Embed Size (px)
Citation preview

Ángel Carmelo Prieto Colorado
Física de la Materia Condensada, Cristalografía y Mineralogía.Facultad de Ciencias.
Universidad de Valladolid.
Síntesis y Caracterización Estructural de los Materiales

©A. Carmelo Prieto Colorado
Técnicas espectroscópicasTema 19. Espectroscopía vibracional. Fundamento molecular de la espectroscopía.
Fenómenos de absorción y de dispersión de la radiación por la materia. Reglas de Selección: IR y Raman. Número y simetría de los modos normales de vibración (MNV). Predicción de los MNV mediante métodos teóricos “ab initio”.
Tema 20. Espectroscopía vibracional de absorción Infrarroja. Espectroscopía IR en sólidos cristalinos. Disposición experimental IR. Equipos dispersivos y mediante transformada de Fourier. Preparación de las muestras. Tratamiento de la señal en espectros IR. Espectroscopía mediante A.T.R. Aplicaciones de la espectroscopía infrarroja.
Tema 21. Espectro de dispersión Raman. Historia del Efecto Raman. Fundamentos. Polarización en sólidos cristalinos. Raman Resonante y fluorescencia. Disposición experimental Raman. Equipos microRaman y nanoRaman. Equipos portátiles y teleRaman. Tratamiento de la señal espectral Raman. Imagen y cartografía Raman. Aplicaciones de la espectroscopía Raman. Similitudes y diferencias entre las espectroscopias IR y Raman.
Tema 22. Espectroscopía óptica: Ultravioleta – Visible. Espectroscopia de ruptura dieléctrica asistida por láser: LIBS. Determinación química elemental cualitativa y cuantitativa. Espectroscopía Mössbauer. Raman acoplado con técnicas de análisis instrumental: SEM, DRX, LIBS.

©A. Carmelo Prieto Colorado
Técnicas espectroscópicas
Tema 19 Espectroscopía vibracional.Fundamento molecular de la espectroscopía.Fenómenos de absorción y de dispersión de la radiación por la materia.Reglas de Selección: IR y Raman.Número y simetría de los modos normales de vibración (MNV).Predicción de los MNV mediante métodos teóricos “ab initio”.
©A. Carmelo Prieto Colorado
Espectroscopía Vibracional
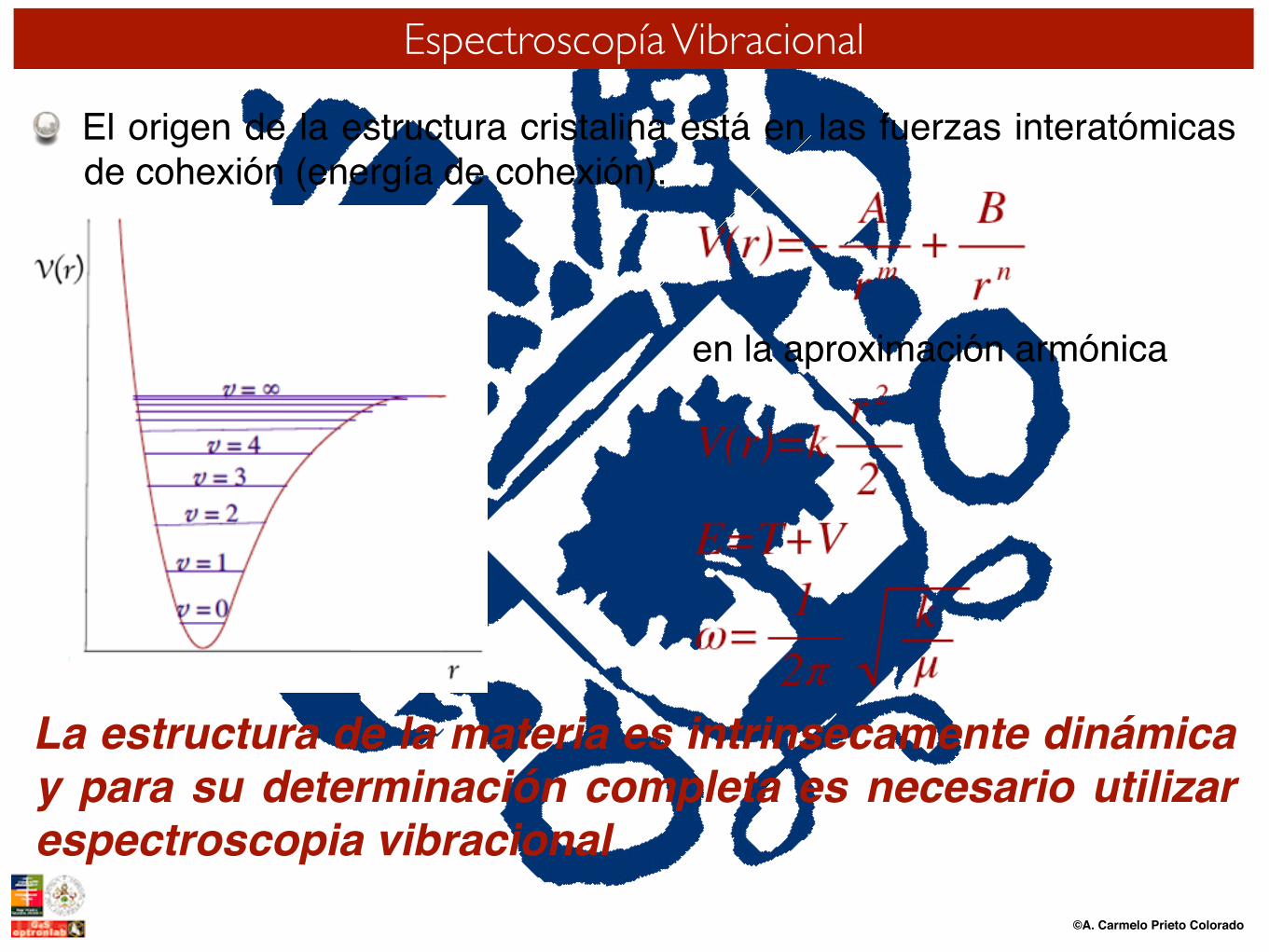
El origen de la estructura cristalina está en las fuerzas interatómicas de cohexión (energía de cohexión).
La estructura de la materia es intrinsecamente dinámica y para su determinación completa es necesario utilizar espectroscopia vibracional
en la aproximación armónica
©A. Carmelo Prieto Colorado
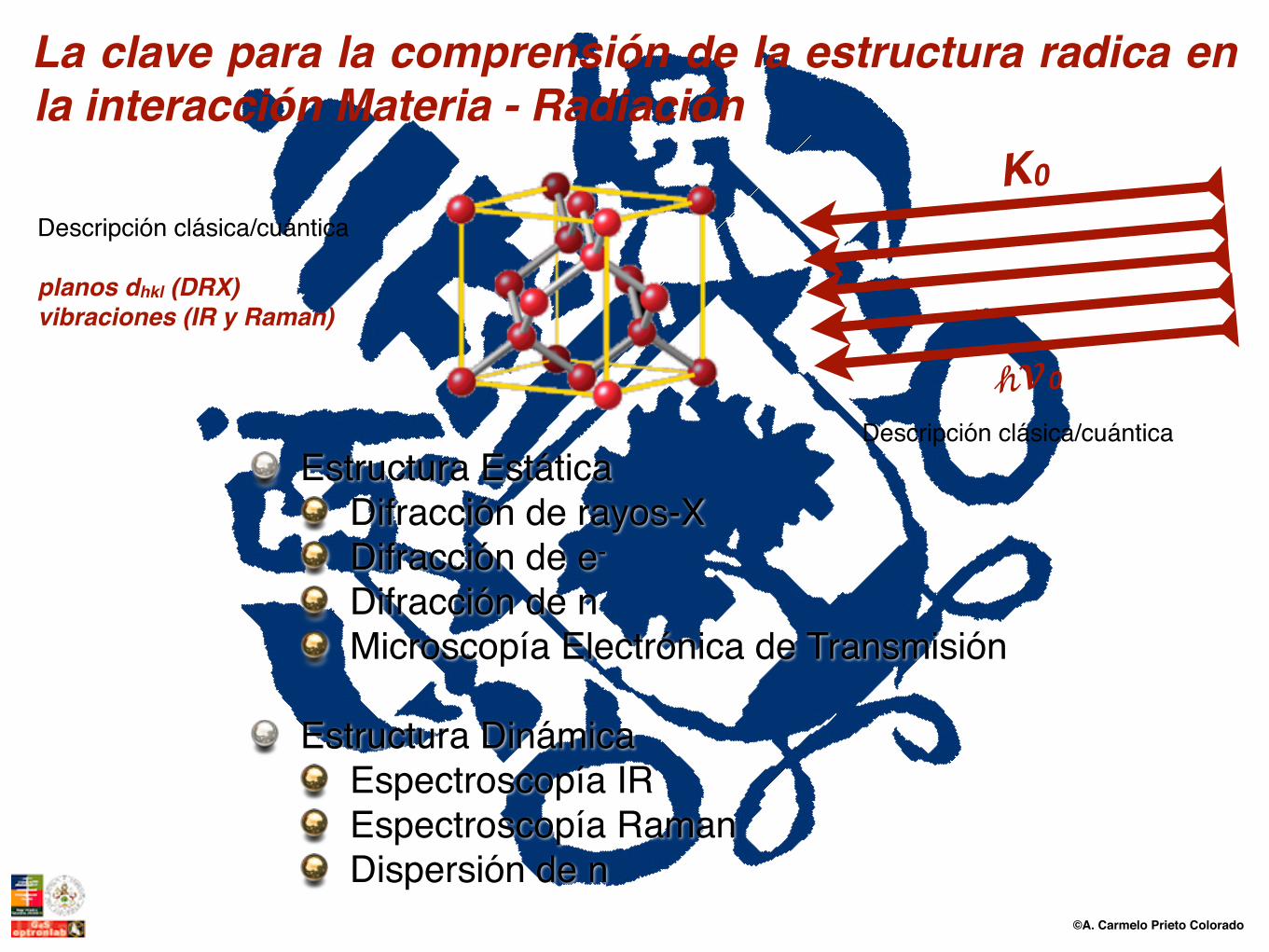
Estructura EstáticaDifracción de rayos-XDifracción de e-
Difracción de nMicroscopía Electrónica de Transmisión
Estructura DinámicaEspectroscopía IREspectroscopía RamanDispersión de n
La clave para la comprensión de la estructura radica en la interacción Materia - Radiación
K0
𝒽𝓥0
Descripción clásica/cuántica
Descripción clásica/cuántica
planos dhkl (DRX)vibraciones (IR y Raman)
©A. Carmelo Prieto Colorado
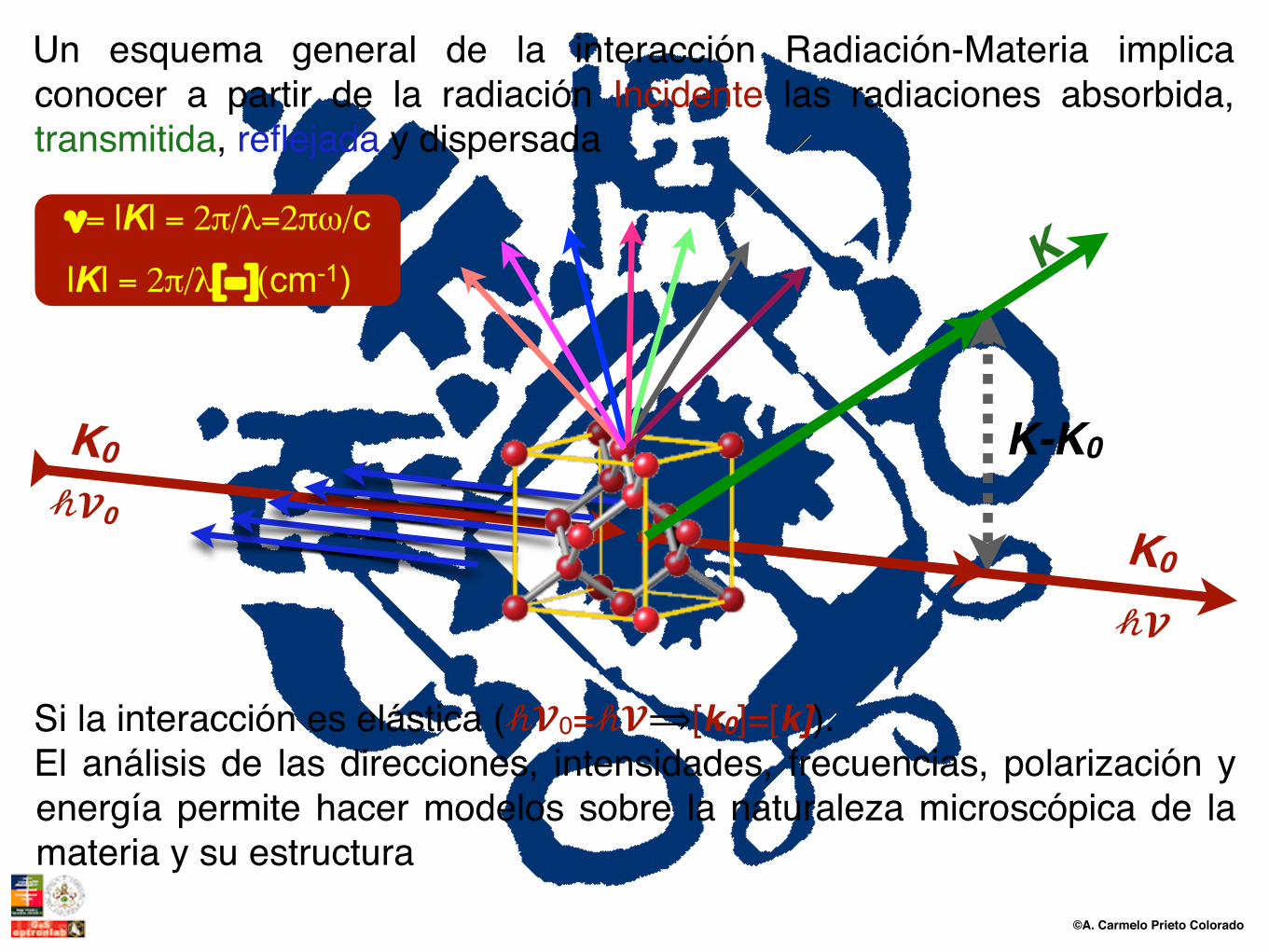
Un esquema general de la interacción Radiación-Materia implica conocer a partir de la radiación Incidente las radiaciones absorbida, transmitida, reflejada y dispersada
lKl = 2π/λ[=](cm-1)
ν= lKl = 2π/λ=2πω/c
K0𝒽𝓥0
K-K0
Si la interacción es elástica (𝒽𝓥0=𝒽𝓥⟹[k0]=[k]).El análisis de las direcciones, intensidades, frecuencias, polarización y energía permite hacer modelos sobre la naturaleza microscópica de la materia y su estructura
K0
𝒽𝓥
K
©A. Carmelo Prieto Colorado
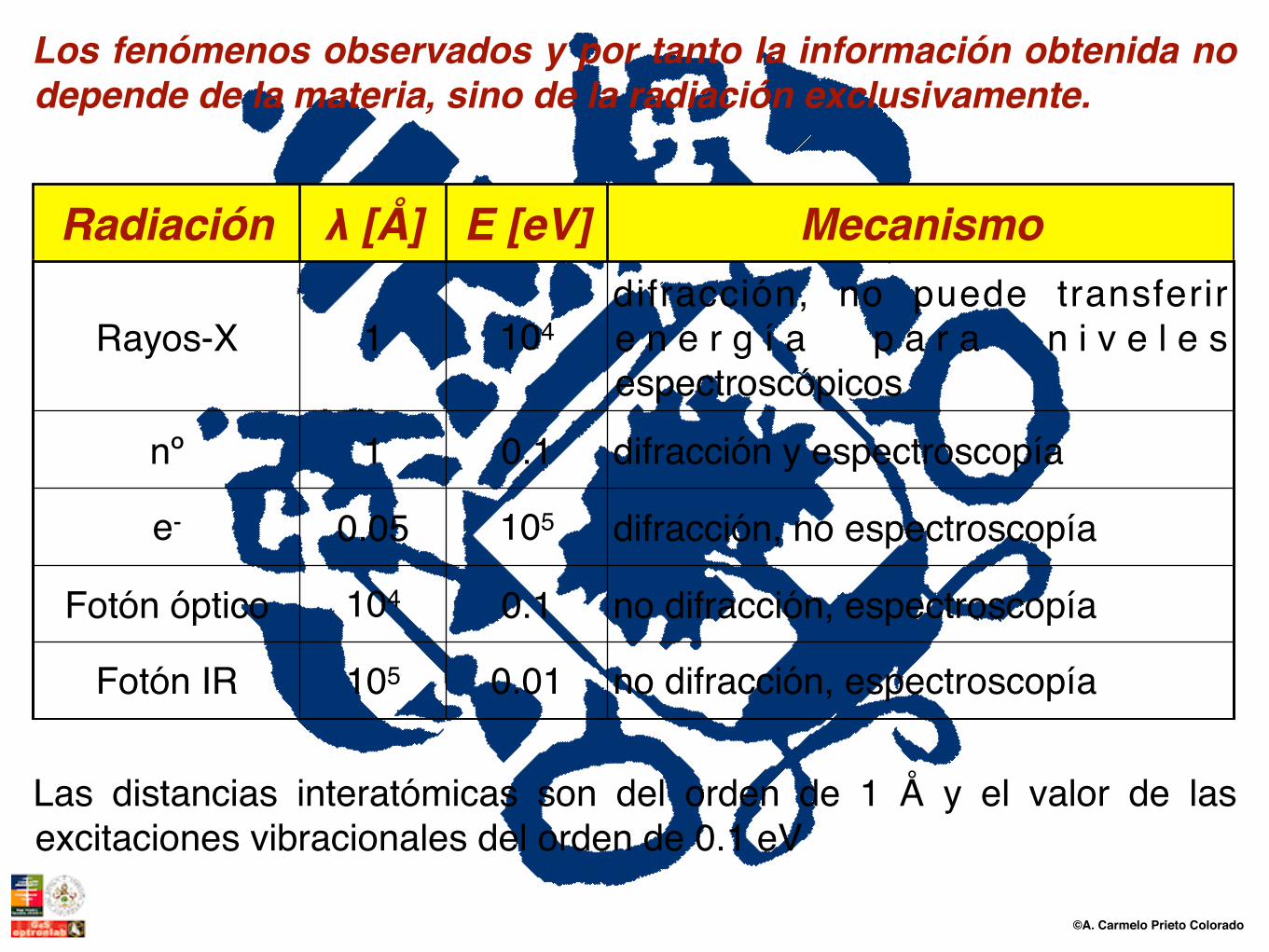
Radiación λ [Å] E [eV] Mecanismo
Rayos-X 1 104difracción, no puede transferir e n e r g í a p a r a n i v e l e s espectroscópicos
nº 1 0.1 difracción y espectroscopía
e- 0.05 105 difracción, no espectroscopía
Fotón óptico 104 0.1 no difracción, espectroscopía
Fotón IR 105 0.01 no difracción, espectroscopía
Los fenómenos observados y por tanto la información obtenida no depende de la materia, sino de la radiación exclusivamente.
Las distancias interatómicas son del orden de 1 Å y el valor de las excitaciones vibracionales del orden de 0.1 eV

©A. Carmelo Prieto Colorado
La Difracción de rayos-X es una técnica estructural macroscópica.
La Microscopia Electrónica de Transmisión es una técnica estructural microscópica.
Las Espectroscopías IR y Raman son técnicas estructurales microscópicas o macroscópicas, dando información atómico molecular.
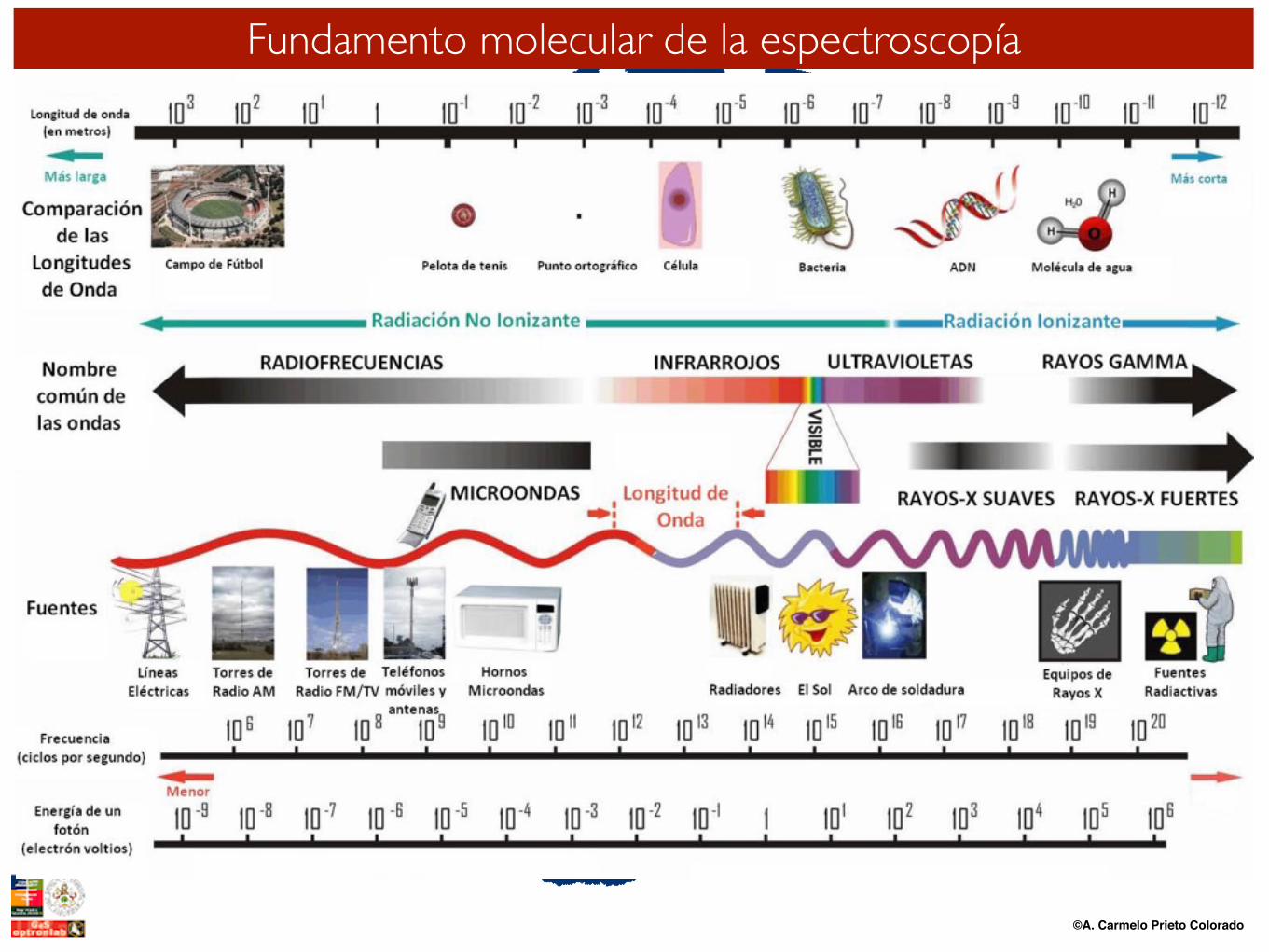
©A. Carmelo Prieto Colorado
Fundamento molecular de la espectroscopía

©A. Carmelo Prieto Colorado
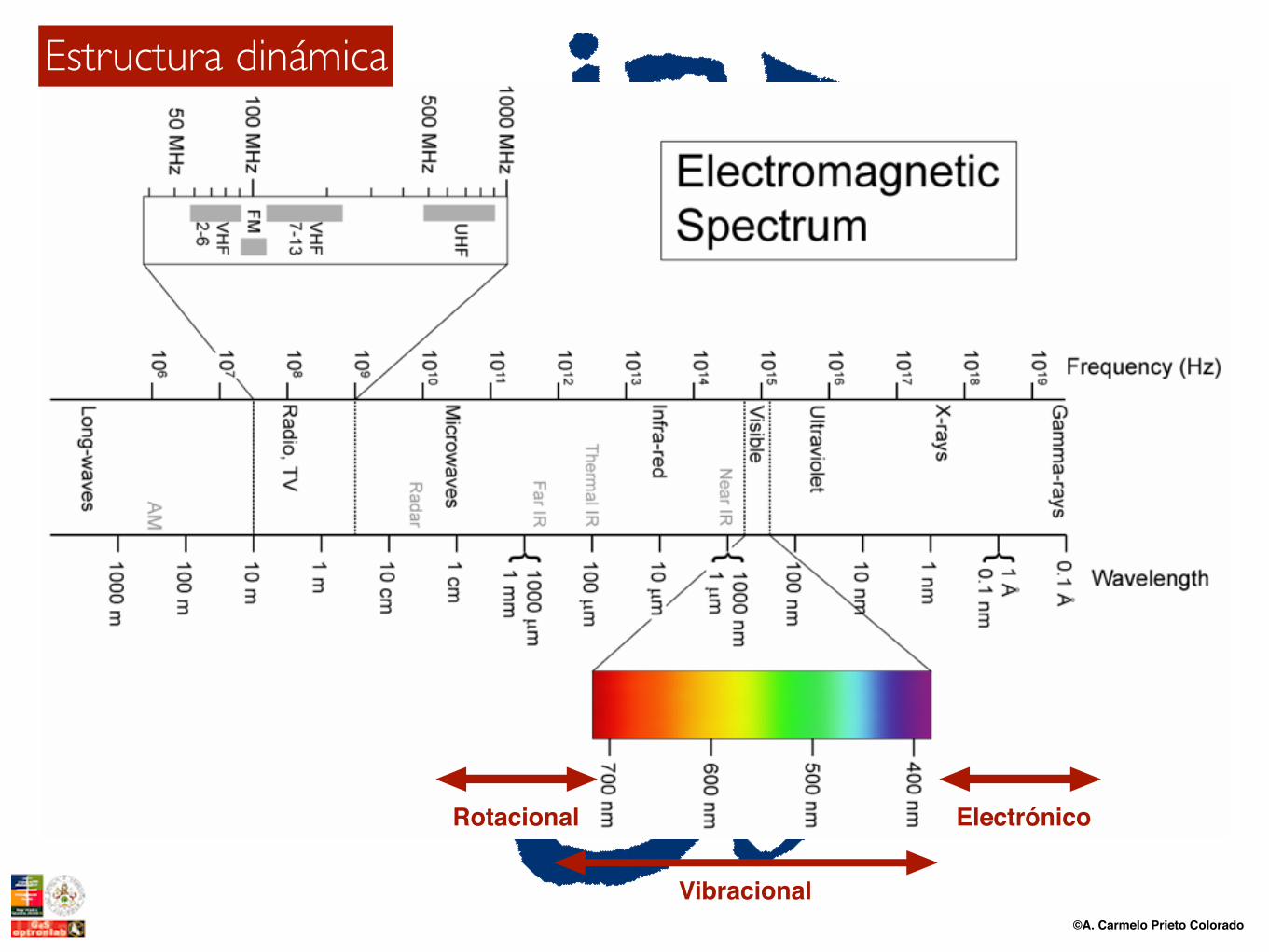
©A. Carmelo Prieto Colorado
Estructura dinámica
Rotacional
Vibracional
Electrónico
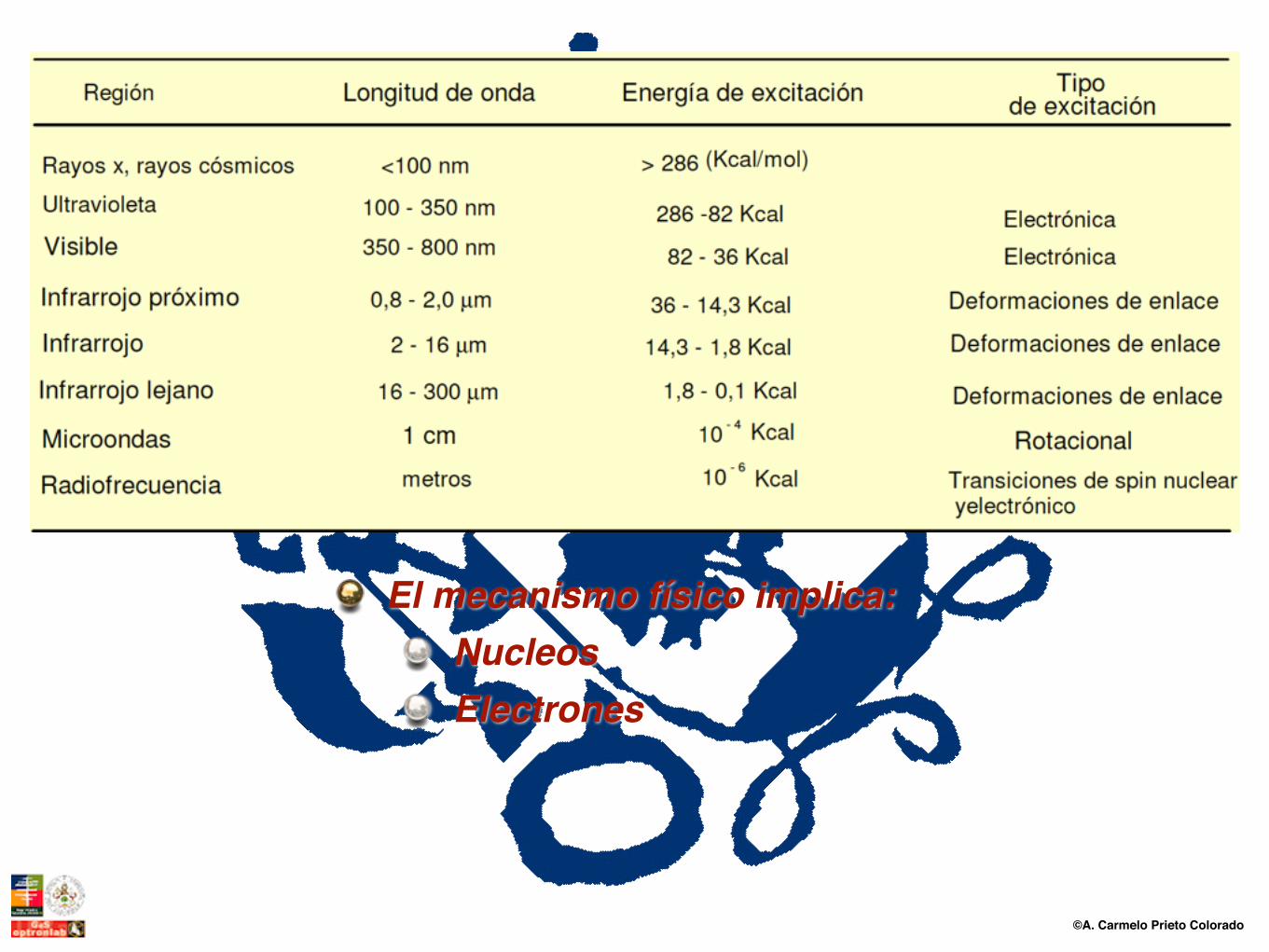
©A. Carmelo Prieto Colorado
El mecanismo físico implica:NucleosElectrones
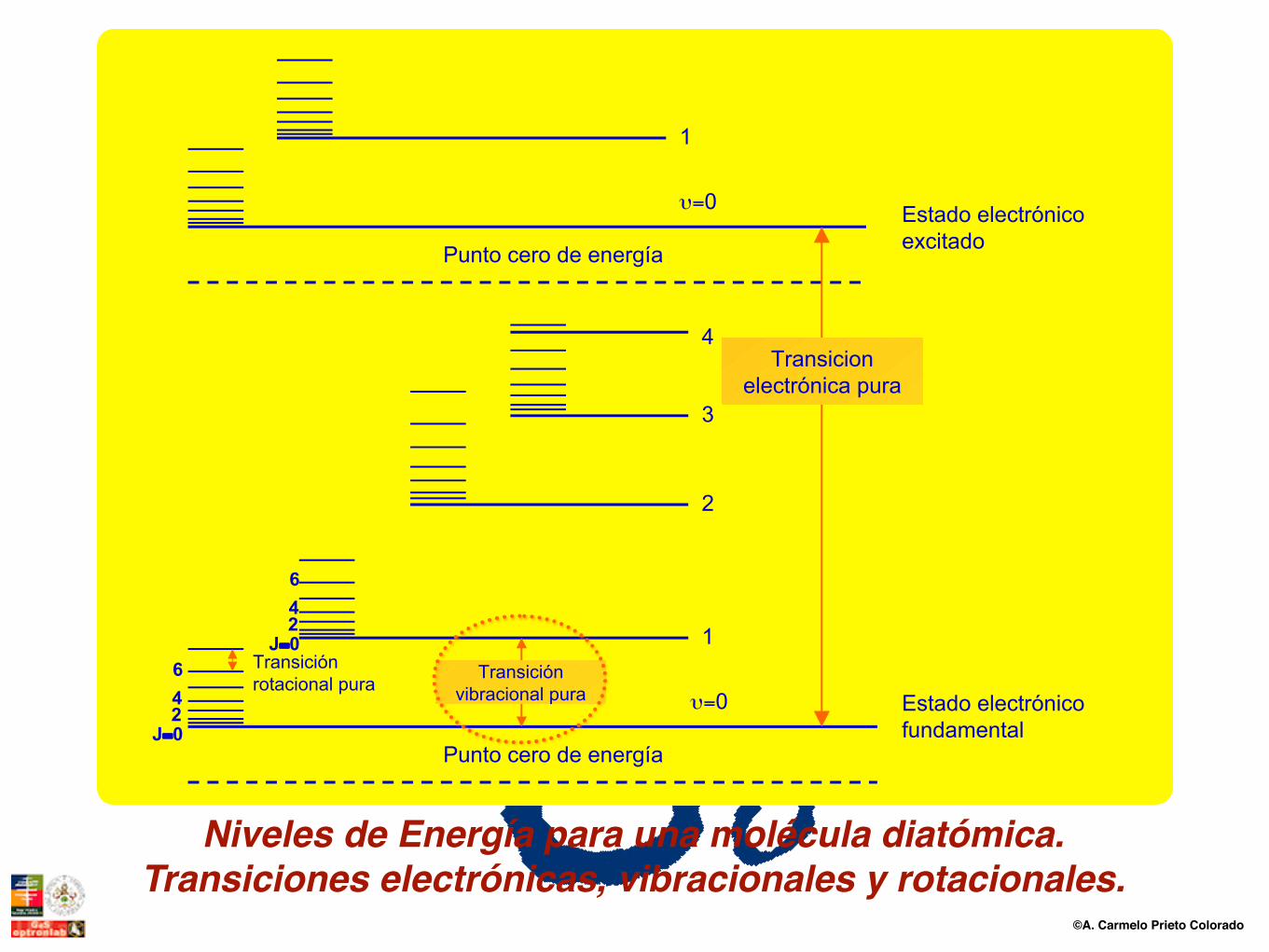
©A. Carmelo Prieto Colorado
Transicionelectrónica pura
Estado electrónicofundamental
Estado electrónicoexcitado
Punto cero de energía
Punto cero de energía
υ=0
υ=0
1
2
3
4
1
J=0246 Transición
rotacional puraTransición
vibracional pura
J=0246
Niveles de Energía para una molécula diatómica.Transiciones electrónicas, vibracionales y rotacionales.

©A. Carmelo Prieto Colorado
Fenómenos de absorción y de dispersión de la radiación por la materia
Básicamente la in teracc ión de la rad iac ión electromagnética con la materia consiste en la excitación, por parte del campo eléctrico de la onda, bien del momento dipolar permanente (caso de la espectroscopía rotacional) bien del momento dipolar dinámico (caso de las espectroscopías en el infrarrojo), bien de la polarizabilidad inducida (caso de la difusión Raman) de las moléculas de la muestra.

©A. Carmelo Prieto Colorado
Absorción InfrarrojaCuando una molécula absorbe un fotón, salta de un estado fundamental a un estado excitado y da lugar a una vibración y debida a la posibilidad de transiciones entre estados de energía vibracionales y rotacionales de las moléculas y a que la molécula puede absorber la energía de fotones en el rango energético de IR tendremos espectroscopía IR, si:
a. Se produce un cambio en el momento dipolar de la molécula durante un movimiento vibracional o rotacional y,
b. La frecuencia asociada con el fotón coincide con la frecuencia natural del movimiento vibracional.

©A. Carmelo Prieto Colorado
Cuando las moléculas vibran dependen de las masas de sus átomos (mA y mB en el caso de moléculas diatómicas A-B, con enlace covalente) y de la fuerza del enlace (constante de fuerza del enlace k), de modo que aplicando la ley de Hooke:
©A. Carmelo Prieto Colorado
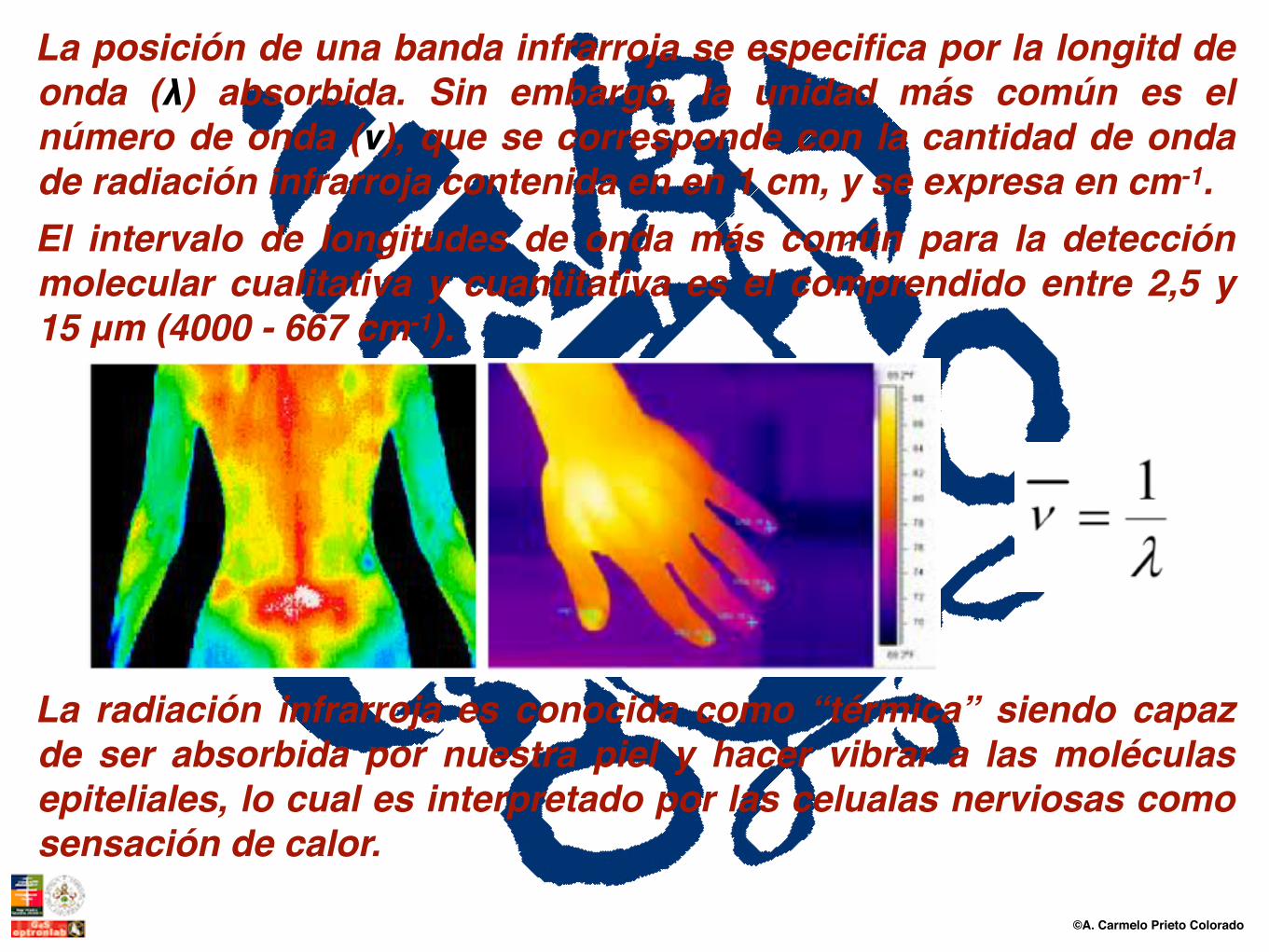
La posición de una banda infrarroja se especifica por la longitd de onda (λ) absorbida. Sin embargo, la unidad más común es el número de onda (ν), que se corresponde con la cantidad de onda de radiación infrarroja contenida en en 1 cm, y se expresa en cm-1. El intervalo de longitudes de onda más común para la detección molecular cualitativa y cuantitativa es el comprendido entre 2,5 y 15 μm (4000 - 667 cm-1).
La radiación infrarroja es conocida como “térmica” siendo capaz de ser absorbida por nuestra piel y hacer vibrar a las moléculas epiteliales, lo cual es interpretado por las celualas nerviosas como sensación de calor.
©A. Carmelo Prieto Colorado
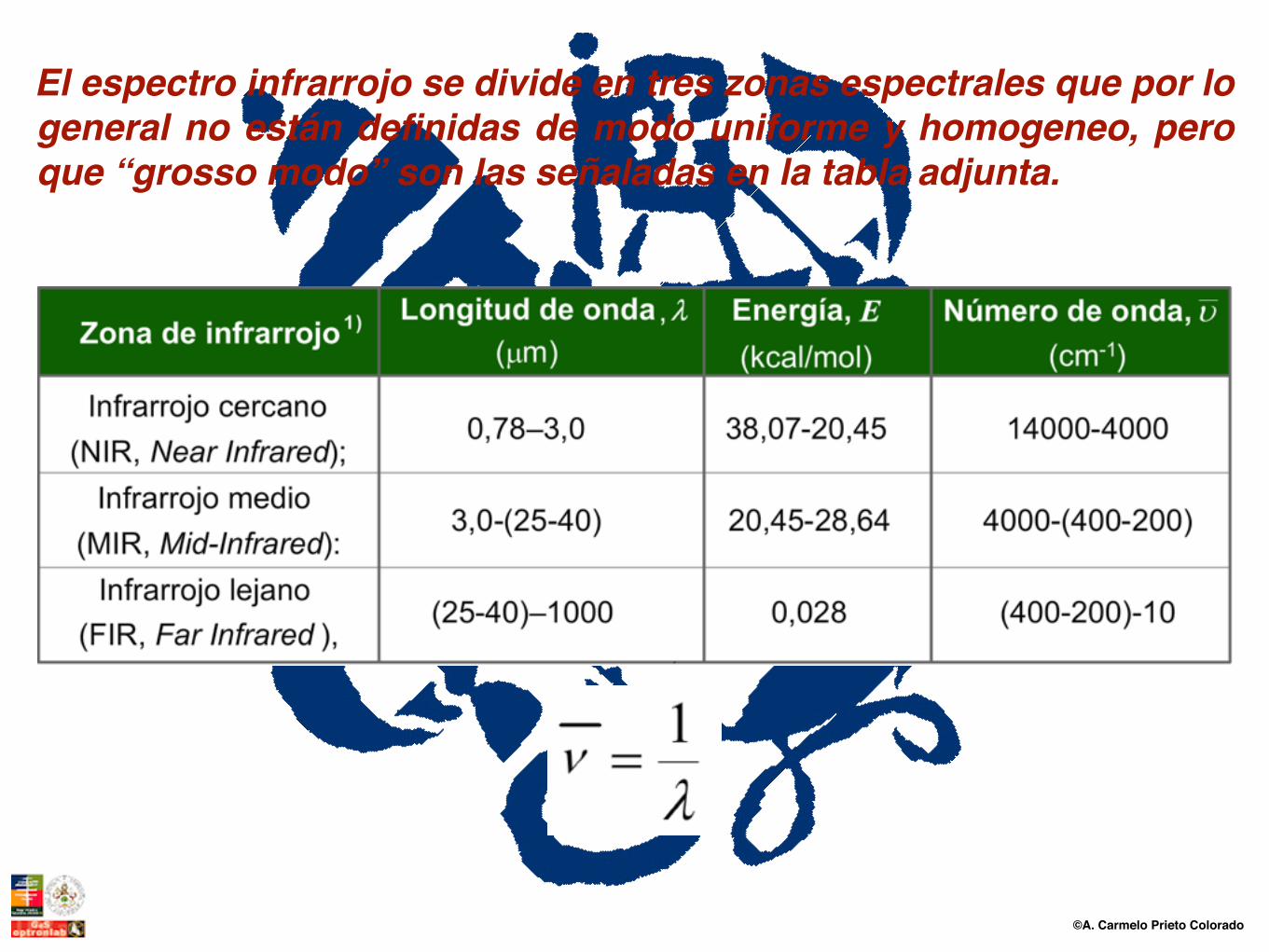
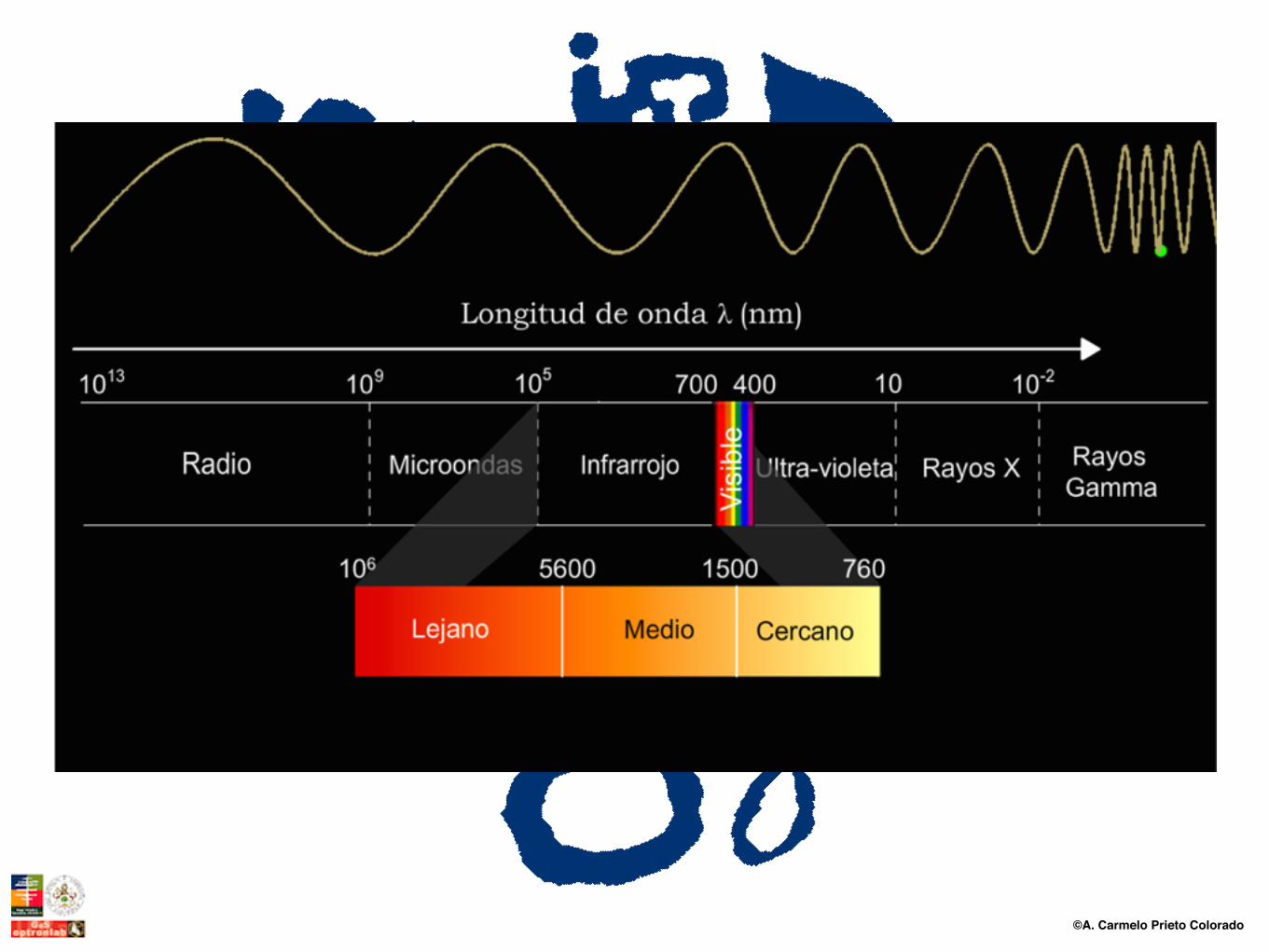
El espectro infrarrojo se divide en tres zonas espectrales que por lo general no están definidas de modo uniforme y homogeneo, pero que “grosso modo” son las señaladas en la tabla adjunta.
©A. Carmelo Prieto Colorado
©A. Carmelo Prieto Colorado
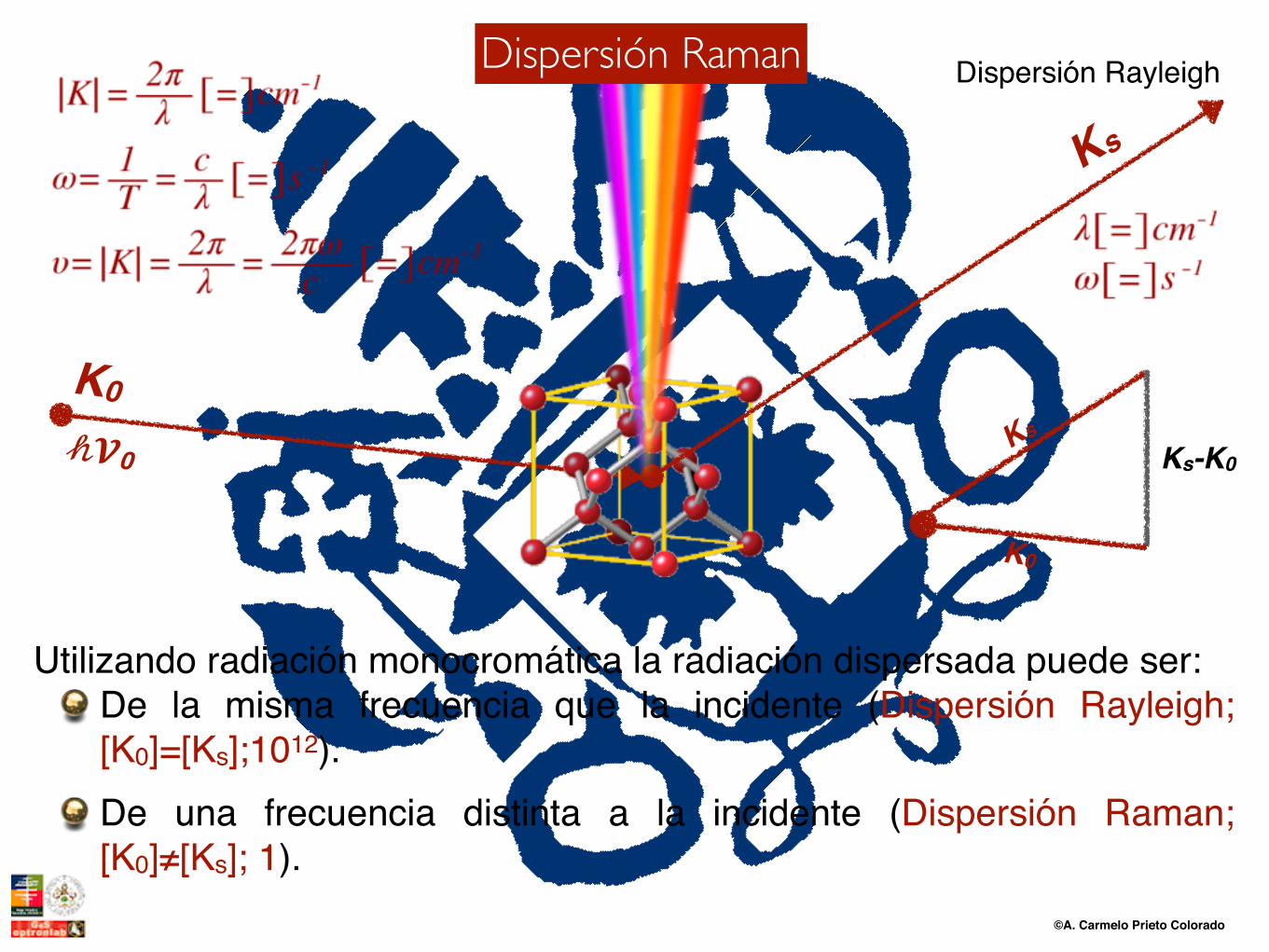
Utilizando radiación monocromática la radiación dispersada puede ser:De la misma frecuencia que la incidente (Dispersión Rayleigh; [K0]=[Ks];1012).De una frecuencia distinta a la incidente (Dispersión Raman; [K0]≠[Ks]; 1).
K0
𝒽𝓥0 Ks-K0
K0
Ks
Ks
Dispersión RayleighDispersión RamanDispersión Raman
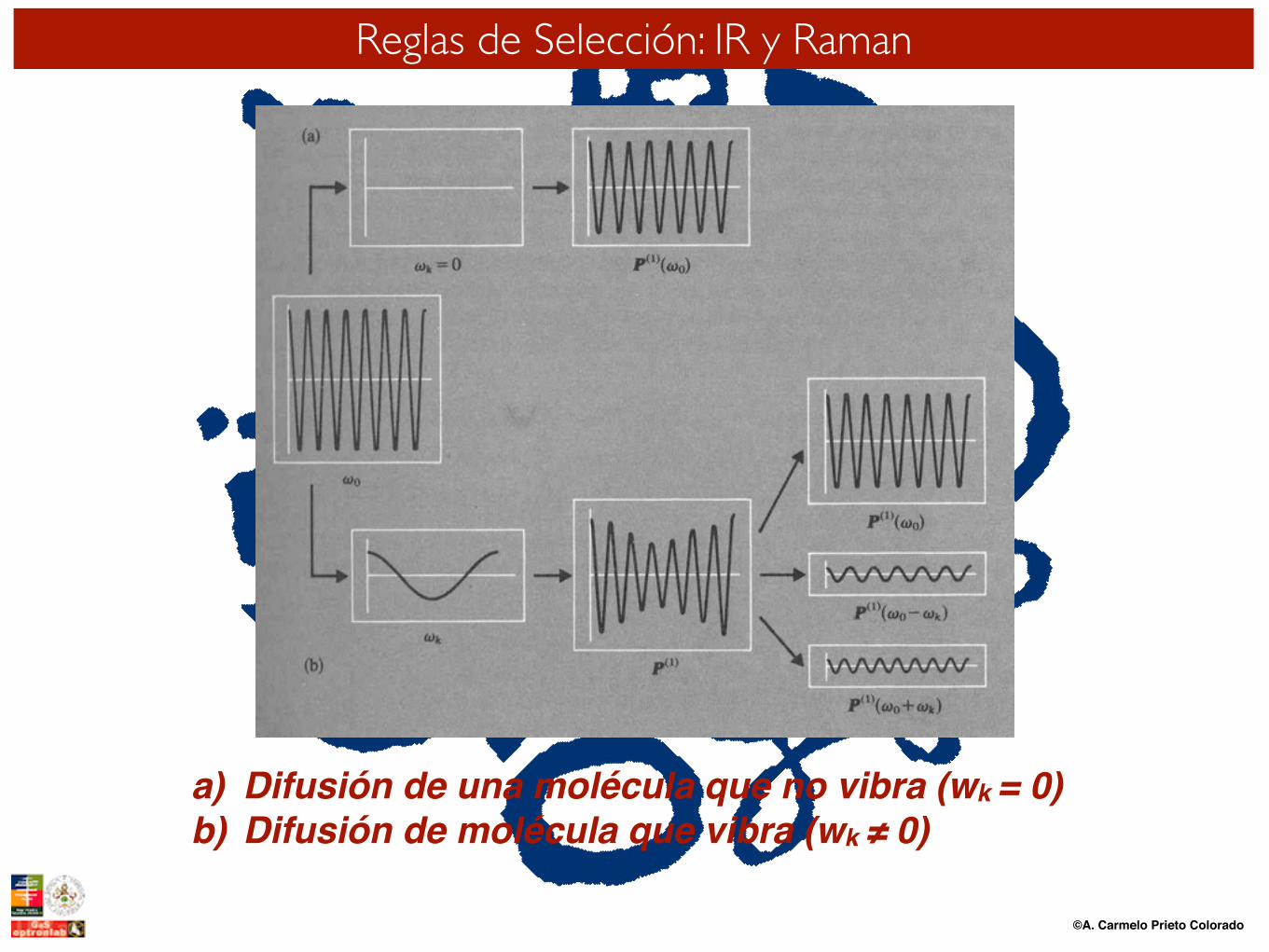
©A. Carmelo Prieto Colorado
a) Difusión de una molécula que no vibra (wk = 0)b) Difusión de molécula que vibra (wk ≠ 0)
Reglas de Selección: IR y Raman
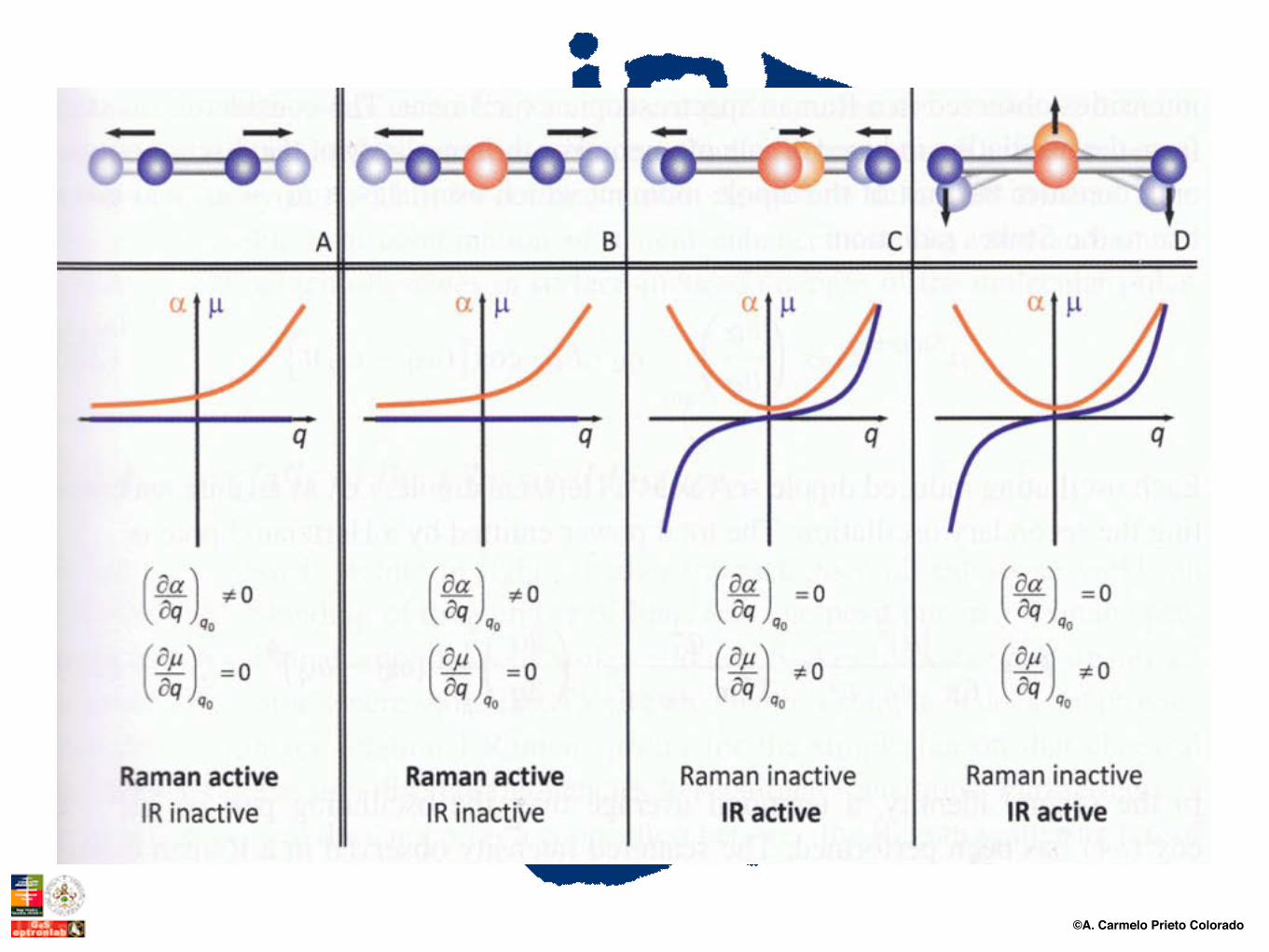
©A. Carmelo Prieto Colorado

©A. Carmelo Prieto Colorado
Número y simetría de los modos normales de vibración (MNV)
El problema dinámico radica en describir las vibraciones. Ello se hace a través de las 3N coordenadas de desplazamiento y de las 3N-6 de movimiento (3N-5, en moléculas lineales) o vibracionales.

©A. Carmelo Prieto Colorado
De las 3N coordenadas de movimento, 6 son movimientos globales o de conjunto, 3 son de traslación global (Tx,Ty,Tz), 3 de rotación global, (Rx, Ry, Rz) y las restantes 3N-6 son vibracionales.
Para describir el movimiento debemos considerar:1. Que cada átomo se mueve con diferente frecuencia, ωi 2. Que para cada frecuencia el conjunto se mueve
sincrónicamente pero con diferentes amplitudes
Ello permite introducir el concepto de coordenada normal de vibración, con 3N-6 Qi, atribuidas a las ωi.

©A. Carmelo Prieto Colorado
Por consiguiente se puede describir el movimiento partiendo de la siguiente transformacion de coordenadas
1. 3N coordenadas cartesianas (xi, yi, zi).2. Coordenadas internas (Δr, δφ, δτ), que dan sentido físico al
movimiento, se pueden relacionar con los potenciales del enlace químico y reducen los 6 movimientos de conjunto.
3. Coordenadas de simetría (Si), que reducen la ecuación secular, describen la simetría del movimiento, permiten establecer las reglas de selección (IR, Raman) y se corresponden biunivocamente con las normales
4. Se puede usar el formalismo de la teoría de grupos
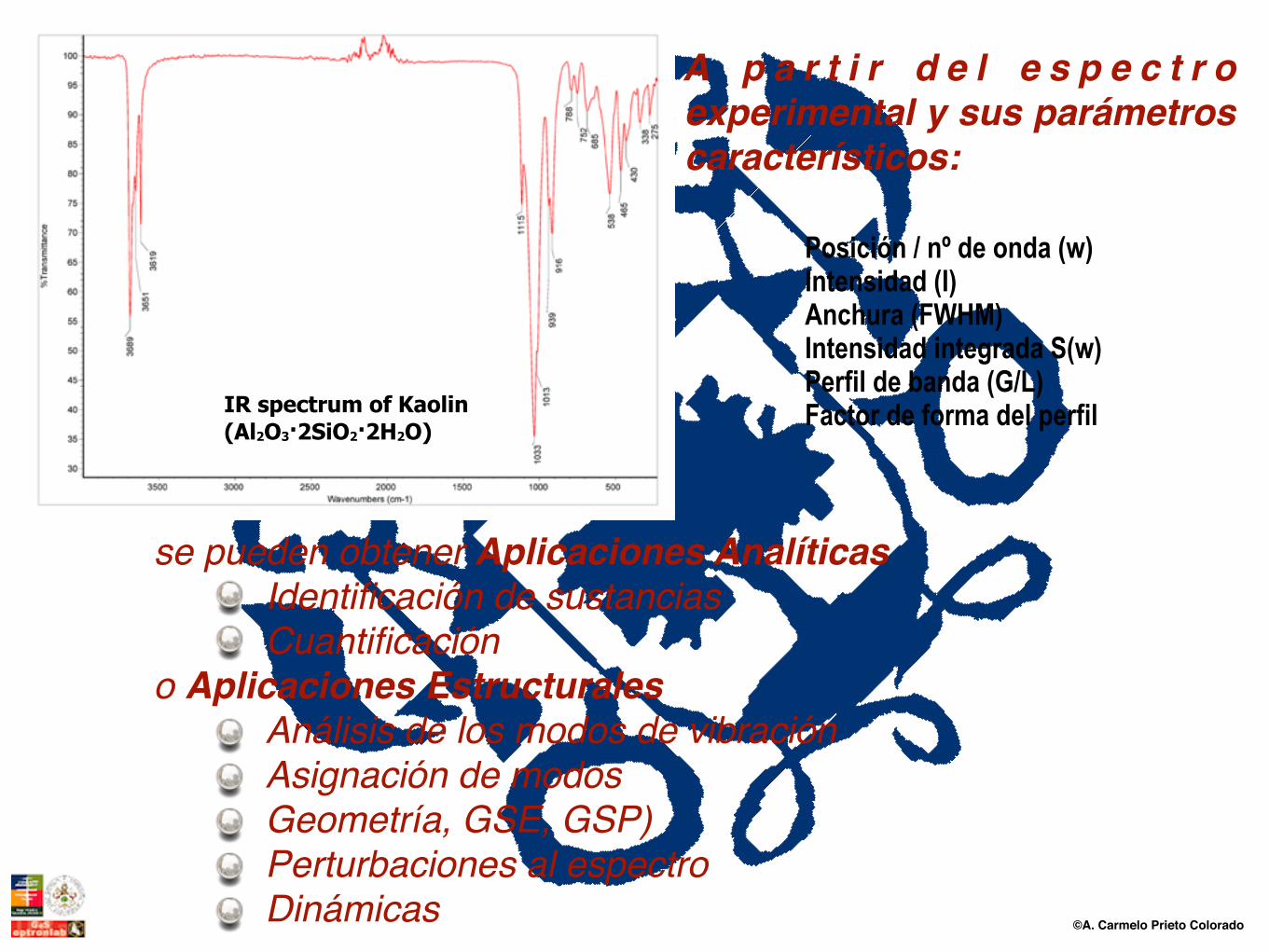
©A. Carmelo Prieto Colorado
se pueden obtener Aplicaciones Analíticas Identificación de sustanciasCuantificación
o Aplicaciones EstructuralesAnálisis de los modos de vibración Asignación de modos Geometría, GSE, GSP)Perturbaciones al espectroDinámicas
A p a r t i r d e l e s p e c t r o experimental y sus parámetros característicos:
IR spectrum of Kaolin (Al2O3·2SiO2·2H2O)
Posición / nº de onda (w) Intensidad (I) Anchura (FWHM) Intensidad integrada S(w) Perfil de banda (G/L) Factor de forma del perfil

©A. Carmelo Prieto Colorado

©A. Carmelo Prieto Colorado
Predicción de los MNV mediante métodos teóricos “ab initio”
Elegido un método de calculo de la estructura electrónica, es posible predecir la estructura de una molécula mediante una optimización geométrica. La optimización de geometría es simplemente la determinación de la estructura molecular en la cual la energía obtenida mediante el cálculo de estructura electrónica es mínima.
Para encontrar ese mínimo se emplean distintos algoritmos matemáticos de minimización de funciones de muchas variables (en este caso las distancias y ángulos de enlace de la molécula). Los métodos semiempíricos son muy eficientes para tal fin, en especial para moléculas orgánicas, siendo los errores cometidos del orden de 0.01 Å en distancias de enlace y de 1-2º en ángulos de enlace.
Los modos normales de vibración se pueden determinar mediante el cálculo de la matriz Hessiana. Esta matriz contiene las derivadas segundas de la energía molecular (calculada con algún método de estructura electrónica) con respecto a las coordenadas moleculares.

©A. Carmelo Prieto Colorado
Diagonalizando la matriz hessiana se obtienen:1. Los modos normales: combinaciones de las coordenadas
atómicas que permiten describir de una manera sencilla el movimiento vibracional de la molécula, ya que la energía potencial asociada con las vibraciones se puede descomponer como una suma de términos armónicos, uno por cada modo normal.
2. Las frecuencias de vibración, asociadas con cada modo normal.
Estas frecuencias pueden asignarse a las frecuencias observadas en espectros IR o Raman. Utilizando las frecuencias de vibración se puede obtener la energía vibracional en el 0K, denominada energía de punto cero, ZPE extendiendo la suma a todos los modos normales asociados a vibraciones.

Ángel Carmelo Prieto Colorado
Física de la Materia Condensada, Cristalografía y Mineralogía.Facultad de Ciencias.
Universidad de Valladolid.





























