Embed Size (px)
Citation preview

Simple quantification of complement factors C3and C3b using separation by isotachophoresisThe separation of complement factors C3 and C3b by isotachophoresis in 1% agarosegel followed by immunoprecipitation and quantification is presented. Glycine was usedas spacer in a nonequilibrium isotachophoresis (Acevedo, F., J. Chromatogr. 1991,545, 391±396). Tricine, b-alanine and Tris were the leading ion, terminating ion andcounter ion, respectively. After electrophoresis the gel was incubated in rabbit anti-human complement factor C3c. The amounts of C3 and C3b in the sample weremeasured by optical densitometry of the Coomassie Brilliant blue-stained immuno-precipitates in the agarose gel. The correlation coefficient obtained for the logarithm ofthe integrated densitometric measurement vs. the logarithm of the amount of appliedC3 was higher than 0.98 in calibration experiments. The extent of complement factorC3 activation is calculated as the ratio between the amount of C3b and the amount ofC3b plus C3 and expressed as percent. The progress of complement activation fromhuman blood plasma samples induced by Mg2+ and zymosan are presented asexamples.
Keywords: Complement / C3, C3b / Isotachophoresis / Quantification EL3324
Fernando Acevedo
Department of OccupationalMedicine, National Institutefor Working Life, Solna,Sweden
1 Introduction
Complement factor C3 is the central factor of thecomplement system and it plays a pivotal role in theimmunological defense against pathogens. Virus, bac-teria, lipopolysaccharides and several other substancesstimulate activation of complement C3. Fragmentsgenerated during the activation of C3 have importantbiological functions; for instance, they influence release ofinterleukins and modulate lymphocyte growth, differ-entiation and adhesion, opsonization and fagocytosis[1]. The activation of complement in vivo can take placeby three different pathways [2±4]. The three activationpathways converge in the conversion of C3 to C3b, beingthus the central reaction in activation of the complementsystem. A simple and rapid method for the measurementof the extent of this particular reaction is highly desirable.
The C3 molecule is a 185 kDa protein consisting of an a-chain (115 kDa) and a b-chain (75 kDa) linked by disulfidebonds. C3 is specifically activated by the convertases ofthe classical or the alternative pathway. The anaphyla-toxin C3a (9 kDa) is then released from the a-chain with aconformational change in the remaining C3 molecule. Athioester bond on the a-chain becomes accessible to H2Oor to nucleophiles on the target surface. Upon H2O attackof the thioester bond, a glutamic acid residue is formed
and C3b remains in the aqueous phase. The consequentincrease in net charge of the C3 molecule leads to achange in its electrophoretic mobility. The thioester bondin native C3 can be subjected to nucleophilic attack evenwithout proteolytic cleavage and generate iC3, alsotermed C3 (H2O), which is, in effect, an activated formof C3 possessing C3b-like functions [5]. The fact that thefragment C3b has a higher electrophoretic mobilitytowards the anode than C3 and that both C3 and C3bare recognized by antibodies against the fragment C3c [6]can be exploited to measure the extent of C3 activation.
The separation of proteins by isotachophoresis (ITP) inagarose gel is simple to perform and rapid [7, 8]. Whenweak ions are used as discrete spacers for theisotachophoretic separation of proteins, some proteinsmigrate at nonequilibrium in the zone of a particularspacer [9]. This fact can be utilized for the separation ofC3 and C3b using a single spacer, which simplifies theprocedure. In addition, the use of agarose gel has theadvantage that different proteins can be specificallyimmunoprecipitated in the gel for their recognition [8]and quantification. C3 and C3b can be separated byordinary agarose gel electrophoresis; however, theseparation distance is small, in addition to the bandsbroadening by diffusion. An isotachophoretic system forthe separation of C3 and C3b in 1% agarose gel usingglycine as single spacer is presented in this paper. Thetechnique allows kinetic studies of the first step in theactivation of C3. The effect of Mg2+ and zymosan, knownactivators of complement factor C3, are used to illustratethe new technique.
Correspondence: Fernando Acevedo, Department of Occupa-tional Medicine, National Institute for Working Life, Ekelunsvägen16, S-17184 Solna, SwedenE-mail: [email protected]
� WILEY-VCH Verlag GmbH, 69451 Weinheim, 1999 0173-0835/99/0303-0469 $17.50+.50/0
Electrophoresis 1999, 20, 469±472 469
Gen
eral

2 Materials and methods
2.1 Materials
The Blood Center at the Karolinska Hospital in Stockholmsupplied heparin and EDTA-human blood samples.Plasma, obtained by centrifugation of the freshly drawnsamples, was aliquoted and frozen at ±70oC until use.Agarose Isogel� and Gel Bond� were from FMCBioproducts (Maryland, MN, USA); electrode strips, 6 �10 mm, were Cat. No. 19-3664-01 from Pharmacia LKB(Uppsala, Sweden). Human serum protein calibrator andrabbit anti-human C3c complement factor were from DakoA/S (Glostrup, Denmark). The desk scanner was a StudioStar� from Agfa-Gevaert N.V. (Mortsel, Belgium) and thecomputer program QuantiScan was from Biosoft (Cam-bridge, England). All chemicals were of analytical quality.
2.2 Methods
2.2.1 Activation of complement factor C3 byzymosan and Mg2+
The plasma samples were diluted 1:10 in 25 mM Tris-HClbuffer in 150 mM NaCl, pH 7.2, (TBS) containing variousconcentrations of zymosan and/or Mg2+ and incubatedvarious times at 37oC. The incubation was stopped by theaddition of ice-cold 100 mM EDTA-NaOH, pH 7.4, to afinal concentration of 10 mM and cooling on an ice-waterbath.
2.2.2 Preparation of the gels
Agarose (300 mg) was dispersed in 30 mL of a watersolution of 200 mM Tris, 50 mM Tricine and 150 mM
glycine. The suspension was boiled for 3 min and pouredon the hydrophilic side of a 24 � 12 cm GelBond filmplaced on a warm, horizontally level surface. The gelformed upon cooling was kept at 5oC for at least 1 hbefore use. The anode solution, 10 mL, was 1 M Tris,0.5 M acetic acid. The cathode solution, 10 mL, was 1 M b-alanine, 0.5 M Tris. The electrode solutions wereembedded in the electrode strips.
2.2.3 Isotachophoresis
The run was done as described [9] on a Multiphor II unit(Pharmacia LKB). The power settings were 1000 V,150 mA and 15 W at 15oC. The gel was prerun until theITP front reached 3 cm from the cathode, which waseasily visualized by using bromophenol blue in thecathode strip. Next, the sample applicator strips (Cat.No. 18-1002-74 from Pharmacia LKB) was placed 0.5 cmfrom the cathode and the samples (10 mL) were applied.Electrophoresis was continued until the ITP front reachedthe anode.
2.2.4 Calibration curve
A series of dilutions of human protein calibrator in TBSwere prepared. The concentrations of C3 varied from 1 to~250 mg/mL. Samples of 10 mL of the dilutions wereapplied to the gel.
2.2.5 Immunofixation of complement factors C3and C3b
Immediately after the run the gel was transferred to astaining tray (Cat. No. 18-18-08, Pharmacia LKB). Inorder to precipitate the complement proteins, 100 mL ofTBS containing 200 mL of rabbit anti-human C3c wereadded. The gel was gently shaken for at least 1 h at 37oC.This incubation could also be done overnight at roomtemperature. The gel was washed three times with100 mL TBS for 15 min each. TBS in the gel wasremoved by washing once with 300 mL of water beforedrying the gel. The gel was dried at 50oC or at roomtemperature under an air stream.
2.2.6 Staining of the precipitated proteins
The dried gel was washed for 15 min with 100 mL of asolution containing 40% ethanol and 10% acetic acid inwater (destaining solution) followed by 30 min stainingwith 100 mL of 0.25% Coomassie Brilliant Blue R-250(CBB) dissolved in destaining solution. Then the gel waswashed to obtain a clear background with 100 mL ofdestaining solution and allowed to dry in an air stream.
2.2.7 Scanning and quantification of thestained bands
The gel was scanned with the desktop scanner using agrey scale of 256 values and a resolution of 300 pixels perinch. Data were processed with the program QuantiScanfrom Biosoft (Cambridge, England). The sample lanesand the peaks in each lane on the scanned gel wereselected. The width of each peak was adjusted manuallyto the inflexion point of the baseline at the neighborhoodof the peak. The integrated value in each peak on the lanewas measured using an interpolated minimum baselinefor the whole lane. The integrated value for the peaks in alane was plotted against the amount of C3 applied for thecalibration lanes to generate a calibration curve. Theintegrated values from the peak or peaks in a lane withunknown C3 and/or C3b were interpolated in thecalibration curve. The percentage of C3 conversion wascalculated from the ratio between C3b and C3 plus C3band expressed as percent.
470 F. Acevedo Electrophoresis 1999, 20, 469±472
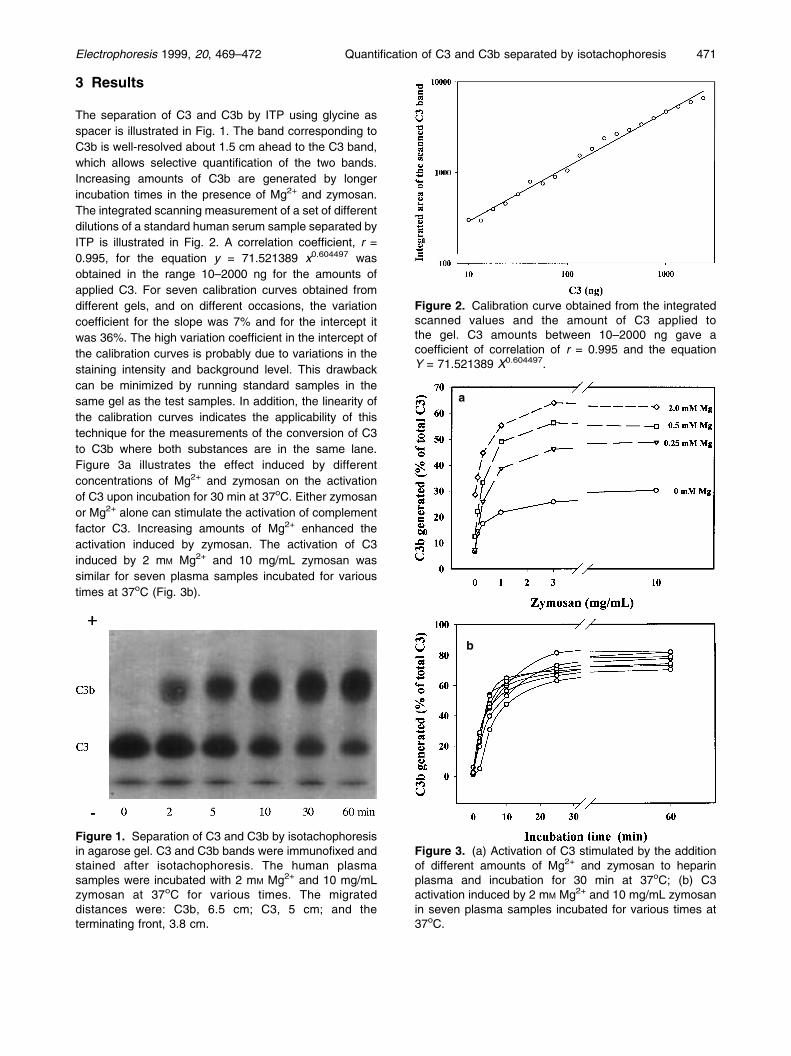
3 Results
The separation of C3 and C3b by ITP using glycine asspacer is illustrated in Fig. 1. The band corresponding toC3b is well-resolved about 1.5 cm ahead to the C3 band,which allows selective quantification of the two bands.Increasing amounts of C3b are generated by longerincubation times in the presence of Mg2+ and zymosan.The integrated scanning measurement of a set of differentdilutions of a standard human serum sample separated byITP is illustrated in Fig. 2. A correlation coefficient, r =0.995, for the equation y = 71.521389 x0.604497 wasobtained in the range 10±2000 ng for the amounts ofapplied C3. For seven calibration curves obtained fromdifferent gels, and on different occasions, the variationcoefficient for the slope was 7% and for the intercept itwas 36%. The high variation coefficient in the intercept ofthe calibration curves is probably due to variations in thestaining intensity and background level. This drawbackcan be minimized by running standard samples in thesame gel as the test samples. In addition, the linearity ofthe calibration curves indicates the applicability of thistechnique for the measurements of the conversion of C3to C3b where both substances are in the same lane.Figure 3a illustrates the effect induced by differentconcentrations of Mg2+ and zymosan on the activationof C3 upon incubation for 30 min at 37oC. Either zymosanor Mg2+ alone can stimulate the activation of complementfactor C3. Increasing amounts of Mg2+ enhanced theactivation induced by zymosan. The activation of C3induced by 2 mM Mg2+ and 10 mg/mL zymosan wassimilar for seven plasma samples incubated for varioustimes at 37oC (Fig. 3b).
Electrophoresis 1999, 20, 469±472 Quantification of C3 and C3b separated by isotachophoresis 471
Figure 1. Separation of C3 and C3b by isotachophoresisin agarose gel. C3 and C3b bands were immunofixed andstained after isotachophoresis. The human plasmasamples were incubated with 2 mM Mg2+ and 10 mg/mLzymosan at 37oC for various times. The migrateddistances were: C3b, 6.5 cm; C3, 5 cm; and theterminating front, 3.8 cm.
Figure 2. Calibration curve obtained from the integratedscanned values and the amount of C3 applied tothe gel. C3 amounts between 10±2000 ng gave acoefficient of correlation of r = 0.995 and the equationY = 71.521389 X0.604497.
b
Figure 3. (a) Activation of C3 stimulated by the additionof different amounts of Mg2+ and zymosan to heparinplasma and incubation for 30 min at 37oC; (b) C3activation induced by 2 mM Mg2+ and 10 mg/mL zymosanin seven plasma samples incubated for various times at37oC.
a

4 Discussion
The separation of C3 and C3b by conventional zoneelectrophoresis in agarose gel gives too poor a resolutionto allow quantification after immunoprecipitation of C3 andC3b. A rough estimation of the extent of C3 conversion toC3b is possible after crossed immunoelectrophoresis intoa gel containing anti C3c. This technique is not simple toperform and only one sample can be analyzed at a time ineach gel. Alternatively, the activation of C3 can bemeasured indirectly by the amount of C3d produced, afragment of C3b, which is generated by the action ofserum proteases and factor I on C3b. A major disadvant-age is that the generation of C3d from C3 proceeds at aslower rate than the conversion of C3 to C3b and includesfurther reaction steps. Monoclonal antibodies recognizingC3-neoepitopes may be used in ELISA techniques tomeasure the extent of complement activation [10];however, the conformational changes required for theexpression of such neoepitopes may not be concomitantwith the charge change due to the hydrolysis of thethiolester group.
The normal concentration of complement factor C3 inplasma is about 1 mg/mL and an undiluted sample wouldallow the detection of about 0.1% conversion to C3b. Anincreased resolution can be easily achieved by usingenzyme-labeled antibodies; however, this is outside thescope of this paper. The technique presented here takesadvantage of the rapid separation of C3 and C3b by ITPfollowed by simple immunoprecipitation in the gel. Thistechnique allows the quantification of the extent of C3activation stimulated by different substances and facili-tates studies of the mechanism of C3 activation inplasma. The whole analytical procedure, from preparingthe gels to collecting results, can be completed within oneworking day. The two substances zymosan and Mg2+,well-known stimulators of complement activation, wereused to illustrate the potential of the techniques presented
here. Further studies on the stimulatory effect of varioussubstances on the complement system are in progress atour laboratory. Different spacer ions can be used for theisotachophoretic separation of C3 and C3b; in that case,however, the pH and composition of the system has to bechanged accordingly [9]. An advantage of the ITP systempresented here is that the ions used (glycine, Tricine, Trisand b-alanine) are nontoxic and inexpensive.
Professor Olof Vesterberg is gratefully acknowledged forvaluable comments. The Environmental & Climateresearch program of the DGXII of the European Com-mission supported this work (contract # ENV4-CT95-0167).
Received September 15, 1998
5 References
[1] Sahu, A., Sunyer, J. O., Moore, W. T., Sarrias, M. R.,Soulika, A. M., Lambris, J. D., Immunol. Res. 1998, 17,109±121.
[2] Reid, K. B. M., in: Rother, K., Til, G. O., Hänsch, G. M.(Eds.), The Complement System, Springer Verlag, Heidel-berg 1998, pp. 68±85.
[3] Reid, K. B. M., in: Rother, K., Til, G. O., Hänsch, G. M.(Eds.), The Complement System, Springer Verlag, Heidel-berg 1998, pp. 86±92.
[4] Pangburn, M. K., in: Rother, K., Til, G. O., Hänsch, G. M.(Eds.), The Complement System, Springer Verlag, Heidel-berg 1998, pp. 93±115.
[5] Pangburn, M. K., Schreiber, R. D., Müller-Eberhard, H. L.,J. Expl. Med. 1981, 154, 856±867.
[6] Acevedo, F., Hammar, H., Br. J. Dermatol. 1989, 121,329±335.
[7] Acevedo, F., in: Schafer-Nielsen (Ed.), Electrophoresis ©88,VCH, Weinheim 1988, pp. 112±119.
[8] Acevedo, F., J. Chromatogr. 1989, 470, 407±414.
[9] Acevedo, F., J. Chromatogr. 1991, 545, 391±396.
[10] Garred, P., Mollnes, T. E., Lea, T., Scand J. Immunol. 1988,27, 329±335.
472 F. Acevedo Electrophoresis 1999, 20, 469±472





























