Embed Size (px)
Citation preview

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 1/26
Epigenetic control of cellular
senescence in disease: opportunities
for therapeutic intervention
Stuart P. Atkinson and W. Nicol Keith*
Understanding how senescence is established and maintained is an important
area of study both for normal cell physiology and in tumourigenesis.
Modifications to N-terminal tails of histone proteins, which can lead to
chromatin remodelling, appear to be key to the regulation of the senescence
phenotype. Epigenetic mechanisms such as modification of histone proteinshave been shown to be sufficient to regulate gene expression levels and
specific gene promoters can become epigenetically altered at senescence.
This suggests that epigenetic mechanisms are important in senescence
and further suggests epigenetic deregulation could play an important role in
the bypass of senescence and the acquisition of a tumourigenic phenotype.
Tumour suppressor proteins and cellular senescence are intimately linked and
such proteins are now known to regulate gene expression through chromatin
remodelling, again suggesting a link between chromatin modification and
cellular senescence. Telomere dynamics and the expression of the telomerase
genes are also both implicitly linked to senescence and tumourigenesis, and
epigenetic deregulation of the telomerase gene promoters has been identified
as a possible mechanism for the activation of telomere maintenance
mechanisms in cancer. Recent studies have also suggested that epigenetic
deregulation in stem cells could play an important role in carcinogenesis, and
new models have been suggested for the attainment of tumourigenesis
and bypass of senescence. Overall, proper regulation of the chromatin
environment is suggested to have an important role in the senescence
pathway, such that its deregulation could lead to tumourigenesis.
Centre for Oncology and Applied Pharmacology, University of Glasgow, Cancer Research UKBeatson Laboratories, Bearsden, Glasgow, G61 1BD, UK.
*Corresponding author: W. Nicol Keith, Centre for Oncology and Applied Pharmacology,University of Glasgow, Cancer Research UK Beatson Laboratories, Alexander Stone Building,Garscube Estate, Switchback Rd, Bearsden, Glasgow, G61 1BD, UK. Tel: +44 (0)141 330 4811;Fax: +44 (0)141 330 4127; E-mail: [email protected]
expert reviewshttp://www.expertreviews.org/ in molecular medicine
1 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 2/26
Cellular senescence provides a barrier toexcessive cellular growth and is triggered bythree main mechanisms. Telomere attritionduring extended cell proliferation leadseventually to cellular senescence (Ref. 1) when
critically short telomeres are recognised asDNA damage, thereby triggering thesenescence arrest. Although this proliferative
barrier provides a limit to the outgrowth of acell, senescence can also be triggered before thislimit in response to cellular damage (such asDNA damage) or stress and oncogenic signalling.As senescent cells do not proliferate, it has beenproposed that cellular senescence is a major
barrier to cancerous transformation (Refs 2, 3)and premature induction of cellular senescencecan therefore be targeted for anti-cancer therapyin tumourigenic cells (Refs 4, 5) (Fig. 1).
The senescence phenotype is associated withmultiple cellular changes including alteredcellular morphology, size and, importantly,expression of numerous genes involved inmany aspects of cell physiology (Refs 6, 7, 8, 9,10). The altered regulation of genes and thecomplex phenotype suggest that epigeneticmechanisms may be involved. Indeed, it is nowapparent that, in addition to multiple geneticpathways, epigenetics plays a major contributingrole in senescence (Refs 11, 12) as well as in the
bypass of senescence leading to tumourigenicity(Refs 13, 14, 15). Therefore, to understand
senescence and tumourigenesis, both geneticand epigenetic mechanisms regulating cellularsenescence must be appreciated.
Epigenetic regulation of gene expressionEpigenetic mechanisms are defined as processesin which changes in gene function can occurwithout a change in the DNA sequence. Suchmechanisms include covalent modifications of histones, DNA methylation, ATP-dependentchromatin remodelling, incorporation of varianthistones, looping and changes in the local
conformation of DNA, and alterations of higher-order chromatin structure. It isunderstood that epigenetic processes such asDNA methylation and post-translationalmodification of histones are fundamental to theregulation of gene expression and therefore tothe regulation of multiple cellular processes.For these reasons it is plausible to suggest thatchanges in the chromatin environment can playa role in the changes in gene expression
involved in senescence and also in the bypassof senescence towards tumourigenesis.
The binding of DNA to histone proteins isrequired for efficient packaging of DNA intothe nucleus of the cell, but this tight binding
creates a barrier for factors that need access toDNA to regulate various cellular processes.Histone proteins bind together to form ahistone octamer comprising two copies each of histones H2A, H2B, H3 and H4 to which DNAwraps round. Each member of the octamer can
be post-translationally modified and onefunction of these modifications is to mediate
On-rampTelomerase
gene expression
O n - r a m p
N o t e l o m e r a s e a c t i v i t y
A L T p a t h w a y
r e c o m b i n a t i o n
Telomerase activity
Immortality
Senescence
E x i t r o u t e
E x i t r o u t e
Normal cell
NeoplasiaImmortality
Routes to immortality
Expert Reviews in Molecular Medicine
© 2007 Cambridge University Press
Figure 1. Routes to immortality. Cellular
senescence (forward and reverse exit routes)
provides a barrier to excessive cellular growth and
so is considered to be a tumour suppressor
mechanism. A normal cell can bypass senescence
and move towards neoplasia through
immortalisation by two means, or two different ‘on-
ramps’. First, cells can maintain their telomeres
through telomerase activation (horizontal on-ramp).
Second, cells can engage the alternative
lengthening of telomeres (ALT) pathway (vertical on-
ramp), which allows telomere elongation through
intra-telomeric recombination processes. The
induction of premature cellular senescence of
neoplastic cells (reverse exit route) may therefore be
a strategy for anti-cancer therapeutics.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
2 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 3/26
the binding of the histone proteins to DNAand therefore to regulate DNA access andthe transcriptional status of DNA therein.Modifications occur mainly on the histoneN-terminal tail, but also occur on the
histone globular domain, and includeacetylation, methylation, phosphorylation,SUMOylation (for ‘small ubiquitin-relatedmodifier’), ubiquitination and ADP ribosylation.It has been hypothesised that one of the roles of these modifications is to act as a ‘histone code’(Refs 16, 17) in which combinations of histonemodifications can be read as a code andtranslated into an output signal; thus, distinctpatterns of histones may be a signal for genetranscription to be turned on or off.Furthermore, levels of modifications mightfunction to regulate levels of transcription (and
therefore high or low levels of gene expression).In general, silenced or repressed chromatin
(heterochromatin) is characterised by hypo-acetylated histones (histones H3 and H4),methylation of residues Lys9 and Lys27 of histone H3 (K9 H3 and K27 H3) andmethylation of Lys20 of histone H4 (K20 H4).Active, de-repressed chromatin (euchromatin)is characterised by hyper-acetylated histonesand can have methylation of Lys4, Lys36and Lys79 of histone H3 (K4 H3, K36 H3 andK79 H3) (Refs 18, 19). In addition, DNAhypermethylation tends to be associated with
silent chromatin whereas hypomethylationgenerally associates with active chromatin.
Senescence, chromatin and diseaseTheinduction of premature senescence of tumourcells by chemotherapeutics is utilised in anti-cancer therapy. However, it is important torealise that cellular senescence contributes tothe ageing process, can lead to the dysfunctionof tissues such as the heart, lungs and brain(Ref. 20), and is also linked to the pathogenesisof multiple disease states (Table 1) such
as smoking-related lung disease, transplantrejection and liver cirrhosis. Interestingly, it hasalso been demonstrated that senescent cellsmay promote tumourigenesis by providinga permissive tissue microenvironment fortumourigenic cell growth (Refs 21, 22, 23).Therefore, although senescence is an important
barrier to cancer progression, this benefit might be offset by the contribution of cell arrest toother disease states in later life.
In addition, similarly deregulated epigeneticmechanisms have been shown to contributeto ageing, cardiovascular disease, multipleinherited disorders and also tumourigenesis(Table 2). Furthermore, the epigenetic basis of tumourigenicity is an intensely studied area(Refs 13, 14, 24, 25, 26) and it is now wellestablished that the deregulation of epigeneticmodifying enzymes, leading to deregulatedgene expression, plays an important role intumourigenesis (Ref. 14). Such studies have ledto the establishment of an epigenetic model of
cancer to complement, or even replace, thegenetic model for cancer and hypothesises thatepigenetic modifications can play a role intumour initiation and progression (Ref. 26).Another interesting model suggests thatepigenetics is at the very root of tumourigenicity,
by disrupting gene expression at the stem celllevel (Ref. 27). Epigenetic disruption is thereforean important mechanism in the key early eventsin tumourigenesis.
How regulation of the chromatinenvironment affects senescence
Heterochromatin formation and senescenceInitial studies previously established thatchromatin becomes reorganised with increasedpopulation doublings, with large-scale effects
being evident upon senescence (Ref. 28). It hasnow been shown that the formation of heterochromatin at specific foci plays asignificant role in senescence in response tooncogenic insult and telomere attrition, and thatthis senescence arrest as a cellular response to
Table 1. Senescence and disease
Senescence-associateddisease
Refs
Tumourigenesis 21, 22, 23
Wound healing 173Neurodegeneration 174, 175
Alzheimer’s disease 176
Chronic obstructive
pulmonary disease (COPD)
177
Emphysema 178
Kidney failure 179
Cardiovascular disease 180
Chronic inflammation 181, 182
Transplant rejection 183
Osteoarthritis 184
Liver cirrhosis 185
Atherosclerosis 172
expert reviewshttp://www.expertreviews.org/ in molecular medicine
3 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n
8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 4/26
stress might act as a tumour suppressor
mechanism (Refs 2, 3). In addition, it has also been observed that many genes upregulated atsenescence are actually physically clustered(Ref. 29), again indicating that re-organisation of chromatin domains are important in senescence.Therefore, formation of heterochromatin may beviewed as a tumour suppressor mechanism.
Early studies showed that expression of oncogenic Ras induced the accumulation of p16INK4a (also known as cyclin-dependent kinase
inhibitor 2A) and hypophosphorylated pRB
(retinoblastoma protein) with a senescence-likearrest in normal human fibroblasts (Ref. 30). Itwas also noted that this senescence-like arrestwas associated with the formation of distinctheterochromatic structures, which weredesignated senescence-associated heterochromatinfoci (SAHF) (Ref. 30). Importantly, SAHF wereshown to be formed by newly depositedheterochromatin rather than from redistributedheterochromatin, and were concentrated for
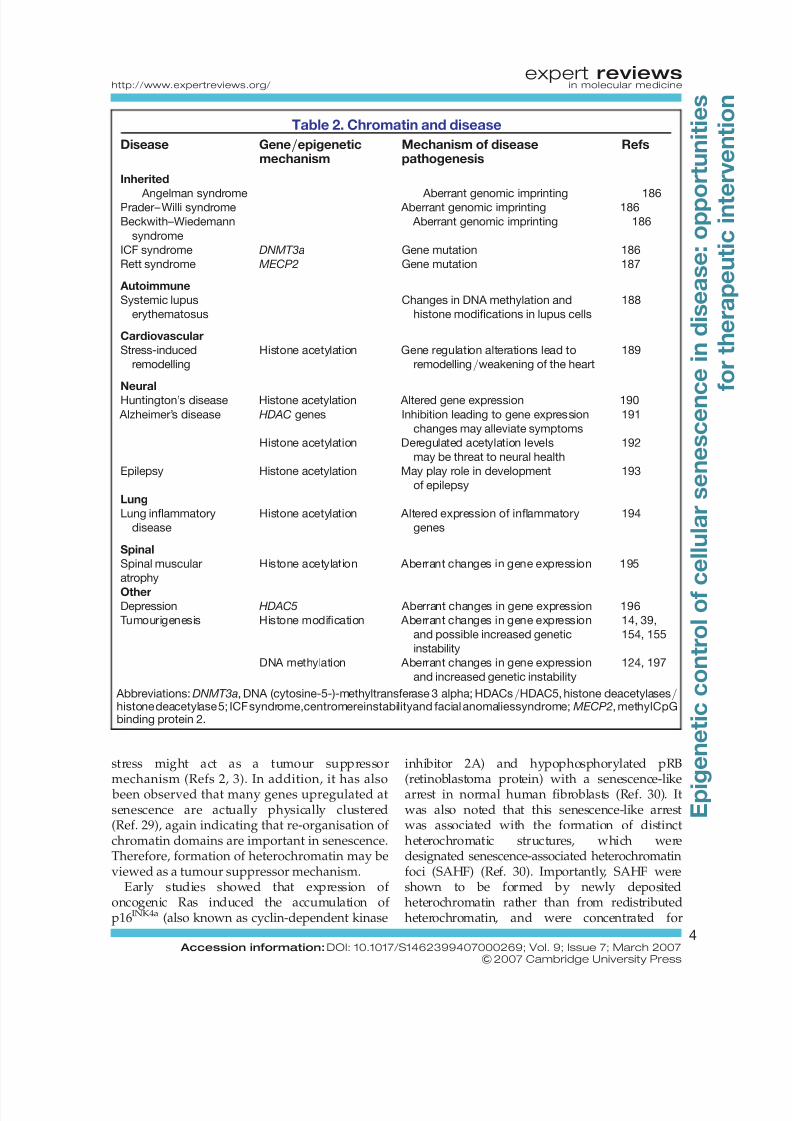
Table 2. Chromatin and disease
Disease Gene/epigeneticmechanism
Mechanism of diseasepathogenesis
Refs
Inherited
Angelman syndrome Aberrant genomic imprinting 186Prader– Willi syndrome Aberrant genomic imprinting 186
Beckwith–Wiedemann
syndrome
Aberrant genomic imprinting 186
ICF syndrome DNMT3a Gene mutation 186
Rett syndrome MECP2 Gene mutation 187
Autoimmune
Systemic lupus
erythematosus
Changes in DNA methylation and
histone modifications in lupus cells
188
Cardiovascular
Stress-induced
remodelling
Histone acetylation Gene regulation alterations lead to
remodelling/weakening of the heart
189
Neural
Huntington’s disease Histone acetylation Altered gene expression 190
Alzheimer’s disease HDAC genes Inhibition leading to gene expression
changes may alleviate symptoms
191
Histone acetylation Deregulated acetylation levels
may be threat to neural health
192
Epilepsy Histone acetylation May play role in development
of epilepsy
193
Lung
Lung inflammatory
disease
Histone acetylation Altered expression of inflammatory
genes
194
Spinal
Spinal muscular
atrophy
Histone acetylation Aberrant changes in gene expression 195
OtherDepression HDAC5 Aberrant changes in gene expression 196
Tumourigenesis Histone modification Aberrant changes in gene expression
and possible increased genetic
instability
14, 39,
154, 155
DNA methylation Aberrant changes in gene expression
and increased genetic instability
124, 197
Abbreviations:DNMT3a, DNA (cytosine-5-)-methyltransferase 3 alpha; HDACs/HDAC5, histone deacetylases/histone deacetylase 5; ICF syndrome,centromereinstabilityand facial anomaliessyndrome;MECP2, methylCpGbinding protein 2.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
4 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 5/26
repressive histone modifications such asmethylation of K9 H3 and heterochromatinprotein 1 (HP1), whereas permissivemodifications, such as methylated K4 H3 andacetylated K9 H3, were excluded (Fig. 2). It was
also established that active transcription was notoccurring at the sites of SAHF formation and thatchromatin features indicative of SAHF formed onE2F-responsive promoters of genes known to bedownregulated at senescence. E2F is atranscription factor important for regulation of genes associated with the cell cycle and was firstidentified as a cellular factor that had the ability
to bind the promoter for the gene encodingadenovirus E2. The genes that gainedheterochromatic features and becamedownregulated included the genes encodingcyclin A and proliferating cell nuclear antigen
(PCNA), and their promoters were bound by pRB,thereby linking pRB binding to the formation of SAHF. It is known that the upregulation of p16INK4a during senescence can inhibit thephosphorylation of pRB, causing it to bind to E2Fto cause a permanent growth arrest by silencinggrowth regulatory genes through altering thechromatin state of the promoter sequences. pRBhas been shown to interact with factors involvedin the formation of heterochromatin, includinghistone deacetylases (HDACs) (Refs 31, 32, 33),the K9 methyltransferase SUV39H1 (Ref. 34) andHP1 proteins (Ref. 35); thus, pRB binding to
regulatory sequences could be sufficient to formSAHF in order to silence gene expression.Importantly, the authors showed in the samestudy that promoters of genes known to beupregulated at senescence gained euchromaticpatterns at their promoters at the same time asSAHF were being formed on other promoters,
Figure 2. The formation of senescence-
associated heterochromatin foci (SAHF) in
response to oncogenic insult. In this model, (a)
an active gene promoter (e.g. cyclin D1) shows
methylation of Lys4 (K4) H3 and acetylation of
H3 and H4, and its chromatin has an open(euchromatic) structure. Under these conditions,
the transcription factor E2F mediates transcription,
whereas retinoblastoma protein (pRB) remains in a
phosphorylated state as a result of low p16INK4a
levels and therefore cannot bind to E2F to repress
transcription. (b) Upon oncogenic insult
(e.g. aberrant Ras signalling), pRB becomes
hypophosphorylated, mediated by increased levels
of p16INK4a. Hypophosphorylated pRB is
sequestered to the promoter along with several
other proteins, such as heterochromatin protein 1
(HP1), histone deacetylase (HDAC) and the Lys9
(K9) H3 methyltransferase SUV39H1, which can
bind pRB. (c) This leads to the silencing of gene
expression by altering the chromatin architecture
towards a heterochromatic state by promoting
K9 H3 methylation and removing/inhibiting
euchromatic modifications, as is observed in SAHF
formation. SAHF formation as a mode of gene
expression regulation in response to oncogenic
insult might lead to the onset of premature
senescence and might therefore inhibit
tumourigenic growth.
P
P
PP RB
RB
HP1
E2F
Openchromatinstructure
Closedchromatinstructure
Oncogenic insulte.g. aberrantRas signalling
Increased p16expression
a
b
c
Geneexpression
Generepression
E2F
SAHF formation
SUV39H1 HDAC
RB
HP1HP1
E2F
SUV39H1 HDAC
H3 and H4acetylation
K4 H3methylation
K9 H3methylation
The formation of senescence-associated heterochromatin foci(SAHF) in response to oncogenicinsult
Expert Reviews in Molecular Medicine
© 2007 Cambridge University Press
expert reviewshttp://www.expertreviews.org/ in molecular medicine
5 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 6/26
suggesting that the formation of SAHFs might bedirected towards certain genes and not anindiscriminate global effect. This indicates thatmodification of the chromatin environment of gene promoters is important for the stable
senescence-like arrest attained. It also suggests amodel in which SAHF formation accompanies thesenescence process and indicates a role for pRB-mediated heterochromatin formation at growthregulatory genes during senescence, leading tothe stable repression of these genes. From this itwas further proposed that failure of certainprocesses in these cells (e.g. mutations in genesencoding p16INK4a or pRB, or silencing events)could lead to bypass of senescence andprogression to cancer as a result of the inhibitionof SAHF formation (Ref. 30).
Several other papers have provided more
evidence of the link between chromatinremodelling and senescence. Braig et al. havedemonstrated that oncogenic Ras could inducea senescence-like arrest in mouse primarylymphocytes by a SUV39H1-dependent and K9H3 methylation-related mechanism (Ref. 36). Inthe same study, the deletion of SUV39H1 inprimary lymphocytes led to the impropermethylation of K9 H3 and the promotion of Ras-driven lymphomagenesis. This study alsocorrelates with other work demonstrating thatloss of methylation of K9 H3 and also K20 H4resulted in genomic instability in a variety of
normal cells and was associated with increasedtumour risk (Ref. 37). Decreased expression of SUV39H1 has also been observed in the earlypremalignant stage of tumour development inthe context of hepatocarcinogenesis induced bymethyl deficiency in rats, whereas loss of tri-methylation of K20 H4 was also observed at
both preneoplastic and tumour stages of livercancer and corresponded to a decrease in theK20 H4 histone methyltransferase, SUV420H2(Ref. 38). Interestingly, loss of tri-methylation of K20 H4, as well as loss of acetylated K16 H4
and DNA hypomethylation, has been shown to be a common epigenetic change in tumourigeniccells (i.e. an ‘epigenetic signature’ of cancer)(Ref. 39). These studies provide evidence thatimproper heterochromatin maintenance mightlead to genomic instability and progression totumourigenicity.
Michaloglou et al. have also demonstratedthat BRAFE600-mediated oncogenic signallingin melanocytes induces a senescence-like
phenotype with the formation of SAHF (Ref. 40).BRAF is a protein kinase and downstreamregulator of Ras, and the V600E mutation in thegene encoding BRAF is an oncogenic mutationobserved frequently in human naevi (moles). A
further study focusing on Ras utilised a mousemodel with a conditional oncogenic K-rasV12allele activated by the Cre recombinase (Ref. 41).Upon activation of oncogenic Ras, the micedeveloped multiple lung adenomas (pre-malignant) and a few lung adenocarcinomas(malignant), and markers for senescence wereevident in the adenomas but not in thecarcinomas. Interestingly, HP1g was stronglypositive in the adenomas, whereas thecarcinomas were negative. As the HP1 proteinscan bind methylated K9 H3 to mediate genesilencing, this study further reinforces the role
of epigenetics in senescence. The formation of SAHF has also been shown in response tooverexpression of E2F3 (Ref. 42) and repressionof the E1A (adenovirus 5 early region 1Aprotein)-associated protein p400 (Ref. 43). In thefirst study, which analysed whether deregulatedE2F activity would be sufficient to mimic loss of pRB, short-term deregulation of E2F3 led tohyperplasia of quiescent melanotrophs but didnot lead to tumourigenicity due to the inductionof senescence with the appearance of SAHF(Ref. 42). p400, a protein belonging to the SWI2/SNF2 family of chromatin-remodelling proteins
and postulated to be a transforming agent, wasstudied in relation to the p53/p21 senescencepathway (Ref. 43). When p400 expression wasrepressed in normal human fibroblasts and inhuman fibroblasts transduced with humantelomerase reverse transcriptase (hTERT),senescence was induced along with theappearance of SAHF, suggesting that p400expression could repress p53 induction of p21expression, possibly allowing the bypass of thesenescence arrest.
Further studies into the formation of SAHF
have also indicated roles for the chromatinregulators histone regulating A (HIRA) andanti-silencing factor 1 (ASF1), which wereshown to be rate limiting for the formation of SAHF and were required for senescence(Ref. 44), whereas the high-mobility group A(HMGA) proteins have been shown to be majorstructural components of SAHFs (Ref. 45).HMGA proteins are usually associated withthe formation of chromatin environments
expert reviewshttp://www.expertreviews.org/ in molecular medicine
6 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 7/26
permissive for gene transcription, but have beenshown to be vital for SAHF formation and generepression in Ras-induced senescence of normalhuman fibroblasts (Ref. 45). Also, similar to therole of pRB, the growth regulatory protein
prohibitin, which has previously been linked tothe regulation of senescence (Ref. 46), has also been shown to localise to SAHFs induced byDNA-damaging agents (Ref. 47). It was shownthat prohibitin plays an integral role in thestability of the senescent phenotype bymaintaining the repression of E2F-responsivepromoters through the maintenance of arepressive chromatin environment by interactionwith HP1g. Reduction in the cellular levels of prohibitin led to a reduction in the numberof SAHFs and impaired the ability of the cells toundergo senescence, further indicating an
integral role for epigenetics in cellular senescence.The formation of SAHF in senescence arrest has
also been studied in vivo in ageing primates(Ref. 48). Increases in the presence of telomere-dysfunction-induced DNA damage foci werecorrelated to increasing age in dermal fibroblastsand, importantly, this was further correlated toSAHF formation. This study indicates thattelomere dysfunction, occurring as a result of telomere attrition with increased cellularproliferation, also induces SAHF formation.Whether this occurs in human cells with ageingis as yet unknown, but this study does indicate
that SAHF formation at senescence is not only aresponse to oncogenic stress, but might also be acommon mechanism in cellular senescence.
In summary, multiple studies have now shownthe strong correlation between the formationof heterochromatin and the attainment andmaintenance of the senescence phenotype.Repression of proliferation-associated genes bythe formation of heterochromatin serves to
block cellular division and so acts as a potenttumour suppressor mechanism. Furthermore,improper regulation of heterochromatin is
associated with deregulated cellular proliferationand tumourigenesis.
Epigenetic modifications by thesenescence mediators p53 and INGSimilar to pRB, other tumour suppressor proteinshave also been shown to interact with histone-modifying enzymes to regulate transcription(Ref. 49). p53 is an important mediator of senescence and one of its main transcriptional
targets is p21. It is known that p53 binds to andactivates the p21 promoter and is mediated, inpart, by increased promoter-proximal histoneacetylation (Ref. 50) through the recruitmentof histone acetyltransferases (HATs) (Ref. 51)
and the displacement of HDAC1 (Ref. 52).Interactions between p53 and the HAT p300 andprotein arginine methyltransferases, which canalso mediate transcriptional activation by post-translational modifications of histones, have also
been reported to be required for p53-dependentgene activation (Ref. 53). However, p53 alsointeracts with histone deacetylases to mediategene repression as it has been shown that p53
binding to promoters was concomitant withincreases in levels of K9 methylation and loss of acetylation of K9 H3 (Refs 54, 55, 56). Given thatepigenetics plays a key role in transcriptional
control by p53, it is important to note thatderegulation of epigenetic mechanisms couldaffect p53-mediated senescence or apoptosis.
Theinhibitor of growth (ING) familyof proteinsare also important regulators of cellularsenescence through the regulation of geneexpression (Ref. 57). Like p53, HATs and HDACshave both been shown to interact with the INGproteins to mediate chromatin-remodellingactivities and gene expression (Ref. 58). Studieshave shown a direct interaction of an ING familymember with chromatin upon senescence,suggesting that ING proteins might be important
mediators of senescence through epigeneticcontrol of gene expression (Refs 59, 60).Furthermore, it has been shown that ING2 can
bind di- and tri-methylated K4 H3 to mediategene repression through histone deacetylation
by associated factors in the mSin3a–HDACrepressor complex (Ref. 61), allowing subsequentrepressive lysine methylation required for theformation of heterochromatin. This reversal of chromatic states (from euchromatic to hetero-chromatic) is an important mechanism to repressproliferation-associated genes at senescence, and
one method to realise such a reversal could be the sequestering to chromatin of ING2-mediated histone deacetylation by a modificationassociated with active gene expression(methylation of K4 H3).
Reversing the irreversible: a newepigenetic mechanism uncoveredMethylation of K9 H3 plays an important role inthe maintenance of gene repression and in
expert reviewshttp://www.expertreviews.org/ in molecular medicine
7 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 8/26
SAHF-mediated gene repression at senescence.Methylation of K9 H3 was generally understoodto be a stable, long-lasting modification, allowingfor effective gene silencing. However, multiplestudies have now shown that methylation of K9
H3 is readily reversible (Refs 62, 63, 64, 65, 66)and it has been suggested that enzymes thatmediate K9 H3 demethylation could also affecttumourigenesis (Ref. 65). GASC1 (for ‘geneamplified in squamous cell carcinoma 1’; alsoknown as JMJD2C, for ‘jumonji domaincontaining 2 family member C’) is one suchprotein found with the ability to demethylate di-and tri-methylated K9 H3 (Ref. 65). This,coupled with previous findings that GASC1 wasamplified in several carcinoma cell lines and thatinhibition of the protein led to a decrease incellular proliferation, led to the suggestion
that GASC1 could act as an oncogene.Hypothetically, overexpression of GASC1 orother K9 H3 demethylases could lead to thedissolution of SAHF and to the bypass of senescence by the de-repression of proliferation-associated genes. In such a model, the formationof SAHF in response to a senescence-inducingsignal would ‘force’ cells to undergo prematuresenescence and stop possible tumourigenicity.However, if SAHF formation was inhibited bythe overexpression or aberrant localisation of aK9 H3 demethylase, this could allow cells tomove preferentially towards tumourigenesis and
bypass senescence (Fig. 3).
Polycomb group proteins, stem cellsand senescenceAlthough it has been shown that the gain of heterochromatin by increased levels of repressive histone modifications is linked tosenescence, the gain of repressive histone-modifying activity by Polycomb group (PcG)proteins and gene silencing has also beenobserved to allow extension of lifespan and isinvolved in tumourigenesis. There are two
distinct PcG complexes, Polycomb repressivecomplex 1 and 2 (PRC1 and PRC2); thesemultiprotein complexes can repress target geneexpression by catalysing the addition of repressive modifications of histones, such asK27 H3 methylation, and allowing thecompaction and maintenance of chromatin intoa heterochromatic structure.
Loss of function of the PcG protein BMI1 (for‘B-cell-specific Moloney murine leukaemia
virus integration site 1’) was shown to impairthe progression of mouse embryonic fibroblastsinto S-phase; indeed, these cells underwentpremature senescence, thereby linkingtranscriptional repression by PcG proteins with
cell-cycle control and senescence (Ref. 67).Additionally, BMI1 has been shown to bedownregulated in senescing human fibroblasts(Ref. 68), whereas overexpression has beenobserved in tumours and tumour cell lines(Refs 69, 70, 71, 72) and BMI1 is required for theshort-term survival of cancer cells (Ref. 73).
The role of senescence-associatedheterochromatin foci (SAHF) insenescence and tumourigenesis
Expert Reviews in Molecular Medicine
© 2007 Cambridge University Press
‘Normal’cell
SAHF formation
Senescence signala
‘Normal’cell
SAHF formation
Senescence signalb
Senescence Tumourigenesis
Figure 3. The role of senescence-associated
heterochromatin foci (SAHF) in senescence and
tumourigenesis. (a) Formation of SAHF after
oncogenic stress in a ‘normal’ cell might promote
the induction of senescence over tumourigenesis
by the repression of specific genes. (b) However,
if SAHF formation is inhibited, potentially by
the reversal of Lys9 (K9) H3 methylation,
tumourigenesis may be promoted owing to
‘blockage’ of the route towards senescence.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
8 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 9/26
Interestingly, additional studies have shown thatoverexpression of BMI1 allows immortalisationand transformation of some human cells(Refs 67, 74, 75). Importantly, the INK4a locusfrom which the cyclin-dependent kinase
inhibitors p16INK4a
and p14ARF
are transcribedare crucial downstream targets for BMI1 withregard to its effects on cell proliferationand senescence (Ref. 68). Therefore, throughinhibiting the expression of these cyclin-dependent kinase inhibitors, BMI1 can lead tothe promotion of cell-cycle progression.
EZH2 (for ‘enhancer of zeste homolog 2’),another PcG protein, has been shown tomethylate K27 H3 (Ref. 76) and is alsooverexpressed in some cancers, again linking theformation of heterochromatin to the extension of lifespan (Refs 77, 78, 79). EZH2 expression is
also known to be repressed by pRB andactivated p53, suggesting a possible mechanismfor EZH2 overexpression in cancers (Ref. 79)and is further observed to be specificallydownregulated in senescent cells (Ref. 80). Lossof K27 H3 histone methyltransferase activity atsenescence might correspond to loss of heterochromatin and activation of genes suchas those encoding p16INK4a and p14ARF, leadingto cellular senescence (Ref. 81). By contrast,overexpression of EZH2 in cancers might lead tothe proper maintenance of heterochromatin atthe INK4a locus, allowing the elongation of
lifespan in cancer cells by bypassing senescencethrough the repression of expression of genessuch as those encoding p16INK4a and p14ARF.
Complementing these reports have been recentstudies on epigenetics in embryonic stem cells(ESCs) and how epigenetics affects pluripotency;these studies have suggested another route bywhich a normal cell might bypass senescence andgain tumourigenic properties. The chromatinenvironment of ESCs has been shown to behyperdynamic and therefore in a highlypermissive state for transcription. However, as a
cell differentiates along various lineages, specificgenes will become repressed, whereas others arefurther activated, leading to a less dynamic andtherefore less permissive chromatin environment(Refs 82, 83, 84). PRC1 and PRC2 have now
been shown to be integral in maintainingpluripotency by initiating and maintaining thesilencing of genes involved in differentiation bymarking such genes with K27 H3 methylation(Refs 85, 86, 87, 88). Therefore, if any of the
components of PRC1 and PRC2 becomeoverexpressed in a normal cell, genes involved indifferentiation could become epigeneticallysilenced and perhaps allow the reversion of anormal cell towards a more pluripotent state
(Fig. 4). This could allow pluripotent stem-likecells to arise in normal tissues and nucleatecancer growth. Supporting this, as mentionedpreviously, it has been shown that genes suchas those encoding BMI1 and EZH2 areoverexpressed in some cancers (Refs 69, 70, 71, 72,77, 78, 79).
These studies firmly establish a link betweenPcG group proteins, senescence and themodulation of the chromatin environment.Supporting this hypothesis is a recent study of PcG group proteins in prostate cancer (Ref. 89).This study showed that increased expression of
BMI1 and EZH2, with increases in K27 H3methylation and gene silencing, were linked toincreased malignant potential and metastasis inprostate cancer and were also linked to failurein therapy of prostate cancer. Prior work bythe same group also identified an 11-genesignature that was common between normalstem cells with normal self-renewal capacityand malignant tumour cells (Ref. 90). Thesepapers suggest that PcG-mediated genesilencing can mediate increased malignancyperhaps by bestowing stem cell properties topreviously non-metastatic cancer cells with low
malignant potential; this could occur throughthe repression of multiple developmental anddifferentiation pathways by epigenetic changesto gene expression. Thus, PcG-mediatedpathways are now known to be involved in bothnormal stem cell function and highly malignanttumour cells. This reinforces the concept thatregulation of the chromatin environment andgene regulation by the PcG pathway is veryimportant to proper stem cell function and toprevent highly malignant metastatic tumoursfrom arising.
Support for the ‘reversion’ theory has beenshown in a study of committed progenitor cellsexpressing the mixed lineage leukaemia (MLL)–AF9 fusion protein known to impart leukaemiastem cell (LSC) properties (Ref. 91). These cellsgained the expression of a subset of geneshighly expressed in haematopoietic stem cells(HSCs) and also gained self-renewal properties.The MLL protein is a common fusion partnerin leukaemias and is a trithorax group
expert reviewshttp://www.expertreviews.org/ in molecular medicine
9 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 10/26
(Trx-G) member involved in the transcriptionalactivation of gene expression; MLL is known
to mediate methylation of K4 H3 (Ref. 92).This study is interesting as it links the action of an epigenetic modifier with the reversion of amore committed cell towards a more primitivestem cell type and tumourigenesis.
These studies also raise questions against therising popularity of the stem cell theory of cancer (Ref. 93), by suggesting that somecancers might arise from the gain of pluripotency by deregulation of the chromatinenvironment in normal somatic cells. However,lending support to the cancer stem cellhypothesis is the premise that epigeneticdisruption of stem cells might be a commonunifying theme in cancer aetiology (Ref. 27),which is an attractive proposition. This so-called ‘epigenetic progenitor model’ might helpto explain many different aspects about cancer,
including initiation, progression, and geneticand epigenetic instability. To understand
Figure 4. Attainment of pluripotency in normal
cells: a possible path towards the cancer stem
cell. (a) In embryonic stem cells (ESCs), Polycomb
group (PcG) complexes [Polycomb repressive
complex 1 and 2 (PRC1 and PRC2)] function to
repress differentiation-associated genes by
repressive histone modifications [Lys9 (K9) H3 and
Lys27 (K27) H3 methylation], whereas pluripotency
genes are expressed with the presence of
permissive histone modifications [acetylation of
histones H3 and H4 and methylation of Lys4 (K4)H3]. This allows for the self-renewal capacity
characteristic of stem cells. (b) Loss of the PcG-
mediated repressive modifications leads to the
expression of lineage-specific genes upon
differentiation of ESCs, leading to the cell
becoming more committed to a certain lineage.
Pluripotency genes also become repressed and
therefore these cells lose some ESC
characteristics. (c) Overexpression of PcG proteins
is frequently observed in cancer cells and it is
possible that overexpression of PcG proteins in
normal cells could allow the silencing of lineage-
specific genes involved in differentiation by
repressive histone modifications. (d) This might
also allow the cell to re-express pluripotency-
associated genes and revert back to a pluripotent
state, undergoing deregulated cell proliferation. This
may be an epigenetic mechanism by which normal
cells can bypass senescence and grow without
limit, allowing sustained cell proliferation and self-
renewal. Therefore, this could represent a new
mechanism by which cancer stem cells can arise
and nucleate cancer growth.
Attainment of pluripotency in normalcells: a possible path towards thecancer stem cell
Expert Reviews in Molecular Medicine
© 2007 Cambridge University Press
Cancerstemcells?
a
b
c
d
Embryonic stem cells
Lineage-specified cells
Deregulated lineage-specified cells
Pluripotent cancer cell
Differentiation
Tumourigenic event
Attainment ofpluripotency
K27 H3methylation
K9 H3methylation
Pluripotencygenes
Differentiationgenes
H3 and H4acetylation
K4 H3methylation
PRC1/ PRC2
Normal
cells
expert reviewshttp://www.expertreviews.org/ in molecular medicine
10 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 11/26
whether one of these models is predominant orwhether they work in conjunction to fostertumourigenesis is an important goal.
Histone acetylation and senescence
As previously mentioned, histone acetylationmight also have an important role in mediatingthe senescence phenotype. The HATs p300and CREB-binding protein (CBP) were bothstudied in senescing human melanocytes and,interestingly, levels of both proteins wereobserved to decrease as these cells underwentreplicative senescence (Refs 94, 95). Thedecrease in the levels of these HATs led todramatic decreases in total histone H4acetylation levels. Specifically, the promoter forthe gene encoding cyclin E was shown to loseH4 acetylation and become repressed upon
senescence, whereas cyclin E was highlyexpressed and H4 acetylation was high inproliferating cells. This parallels previous studiesof the cyclin E promoter during senescence,which showed that pRB and HDACs bound tothe promoter to repress transcription, linking thesenescent phenotype to the pRB/p16INK4a
pathway and the formation of SAHF (Ref. 30). Asmentioned previously, p53 also interacts withenzymes that control histone acetylation,indicating a role for two main senescencemediatorsin the modulationof histoneacetylation.
However, promotion of histone acetylation bythe use of HDAC inhibitors can induce asenescence-like phenotype in human fibroblasts(Refs 96, 97, 98). Furthermore, in senescingnormal human fibroblasts, HDAC1 becomesdownregulated (Ref. 98), indicating that overallthe promotion of acetylation leads to activationof the senescence phenotype. Interestingly,histone deacetylation has also been shown tohave a role in the extension of cellular lifespan.The yeast protein Sir2 (for ‘silent mating typeinformation regulation 2’), identified as aNADþ-dependent HDAC involved in
ribosomal DNA silencing, contributes to thereplicative lifespan of yeast (Refs 99, 100).Enhanced activity of the gene encoding Sir2was also found to be associated with increasedlongevity in Caenorhabditis elegans (Ref. 101),Drosophila (Ref. 102) and rodents (Ref. 103).Drosophila Sir2 has also been shown to beinvolved in epigenetic gene silencing by thePcG proteins and is physically associated with acomplex containing the Drosophila homologue
of EZH2. This further implicates Sir2 in thecontrol of senescence (Ref. 104) and suggests arole for Sir proteins in PcG-mediated generepression that might affect cellular proliferationand lifespan as discussed above. A human
homologue of yeast Sir2, SIRT1, has also beenassociated with longevity and is believed to actprimarily by inhibiting cellular senescence(Refs 105, 106, 107) and overexpression has beenobserved in leukaemia cells (Ref. 108). SIRT1 andother Sir2 homologues are able to deacetylatetranscription factors and p53, and also have K16H4 deacetylase activity (Ref. 109). Recently, aSIRT1 inhibitor, Sirtinol, was shown to induce asenescence-like growth arrest in human cancercells (Ref. 110) in agreement with previous dataindicating that the use of HDAC inhibitors couldlead to induction of the senescence phenotype.
Additionally, loss of histone acetylation has been linked to tumourigenesis throughdecreased K16 H4 acetylation, as well asdecreased tri-methylation of K20 H4 and DNAhypomethylation, as demonstrated in cancercells and tissues (Ref. 39).
These studies suggest that theincreases in globalhistoneacetylation can lead to senescence, perhapsmediated by the re-expression of genes previouslysilenced by heterochromatin that, when activated,promote senescence. By contrast, de-acetylationmight serve to repress genes that contribute tothe senescence phenotype. Again, this shows
that regulation of the chromatin environment iscrucial for proper cellular control.
DNA methylation and senescenceGenerally, histone modifications and DNAmethylation are often discussed separately inrelation to their effects on gene expression,although it is now known that they are in factintimately linked (Refs 111, 112, 113, 114), withmultiple interactions now known betweenproteins associated with histone modificationsand those associated with DNA methylation.
Generally, DNA methylation of CpG islands(regions of adjacent cytosine and guanineresidues linked by phosphodiester bonds) atgene promoters is correlated with generepression, whereas expression is linked to thelack of methylation. Global DNA methylationhas been shown to decrease with age (Ref. 115)and this loss had been hypothesised to functionas a counting mechanism for senescence, suchthat senescence would occur when a cell loses a
expert reviewshttp://www.expertreviews.org/ in molecular medicine
11 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 12/26
certain degree of DNA methylation (Refs 115,116). Treatment with 5-azadeoxycytidine(5-azadC), a DNA demethylating agent, has
been shown to reduce the replicative lifespan of normal human fibroblasts, supporting the DNA
methylation counting mechanism hypothesis(Refs 117, 118). Also in agreement with thishypothesis are the findings that DNAmethyltransferase activity is elevated in cancercells in vitro, in tumours in vivo (Ref. 119) andin transformed cells (Refs 120, 121), whereasDNA methyltransferase activity decreases inserially passaged normal human fibroblasts(Ref. 122). Therefore, the loss of DNAmethylation with age might eventually havedeleterious consequences, such as deregulationof gene expression. Consistent with this view, ithas been hypothesised that DNA methylation
might repress a set of growth inhibitory genes(Refs 24, 123) and, as the number of cellpopulation doublings increases and DNAmethylation decreases, the repression of suchgrowth inhibitory genes might become lessstringent, leading to their eventual expressionand contributing to senescence. However,it has also been shown that a striking feature of some tumours (both benign and malignant) isthat global DNA methylation is reduced(Refs 124, 125). This loss of DNA methylationis also linked to chromosomal instability,which can contribute to tumourigenesis
(Refs 126, 127). Thus, paradoxically, it has beenshown that the loss of DNA methylation cancontribute to senescence and tumourigenicity.However, it is acknowledged that gene-specificmethylation is variable in cancer and that manytumour suppressor genes show promoter-specific DNA hypermethylation leading togene silencing, including the genes encodingpRB (Ref. 128) and p16INK4a (Ref. 129), whereasthe genome is hypomethylated overall.Interestingly, recent data have shown thatp16INK4a is crucially involved in DNA
methylation, leading to gene silencing and breasttumourigenesis (Ref. 130). Loss of p16INK4a leadsto the increased expression of key PcG proteins,resulting in a specific pattern of DNAhypermethylation and importantly the silencingof the gene encoding HOXA9, a transcriptionfactor that is epigenetically silenced in breasttumourigenesis. Therefore, it is likely thatglobal loss of DNA methylation leading tochromosomal instability could trigger cellular
senescence and that promoter-specific alterationsin key tumour suppressor genes might allow the
bypass of senescence and lead to tumourigenicity.
Senescence and DNA damage detection
by histone modificationHistone modifications can also play a role in thedetection and repair of DNA damage that, aspreviously mentioned, can lead to prematuresenescence. DNA damage can be incurredthrough the actions of various types of radiation(Ref. 131), DNA-damaging drugs (many of which are used as anti-tumourigenic therapies)(Ref. 132) and oxidative stress (Ref. 133), andhistone phosphorylation is now know to play arole in mediating cellular responses to this DNAdamage. Signalling kinases, notably ATM (for‘ataxia telangiectasia mutated’) and ATR (for
‘ataxia telangiectasia and Rad3 related’), arerecruited to the site of damage and are activated,leading to phosphorylation of histone H2AXmolecules (g-H2AX) (Ref. 134). Histone H2AX isa variant of the H2A histone that makes up partof the histone octamer. The phosphorylation of histone H2AX facilitates the recruitment of checkpoint and DNA repair factors includingp53-binding protein 1 (153BP1), and promotesthe activation of checkpoint kinase 1 (Chk1) andChk2, which can activate p53 and therebymediate cellular senescence. Furthermore, it isknown that the uncapping of telomeres can leadto senescence through a DNA damage responsemediated by a similar mechanism (Refs 135, 136).Phosphorylated histone H2AX also serves torecruit histone modifiers and ATP-dependentchromatin remodellers to these sites of DNAdamage and aid the repair of these sites (Ref. 137).
Two recent studies have uncovered a link between histone lysine methylation and theDNA damage checkpoint and double-strand
break (DSB) repair proteins 53BP1 in mammalsand Schizosaccharomyces pombe putative homologCrb2 (for ‘Crumbs-like protein 2’): the
relocalisation of 53BP1 and Crb2 to DNA DSBshas been found to require histone methylation(Refs 138, 139). It was proposed that 53BP1senses DSBs through changes in higher-orderchromatin structure that occur upon exposureof the 53BP1 binding site [methylated Lys79 H3(K79 H3), which lies in the globular domain of histone H3] (Ref. 138). In S. pombe, a similarmechanism occurs with Crb2 binding tomethylated K20 (Ref. 139). However, recent
expert reviewshttp://www.expertreviews.org/ in molecular medicine
12 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 13/26
studies have shown that in fact 53BP1 and Crb2 bind to the same modified lysine residue, di-methylated K20 H4 (Ref. 140).
Epigenetic regulation of telomerase
gene expression and senescenceTelomerase and telomeres in senescenceIn adult humans, most normalcells have lowor nodetectable telomerase activity (Ref. 141). The lackof telomerase activity, or other telomeremaintenance mechanisms, leads to the loss of around 50–100 bp of telomeric sequence at eachcell division (Refs 142, 143). The loss of telomeric tract during cell division presents alimit to the maximum number of divisions acell can undertake before entering senescenceand therefore provides a limit to its lifespan(Ref. 144). Cellular senescence as a response to
telomere attrition is thought to have evolved asa tumour suppressive barrier against excessiveclonal expansion. However, the detection of telomerase activity in an estimated 85% of malignant tumours, together with the stronglinks between telomerase activity and malignantcells (Refs 141, 145), indicates the importance of telomerase activity in tumourigenesis and alsosuggests that telomerase activation is the mostcommon mechanism for telomere maintenancein cancer cells. However, several immortal celllines and tumours lack telomerase activity, yethave the ability to elongate their telomeres.Such non-telomerase-dependent mechanisms of telomere maintenance include the alternativelengthening of telomeres (ALT) pathway(Ref. 146) (Fig. 1).
In cells where ALT is utilised, telomere length ismaintained by a recombination-based DNAreplication mechanism (Ref. 147) and leads toheterogeneity in telomere lengths (Ref. 148).Also associated with use of the ALT mechanismis the presence of ALT-associated promyelocyticleukemia protein (PML) bodies, called APBs(Ref. 149), which contain the protein PML,
telomeric DNA, the telomeric-binding proteinstelomere repeat factor 1 (TRF1) and TRF2, andproteins involved in DNA recombination andreplication. The utilisation of ALT in tumourcells is much rarer than the utilisation of telomerase activity (Ref. 148) and it has beenhypothesised that ALT might normally occurunder selective pressure when expression of telomerase is prevented. ALT is commonlyactivated in tumours of mesenchymal origin,
including osteosarcomas, soft tissue sarcomasand glioblastoma multiforme (Refs 150, 151,152, 153). Furthermore, in normal mesenchymaltissues, telomerase activity is more tightlyrepressed that in epithelial cells, possibly leading
to the selective pressure for ALT activation overtelomerase activity in these cell types.
Epigenetic regulation of telomeremaintenance mechanismsExpression of the human telomerase RNAcomponent (hTR) and hTERT are understood to
be the minimal requirements for telomeraseactivity. Studies have shown that specificepigenetic modifications at the hTR and hTERTpromoters correlate with expression orrepression of hTR and hTERT (Refs 154, 155).Lack of expression of hTR in ALT cell lines, and
hTERT in ALT and normal cell lines, isassociated with a repressive chromatinenvironment at the appropriate gene promoter.Conversely, expression of hTR in ALT andtelomerase-positive cell lines, and hTERTexpression in telomerase-positive cell lines, isassociated with a more permissive chromatinenvironment at the appropriate gene promoter.Moreover, in the pre-senescent normal fibroblastcell line WI38, which has no detectable hTERTexpression and low hTR expression, thetelomerase gene promoters contain no greatquantities of either positive or negativechromatin modifications, and therefore thechromatin environment of the promoter remainsin a neutral chromatin configuration (Fig. 5a).
Furthermore, upon comparison of the hTERTpromoter chromatin environment in ALT celllines (including a cell transformed from theparental WI38 cell line WI38-SV40) withthe normal WI38 cell line, an increase in theamounts of repressive modifications is evidentand links the heavy repression of hTERTexpression by the chromatin environment to theALT phenotype. In the telomerase-positive
tumour cell lines, permissive modifications aregained at the hTERT promoter relative to theWI38 cell line. Similarly, heavy repression of hTR is evident in some ALT cell lines with nohTR expression, whereas high levels of permissive chromatin modifications are linkedto hTR expression. Additionally, treatment of normal and ALT cells with 5-azadC incombination with the HDAC inhibitortrichostatin A (TSA) caused chromatin
expert reviewshttp://www.expertreviews.org/ in molecular medicine
13 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 14/26
remodelling of both promoters and led to theformation of a more permissive chromatinenvironment, as well as reactivation orincreased expression of hTR and hTERTexpression. This suggests a model by which
gain of permissive modifications at thetelomerase gene promoters leads to telomeraseactivity by the expression of hTR and hTERT,whereas the gain of repressive modificationsleads to activation of the ALT mechanism(Fig. 5b). Therefore, epigenetic deregulation of
Figure 5. Epigenetic regulation of telomere
maintenance mechanisms. (a) Histograms
showing a simplistic view of what we know about
the relationship between the relative levels of
repressive and permissive chromatin modifications
at the telomerase gene promoters in telomerase-
expressing cancer cells, pre-senescent normalcells and cells that maintain their telomeres by the
alternative lengthening of telomeres (ALT)
mechanism. The presence of permissive histone
modifications at the telomerase gene promoters is
linked to telomerase activity, whereas the presence
of repressive histone modifications is linked to the
activation of ALT. Both of these scenarios are in
contrast to the chromatin environment of pre-
senescent normal cells, in which permissive and
repressive modifications are at a minimum. This
suggests a model by which epigenetic deregulation
of the telomerase gene promoters could lead to
aberrant telomere maintenance. (b) Model depicting
how chromatin modifications might influencetelomere maintenance mechanisms. In a ‘normal’
cell that does not have telomerase activity and has
no or low levels of expression of the telomerase
genes, few chromatin modifications are present at
the gene promoters. The choice of telomere
maintenance mechanism may be influenced
through epigenetic mechanisms and changes
in histone modifications, possibly during
tumourigenesis. In the left-hand pathway shown,
telomerase gene expression is linked to the
accumulation of permissive histone modifications
[acetylation of histones H3 and H4 and methylation
of Lys4 (K4) H3], which leads to telomerase activity
and the elongation/maintenance of telomerelength. However, in the right-hand pathway shown,
heavy repression of the telomerase gene promoters
by the accumulation of repressive histone
modifications [methylation of Lys20 (K20) H4 and/or methylation of Lys9 (K9) H3] could lead to
utilisation of the ALT mechanism rather than
telomerase activity for telomere elongation/maintenance owing to the repression of telomerase
gene expression.
Epigenetic regulation of telomeremaintenance mechanisms
Expert Reviews in Molecular Medicine
© 2007 Cambridge University Press
Telomeraseactivity
Pre-senescentnormal cell
Normal
Epigeneticmechanisms
Telomeraseactivity
Cancer ALT
Telomerase generepression
ALT
Permissivemodifications
Repressivemodifications
Telomerase generepression
Telomerase geneexpression
b
a
K20 H4methylation K9 H3methylation
H3 and H4acetylation
K4 H3methylation
expert reviewshttp://www.expertreviews.org/ in molecular medicine
14 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 15/26
the telomerase gene promoters might allow theactivation of a telomere maintenancemechanism, allowing bypass of senescence.
Epigenetic deregulation and
tumourigenicity in adult stem cellsFurther studies into the epigenetic regulation of the telomerase gene promoters utilised humanmesenchymal stem cells (hMSCs) (Ref. 156).These cells do not show telomerase activity buthave been shown to be the target for neoplastictransformation after transduction with hTERT(Ref. 155). However, by supplying hTERT, themolecular events required to upregulate hTERTexpression in cancer stem cell development aremissed. Therefore, hMSCs are ideal for theidentification of molecular mechanismsregulating telomerase gene expression in stem
cells.It has now been shown that repression of
hTERT expression in hMSCs is a result of promoter-specific histone hypo-acetylationcoupled with low RNA polymerase II (RNAPII)and transcription factor IIB (TFIIB) tracking.This repression can be overcome by treatmentof hMSCs with TSA, concomitant with increasesin promoter-specific histone acetylation andincreases in RNAPII and TFIIB tracking. hTRexpression is also increased in TSA-treatedhMSCs, concomitant with changes in RNAPIIand TFIIB dynamics. These data suggestthat deregulation of chromatin modificationsat the hTERT and hTR promoter mightfacilitate tumourigenesis in hMSCs and supportsthe hypothesis that tumourigenesis can arise inepigenetically deregulated stem cell populations.
Epigenetic regulation of telomere lengthIt has been shown that epigenetic modificationscan also regulate telomere length through themodulation of the chromatin environment of the telomeres themselves. Telomeres are rich inheterochromatin features (Ref. 157) and
disruption of this heterochromatin structure has been shown to lead to altered telomere lengthin mice, established by knockout of theSUV39H1 and SUV39H2 histone methyltransferases in mice (Ref. 158) and by theknockout of either DNA methyltransferase 1(DNMT1) or both DNMT-3a and -3b in mouseESCs (Ref. 159). In the latter study, the lack of DNMTs led to increased intra-telomericrecombination and increased the levels of other
markers of the ALT telomere maintenancemechanism, further suggesting the role forepigenetics in the choice of telomeremaintenance mechanism used in cancer cells.This also shows that epigenetic deregulation can
lead to immortalisation events and mightfacilitate tumourigenesis.
Therapeutic aspects of chromatin-mediated senescence
The intimate links between senescence andchromatin that have been uncovered mayprovide therapeutic targets for anti-cancertreatment. As induction of prematuresenescence in tumour cells is targeted as ananti-cancer therapy (Refs 4, 5), one therapeuticavenue may be to induce premature cellularsenescence through control of the enzymes that
catalyse epigenetic modifications. Intimateknowledge of gene expression changes insenescence might allow targeted upregulationor downregulation of such genes by epigenetictherapies. Also, epigenetic therapy might allowthe reversal of senescence in certain cell typesassociated with senescence-related disease.However, it must be remembered that cellularsenescence also plays roles in multiple otherdisease states and might potentially nurturetumourigenic growth. Overall, it can beappreciated that this mode of therapy wouldrequire strict control.
Current epigenetic therapies include HDACand DNMT inhibitors (Table 3), which promotere-expression of silenced genes and are widelyused in cancer therapies to facilitate there-expression of tumour suppressor genes.However useful these drugs are, other more-targeted means of enzyme function ormodulating gene expression would beadvantageous. As these drugs are not targetedthey will have pan-genomic effects and couldnurture tumour growth by aberrant activation of growth-promoting genes or cancer-associated
genes, further indicating a requirement for strictcontrol for such epigenetic therapies. The use of therapeutic RNA interference (RNAi) can allowgreater targeting of specific enzymes involved inepigenetic processes, whereas changes in specificgene expression by the use of engineeredtranscription factors is an exciting newmechanism of gene control (Ref. 160). By fusingepigenetic modifiers to engineered transcriptionfactors that can bind to specific gene promoters,
expert reviewshttp://www.expertreviews.org/ in molecular medicine
15 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 16/26
gene expression can be altered through epigeneticmechanisms. However, the ultimate therapeuticaim is to find small molecules that could besuccessfully moved into the clinical arena. Thisapproach has recently been successfully used inone study that screened for compounds thatwere able to modulate cellular senescence(Ref. 161) and could allow further detailedanalysis of the senescence pathway andepigenetic mechanisms important to it.
Epigenetic therapy might also allow the
targeting of cancer stem cells as current cancertherapeutics may not efficiently target such cellsdue to their unique cellular physiology. Recentstudies demonstrating the importance of epigenetic silencers in the maintenance of pluripotency and a proposed role intumourigenesis suggests that it may be possibleto target such cells more efficiently throughtargeting epigenetic silencers. Furthermore,studies have shown that the PcG proteins may
be regulated by signalling pathways unique topluripotent cells and targeting of thesepathways might allow modulation of epigeneticsilencing and hence pluripotency, and thereforemay have direct relevance to the treatment of cancer stem cells (Refs 162, 163, 164). Molecularsignalling pathways such as PTEN (for‘phosphate and tensin homologue’), Wnt,Hedgehog and Notch have been identified astargets for therapies aimed at cancer stem cellsand therefore targeted gene expression changes
of members of these pathways by epigenetictherapies may allow for selectivity (Ref. 165).However, effects on normal stem cells mightalso become compromised. Studies havesuggested other signalling pathways that couldaffect epigenetic modifications and thereforeprovide suitable therapeutic targets. A recentstudy into the Janus kinases/signal transducersand activators of transcription (JAK/STAT)pathway in Drosophila, a pathway whose
Table 3. Current drugs against epigenetic modifiers
Epigenetic therapy Mode of action
S-adenosylhomocysteine Inhibits S-adenosyl-L-methionine (SAM)-dependent
methyltransferases
5-azacytidine (azacytidine) DNA methyltransferase (DNMT) inhibitor (nucleosideanalogue)
5-aza-20-deoxycytidine (decitabine) DNMT inhibitor (nucleoside analogue)
1-( b-D-ribofuranosyl)-1,2-
dihydropyrimidin-2-one (zebularine)
DNMT inhibitor (nucleoside analogue)
(-)Epigallocatechin-3 gallate (EGCG) DNMT inhibitor (nucleoside analogue)
Procaine DNMT inhibitor (nucleoside analogue)
Procainamide DNMT inhibitor (nucleoside analogue)
Hydralazine DNMT inhibitor (nucleoside analogue)
MG98 DNMT1 inhibitor (second-generation phosphorothioate
antisense oligodeoxynucleotide)
Trichostatin A (TSA) Hydroxamate histone deacetylase (HDAC) inhibitor
Suberoylanilide hydroxamic acid (SAHA) Hydroxamate HDAC inhibitor
m-Carboxycinnamic acid bishydroxamide
(CBHA)
Hydroxamate HDAC inhibitor
LAQ824 Hydroxamate HDAC inhibitor
PXD101 Hydroxamate HDAC inhibitor
Depsipeptide Cyclic peptide HDAC inhibitor
Apicidin Cyclic peptide HDAC inhibitor
Cyclic hydroxamic-acid-containing
peptides (CHAPs)
Cyclic peptide HDAC inhibitor
Valproic acid Aliphatic acid HDAC inhibitor
Phenylbutyrate Aliphatic acid HDAC inhibitor
MS-275 Benzamide HDAC inhibitor
CI-994 Benzamide HDAC inhibitor
Trifluoromethyl ketones Electrophilic ketone HDAC inhibitor
Alpha-ketoamides Electrophilic ketone HDAC inhibitor
expert reviewshttp://www.expertreviews.org/ in molecular medicine
16 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 17/26
deregulation has been implicated in human cancer(Ref. 166), has shown that over-activation of JAKkinase disrupted heterochromatin formation andchanged the expression of genes through STAT-independent routes. Further understanding of
pathways that control epigenetic modifiers couldrepresent a wealth of opportunity for discovery of therapeutic targets.
Data establishing epigenetic processes asimportant regulators of senescence andtumourigenesis also suggest that such epigeneticmodifications may be useful as prognosticmarkers. Expression of genes involved inepigenetic mechanisms have been shown to beassociated with therapy failure in prostate cancer(Ref. 89) and the presence of certain patterns of global histone modifications have been linked torecurrence of prostate cancer (Ref. 167). Loss of
lysine acetylation and methylation on histone H4has also been identified as an epigeneticsignature of human cancer (Ref. 39), whereasother studies have identified the loss of repressive lysine methylation and the enzymesthat catalyse the addition of these modificationsas early markers of tumourigenesis (Refs 37, 38).Further levels of methylation status of K27 H3might allow the differentiation between weaklyand strongly malignant tumour cells (Ref. 89).Therefore, analysis and detection of suchmodifications and enzymes could be importantin early cancer detection and might also allow
tailored epigenetic therapies. Furthermore, aschemotherapeutics often promote prematurecellular senescence as an anti-cancer therapy andsenescence-associated chromatin changes arenow understood, such changes might be usedas markers of response to chemotherapeutictherapies (Ref. 168).
Conclusions and summary It is now accepted that epigenetic mechanisms arevital regulators of gene expression and thederegulation of epigenetic mechanisms has been
linked to cancer in multiple studies. Pathwaysand mechanisms important to the senescencephenotype have all been shown to regulate or beregulated by epigenetic mechanisms, underlyingthe importance of this level of control.Epigenetics provides the perfect regulatorymechanism for dynamic changes in globalgene expression required for multiple cellularprocesses such as senescence. Deregulation of epigenetic modifications may therefore promote
tumourigenesis through the disruption of thesenescence phenotype and the deregulation of gene expression.
How does the research now proceed? The roleepigenetics has to play in cellular processes such
as senescence, tumourigenesis and stem cellphenotypes has only recently been uncoveredin any great detail. In addition, epigeneticregulation is key to many other cellularprocesses, whereas deregulation is an importantfactor in numerous disease states. Furtheranalysis of global states of chromatin indifferent cell types and comparisons betweennormal and diseased tissues might allowgreater understanding of the causes of thedisease and also potentially lead to thediscovery of therapeutic targets. Such studieshave already begun, as mentioned, in ESCs
(Refs 85, 86, 87, 88) and also in cancer,including the exciting prospect of the humanepigenome project, an international projectaiming to identify all the chemical changes andrelationships among chromatin constituents thatprovide function to the DNA code (Refs 39, 167,169, 170, 171). Such studies will allow us tounderstand how chromatin modificationsmight control distinct cellular processes andwhat enzymes mediate such changes. Fullunderstanding of the signalling pathways thatmodulate enzymatic control is also animportant step to begin to uncover how the
deregulation of these pathways affects theepigenetic control of gene expression andcellular functions, and also potentially to enablethe discovery of novel therapeutic targets.
Acknowledgements and fundingResearch in the authors’ laboratory is supported
by Cancer Research UK, University of Glasgowand European Community grant LSHC-CT-2004-502943. The authors thank the anonymouspeer reviewers for their invaluable comments.
References1 Hayflick, L. (1965) The Limited in Vitro Lifetime
of Human Diploid Cell Strains. Exp Cell Res 37,
614-636
2 Campisi, J. (2001) Cellular senescence as a tumor-
suppressor mechanism. Trends Cell Biol 11, S27-31
3 Mathon, N.F. and Lloyd, A.C. (2001) Cell
senescence and cancer. Nat Rev Cancer 1, 203-213
expert reviewshttp://www.expertreviews.org/ in molecular medicine
17 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 18/26
4 Shay, J.W. and Roninson, I.B. (2004) Hallmarks of
senescence in carcinogenesis and cancer therapy.
Oncogene 23, 2919-2933
5 Roninson, I.B. (2003) Tumor cell senescence in
cancer treatment. Cancer Res 63, 2705-2715
6 Shelton, D.N. et al. (1999) Microarray analysis of replicative senescence. Curr Biol 9, 939-945
7 Hardy, K. et al. (2005) Transcriptional networks
and cellular senescence in human mammary
fibroblasts. Mol Biol Cell 16, 943-953
8 Yoon, I.K. et al. (2004) Exploration of replicative
senescence-associated genes in human dermal
fibroblasts by cDNA microarray technology. Exp
Gerontol 39, 1369-1378
9 Guo, S., Zhang, Z. and Tong, T. (2004) Cloning and
characterization of cellular senescence-associated
genes in human fibroblasts by suppression
subtractive hybridization. Exp Cell Res 298, 465-472
10 Mason, D.X., Jackson, T.J. and Lin, A.W. (2004)Molecular signature of oncogenic ras-induced
senescence. Oncogene 23, 9238-9246
11 Bandyopadhyay, D. and Medrano, E.E. (2003)
The emerging role of epigenetics in cellular
and organismal aging. Exp Gerontol 38,
1299-1307
12 Young, J. and Smith, J.R. (2000) Epigenetic
aspects of cellular senescence. Exp Gerontol 35,
23-32
13 Esteller, M. (2006) Epigenetics provides a new
generation of oncogenes and tumour-suppressor
genes. Br J Cancer 94, 179-183
14 Santos-Rosa, H. and Caldas, C. (2005) Chromatinmodifier enzymes, the histone code and cancer.
Eur J Cancer 41, 2381-2402
15 Baylin, S.B. and Ohm, J.E. (2006) Epigenetic gene
silencing in cancer - a mechanism for early
oncogenic pathway addiction? Nat Rev Cancer 6,
107-116
16 Jenuwein, T. and Allis, C.D. (2001) Translating the
histone code. Science 293, 1074-1080
17 Strahl, B.D. and Allis, C.D. (2000) The language
of covalent histone modifications. Nature 403,
41-45
18 Martin, C. and Zhang, Y. (2005) The diverse
functions of histone lysine methylation. Nat Rev
Mol Cell Biol 6, 838-849
19 Schubeler, D. et al. (2004) The histone modification
pattern of active genes revealed through genome-
wide chromatin analysis of a higher eukaryote.
Genes Dev 18, 1263-1271
20 Campisi, J. (2005) Senescent cells, tumor
suppression, and organismal aging: good citizens,
bad neighbors. Cell 120, 513-522
21 Krtolica, A. et al. (2001) Senescent fibroblasts
promote epithelial cell growth and tumorigenesis:
a link between cancer and aging. Proc Natl Acad
Sci U S A 98, 12072-12077
22 Campisi, J. et al. (2001) Cellular senescence, cancer
and aging: the telomere connection. Exp Gerontol36, 1619-1637
23 Campisi, J. (2000) Cancer, aging and cellular
senescence. In Vivo 14, 183-188
24 Jones,P.A.and Baylin,S.B. (2002) Thefundamental
role of epigenetic events in cancer. Nat Rev Genet
3, 415-428
25 Hake, S.B., Xiao, A. and Allis, C.D. (2004) Linking
the epigenetic ‘language’ of covalent histone
modifications to cancer. Br J Cancer 90, 761-769
26 Feinberg, A.P. (2004) The epigenetics of cancer
etiology. Semin Cancer Biol 14, 427-432
27 Feinberg, A.P., Ohlsson, R. and Henikoff, S. (2006)
The epigenetic progenitor origin of human cancer.Nat Rev Genet 7, 21-33
28 Macieira-Coelho, A. (1991) Chromatin
reorganization during senescence of proliferating
cells. Mutat Res 256, 81-104
29 Zhang, H., Pan, K.H. and Cohen, S.N. (2003)
Senescence-specific gene expression fingerprints
reveal cell-type-dependent physical clustering of
up-regulated chromosomalloci. ProcNatl Acad Sci
U S A 100, 3251-3256
30 Narita, M. et al. (2003) Rb-mediated
heterochromatin formation and silencing of
E2F target genes during cellular senescence.
Cell 113, 703-71631 Luo, R.X., Postigo, A.A. and Dean, D.C. (1998) Rb
interacts with histone deacetylase to repress
transcription. Cell 92, 463-473
32 Magnaghi-Jaulin, L. et al. (1998) Retinoblastoma
protein represses transcription by recruiting
a histone deacetylase. Nature 391, 601-605
33 Brehm, A. et al. (1998) Retinoblastoma protein
recruits histone deacetylase to repress
transcription. Nature 391, 597-601
34 Vandel, L. et al. (2001) Transcriptional repression
by the retinoblastoma protein through the
recruitment of a histone methyltransferase. Mol
Cell Biol 21, 6484-6494
35 Nielsen, S.J. et al. (2001) Rb targets histone H3
methylation and HP1 to promoters. Nature 412,
561-565
36 Braig, M. et al. (2005) Oncogene-induced
senescence as an initial barrier in lymphoma
development. Nature 436, 660-665
37 Peters,A.H. et al.(2001)Loss of theSuv39 h histone
methyltransferases impairs mammalian
expert reviewshttp://www.expertreviews.org/ in molecular medicine
18 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 19/26
heterochromatin and genome stability. Cell
107, 323-337
38 Pogribny, I.P. et al. (2006) Histone H3 lysine 9 and
H4 lysine 20 trimethylation and the expression of
Suv4–20h2 and Suv-39h1 histone
methyltransferases in hepatocarcinogenesisinduced by methyl deficiency in rats.
Carcinogenesis 27, 1180-1186
39 Fraga, M.F. et al. (2005) Loss of acetylation at
Lys16 and trimethylation at Lys20 of histone H4
is a common hallmark of human cancer. Nat
Genet 37, 391-400
40 Michaloglou, C. et al. (2005) BRAFE600-associated
senescence-like cell cycle arrest of human naevi.
Nature 436, 720-724
41 Collado, M. et al. (2005) Tumour biology:
senescence in premalignant tumours. Nature
436, 642
42 Lazzerini Denchi, E. et al. (2005) Deregulated E2Factivity induces hyperplasia and senescence-like
features in themouse pituitary gland.Mol Cell Biol
25, 2660-2672
43 Chan, H.M. et al. (2005) The p400 E1A-associated
protein is a novel component of the p53 ! p21
senescence pathway. Genes Dev 19, 196-201
44 Zhang, R. et al. (2005) Formation of MacroH2A-
containing senescence-associated heterochromatin
foci and senescence driven by ASF1a and HIRA.
Dev Cell 8, 19-30
45 Narita, M. et al. (2006) A novel role for
high-mobility group a proteins in cellular
senescence and heterochromatin formation. Cell126, 503-514
46 Liu, X.T. et al. (1994) Prohibitin expression during
cellular senescence of human diploid fibroblasts.
Biochem Biophys Res Commun 201, 409-414
47 Rastogi, S. et al. (2006) Prohibitin facilitates
cellular senescence by recruiting specific
corepressors to inhibit E2F target genes.
Mol Cell Biol 26, 4161-4171
48 Herbig, U. et al. (2006) Cellular senescence in aging
primates. Science 311, 1257
49 Allison, S.J. and Milner, J. (2004) Remodelling
chromatin on a global scale: a novel protective
function of p53. Carcinogenesis 25, 1551-1557
50 Espinosa, J.M. and Emerson, B.M. (2001)
Transcriptional regulation by p53 through intrinsic
DNA/chromatin binding and site-directed
cofactor recruitment. Mol Cell 8, 57-69
51 Barlev, N.A. et al. (2001) Acetylation of p53
activates transcription through recruitment of
coactivators/histone acetyltransferases. Mol Cell
8, 1243-1254
52 Lagger, G. et al. (2003) The tumor suppressor p53
and histone deacetylase 1 are antagonistic
regulators of the cyclin-dependent kinaseinhibitor
p21/WAF1/CIP1 gene. Mol Cell Biol 23, 2669-2679
53 An, W., Kim, J. and Roeder, R.G. (2004) Ordered
cooperative functions of PRMT1, p300, andCARM1 in transcriptional activation by p53. Cell
117, 735-748
54 Murphy, M. et al. (1999) Transcriptional repression
by wild-type p53 utilizes histone deacetylases,
mediated by interaction with mSin3a. Genes Dev
13, 2490-2501
55 Nguyen, T.T. et al. (2005) Transcription factor
interactions and chromatin modifications
associated with p53-mediated, developmental
repression of the alpha-fetoprotein gene. Mol Cell
Biol 25, 2147-2157
56 Cui, R. et al. (2005) Family members p53 and p73
act together in chromatin modification and directrepressionof alpha-fetoprotein transcription. J Biol
Chem 280, 39152-39160
57 Russell, M. et al. (2006) Grow-ING, Age-ING and
Die-ING: ING proteins linkcancer, senescence and
apoptosis. Exp Cell Res 312, 951-961
58 Feng, X.,Hara, Y. and Riabowol, K. (2002) Different
HATS of the ING1 gene family. Trends Cell Biol
12, 532-538
59 Vieyra, D. et al. (2002) Human ING1 proteins
differentially regulate histone acetylation. J Biol
Chem 277, 29832-29839
60 Vieyra, D. et al. (2002) ING1 isoforms differentially
affect apoptosis in a cell age-dependent manner.Cancer Res 62, 4445-4452
61 Shi, X.et al. (2006) ING2 PHDdomain links histone
H3 lysine 4 methylation to active gene repression.
Nature 442, 96-99
62 Yamane, K. et al. (2006) JHDM2A, a JmjC-
containing H3K9 demethylase, facilitates
transcription activation by androgen receptor. Cell
125, 483-495
63 Whetstine, J.R. et al. (2006) Reversal of histone
lysine trimethylation by the JMJD2 family of
histone demethylases. Cell 125, 467-481
64 Klose,R.J. et al.(2006) Thetranscriptionalrepressor
JHDM3A demethylates trimethyl histone H3
lysine 9 and lysine 36. Nature 442, 312-316
65 Cloos, P.A. et al. (2006) The putative oncogene
GASC1 demethylates tri- and dimethylated lysine
9 on histone H3. Nature 442, 307-311
66 Metzger, E. et al. (2005) LSD1 demethylates
repressive histone marks to promote androgen-
receptor-dependent transcription. Nature 437,
436-439
expert reviewshttp://www.expertreviews.org/ in molecular medicine
19 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 20/26
67 Jacobs, J.J. et al. (1999) The oncogene and
Polycomb-group gene bmi-1 regulates cell
proliferation and senescence through the ink4a
locus. Nature 397, 164-168
68 Itahana, K. et al. (2003) Control of the replicative
life span of human fibroblasts by p16 and thepolycomb protein Bmi-1. Mol Cell Biol 23, 389-401
69 Bea, S. et al. (2001) BMI-1 gene amplification and
overexpression in hematological malignancies
occur mainly in mantle cell lymphomas. Cancer
Res 61, 2409-2412
70 Vonlanthen, S. et al. (2001) The bmi-1 oncoprotein
is differentially expressed in non-small cell lung
cancer and correlates with INK4A-ARF locus
expression. Br J Cancer 84, 1372-1376
71 Nowak, K. et al. (2006) BMI1 is a target gene of
E2F-1 and is strongly expressed in primary
neuroblastomas. Nucleic Acids Res 34, 1745-1754
72 Sawa, M. et al. (2005) BMI-1 is highly expressed inM0-subtype acutemyeloid leukemia. Int J Hematol
82, 42-47
73 Liu, L., Andrews, L.G. and Tollefsbol, T.O. (2006)
Loss of the human polycomb group protein BMI1
promotes cancer-specific cell death. Oncogene 25,
4370-4375
74 Leung, C. et al. (2004) Bmi1 is essential for
cerebellar development and is overexpressed in
human medulloblastomas. Nature 428, 337-341
75 Dimri, G.P. et al. (2002) The Bmi-1 oncogene
induces telomerase activity and immortalizes
human mammary epithelial cells. Cancer Res 62,
4736-474576 Cao, R. and Zhang, Y. (2004) The functions of E(Z)/
EZH2-mediated methylationof lysine27 in histone
H3. Curr Opin Genet Dev 14, 155-164
77 Varambally, S. et al. (2002) The polycomb group
protein EZH2is involved in progressionof prostate
cancer. Nature 419, 624-629
78 Kleer, C.G. et al. (2003) EZH2 is a marker of
aggressive breast cancer and promotes neoplastic
transformation of breast epithelial cells. Proc Natl
Acad Sci U S A 100, 11606-11611
79 Bracken, A.P. et al. (2003) EZH2 is downstream
of the pRB-E2F pathway, essential for proliferation
and amplified in cancer. Embo J 22, 5323-5335
80 Tang, X. et al. (2004) Activated p53 suppresses the
histone methyltransferase EZH2 gene. Oncogene
23, 5759-5769
81 Villeponteau, B. (1997) The heterochromatin loss
model of aging. Exp Gerontol 32, 383-394
82 Meshorer, E. et al. (2006) Hyperdynamic plasticity
of chromatin proteins in pluripotent embryonic
stem cells. Dev Cell 10, 105-116
83 Meshorer, E. and Misteli, T. (2006) Chromatin in
pluripotent embryonic stem cells and
differentiation. Nat Rev Mol Cell Biol 7, 540-546
84 Szutorisz, H. and Dillon, N. (2005) The epigenetic
basis for embryonic stem cell pluripotency.
Bioessays 27, 1286-129385 Boyer, L.A. et al. (2006) Polycomb complexes
repress developmental regulators in murine
embryonic stem cells. Nature 441, 349-353
86 Bernstein, B.E. et al. (2006) A bivalent chromatin
structure marks key developmental genes in
embryonic stem cells. Cell 125, 315-326
87 Azuara, V. et al. (2006) Chromatin signatures of
pluripotent cell lines. Nat Cell Biol 8, 532-538
88 Lee, T.I. et al. (2006) Control of developmental
regulators by Polycomb in human embryonic stem
cells. Cell 125, 301-313
89 Berezovska, O.P. et al. (2006) Essential role for
activation of the polycomb group (PcG) proteinchromatinsilencing pathway in metastatic prostate
cancer. Cell Cycle 5, 1886-1901
90 Glinsky, G.V., Berezovska, O. and Glinskii, A.B.
(2005) Microarray analysis identifies a death-from-
cancer signature predicting therapy failure in
patients withmultiple types of cancer. J Clin Invest
115, 1503-1521
91 Krivtsov, A.V. et al. (2006) Transformation from
committed progenitor to leukaemia stem cell
initiated by MLL-AF9. Nature 442, 818-822
92 Daser, A. and Rabbitts, T.H. (2005) The versatile
mixed lineage leukaemia gene MLL and its many
associations in leukaemogenesis. Semin CancerBiol 15, 175-188
93 Anonymous (2006) European Journal of Cancer
Special Issue on Cancer Stem Cells: Opportunities
for Novel Diagnostics and Drug Discovery. 42,
1298-1308
94 Bandyopadhyay, D. et al. (2002) Down-regulation
of p300/CBP histone acetyltransferase activates a
senescence checkpoint in human melanocytes.
Cancer Res 62, 6231-6239
95 Yao, T.P. et al. (1998) Gene dosage-dependent
embryonic development and proliferation defects
in mice lacking the transcriptional integrator
p300. Cell 93, 361-372
96 Ogryzko, V.V. et al. (1996) Human fibroblast
commitment to a senescence-like state in response
to histone deacetylase inhibitors is cell cycle
dependent. Mol Cell Biol 16, 5210-5218
97 Munro, J. et al. (2004) Histone deacetylase inhibitors
induce a senescence-like state in human cells by a
p16-dependent mechanism that is independent of a
mitotic clock. Exp Cell Res 295, 525-538
expert reviewshttp://www.expertreviews.org/ in molecular medicine
20 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 21/26
98 Place, R.F., Noonan, E.J. and Giardina, C. (2005)
HDACs and the senescent phenotype of WI-38
cells. BMC Cell Biol 6, 37
99 Kaeberlein, M., McVey, M. and Guarente, L.
(1999) The SIR2/3/4 complex and SIR2 alone
promote longevity in Saccharomyces cerevisiae bytwo different mechanisms. Genes Dev 13,
2570-2580
100 Imai, S. et al. (2000) Transcriptional silencing and
longevity protein Sir2 is an NAD-dependent
histone deacetylase. Nature 403, 795-800
101 Hekimi, S. and Guarente, L. (2003) Genetics
and the specificity of the aging process. Science
299, 1351-1354
102 Rogina, B. and Helfand, S.L. (2004) Sir2 mediates
longevity in the fly through a pathway related to
calorie restriction. Proc Natl Acad Sci U S A 101,
15998-16003
103 Cohen, H.Y. et al. (2004) Calorie restrictionpromotes mammalian cell survivalby inducing the
SIRT1 deacetylase. Science 305, 390-392
104 Furuyama, T. et al. (2004) SIR2 is required for
polycomb silencing and is associated with an E(Z)
histone methyltransferase complex. Curr Biol 14,
1812-1821
105 Yeung, F. et al. (2004) Modulation of NF-
kappaB-dependent transcription and cell
survival by the SIRT1 deacetylase. Embo J 23,
2369-2380
106 Brunet, A. et al. (2004) Stress-dependent regulation
of FOXO transcription factors by the SIRT1
deacetylase. Science 303, 2011-2015107 Vaziri, H. et al. (2001) hSIR2(SIRT1) functions as
an NAD-dependent p53 deacetylase. Cell 107,
149-159
108 Bradbury, C.A. et al. (2005) Histone deacetylases in
acute myeloid leukaemia show a distinctive
pattern of expression that changes selectively in
response to deacetylase inhibitors. Leukemia 19,
1751-1759
109 Michishita, E. et al. (2005) Evolutionarily
conserved and nonconserved cellular localizations
and functions of human SIRT proteins. Mol Biol
Cell 16, 4623-4635
110 Ota, H. et al. (2005) Sirt1 inhibitor, Sirtinol, induces
senescence-like growth arrestwith attenuated Ras-
MAPK signaling in human cancer cells. Oncogene
25, 176-185
111 Stancheva, I. (2005) Caught in conspiracy:
cooperation between DNA methylation and
histone H3K9 methylation in the establishment
and maintenance of heterochromatin. Biochem
Cell Biol 83, 385-395
112 Lachner, M. and Jenuwein, T. (2002) The many
faces of histone lysine methylation. Curr Opin Cell
Biol 14, 286-298
113 Vire, E. et al. (2006) The Polycomb group protein
EZH2 directly controls DNA methylation. Nature
439, 871-874114 Esteve, P.O. et al. (2006) Direct interaction between
DNMT1 and G9a coordinates DNA and histone
methylation during replication. Genes Dev 20,
3089-3103
115 Wilson, V.L. and Jones, P.A. (1983) DNA
methylation decreasesin aging but notin immortal
cells. Science 220, 1055-1057
116 Hoal-van Helden,E.G. and vanHelden, P.D. (1989)
Age-related methylation changes in DNA may
reflect the proliferative potential of organs. Mutat
Res 219, 263-266
117 Holliday, R. (1986) Strong effects of 5-azacytidine
on the in vitro lifespan of human diploidfibroblasts. Exp Cell Res 166, 543-552
118 Fairweather, D.S., Fox, M. and Margison, G.P.
(1987) The in vitro lifespan of MRC-5 cells is
shortened by 5-azacytidine-induced
demethylation. Exp Cell Res 168, 153-159
119 Belinsky, S.A. et al.(1996) Increasedcytosine DNA-
methyltransferase activity is target-cell-specific
and an early event in lung cancer. Proc Natl Acad
Sci U S A 93, 4045-4050
120 Slack, A. et al. (1999) DNA methyltransferase is a
downstream effector of cellular transformation
triggered by simian virus 40 large Tantigen. J Biol
Chem 274, 10105-10112121 Rouleau, J., MacLeod, A.R. and Szyf, M. (1995)
Regulation of the DNA methyltransferase by the
Ras-AP-1 signaling pathway. J Biol Chem 270,
1595-1601
122 Issa, J.P. et al. (1994) Methylation of the oestrogen
receptor CpG island links ageing and neoplasia in
human colon. Nat Genet 7, 536-540
123 Young, J.I. and Smith, J.R. (2001) DNA
methyltransferase inhibition in normal human
fibroblasts induces a p21-dependent cell cycle
withdrawal. J Biol Chem 276, 19610-19616
124 Feinberg, A.P. and Vogelstein, B. (1983)
Hypomethylation distinguishes genes of some
human cancers from their normal counterparts.
Nature 301, 89-92
125 Goelz, S.E. et al. (1985) Hypomethylation of DNA
from benign and malignant human colon
neoplasms. Science 228, 187-190
126 Eden, A. et al. (2003) Chromosomal instability and
tumors promoted by DNA hypomethylation.
Science 300, 455
expert reviewshttp://www.expertreviews.org/ in molecular medicine
21 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 22/26
127 Gaudet, F. et al. (2003) Induction of tumors in
mice by genomic hypomethylation. Science 300,
489-492
128 Sakai, T. et al. (1991) Allele-specific
hypermethylation of the retinoblastoma tumor-
suppressor gene. Am J Hum Genet 48, 880-888129 Gonzalez-Zulueta, M. et al. (1995) Methylation of
the 50
CpG island of the p16/CDKN2 tumor
suppressor gene in normal and transformed
human tissues correlates with gene silencing.
Cancer Res 55, 4531-4535
130 Reynolds, P.A. et al. (2006) Tumor suppressor
p16INK4A regulates polycomb-mediated DNA
hypermethylation in human mammary epithelial
cells. J Biol Chem 281, 24790-24802
131 Herskind, C. and Rodemann, H.P. (2000)
Spontaneous and radiation-induced differentiation
of fibroblasts. Exp Gerontol 35, 747-755
132 Robles, S.J. and Adami, G.R. (1998) Agents thatcause DNA double strand breaks lead to
p16INK4a enrichment and the premature
senescence of normal fibroblasts. Oncogene 16,
1113-1123
133 von Zglinicki, T. et al. (1995) Mild hyperoxia
shortens telomeres and inhibits proliferation of
fibroblasts: a model for senescence? Exp Cell Res
220, 186-193
134 Shroff, R. et al. (2004) Distribution and
dynamics of chromatin modification induced by a
defined DNA double-strand break. Curr Biol 14,
1703-1711
135 Karlseder, J., Smogorzewska, A. and de Lange, T.(2002) Senescence induced by altered telomere
state, not telomere loss. Science 295, 2446-2449
136 d’Adda di Fagagna, F. et al.(2003) A DNA damage
checkpoint response in telomere-initiated
senescence. Nature 426, 194-198
137 van Attikum, H. and Gasser, S.M. (2005) The
histone code at DNA breaks: a guide to repair?
Nat Rev Mol Cell Biol 6, 757-765
138 Huyen, Y. et al. (2004) Methylated lysine 79 of
histone H3 targets 53BP1 to DNA double-strand
breaks. Nature 432, 406-411
139 Sanders, S.L. et al.(2004)Methylationof histone H4
lysine 20 controls recruitment of Crb2 to sites of
DNA damage. Cell 119, 603-614
140 Botuyan, M.V. et al. (2006) Structural basis for the
methylation state-specific recognition of histone
H4-K20by53BP1andCrb2inDNArepair.Cell127,
1361-1373
141 Shay, J.W. and Bacchetti, S. (1997) A survey of
telomerase activity in human cancer. Eur J Cancer
33, 787-791
142 Harley, C.B.,Futcher, A.B. and Greider, C.W. (1990)
Telomeres shorten during ageing of human
fibroblasts. Nature 345, 458-460
143 Hastie, N.D. et al. (1990) Telomere reduction in
human colorectal carcinoma and with ageing.
Nature 346, 866-868144 Allsopp, R.C. et al. (1992) Telomere length predicts
replicative capacity of humanfibroblasts. ProcNatl
Acad Sci U S A 89, 10114-10118
145 Kim, N.W. et al. (1994) Specific association of
human telomerase activity withimmortalcells and
cancer. Science 266, 2011-2015
146 Reddel, R.R. (2003) Alternative lengthening of
telomeres, telomerase, and cancer. Cancer Lett
194, 155-162
147 Dunham, M.A. et al. (2000) Telomere maintenance
by recombination in human cells. Nat Genet 26,
447-450
148 Bryan, T.M. and Reddel, R.R. (1997) Telomeredynamics and telomerase activity in in vitro
immortalised human cells.Eur J Cancer33, 767-773
149 Yeager, T.R. et al. (1999) Telomerase-negative
immortalized human cells contain a novel type of
promyelocytic leukemia (PML) body. Cancer Res
59, 4175-4179
150 Henson, J.D. et al. (2005) A robust assay for
alternative lengthening of telomeres in tumors
shows the significance of alternative lengthening
of telomeres in sarcomas and astrocytomas. Clin
Cancer Res 11, 217-225
151 Ulaner, G.A. et al. (2003) Absence of a telomere
maintenance mechanism as a favorable prognosticfactor in patients with osteosarcoma. Cancer Res
63, 1759-1763
152 Hakin-Smith, V. et al. (2003) Alternative
lengthening of telomeres and survival in
patients with glioblastomamultiforme.Lancet 361,
836-838
153 Johnson, J.E. et al. (2005) Multiple mechanisms of
telomere maintenance exist in liposarcomas. Clin
Cancer Res 11, 5347-5355
154 Atkinson, S.P. et al. (2005) Lack of telomerase gene
expression in alternative lengthening of telomere
cells is associated with chromatin remodeling of
the hTR and hTERT gene promoters. Cancer Res
65, 7585-7590
155 Serakinci, N. et al. (2006) Telomerase promoter
reprogramming and interaction with general
transcription factors in the human mesenchymal
stem cell. Regenerative Medicine 1, 125-131
156 Serakinci, N. et al. (2004) Adult human
mesenchymal stem cell as a target for neoplastic
transformation. Oncogene 23, 5095-5098
expert reviewshttp://www.expertreviews.org/ in molecular medicine
22 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 23/26
157 Blasco, M.A. (2004) Carcinogenesis Young
Investigator Award. Telomere epigenetics: a
higher-order control of telomere length in
mammalian cells. Carcinogenesis 25, 1083-1087
158 Garcia-Cao, M. et al. (2004) Epigenetic regulation
of telomere length in mammalian cells by theSuv39h1 and Suv39h2 histone methyltransferases.
Nat Genet 36, 94-99
159 Gonzalo, S. et al. (2006) DNA methyltransferases
control telomerelength andtelomererecombinationin
mammalian cells. Nat Cell Biol 8, 416-424
160 Papworth, M., Kolasinska, P. and Minczuk, M.
(2006) Designer zinc-finger proteins and their
applications. Gene 366, 27-38
161 Won, J. et al. (2006) Small molecule-based
reversible reprogramming of cellular lifespan.
Nat Chem Biol 2, 369-374
162 Galmozzi,E., Facchetti,F.and La Porta, C.A. (2006)
Cancer stem cells and therapeutic perspectives.Curr Med Chem 13, 603-607
163 Fuchs, E., Tumbar, T. and Guasch, G. (2004)
Socializing withthe neighbors: stem cells and their
niche. Cell 116, 769-778
164 Zheng, X. et al. (2004) Gamma-catenin contributes
to leukemogenesis induced by AML-associated
translocation products by increasing the
self-renewal of very primitive progenitor cells.
Blood 103, 3535-3543
165 Massard, C., Deutsch, E. and Soria, J.C. (2006)
Tumour stem cell-targeted treatment: elimination
or differentiation. Ann Oncol 17, 1620-1624
166 Shi, S. et al. (2006) JAK signaling globallycounteracts heterochromatic gene silencing. Nat
Genet 38, 1071-1076
167 Seligson, D.B. et al. (2005) Global histone
modification patterns predict risk of prostate
cancer recurrence. Nature 435, 1262-1266
168 Collado, M. and Serrano, M. (2006) The power and
the promise of oncogene-induced senescence
markers. Nat Rev Cancer 6, 472-476
169 Fraga, M.F. and Esteller, M. (2005) Towards the
human cancer epigenome: a first draft of histone
modifications. Cell Cycle 4, 1377-1381
170 Esteller, M. (2006) The necessity of a human
epigenome project. Carcinogenesis 27, 1121-1125
171 Jones, P.A. and Martienssen, R. (2005) A blueprint
for a Human Epigenome Project: the AACR
Human Epigenome Workshop. Cancer Res 65,
11241-11246
172 Matthews, C. et al. (2006) Vascular smooth muscle
cells undergo telomere-based senescence in
human atherosclerosis: effects of telomerase and
oxidative stress. Circ Res 99, 156-164
173 Henderson, E.A. (2006) The potential effect of
fibroblast senescence on wound healing and the
chronic wound environment. J Wound Care 15,
315-318
174 Streit, W.J. (2006) Microglial senescence: does the
brain’s immune system have an expiration date?Trends Neurosci 29, 506-510
175 Shimada, A. et al. (2002) Age-related progressive
neuronal DNA damage associated with cerebral
degeneration in a mouse model of accelerated
senescence. J Gerontol A Biol Sci Med Sci 57,
B415–421
176 Flanary, B. (2005) The role of microglial cellular
senescence in the aging and Alzheimer diseased
brain. Rejuvenation Res 8, 82-85
177 Nyunoya, T. et al. (2006) Cigarette smoke induces
cellular senescence. Am J Respir Cell Mol Biol
35, 681-688
178 Muller, K.C. et al. (2006) Lung fibroblasts frompatients with emphysema show markers of
senescence in vitro. Respir Res 7, 32
179 Jimenez, R. et al. (2005) Replicative senescence in
patients with chronic kidney failure. Kidney Int
Suppl S11– 15
180 Erusalimsky, J.D. and Kurz, D.J. (2005) Cellular
senescence in vivo: its relevance in ageing and
cardiovascular disease. Exp Gerontol 40, 634-642
181 van Baarle, D. et al. (2005) Significance of senescence
for virus-specific memory Tcell responses: rapid
ageing during chronic stimulation of the immune
system. Immunol Lett 97, 19-29
182 Vallejo, A.N., Weyand, C.M. and Goronzy, J.J.(2004) T-cell senescence: a culprit of immune
abnormalities in chronic inflammation and
persistent infection. Trends Mol Med 10, 119-124
183 Joosten, S.A. et al. (2003) Telomere shortening
and cellular senescence in a model of chronic
renal allograft rejection. Am J Pathol 162,
1305-1312
184 Martin, J.A. and Buckwalter, J.A. (2002) Aging,
articular cartilage chondrocyte senescence and
osteoarthritis. Biogerontology 3, 257-264
185 Wiemann, S.U. et al. (2002) Hepatocyte telomere
shortening and senescence are general markers of
human liver cirrhosis. Faseb J 16, 935-942
186 Lu, Q. et al. (2006) Epigenetics, disease, and
therapeutic interventions. Ageing Res Rev
187 Moretti, P. and Zoghbi, H.Y. (2006) MeCP2
dysfunction in Rett syndrome and related
disorders. Curr Opin Genet Dev 16, 276-281
188 Ballestar, E., Esteller, M. and Richardson, B.C.
(2006) The epigenetic face of systemic lupus
erythematosus. J Immunol 176, 7143-7147
expert reviewshttp://www.expertreviews.org/ in molecular medicine
23 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 24/26
189 McKinsey, T.A. and Olson, E.N. (2005) Toward
transcriptional therapies for the failing heart:
chemical screens to modulate genes. J Clin Invest
115, 538-546
190 Sadri-Vakili, G. and Cha, J.H. (2006) Mechanisms
of disease: Histone modifications in Huntington’sdisease. Nat Clin Pract Neurol 2, 330-338
191 Beglopoulos, V. and Shen, J. (2006) Regulation
of CRE-dependent transcription by
presenilins: prospects for therapy of Alzheimer’s
disease. Trends Pharmacol Sci 27, 33-40
192 Langley, B. et al. (2005) Remodeling chromatin
and stress resistance in the central nervous
system: histone deacetylase inhibitors as
novel and broadly effective neuroprotective
agents. Curr Drug Targets CNS Neurol Disord 4,
41-50
193 Taniura, H., Sng, J.C. and Yoneda, Y. (2006)
Histone modifications in status epilepticus
induced by kainate. Histol Histopathol 21,
785-791
194 Adcock, I.M. and Lee, K.Y. (2006) Abnormal
histone acetylase and deacetylase expression andfunction in lung inflammation. Inflamm Res 55,
311-321
195 Hirtz, D. et al. (2005) Challenges and opportunities
in clinical trials for spinal muscular atrophy.
Neurology 65, 1352-1357
196 Bredy, T.W. (2007) Behavioural epigenetics and
psychiatric disorders. Med Hypotheses 68, 453
197 Herman, J.G. and Baylin, S.B. (2003) Gene
silencing in cancer in association with
promoter hypermethylation. N Engl J Med 349,
2042-2054
expert reviewshttp://www.expertreviews.org/ in molecular medicine
24 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 25/26
Further reading, resources and contacts
The Cancer Research UK Laboratories in Glasgow, UK, houses nearly 20 research groups working towards
understanding cancer growth and its treatment:
http://www.beatson.gla.ac.uk /
Antibody and protocol resources
Both Upstate and Abcam are providers of ChIP-grade antibodies and are widely used in the chromatin field
http://www.abcam.com
http://www.upstate.com
Upstate also hosts a new informational resource that details significant developments within the field of
chromatin research:
http://www.histone.com
Chromatin resources
The Chromatin Structure and Function website has excellent links to researchers, journals, search tools and
other related topics. It also includes a mailing list for recent chromatin papers from all the major journals:
http://www.chromatin.us/chrom.html
The Human Epigenome Project homepage:
http://www.epigenome.org/
Chromatin.com keeps up to date with allthe recent high-impact journal articles in thefield of chromatin andalso
with chromatin-related meetings:
http://www.chromatin.com
Other useful resources
The biopharmaceutical company Geron is involved in drug discovery for regenerative medicine and oncology:
http://www.geron.com/
Various laboratory homepages with excellent links on senescence, telomerase and telomeres, further resources
and journal articles:
Jerry Shay’s and Woodring Wright’s laboratory homepage:
http://www4.utsouthwestern.edu/cellbio/shay-wright/index.html
Igor Roninson’s laboratory homepage
http://www.ordwayresearch.org/profile_roninson.html
Judith Campisi’s laboratory homepage
http://www.lbl.gov/lifesciences/CMB/Campisi.html
expert reviewshttp://www.expertreviews.org/ in molecular medicine
25 Accession information: DOI: 10.1017/S1462399407000269; Vol. 9; Issue 7; March 2007
&2007 Cambridge University Press
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n

8/3/2019 Senescence in Disease
http://slidepdf.com/reader/full/senescence-in-disease 26/26
Features associated with this article
Figures
Figure 1. Routes to immortality.
Figure 2. The formation of senescence-associated heterochromatin foci (SAHF) in response to oncogenic insult.
Figure 3. The role of senescence-associated heterochromatin foci (SAHF) in senescence and tumourigenesis.Figure 4. Attainment of pluripotency in normal cells: a possible path towards the cancer stem cell.
Figure 5. Epigenetic regulation of telomere maintenance mechanisms.
Tables
Table 1. Senescence and disease.
Table 2. Chromatin and disease.
Table 3. Current drugs against epigenetic modifiers.
Citation details for this article
Stuart P. Atkinson and W. Nicol Keith (2007) Epigenetic control of cellular senescence in disease: opportunities
for therapeutic intervention. Expert Rev. Mol. Med. Vol. 9, Issue 7, March 2007, DOI: 10.1017/ S1462399407000269
expert reviewshttp://www.expertreviews.org/ in molecular medicine
26
E p i g e n e t i c
c o n t r o l o f
c e l l u l a r s e n e s c e n c
e
i n
d i s e a s e : o p p o
r t u n i t i e s
f o r t h e r a p e u t i c
i n t e
r v e n t i o n




![Review Article The Emerging Role of Senescence in Ocular Disease · 2020-04-07 · aggravate age-related diseases [7]. “Chronic” senescence develops gradually because of progressive](https://img.pdfslide.net/doc/110x75/5f0c5c5c7e708231d43504a9/review-article-the-emerging-role-of-senescence-in-ocular-disease-2020-04-07-aggravate.jpg)