Embed Size (px)
Citation preview

Shell Scripting Basics
Arun Sethuraman

What’s a shell?
• Command line interpreter for Unix• Bourne (sh), Bourne-again (bash), C shell (csh,
tcsh), etc• Handful of commands• Text mining made easy!

Before we get started
• Unix/Mac Users: Open a terminal• Windows Users: Should have installed
VMware Player, and downloaded the virtual machine with Unix pre-loaded on it (else do it now!)

VMware Player Basics• Allows creating/playing virtual machines• We will use a standalone version of GNU/Linux called SliTaz,
which is very minimalist (< 40 mb), but should work for all our exercises.
• Download all example files from my website: www.sites.google.com/site/arunsethuraman1/teaching instead of from Blackboard.
• Save state of virtual machine, suspend, restart, etc.• Switch environments using CTRL+ALT• File sharing is a little complicated – so before you submit your
assignment for next week, VMware users please email me and stop by my office with your laptop to submit it (unless you can get Gmail to work without any glitches inside Midori).

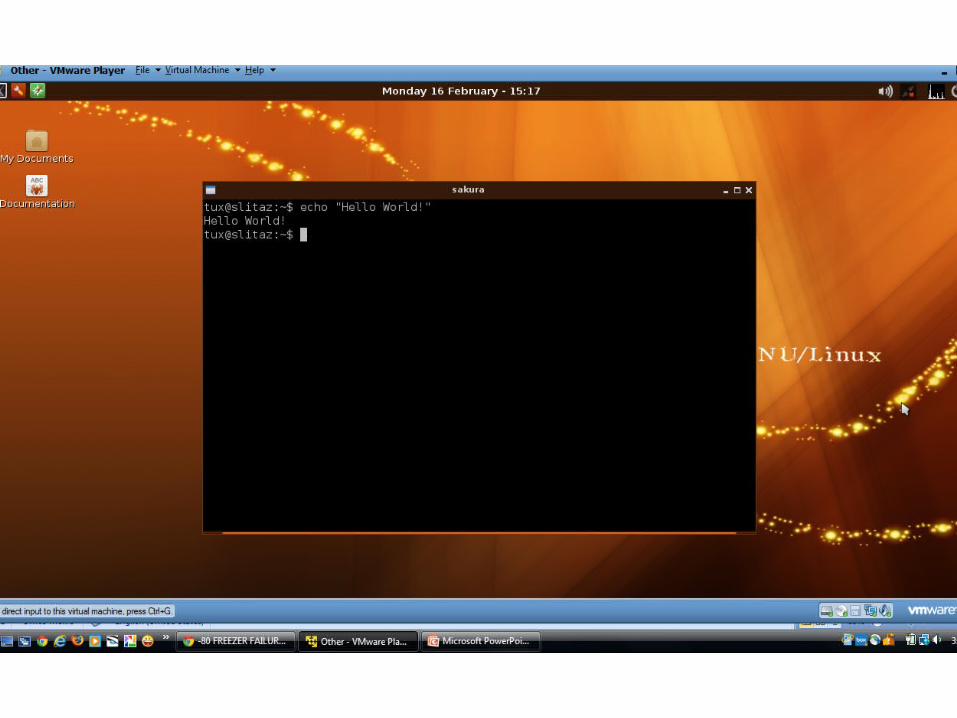

Working at the prompt
• The ‘prompt’ refers to Unix’s native command line interface.
• Your prompt should look something like:username@prompt:~$• Prompt commands are similar to python
scripts – can specify variables, run one-liner commands, specify entire program flows, etc.

Unix 101
Try:• man• ls• pwd• clear• Ctrl+C• echo• ps• cat• tail• head
• cd• mkdir• rm• cp• mv• cal• kill• vi/vim• find• set• who

Piping
• Piping (|) refers to sequentially running multiple commands at one go.
• For eg. Say I want to read a file, then print only the last line of the file, try:
cat example1.txt | tail –n 1 ls | grep “exam” cat example4.txt | head • Important: Piped commands only work on the output of the
previous command!

Regular Expressions
• Describe a pattern (sequence of characters)• [A-Z]*, [a-z]*• [0-9]*, [0-9]\{n\}• Escape (special) characters – start with \• ^ - start of a line• $ - end of a line

Examples
• Eg. {bicycle, bidirectional, biology, binary, bigotry, bill, big, bin, bionic, …}
• Eg. {Sunday, Monday, …, Saturday}
• Eg. {121, 123, 124, …, 129}

Examples
• TATAAA – TATA box, 25 bases upstream of transcription start site
• Telomeric repeat - (TTAGGG)n

Example 1 – grepSyntax: grep ‘pattern’ <filename>
• Create a new directory.• Copy file “example1.txt” from /usr/home/shellbasics to
your folder• Explore contents of the file using cat/head/tail/vi• Explore grep - copy first line of the file into another file
(use –n flag)• Copy 14th line/last line/last 4 lines into another file• Look for the word “Poe” in example1.txt, paste all
instances into another file (name it <yourname1.txt>)• Look for all numbers in the file – what’s wrong?

Example 2 – sedSyntax: sed ‘s/<find>/<replace>/g’
• Stream Editor – substituting text• Substitute all words that are “old” with “new”
in example1.txt.• Substitute all “a” with “A”, and all “b” with “B”
in one line.

Example 3 – Your first shell script!
• Copy example3.sh to your folder.• Explore its contents:
#!/bin/sh
sed ‘
s/a/A/g
s/b/B/g
‘ example2.txt > example3.txt
• Execute this script using ./example3.sh• Oops – what happened here?

Permissions in Unix
• Unix has three permission/file access modes for all files – read (r), write (w), and execute (x).
• Need to specify permissions explicitly for executables.
• Try chmod +x example3.sh, then try ./example3.sh

Example 3 – contd.
• Add script to change all small letters to capital letters in example2.txt and save it as a new file, example3.txt
• Execute it in the command line.• Write a script to change find all numbers, and
replace them with “[ref]”.

Example 4 – awkSyntax: awk ‘{<action>}’
• Used to mine column formatted data.• Columns denoted by $<column number>• Copy example4.txt to your folder• awk to print only the third column of the file
and save it to <yourname5.txt>• awk to print the 4th and 5th columns, separated
by a tab character to a new file <yourname6.txt>

Example 5 – a FASTA file
• Copy example5.fasta from /usr/home to your folder• Explore its contents – what is the FASTA file format?
What does it contain? Do you see a pattern?• Now use any of the commands we just learned to
extract only the gene-ID from the FASTA file. Print it.• Count the number of “AC” repeats, save to a file
<yourname7.txt>• Save only the first 5 lines in example5.fasta to
<yourname8.fasta>

Example 6 – Executing commands in Shell
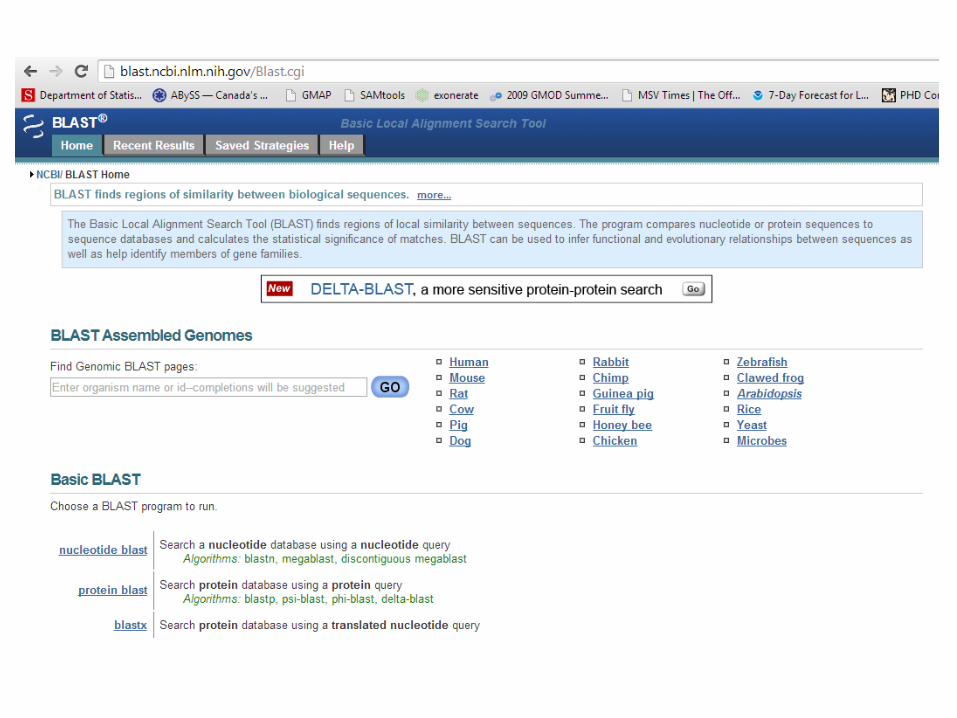
• What is BLAST?• Write a shell script to:• BLAST <yourname8.fasta> against all nucleotide BLAST
databases.• Save output of BLAST to a separate file – call it <yourname9.txt>• What hits do you get?• Explore the BLAST output, pull out only gene ID’s for all your hits
with ‘e’ value = 0.0, and with Genbank accessions (gb), save it to a new file <yourname10.txt>
• HINT: You’ll notice that there are multiple ID’s, separated by “|” – to tell awk to use this as a delimiter, use awk ‘BEGIN { FS=“|”};…’
• HINT: To sort a list, use “sort” function

Example 7 – Advanced scripts (Assignment)
• Write a python script to pull all gene ID’s from <yourname10.txt>, look for these gene ID’s against NCBI and obtain all hits, save it to a file.
• Execute this python script, then parse out only protein id’s (gene/protein=) values from it using a shell script into a separate file.
• Copy all these protein ID’s (they should be Genbank accession ID’s), paste into the query at www.pantherdb.org, select all species on the list, add PANTHER-GO-Slim Biological Process to your columns.
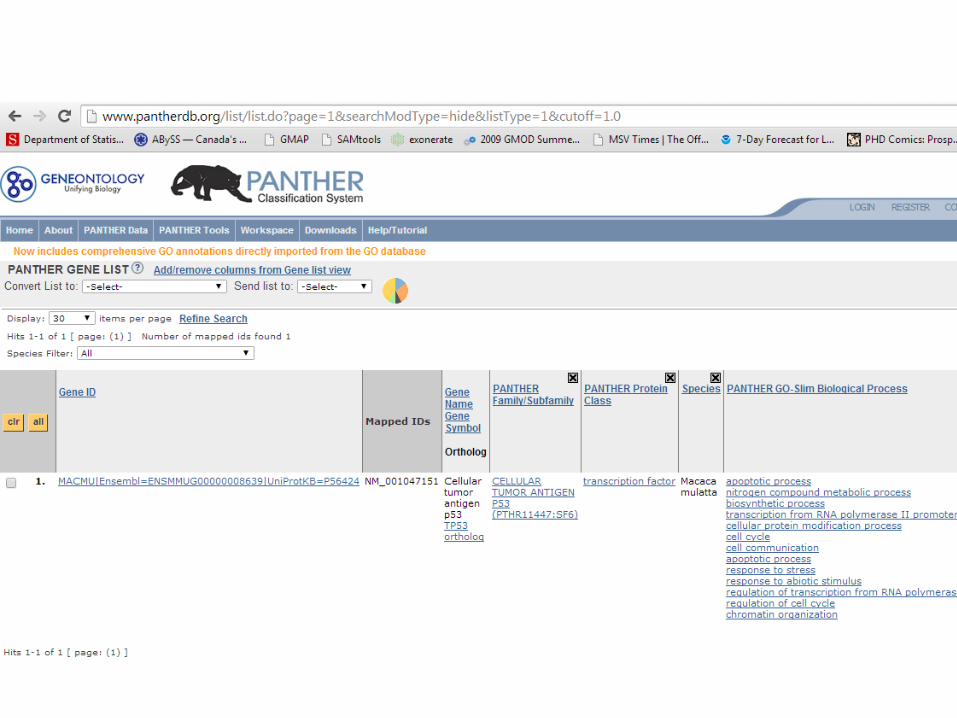

Assignment (contd.)
• Save the output of PANTHER as a file. Now parse this file using grep/sed/awk to print only the GO terms – they should be separated by ;
• Make a unique list of these GO terms by using the ‘uniq’ function, save this to a final assignment submission file.
• HINT: Prior to pulling unique values, try replacing the “;” values with something else, say a newline character “\n”.





























