Embed Size (px)
Citation preview

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 1/34
THE ROLE OF BIOINFORMATICSIN PROTEIN STRUCTURE STUDY
Jennie Varghese
09BIF089
8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 2/34
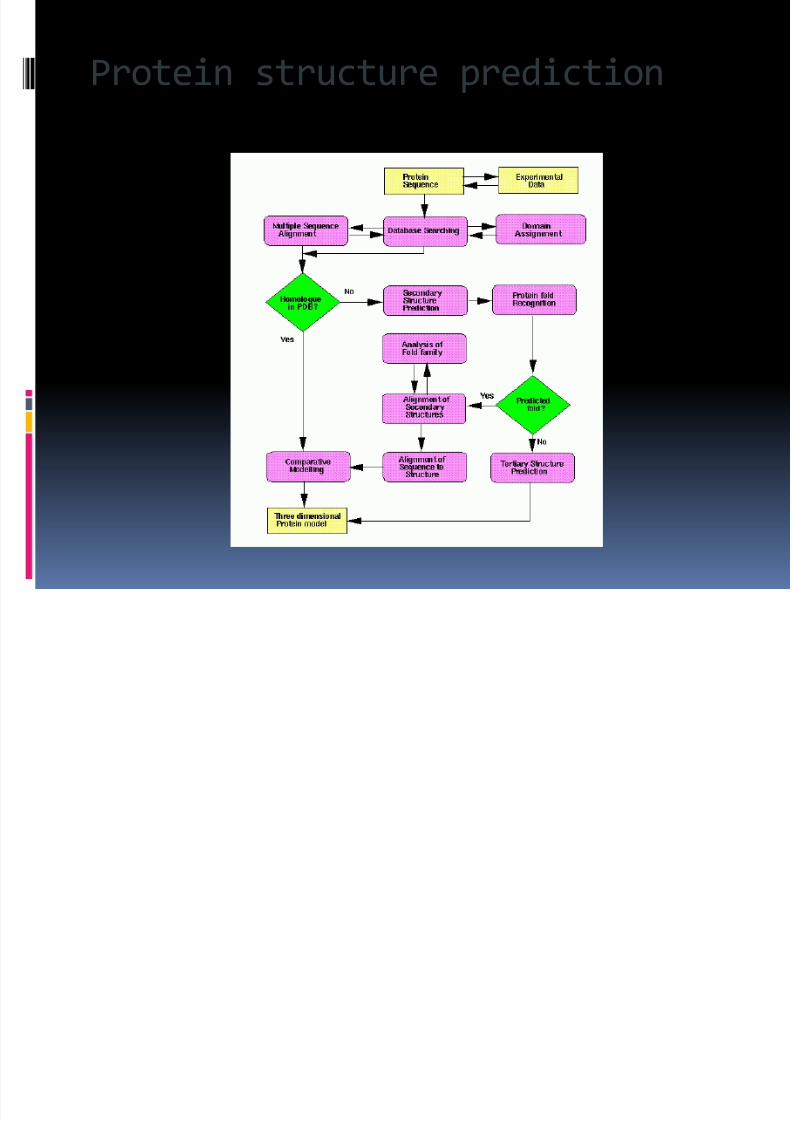
Protein structure prediction

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 3/34
Introduction
Protein structure prediction is anotherimportant application of bioinformatics.The amino acid sequence of a protein, the so-called primary structure, can be easilydetermined from the sequence on the genethat codes for it. In the vast majority of cases,
this primary structure uniquely determines astructure in its native environment.

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 4/34
Introduction
For lack of better terms, structuralinformation is usually classified as oneof secondary , tertiary andquaternary structure. A viable generalsolution to such predictions remains an openproblem. As of now, most efforts have been
directed towards heuristics that work most of the time.

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 5/34
Introduction
One of the key ideas in bioinformatics is thenotion of homology. In the genomic branch of bioinformatics, homology is used to predict the
function of a gene: if the sequence of gene A,whose function is known, is homologous to thesequence of gene B, whose function is unknown,one could infer that B may share A's function. In
the structural branch of bioinformatics,homology is used to determine which parts of aprotein are important in structure formation andinteraction with other proteins

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 6/34
Introduction
. In a technique called homology modeling,this information is used to predict thestructure of a protein once the structure of ahomologous protein is known. This currentlyremains the only way to predict proteinstructures reliably. techniques for predicting
protein structure include protein threadingand de novo (from scratch) physics-basedmodeling.

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 7/34
Protein structure prediction

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 8/34
A good protein structure
• Minimizes disallowedtorsion angles
• Maximizes number of
hydrogen bonds• Minimizes interstitial
cavities or spaces
• Minimizes number of “bad” contacts
• Minimizes number of buried charges

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 9/34
Protein structure prediction
– Secondary structure
– 3D structure• Modeling by homology (Comparative modeling)
• Fold recognition (Threading)• Ab initio prediction– Rule-based approaches– Lattice models– Simulating the time dependence of folding
• Refinement• Exploring the effect of single amino acid substitutions• Ligand effects on protein structure and dynamics
(induced fit)

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 10/34
Modeling by Homology
(Comparative Modeling) Comparative modeling predicts the three-dimensional structure of a given
protein sequence (target) based primarily on its alignment to one or more proteins
of known structure (templates).
The prediction process consists of
• fold assignment,
• target template alignment,• model building, and
• model evaluation and refinement.
The number of protein sequences that can be modeled and the accuracy of
the predictions are increasing steadily because of the growth in the number of
known protein structures and because of the improvements in the modeling
software.
Further advances are necessary in recognizing weak sequence structure
similarities, aligning sequences with structures, modeling of rigid body shifts,
distortions, loops and side chains, as well as detecting errors in a model.
Despite these problems, it is currently possible to model with useful accuracy
significant parts of approximately one third of all known protein sequences.

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 11/34
Modeling by Homology
(Comparative Modeling)

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 12/34
Modeling by Homology
(Comparative Modeling)

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 13/34
Modeling by Homology
(Comparative Modeling)

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 14/34
Modeling by Homology
(Comparative Modeling)
8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 15/34
Modeling by Homology
(Comparative Modeling)

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 16/34
Fold Recognition (Threading)
Methods of protein fold recognition attempt to detect similaritiesbetween
protein 3D structure that are not accompanied by any significantsequence similarity.
The unifying theme of these appraoches is to try and find folds that are
compatible with a particular sequence. Unlike sequence-onlycomparison,
these methods take advantage of the extra information made availableby
3D structure information.
Rather than predicting how a sequence will fold, they predict how well afold
will fit a sequence.

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 17/34
Fold Recognition (Threading) –
2 Kinds
2D Threading or Prediction Based Methods(PBM) Predict secondary structure (SS) or ASA of query
Evaluate on basis of SS and/or ASA matches
3D Threading or Distance Based Methods (DBM) Create a 3D model of the structure
Evaluate using a distance-based “hydrophobicity” orpseudo-thermodynamic potential

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 18/34
Fold Recognition (Threading)

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 19/34
Fold Recognition
Database of 3D structures and sequences
Protein Data Bank (or non-redundant subset)
Query sequence
Sequence < 25% identity to known structures
Alignment protocol
Dynamic programming
Evaluation protocol Distance-based potential or secondary structure
Ranking protocol

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 20/34
Fold Recognition

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 21/34
Ab Initio Prediction
• Predicting the 3D structure without any “priorknowledge”
• Used when homology modelling or threadinghave failed (no homologues are evident)
• Equivalent to solving the “Protein FoldingProblem”
• Still a research problem

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 22/34
Ab Initio Prediction

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 23/34
Ab Initio Prediction

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 24/34
Ab Initio Prediction

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 25/34
Combining Prediction Procedures

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 26/34
Structure Validation
A structure can (and often does) havemistakes
A poor structure will lead to poor models of mechanism or relationship
Unusual parts of a structure may indicatesomething important (or an error)

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 27/34
Structure Validation
Assess experimental fit look at Resolution, R-Factor or RMSD
Assess correctness of overall fold look at disposition of hydrophobic residues
Assess structure quality packing
stereochemistry contacts

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 28/34
Structure Validation
Servers WHAT IF
http://swift.cmbi.kun.nl/WIWWWI/
Verify3D http://www.doe-mbi.ucla.edu/Services/Verify_3D/
VADAR http://redpoll.pharmacy.ualberta.ca

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 29/34

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 30/34

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 31/34
Structure Validation
Programs PROCHECK
http://www.biochem.ucl.ac.uk/~roman/procheck/procheck.html
VADAR http://www.pence.ca/software/vadar/latest/vadar.html
DSSP http://www.cmbi.kun.nl/gv/dssp

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 32/34
Procheck

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 33/34
Conclusions
Protein structures are now sufficientlyabundant and well defined that they can beclassified using well-developed rules of taxonomy
Distant relationships and common rules of folding can be uncovered through fold
classification & comparison

8/3/2019 Smm -Protein Structure Prediction 2
http://slidepdf.com/reader/full/smm-protein-structure-prediction-2 34/34
Conclusions
Structure prediction is still one of the keyareas of active research in bioinformatics andcomputational biology
Significant strides have been made over thepast decade through the use of largerdatabases, machine learning methods and
faster computers





























