leve de la talasemia beta produce glóbulos rojos pequeños y no causa síntomas. Los
factores de riesgo incluyen antecedentes familiares de talasemia y un antecedente étnico que haya
mostrado susceptibilidad a la enfermedad.
Las talasemias beta se presentan en personas de origen mediterráneo y, en menor grado, en
individuos chinos, otros asiáticos y negros.
Las talasemias alfa se presentan más comúnmente en habitantes del sudeste de Asia y China y son
causadas por la eliminación de uno o más genes de la cadena de globina alfa. La forma más severa
de estas talasemias causa un mortinato (muerte de un feto antes del parto).
1.4.-SÍNTOMAS
Fatiga
Dificultad respiratoria
Ictericia
Deformidades en los huesos de la cara
.

9
2.-RETINOBLASTOMA
2.1.-NOMBRES ALTERNATIVOS
Cáncer de retina; Tumor de retina
2.2-DEFINICIÓN
Es un tumor maligno (cáncer) de la retina (parte del ojo) que generalmente afecta a niños menores
de 6 años. Se diagnostica con mayor frecuencia en niños en edades comprendidas entre 1 y 2 años.
2.3.-CAUSAS, INCIDENCIA Y FACTORES DE RIESGO
El retinoblastoma se presenta cuando una célula de la retina en crecimiento sufre una mutación,
haciendo que dicha célula crezca sin control y se vuelva cancerosa.
Algunas veces, esta mutación se desarrolla en un niño cuya familia nunca ha tenido cáncer en el
ojo, pero otras veces, la mutación está presente en varios miembros de la familia. Si la mutación es
un mal de familia, existe un 50% de probabilidades de que los hijos de la persona afectada también
tengan la mutación y, por lo tanto, tendrán un alto riesgo de desarrollar retinoblastoma.
Uno o ambos ojos pueden estar afectados y se puede presentar una blancura visible en la pupila.
La ceguera puede ocurrir en el ojo afectado y los ojos pueden parecer bizcos o con estrabismo
convergente. Este tumor se puede diseminar a la órbita a través del nervio óptico e igualmente al
cerebro, los pulmones y los huesos. Se trata de un tumor poco común, excepto en las familias
portadoras de la mutación del gen RB.
2.4.-SÍNTOMAS
Un brillo blanco en el ojo que con frecuencia se observa en las fotografías tomadas con
flash; en lugar del típico "ojo rojo" del flash, la pupila puede parecer blanca o distorsionada.
Manchas blancas en la pupila
Estrabismo convergente
Enrojecimiento y dolor en el ojo
Visión deficiente
Iris que puede ser de diferente color en cada ojo
3.-ENFERMEDAD DE HUNTINGTON
3.1.-DESCRIPCIÓN:
En 1872, el Dr. George Huntington describió claramente tanto la naturaleza familiar de la
enfermedad como las características físicas de la misma, lo que aún provee las bases de su
diagnóstico clínico. Uno de los primeros nombres fue “corea” que es la denominación griega de la

10danza. Este término describe cómo, las personas afectadas, se mueven descontroladamente, como
si danzaran.
La Enfermedad de Huntington (HD) es un desorden neurodegenerativo raro, progresivo y fatal, de
origen autosómico dominante, (sólo la presencia de una copia mutada del gen es suficiente para el
desarrollo de la enfermedad), que afecta típicamente a adultos. Es consecuencia de una
degeneración genética programada de las neuronas en ciertas regiones del cerebro. Esto causa
movimientos incontrolados, pérdidas de facultades intelectuales y disturbios emocionales.
La prevalencia en Estados Unidos y la mayoría de Europa es de aproximadamente 5 casos cada
100.000 individuos, con algunas regiones pequeñas que presentan una prevalencia mucho mayor.
Como se mencionó anteriormente, la Enfermedad de Huntington está asociada a la degeneración
progresiva de neuronas en ciertas regiones del cerebro y la presencia de astrocitos que se
acumulan debido a la destrucción de las neuronas cercanas (gliosis). Estos cambios ocurren
primariamente dentro del núcleo caudado y el putamen, subestructuras del ganglio basal que se
conocen colectivamente como el striatum. También se caracteriza por degeneración neuronal en
los lóbulos frontal y temporal de la corteza cerebral. Esta parte del cerebro es la responsable de la
integración de funciones mentales, movimientos y sensaciones.
Algunos estudios han demostrado que la disminución del neurotransmisor dopamina dentro del
striatum potencialmente juega un rol en el origen de los movimientos coreicos. Asimismo, algunas
investigaciones indican que el deterioro del metabolismo energético (disfunción mitocondrial),
resulta en una excesiva o prolongada activación (exocitotoxicidad) de neurotransmisores, tales
como glutamato o Nmetil- D-aspartato. Esto causa daño y pérdida de las células nerviosas
(apoptosis). Se suma esto la actividad metabólica del cerebro que produce componentes tóxicos
llamados radicales libres.

113.2.-SÍNTOMAS:
Cambios de humor, irritables, apáticos, pasivos, depresivos o cansados.
Se ve afectado el juicio.
Funciones cognitivas.
Problemas para manejarse.
Dificultades para el aprendizaje de cosas nuevas, recordar, contestar preguntas.
En algunas personas la enfermedad comienza con movimientos incontrolados de sus dedos,
pies, cara o tronco generalmente se intensifican cuando la persona se pone ansiosa.
La enfermedad llega a su punto máximo cuando el habla se vuelve farfullante y las
funciones vitales tales como tragar, comer, hablar y, especialmente, caminar, continúan
declinando.
La causa más común de muerte es la infección (mayormente neumonía),
Los movimientos que se ven en la forma juvenil se asemejan más a los de la Enfermedad
de Parkinson
Rigidez muscular y bradicinesia
4.-ACONDROPLASIA
4.1.-DESCRIPCIÓN:
La acondroplasia es un trastorno óseo genético (hereditario) que se presenta en uno de cada
25.000 niños que nacen vivos. La acondroplasia es el tipo más frecuente de enanismo, en la cual
los brazos y las piernas del niño son cortas en proporción a la longitud corporal. Además, con
frecuencia, la cabeza es de un tamaño mayor y el tronco, de tamaño normal. La estatura promedio
de los adultos hombres con acondroplasia es de 1,32m. La estatura promedio de las mujeres
adultas con acondroplasia es de 1,25 m.
La acondroplasia se hereda mediante un gen autosómico dominante que causa una formación
anormal de los cartílagos. La herencia autosómica dominante significa que el gen está ubicado en
uno de los autosomas (pares de cromosomas 1 a 22). Esto significa que afecta a hombres y
mujeres por igual. Dominante significa que un solo gen es necesario para tener el rasgo. Cuando
uno de los padres tiene la característica dominante, hay un 50 por ciento de posibilidades de que
cualquiera de sus hijos también herede ese rasgo.
Por lo tanto, en algunos casos, el niño hereda la acondroplasia de un padre con acondroplasia. La
mayoría de los casos de acondroplasia (80 por ciento) son consecuencia de una nueva mutación en
la familia, los padres tienen una estatura promedio y no poseen el gen anormal.
Como ya se dijo, las personas con acondroplasia tienen un 50 por ciento de posibilidades de
transmitir el gen a un niño, causando esta condición. Si ambos padres padecen de acondroplasia,
en cada embarazo, las posibilidades de dar a luz a un niño con acondroplasia ascienden a un 50 por
ciento, la posibilidad de que el niño no herede el gen y alcance una estatura promedio es de un 25
Se observa degeneración de células neuronales del cuerpo estriado

12por ciento y la posibilidad de que el niño herede un gen anormal de cada padre asciende a un 25
por ciento. En este último caso, pueden presentarse problemas severos en el sistema esquelético
que, con frecuencia, pueden causar la muerte a una edad temprana.
4.2.- SÍNTOMAS:
Brazos y piernas cortos, con la parte superior de los brazos más cortos que los antebrazos
y los muslos más cortos que la parte inferior de las piernas.
Cabeza de gran tamaño, frente prominente y tabique nasal aplanado.
Dientes mal alineados o montados.
Parte baja de la columna vertebral curvada - condición también denominada lordosis (o
"corcova") que puede ocasionar cifosis o la formación de una pequeña corcova cerca de los
hombros que generalmente desaparece una vez que el niño comienza a caminar.
Parte inferior de las piernas curvada.
Pie plano, corto y ancho.
Espacio excesivo entre los dedos medio y anular (también denominado mano tridente).
Falta de tonicidad muscular y articulaciones flojas.
Infecciones frecuentes en el oído medio que pueden provocar la pérdida de la audición.
Inteligencia normal.
Retrasos en los avances principales del desarrollo tales como caminar (que puede ocurrir
entre los 18 y 24 meses en vez de darse alrededor del primer año de edad).
5.-DISTROFIA MUSCULAR DE DUCHENNE
5.1.-DESCRIPCIÓN
La enfermedad de Duchenne, es una de las distrofias más comunes y más graves que afectan al
ser humano. Se hereda de forma recesiva ligada al sexo, el gen que la determina está ubicado en

13el brazo corto del cromosoma X, este gen es incapaz de codificar la proteína distrofina, lo que se
traduce en un deterioro progresivo de las fibras musculares.
5.2.-¿QUÉ LE OCURRE AL MÚSCULO EN LA ENFERMEDAD DE DUCHENNE?
En La distrofia muscular de Duchenne, DMD, el tejido muscular deja de funcionar adecuadamente y
es sustituido lentamente por el tejido graso. Los músculos del cuerpo están formados por un
número de fibras musculares de diferentes tipos, cuya proporción y cantidad varían según el tipo
de actividad que el músculo normalmente realiza. Es importante tener en cuenta esto porque
ayuda a comprender por qué algunos músculos se debilitan más que otros o se dañan antes.
La progresiva pérdida de fuerza muscular hace cada vez más difícil andar y vestirse, hay medios
para que el paciente mantenga al máximo sus posibilidades y de esta forma, sea lo más
independiente posible. Es importante recordar que en todo el mundo se está haciendo un esfuerzo
por encontrar, la curación de esta enfermedad. Uno de los modelos de intervención es la
fisioterapia. Sus principales objetivos son:
· Reducir el desarrollo de contracturas por medio de ejercicios de estiramiento.
· Prolongar la fuerza de los músculos a través de ejercicio.
· Prolongar las posibilidades de movimiento por medio de los aparatos que le recomiende el
especialista.
La distrofia muscular de Duchenne, se debe a una deficiencia de la distrofina, una proteína
fundamental para el mantenimiento del tejido muscular. Investigadores británicos experimentaron
con la utrofina, una proteína similar. Utilizaron técnicas de ingeniería genética para engendrar
ratones con una carencia de distrofina, pero con una capacidad para producir más utrofina de lo
normal. El experimento fue todo un éxito ya que el tejido muscular de estos ratones se mantuvo
relativamente sano, a pesar de la falta de distrofina.
Ahora el gran desafío es desarrollar alguna terapia para incrementar el nivel de utrofina en los
enfermos (DMD), aunque es difícil parece factible.
“No obstante, hoy por hoy no existe tratamiento”
6.-HEMOFILIA A
6.1.-DEFINICIÓN:
Es un trastorno hemorrágico hereditario causado por la falta del factor de coagulación sanguínea
VIII.
6.2.-CAUSAS E INCIDENCIA:
La hemofilia A es el resultado de la deficiencia del factor VIII de la coagulación. El trastorno es
causado por un rasgo hereditario recesivo ligado al cromosoma X, con el gen defectuoso localizado
en dicho cromosoma. Eso significa que el trastorno se presenta principalmente en las mujeres.

14Éstas portan dos copias del cromosoma X, de tal forma que si el gen del factor VIII en un
cromosoma no funciona, el gen en el otro cromosoma puede compensar. Los hombres, sin
embargo, portan sólo un cromosoma X, de tal manera que si el gen del factor VIII en ese
cromosoma está defectuoso, el niño desarrollará la hemofilia A.
Si una mujer tiene el gen defectuoso del factor VIII, se considera una portadora y puede
transmitirles dicho gen a sus hijos. La mitad de los bebés de sexo masculino nacidos de mujeres
portadoras del gen defectuoso desarrollan la enfermedad y la mitad de los bebés de sexo femenino
nacidos también de estas mujeres son portadores. Así mismo, todas las hijas de hombres
hemofílicos son portadoras del gen defectuoso.
6.3.-SÍNTOMAS:
Hematomas
Sangrado espontáneo
Sangrado al interior de las articulaciones y el
correspondiente dolor y edema
Hemorragias del tracto gastrointestinal y urinario
Sangre en la orina o en las heces
Sangrado prolongado producido por heridas,
extracciones dentales y cirugía
7.-SINDROME DE MARFAN
7.1.-DEFINICIÓN
Se trata de un trastorno hereditario del tejido conectivo, que es el encargado de mantener unidos
los tejidos del cuerpo. Se pueden producir un número variable de alteraciones que pueden afectar
al corazón, los vasos sanguíneos, los pulmones, a los huesos y a los ligamentos.
En general, son personas muy altas, con los miembros desproporcionadamente largos con respecto
al torso.
7.2.-CAUSAS
El Síndrome de Marfan es de origen genético y se debe a un anormal comportamiento del gen
FBN1, que determina la producción de una proteína denominada fibrilina. Este gen reside en el
cromosoma 15; hay gran variedad de cambios (mutaciones) en este gen que puede causar el
síndrome, lo que explicaría la gran variabilidad de afectación de las personas.
El síndrome de Marfan se hereda como rasgo autosómico dominante y, por tanto, cada niño con un
padre con el gen tienen el 50% de probabilidades de heredarlo. Sin embargo, hasta el 30% de los
casos no tienen historia familiar y se les denominan casos "esporádicos".
La fibrilina es un componente fundamental del tejido conectivo contribuyendo a su fuerza y
elasticidad. La fibrilina es especialmente abundante en la arteria aorta, en los elementos de sostén

15del ojo y en los huesos. Las personas con Síndrome de Marfan tienen poca cantidad de fibrilina o
ésta es de escasa calidad.
7.3.-SÍNTOMAS
Sistema osteoarticular
Hay una importante hiperlaxitud, con escasa masa muscular en la mayoría de las personas.
Articulaciones muy flexibles.
Los brazos y las piernas suelen ser más largos de lo normal en relación con el torso.
Con frecuencia pies planos
Curvatura anormal de la columna (escoliosis). En los casos extremos se presenta en el
nacimiento Espina Bífida.
El esternón puede sobresalir (tórax en quilla) o estar hundido (tórax en embudo).
Dedos largos, como de araña (aracnodactilia).
Cara fina y delgada.
Mandíbula pequeña.
Problemas oftalmológicos
Luxación o subluxación del cristalino
Es la lente de nuestro ojo y se encuentra suspendido por tejido conectivo. Cuando estas fibras de
tejido conectivo se debilitan o se rompen, el cristalino desplaza su eje respecto al sistema óptico
que permite la entrada de luz a nuestro ojo, provocando que se vean las imágenes distorsionadas;
la consecuencia inmediata es una miopía más o menos severa.
Desprendimiento de retina: amenaza gravemente la vista.
Catarata.
8.-ALBINISMO
8.1.-DESCRIPCIÓN GENERAL
El albinismo es un conjunto de condiciones congénitas (heredadas) que afectan a los humanos (y al
resto de animales) y que globalmente se caracterizan por la ausencia o disminución de pigmento
(melanina) en la piel, los ojos o el pelo.
Una de cada 17,000 personas en los Estados Unidos tiene algún tipo de albinismo. El albinismo
afecta a personas de todas las razas. La mayoría de niños con albinismo tienen padres con pelo y
ojos normales típicos de su raza.
En realidad, hay diferentes tipos de albinismo,
y la cantidad de pigmento en los ojos varía.

168.2.-LOS GENES DEL ALBINISMO
Para casi todos los tipos de albinismo, los dos padres tienen que tener un gen para albinismo para
tener un hijo con albinismo. Como el cuerpo tiene dos pares enteros de genes, una persona puede
que se vea normal, pero puede contener los genes para el albinismo. Si una persona tiene un par
de genes normales y un par de genes con albinismo, el ó ella tienen la información genética
suficiente para hacer pigmento normal. El gen del albinismo es "recesivo" y no resultará en
una persona con albinismo a menos que los dos pares de genes contengan albinismo y no hay
copia del gen que tiene pigmento normal.
Cuando los dos padres tienen el gen pero ninguno de los dos tienen albinismo, existe una
probabilidad de 25% en cada embarazo de que el bebé nazca con albinismo. Este tipo de herencia
se llama " herencia recesiva autosomal".
8.3.-PROBLEMAS MÉDICOS
Las condiciones comunes en el albinismo incluyen:
Nistagmus, movimiento irregular del ojo.
Estrabismo, descontrol de los músculos de los ojos ("ojos cruzados" o sin coordinación).
Sensibilidad a luces brillantes ó claras. Las personas con albinismo pueden tener miopía
(mala vista de lejos ó de cerca) y frecuentemente tienen astigmatismo (imágenes
distorsionadas).
Estos problemas resultan de un desarrollo anormal del ojo porque no hay suficiente pigmento. La
retina, la superficie dentro del ojo que recibe luz, no se desarrolla normalmente antes de nacer y en
la infancia. Las señales del nervio de la retina al cerebro no siguen los caminos usuales.
El iris, la parte de color en el centro del ojo, no tiene pigmento suficiente para protegerse de los
rayos de luz que entran al ojo. (La luz normalmente entra al ojo por la pupila, la parte oscura del
ojo, pero en personas con albinismo la luz puede pasar por la muralla del iris también.)
9.-GALACTOSEMIA

179.1.-NOMBRES ALTERNATIVOS
Deficiencia de galactosa-1-fosfatouridil transferasa; Deficiencia de galactocinasa; Deficiencia de
galactosa-6-fosfato epimerasa
9.2.-DEFINICIÓN
Es la incapacidad del organismo para utilizar (metabolizar) el azúcar simple galactosa, ocasionando
la acumulación de galactosa 1-fosfato en el cuerpo, lo cual causa daño al hígado, al sistema
nervioso central y a otros sistemas del organismo.
9.3.-CAUSAS, INCIDENCIA Y FACTORES DE RIESGO
La galactosemia es una enfermedad enzimática hereditaria, transmitida como un rasgo autosómico
recesivo y cuya ocurrencia es aproximadamente de 1 por cada 60.000 nacimientos entre personas
de raza blanca, mientras que la tasa es diferente para otros grupos.
Existen 3 formas de la enfermedad: deficiencia de galactosa-1-fosfatouridil transferasa
(galactosemia clásica, la forma más común y la más grave), deficiencia de galactosa cinasa y
deficiencia de galactosa-6-fosfato epimerasa.
Las personas con galactosemia son incapaces de descomponer completamente el azúcar simple
galactosa, que compone la mitad de la lactosa, el azúcar que se encuentra en la leche.
La lactosa es un disacárido (di significa 2 y sacárido significa azúcar) debido a que está compuesto
de dos azúcares, galactosa y glucosa, enlazados.
Si a un bebé con galactosemia se le da leche, los derivados de la galactosa se acumulan en el
sistema del bebé, causando daño al hígado, al cerebro, a los riñones y a los ojos. Los individuos con
galactosemia no pueden tolerar ninguna forma de leche (ni humana ni animal) y deben vigilar
cuidadosamente la ingesta de otros alimentos que contengan galactosa. La exposición a los
productos lácteos puede ocasionar daño hepático, retardo mental, formación de cataratas e
insuficiencia renal.
Después de tomar leche durante algunos días, un neonato con galactosemia se rehusará a comer y
desarrollará ictericia, vómitos, letargo, irritabilidad y convulsiones. Asimismo, se presentará
agrandamiento del hígado y el azúcar puede estar bajo. La alimentación continua con productos
lácteos lleva a que se presente cirrosis hepática, formación de cataratas en el ojo (que puede
ocasionar ceguera parcial) y retardo mental.
9.4.-SÍNTOMAS
Ictericia (coloración amarillenta de la piel y de la esclerótica)
Vómitos
Alimentación deficiente (el bebé se niega a beber fórmula que contenga leche)
Poco aumento de peso
Letargo

18 Irritabilidad
Convulsiones
10.-FIBROSIS QUÍSTICA
10.1.-DEFINICIÓN
Es una enfermedad hereditaria que provoca la
acumulación de moco espeso y pegajoso en los
pulmones y el tubo digestivo. Es el tipo de enfermedad
pulmonar crónica más común en niños y adultos jóvenes,
y puede ocasionar la muerte prematura.
10.2.-CAUSAS, INCIDENCIA Y FACTORES DE RIESGO
La fibrosis quística (FQ) es causada por un gen defectuoso que le indica al cuerpo que produzca un
fluido anormalmente espeso y pegajoso llamado moco. Este moco se acumula en las vías
respiratorias de los pulmones y en el páncreas, el órgano que ayuda a descomponer y absorber los
alimentos.
Esta acumulación de moco pegajoso ocasiona infecciones pulmonares potencialmente mortales y
serios problemas digestivos. Esta enfermedad también puede afectar las glándulas sudoríparas y el
aparato reproductor masculino.
A la mayoría de los niños se les diagnostica fibrosis quística a los dos años. Sin embargo, a un
pequeño número no se le diagnostica la enfermedad hasta los 18 años o más. Estos pacientes
generalmente padecen una forma más leve de la enfermedad.
10.3.-SÍNTOMAS
Ausencia de deposiciones durante las primeras 24 a 48 horas de vida
Heces pálidas o color arcilla y con olor fétido o heces flotantes
Es posible que los bebés tengan la piel salada
Infecciones respiratorias recurrentes, como neumonía o sinusitis
Tos o sibilancias
Pérdida de peso o ausencia de aumento de peso normal en la niñez
Diarrea
Retraso en el crecimiento
Fatiga

19
B.1.-DELECCIONES
El término "delección" significa simplemente que una parte del cromosoma se perdió o se
"eliminó". Un pieza muy pequeña de un cromosoma puede contener muchos genes diferentes.
Cuando hay pérdida de material genético, puede haber errores en el desarrollo del bebé, como
consecuencia de la pérdida de algunas de las "instrucciones".
En las roturas y reparaciones de los cromosomas, pueden perderse parte de los mismos. Si la parte
perdida es muy grande, la situación es incompatible con la vida. En un 85% de los casos, las
delecciones provienen de novo y las restantes son hereditarias, representando una monosomía o
trisomía parciales.
Los fenotipos dependen del cromosoma y de la región afectada, como por ejemplo en el caso del
síndrome por maullido de gato, en el que se ha producido una deleción del cromosoma 5. Este
síndrome se caracteriza por retraso mental, microcefalia y cara de media luna que se va alargando
con la edad. Otro síndrome, la oligospermia o la azoospermia están asociados a Delecciones en el
cromosoma Y en la región donde están situados los genes para el factor AZF (factor de la
azoospermia).
Cuando se producen roturas a ambos lados del centrómero y posteriormente se unen los extremos,
se forma un cromosoma en anillo. Este tipo de cromosoma anular es bastante frecuente en
carcinomas sarcomas y leucemias y raras veces son detectados al nacer. El síndrome de
cromosoma 15 en anillo, que resulta de la delección de la parte del brazo corto del cromosoma 15
(15q25-26), se caracteriza por crecimiento retartado, estatura corta y anomalías faciales distintivas
parecidas a las del síndrome de Russell-Silver.
1.-EL SÍNDROME DEL CRI-DU-CHAT.
Éste síndrome está asociado con la perdida de aproximadamente la mitad del brazo corto del
cromosoma 5. Su constitución genética se puede designar como 46, -5p, indicando que tal
individuo tiene los 46 cromosomas, pero que algo del brazo p (el brazo corto) del cromosoma se ha

20perdido. En la especie humana no se han documentado monosomías autosómicas más allá del
nacimiento; sin embargo hay ejemplos de supervivientes con monosomías parciales, en donde se
ha perdido solo parte de un cromosoma, casos a los que denominamos delecciones segmentales,
uno de cuyos primeros casos fue documentado por Jêrome Lejeune cuando describió los síntomas
clínicos del síndrome del cri-du-chat ( maullido del gato).
1.1.-CAUSAS, INCIDENCIA Y FACTORES DE RIESGO
El síndrome del maullido del gato es poco común y ocurre cuando falta una parte de la información
en el cromosoma 5. Es probable que se supriman múltiples genes en dicho cromosoma. Uno de los
genes suprimidos, llamado TERT (transcriptasa inversa de la telomerasa), está involucrado en el
control del crecimiento celular y puede jugar un papel en la forma como se desarrollan algunas de
las características de este síndrome.
Se cree que la mayoría de los casos ocurren durante el desarrollo de un óvulo o de un
espermatozoide. Una minoría de estos casos se debe a que uno de los padres es portador de una
reordenación del cromosoma 5 denominada traslocación.
Entre 1 en 20.000 y 1 en 50.000 bebés se ven afectados por este síndrome que puede ser
responsable de hasta el 1% de individuos con retardo mental severo.
1.2.-SÍNTOMAS
Llanto de tono alto similar al de un gato
Inclinación de los ojos hacia abajo Bajo peso al nacer y crecimiento lento Orejas de implantación baja o de forma anormal Retardo mental
Dedos de las manos y pies parcialmente unidos por membranas Una sola línea en la palma de la mano (pliegue simiesco) Papilomas cutáneos justo delante de la oreja Desarrollo lento o incompleto de las habilidades motoras Cabeza pequeña (microcefalia) Quijada pequeña (micrognacia) Ojos separados
1.3.-SIGNOS Y EXÁMENES
Además de los otros síntomas, el examen físico puede mostrar:
Hernia inguinal
Diastasis de rectos (músculos abdominales separados)
Bajo tono muscular
Epicanto, un pliegue extra de piel sobre el ángulo interior del ojo
Orejas con pliegues anormales o incompletos
B.2.-TRANSLOCACIONES

21Una translocaciones del intercambio de material genético entre cromosomas no homólogos. Las
translocaciones balanceadas constituyen una de las cromosomopatias mas frecuentes en el ser
humano con un prevalecía de al menos 1/500 - 1000 individuos .Existiendo dos tipos:
1.-LEUCEMIA PROMIELOCÍTICA AGUDA
Conocida como leucemia mieloide-3 (o M3 de la clasificación FAB), Es un tipo de cáncer que se
caracteriza por un predominio de promielocitos malignos que muestran una translocación recíproca
entre los brazos largos de los cromosomas 15 y 17, t(15;17)(q22;q11.2-q12) que producen la
proteína RARα, la que únicamente puede responder a ácido retinoico (ATRA).
[]Como consecuencia de esta translocación se produce una fusión del gen situado en el locus 15q22
(que recibe el nombre de gen PML - iniciales de ProMyelocytic Leukaemia-) con el gen para el ácido
retinoico (RAR), este último localizado en 17q12-21. De esta manera, se forma un gen híbrido
PML-RAR, que está presente en la mayoría de los casos de leucemia promielocítica aguda y su
recíproco, RAR-PML que se presenta en el 60% de los casos.
1.1.-Incidencia
Alrededor de 50 niños, de un total estimado de 500 a los que se les diagnostica LMA cada
año en los Estados Unidos, se diagnosticar con LPA.
La LPA representa alrededor del 1% de la leucemia infantil.
La LPA se encuentra con mayor frecuencia en los niños de entre dos y tres años; no
obstante, también se observa en niños mayores de esa edad, y en adolescentes.
1.2.-Caracteristicas morfológicas e inmunológicas
Se caracteriza por promielocitos (una forma de glóbulos blancos) anormales, fuertemente
granulados. La LPA favorece una acumulación de estos promielocitos atípicos en la médula ósea y
la sangre periférica, y reemplaza los glóbulos normales.
Se conocen dos variantes morfológicas de la leucemia promielocítica aguda, la APL hipergranular
que representa la mayor parte de los casos y la variante microgranular denominada M3v que
supone el 15-20% de los casos.
La forma clásica se caracteriza por la presencia de promielocitos patológicos con abundante
coloración azurófila. Los núcleos tienen una forma irregular, presentando escotaduras o
lobulaciones. Es característica la presencia de abundantes cuerpos de Auer formando astillas
apiladas. En muchas ocasiones, la simple observación de los mismos con el microscopio óptico es
suficiente para el diagnóstico. La variante microgranular presenta las mismas caracteristicas de la
forma de los núcleos pero los gránulos son muy pequeños y no se pueden siempre ver con el
microscopio óptico. Esta variante suele presentarse acompañada de hiperleucocitosis.
B.3) DUPLICACIONES
Cuando se encuentra cualquier parte del material genético -un locus o un fragmento grande de un
cromosoma- más de una vez en el genoma, nos referimos a ellos como una duplicación.

22
Duplicación en Tándem: La región duplicada se encuentra inmediatamente adyacente al segmento
original y en el mismo orden que la región original.
Duplicación Desplazada: Si el segmento duplicado se localiza a cierta distancia del segmento
original, sea en el mismo cromosoma o en un cromosoma diferente.
Duplicaron Inversa: Se da cuando el segmento duplicado gira en 180 grados.
En realidad las secuencias genéticas, no se alteran por las duplicaciones y no hay pérdida de
información genética; el único cambio que se produce es la presencia de copias adicionales de
secuencias normales.
Si bien las duplicaciones pueden tener consecuencias graves cuando el equilibrio preciso de un
producto génico es esencial para la función celular, con frecuencia se originan duplicaciones
durante la evolución de numerosos organismos eucariontes y constituyen una fuente de nuevos
genes que pueden proporcionar nuevas funciones.
B.4) INVERSIONES:
Es la rotura por 2 partes de un cromosoma, obteniendo un fragmento que si llega a rotar 180º,
puede cambiar de sentido y volver a unirse a la estructura. Es importante hacer notar que, por la
naturaleza antiparalela de las hélices, aparte de rotar 180º en horizontal, debe girar otros 180º en
perpendicular, para restablecer la polaridad entre las dos cadenas.

23Podemos considerar las inversiones como un mecanismo supresor de la recombinación
genética, pues los individuos heterocigotos para una inversión, suelen tener problemas mecánicos
para aparear en la región de la inversión; esto reduce la frecuencia de entrecruzamiento y, por
consiguiente, la frecuencia de recombinación en la región.
Cuando se produce una inversión, la combinación genética presente en los loci incluidos en la
misma, presentan una fuerte tendencia a mantenerse constante, constituyendo un supergen; un
grupo de genes ligados que tienden a ser transmitidos como una unidad hereditaria y que se
mantienen juntos en el cromosoma.
Las inversiones suelen estar presentes en los cariotipos de los humanos, en aproximadamente un
2% de los casos, de forma que 2 de 100 individuos aproximadamente, pueden sufrir inversiones.
C) MUTACIONES GENÓMICAS
1. Anomalías Cromosómicas Autosómicas Numéricas
Nos referimos con anomalías cromosómicas autosómicas a aquellas alteraciones en el número
de copias de alguno de los cromosomas no sexuales. En humanos, no todas las aneuploidias
numéricas son viables, pero existen y generan alteraciones en el fenotipo de los humanos.
Entre las más frecuentes destacan:
Trisomía del cromosoma 21 más conocida como Síndrome de Down
Trisomía del cromosoma 18 más conocida como Síndrome de Edwards
Trisomía del cromosoma 13 más conocida como Síndrome de Patau
SÍNDROME DE DOWN
El síndrome de Down (SD) es un trastorno genético causado por la presencia de una copia extra
del cromosoma 21 (o una parte del mismo), en vez de los dos habituales (trisomía del par 21),
caracterizado por la presencia de un grado variable de retraso mental y unos rasgos físicos
peculiares que le dan un aspecto reconocible. Es la causa más frecuente de discapacidad psíquica
congénita y debe su nombre a John Langdon Haydon Down que fue el primero en describir esta
alteración genética en 1866, aunque nunca llegó a descubrir las causas que la producían. En julio
de 1958 un joven investigador llamado Jérôme Lejeune descubrió que el síndrome es una
alteración en el mencionado par de cromosomas. No se conocen con exactitud las causas que
provocan el exceso cromosómico, aunque se relaciona estadísticamente con una edad materna
superior a los 35 años. Las personas con Síndrome de Down tienen una probabilidad algo superior a
la de la población general de padecer algunas patologías, especialmente de corazón, sistema
digestivo y sistema endocrino, debido al exceso de proteínas sintetizadas por el cromosoma de
más. Los avances actuales en el descifrado del genoma humano están desvelando algunos de los
procesos bioquímicos subyacentes al retraso mental, pero en la actualidad no existe ningún
tratamiento farmacológico que haya demostrado mejorar las capacidades intelectuales de estas
personas.
INCIDENCIA
El SD ocurre con una frecuencia de alrededor de 1 en 700 recién nacidos vivos (RNV) y 1 en 150
concepciones, con una estimada relación varón/mujer al nacimiento de 1,5. Al igual que otras

24anomalías cromosómicas, las concepciones con T21 son altamente inviables y alrededor del 80%
abortan espontáneamente.
A semejanza de lo que ocurre con las anomalías cromosómicas en general, el nacimiento de un
niño con SD es un hecho esporádico dentro de una familia, ya que sólo una minoría de los casos es
de origen familiar. El riesgo de ocurrencia de acuerdo a la edad materna es entre los 15 y los 24
años.
Actualmente, la frecuencia de las anomalías cromosómicas en los RNV está cambiando; entre otras
razones, por el impacto de las técnicas de detección prenatal y la estructura de la edad materna en
diferentes poblaciones.
SIGNOS Y SÍNTOMAS
Los síntomas del síndrome de Down varían de una persona a otra y
pueden ir de leves a severos. Sin embargo, los niños con síndrome
de Down tienen una apariencia característica ampliamente
reconocida.
La expresividad de los rasgos propios del síndrome es sumamente amplia y difiere de un sujeto a
otro por la interacción compleja entre factores genéticos
intrínsecos y medioambientales.
Los signos físicos comunes abarcan:
Disminución del tono muscular al nacer
Exceso de piel en la nuca
Nariz achatada
Suturas separadas (articulaciones entre los huesos del
cráneo)
Pliegue único en la palma de la mano
Orejas pequeñas
Boca pequeña
Ojos inclinados hacia arriba
Manos cortas y anchas con dedos cortos
Manchas blancas en la parte coloreada del ojo (manchas de Brushfield)

25
Los principales síntomas son:
Cabeza anormalmente grande, pequeña
o deformada
Ojos, cara u otras partes del cuerpo de
aspecto raro
Mano cortas, anchas, posiblemente con sólo
un pliegue en la palma; dedos cortos,
posiblemente con una articulación
A pesar de que hay más de 50 síntomas reconocidos del síndrome de Down, es raro encontrar una
persona con todos o una gran cantidad de éstos.
Los únicos rasgos presentes en todos los casos son la atonía muscular generalizada (falta de un
tono muscular adecuado, lo que dificulta el aprendizaje motriz) y el retraso mental aunque en
grados muy variables.

26Los individuos con síndrome de Down típicamente son más pequeños que sus compañeros
normales, y su desarrollo físico e intelectual es más lento. Aparte de un distintivo aspecto físico, los
niños con síndrome de Down frecuentemente experimentan problemas relacionados a la salud.
Muchas afecciones médicas diferentes se observan en los bebés nacidos con síndrome de Down,
incluyendo:
Anomalías congénitas que comprometen el corazón, como la comunicación interauricular y la
comunicación interventricular
Problemas de los ojos como cataratas
Obstrucciones gastrointestinales como atresia esofágica y atresia duodenal
Problemas auditivos
Dislocación de la cadera
Apnea del sueño
Actividad deficiente de la tiroides (hipotiroidismo)
SÍNDROME DE EDWARDS
Consiguió su nombre después del doctor famoso, el Dr. Juan Edward, descrita en 1960. Es un
desorden cromosómico genético causado por un error en la división de célula resultando en el
tercer cromosoma adicional 18. El síndrome, un resultado de uno de los desórdenes genéticos y
más el campo común de Edward después abajo del síndrome, ocurre en aproximadamente uno
entre 3000 a 6000 nacimientos.
Las células del cuerpo humano contienen 23 pares de los cromosomas que heredan de sus padres.
Las células reproductivas humanas, las células de la esperma en varón y el ovum en hembras cada
uno tienen 23 cromosomas individuales, conocidos como XX en hembras y XY en varones y
numeran uno a 22. El material adicional del cromosoma 18 obtuvo después de que la fertilización
del huevo sea responsable de causar el síndrome de Edward.

27
Ideograma de una célula humana de una persona con Síndrome de Edwars, trisomía del par 18.
INCIDENCIAS
Su frecuencia se calcula entre 1/6000-1/13000 nacidos vivos. Se da en todas las razas y zonas
geográficas. Es la segunda trisomía en orden de frecuencia, la trisomía 18 aumenta su incidencia
con la edad materna avanzada y muestra una tasa de letalidad intrauterina mayor.
SIGNOS Y SÍNTOMAS
- Retraso de crecimiento pre y postnatal(Peso medio al nacer: 2340 g)
- Panículo adiposo y masa muscular escasa al nacer
- Hipotonía inicial que evoluciona a hipertonía
- Orejas de implantación baja
- Craneofacial: microcefalia, fontanelas amplias, occipucio prominente con diámetro bifrontal
estrecho, defectos oculares (opacidad corneal, catarata, microftalmía, coloboma de iris), fisuras
palpebrales cortas, orejas displásicas de implantación baja, micrognatia, boca pequeña, paladar
ojival, labio/paladar hendido
- Extremidades: mano trisómica (posición de las manos característica con tendencia a puños
cerrados, con dificultad para abrirlos, y con el segundo dedo montado sobre el tercero y el
quinto sobre el cuarto), uñas de manos y pies hipoplásicas, limitación a la extensión (>45º) de
las caderas, talón prominente con primer dedo del pie corto y en dorsiflexión, hipoplasia/
aplasia radial, sindactilia 2º-3er dedos del pie, pies zambos
- Tórax-Abdomen: mamilas hipoplásicas, hernia umbilical y/ó inguinal, espacio intermamilar
aumentado, onfalocele
- Urogenital: testes no descendidos, hipoplasia labios mayores con clítoris prominente,
malformaciones uterinas, hipospadias, escroto bífido
- Malformaciones renourológicas: riñón en herradura, ectopia renal, hidronefrosis, duplicidad
ureteral, riñón poliquístico

28- Cardiovascular: cardiopatía congénita presente en 90% de casos (comunicación interventricular
con afectación valvular múltiple, conducto arterioso persistente, estenosis pulmonar, coartación
de aorta, transposición de grandes arterias, tetralogía de Fallot, arteria coronaria anómala)
- Tracto gastrointestinal: divertículo de Meckel, páncreas ectópico, fijación incompleta del colon,
ano anterior, atresia anal
- Sistema Nervioso Central: hipoplasia/ Aplasia de cuerpo calloso, agenesia de septum
pellucidum, circunvoluciones cerebrales anómalas, hidrocefalia, espina bífida
- Piel: cutis marmorata, hirsutismo en espalda y frente
- Signos radiológicos: esternón corto con núcleos de osificación reducidos, pelvis pequeñas,
caderas luxadas
Los dedos de manos y pies pueden fusionarse (sindactilia) o estar unidos entre sí por membranas
(membranas interdigitales) que se extienden hacia los mismos dedos. La sindactilia a menudo se
presenta entre el segundo y tercer dedos de los pies y por lo general se asocia con algún síndrome.

29
Pie en mecedora en un feto afecto de trisomía 18

30
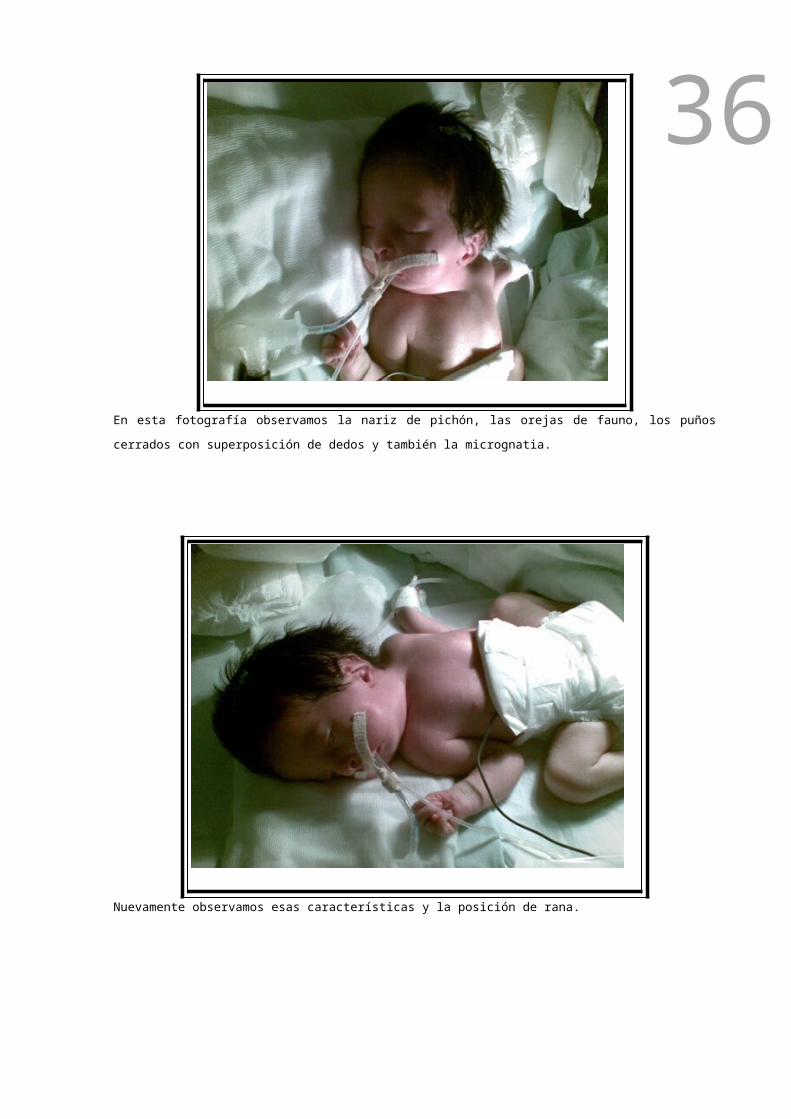
En esta fotografía observamos la nariz de pichón, las orejas de fauno, los puños cerrados con
superposición de dedos y también la micrognatia.
Nuevamente observamos esas características y la posición de rana.

31
Observamos la superposición de dedos en la extremidad inferior también. El pronóstico para estos
niños es muy malo, generalmente no sobreviven por arriba de los 3 meses de vida. Debido a fallo
de múltiples órganos.
SÍNDROME DE PATAU
El síndrome de Patau es una enfermedad congénita originada por una trisomía del cromosoma 13.
La supervivencia es escasa, pocos llegan a cumplir el año de vida y junto a las distintas
malformaciones siempre hay un retraso psicomotor severo.
Ideograma de una célula humana de una persona con Síndrome de Patau, trisomía del par 13.

32INCIDENCIAS
El síndrome de Patau es la tercera cromosomopatía en orden de frecuencia y a su vez la más letal
de las trisomías autosómicas viables
Actualmente la trisomía 13 se presenta en aproximadamente 1 de cada 5.000 nacidos vivos y es un
síndrome con múltiples anomalías, muchas de las cuales no son compatibles con la vida. De hecho,
más del 80% de los niños con trisomía 13 mueren en el primer mes. Predomina ligeramente en el
sexo femenino.
Supervivencia: similar a la de la trisomía 18.
La edad promedio de los padres de los niños afectados es mayor que la de los padres de los niños
normales, pero no tan alta como la edad promedio materna en los casos de síndrome de Down.
Ambos padres tienen una edad promedio de 32 años cuando nace el niño afectado.
Debido a que este caso es muy raro no se sabe si el origen del cromosoma extra es más a menudo
de origen materno o paterno, o si surge de ambos padres con igual frecuencia.
SIGNOS Y SÍNTOMAS
En el recién nacido, las primeras malformaciones que se observan suelen ser el labio leporino (con
o sin fisura palatina), el onfalocele, y la antes mencionada holoprosencefalia. También son
habituales la polidactilia, la criptorquidia (en varones, testículo no descendido) y la arteria umbilical
única. Además de las mencionadas pueden asociarse otras malformaciones con menor frecuencia.
El retraso psicomotor grave se hace patente ya desde los primeros meses de vida. Normalmente no
llegan a adquirir las funciones básicas (sedestación, marcha, lenguaje, etc.). Y el cociente
intelectual ya de por sí bajo continua disminuyendo con la edad. Pese a todo en ocasiones llegan a
aprender algunas capacidades como la marcha o pedir sus necesidades.
Síntomas
o Retraso mental severo
o Convulsiones
o Cabeza pequeña (microcefalia)
o Defectos del cuero cabelludo (piel ausente)
o Ojos pequeños (microftalmia)
o Labio leporino y/o paladar hendido
o Ojos muy juntos (hipotelorismo); los ojos pueden realmente fusionarse en uno
o Defectos del iris (coloboma)
o Anomalías del pabellón de la oreja y orejas de implantación baja
o Pliegue simiano
o Polidactilia
o Uñas hiperconvexas

33o Calcáneo prominente
o Hernias: hernia umbilical, hernia inguinal
o Criptorquidia: testículo no descendido
o Útero bicorne(útero de forma anormal, que adopta forma de dos cuernos)
o Hidronefrosis (acumulo anormal de orina en los riñones) y riñón poliquístico
o Hipotonía
o Micrognatia
o Anomalías esqueléticas (extremidades)
Signos
o El bebé puede presentar una arteria umbilical única al nacer y existen a menudo signos de
enfermedad cardíaca congénita:
o Defecto del tabique ventricular (VSD)
o Defecto del tabique auricular (ASD)
o Conducto arterioso persistente (PDA)
o Ubicación anormal del corazón (dextroversión o ubicación del corazón hacia el lado derecho
del pecho, en vez del izquierdo)

34
PolidactiliaSe observa en la paciente una micrognatia como
única malformación craneofacial presente en la
evaluación inicial. Posteriormente se describió
talipes y pliegue palmar único.
2. Anomalías Cromosómicas Sexuales Numéricas
Nos referimos a aquellas alteraciones en el número de copia de alguno de los dos cromosomas
sexuales humanos. Las aneuploidias en este caso suelen ser viables. Entre las más frecuentes
destacan:

35 Síndrome de Klinefelter (trisomía de los cromosomas sexuales: 47,XXY)
Síndrome de Turner (monosomía de los cromosomas sexuales: 45,X)
Síndrome del doble Y (llamado a veces síndrome del supermacho: 47,XYY)
Síndrome del triple X (llamado a veces síndrome de la superhembra: 47,XXX)
SÍNDROME DE KLINEFELTER
Es la presencia de un cromosoma X extra en un hombre, éste síndrome asociado con el cariotipo
47, XXY fue descrito por Harry Klinefelter en 1942. Los varones con este síndrome tienden hacer
más altos que la media, con brazos y piernas desproporcionalmente largos. En los adolescentes y
adultos los niveles de testosterona son bajos, aunque hay un desarrollo masculino, el desarrollo
sexual femenino no está totalmente suprimido, por ejemplo la ginecomastia (desarrollo de las
mamas) se observa en alrededor de un tercio de los varones afectados. Éste desarrollo sexual
ambiguo se denomina intersexual, y puede dar lugar a un desarrollo social anormal Típicamente
en los varones después de la pubertad es bello corporal es escaso y la masa corporal suele estar
disminuida. Además existe la predisposición a las alteraciones del aprendizaje y a una reducción
en el CI verbal.
El cromosoma extra deriva de la madre en el 50 % de los pacientes y la incidencia aumenta con el
aumento de la edad de la materna, también se calcula que al menos de la mitad de las
fecundaciones 47, XXY sufren aborto espontáneo.
También se han comunicado individuos con cariotipos 48, XXXY; 48, XXYY; 49, XXXXY; 49,
XXXYY; los cuales son similares fenotípicamente al 47, XXY. Dado que tienen un cromosoma Y ,
todavía presentan un fenotipo masculino, pero el grado de retraso mental y las malformaciones
físicas aumentan con cada cromosoma adicional.
SÍNTOMAS
El síntoma más común es la infertilidad. Otros síntomas pueden abarcar:
Pene pequeño
Testículos pequeños ( < 10 ml ) y firmes
Atrofia de los túbulos seminiferos
Vello púbico, axilar y facial escaso
Problemas sexuales
Agrandamiento de las mamas (ginecomastia)
Estatura alta
Proporciones corporales anormales (piernas largas, tronco corto)
SIGNOS Y EXÁMENES
Los adultos pueden acudir al médico debido a la infertilidad, y a los niños en edad escolar se los
puede llevar y evaluar las dificultades de aprendizaje.
Se pueden encontrar los siguientes resultados en los exámenes:
36 Cariotipo que muestra 47 XXY
Conteo de semen bajo
Nivel de testosterona en suero bajo
Nivel de hormona luteinizante en suero alto
Nivel de hormona foliculoestimulante en suero alto
Niveles de estradiol en suero (un tipo de estrógeno) alto
SINDROME DE TURNER
Trastorno de la diferenciación sexual, derivado de la ausencia de un cromosoma X. El resultado es
un fenotipo (características externas debidas a la interacción de una determinada carga genética
con los factores ambientales) femenino. A esta anomalía cromosómica también se la denomina
disgenesia gonadal y está presente en aproximadamente 1 de cada 2.500-3.000 nacimientos de
niñas vivas.
Las gónadas están representadas por unas cintillas fibrosas, en las que es difícil observar folículos.
Clínicamente se caracteriza por la presencia de una amenorrea primaria (ausencia de
menstruación), infantilismo sexual, talla corta y anomalías congénitas múltiples. Normalmente el
diagnóstico se establece en la pubertad ante la ausencia de desarrollo sexual. En otras ocasiones,
son otras anomalías asociadas al cuadro las que permiten el diagnóstico en el momento del
nacimiento.
En 1959 se determinaron, independientemente, cariotipos de individuos con estos síndromes y se
vio que eran anormales en relación con los cromosomas sexuales.los individuos con el síndrome de
klinefelter era muy a menudo triso micos y tenían, además de los autosomas, un complemento
XXY , las personas con este cariotipo se designan 47,XXY.los individuos con el síndrome de Turner
son, muy a menudo, monosomicos y tienen solo 45 cromosomas, incluido un único cromosoma X.
se designan 45,X Advierta la convención utilizada para designar las constituciones cromosómicas
anteriores. EL número indica cuantos cromosomas hay y la información después de la coma indica
la desviación respecto del contenido diploide normal. Ambas situaciones son consecuencia de una
no disyunción de los cromosomas X en la meiosis. Estos cariotipos y sus correspondientes fenotipos
sexuales nos permiten concluir con el cromosoma y determina la masculinidad en la especie
humana. En ausencia del Y, el sexo del individuo es femenino, incluso si solo hay presente un
cromosoma x. la presencia del cromosoma Y el individuo con el síndrome del klinefelter es un
suficiente para determinar su masculinidad, aun cuando su expresión no sea completa.
Igualmente, en la ausencia de un cromosoma Y, como el caso de los individuos con el síndrome de
Turner, no sea da la masculinización.
Cariotipos distintos al 45,X también dan lugar al síndrome de Turner. Incluyen individuos que
presentan en sus tejidos dos líneas celulares genéticamente diferentes, cada una con un cariotipo
distinto tales individuos se denominan mosaicos.las dos líneas celulares son el resultado de errores
en la mitosis en las primeras etapas del desarrollo, siendo las combinaciones cromosómicas mas
corrientes45,X /46,XY y 45,X /46XX.Asi un embrión que comienza su desarrollo con un cariotipo
normal puede dar lugar a un individuo cuya célula presenten una mescla de cariotipo y expresen
este síndrome .

37El síndrome de Turner se observa en uno de cada 3.000 nacimientos femeninos, una secuencia
mucho mas baja que el síndrome klinifelder. Una explicación de esta diferencia es el hecho de que
una proporción importante de fetos 45,x muere en el útero y se abortan espontáneamente. Por ello
podría darse una frecuencia similar de los dos síndromes en el momento de la fecundación
SÍNDROME DEL XYY
El síndrome XYY (también llamado síndrome del superhombre, entre otros nombres) es una
anomalía (específicamente una trisomía) de los cromosomas sexuales donde el hombre recibe un
cromosoma Y extra, produciendo el cariotipo 47,XYY.
Algunos médicos genetistas cuestionan si el uso del término "síndrome" es apropiado para ésta
condición, porque el fenotipo es normal, ya que la gran mayoría (estimando que un 97% en el
Reino Unido) de hombres con 47,XYY no conocen su cariotipo
.
[][]Rasgos físicos
Con gran frecuencia, esta alteración cromosómica no causa características físicas inusuales o
problemas médicos. Los jóvenes y adultos con 47,XYY son regularmente algunos centímetros más
altos que sus padres y hermanos.
En muy pocos casos se ha reportado acné severo, pero dermatólogos especialistas en este campo
manifiestan que no existe evidencia que se relacione con 47,XYY.[]
Los niveles de testosterona (prenatal y postnatal) son normales en hombres con 47,XYY. []La
mayoría de los hombres con 47,XYY tienen un desarrollo sexual normal y por lo regular son fértiles.

38El XYY no ha sido identificado por las características físicas, la condición es usualmente detectada
sólo durante el análisis genético, solicitado por razones distintas.
Comportamiento característico
Los jóvenes con 47,XYY tienen mayor riesgo de padecer problemas de aprendizaje (por encima del
50%) y retardo en el desarrollo del lenguaje[][][][]. [][][][]En este contexto, estudios reportan que el 10%
de todos los jóvenes tenían un problema de aprendizaje.[]
Como los niños con síndrome de Klinefelter (XXY) y las niñas con síndrome del triple X (XXX), la
puntuación de cociente intelectual de jóvenes con 47,XYY son en promedio 10–15 puntos por
debajo de sus hermanos. [][][][]Es importante resaltar que esta variación —tiene en promedio una
diferencia de 12 puntos de CI —y que ocurre normalmente entre niños de la misma familia. [] En 14
diagnósticos prenatales que arrojaron 47,XYY de niños de familias con alto estatus socioeconómico,
el CI disponible para 6 jóvenes tuvieron rangos entre 100–147 con una media de 120. Para 11
muchachos con sus hermanos, en 9 casos sus hermanos tenían mejor desempeño académico, en
un caso el desempeño fue igual y en otro caso superior a sus hermanos y hermanas.[]
El retraso en el desarrollo y los problemas de comportamiento también son posibles.[] []La agresión
no es vista frecuentemente en hombres con 47,XYY.[] [] [][][]
Causa
El 47,XYY no es heredable, pero usualmente ocurre como evento aleatorio durante la formación del
espermatozoide. Un error en la división celular durante la metafase II de la meiosis, llamada no
disyunción meiótica, puede dar como resultado un espermatozoide con una copia extra del
cromosoma Y. Si uno de estos espermatozoides atípicos contribuye a la formación genética del
niño, éste tendrá un cromosoma Y extra en cada célula de su cuerpo.[] []
En algunos casos, la adición del cromosoma Y extra resulta de la no disyunción durante la división
celular postcigótica (mitosis) en el desarrollo embrionario temprano. Esto puede producir
46,XY/47,XYY, es decir, mosaico genético (mosaicismo).[] []

39Incidencia
Cerca de 1 de cada 1.000 niños nacen con cariotipo 47,XYY. La incidencia de 47,XYY no es afectada
por la edad avanzada paternal ni maternal.[][][]
SÍNDROME 47, XXX
La presencia de tres cromosomas X junto con la dotación normal de autosomas (47,xxx)da lugar a
diferenciación femenina .Este síndrome, que se estima ocurre en 1de cada 1.200 nacimientos
femeninos ,es muy variable en su expresión. Frecuentemente las mujeres47, XXX son
perfectamente normales. En otros casos pueden darse un menor desarrollo de las características
sexuales segundarias, esterilidad y retraso mental en caso s raros se han encontrado cariotipos 48,
XXXX y 49, XXXXX.los síndromes asociados con estos cariotipos son similares al del 47,xxxx pero
más pronunciados. Así en muchos casos, la presencia de cromosomas X extras parece romper el
delicado equilibrio de la información genética esencial para un desarrollo femenino normal.
Los individuos con este síndrome son del sexo femenino con órganos genitales atrofiados y
fertilidad limitada y retaso mental frecuente
Las personas triple XXX son mujeres fenotípicamente normales y fértiles, sus meiosis son las de
una mujer normal XX produciendo gametos que llevan un solo x
DISOMÍAS
Una disomía es la presencia de un par de cromosomas. En los organismos diploides como los
humanos, esta es la condición normal. Para aquellos organismos haploides (como los gametos
humanos), triploides o poliploides, la disomía constituye una aneuploidía. En la disomía uniparental,
la disomía hace referencia a que ambas copias del cromosoma provienen únicamente de uno de los
progenitores.
SÍNDROME DE PRADER - WILLI
Se trata de un desorden neuroendocrino descrito por primera vez en 1956 por Prader, Labhart y
Willi. Su incidencia global se sitúa entre un 1/10.000 y un 1/25.000 recién nacidos vivos.
Clínicamente, presenta una evolución clínica muy característica. Se trata de recién nacidos de
ambos sexos en cuyo embarazo la percepción de movimientos fetales se ha retrasado o no ha
existido.
A menudo son productos de un parto difícil con presentaciones fetales anormales. Durante los
dos primeros años de vida predomina la hipotonía muscular que conlleva a patología respiratoria
más o menos grave, a un llanto débil y a graves problemas en la alimentación; todo ello les origina
hospitalizaciones frecuentes. Posteriormente, el tono muscular de estos pacientes va mejorando,
evidenciándose entonces su sintomatología clínica más florida, consistente en una obesidad
progresiva que se acompaña de una actitud compulsiva frente al alimento, retraso mental variable,
trastornos en el aprendizaje y en la articulación del lenguaje y un hipogonadismo normalmente
hipogonadotropo.

40Síntomas
Este síndrome altera el funcionamiento del hipotálamo, una sección del diencéfalo cuyas funciones
incluyen, entre otras, el control del apetito: carecen de sensación de saciedad. La observación
clínica y algunos trabajos de investigación, han demostrado una diferencia entre “sensación de
hambre” y “falta de saciedad”. Un error muy común es pensar que la búsqueda incesante de
“comida” se debe a un “hambre excesiva”. La alimentación de las personas con SPW necesita estar
supervisada constantemente, además de seguir una estricta dieta.
Para agravar el problema del control alimenticio, el síndrome también provoca deficiencia del tono
muscular, un alto porcentaje de grasa en el organismo y falta de energía. Todas estas condiciones
reducen las necesidades calóricas de los niños y adultos que tienen este síndrome, a dos tercios de
la necesidad calórica estándar.
Si bien el trastorno alimenticio es el síntoma más evidente y el que demanda más tiempo, además
de su mayor riesgo vital, es sólo un aspecto de esta compleja dolencia. Al principio, los bebés que
tienen este síndrome se alimentan deficientemente y no aumentan de peso, ya que la debilidad de
su tono muscular reduce su capacidad de succión.
Causas
En el origen del SPW se han constatado varios mecanismos genéticos de origen desconocido. En la
década de los 80 fue objetivado su origen genético y en la actualidad se sabe que el 70% de los
casos consiste en una ausencia genes (deleción del brazo largo del cromosoma 15 paterno 15q 11-
13, 1981), un 30% de casos presentan la herencia de dos copias de origen materno (disomia
uniparetal materna, 1989). Puede existir asimismo en 1 al 2% de los casos un defecto en el
imprinting o una reorganización cromosómica en forma de translocaciones o inversiones (alteración
de la impronta o imprinting). En 1993, Holm y col. establecieron los criterios de diagnóstico clínico
del SPW para recomendar el test genético. En 2001, en Pediatrics, se publicó una revisión
consensuada por varios especialistas de dichos criterios.
SÍNDROME DE ANGELMAN
El síndrome de Angelman fue descrito por este autor en 1965 y presenta una incidencia de
1/20.000 recién nacidos vivos. Clínicamente, son individuos con retraso mental grave, epilepsia
severa y que presentan de forma muy característica episodios paroxísticos de risa, ausencia del
lenguaje y microcefalia.
El síndrome de Prader-Willi y el de Angelman han constituido en los últimos años el ejemplo más
claro de “imprinting” genómico en procesos neurológicos y en los que la falta de la misma región
cromosómica del cromosoma 15 se manifestaba como dos situaciones fenotípicas muy diferentes,
dependiendo de la procedencia materna o paterna de la alteración genética. Mientras que en el
primero la falta de información genética es de origen paterno, en el síndrome de Angelman se trata
de una pérdida de información de genes que se expresan habitualmente en el cromosoma 15
materno.

41TETRASOMÍAS Y PENTASOMÍAS
Tetrasomía y pentasomía hacen referencia a la presencia en una célula de cuatro o cinco copias de
un cromosoma, respectivamente. Son casos extremadamente raros pero han sido documentados
diversos casos en humanos que presentaban los siguientes cariotipos: XXXX (síndrome XXXX),
XXYY, XXXY, XYYY, XXXXX, XXXXY, XXXYY, XYYYY, XXYYY.
Los sindromes denominados Tetrasomia X y Pentasomia X son llamados también: 48, XXXX; 49,
XXXXX; 48xxxx; 49xxxx; Tetra X, Penta X, XXXX, XXXXX, o Cuadruple X.
El ser humano normal tiene 23 pares de cromosomas, o sea un total de 46 cromosomas. El último
par se denomina cromosoma sexual que en la mujer consta de dos cromosomas X. En cambio en el
Hombre hay uno X y otro Y.
En los Sindromes Tetra X, las mujeres tienen cuatro cromosomas X en lugar de dos. Así, en lugar de
un total de 46 hay 48.
En el caso del Sindrom Penta, hay cinco cromosomas, total 49, producto de cinco cromosomas X.
Las Tetra y Pentasomia X ocurren solo en mujeres.
Hay aproximadamente 60 casos conocidos en todo el mundo, con esta rara condicion. Se cree que
hay otro más pero sin diagnosticar. Esta condicion fue identificada por primera vez en 1961.
Los síntomas pueden ser relativamente menores o bien todo lo contrario muy severos.
Ellos pueden incluir desafíos en el aprendizaje, dificultades sociales, defectos cardiacos congenitos
y varios trastornos fisicos comunes a muchos sindromes.
Se ha notado que las niñas afectas de sindromes tetra/penta X van al colegio, disfrutan deportes y
desarrollan relaciones personales duraderas.
Características y desarrollo fisico:
Este resumen de ninguna manera es un análisis exhaustivo de todas las características asociadas
con éste diagnóstico. Puede haber manifestaciones menos severas que no estén incluidas.
La información siguiente ha sido recopilada de los casos de 16 familias con hijas con tetra o penta
X.
· LEUCEMIA NEUTROFÍLICA CRÓNICA CON TETRASOMÍA 8*
La leucemia neutrofílica crónica es un desorden mieloproliferativo poco frecuente (sólo se han
descrito 143 casos) que se caracteriza por esplenomegalia, leve leucocitosis, marcada neutrofilia
sin basofilia, cromosoma Filadelfia BCR/ABL negativos y anormalidades cromosómicas hasta en el
10% de los casos; en el caso clínico descrito se encontró tetrasomía 8. El diagnóstico diferencial de
la leucemia neutrofílica crónica es difícil e incluye la reacción leucemioide, la leucemia mieloide
crónica y la policitemia vera.

42_____________________________________
(*) En junio de 1997 a junio de 2003, en el Instituto Nacional de Cancerología se
diagnosticaronn318 casos de leucemia mieloide crónica. Entre los pacientes afectados se encontró
uno que cumplió con todos los criterios clínicos, genéticos y patológicos para ser clasificado como
víctima de una leucemia neutrofílica crónica.
III. BIBLIOGRAFIA
ROBBINS, ‘‘Patología funcional y estructural’’, Editorial Mcgraw-Hill sexta edición, Cap. 6
páginas 150-192
DE ROBERTIS, Eduardo ‘‘Biología celular y molecular’’, Editorial el ateneo, duodécima
edición, Pág. 307-425
PIERCE, Benjamín A., “Genética, un enfoque conceptual”, Editorial médica Panamericana,
Segunda Edición – 2006
CAREY, Jorde; WHITE, Bamshad, “Genética Médica” Editorial Elsevier, Tercera edición –
2005
GRIFFITHS, Anthony; MILLER, Jeffrey, “Genética” Editorial McGraw Hill Interamericana,
Sétima edición - 2002


