Embed Size (px)
Citation preview

製品コード 6090製品コード 6091
説 明 書
Site-directed mutagenesis system
Mutan®-Express KmEnzyme/Oligo Set( 製品コード 6090 )
Vector/Host Set( 製品コード 6091 )
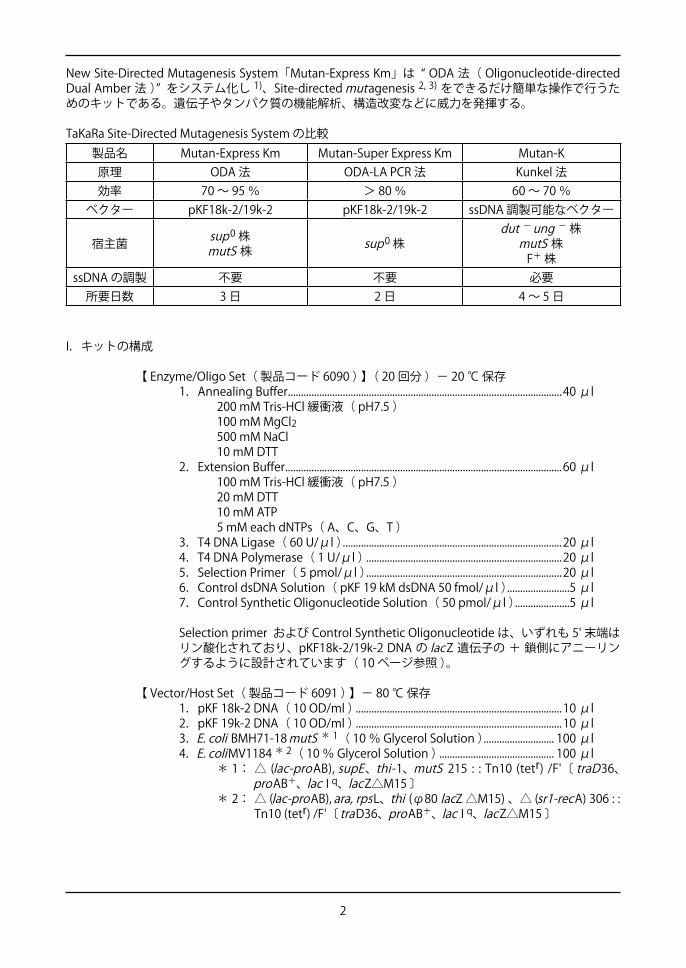
New Site-Directed Mutagenesis System「Mutan-Express Km」は “ ODA 法( Oligonucleotide-directed Dual Amber 法 )” をシステム化し 1)、Site-directed mutagenesis 2, 3) をできるだけ簡単な操作で行うためのキットである。遺伝子やタンパク質の機能解析、構造改変などに威力を発揮する。
TaKaRa Site-Directed Mutagenesis System の比較製品名 Mutan-Express Km Mutan-Super Express Km Mutan-K
原理 ODA 法 ODA-LA PCR 法 Kunkel 法効率 70 ~ 95 % > 80 % 60 ~ 70 %
ベクター pKF18k-2/19k-2 pKF18k-2/19k-2 ssDNA 調製可能なベクター
宿主菌 sup0 株mutS 株 sup0 株
dut - ung - 株mutS 株
F+ 株ssDNA の調製 不要 不要 必要
所要日数 3 日 2 日 4 ~ 5 日
I.キットの構成
【 Enzyme/Oligo Set( 製品コード 6090 ) 】( 20 回分 )- 20 ℃ 保存1.Annealing Buffer .........................................................................................................40 μl
200 mM Tris-HCl 緩衝液( pH7.5 )100 mM MgCl2500 mM NaCl10 mM DTT
2.Extension Buffer ..........................................................................................................60 μl100 mM Tris-HCl 緩衝液( pH7.5 )20 mM DTT10 mM ATP5 mM each dNTPs( A、C、G、T )
3.T4 DNA Ligase( 60 U/μl ) ....................................................................................20 μl4.T4 DNA Polymerase( 1 U/μl ) ...........................................................................20 μl5.Selection Primer( 5 pmol/μl ) ...........................................................................20 μl6.Control dsDNA Solution( pKF 19 kM dsDNA 50 fmol/μl )........................5 μl7.Control Synthetic Oligonucleotide Solution( 50 pmol/μl ) .....................5 μl
Selection primer および Control Synthetic Oligonucleotide は、いずれも 5' 末端はリン酸化されており、pKF18k-2/19k-2 DNA の lacZ 遺伝子の + 鎖側にアニーリングするように設計されています( 10 ページ参照 )。
【 Vector/Host Set( 製品コード 6091 ) 】- 80 ℃ 保存1.pKF 18k-2 DNA( 10 OD/ml ) ...............................................................................10 μl2.pKF 19k-2 DNA( 10 OD/ml ) ...............................................................................10 μl3.E. coli BMH71-18 mutS * 1( 10 % Glycerol Solution ) ........................... 100 μl4.E. coliMV1184 * 2( 10 % Glycerol Solution ) ............................................ 100 μl
* 1: △ (lac-proAB), supE、thi -1、mutS 215 : : Tn10 (tetr) /F'〔 traD36、proAB+、lac I q、lacZ△M15 〕
* 2: △ (lac-proAB), ara, rpsL、thi (φ80 lacZ △M15) 、△ (sr1-recA) 306 : : Tn10 (tetr) /F'〔 traD36、proAB+、lac I q、lacZ△M15 〕
2

図 1.pKF18k-2/19k-2 のクローニングサイト
pKF 18k-2 / 19k-22,204 bp
P O
ori
lacZ
lacZ
km'am
2
5' GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGC 3'
Hind IIISph I
Pst I
Sse8387 ISal IAcc I
Hinc II
Xba IBamH IKpn I Sma I
Xma ISac I
EcoR I
5' GCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC 3'
Xba ISal IAcc I
Hinc IISse8387 I
Sph IHind III EcoR I
Sac IKpn I
Sma IXma I
BamH IPst I
Multi cloning sltespkF 18k-2
pkF 19k-2lacZ
GenBank の Accession No.pKF18k * D63846pKF19k * D63847
*:なお、pKF18k と pKF19k については、最新のデータベースには、マルチクローニングサイト近くにある Nde I サイトの前の C が欠失したものが登録されています。弊社が販売している製品は欠失していない 2,204 bp のプラスミドです。
● キット以外に必要な試薬類・M9 glucose 培地: Na2HPO4 6 g/l KH2PO4 3 g/l NaCl 0.5 g/l NH4Cl 1 g/l thiamine * 1 mg/l MgSO4 * 1 mM CaCl2 * 0.1 mM glucose * 0.2 %
・M9 glucose plate: M9 glucose 培地に agar を 1.5 % 加える。・LB 培地( pH7.0 ): Bacto tryptone 10 g/l Bacto yeast extract 5 g/l NaCl 5 g/l
・LB plate: LB 培地に agar を 1.5 %加える。・テトラサイクリン・ストレプトマイシン・カナマイシン・TE Buffer( pH8.0 ): 10 mM Tris-HCl( pH8.0 )、1 mM EDTA・滅菌水・変異導入用合成オリゴヌクレオチド:
DNA プライマー合成の依頼はタカラバイオドラゴンジェノミクスセンターで承っております( TEL 077-543-7331、FAX 077-543-7225 )。また、ホームページからもご注文いただけます。
3

図 2.pKF18k-2/19k-2 の制限酵素認識部位とプライマー
・各種 Mini-prep に必要な試薬 Miniprep DNA Purification Kit( 製品コード 9085 )など
・0.2 M EDTA( pH8.0 )・E. coli BMH71-18 mutS Competent Cells 4, 5, 7) ( 製品コード 9054 )・E. coli MV1184 Competent Cells 4, 5, 7) ( 製品コード 9055 )・E. coli MV1184 Electro-Cells 5, 6, 7) ( 製品コード 9025 ):
コンピテントセル・エレクトロセルの形質転換効率が低いと十分な数のコロニーが得られないので、できるだけ効率のよいもの( > 106 cfu/μg DNA )を使う必要がある。
・SOC 培地 Bacto-tryptone 20 g/l Bacto yeast extract 5 g/l NaCl 10 mM KCl 2.5 mM MgSO4 * 10 mM MgCl2 * 10 mM glucose * 20 mM
*:これらは別々にオートクレーブし、無菌的に混合する。・pKF18k-2/19k-2 は pUC 系ベクターであり、変異導入後のシーケンス解析には以
下のプライマーをお勧めする。M13Primer M1 ( 製品コード 3810 ) 150 pmolM13Primer M2 ( 製品コード 3820 ) 150 pmolM13Primer M3 ( 製品コード 3831 ) 150 pmolM13Primer M4 ( 製品コード 3832A ) 150 pmol ( 製品コード 3832B ) 3,000 pmolM13Primer RV-N ( 製品コード 3833 ) 150 pmol
5' - TGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACACATATGACCATGATTAC3' - ACACCTTAACACTCGCCTATTGTTAAAGTGTGTCCTTTGTGTATACTGGTACTAATG
シーケンシングの方向
マルチクローニングサイト
SD配列 Nde I
M13 Primer RV-N5'... - 3'TGTGGAATTGTGAGCGG
lacZ5' GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGC 3'
Hind IIISph I
Pst I
Sse8387 ISal IAcc I
Hinc II
Xba IBamH IKpn I Sma I
Xma ISac I
EcoR I
5' GCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC 3'
Xba ISal IAcc I
Hinc IISse8387 I
Sph IHind III EcoR I
Sac IKpn I
Sma IXma I
BamH IPst I
pkF 18k-2
pkF 19k-2lacZ
ACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAAC...3'TGACCGGCAGCAAAATGTTGCAGCACTGACCCTTTTG...5'
5'...CAGGAAACACATATGACCATGATTAC3'...GTCCTTTGTGTATACTGGTACTAATG
マルチクローニングサイト
← TGACCGGCAGCAAAATG M3
← ATGTTGCAGCACTGA M1
← TTGCAGCACTGACCC M2
← CAGCACTGACCCTTTTG M4
Nde I
4
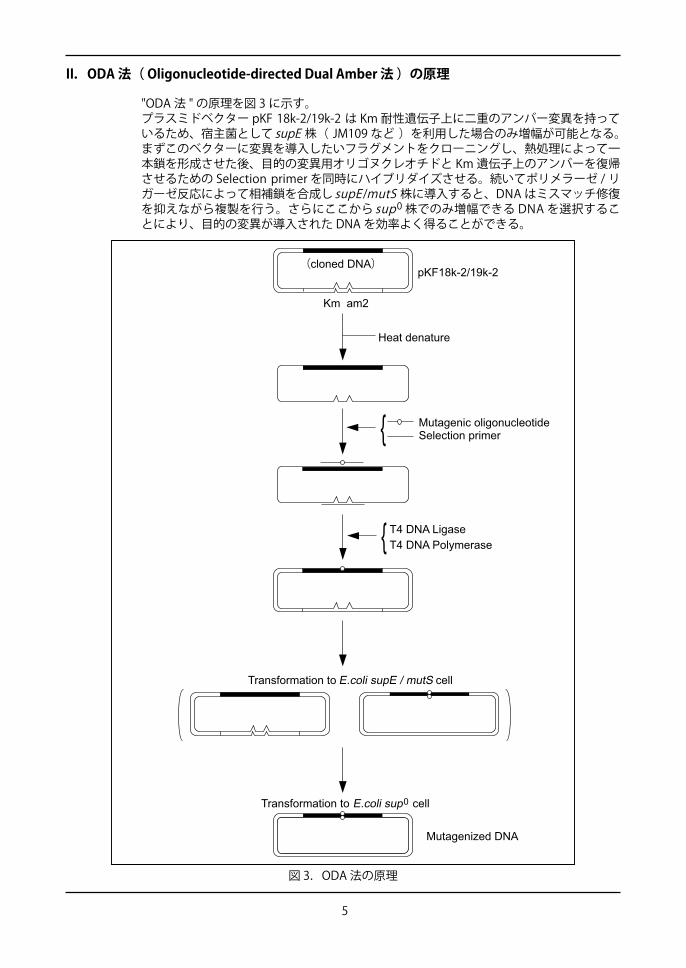
図 3.ODA 法の原理
II.ODA 法( Oligonucleotide-directed Dual Amber 法 )の原理
"ODA 法 " の原理を図 3 に示す。プラスミドベクター pKF 18k-2/19k-2 は Km 耐性遺伝子上に二重のアンバー変異を持っているため、宿主菌として supE 株( JM109 など )を利用した場合のみ増幅が可能となる。まずこのベクターに変異を導入したいフラグメントをクローニングし、熱処理によって一本鎖を形成させた後、目的の変異用オリゴヌクレオチドと Km 遺伝子上のアンバーを復帰させるための Selection primer を同時にハイブリダイズさせる。続いてポリメラーゼ / リガーゼ反応によって相補鎖を合成し supE /mutS 株に導入すると、DNA はミスマッチ修復を抑えながら複製を行う。さらにここから sup0 株でのみ増幅できる DNA を選択することにより、目的の変異が導入された DNA を効率よく得ることができる。
(cloned DNA)
Transformation to E.coli supE / mutS cell
Transformation to E.coli sup cell0
Mutagenized DNA
Km am2
Heat denature
Mutagenic oligonucleotideSelection primer
pKF18k-2/19k-2
{
T4 DNA LigaseT4 DNA Polymerase{
5

III.菌株の保存
BMH71-18mutS * 1
M9 glucose plate( テトラサイクリン 10 μg/ml を含む )上で 37 ℃ で一晩培養し、シングルコロニーを形成させる。コロニーを M9 glucose 培地( テトラサイクリン 10 μg/ml を含む )で 37 ℃ で一晩培養し、培養液 1.5 ml を、1.5 ml の 20 % グリセロールと混合し - 80 ℃ で保存する。
MV1184 * 2
BMH71-18mutS と同様に保存する。ただし、M9 glucose 培地にはテトラサイクリンを 10 μg/ml、ストレプトマイシンを 30 μg/ml となるように加える。
* 1:mutS のためミスマッチの修復能が低下している株で、合成オリゴヌクレオチドにより変異を導入した DNA を、ミスマッチを修復せずに固定するための host strain である。
* 2:mutS 株により固定化された変異導入 DNA と、テンプレート DNA をセレクションするための sup0 株である。
IV.変異導入用合成オリゴヌクレオチドの設計
変異導入用合成オリゴヌクレオチド( Mutagenic oligonucleotide )は Selectionprimer と同じ鎖にアニーリングさせなければならないので、lacZ 遺伝子の + 鎖と相補的な合成 DNA を使用する( 図 4 )。変異の導入にはシークエンスプライマーグレードの合成 DNA( 5'- 末端リン酸化済み )を使用する。変換したい塩基を中心にして、前後に 8 ~ 10 base、全体で約 20 base 程度の長さのものがよい。ただし欠失挿入または多数の塩基置換を行う場合は、ミスマッチ部分より前後 15 ~ 20 base の長さのもので 3' 末端が G、C の方が望ましい。合成 DNA が変異導入に適しているかどうかは、dideoxy 法によるシーケンスに使用して明瞭なラダーが得られるかどうかで検定すればよい。合成オリゴヌクレオチドは - 20 ℃ で保存し、凍結融解はできるだけ避ける。
V.操作
1.Template ds DNA の調製および Competent Cells の調製pKF18k-2 DNA( または pKF19k-2 DNA )に目的の DNA 断片を組み込んだ tem-plate DNA を調製する。この際の宿主は supE 株( JM109 など )を使用し、カナマイシン耐性培地で X-Gal、IPTG によるカラーセレクションにより組換え体を選択する。組換え体は Mini-Prep 法で精製する。また、Vector/Host Set に含まれている E. coli BMH71-18 mutS および E. coli MV1184 より通常の方法により Competent Cells を調製する。本法の場合、形質転換効率が低いと充分なコロニーが得られないので、できるだけ効率のよい Com-petent Cells( > 106 cfu/μg DNA )を使う必要がある。また、すでに調製済の Competent Cells も別途発売している。
E. coli Competent Cells BMH71-18 mutS( 製品コード 9054 )E. coli Competent Cells MV1184( 製品コード 9055 )E. coli Electro-Cells MV1184( 製品コード 9025 )
6

2.相補鎖の合成Template DNA 50 fmolSelection primer( 5 pmol/μl ) 1 μlMutagenic oligonucleotide 50 pmolAnnealing Buffer 2 μl滅菌水 X μlTotal 20 μl
( 1 ) 上記の反応液を用意し、100 ℃ で 3 分間、続いて氷水中で 5 分間静置する*。( 2 ) 3 μl の Extension Buffer、1 μl の T4 DNA Ligase、1 μl の T4 DNA
Polymerase および 5 μl の滅菌水を加えて 25 ℃ で 2 時間( または 37 ℃ で 1.5 時間 )静置する。
( 3 ) 3 μl の 0.2 M EDTA( pH8.0 )溶液を加え、65 ℃ で 5 分間静置する( すぐにトランスフォーメーションしないときは、- 20 ℃ で保存し凍結融解の繰り返しは避ける )。
* 冷却は、必ず氷に水をはった状態で行う。また、合成オリゴヌクレオチドの配列・鎖長などにより、アニーリングの温度を調整した方がよい場合がある。欠失・挿入などミスマッチが多くなるほどアニーリング条件は重要となる。
3.トランスフォーメーション-( 1 )変異の固定( 1 ) E.coli BMH71-18 mutS Competent Cells 100 μl と、相補鎖合成を行った DNA
溶液 10 μl を混合し、0 ℃ で 30 分間、42 ℃ で 45 秒間、0 ℃ で 1 ~ 2 分間静置する。
( 2 ) 37 ℃ の LB 培地( または SOC 培地 )を加えて 1 ml とし、37 ℃ で 1 時間振とうする。
( 3 ) LB 培地( または SOC 培地 )を 2 ml 追加し、カナマイシンを 100 μg/ml になるよう加えて一晩培養する。
4.DNA の精製 8, 9)( Mini-prep )アルカリ SDS 処理法、Boiling 法、リチウム法などの各種 Mini prep 法を参考に、3. の培養液から DNA を精製する。Miniprep DNA Purification Kit( 製品コード 9085 )の使用が便利である。
5. トランスフォーメーション -( 2 )変異導入 DNA の選別( 1 )E.coli MV1184 Competent Cells 100 μl * 1 と 4. の DNA 溶液 1 ~ 10 μl( 約
10 ng )を混合し、3. と同様に行なう* 2。( 2 )37 ℃ の LB 培地( または SOC 培地 )を加えて 1 ml とし、37 ℃ で 1 時間振
とうする* 3。( 3 )LB plate( カナマイシン 100 μg/ml を含む* 4 )上に培養液を適当量塗布し、
37 ℃ で一晩静置する。( 4 )できたコロニーのいくつかから DNA を調製し、シーケンスを行なってミュー
タントを確認する。* 1: Electro-Cells を用いた系で行なってもよい。* 2: この時過剰の DNA を加えるとバックグラウンド( 変異の導入された
プラスミドとそうでないプラスミドが共存する )がでる場合がある。* 3: トランスフォーメーションの効率が悪いコンピテントセルを使用す
る場合は、2 ~ 3 時間振とうする。* 4: カナマイシンの濃度が低いと、バックグラウンドがでる場合がある。
7

図 4.Mutan-Express Km のフローチャート
< 1 日目 > Reaction Mixture
100 ℃ 3 分氷水中 5 分
3 μl Extension buffer1 μl T4 DNA Ligase( 60 U )1 μl T4 DNA Polymerase( 1 U )5 μl 減菌水
25 ℃ , 2 時間( または 37 ℃ , 1.5 時間 )3 μl 0.2 M EDTA( pH8.0 )65 ℃ , 5 分
Sample DNA 溶液
100 μl E.coli BMH71-18 mutS コンピテントセル0 ℃ , 30 分42 ℃ , 45 秒0 ℃ , 1 ~ 2 分890 μl LB 培地( または SOC 培地 )37 ℃ , 1 時間振とう2 ml LB 培地 , 100 μg/ml カナマイシン37 ℃ , 1 晩振とう
< 2 日目 >
DNA 溶液
100 μl E. coli MV1184 コンピテントセル0 ℃ , 30 分42 ℃ , 45 秒0 ℃ , 1 ~ 2 分900 μl SOC 培地37 ℃ , 1 時間振とう
37 ℃ , 1 晩
< 3 日目 >
Transformation 2( 変異導入 DNA の選別 )
Mini Prep
Transformation 1( 変異の固定 )
Annealing-Extension
Template DNA 50 fmolSelection primer 1 μlMutagenic oligonucleotide 50 pmolAnnealing buffer 2 μl減菌水
20μl
30 μl
1 ~ 10 μl
適当量
LB plate( + Km )
コロニー
シーケンス解読
10 μl
ミュータントの確認
8
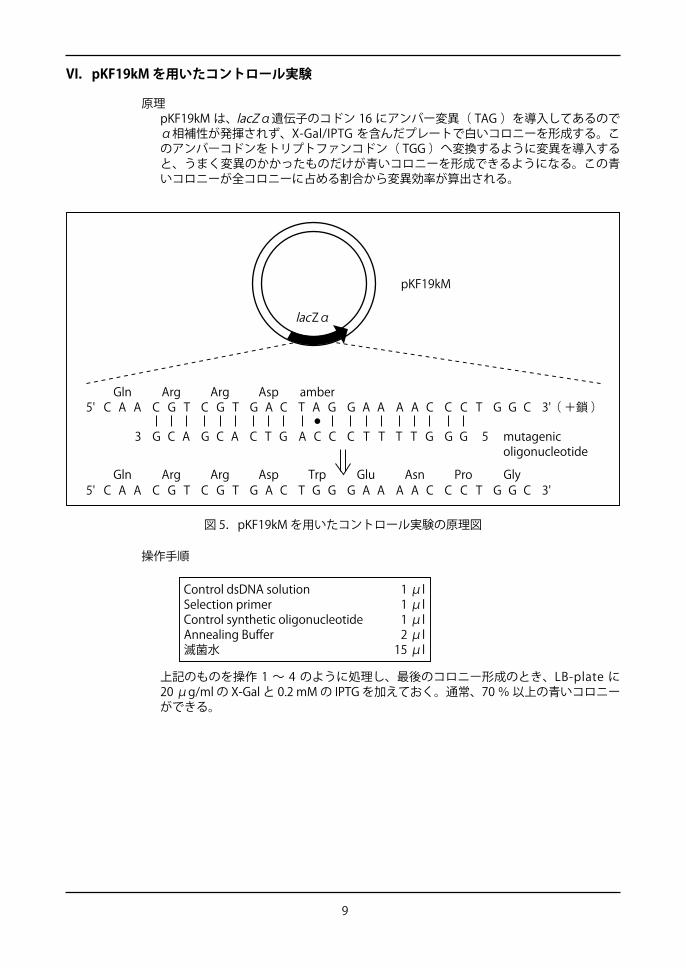
図 5.pKF19kM を用いたコントロール実験の原理図
VI.pKF19kM を用いたコントロール実験
原理pKF19kM は、lacZα遺伝子のコドン 16 にアンバー変異( TAG )を導入してあるのでα相補性が発揮されず、X-Gal/IPTG を含んだプレートで白いコロニーを形成する。このアンバーコドンをトリプトファンコドン( TGG )へ変換するように変異を導入すると、うまく変異のかかったものだけが青いコロニーを形成できるようになる。この青いコロニーが全コロニーに占める割合から変異効率が算出される。
操作手順
Control dsDNA solution 1 μlSelection primer 1 μlControl synthetic oligonucleotide 1 μlAnnealing Buffer 2 μl滅菌水 15 μl
上記のものを操作 1 ~ 4 のように処理し、最後のコロニー形成のとき、LB-plate に 20 μg/ml の X-Gal と 0.2 mM の IPTG を加えておく。通常、70 % 以上の青いコロニーができる。
lacZα
5' 3'C A A C G T C G T G A C T A G G A A A A C C C T G G C
53 G GT T GC T TA C CC T GG C AG C A
5' 3'C A A C G T C G T G A C T G G G A A A A C C C T G G C
mutagenicoligonucleotide
Gln Arg Asp amberArg
Gln Arg Arg Asp Trp AsnGlu Pro Gly
( +鎖 )
pKF19kM
9
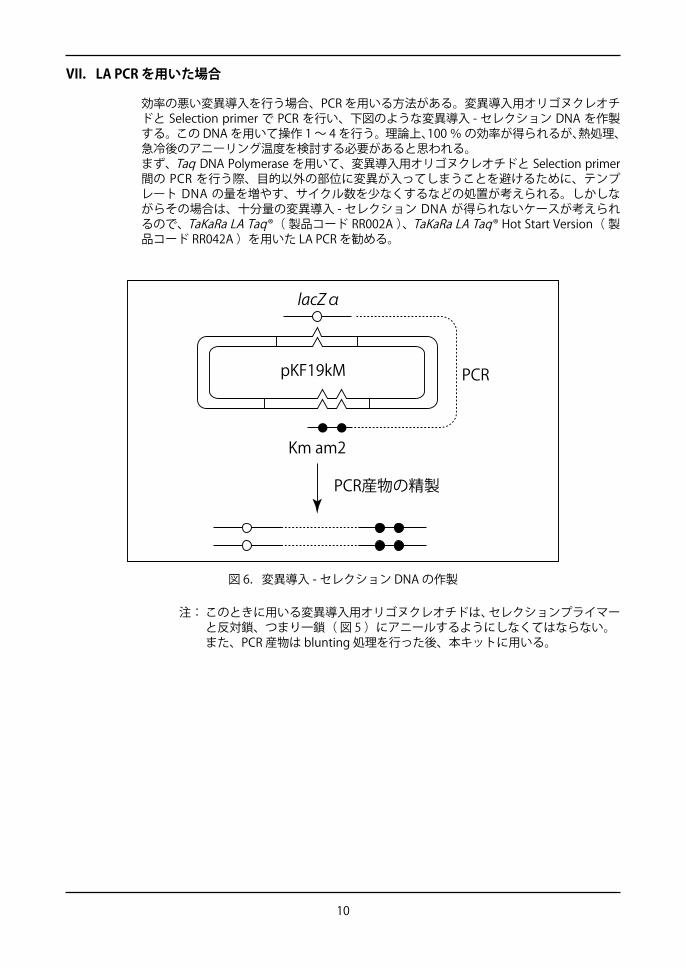
VII.LA PCR を用いた場合
効率の悪い変異導入を行う場合、PCR を用いる方法がある。変異導入用オリゴヌクレオチドと Selection primer で PCR を行い、下図のような変異導入 - セレクション DNA を作製する。この DNA を用いて操作 1 ~ 4 を行う。理論上、100 % の効率が得られるが、熱処理、急冷後のアニーリング温度を検討する必要があると思われる。まず、Taq DNA Polymerase を用いて、変異導入用オリゴヌクレオチドと Selection primer 間の PCR を行う際、目的以外の部位に変異が入ってしまうことを避けるために、テンプレート DNA の量を増やす、サイクル数を少なくするなどの処置が考えられる。しかしながらその場合は、十分量の変異導入 - セレクション DNA が得られないケースが考えられるので、TaKaRa LA Taq®( 製品コード RR002A )、TaKaRa LA Taq® Hot Start Version( 製品コード RR042A )を用いた LA PCR を勧める。
図 6.変異導入 - セレクション DNA の作製
lacZα
pKF19kM
Km am2
PCR産物の精製
PCR
注: このときに用いる変異導入用オリゴヌクレオチドは、セレクションプライマーと反対鎖、つまり一鎖( 図 5 )にアニールするようにしなくてはならない。また、PCR 産物は blunting 処理を行った後、本キットに用いる。
10

VIII.Q & A
Q1: スキーム最終段階での変異の入ったコロニーが得られないが?A1: ( 1 )Extension の条件を 37 ℃、1.5 時間に変更する。 ( 2 )Selection Primer の量を 0.5 μl に減らす。 ( 3 )変異導入用プライマーの量を 100 ~ 200 pmol くらいに増やす。 ( 4 )template 量を半分程度にまで減らす。
なお、これらの操作でも変異が導入されない場合は、目的のインサート DNA をサブクローニングした pKF18k-2 DNA( あるいは pKF19k-2 DNA )を MV1184 に形質転換し、100 μg/ml のカナマイシンを含む LB plate 上で生育してこないことをご確認ください。この操作で系の確認を行うことができます。
Q2: 同時に複数か所に変異導入したいが可能か?A2: デザインにもよりますが、1 つの変異プライマーに 2、3 か所のミスマッチができ
る程度のものであれば可能ですが、その場合変異導入用のオリゴヌクレオチドを過剰に加えるなどの工夫が必要です。また 2 種類以上の変異プライマーの使用は変異のかかっていないバックグラウンドが上昇するため、お勧めできません。
Q3: pKF18k-2、19k-2 に DNA を組み込む際の宿主は何を使用したらよいか?A3: E. coli JM109( supE 44 )の使用をお勧めします。
Q4: シーケンスを行う際の注意点は?A4: pUC 系のシーケンスプライマーを利用していただくことになりますが、M13
Primer RV は Nde I サイトの関係上使用できません。M13 Primer RV-N( 製品コード 3833 )をご使用ください。
IX.参考文献
1) Hashimoto -Gotoh, T., et al. (1995) Gene 152, 271-275.2) Zoller, M.J. and Smith, M. (1983) Methods in Enzymology 100, 468.3) 広瀬 進 (1986) 続生化学実験講座 1「遺伝子研究法 II」105.4) Hanahan, D. (1983) J.Mol. Biol. 166, 557.5) Sambrook, J., et al . (1989) Molecular Cloning 2nd. Edition 1.74-1.84.6) Dower, W. J., et al . (1988) Nucl. Acids Res.16, 61277) Ausubel, F. M., et al . (1987) Current Protocols In Molecular Biology. 1.8.1-1.8.8.8) Sambrook, J., et al . (1989) Molecular Cloning 2nd. Edition 1. 25-1.319) Ausubel, F. M., et al . (1987) Current Protocols In Molecular Biology 1. 6.1-1. 6.10.
X.関連商品
pKF18k-2 DNA( 製品コード 3101 )pKF19k-2 DNA( 製品コード 3102 )T4 DNA Ligase( 製品コード 2011A/B )T4 DNA Polymerase( 製品コード 2040A/B )E. coli BMH71-18 mutS Competent Cells( 製品コード 9054 )E. coli MV1184 Competent Cells( 製品コード 9055 )E. coli MV1184 Electro-Cells( 製品コード 9025 )Miniprep DNA Purification Kit( 製品コード 9085 )
11

NOTICE TO PURCHASER : LIMITED LICENSE
[M22] Site-directed mutagenesisThis product is covered by the claims of Japanese Patent No.3743525.
XI.注意
・本製品は研究用として販売しております。ヒト、動物への医療、臨床診断用 には使用しないようご注意ください。また、食品、化粧品、家庭用品等として使用しないでください。
・タカラバイオの承認を得ずに製品の再販・譲渡、再販・譲渡のための改変、商用製品の製造に使用することは禁止されています。
200811Da





























