Embed Size (px)
Citation preview

Smith Group Research
Invention of Catalytic Reactions
Materials From Renewable Resources
H Z
Z = BX2, aryl, OH, ...
Catalyst
CH3
OO
OO CH3
OHHO2C
n
Polylactic Acid
Commodity Materials Biological Applications
Energy
Developing New Chemistry for Old Fuels

$upport

C-H Activation: A Chemical “Holy Grail”
HNO3H NO2
Faraday, M. Philos. Trans. Roy. Soc. (London) 1825, 115, 440-466.
!
NO2H2N
OMe
NO2Br
OMe
Br
OH
7. Sn, HCl 8. H2SO4
9. NaNO2, HCl10. CuCl
5.NaNO2, HCl
6. CuBr
1. H2SO4, Na2Cr2O72. HOAc, heat
3. KOCN, MeOH4. Na2S, EtO
Cl
Hodgson, H. H.; Wignall, J. S. J. Chem. Soc. 1926, 2077.
BO
O+ + H2
150°C, 60 h 53% isolated yieldIverson and Smith JACS 1999, 121, 7696.
H H BPin
IrMe3P
H
20 mol %

Great Things Can Have Modest Beginnings
Cho, J.-Y.; Iverson, C. N.; and Smith, M. R., III JACS 2000, 122, 12868.Cho, J.-Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R., III Science 2002, 295, 305-308.Maleczka, R. E., Jr.; Shi, F.; Holmes, D.; Smith, M. R., III JACS 2003, 125, 7792-7793.Chotana, G. A.; Rak, M. A.; Smith, M. R., III JACS 2005, 127, 10539-10544.Paul, S.; et al. JACS 2006, 128, 15552-15553. Holmes, D.; Chotana, G. A.; Maleczka, R. E.; Smith, M. R., III Org. Lett. 2006, 8, 1407-1410.Shi, F.; Smith, M. R., III; Maleczka, R. E., Jr. Org. Lett. 2006, 8, 1411-1414.Chotana, G. A.; Kallepalli, V. A.; Maleczka, R. E., Jr.; Smith, M. R., III Tetrahedron 2008, 64, 6103-6114.Chotana, G. A.; Chem. Commun. 2009, 5731-5733.Vanchura, B. A., et al. Chem. Commun. 2010, 46, 7724-7726.Roosen, P. C., et al. JACS 2012, 134, 11350-11353.
R H R BPin
H H H BPin
[Ir(OMe)(COD)]2,IrIII/IrV cycle
50-5000 turnovers
BPinLL
H BPinIrBPin
R
BPinLL
H BPinIrBPin
H
Ir BPinLBPin
LBPin
Ir HLBPin
LBPin[IrCl(COD)]2, or
Ir(COD)
L LHBPin,
L L = bisphosphine or bpy

Boron is a Portal to Molecular Diversity
Cl
Br 1. H-Bpin, 0.02 eq. (Ind) Ir(COD), dmpe, 150 °C, 3 h
Cl
Br
OHone-pot, 79% yield
2. aqueous Oxone, acetone, 25 °C, 10 min
previous synthesis,10 steps from TNT
Maleczka, R. E.; Shi, F.; Holmes, D.; Smith, M. R. J. Am. Chem. Soc. 2003, 125, 7792-7793.
Aryl H H B(OR)2+
Aryl OH
Aryl NR2 Aryl X
Aryl SR
Aryl B(OR)2Aryl R

Broad Scope of C–H Functionalization
E CCH
Bpin
MeOOMe
Cl
Bpin
MeO2C
Cl CO2Me
pinB
CN
Br
Bpin
HN Bpin
N
NSO2NMe2
Bpin
Cl
MeO2CNHBoc
Bpin
O O
NH
SClBpin

Power to the People

Pyrrole Is The Biggest Outlier

N...H...O Bonding in the Transition State

Calculated Transition State
selectivity with more acidic N−H bonds were unsuccessful. Forexample, N-aryl triflamides gave N-borylation. Since solutionscontaining both HBpin and N-aryl triflamides are stable, an Ircomplex catalyzes N-borylation. In a plausible mechanism, σ-bond metathesis of the Ir−B bond with the highly acidictriflimide N−H bond generates intermediate 14 (Scheme 3) andsubsequent N−B elimination yields the N-borylated triflamide.An experimental result supporting the H-bonding mechanism
of 9 was the observation that increased basicity of the dipyridylligands enhances ortho selectivity (Figure 3). We propose that
the pinacolate oxygens in complexes with more electron-richdipyridyl ligands are more basic, and this accounts for theincreased ortho selectivity. While more electron-rich dipyridylligands have been shown to increase borylation rates,13 this is thefirst case where dipyridyl electronic effects significantly alterregioselectivities.Themechanism of the ortho direction and an explanation of its
failure with 12c was clarified by theoretical calculations. In M06/SDD(Ir)/6-31+G**(C H O N B) calculations, a series of TSswere located for C−H activation of PhNHCO2Me by the modelcomplex (bpy)Ir(Beg)3 (15, eg = ethyleneglycolate). Thesecalculations predict the ortho borylation to be strongly favored,and the TS (Figure 4) shows a clear NH−O H-bond. The
predicted o:m:p ratio (based on the lowest-energy TSs for eachpossibility) is 88:8:4, which is very close to the experimentalvalue for borylation of PhNHCO2Me by 2 (o:m:p = 90:5:5 fromGC-FID). The experimental ratio is a direct measurement ofrelative rates for directed and nondirected borylation. Theagreement between theory and experiment strongly supports theNH−O H-bonding mechanism. The calculated ortho TS alsosuggests a steric explanation for the failure of H-bondingdirection in 12c; when the OCH3 of the carbamate is replaced byan ethyl group and the Beg groups are replaced by BPin groups,
there is a tight steric interaction (with H−H distances <1.0 Å)that would preclude the ortho structure. An interestingobservation is that the ortho transition structure is very stronglyfavored enthalpically, but the restricted motion associated withthe H-bond is disfavored entropically, so the final selectivity islimited by entropy/enthalpy compensation.For Boc-protected anilines with a single meta substituent,
there is a trade-off between H-bond direction and the usualpreference for reaction at the least hindered position (Table 1,
entry 1). This is not an issue for 4-substituted substrates wherethe ortho selectivity is high. Except for the fluorine-substitutedsubstrates in entries 5 and 6, these products were isolated assingle regioisomers. For entries 11 and 12, 3 equiv of arene wereused tominimize diborylation. Converting Bpin products to theirBF3K salts can in cases ease isolation, leading to the higher yieldin entry 12 versus 11. All regiochemical assignments were basedon NMR spectroscopy and confirmed by X-ray crystallographyfor entries 4 and 7−9.Entry 2 in Table 1 shows that substrates with more acidic
protons are viable through in situ protection as borates by excesspinacolborane. Entries 1, 3−7, and 10 produce boronates withhalogen groups that can be further manipulated. Entries 11 and12 are noteworthy because the selectivity diverges from that ofthe acetamide analog, which borylates ortho to CN exclusively.14
The comparison with DoM in Scheme 4 highlights the newselectivity offered by H-bond direction. When there is adistinguishable choice among ortho C−H bonds, DoM isdominated by acidity. Thus, for substrates 16 and 17 (entries 5and 7 in Table 1), the more acidic C−H bonds flanked byNHBoc and a halogen will metalate first.15 In contrast the Ir-catalyzed process is selective for the less hindered C−H bondortho to NHBoc. For substrates 18 and 19, the O-aryl carbamateis a stronger director for DoM thanNHBoc, but it provides noH-bond. Consequently, the preferred site for C−H borylation is the
Figure 3. Plot of the ortho/meta product ratios using 4,4′-di(R1)-2,2′-dipyridyl catalysts vs pKa's of 4-R
1-pyridium ions.
Figure 4. Lowest energy TS for C−H activation of PhNHCO2Me bymodel complex 15. The o:m:p ratios predicted by theory and thosefound for borylation catalyzed by 2 are given.
Table 1. ortho-Borylation of N-(Boc)-Anilinesa
aSee Supporting Information for details on conditions. Yields refer toisolated material except as noted. bYield determined by 1H NMR.cYield is for the major isomer. dIsolated as a 92:8 mixture.
Journal of the American Chemical Society Communication
dx.doi.org/10.1021/ja303443m | J. Am. Chem. Soc. 2012, 134, 11350−1135311352

Reaction ScopeNH2
R1
R2
NH2
Cl
Bpin
CN
0.25 mol% [Ir(OMe)COD]21.0 mol% tmphen
THF,16h, 80 °C+
3.0 equiv
90%
NH2
Cl
Bpin
Br
NH2Bpin
Br
88% 92%
NH2
Cl
Bpin
OH
63%
NH2Bpin
Cl
93%
NH2
R1
Bpin
R2
NH2Bpin
74%
ClOMe
NH2
OHNCl
NH2
Bpin
73% 90%70%
NH2
F
Bpin
Cl
BpinNH2
CN
Bpin
H2B
89%
HBpin
Sean Preshlock and Don Plattner

Poly(lactic) Acid: A Renewable Commodity Plastic
RO
OO
O
O
H
CH3
CH3
n
RO
OO
O
O
H
CH3
CH3
n
poly(rac-lactide) (PLA)poly(L-lactide) (PLLA)crystalline (Tm = 180 °C) glassy Tg (~55 °C):
OH
O
L-Lactic Acid
H3C
OH
O
O
H3C
O CH3
O
L-Lactide
Dehydration and Thermal Cracking
Hydrolysis
PLLAn
OO
OHR
O
OCH3
CH3
Metal catalystROHFermentation
PLLAn
OO
OHR
O
OCH3
CH3
Metal catalystROH

Degradable Single Molecule Micelles
7
the resulting polymers 4 were comparable to the ratio of the azides in the feed. After isolating the polymer and purification by dialysis, we used cloud point measurements to screen samples for LCST behavior. Samples were dissolved in Milli-Q water and solution turbidity was monitored by measuring absorbance at 450 nm as a function of temperature. At the LCST, the “apparent absorbance” increases as gel formation causes a dramatic increase in scattering. Significantly, the relationship between the cloud point temperature and the mole fraction of mDEG chains in the polymer is linear, as shown in Figure 15. This allows for simple and exquisite adjustment of the LCST in a degradable material, where the mDEG/alkyl ratio serves as a “molecular thermostat” for the LCST. These results are preliminary and it may be possible to expand the LCST window to beyond the 25-65 !C range shown in Figure 15 by varying the lengths of the alkyl and PEG side chains.
Preliminary degradation studies have also been carried out in Milli-Q water at 36 °C and data for polymer 4 where the mDEG incorporation is 70% is shown in Figure 16. The degradation rates are much faster than that for PLA, which shows no molecular weight loss under similar conditions. Hence, the hydrophobic chains are clearly accelerating the hydrolysis rates. We expect that we will be able modulate the degradation rates by varying the pH, altering the side chain composition, and by preparing copolymers with lactide.
C3. Encapsulation of molecules and macromolecules in polymer nanomicelles. With their hydrophobic and hydrophilic chains, polymers 4 are amphipols. We attempted to form micellular assemblies of 4, but we observed no increase in the hydrodynamic radius by dynamic light scattering over a broad concentration. The reason for this was clear when the materials were examined by TEM. As shown in Figure 17, amphipols 4 form discrete unimolecular micelles? This behavior is independent of molecular weight, which means that the dimensions of the polymer nanomicelles can be adjusted by changing molecular weight. These nanomicelles have good dimensional stability as freeze-dried materials were little changed when reexamined one month later.
To test whether our nanomicelles will mirror the behavior of micelles, we have performed two experiments with amphipol 4. As shown in Figure 18, it is possible to drive hydrophobic, azo-tagged PMMA into amphipol 4 by increasing the solvent polarity. Similarly, Rhodamine B, which is hydrocarbon insoluble, can be entrapped when nanomicelle 4 is reversed. After the solvent was evaporated, the reversed nanomicelles can be
Figure 16. Degradation of polymer 4 (70% mDEG/30% decyl side chains) in Milli-Q water at 36 °C.
a b
Figure 17. TEM images of polymer nanomicelles (a) Mn ~ 43,0000, diameter ~ 10 nm (b) Mn~ 260,000, diameter ~ 30 nm.
Figure 18. Encapsulations of an azo-tagged PMMA (Mn ~ 10,000) in nanomicelle 4 and the dye Rhodamine B in reversed nanomicelle 4. When the solutions were evaporated and the nanomicelles were redissolved, they retained their trapped guests.
RO OO n
H
OH
ORO
1-x
OO
Degradable Amphiphilic Polymer
x n
NNN
N3
NNNOO
N3O Ox
1–x
+
O
O"Click"
+

Functional Nanoparticle Applications
Drug Screening(Labeling)
Liu, Y. Y.; Miyoshi, H.; Nakamura, M. Int. J. Cancer 2007, 120, 2527-2537.
Gene Delivery(Transfection--siRNA or DNA)
Diagnosis(Devices and Labeling)
Drug Delivery(Therapy)
Detection(Imaging)
Diagnosis/Monitoring(Disease Markers)
Well-defined
Stimuli-responsive
Cross-linkable
Biocompatible
Degradable
Controllable Release
Easily Synthesized

From Degradable to Nondegradable Nanomicelles
ROO
O
O
O
Hn
N3R
"Click"
ROO
O
O
O
Hn
NNRN
RNN N
H2O HOOH
O
NNRN
2n
degradable
RO O Hn+1
ON3
R
"Click"
RO O Hn+1
O
RNN N
stable
HO O Hn+1
O
BF3·OEt2O
O
R R
O
Cl HO
R R+
Marie Le Gaillard
37-66 % yield, 20 g scale
NaOH, H2O5 mol% [NBu4][HSO4–]
0 °C to r.t., 3 h
4 equivR = Me, H

A Simple Market Analysis
Membrane proteins are projected to have a $30 billion annual market...
... That buys a lot of beer.
Tween-80
CO2–
Na+
OCNH
OCNH
x y z
Amphipol
Poorly degradable

Encapsulation of Non-Polar and Polar Species in Nanomicelles
Enhancing Protein Solubility with Degradable and Tailorable Micelles
! "!
#$%&'()*)+)%,(-'..'! -%,/.'0+&(%)!1(..! 2'! 34..5! ()&'$$%6+&'7! 2'-+48'! (&! (8!-$(&(-+.! &9+&! 1'! +$'! +2.'! &%! 7'&'$,()'!19'&9'$! &9'! /$%&'()! -%,/.'0! 3%$,8:';')!19')!/$%&'()!+88+58!6(;'!)'6+&(;'!$'84.&8<! =4$! /$(,+$5! 8&$+&'65! 1(..!();%.;'! .+2'.()6! &9'! )+)%,(-'..'8!+)7>%$! &9'! /$%&'()! 1(&9! 8/'-&$%8-%/(-!&+68! 8%! -%,/.'0+&(%)! -+)! 2'! +)+.5?'7!25! 6'.! /'$,'+&(%)! -9$%,+&%6$+/95!@A#BC<! D+2'.()6! &9'! )+)%,(-'..'8!89%4.7! 2'! 8&$+(69&3%$1+$7! 8()-'!E-.(-F+2.'G! 75'! &+68! 9+;'! 2'')!7';'.%/'7! 3%$! /$%&'()*.(6+)7! 2()7()6!+88+58<HI! J948K! 1'! -+)! E-.(-FG! $'8(74+.!+.F5)'! 6$%4/8! 1(&9! +?(7'! 75'8! +8!89%1)! ()! L(64$'! 21<! J9'$'! +$'! 8';'$+.!,'&9%78! 3%$! .+2'.()6! &9'! /$%&'()8! 1(&9!8/'-&$%8-%/(-! &+68<! L%$! BOPQK! $'&()%(-!+-(7K! +28%$2()6! +&! 3"I! ),K! (8! 2%4)7!S4(&'! &(69&.5! @2! )T! 7(88%-(+&(%)!-%)8&+)&C! &%! &9'! /$%&'()! ()&'$(%$<! ! U'!+.8%! 9+;'! +--'88! &%! ,4&+)&8! 19'$'!$'&()%(-!+-(7!(8!-%;+.')&.5!.()F'7K!19(-9!1(..!')84$'!&9+&!&9'!&+6!(8!)%&!$'.'+8'7!(3!&9'!/$%&'()!,(83%.78<!L%$!%&9'$!/$%&'()8K!,%$'! 6')'$+.! .+2'.()6!,'&9%78!1(..! 2'!48'7:&9'! -%;+.')&! 3.4%$'8-')&! /$%2'8!7';'.%/'7! 25! J8(')! +)7! -%1%$F'$8!2'()6! /+$&(-4.+$.5! +&&$+-&(;'<H1! U'! -+)!,+F'! &9'! )'-'88+$5! ,4&+)&8! (3!)'-'88+$5! 3%$! &9+&! +8!1'..<! ! Q5! .+2'.()6!&9'!)+)%,(-'..'8!+)7!/$%&'()8!1(&9!&+68!&9+&! +28%$2! +&! 7(33'$')&! 1+;'.')6&98K!2()7()6!-+)!2'!+88+5'7!25!48()6!+!A#B!1(&9! +! VW*;(8! 7'&'-&%$<! X4--'8834.!-%,/.'0+&(%)!1(..! 2'! ';(7')&!19')! &9'!A#B! &$+-'8! +$'! (7')&(-+.! 19')!,%)(&%$'7! +&! !,+0! 3%$! '+-9!-9$%,+/9%$'<! J9'! +88%-(+&(%)! %3!,4.&(/.'! )+)%,(-'..'8! 1(&9! +! 8()6.'!/$%&'()K! 19(-9! ,+5! %--4$! 1(&9! .+$6'$!/$%&'()8!.(F'!#AXY*1K!1(..!2'!%2;(%48!3$%,!A#B!+)+.58(8!2'-+48'!%3!&9'!.+$6'!()-$'+8'!()!,%.'-4.+$!1'(69&<!
B9',(-+.!+88+58!1(..!2'!/+$&(-4.+$.5!(,/%$&+)&!3%$!7'&'$,()()6!19'&9'$!&9'!/$%&'()Z8!+-&(;(&5!(8!,+()&+()'7!%)-'! (&! (8! 8'S4'8&'$'7!1(&9()! &9'! )+)%,(-'..'8<! L(64$'! 22! 89%18! &9'! Q'$6')()! &$+)8'8&'$(3(-+&(%)!1(&9! ;()5.!24&5$+&'K!19(-9! (8! +! -%);')(')&! -9',(-+.! +88+5! 3%$!XB<1[! X()-'!+-&(;(&5!%3! XB! (8! +-&4+..5! ')9+)-'7! ()!%$6+)(-!8%.;')&8K! &9(8! +88+5! 1(..! 2'! /+$&(-4.+$.5! 48'34.! 3%$! 7'&'$,()()6! &9'! '33'-&8! %3! 8428'S4')&! -9',(-+.!&$+)83%$,+&(%)8! %)! &9'! )+)%,(-'..'K! %)-'! &9'! /$%&'()! 9+8! 2'')! -%,/.'0'7<! =)'! '0+,/.'! %3! +! /%8&*2()7()6!,%7(3(-+&(%)!1'!9+;'!/.+))'7!(8!&9'!E-.(-FG!$'+-&(%)!&9'!/$%&'()*)+)%,(-'..'!1(&9!2(8+?(7'8!3$%,!L(64$'!1"<!P8!89%1)! ()! L(64$'! 23! -$%88.()F()6! 89%4.7! $'8&$(-&! 7(3348(%)! %3! &9'! /$%&'()! 3$%,! &9'! -%$'K! 5'&! (&! 89%4.7! 8&(..! 2'!/%88(2.'!3%$!8,+..!,%.'-4.'8!&%!7(3348'!()&%!)+)%/+$&(-.'8<!P6+()K!1'!-+)!48'!-9',(-+.!+88+58!&%!7'&'$,()'!&9'!'33'-&8!%3!&9'!,%7(3(-+&(%)!%)!/$%&'()!+-&(;(&5<!
B$%88.()F()6!,+5!+.8%!+88(8&! ()! &$+)8/%$&!%3! &9'!)+)%,(-'..'*/$%&'()!+88',2.5! ()&%!%$6+)(-!8%.;')&8<!=)'!$'+8%)!&9+&!1'!+$'!()&'$'8&'7!()!();'$&()6!&9'!-$%88.()F'7!,(-'..'!(8!89%1)!()!L(64$'!2\<!X/'-(3(-+..5K!1'!9%/'!&%! ();'$&! &9'! )+)%,(-'..'! ()! +)! %$6+)(-! 8%.;')&! +)7! &9')! +77! +! .+$6'$! +,/9(/%.<! Q5! ()-$'+8()6! &9'! 8%.;')&!
!"#$%e 21.! X&$+&'6('8! 3%$! .+2'.()6! )+)%,(-'..'8! &%! +88+5! /$%&'()*)+)%,(-'..'!-%,/.'0+&(%)<!!
!
!
!"#$%e 22.! J9'! Q'$6')()! &$+)8'8&'$(3(-+&(%)! 1(&9! ;()5.! 24&5$+&':+! -%);')(')&!-9',(-+.!+88+5!3%$!X42&(.(8()!B+$.82'$6!@XBC<!
!
!"#$%e 2+.! EB.(-FG! -$%88*.()F()6! %3! /$%&'()*)+)%,(-'..'! -%,/.'0'8<! ]33'-&8! %3! &9'!,%7(3(-+&(%)!%)!&9'!/$%&'()!-+)!2'!7'&'$,()'7!25!-9',(-+.!+88+58<!
!
! "!
#$%&'()*)+)%,(-'..'! -%,/.'0+&(%)!1(..! 2'! 34..5! ()&'$$%6+&'7! 2'-+48'! (&! (8!-$(&(-+.! &9+&! 1'! +$'! +2.'! &%! 7'&'$,()'!19'&9'$! &9'! /$%&'()! -%,/.'0! 3%$,8:';')!19')!/$%&'()!+88+58!6(;'!)'6+&(;'!$'84.&8<! =4$! /$(,+$5! 8&$+&'65! 1(..!();%.;'! .+2'.()6! &9'! )+)%,(-'..'8!+)7>%$! &9'! /$%&'()! 1(&9! 8/'-&$%8-%/(-!&+68! 8%! -%,/.'0+&(%)! -+)! 2'! +)+.5?'7!25! 6'.! /'$,'+&(%)! -9$%,+&%6$+/95!@A#BC<! D+2'.()6! &9'! )+)%,(-'..'8!89%4.7! 2'! 8&$+(69&3%$1+$7! 8()-'!E-.(-F+2.'G! 75'! &+68! 9+;'! 2'')!7';'.%/'7! 3%$! /$%&'()*.(6+)7! 2()7()6!+88+58<HI! J948K! 1'! -+)! E-.(-FG! $'8(74+.!+.F5)'! 6$%4/8! 1(&9! +?(7'! 75'8! +8!89%1)! ()! L(64$'! 21<! J9'$'! +$'! 8';'$+.!,'&9%78! 3%$! .+2'.()6! &9'! /$%&'()8! 1(&9!8/'-&$%8-%/(-! &+68<! L%$! BOPQK! $'&()%(-!+-(7K! +28%$2()6! +&! 3"I! ),K! (8! 2%4)7!S4(&'! &(69&.5! @2! )T! 7(88%-(+&(%)!-%)8&+)&C! &%! &9'! /$%&'()! ()&'$(%$<! ! U'!+.8%! 9+;'! +--'88! &%! ,4&+)&8! 19'$'!$'&()%(-!+-(7!(8!-%;+.')&.5!.()F'7K!19(-9!1(..!')84$'!&9+&!&9'!&+6!(8!)%&!$'.'+8'7!(3!&9'!/$%&'()!,(83%.78<!L%$!%&9'$!/$%&'()8K!,%$'! 6')'$+.! .+2'.()6!,'&9%78!1(..! 2'!48'7:&9'! -%;+.')&! 3.4%$'8-')&! /$%2'8!7';'.%/'7! 25! J8(')! +)7! -%1%$F'$8!2'()6! /+$&(-4.+$.5! +&&$+-&(;'<H1! U'! -+)!,+F'! &9'! )'-'88+$5! ,4&+)&8! (3!)'-'88+$5! 3%$! &9+&! +8!1'..<! ! Q5! .+2'.()6!&9'!)+)%,(-'..'8!+)7!/$%&'()8!1(&9!&+68!&9+&! +28%$2! +&! 7(33'$')&! 1+;'.')6&98K!2()7()6!-+)!2'!+88+5'7!25!48()6!+!A#B!1(&9! +! VW*;(8! 7'&'-&%$<! X4--'8834.!-%,/.'0+&(%)!1(..! 2'! ';(7')&!19')! &9'!A#B! &$+-'8! +$'! (7')&(-+.! 19')!,%)(&%$'7! +&! !,+0! 3%$! '+-9!-9$%,+/9%$'<! J9'! +88%-(+&(%)! %3!,4.&(/.'! )+)%,(-'..'8! 1(&9! +! 8()6.'!/$%&'()K! 19(-9! ,+5! %--4$! 1(&9! .+$6'$!/$%&'()8!.(F'!#AXY*1K!1(..!2'!%2;(%48!3$%,!A#B!+)+.58(8!2'-+48'!%3!&9'!.+$6'!()-$'+8'!()!,%.'-4.+$!1'(69&<!
B9',(-+.!+88+58!1(..!2'!/+$&(-4.+$.5!(,/%$&+)&!3%$!7'&'$,()()6!19'&9'$!&9'!/$%&'()Z8!+-&(;(&5!(8!,+()&+()'7!%)-'! (&! (8! 8'S4'8&'$'7!1(&9()! &9'! )+)%,(-'..'8<! L(64$'! 22! 89%18! &9'! Q'$6')()! &$+)8'8&'$(3(-+&(%)!1(&9! ;()5.!24&5$+&'K!19(-9! (8! +! -%);')(')&! -9',(-+.! +88+5! 3%$!XB<1[! X()-'!+-&(;(&5!%3! XB! (8! +-&4+..5! ')9+)-'7! ()!%$6+)(-!8%.;')&8K! &9(8! +88+5! 1(..! 2'! /+$&(-4.+$.5! 48'34.! 3%$! 7'&'$,()()6! &9'! '33'-&8! %3! 8428'S4')&! -9',(-+.!&$+)83%$,+&(%)8! %)! &9'! )+)%,(-'..'K! %)-'! &9'! /$%&'()! 9+8! 2'')! -%,/.'0'7<! =)'! '0+,/.'! %3! +! /%8&*2()7()6!,%7(3(-+&(%)!1'!9+;'!/.+))'7!(8!&9'!E-.(-FG!$'+-&(%)!&9'!/$%&'()*)+)%,(-'..'!1(&9!2(8+?(7'8!3$%,!L(64$'!1"<!P8!89%1)! ()! L(64$'! 23! -$%88.()F()6! 89%4.7! $'8&$(-&! 7(3348(%)! %3! &9'! /$%&'()! 3$%,! &9'! -%$'K! 5'&! (&! 89%4.7! 8&(..! 2'!/%88(2.'!3%$!8,+..!,%.'-4.'8!&%!7(3348'!()&%!)+)%/+$&(-.'8<!P6+()K!1'!-+)!48'!-9',(-+.!+88+58!&%!7'&'$,()'!&9'!'33'-&8!%3!&9'!,%7(3(-+&(%)!%)!/$%&'()!+-&(;(&5<!
B$%88.()F()6!,+5!+.8%!+88(8&! ()! &$+)8/%$&!%3! &9'!)+)%,(-'..'*/$%&'()!+88',2.5! ()&%!%$6+)(-!8%.;')&8<!=)'!$'+8%)!&9+&!1'!+$'!()&'$'8&'7!()!();'$&()6!&9'!-$%88.()F'7!,(-'..'!(8!89%1)!()!L(64$'!2\<!X/'-(3(-+..5K!1'!9%/'!&%! ();'$&! &9'! )+)%,(-'..'! ()! +)! %$6+)(-! 8%.;')&! +)7! &9')! +77! +! .+$6'$! +,/9(/%.<! Q5! ()-$'+8()6! &9'! 8%.;')&!
!"#$%e 21.! X&$+&'6('8! 3%$! .+2'.()6! )+)%,(-'..'8! &%! +88+5! /$%&'()*)+)%,(-'..'!-%,/.'0+&(%)<!!
!
!
!"#$%e 22.! J9'! Q'$6')()! &$+)8'8&'$(3(-+&(%)! 1(&9! ;()5.! 24&5$+&':+! -%);')(')&!-9',(-+.!+88+5!3%$!X42&(.(8()!B+$.82'$6!@XBC<!
!
!"#$%e 2+.! EB.(-FG! -$%88*.()F()6! %3! /$%&'()*)+)%,(-'..'! -%,/.'0'8<! ]33'-&8! %3! &9'!,%7(3(-+&(%)!%)!&9'!/$%&'()!-+)!2'!7'&'$,()'7!25!-9',(-+.!+88+58<!
!
"
#$%&''()*$% &+ ,()-$+$ .&,/ +( 0#$1&0&,2,&(+ (3 4/(%25&+$ 67 8/$'$ $90$#&5$+,' 0#(*&%$ :0#((3 (3 0#&+1&0)$; 3(# (-# 0#(0('$% #$'$2#1/7
!"# $e&e'(c*#!e&+,-#'-.#Me0*o.&#
!"#$ %&'(n*in,$ -.e$ 0c2ic34$ -oo26o&$ 7o8$ 9:69-i-:-e*$ 'o2;,2;co2i*e9# <+ (#%$# ,( ',2=&)&>$ 0#(,$&+'? .$ .&)) )&@$)A +$$% ,( 2%(#+ 0()AB)A1()&%$' .&,/ 2+ 2##2A (3 '&%$ 1/2&+'7 C)1(/() '&%$ 1/2&+' '/(-)% /$)0 ',2=&)&>$ /A%#2,$% 1(-+,$#&(+' 3(# 1/2#B$% #$'&%-$' 2+% 52@$ %&#$1, /A%#(B$+ =(+%' ,( ,/$ 0#(,$&+D' '-#321$7 E2#=(9A)&1 21&%' 2+% 25&+$' .&)) =$ &(+&>$% 2, 0/A'&()(B&12))A #$)$*2+, 0F 2+% 52A 2)'( 2''&', &+ ',2=&)&>&+B 1/2#B$% 0$0,&%$ '&%$ 1/2&+'7 G/('0/2,&%A) 1/()&+$ 2>&%$' 12+ 0(,$+,&2))A 5&5&1 1$))-)2# 5$5=#2+$'7 6&'2>&%$' .&)) $+2=)$ 1#('')&+@&+B (3 +2+(5&1$))$' ,/2, 1(+,2&+ #$'&%-2) 2)@A+$ 5(&$,&$'7 8/&' '/(-)% /$)0 &50#(*$ ',2=&)&,A 2+% 2)'( =$,,$# 1(+3&+$ B-$',' '$H-$',$#$% &+ ,/$ +2+(5&1$))$ &+,$#&(#7 <+ ,/$ &55$%&2,$ 3-,-#$ .$ 0)2+ ,( 'A+,/$'&>$ ,/$ 2>&%$' &+ I&B-#$ JK? 2)) (3 ./&1/ 2#$ @+(.+ ,( ,/$ )&,$#2,-#$7L"MNO I(# 2>&%$' ,/2, 2#$ -+@+(.+? 0#$02#2,&(+ 3#(5 ,/$ 1(##$'0(+%&+B 2)@A) =#(5&%$' 2+% '(%&-5 2>&%$ '/(-)% =$ ',#2&B/,3(#.2#%7
P/&)$ 5(', (3 (-# .(#@ .&)) 3(1-' (+ ,/$ 0()A5$# 2+% 1(0()A5$#' (3 0#(02#BA) B)A1()&%$ !? 2)@A+$ '-=',&,-,$% /A%#(9A =-,A#&1 21&%' 2#$ @+(.+7NQ R&+1$ ,/$ $+>A5$' ,/2, 'A+,/$'&>$ 0()AS/A%#(9A=-,A#&1T 21&% 12+ 2)'( 0#(%-1$ (,/$# 0()AS/A%#(9A2)@2+(&1T 21&%'? 0()A5$#&>2,&(+ (3 ,/$ 2)@A+A) /A%#(9A 21&% " &' 2+ &+,#&B-&+B 0(''&=&)&,A '&+1$ ,/$ #$'-),&+B 0()A5$# # .(-)% =$ ',$#$(#$B-)2# 2+% 25$+2=)$ ,( :1)&1@; 3-+1,&(+2)&>2,&(+7 P/&)$ .$ %( +(, 2+,&1&02,$ =&()(B&12) &+1(502,&=&)&,A 2#&'&+B 3#(5 ,/$ ,#&2>()$ B#(-0'? 2),$#+2,&*$ 0(',M0()A5$#&>2,&(+ 5(%&3&12,&(+' (3 GGUV 52A =$ 0(''&=)$7 I(# $9250)$? .$ 12+ 0(,$+,&2))A 1(+*$#, GGUV ,( &,' 2))A) 2+2)(B $ =A '$)$1,&*$ #$%-1,&(+ (3 ,/$ ,$#5&+2) 2)@A+$7NL?NN P$ /2*$ '/(.+ ,/2, 2))A)M'-=',&,-,$% 0()AB)A1()&%$' 12+ =$ '$)$1,&*$)A 3-+1,&(+2)&>$% -'&+B ()$3&+ 5$,2,/$'&'NW 2+% (,/$# ,#2+'3(#52,&(+'7N"?NK
!2#$ P8o-ein$ enc('9:2(-ion$ in$ n(no>ice22e9#$ 8/$ 321, ,/2, +2+(5&1$))$' % 12+ $+120'-)2,$ /A%#(0/&)&1 (# /A%#(0/(=&1 B-$',' 52@$' ,/$ 0#('0$1,' 3(# &+1(#0(#2,&+B 0#(,$&+' =#&B/,7 P$ .&)) '-#*$A '$)$1,$% 0#(,$&+' ,( 2''$'' ,/$ 0#('0$1,' 3(# 0#(,$&+ '()-=&)&>2,&(+ 2+% ',2=&)&>2,&(+7 V(B&12) ',2#,&+B 0(&+,' 2#$ 0#(,$&+' ,/2, /2*$ =$$+ '/(.+ ,( #$,2&+ ,/$&# 21,&*&,A ./$+ '()-=&)&>$% &+ (#B2+&1 '()*$+,'7JL X3 ,/$'$? R-=,&)&'&+ E2#)'=$#B SRET &' 2+ &%$2) 12+%&%2,$ =$12-'$ &, &' 1(55$#1&2))A 2*2&)2=)$? &,' ',#-1,-#$ &' @+(.+? 2+% &, &' ',2=)$ &+ 21$,(+&,#&)$7 <,' 5()$1-)2# .$&B/, &' YW @Z .&,/ ,/$ )(+B$', %&5$+'&(+ &+ &,' [M#2A ',#-1,-#$' =$&+B \ Q] ^ SQ +5T7 8/$ ',#-1,-#$ (3 RE? 2' .$)) 2' ,/('$ (3 E4C6 2+% GUFRMJ? &' '/(.+ &+ I&B-#$ Y]7 <, &' '52)) &+ 1(502#&'(+ ,( ,/$ ,A0&12) %&5$+'&(+' (3 (-# +2+(5&1$))$'7 F$+1$? &, &' 1(+1$&*2=)$ ,/2, &, 12+ =$ $+120'-)2,$% $+,&#$)A .&,/&+ 2 +2+(5&1$))$7
H2N N$
N$O
HO
&
O
O
ON$
OOP
O
O O
Me$N
*+,-o/en12on,in/
4*o54*o6i4i,17n76o/
Me2N N$
N$HO
O
7ci,ic
275ic
N$O
OO
N$N$ N$
c-o556in9in/ !
&'()*e, !-. 82#B$,$% 2>&%$' 3(# :1)&1@; 3-+1,&(+2)&>2,&(+ (3 0#(02#BA)M'-=',&,-,$% B)A1()&%$'7
HOCO2H
Pol()e+,-e
O
O
OHH
-.e+eo+e/0l,+123clic6,7le328ol(9:(;+o<(,l6,noic>2,ci;
!
?OO
On
H
PP@A
?OO
On
H
"
H212B0inoline1Ain;l,+C-2c,.,l(-.
oleDin2)e.,.:e-i-
OO
E
CFGH2F
?OO
O<
H
CFGH2F
O
O#
OH
OE
(
$
,ce.one
!
&'()*e,20. G#(0('$% :1)&1@2=)$; 0()AS/A%#(9A2)@2+(&1T 21&% # (=,2&+$% 3#(5 $+>A52,&1 0()A5$#&>2,&(+ (3 "? 2+% 0(,$+,&2) 1(+*$#'&(+ (3 GGUV ,( 2))A)M'-=',&,-,$% B)A1()&%$ $7 X)$3&+ 1#(''M5$,2,/$'&' 1(-)% 233(#% 2+2)(B(-' 250/&0()' '-1/ 2' 17
!
!
&'()*e, 20. 4$)2,&*$ '&>$' (3 +2+(5&1$))$' 2+% 0#(,$&+' ,( =$ ',-%&$%7

By 1853 New York omnibuses carried 120,000 passengers per day.
In 2009, the New York Subway carries 4,300,000 passengers per day
Sustainability is an Old Problem

Fossil Fuels: Key to 20th Century Sustainability
Growth is the biggest challenge to sustainability

The Energy Problem
By 2050 Earth’s population is predicted to increase by 50% while energy demand is predicted to increase by at least 100%

The Problem with CO2 Recycling
3 Billion Years Ago
Today

Carbon-Free Fuels
Disadvantages of carbon-based fuels
CO2 recycling required for carbon neutrality
Low CO2 concentration hampers atmospheric recovery
Onboard CO2 recovery impractical for transportation applications
One possible solution: Avoid carbon!

Carbon-Free Fuels

Carbon Free Fuels: Research TeamMSU Research TeamProfessor Thomas Hamann-Energy conversion chemistryProfessor Aaron Odom-Nitrogen chemistry, catalysisProfessor Viktor Poltavets-Solid-state chemistry, catalysisProfessor Milton Smith-CatalysisProfessor Daniel Nocera (Harvard/MSU)-Energy science
Dr. James Boncella (Los Alamos)-Fuel cells, nitrogen chemistry
Targeted agencies for center funding: DOE (ARPA-E), NSF
CatalystsHamann, Poltavets, Odom, and Smith
Fuel CellsChemistry and Engineering
Automotive ResearchExperimental Station

Ammonia: A Hydrogen Dense FuelAdvantages
Haber-Bosch process is well-established and is the second largest industrial chemical process. Approximately 1-2% of global energy is dedicated to ammonia production.
By mass, liquid NH3 energy density is 40% of gasoline, 94% of methanol
Liquid NH3 is an efficient H2 carrier
1/2 N2 + 3/2 H2 NH3ΔH°rxn = -11 kcal/mol, ΔS°rxn = -24 cal/mol·K
T (°C) Keq
25 7.1160 1.0450 0.01
2 NH3 + 3/2 O2 N2 + 3 H2OReleases 87% of the energy of H2 oxidation!
NH3 1/2 N2 + 3/2 H2 0.077 V

Ammonia: A Hydrogen Dense Fuel

Ammonia: A Hydrogen Dense Fuel
AdvantagesSynthesized from nitrogen, the most abundant gas in the atmosphere
Can be used as fuel in internal combustion engines
Produces less NOx emissions than gasoline
Ammonia is non-flammable

Ammonia: A Hydrogen Dense Fuel
ChallengesCarbon-neutral H2 production does not meet energy needs
Current Haber-Bosch process requires high pressure of N2 and H2.
Like gasoline, ammonia is toxic.
Ammonia cracking to N2 and H2 is inefficient
Fundamental research is needed to address these challenges

Current Cracking Technology: Reverse Haber-Bosch
1/2 N2 + 3/2 H2NH3T > 500 °Ccatalyst

Carbon Free Fuels: Proposed Research
New catalysts for ammonia synthesis
Improve current Haber-Bosch by synthesizing nano-structures that increase active sites for catalysis.
Develop ammonia synthesis where water is the hydrogen source instead of hydrogen gas.
N2 + 3 H2O + 2 NH3 + 3/2 O2electricalenergy

Ammonia Synthesis

Computational Studies of Haber-Bosch
Ammonia: The liquid fuel of the future.
! 10
have been observed and serve as a guideline as to how the catalysis might be accomplished.
Another approach for extracting the chemical energy in NH3 is crack NH3 to N2 and H2 by reversal of HB synthesis. The H2 produced can then be converted to electrical energy in an HFC. Figure 11 shows equilibrium between N2, H2, and NH3 as a function of temperature. At room temperature, NH3 is favored. Consequently, cracking to N2 and H2 will most likely require elevated temperatures, although extremely high temperatures should not be required since separation of NH3 and H2 is straightforward.
One of the most efficient HB catalyst systems is based on Ru and calculations suggest that scission of coordinated N2 to form two surface bound nitrides is exothermic by ~ 20 kcal/mol.23 Only a few metal nitrides and oxides were tested for NH3 decomposition: Fe4N, Mo2N, MoO3, Cr2O3, MnO2, Mn2O3 and MnO. Of pure metal catalysts, it has been established that Ru metal is by far the best NH3 decomposition catalyst. Nitrides for less electron rich metals like Fe, Co, Rh, Ni, and Pd may be less stable and these materials will be examined for reverse HB generation of H2 from NH3.
Ammonia synthesis. One of the most efficient HB catalyst systems is based on Ru and it has been particularly well studied under catalytically relevant conditions.24 Optimal activity is found when metal crystal size is ~ 2 nm where it is estimated that ~9% of the surface sites are active, which corresponds to only ~3 % of the Ru atoms in the nanocrystal. While catalytic activity has been improved by adding BaO, the primary limitation to catalysis is the low number of catalytic active sites. The reason for this can be appreciated when one considers the currently accepted mechanism for catalysis.
It is believed that reactions occur at terrace steps between Ru (0001) planes. Calculations on 11, 15, and 21-atom clusters support a mechanism where N2 coordination and cleavage is mediated by 5 Ru atoms at the step as shown in Figure 12.23 Note the critical feature that the angle between the step edges is acute. When one considers that the angles between facets of nanocrystals are oblique, it is not surprising that few sites are active.
We propose synthesis of new Ru geometries with concave edges using ALD to make core-shell nanostructures. As shown in Figure 13, nanorods (most likely Si) will be grown from a surface.25 Ru metal will be deposited onto the nanorods and the void between the core shell structures will be filled with an inert support, like BN, which can be prepared from polyborazene precursors. At this point, the Si will be removed with an HF etch leaving Ru nanobelts where the angles between the interior faces will be acute. We anticipate the materials like this could exhibit significantly higher activity for HB synthesis. Consequently, it may be possible to have similar of ammonia production rates using lower temperatures, lower pressures, and lower
!Figure 11. Thermodynamics of Haber-Bosch synthesis
!Figure 12. An ammonia fuel cell with a metal amide supporting electrolyte.8
!Figure 13. An approach to synthesis of new HB catalyst morphologies with higher catalytic activities.
Cao, Z. X.; Wan, H. L.; Zhang, Q. N., J. Chem. Phys. 2003, 119, 9178-9182.

Carbon Free Fuels: Proposed Research
New catalysts for ammonia synthesis
Improve current Haber-Bosch by synthesizing nano-structures that increase active sites for catalysis.

Carbon Free Fuels: Proposed Research
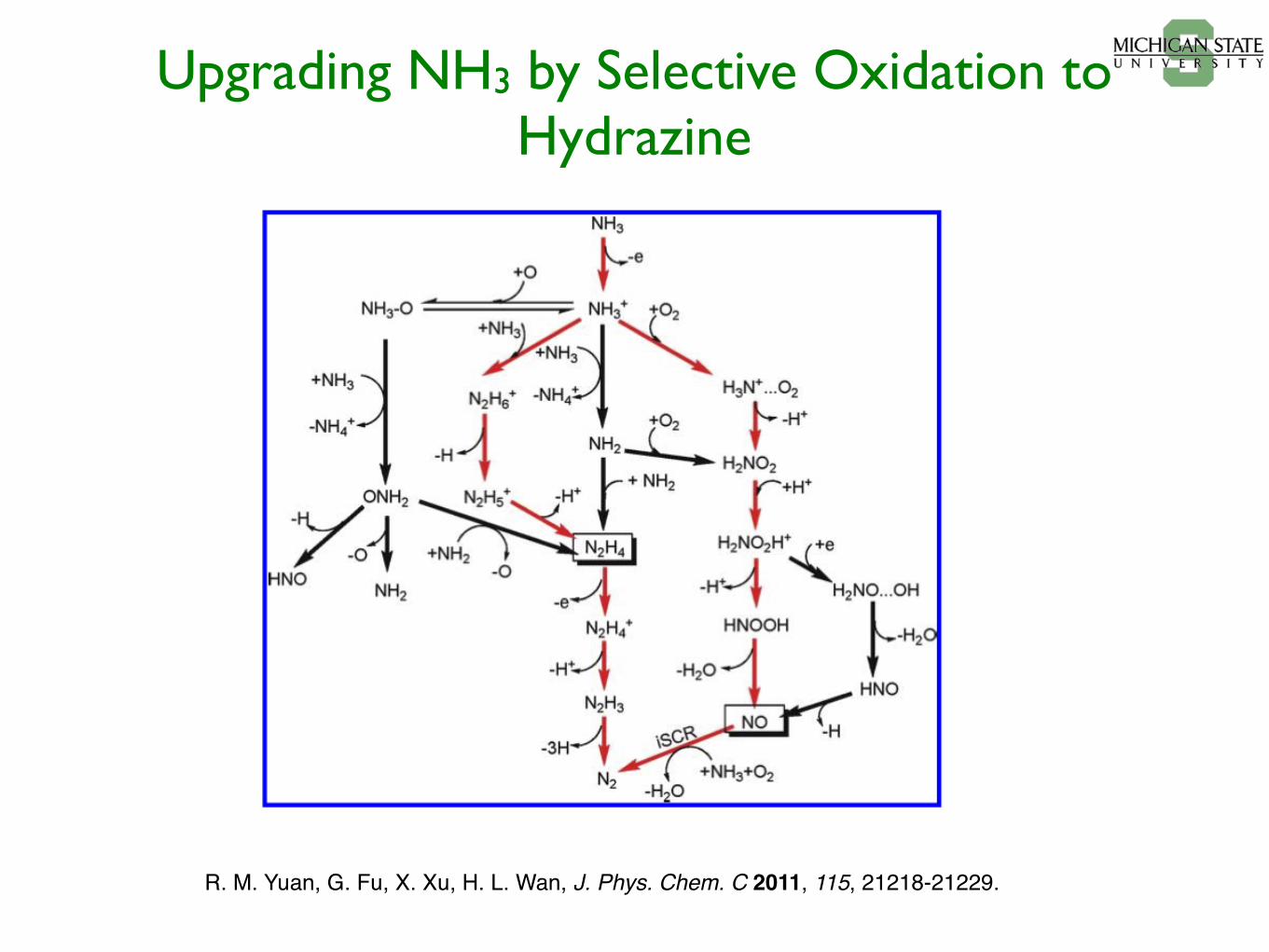
Upgrading NH3 by Selective Oxidation to Hydrazine
R. M. Yuan, G. Fu, X. Xu, H. L. Wan, J. Phys. Chem. C 2011, 115, 21218-21229.

R. M. Yuan, G. Fu, X. Xu, H. L. Wan, J. Phys. Chem. C 2011, 115, 21218-21229.

Upgrading NH3 by Selective Oxidation to Hydrazine

Proposed Research: Ammonia to Energy
Develop catalysts for ammonia oxidation
Design new ammonia fuel cells
1/2 O2 + 2 H+ + 2 e–H2OPS-II
1/2 N2 + 3 H+ + 3 e–NH3
2 NH3
N2 +6 H+ H+
motor
reductioncatalyst
ammoxcatalyst
3 H2O
3/2 O2 +6 H+
e– e–

Proposed Research: Ammonia to Energy
Electrolysis at 2 V potential!
Hanada, N.; Hino, S.; Ichikawa, T.; Suzuki, H.; Takai, K.; Kojima, Y., Chem. Commun. 2010, 46, 7775-7777.

A synthetic water splitting catalyst:
Nocera and coworkers, J. Am. Chem. Soc. 2011, 133, 5174–5177
Vol. 45, No. 5 ’ 2012 ’ 767–776 ’ ACCOUNTS OF CHEMICAL RESEARCH ’ 771
The Artificial Leaf Nocera
energybarrier of >0.16V imposedbyprotonationof theCo(IV)species. This result suggests the charge propagation mech-anism as hole hopping through catalyst films.
Electrokinetic studies show that the oxidation of Co(III) toCo(IV) is coupled to the loss of a proton.36 In the absence of
phosphate buffer, the overall activity of theCo-OEC is greatly
diminished. Together, these studies suggest the PCET mech-
anism shown in Figure 7. As in PSII-OEC, the redox potential
of the Co-OEC is increased by four hole equivalents akin to
the S-state pumpingof theKok cycle. The critical step is at the
equivalent of the S3 to S4 transition of the Kok cycle. A rapid
one-electron, one-proton equilibrium between CoIII!OH
and CoIV!O in which a phosphate species is the proton
acceptor is followed by a chemical turnover-limiting process
involving oxygen!oxygen bond coupling. Enriched 18O cata-
lyst operated in unenriched water exhibits extrusion of 18O in
34O2, indicating the participation of a bridging O atom of the
catalyst core in the O2 evolution pathway.36 Mechanistic de-
tails of how theO!Obond forms and the precise nature of the
turnover-limiting elementary step remain to be determined.Water is a poor proton acceptor under neutral conditions.
Hence, the activation of oxygen in water requires a bidirec-tional PCET involving phosphate as a proton acceptor, as hasbeen established in model PCET systems.37,38 In the Co-OECmechanism ofwater splitting, the phosphate plays the essen-tial role of maintaining this PCET equilibrium by facilitatingrapid proton transferwith the concomitant oxidation of Co(III)to Co(IV) to furnish the active O2 producing “S4” state.
A Self-Healing OEC Catalyst. Like PSII-OEC, metal-oxidecatalysts are unstable in neutral water and corrode whenwater splitting is performed under benign conditions. Inneutral water, the best base is the metal oxide itself, and
FIGURE6. CWX-band EPR spectra of (left top) frozen electrolysis solution (;), Co3(PO4)2 ( 3 3 3 ), Co3O4 (!!!), (leftmiddle) CoPi catalyst films depositedat 1.34 V, and (left bottom) the Co!oxo cubane [Co4O4(C5H5N)4(CH3CO2)4](ClO4), which exhibits an EPR signal for Co(IV) at g = 2.27. (right) Potentialdependence of EPR spectrum of catalyst films from 1.03 V, at which catalysis does not occur, to 1.34 V, at which catalysis occurs.
FIGURE7. Proposed pathway forwater splitting by Co-OEC. A PCET equilibriumproceeds the turnover-limitingO!Obond-forming step. Curved linesdenote phosphate or terminal oxygen (from water or hydroxide). The oxyl radical in the far right structure is shown for emphasis. If the hole iscompletely localized on oxygen, then the Co oxidation state is Co(III) and not Co(IV).Nocera and coworkers, Acc. Chem. Res., 2011, 45, 767-776.
Published: March 17, 2011
r 2011 American Chemical Society 5174 dx.doi.org/10.1021/ja110908v | J. Am. Chem. Soc. 2011, 133, 5174–5177
COMMUNICATION
pubs.acs.org/JACS
Bidirectional and Unidirectional PCET in a Molecular Model of aCobalt-Based Oxygen-Evolving CatalystMark D. Symes, Yogesh Surendranath, Daniel A. Lutterman, and Daniel G. Nocera*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139-4307, United States
bS Supporting Information
ABSTRACT:The oxidation of water tomolecular oxygen isa kinetically demanding reaction that requires efficientcoupling of proton and electron transfer. The key proton-coupled electron transfer (PCET) event in water oxidationmediated by a cobalt-phosphate-based heterogeneous cata-lyst is the one-electron, one-proton conversion of CoIII-OH to CoIV-O. We now isolate the kinetics of this PCETstep in a molecular Co4O4 cubane model compound.Detailed electrochemical, stopped-flow, and NMR studiesof the CoIII-OH to CoIV-O reaction reveal distinctmechanisms for the unidirectional PCET self-exchangereaction and the corresponding bidirectional PCET. Astepwise mechanism, with rate-limiting electron transfer isobserved for the bidirectional PCET at an electrode surfaceand in solution, whereas a concerted proton-electrontransfer displaying a moderate KIE (4.3 ( 0.2), is observedfor the unidirectional self-exchange reaction.
As global demand for energy continues to grow, the need tofind a carbon-neutral and sustainable energy source for future
generations has become imperative.1-4 An especially attractivesolution is to store solar energy in the form of chemical bonds viathe electrochemical splitting of water to produce hydrogen andoxygen.5-7 In this process, the electrochemical oxidation ofwater to O2 is the bottleneck because it is a kinetically demand-ing four electron-four proton transformation. The intimate cou-pling of electron and proton transfer8-11 thus becomes a keyrequirement for effecting the solar fuels process of water splitting.
We recently reported a heterogeneous Co-Pi catalyst12 that formsspontaneously upon electrolysis of Co(II) in phosphate-bufferedsolutions at neutral pH.13 This catalyst enhances the efficiency ofelectrochemical and photoelectrochemical water oxidation and is ofinterest because it (1) forms in situ under mild conditions on avariety of conductive substrates from inexpensive and earth-abundant materials;13,14 (2) exhibits high activity in naturalwater and seawater at room temperature;15 (3) is self-healing byreversing catalyst corrosion at open circuit upon reapplicationof an anodic potential;16 (4) can be interfaced with light-absorbingand charge-separating materials to enhance photoelectrochemicalwater splitting;17-19 and (5) is a functional and related structuralmodel of the oxygen-evolving complex of Photosystem II (PS II).20
With regard to the latter, XAS studies21,22 have established that theCo-Pi is a structural relative of the oxygen-evolving complex (OEC)of PSII. Shown in Scheme 1 are the structures of the PSII OEC23
and the core structure of Co-Pi as deduced from XAS where a
distribution of cluster sizes is observed, where the seven-atomcluster shown in Scheme 1 predominates in ca. nanometer thinfilms of the catalyst. Both systems have a partial cubane. In theOEC, the cube is completed with a Ca2þ ion; although the alkalimetal ions for Co-Pi have not been located, they likely reside onthe three-fold oxygen triangle to complete the cube structure, as isthe case for cobaltates.24 As highlighted in Scheme 1, Co-Pi is thecorner-sharing head-to-tail dimer of the OEC monomer of PSII.Themetal-metal (d = 2.8 Å) andmetal-oxo (d = 1.9 Å) distancesinCo-Pi and PSIIOEC are similar. Thus,model cubane systems notonly shed light on the PCETwater activation chemistry ofCo-Pi butmay also provide insight into the mechanism of PSII OEC.
XANES22, EPR25 and electrochemical studies26 of Co-Pi have,together, established the proton-coupled electron transfer (PCET)conversion of CoIII-OH to CoIV-O as a key mechanistic step priorto O2 evolution. The same conversion is also believed to be critical toCo-oxide-mediated water oxidation in alkaline media.27-29 Herein,we isolate the kinetics of this key step in a Co4(μ3-O)4 molecularmodel cubane, [Co4O4(CO2Me)2(bpy)4](ClO4)2 (1), originallyreported by Christou and co-workers30 (Figure 1), which is astructural relative of Co-Pi with complete Co residency withinthe cube. In both Co-Pi and this molecular model, μ3-O/OHmoieties link the Co centers. We now report electrochemical,stopped-flow, andNMR-based studies of theCoIII-OHtoCoIV-Oconversion in 1 that reveal that the PCET reaction forwhich electronand proton are transferred to the samemolecule (unidirectional) is aconcerted PCET, whereas transfer of the proton and electron todifferent molecules (bidirectional) occurs by stepwise PCET.
The stepwise and concerted PCET pathways of the cubanemodelsystem are detailed in Figure 1. As synthesized, all Co centers in thecubane, 1, are in the 3þ oxidation state, and the cluster can beprotonated to yield 1-Hþ, which exhibits a pKa of 3.15.
31 The cyclicvoltammogram (CV) of 1 at pH>4 exhibits a reversible one-electronoxidation32 with an E1/2 =þ1.25 V (all potentials are reported vs thenormal hydrogen electrode). This potential is similar to the poten-tials (>þ1.1 V) at which Co(IV) is detected in Co-Pi.25 Thus, the
Scheme 1
Received: December 10, 2010
770 ’ ACCOUNTS OF CHEMICAL RESEARCH ’ 767–776 ’ 2012 ’ Vol. 45, No. 5
The Artificial Leaf Nocera
aqueous electrodeposition conditions permit conformal cat-alyst layers to be deposited at a desired thickness on asurface of any architecture. The self-assembly of the Co-OEC may also be photodriven. The Co-OEC self-assembleson the surfaces of irradiated semiconducting materials suchas Fe2O3,
21!24 ZnO,25 orWO326 immersed in water contain-
ing Co2þ and phosphate.The catalysts films are structurally amorphous and thus
X-ray absorption spectroscopy (XAS)was used to structurallycharacterize Co-OEC. The extended X-ray absorption finestructure (EXAFS) spectra of in situ Co-OEC during catalyticturnover27 and ex situ films28 are consistentwith amolecularcobalt complex consisting of edge-sharing CoO6 octahedra.The average size of the molecular cobaltate cluster is sevenatoms. The core of Co-OEC is a nearly identical structuralcongener of the PSII-OEC as schematically illustrated inFigure 5. Both systems are a partial cubane with identicalmetal!metal (d = 2.82 Å) and metal!oxo (d = 1.89 Å)distances. In OEC, the cube is completed with a Ca2þ ion;though the alkali metal ions for CoPi have not been located,they likely reside on the 3-fold oxygen triangle to completethe cube structure, as is the case for cobaltates.29 The edgeCo atoms of the cluster are likely terminated by the phos-phate ions, which have been shown to be exchangeable.30
AHighly Active PCETWater-Splitting Catalysis. Co-OECperforms water oxidation at neutral pH. The catalyst
operates at 350mV lower overpotential than Pt at all currentdensities and under neutral conditions. Current densities ashigh as 100 mA/cm2 (geometric surface area) have beenachieved at overpotentials as low as 360 mV (67% energyconversion efficiency).31 The high current density is main-tained for days with no discernible drop in activity. TheFaradaic efficiency of the catalyst is 100%; that is, thecatalyst is selective for O2 production from water, and noother product is obtained.
As in PSII-OEC, water splitting in Co-OEC occurs from theþ4 oxidation state. Co-OEC catalyst films exhibit EPR signalscorresponding to populations of both Co(II) and Co(IV).32
Figure 6 shows the EPR spectrum of Co-OEC under deposi-tion conditions compared with Co(II)-containing species (toppanel, left), Co-OEC under an applied voltage at whichcatalysis occurs (middle panel, left), and a Co4O4 cubanecontaining cobalt in the þ4 oxidation state (bottom panel,left). The presence of Co(IV) is clearly established at poten-tials where OER catalysis occurs. As the deposition voltage isincreased into the region where water oxidation prevails(right panel), the population of Co(IV) rises and the popula-tion of Co(II) decreases. The spin distribution of Co(IV) hasbeen examined by EPR for a cobalt!oxo cubane clusterpossessing formal cobalt oxidation states III, III, III, and IV.33
Davies ENDOR, mims 1H ENDOR, and multifrequency 14NESEEM spectra indicate that the unpaired spin is delocalizedalmost equally across the cobalt atoms, a finding corrobo-rated by DFT calculations. These results suggest that thecharge on the Co-OEC catalyst is also highly delocalized.
Insight into the propagation of the catalytically active “holes”of theCo(IV) state through the catalyst filmhasbeenaffordedbymeasuring the CoIII/CoIV self-exchange kinetics of the Co4O4
cubane compound, [Co4O4(O2CMe)2(bpy)4](ClO4)2,34 using
NMR line broadening techniques.35
The Co(IV) center is indicated by the blue color coding andCo(III) by red color coding. The second-order rate constantfor self-exchange is 1.3 # 104 M!1 s!1 at pH 1. At pH 4,where the cluster is deprotonated, the rate constant in-creases by a factor of 10 to 3# 105 M!1 s!1, consistent witha simple electron-transfer step. A KIE of 4.3 is suggestive of aunidirectional concerted proton!electron transfer (CPET). Wehave shown that CPET results in order to avoid the high
FIGURE 4. Dried catalyst films formed from solutions of Co(aq)2þ and Piunder (right) quiescent (0.85 V vs Ag/AgCl) and (left) catalytically active(1.1 V vs Ag/AgCl) conditions on an ITO substrate.
FIGURE 5. (left) Schematic of cubane structure of PSII-OEC. (middle)Structure of the Co-OEC as determined from EXAFS (Pi not shown). Co-OEC is the head-to-tail dimer of the cubane of PSII-OEC. (right) Co-OECstructure rotated by 45! to more clearly shows edge sharing octahedra.The alkali metal ions, which are not shown, likely reside above the3-fold triangle defined by the μ-bridging oxygens.

Proposed Research: Metal Catalyzed Oxidation

Proposed Research: Metal Catalyzed Oxidation

Impacts: Local and Global
Elimination of CO2 emission at the tailpipe of transportation vehicles
Hydrogen storage from Earth abundant feedstocks
Off-grid synthesis of fuel and fertilizer for third-world countries by converting electrical energy (from solar, wind, etc.) to fuel
Complements MSU’s efforts in Biofuels making us the leader in fuel research
Significant IP opportunities in energy and agricultural sectors





























