Embed Size (px)
Citation preview

Session 1b Applications, genome sequencing, RNA Seq, GBS
Applications: RNASeq and GBS (Simon)Making RNA-Seq libraries
Making GBS librariesQuality filtering Next-gen sequence data
Applications for RNA-Seq data: finding differentially-expressed genes.Applications for GBS: genetic map making, Genomic Selection (basic intrduction)
1Sunday, April 14, 13
Introduction to gene expression studies
• Why it is so useful
• What you can learn with it.
2Sunday, April 14, 13
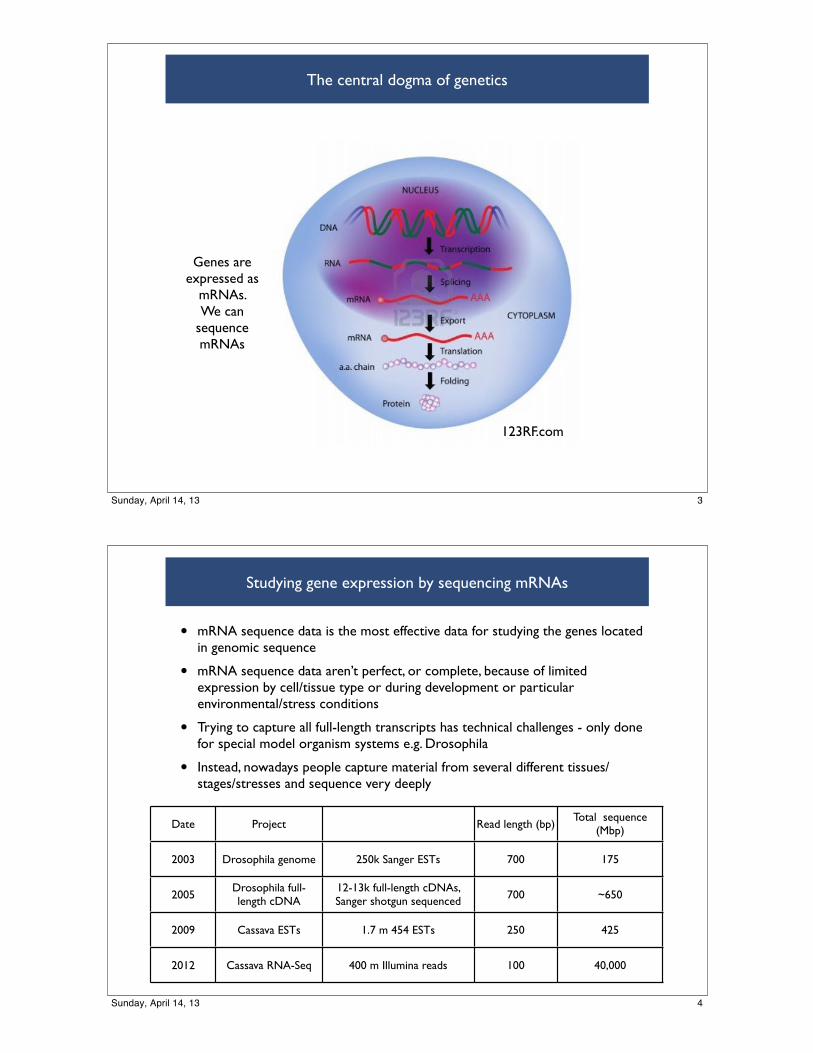
The central dogma of genetics
123RF.com
Genes are expressed as
mRNAs. We can
sequence mRNAs
3Sunday, April 14, 13
Studying gene expression by sequencing mRNAs
• mRNA sequence data is the most effective data for studying the genes located in genomic sequence
• mRNA sequence data aren’t perfect, or complete, because of limited expression by cell/tissue type or during development or particular environmental/stress conditions
• Trying to capture all full-length transcripts has technical challenges - only done for special model organism systems e.g. Drosophila
• Instead, nowadays people capture material from several different tissues/stages/stresses and sequence very deeply
Date Project Read length (bp)Total sequence
(Mbp)
2003 Drosophila genome 250k Sanger ESTs 700 175
2005Drosophila full-length cDNA
12-13k full-length cDNAs, Sanger shotgun sequenced 700 ~650
2009 Cassava ESTs 1.7 m 454 ESTs 250 425
2012 Cassava RNA-Seq 400 m Illumina reads 100 40,000
4Sunday, April 14, 13

DNA and genome sequencing
5Sunday, April 14, 13
Cut plant sample and purify DNA
Fragment DNA into several size
ranges
Assemble sequence reads into contigs by
looking for near-perfect overlaps between reads
contig
reads
Long genomic DNA
molecules
Sequence one or both ends of fragments on 454
platform
Construct scaffolds by joining contigs that
overlap sequence read pairs from both ends of long DNA
fragments
GGATCTAGNNNNNNNNNNNNNNNNNNNGGCTATTTCCGaps in the sequence (usually
representing repeat sequences that could not be crossed) are filled with Ns
Genome sequence
Sequence read
DNA fragment
paired-end readscaffold
Whole genome shotgun sequencing
6Sunday, April 14, 13

The genome of cassava has been sequenced
• JGI CSP pilot 1x coverage from over 700,000 Sanger shotgun reads using plasmid and fosmid libraries
• Main sequencing effort: partnership with 454 Life Sciences (Roche), Steve Rounsley, Dan Rokhsar, Chinnappa Kodira, and Tim Harkins
• 454 GS FLX Titanium platform. Nearly 61 million 454 reads (single and paired-end) were generated and combined with the Sanger data from the pilot project as input for genome assembly
• $1.3 million grant by the Bill & Melinda Gates Foundation
7Sunday, April 14, 13
Cassava assembly v1
• 11,243 scaffolds
• 416Mb (26% N = gaps)
• scaffold N50/L50 514/180kb
• cucumber assemblies N50/L50 ~60 / ~900kb (Therese’s presentation)
• Cassava has an estimated genome size of ~760Mb
• Remaining 300Mb estimated to be non-genic, repetitive
• 1) A large fraction of reads (both Sanger and 454) were not used by the assembly software, and were primarily repetitive in nature.
• 2) 95% of publicly-available cassava ESTs map to the assembly.
8Sunday, April 14, 13
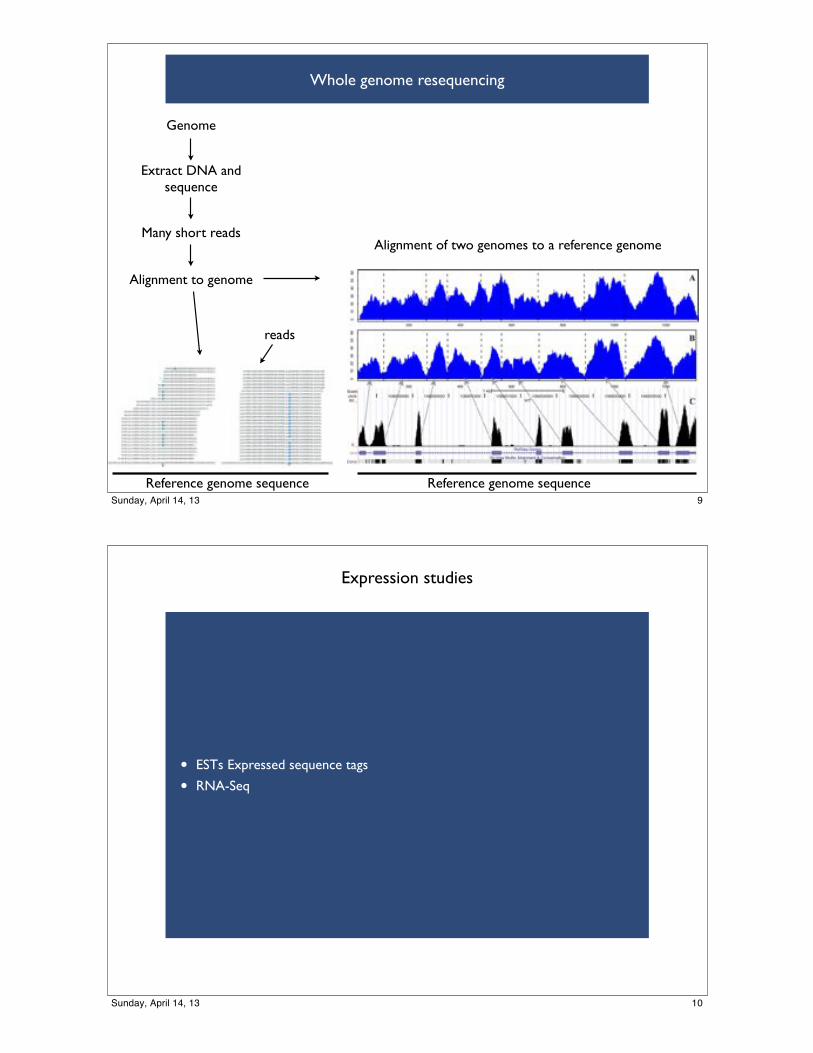
Whole genome resequencing
Genome
Many short reads
Alignment to genome
Alignment of two genomes to a reference genome
Extract DNA and sequence
Reference genome sequenceReference genome sequence
reads
9Sunday, April 14, 13
Expression studies
• ESTs Expressed sequence tags
• RNA-Seq
10Sunday, April 14, 13
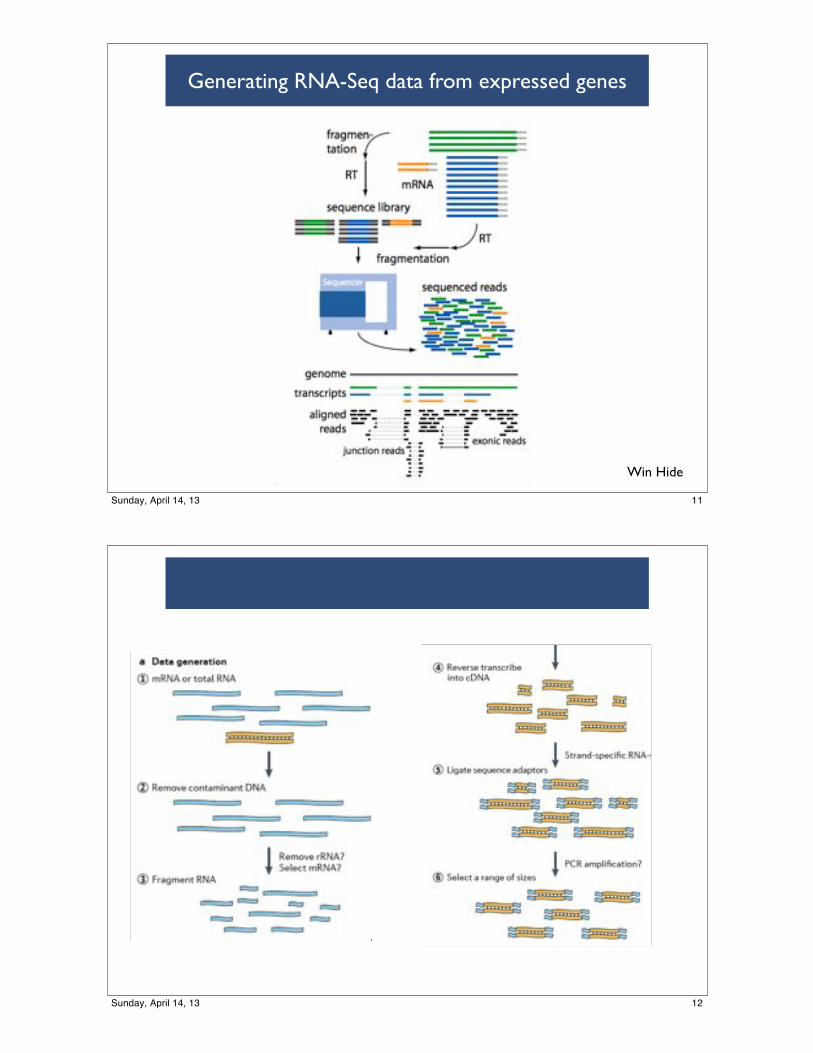
Generating RNA-Seq data from expressed genes
RNA-seq
49
Generate Library(Martin, Wang, 2011)
50
Win Hide
11Sunday, April 14, 13
RNA-seq
49
Generate Library(Martin, Wang, 2011)
50
Ligate and size select
51
Data analysisLow quality reads X
Sequence errors X
52
12Sunday, April 14, 13
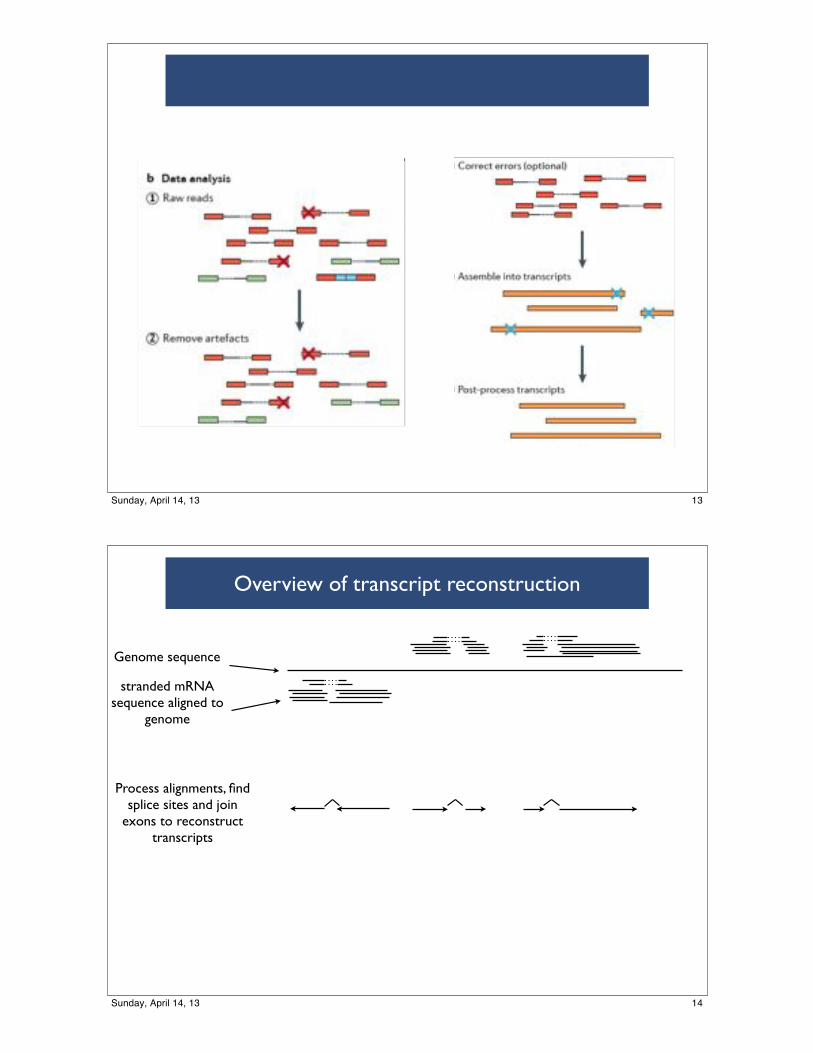
Ligate and size select
51
Data analysisLow quality reads X
Sequence errors X
52
Assemblycorrect assembly errors
53
Estimate expression
54
13Sunday, April 14, 13
Overview of transcript reconstruction
Genome sequence
stranded mRNA sequence aligned to
genome
Process alignments, find splice sites and join
exons to reconstruct transcripts
14Sunday, April 14, 13

Using overlapping k-mers to reconstruct transcripts
!"#$%&'()*&+,,(-./0&1,234&*(&5$6273&8$)9",&
:;8+<=+8=+88<==+8+=8<::&
:;8+<=+8=+& +8=+88<==+8&==+8+=8<::&
:>8+<=>+<=+><=+8>=+8=>+8=+&+8=+>8=+8>=+88>+88<&
==+8>=+8+>+8+=>8+=8>+=8<>:;&>88<=>8<==><==+>==+8&
?(&5$6273&&8$)9"&=#3,%$6@A#3&
BCDEF&
!(G6(3@(&'(@#3,%$6@A#3&50&H)%"&<$)I($,)/& !"#$%&$#&$##%&&$#$&#%!!'
<)$4(%&
'()*,&
Brian Haas
15Sunday, April 14, 13
Generating and analyzing EST sequences
16Sunday, April 14, 13

Sequences obtained for the cassava genome
represented in an assembly of publicly available cassava ESTsequences (http://cassava.igs.umaryland.edu/blast/db/EST_asmbl_and_single.fasta), 96% can be mapped to thegenome assembly. It can be estimated that the remainingportion of the genome is largely repetitive and non-gene-coding. Consistent with this, the fractions of the estimatedgenome size (~31%) and WGS reads (~36%) that do notappear in the assembly are approximately equal, despite lowread error rates. The scaffolds obtained have yet to be assignedto chromosomes, as this requires genetic markers with knownsequence. However, an 88% complete genetic map comprising
23-linkage groups includes the genetic locations of 284 scaf-folds (Sraphet et al. 2011).
With the gene-rich portion of the genome in hand, thenext step is to identify the protein-coding genes and theexons that comprise them. This is achieved computationallyby aligning sequences from mRNA fragments (ESTs) to thegenome, as well as looking for regions with homology toknown proteins from other plant species. The 80,459 SangerESTs from Genbank were augmented by a new set of 2.7million reads from leaf and root libraries, generated by 454Life Sciences using the FLX Titanium platform. While half
b
aFig. 1 a Overview of wholegenome shotgun sequencingand assembly. Starting withplant material, many genomes’worth of DNA is extracted,purified, fragmented, pooledby length and sequenced to ahigh level of redundancy withthe aim of sequencing everyregion of the genome so that thechromosomal sequence canbe generated (assembled) byoverlapping fragments thathave (near-)identical sequences.Longer range, paired-end se-quence information is used tobridge sections of the genomethat are not unique (repeats) andimpossible to resolve by thisapproach b The phytozomegenome browser (http://www.phytozome.net/cassava)provides a portal for accessing,browsing, searching anddownloading all available cas-sava sequence and annotationdata and for comparative plantgenomic analysis
Table 1 Cassava genomic and mRNA sequence data
Sequence type Source Technology Sequencesgenerated
Notes
Genome shotgun Roche 454 Titanium 39,259,112
454 FLX Plus (experimental) 10,785,244
JGI 454 Titanium 21,581,680
Sanger 723,958
U. Maryland & UC Davis Sanger 75,748 BAC-end seq.
Expressed sequence tags (ESTs) Roche 454 Titanium 1.51 M reads (leaf) 0.30 M after removingchloroplast and rDNAsequences
454 Titanium 1.19 M reads (root)
various Sanger 80,459
Tropical Plant Biol. (2012) 5:88–94 91
17Sunday, April 14, 13
ESTs in cassava
• 80,459 ESTs from Manihot esculenta from GenBank database
• Roche 454 sequenced leaf EST library highly redundant
• 1,512,780 reads -> filtered -> 299,509
• Take 10,000 random sequences and group with blastclust 50% length 95% ID
• Four commonest clusters include 30% of reads
• BLASTN &/or TBLASTN a few sequences from each cluster against nr
• Remove 30-50% of ESTS with homology to 18S, 26S rDNA
• seqclean removes short, low-complexity sequences, vector etc
• 1,187,328 root ESTs from 454
gene % EST reads
26S rDNA 48%
18S rDNA 18%
chloroplast 8.5%
Highly redundant leaf EST lib
18Sunday, April 14, 13

Cassava genome at Phytozome
ESTs
Genes
Alternative splice forms
Similar plant proteins
19Sunday, April 14, 13
RNA-Seq Analysis steps
• Filtering
• Constructing transcripts
• Looking at changes in expression
20Sunday, April 14, 13

Tools for processing RNA-Seq data
• fastQC
• trimming and quality filtering tools: fastx toolkit fastx_trimmer, fastx_quality_trimmer.
• For trimming and filtering etc, careful to make sure both reads from Paired-End reads remain in their pairs
• trim adapter with cutadapt or seqPrep
• normalizing reads with diginorm (Titus Brown) or insilinorm (Trinity)
21Sunday, April 14, 13
FastQC generates a detailed report on your sequence fastq file
22Sunday, April 14, 13
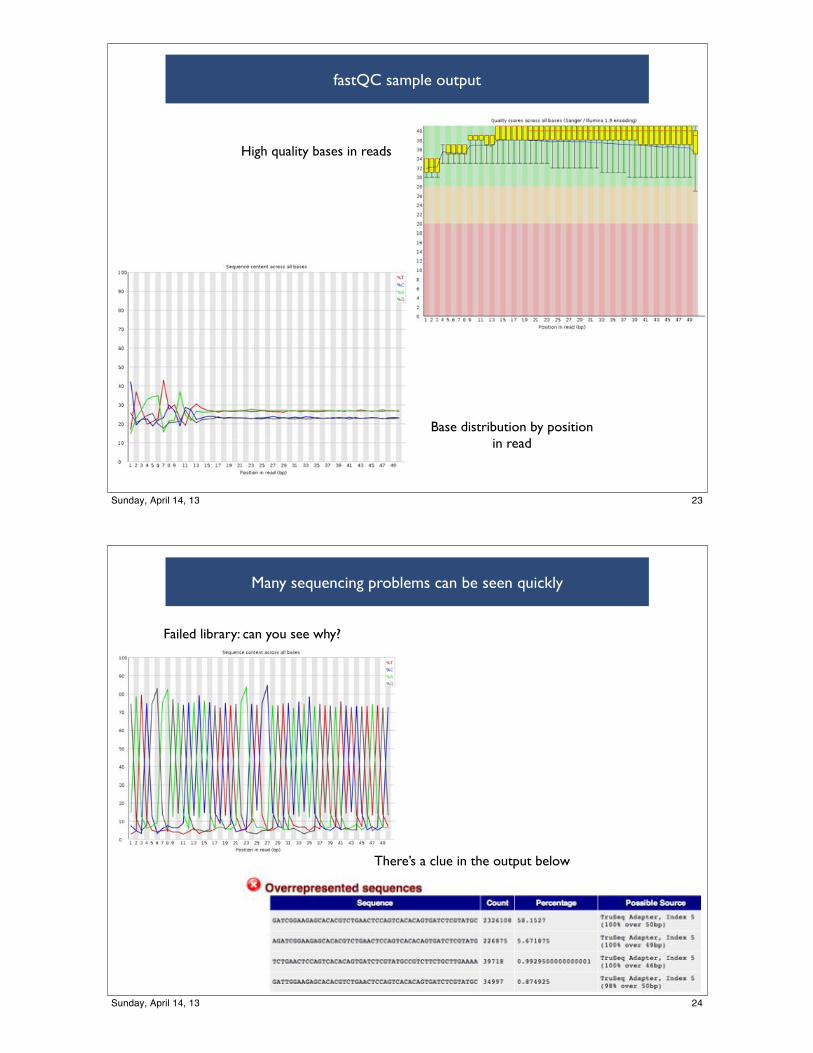
fastQC sample output
Base distribution by position in read
High quality bases in reads
23Sunday, April 14, 13
Many sequencing problems can be seen quickly
Failed library: can you see why?
There’s a clue in the output below
24Sunday, April 14, 13

Digital normalization
Genome sequence
mRNA sequence aligned to genome x
x
x
x
x
x
x
xx
xx x
xx x x
= sequence error. Generates a possible alternative transcript form. This uses up lots of time and memory to process
x
Digital normalization removes all reads
with depth > e.g. 20, leaving a cleaner signal from real
transcripts
25Sunday, April 14, 13
Short read aligners
Win Hide
Bowtie2 maps reads with a single splice site
GSNAP http://research-pub.gene.com/gmap/ fast, accurate short read version of GMAP
Yes
26Sunday, April 14, 13

Trinity overview: Inchworm, Chrysalis, Butterfly
• New in 2012: preprocessing with diginorm (Titus Brown) khmer, screed
• Inchworm builds up k-mers overlapping by 1, starting with the most abundant and using each k-mer exactly once, so alt splice forms are represented by one most complete assembly and partials that overlap k-1 bp.
• Chrysalis collects Inchworm fragments that overlap perfectly by k-1, with minimum number of reads spanning the junction, assigns read regions to appropriate parts of de Bruijn graph thus constructed for each transcript.
• Butterfly resolves ambiguities, constructs alt splice forms, paralogs. First merges adjacent sections and prunes bad edges. Second in DP plausible path scoring reconciles actual read (pair)s with paths.
2 ADVANCE ONLINE PUBLICATION NATURE BIOTECHNOLOGY
complexity of overlaps between variants. Finally, Butterfly (Fig. 1c) analyzes the paths taken by reads and read pairings in the context of the corresponding de Bruijn graph and reports all plausible transcript sequences, resolving alternatively spliced isoforms and transcripts derived from paralogous genes. Below, we describe each of Trinity’s modules.
Inchworm assembles contigs greedily and efficientlyInchworm efficiently reconstructs linear transcript contigs in six steps (Fig. 1a). Inchworm (i) constructs a k-mer dictionary from all sequence reads (in practice, k = 25); (ii) removes likely error-containing k-mers from the k-mer dictionary; (iii) selects the most frequent k-mer in the dictionary to seed a contig assembly, excluding both low-complexity
For transcriptome assembly, each path in the graph represents a possible transcript. A scoring scheme applied to the graph structure can rely on the original read sequences and mate-pair information to discard non-sensical solutions (transcripts) and compute all plausible ones.
Applying the scheme of de Bruijn graphs to de novo assembly of RNA-Seq data represents three critical challenges: (i) efficiently construct-ing this graph from large amounts (billions of base pairs) of raw data; (ii) defining a suitable scoring and enumeration algorithm to recover all plausible splice forms and paralogous transcripts; and (iii) providing robustness to the noise stemming from sequencing errors and other artifacts in the data. In particular, sequencing errors would introduce a large number of false nodes, resulting in a massive graph with millions of possible (albeit mostly implausible) paths.
Here, we present Trinity, a method for the efficient and robust de novo reconstruction of transcriptomes, consisting of three software modules: Inchworm, Chrysalis and Butterfly, applied sequentially to process large volumes of RNA-Seq reads. We evaluated Trinity on data from two well-annotated species—one microorganism (fission yeast) and one mam-mal (mouse)—as well as an insect (the whitefly Bemisia tabaci), whose genome has not yet been sequenced. In each case, Trinity recovers most of the reference (annotated) expressed tran-scripts as full-length sequences, and resolves alternative isoforms and duplicated genes, per-forming better than other available transcrip-tome de novo assembly tools, and similarly to methods relying on genome alignments.
RESULTSTrinity: a method for de novo transcriptome assemblyIn contrast to de novo assembly of a genome, where few large connected sequence graphs can represent connectivities among reads across entire chromosomes, in assembling transcriptome data we expect to encounter numerous individual disconnected graphs, each representing the transcriptional com-plexity at nonoverlapping loci. Accordingly, Trinity partitions the sequence data into these many individual graphs, and then processes each graph independently to extract full-length isoforms and tease apart transcripts derived from paralogous genes.
In the first step in Trinity, Inchworm assembles reads into the unique sequences of transcripts. Inchworm (Fig. 1a) uses a greedy k-mer–based approach for fast and efficient transcript assembly, recovering only a single (best) representative for a set of alternative variants that share k-mers (owing to alterna-tive splicing, gene duplication or allelic varia-tion). Next, Chrysalis (Fig. 1b) clusters related contigs that correspond to portions of alterna-tively spliced transcripts or otherwise unique portions of paralogous genes. Chrysalis then constructs a de Bruijn graph for each cluster of related contigs, each graph reflecting the
cba
>a121:len = 5,845
>a122:len = 2,560
>a123:len = 4,443
>a124:len = 48
>a126:len = 66
k – 1
Read set
Extend in k-merspace andbreak ties
Linear sequences
...
!
A
A
A A
A
CGT
CTC
G
TCGT
T C
T G
T C
T* C
... ... ......
Overlap linearsequences byoverlaps of k – 1to build graphcomponents
De Bruijngraph (k = 5)
Compactgraph
Compact graphwith reads
Transcripts
Compacting
Finding paths
Extracting sequences
ATTCG CTTCG
TTCGC
TCGCA
CGCAA
GCAAT
CAATG CAATC
AATGA AATCA
ATGAT ATCAT
TGATC TCATC
GATCG CATCG
ATCGG
TCGGA
CGGAT
... ...
A C
TTCGCAA...T
ATCGGAT...
CG
... ...
A C
CG
... ...
...CTTCGCAA...TGATCGGAT...
...ATTCGCAA...TCATCGGAT...
k – 1
k – 1
k – 1
k – 1
TTCGCAA...T
ATCGGAT...
Figure 1 Overview of Trinity. (a) Inchworm assembles the read data set (short black lines, top) by greedily searching for paths in a k-mer graph (middle), resulting in a collection of linear contigs (color lines, bottom), with each k-mer present only once in the contigs. (b) Chrysalis pools contigs (colored lines) if they share at least one k – 1-mer and if reads span the junction between contigs, and then it builds individual de Bruijn graphs from each pool. (c) Butterfly takes each de Bruijn graph from Chrysalis (top), and trims spurious edges and compacts linear paths (middle). It then reconciles the graph with reads (dashed colored arrows, bottom) and pairs (not shown), and outputs one linear sequence for each splice form and/or paralogous transcript represented in the graph (bottom, colored sequences).
ART ICL ES
Grabherr et al.
27Sunday, April 14, 13
Transcript reconstruction with a reference genome: the Tuxedo suite by Trapnell et al.
©20
12 N
atur
e A
mer
ica,
Inc.
All
righ
ts r
eser
ved.
PROTOCOL
564 | VOL.7 NO.3 | 2012 | NATURE PROTOCOLS
feel comfortable creating directories, moving files between them and editing text files in a UNIX environment. Installation of the tools may require additional expertise and permission from one’s computing system administrators.
Read alignment with TopHatAlignment of sequencing reads to a reference genome is a core step in the analysis workflows for many high-throughput sequencing assays, including ChIP-Seq31, RNA-seq, ribosome profiling32 and others. Sequence alignment itself is a classic problem in computer science and appears frequently in bioinformatics. Hence, it is per-haps not surprising that many read alignment programs have been developed within the last few years. One of the most popular and to date most efficient is Bowtie33 (http://bowtie-bio.sourceforge.net/index.shtml), which uses an extremely economical data structure called the FM index34 to store the reference genome sequence and allows it to be searched rapidly. Bowtie uses the FM index to align reads at a rate of tens of millions per CPU hour. However, Bowtie is not suitable for all sequence alignment tasks. It does not allow alignments between a read and the genome to contain large gaps; hence, it cannot align reads that span introns. TopHat was created to address this limitation.
TopHat uses Bowtie as an alignment ‘engine’ and breaks up reads that Bowtie cannot align on its own into smaller pieces called seg-ments. Often, these pieces, when processed independently, will align to the genome. When several of a read’s segments align to the genome far apart (e.g., between 100 bp and several hundred kilobases) from one another, TopHat infers that the read spans a splice junction and estimates where that junction’s splice sites are. By processing each ‘initially unmappable’ read, TopHat can build up an index of splice sites in the transcriptome on the fly without a priori gene or splice site annotations. This capability is crucial, because, as numerous RNA-seq studies have now shown, our cata-logs of alternative splicing events remain woefully incomplete. Even in the transcriptomes of often-studied model organisms, new splic-ing events are discovered with each additional RNA-seq study.
Aligned reads say much about the sample being sequenced. Mismatches, insertions and deletions in the alignments can iden-tify polymorphisms between the sequenced sample and the ref-erence genome, or even pinpoint gene fusion events in tumor samples. Reads that align outside annotated genes are often strong evidence of new protein-coding genes and noncoding RNAs. As mentioned above, RNA-seq read alignments can reveal new alter-native splicing events and isoforms. Alignments can also be used to accurately quantify gene and transcript expression, because the number of reads produced by a transcript is proportional to its abundance (Box 2). Discussion of polymorphism and fusion
detection is out of the scope of this protocol, and we address transcript assembly and gene discovery only as they relate to dif-ferential expression analysis. For a further review of these topics, see Garber et al.12.
Transcript assembly with CufflinksAccurately quantifying the expression level of a gene from RNA-seq reads requires accurately identifying which isoform of a given gene produced each read. This, of course, depends on knowing all of the splice variants (isoforms) of that gene. Attempting to quantify gene and transcript expression by using an incomplete or incorrect transcriptome annotation leads to inaccurate expression values8. Cufflinks assembles individual transcripts from RNA-seq reads that have been aligned to the genome. Because a sample may contain reads from multiple splice variants for a given gene, Cufflinks must be able to infer the splicing structure of each gene. However, genes sometimes have multiple alternative splicing events, and there may be many possible reconstructions of the gene model that explain the sequencing data. In fact, it is often not obvious how many splice variants of the gene may be present. Thus, Cufflinks reports a parsi-monious transcriptome assembly of the data. The algorithm reports as few full-length transcript fragments or ‘transfrags’ as are needed to ‘explain’ all the splicing event outcomes in the input data.
TopHat
Cufflinks
Cuffmerge
Finaltranscriptome
assembly
Condition A
Reads
Mappedreads
Assembledtranscripts
Mappedreads
Condition B
Differentialexpression results
Cuffdiff
Expression plots
CummeRbund
Reads
Mappedreads
Assembledtranscripts
Mappedreads
Step 1
Step 2
Steps 3–4
Step 5
Steps 6–18
Figure 2 | An overview of the Tuxedo protocol. In an experiment involving two conditions, reads are first mapped to the genome with TopHat. The reads for each biological replicate are mapped independently. These mapped reads are provided as input to Cufflinks, which produces one file of assembled transfrags for each replicate. The assembly files are merged with the reference transcriptome annotation into a unified annotation for further analysis. This merged annotation is quantified in each condition by Cuffdiff, which produces expression data in a set of tabular files. These files are indexed and visualized with CummeRbund to facilitate exploration of genes identified by Cuffdiff as differentially expressed, spliced, or transcriptionally regulated genes. FPKM, fragments per kilobase of transcript per million fragments mapped.
©20
12 N
atur
e A
mer
ica,
Inc.
All
righ
ts r
eser
ved.
PROTOCOL
NATURE PROTOCOLS | VOL.7 NO.3 | 2012 | 563
TopHat and Cufflinks are both operated through the UNIX shell. No graphical user interface is included. However, there are now commercial products and open-source interfaces to these and other RNA-seq analysis tools. For example, the Galaxy Project18 uses a web interface to cloud computing resources to bring command-line–driven tools such as TopHat and Cufflinks to users without UNIX skills through the web and the computing cloud.
Alternative analysis packagesTopHat and Cufflinks provide a complete RNA-seq workflow, but there are other RNA-seq analysis packages that may be used instead of or in combination with the tools in this protocol. Many alterna-tive read-alignment programs19–21 now exist, and there are several alternative tools for transcriptome reconstruction22,23, quantifica-tion10,24,25 and differential expression26–28 analysis. Because many of these tools operate on similarly formatted data files, they could be used instead of or in addition to the tools used here. For example, with straightforward postprocessing scripts, one could provide GSNAP19 read alignments to Cufflinks, or use a Scripture22 tran-scriptome reconstruction instead of a Cufflinks one before differ-ential expression analysis. However, such customization is beyond the scope of this protocol, and we discourage novice RNA-seq users from making changes to the protocol outlined here.
This protocol is appropriate for RNA-seq experiments on organ-isms with sequenced reference genomes. Users working without a sequenced genome but who are interested in gene discovery should consider performing de novo transcriptome assembly using one of several tools such as Trinity29, Trans-Abyss30 or Oases (http://www.ebi.ac.uk/~zerbino/oases/). Users performing expression ana-lysis with a de novo transcriptome assembly may wish to consider RSEM10 or IsoEM25. For a survey of these tools (including TopHat and Cufflinks) readers may wish to see the study by Garber et al.12, which describes their comparative advantages and disadvantages and the theoretical considerations that inform their design.
Overview of the protocolAlthough RNA-seq experiments can serve many purposes, we describe a workflow that aims to compare the transcriptome pro-files of two or more biological conditions, such as a wild-type versus mutant or control versus knockdown experiments. For simplicity, we assume that the experiment compares only two biological con-ditions, although the software is designed to support many more, including time-course experiments.
This protocol begins with raw RNA-seq reads and concludes with publication-ready visualization of the analysis. Figure 2 highlights the main steps of the protocol. First, reads for each condition are mapped to the reference genome with TopHat. Many RNA-seq users are also interested in gene or splice variant discovery, and the failure to look for new transcripts can bias expression estimates and reduce accuracy8. Thus, we include transcript assembly with
Cufflinks as a step in the workflow (see Box 1 for a workflow that skips gene and transcript discovery). After running TopHat, the resulting alignment files are provided to Cufflinks to generate a transcriptome assembly for each condition. These assemblies are then merged together using the Cuffmerge utility, which is included with the Cufflinks package. This merged assembly provides a uni-form basis for calculating gene and transcript expression in each condition. The reads and the merged assembly are fed to Cuffdiff, which calculates expression levels and tests the statistical signifi-cance of observed changes. Cuffdiff also performs an additional layer of differential analysis. By grouping transcripts into biologi-cally meaningful groups (such as transcripts that share the same transcription start site (TSS)), Cuffdiff identifies genes that are dif-ferentially regulated at the transcriptional or post-transcriptional level. These results are reported as a set of text files and can be displayed in the plotting environment of your choice.
We have recently developed a powerful plotting tool called CummeRbund (http://compbio.mit.edu/cummeRbund/), which provides functions for creating commonly used expression plots such as volcano, scatter and box plots. CummeRbund also han-dles the details of parsing Cufflinks output file formats to con-nect Cufflinks and the R statistical computing environment. CummeRbund transforms Cufflinks output files into R objects suitable for analysis with a wide variety of other packages available within the R environment and can also now be accessed through the Bioconductor website (http://www.bioconductor.org/).
This protocol does not require extensive bioinformatics exper-tise (e.g., the ability to write complex scripts), but it does assume familiarity with the UNIX command-line interface. Users should
Cufflinks package
Cuffcompare Compares transcript assemblies to annotation
Cuffmerge Merges two or more transcript assemblies
Cuffdiff Finds differentially expressed genes and transcripts Detects differential splicing and promoter use
TopHatAligns RNA-Seq reads to the genome using Bowtie
Discovers splice sites
CummeRbundPlots abundance and differential expression results from Cuffdiff
BowtieExtremely fast, general purpose short read aligner
Cufflinks Assembles transcripts
Figure 1 | Software components used in this protocol. Bowtie33 forms the algorithmic core of TopHat, which aligns millions of RNA-seq reads to the genome per CPU hour. TopHat’s read alignments are assembled by Cufflinks and its associated utility program to produce a transcriptome annotation of the genome. Cuffdiff quantifies this transcriptome across multiple conditions using the TopHat read alignments. CummeRbund helps users rapidly explore and visualize the gene expression data produced by Cuffdiff, including differentially expressed genes and transcripts.
28Sunday, April 14, 13

Expression data (RNA-Seq, ESTs) can detect
• New exons, splice forms
• Differentially-expressed genes
29Sunday, April 14, 13
<
Additional 3’ exon predicted by GeneMark and supported by EST evidence. PASA buills this and other transcripts.
30Sunday, April 14, 13

Novel exon discovery
some slides from Win
Win Hide
31Sunday, April 14, 13
!"#$%&'()*+,-.#)//'"0+1%&2)/++• !"#$%&'()*+3"#+4"56+&)0756+"3+56)+5#%0/8#'.5+%0*+5"5%&+*).56+"3+/)92)08'07:+
• !2$4)#+"3+;!<=>)9+!#%7$)05/++++++")#+#'&"4%/)+"3+5#%0/8#'.5+++++++++++++.)#+5"5%&+$'&&'"0+3#%7$)05/+$%..)*+
!"#$%!"5)?+&@AB+C+&)%*/+.)#+D+++'0/5)%*+"3+E#%7$)05/+'/+"F)0+2/)*+G'56+/'07&)=)0*+#)%*/:+
Brian Haas
32Sunday, April 14, 13
!"#$%&'($)*+(,#-#$%.//'*012-#33#"*4-.$35-(263*
• 76.%3%5./*6#363*2#-&8-9#"*8$*&-.)9#$6*58:$63*;$86*<=>?*@./:#3AB*
• C(@#$*8D3#-@#"*-#."*58:$63*&8-*.*6-.$35-(26*($*#.5E*8&*6F8*3.92/#3G*FE.6H3*6E#*2-8D.D(/(6'*6E#'*F#-#*"#-(@#"*&-89*6E#*3.9#*"(36-(D:%8$*;$://*E'286E#3(3AI**;#1B*<(3E#-3*#1.56*6#36A*!&*;=*JK*LBLMAG*3()$(N5.$6/'*"(,#-#$6*
• +8$H6*&8-)#6*68*."O:36*=P@./:#3*":#*68*&./3#*"(358@#-'*-.6#*;<+QA*-#3:/%$)*&-89*-:$$($)*9.$'*;6E8:3.$"3*8&A*36.%3%5./*6#363B**;#1B*:3#*RP@./:#3A*
Brian Haas
33Sunday, April 14, 13
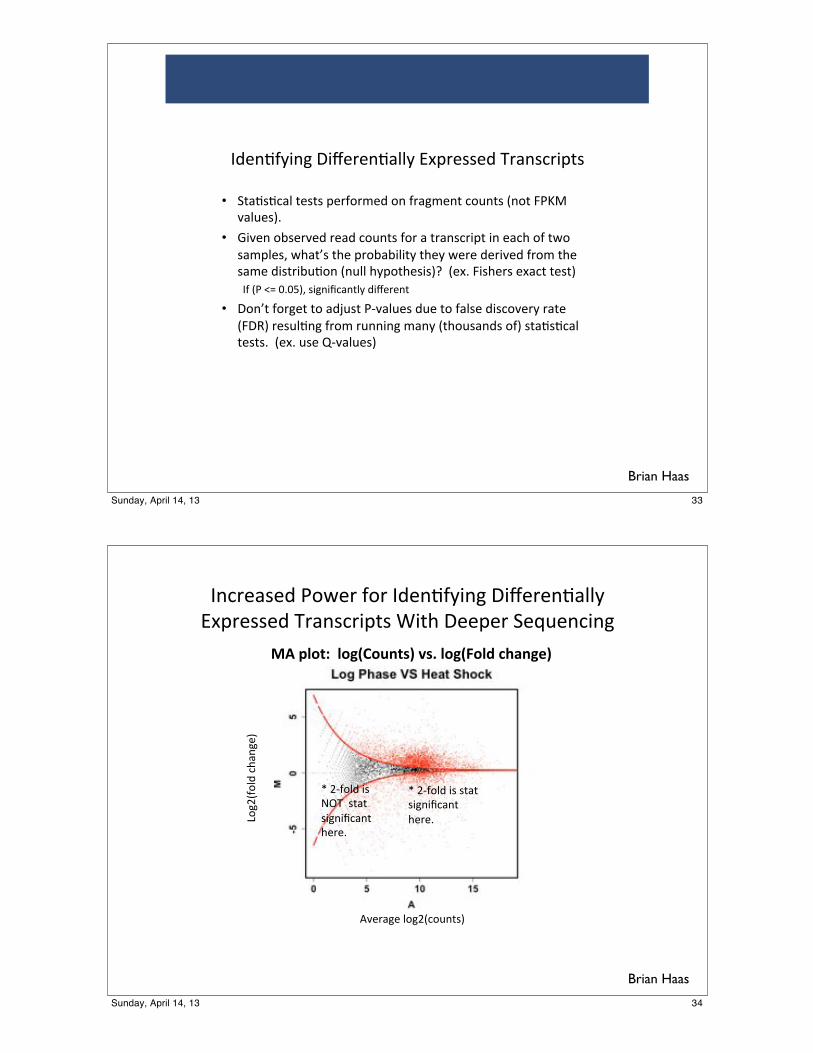
!"#$%&'%()*+,%$)-+$)!(%".-/0"1)203%$%".&44/)
567$%''%()8$&"'#$079'):09;)2%%7%$)<%=>%"#0"1)
?+1@A-+4()#;&"1%B)
CD%$&1%)4+1@A#+>"9'B)
!"#$%&'(##%&)*+&,-'./#0.1#%&)*2&%3#456-)7/#
E)@F-+4()0')'9&9)'01"0G#&"9)
;%$%H)
E)@F-+4()0')IJ8))'9&9)
'01"0G#&"9);%$%H)
Brian Haas
34Sunday, April 14, 13
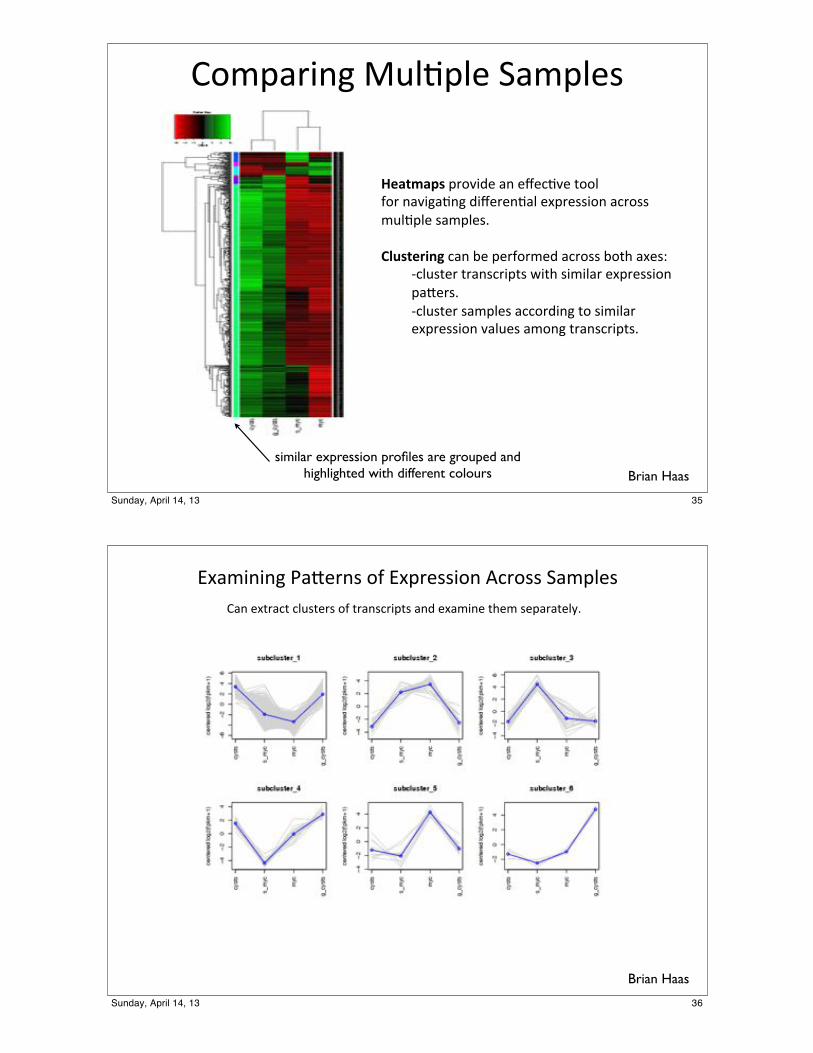
!"#$%&'()*+,-.$-/*0%#$-/1*
!"#$%#&'*$&"2'3/*%(*/4/5.2/*6""-*7"&*(%2')%.()*3'4/&/(.%-*/8$&/11'"(*%5&"11*#,-.$-/*1%#$-/19**()*'$"+,-.*5%(*:/*$/&7"&#/3*%5&"11*:"6;*%8/1<*
*=5-,16/&*6&%(15&'$61*>'6;*1'#'-%&*/8$&/11'"(**$%?/&19**=5-,16/&*1%#$-/1*%55"&3'()*6"*1'#'-%&**/8$&/11'"(*2%-,/1*%#"()*6&%(15&'$619***
Brian Haassimilar expression profiles are grouped and
highlighted with different colours
35Sunday, April 14, 13
!"#$%&%&'()#*+,&-(./(!"0,+--%.&(12,.--(3#$04+-(5#&(+"6,#26(247-6+,-(./(6,#&-2,%06-(#&8(+"#$%&+(69+$(-+0#,#6+4:;(
Brian Haas
36Sunday, April 14, 13

Genotyping by sequencing
• Reduced-representation sequencing
• Genotyping by sequencing and variant analysis
• Genetic mapping
37Sunday, April 14, 13
Making a GBS library: overview
ApeKI: G C W G C C G W C G
W= A or T
1. Digest genomic DNA
3. Pool adapter-ligated DNA 4. Size select: 400–800 bp
5. PCR: 5–10 cyclesTbarcodeCWG CWGbarcodeAAbarcodeGWC GWCbarcodeT
6. Sequence: 100 bp paired-end reads on HiSeq 2000
5’ ACACTCTTTCCCTACACGA
3’ GAGCC
GTAAGGAC
GACTTG
!"#$%&'()*
CGCTCTTCCGATCTbarcode 3’GCGAGAAGGCTAGAbarcodeGWC 5’
+,-,,,'./
0,,'./
+,,'./
+-,,,'./
1
2. Ligate barcoded adapters to DNA
Jessica Lyons
38Sunday, April 14, 13
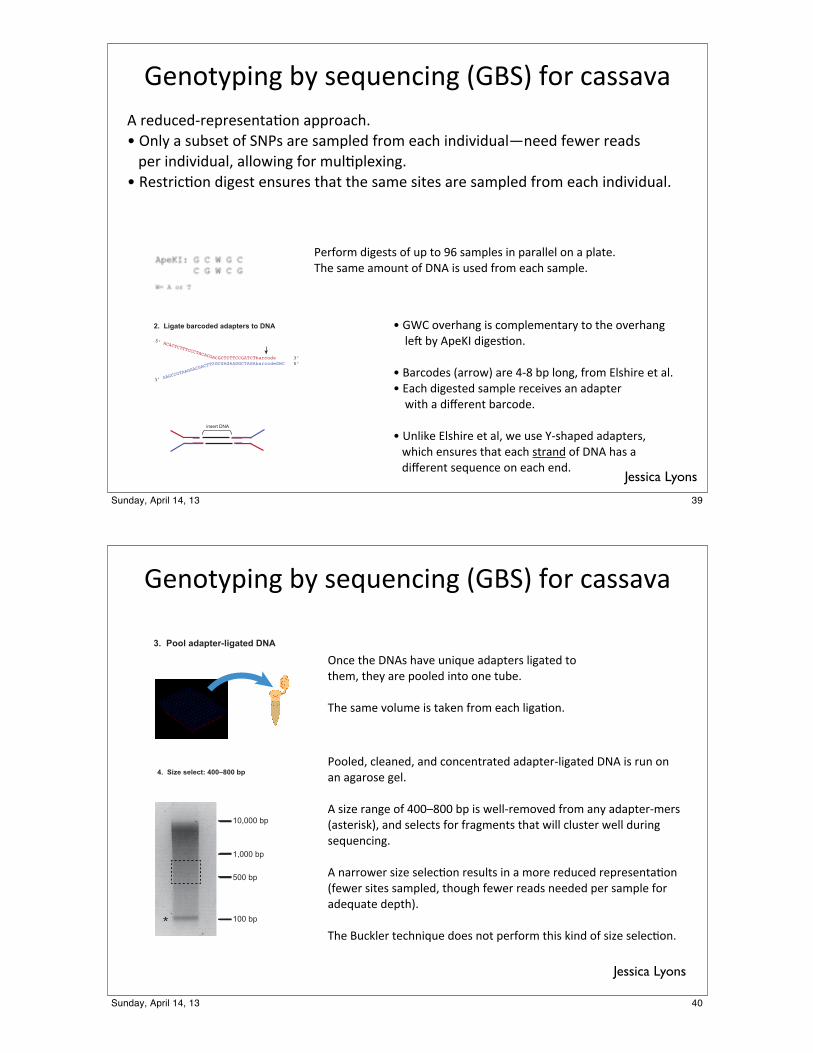
!"#$%&'(#)*+&*,"-."#/(#)*0!123*4$5*/6,,6768*5"9./"9:5"'5","#%6;$#*6''5$6/<=>*?#@&*6*,.+,"%*$4*2AB,*65"*,6C'@"9*45$C*"6/<*(#9(7(9.6@D#""9*4"E"5*5"69,****'"5*(#9(7(9.6@F*6@@$E(#)*4$5*C.@;'@"G(#)=>*H",%5(/;$#*9()",%*"#,.5",*%<6%*%<"*,6C"*,(%",*65"*,6C'@"9*45$C*"6/<*(#9(7(9.6@=
B"54$5C*9()",%,*$4*.'*%$*IJ*,6C'@",*(#*'656@@"@*$#*6*'@6%"=K<"*,6C"*6C$.#%*$4*LA8*(,*.,"9*45$C*"6/<*,6C'@"=
>*!MN*$7"5<6#)*(,*/$C'@"C"#%65&*%$*%<"*$7"5<6#)*****@"O*+&*8'"PQ*9()",;$#=
>*165/$9",*0655$E3*65"*R:S*+'*@$#)F*45$C*T@,<(5"*"%*6@=>*T6/<*9()",%"9*,6C'@"*5"/"(7",*6#*696'%"5*****E(%<*6*9(U"5"#%*+65/$9"=
>*V#@(W"*T@,<(5"*"%*6@F*E"*.,"*X:,<6'"9*696'%"5,F****E<(/<*"#,.5",*%<6%*"6/<*,%56#9*$4*LA8*<6,*6****9(U"5"#%*,"-."#/"*$#*"6/<*"#9=
!"##$%&'()#*'+,-.).#'.'/()+0#(-#123
5’ ACACTCTTTCCCTACACGA
3’ GAGCC
GTAAGGAC
GACTTG
CGCTCTTCCGATCTbarcode 3’GCGAGAAGGCTAGAbarcodeGWC 5’
!"#$%&'()*
Jessica Lyons
39Sunday, April 14, 13
!"#$%&'(#)*+&*,"-."#/(#)*0!123*4$5*/6,,676
?#/"*%<"*LA8,*<67"*.#(-."*696'%"5,*@()6%"9*%$*%<"CF*%<"&*65"*'$$@"9*(#%$*$#"*%.+"=
K<"*,6C"*[email protected]"*(,*%6W"#*45$C*"6/<*@()6;$#=
B$$@"9F*/@"6#"9F*6#9*/$#/"#%56%"9*696'%"5:@()6%"9*LA8*(,*5.#*$#*6#*6)65$,"*)"@=
8*,(Y"*56#)"*$4*RZZ[SZZ*+'*(,*E"@@:5"C$7"9*45$C*6#&*696'%"5:C"5,*06,%"5(,W3F*6#9*,"@"/%,*4$5*456)C"#%,*%<6%*E(@@*/@.,%"5*E"@@*9.5(#)*,"-."#/(#)=
8*#655$E"5*,(Y"*,"@"/;$#*5",.@%,*(#*6*C$5"*5"9./"9*5"'5","#%6;$#*04"E"5*,(%",*,6C'@"9F*%<$.)<*4"E"5*5"69,*#""9"9*'"5*,6C'@"*4$5*69"-.6%"*9"'%<3=
K<"*1./W@"5*%"/<#(-."*9$",*#$%*'"54$5C*%<(,*W(#9*$4*,(Y"*,"@"/;$#=
!"##$%%&#'(')*+,-&./'*+(#012
!"##$%&'#(')'*+,#!--./--#01
!"#"""$%&
'""$%&
!""$%&
!#"""$%&
(
Jessica Lyons
40Sunday, April 14, 13
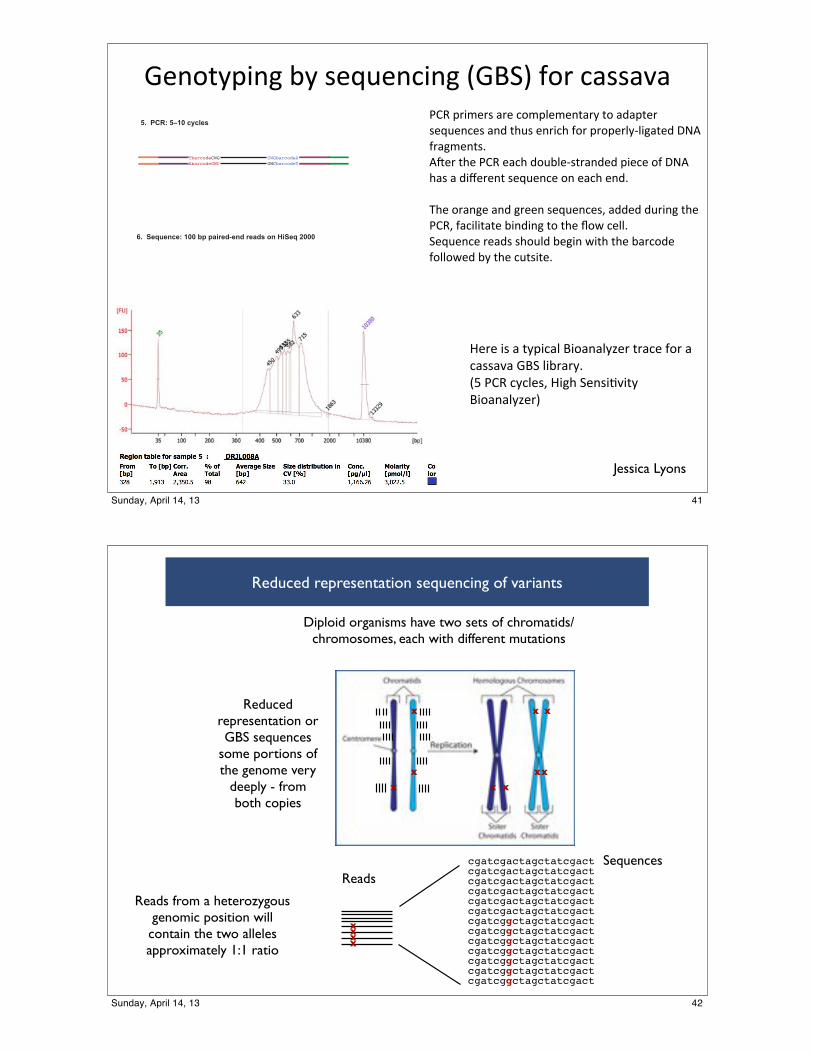
!"#$%&'(#)*+&*,"-."#/(#)*0!123*4$5*/6,,676BNH*'5(C"5,*65"*/$C'@"C"#%65&*%$*696'%"5*,"-."#/",*6#9*%<.,*"#5(/<*4$5*'5$'"5@&:@()6%"9*LA8*456)C"#%,=8O"5*%<"*BNH*"6/<*9$.+@":,%56#9"9*'("/"*$4*LA8*<6,*6*9(U"5"#%*,"-."#/"*$#*"6/<*"#9=**
K<"*$56#)"*6#9*)5""#*,"-."#/",F*699"9*9.5(#)*%<"*BNHF*46/(@(%6%"*+(#9(#)*%$*%<"*\$E*/"@@=2"-."#/"*5"69,*,<$.@9*+")(#*E(%<*%<"*+65/$9"*4$@@$E"9*+&*%<"*/.%,(%"=
]"5"*(,*6*%&'(/6@*1($6#6@&Y"5*%56/"*4$5*6*/6,,676*!12*@(+565&=0^*BNH*/&/@",F*]()<*2"#,(;7(%&*1($6#6@&Y"53
!"##$%&'#!()*#+,+-./
TbarcodeCWG CWGbarcodeAAbarcodeGWC GWCbarcodeT
!"##$%&'%()%*#+,,#-.#./01%23%(2#1%/24#5(#60$%,,,
Jessica Lyons
41Sunday, April 14, 13
Reduced representation sequencing of variants
Diploid organisms have two sets of chromatids/chromosomes, each with different mutations
xx
x
x
x
x
x
x x
Reduced representation or GBS sequences
some portions of the genome very
deeply - from both copies
xxxx
Reads from a heterozygous genomic position will contain the two alleles approximately 1:1 ratio
cgatcgactagctatcgactcgatcgactagctatcgactcgatcgactagctatcgactcgatcgactagctatcgactcgatcgactagctatcgactcgatcgactagctatcgactcgatcggctagctatcgactcgatcggctagctatcgactcgatcggctagctatcgactcgatcggctagctatcgactcgatcggctagctatcgactcgatcggctagctatcgactcgatcggctagctatcgact
ReadsSequences
42Sunday, April 14, 13
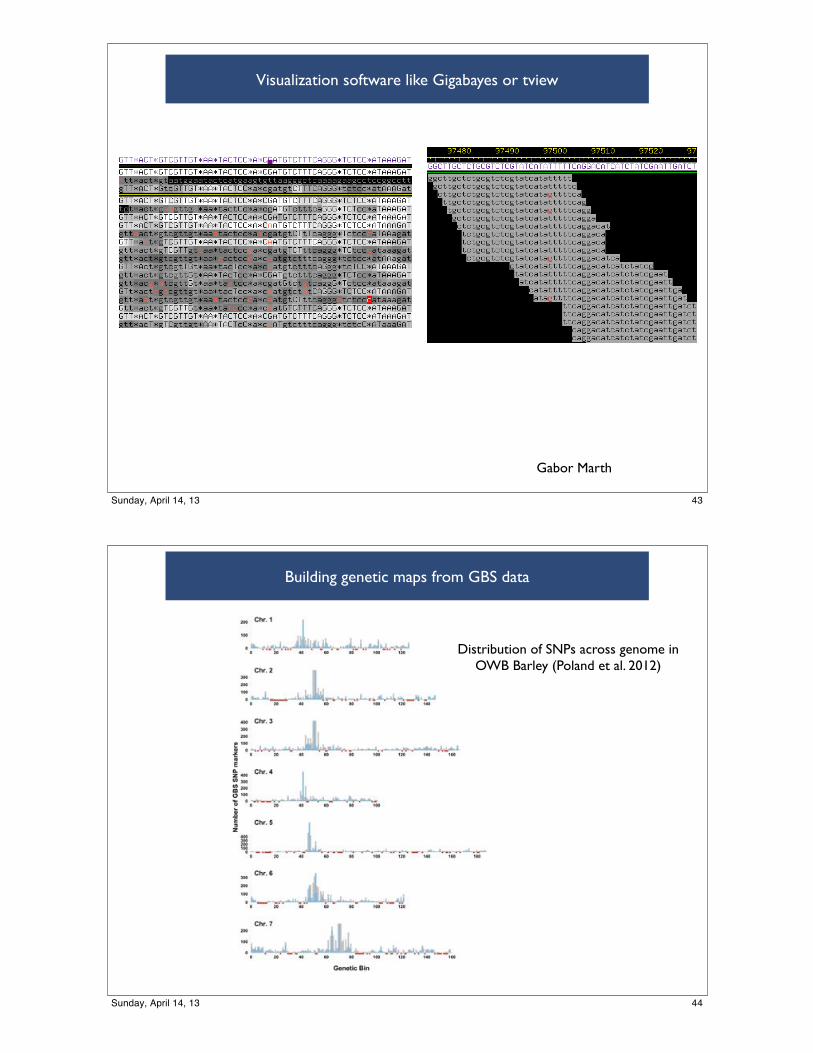
Visualization software like Gigabayes or tview
!"""#$%&'$()*$+,-./$0&123$4(55")6$
Gabor Marth
43Sunday, April 14, 13
Building genetic maps from GBS data
Distribution of SNPs across genome in OWB Barley (Poland et al. 2012)
44Sunday, April 14, 13

Building genetic maps from GBS data with JoinMap
JoinMap encoding of parental genotypes
lm x ll nn x np
Expected Genotype Frequencies in progeny
ll: 0.5
lm: 0.5
mm: 0.0
nn: 0.5
np: 0.5
pp: 0.0
chromosome position SNP type
chr1 354 lm
chr1 4983 lm
chr1 4985 lm
chr1 10876 ll
chr2 765 ll
chr2 1034526 lm
chr2 1034700 ll
chr3 45673 lm
etc
Example SNPs
A/G x A/A C/C x C/A
Data table encoding characters or markers for making the map
45Sunday, April 14, 13
Genomic selection from GBS data
Phenotypic evaluation of cassava is a lengthy process
Make crosses: 10000’s of seedlings
5 to 10 best
500 to 3000 clones
20 to 25 clones
50 to 100 clones
Year 1
Year 2
Year 3
Year 4
Year 5
Year 6 5 best
Martha Hamblin
Evaluation at the Seedling Stageby Genomic Selection
Meuwissen et al. 2001:
Use genome-wide markers to capture effects at many (20-30k) loci
Develop a statistical model to predict breeding value
“Selection on genetic values predicted from markers could substantially increase the rate of genetic gain in animals and plants.”
46Sunday, April 14, 13





























