Embed Size (px)
Citation preview

Washington University in St. LouisWashington University Open Scholarship
Arts & Sciences Electronic Theses and Dissertations Arts & Sciences
Summer 8-15-2018
Smooth as SILK: Translating Discoveries to NewTherapies in Amyotrophic Lateral SclerosisWade SelfWashington University in St. Louis
Follow this and additional works at: https://openscholarship.wustl.edu/art_sci_etds
Part of the Neuroscience and Neurobiology Commons
This Dissertation is brought to you for free and open access by the Arts & Sciences at Washington University Open Scholarship. It has been acceptedfor inclusion in Arts & Sciences Electronic Theses and Dissertations by an authorized administrator of Washington University Open Scholarship. Formore information, please contact [email protected].
Recommended CitationSelf, Wade, "Smooth as SILK: Translating Discoveries to New Therapies in Amyotrophic Lateral Sclerosis" (2018). Arts & SciencesElectronic Theses and Dissertations. 1653.https://openscholarship.wustl.edu/art_sci_etds/1653

WASHINGTON UNIVERSITY IN ST. LOUIS
Division of Biology and Biomedical Sciences
Neurosciences
Dissertation Examination Committee:
Timothy M. Miller, Chair
Randall J. Bateman
Johnathan R. Cirrito
Anne M. Fagan
Conrad C. Weihl
Smooth as SILK: Translating Discoveries to New Therapies in Amyotrophic Lateral Sclerosis
by
Wade Kerner Self
A dissertation presented to
The Graduate School
of Washington University in
partial fulfillment of the
requirements for the degree
of Doctor of Philosophy
August 2018
St. Louis, Missouri

© 2018, Wade Self

ii
Table of Contents List of Figures ................................................................................................................................ iv
List of Tables ................................................................................................................................. vi
Acknowledgments......................................................................................................................... vii
Abstract of the Dissertation ......................................................................................................... xiii
Chapter 1: Towards a New Treatment for ALS ............................................................................ 1
Amyotrophic Lateral Sclerosis: A relentless degenerative disorder ........................................... 4
The Role of SOD1 in Amyotrophic Lateral Sclerosis ................................................................ 7
SOD1 in ALS Pathobiology ........................................................................................................ 8
Controversial Role of SOD1 in Sporadic ALS ......................................................................... 14
Toxicity by Misfolded SOD1: Similarity to other ALS subtypes ............................................. 16
Learning from Past Failures for ALSSOD1
Clinical Trial Design .............................................. 16
Therapeutic Strategies for ALSSOD1
.......................................................................................... 19
Antisense Oligonucleotides for the Treatment of ALSSOD1
...................................................... 23
Considerations for ASO Clinical Trial Design in ALSSOD1
...................................................... 25
Summary of Disse rtation .......................................................................................................... 26
Chapter 2: Defining SOD1 ALS Natural History to Guide Therapeutic Clinical Trial Design . 29
ABSTRACT .............................................................................................................................. 30
INTRODUCTION ..................................................................................................................... 31
METHODS................................................................................................................................ 33
RESULTS.................................................................................................................................. 36
DISCUSSION ........................................................................................................................... 46
ACKNOWLEDGEMENTS ...................................................................................................... 50
Chapter 3: Characterizing SOD1 Protein Kinetics in the Cerebrospinal Fluid of Individuals with
ALS ............................................................................................................................................... 51
INTRODUCTION ..................................................................................................................... 52
METHODS................................................................................................................................ 54
RESULTS.................................................................................................................................. 54
DISCUSSION ........................................................................................................................... 67
ACKNOWLEDGEMENTS ...................................................................................................... 71

iii
Chapter 4: Protein Production is an Early Biomarker for RNA-Targeting Therapies ................ 78
ABSTRACT .............................................................................................................................. 79
INTRODUCTION ..................................................................................................................... 80
METHODS................................................................................................................................ 82
RESULTS.................................................................................................................................. 88
DISCUSSION ........................................................................................................................... 98
ACKNOWLEDGEMENTS .................................................................................................... 101
Chapter 5: Summary and Future Directions.............................................................................. 111
ALSSOD1
clinical outcomes remain unchanged over time ....................................................... 112
Understanding drivers of ALSSOD1
heterogeneity ................................................................... 114
SOD1 protein is long-lived protein in ALS human CSF ........................................................ 115
Analyzing CSF SOD1 protein behavior to elucidate contributions to ALS pathobiology ..... 116
New Protein Production is a viable biomarker for RNA-lowering therapies ......................... 123
Protein Turnover as a measure of pharmacodynamics for SOD1-targeting therapies ............ 124
Understanding the mechanisms of CNS protein deposition into the CSF .............................. 125
Concluding Remarks ............................................................................................................... 126
References ................................................................................................................................... 128
Curriculum Vitae ........................................................................................................................ 148

iv
List of Figures CHAPTER 2
Figure 2.1 Mean age at onset and disease duration for ALSSOD1
.................................................. 42
Figure 2.2 Decreased survival probability from time of disease onset for SOD1A4V
compared to
SOD1Non-A4V
familial ALS ............................................................................................................ 43
Figure 2.3 SOD1A4V
ALS patients exhibit greater decline in ALS-FRS and FVC rates compared
to SOD1Non-A4V
patients ................................................................................................................ 45
CHAPTER 3
Figure 3.1 Representative Kinetic Curve of a participant labeled with 10 g 13
C6-Leucine via 16
hour intravenous infusion. ............................................................................................................ 61
Figure 3.2 Theoretical modeling of CSF SOD1 protein levels after ASO Treatment. ................. 70
Supplementary Figure 3.1. Compartmental model for kinetic data using plasma free 13
C6-Leucine
(input), CSF total protein, plasma total protein, and CSF SOD1 species for each subject to
caclulate protein FTR. ................................................................................................................... 72
CHAPTER 4
Figure 4.1 Lowered hTau protein synthesis precedes protein lowering after ASO treatment in
hTau transgenic mice .................................................................................................................... 89
Figure 4.2 Lowered 13
C6-Leucine-labeled hSOD1 observed in cerebrospinal fluid after ASO
treatment in hSOD1 transgenic rats .............................................................................................. 93
Figure 4.3 A therapeutic window exists to observe lowered protein synthesis pharmacodynamics
in the central nervous system ........................................................................................................ 96
Figure 4.4 A model of the interplay between new protein production and protein concentration
pharmacodynamics in RNA-lowering treatment strategies .......................................................... 99
Supplementary Figure 4.1 No differences in 13
C6-Leucine oral labeling between treatment groups
in all experiments performed. ..................................................................................................... 106
Supplementary Figure 4.2 Correlation between all hTau peptides measured ........................... 107
Supplementary Figure 4.3 Correlation between all hSOD1 peptides measured. ....................... 108
Supplementary Figure 4.4 No pharmacodynamics observed in the periphery after 1000 ug
hSOD1 ASO intracerebroventricular injection. .......................................................................... 109

v
Supplementary Figure 4.5 No protein concentration pharmacodynamics observed in CSF after
1000 ug hSOD1 ASO intracerebroventricular injection at 50 days post-treatment.................... 110

vi
List of Tables CHAPTER 2
Table 2.1 Clinical Characteristics of SOD1A4V
versus SOD1Non-A4V
mutation populations in
familial ALS.................................................................................................................................. 37
Table 2.2 Disease characteristics by mutation type in SOD1 familial ALS ................................. 40
CHAPTER 3
Table 3.1 Participant Demographics ............................................................................................. 60
Table 3.2 Protein Behavior in participants grouped by mutation and disease status .................... 63
Table 3.3 CSF SOD1 protein in participants with SOD1 mutations ............................................ 66
Supplementary Table 3.1 Transition ions used for SOD1 tandem liquid chromatography-mass
spectrometry analysis using GluC digestion protocol. ................................................................. 74
Supplementary Table 3.2 Transition ions used for SOD1 tandem liquid chromatography-mass
spectrometry analysis using LysC/Trypsin digestion protocol for mutant peptide detection ....... 77
CHAPTER 4
Table 4.1. Antisense Oligonucleotides ...................................................................................... 103
Supplementary Table 4.1. Primers and probes used for genotyping and qPCR analysis of hTau
(MAPT), hSOD1 (SOD1), and mouse GAPDH (GAPDH) DNA .............................................. 103
Supplementary Table 4.2. Transition ions used for hSOD1 tandem LC-MS/MS. GLHGFHVHE
peptide measurements were used as representative data in Figures. .......................................... 104
Supplementary Table 4.3 Transition ions used for hTau tandem LC-MS/MS. TPSLPTPPTR
peptide measurements were used as representative data in Figures. .......................................... 105

vii
Acknowledgments
I received financial support from the Washington University Institute of Clinical and
Translational Sciences grant UL1TR002345, sub-award TL1TR002344 from the National Center
for Advancing Translational Sciences (NCATS) to conduct the research described here.
Additionally, I thank the National Institute of Health for funding provided from the following
sources: R01NS09871601, R01NS078398, R25NS065743, U01NS084970, R01NS095773, as
well as support from the Seattle Veterans Affairs Medical Center, the Amyotrophic Lateral
Sclerosis Association, the Harvard NeuroDiscovery Center, the Dr. Anne B. Young
Neuroscience Translational Medicine Fellowship, the Muscular Dystrophy Association, and the
MetLife Foundation Award to support the work performed in the laboratory of Dr. Timothy
Miller and our collaborators.
The arduous journey to finish this body of work would not be realized without the help of
the extraordinary group of individuals I met along the way. Words will fail to articulate the
magnitude of my feelings towards you all. Nonetheless, I will try to express gratitude for those
that have been instrumental throughout my scientific development. More importantly, you all
have made lasting impacts on my life that I will cherish forever.
First, I thank my thesis mentor, Dr. Timothy Miller. Although he granted me the
autonomy to pursue multiple projects in my early years of training, Tim had a knack for gently
guiding my scattered mind back to the path when I strayed too far away from productivity. “We
won’t know until we see the data” was an oft-mentioned phrase in our weekly meetings, and I
appreciate his patience in allowing me to openly express my thoughts. Further, Tim’s optimism
and excitement for new ideas, even in the face of failure, provided the spark to reignite my

viii
motivation for research repeatedly. It also helped that he provided me a healthy work
environment with brilliant lab mates and collaborators who were indispensable for performing
experiments and developing my scientific skill set. Finally, I appreciate Tim’s willingness to
support my career development. I involved myself in many activities outside of laboratory work,
and Tim never gave anything less than his full support. I cannot thank him enough for these
selfless gestures, and I can only hope people will view me half as fondly as I do for Tim as a
mentor, scientist, and human being.
Tim instilled in me the importance of working in teams, as he surrounded me with
additional mentors that were instrumental in cultivating a doctoral-level body of work. I must
first thank Dr. Randy Bateman, as his willingness to allow me as a quasi-member of his research
group was necessary and sufficient for my technical growth as a researcher. Despite being one of
the busiest people at Washington University, I deeply admire Randy’s focus and attention to
detail with every piece of data he comes across, while simultaneously asking questions that push
you towards a higher level of learning. I also thank the rest of my thesis committee, Drs. John
Cirrito, Chris Weihl, and Anne Fagan, for providing valued discussions about my work and
patience when my enthusiasm resulted in convoluted answers to their poignant questions.
I must also thank my mentors as a member of the TL1 Predoctoral Clinical Research
Training Program, Drs. Jay Piccirillo, Jeff Peipert, Susy Stark, Ana Maria Arbelaez, and David
Brody. By exposing me to concepts in clinical research, the TL1 program was instrumental in
helping me understand the need to bridge laboratory research and clinical practice to become a
well-rounded translational scientist. I must also thank my first research mentor, Dr. Jeff
Capadona of Case Western Reserve University, who was the fundamental inspiration and role
model for my career in science.

ix
I thank my lab mates and collaborators for being far more superior scientists than I could
ever dream. Drs. Matt Crisp and Sarah DeVos were senior graduate students when I joined Tim’s
group, and their mentorship in the infancy of my graduate life strongly influenced my outlook on
science. As they continue to excel at every step along their career, I am proud to not only call
them mentors and role models, but my friends. I also thank my laboratory mentors in the
Bateman Lab: Drs. Kwasi Mawuenyega and Jim Bollinger. I thank Kwasi for his patience with
me as he guided a naïve graduate student through the fundamentals of mass spectrometry. After
the retirement of the TSQ-Vantage, I thank Jim Bollinger for grabbing the reins as my mentor in
the SOD1 Xevo and Lumos adventures. Jim’s endless enthusiasm for science and teaching were
the breath of fresh air I needed through the many days of struggle that came with research. I am
deeply indebted to him for all of the time he sacrificed on his primary projects to help me. I also
thank my lab mom, Amy Wegener, whose tough love helped my organization and collaboration
skills blossom from appalling to mediocre. I am also in hours of debt to Tao Shen, who
tremendously increased my quality of life as a graduate student with her incredible work ethic
and gratitude. I also extend a huge acknowledgement to the Miller lab clinical research team who
allowed me to realize the possibility of working with human participants: Jennifer Jockel-
Balsarotti, Debbie King, Jesse Markway, Amber Malcolm, Dr. Bob Bucelli, and Dr. Arun
Varadarchary.
In the 4+ years I spent in the Miller lab trenches, I may have logged the most hours with
Alex Cammack, Dr. Kathleen Schoch, and Dr. Mariah Hoye than anyone else in the city of St.
Louis. From being his “buddy” during candidate interviews, creating a new transgenic mouse
that no one ever used, sharing a love for sports, and being the most reliable friend to grab a beer,
I am proud to call Alex a colleague and friend. Kathleen’s scientific mentorship has been crucial

x
in improving my technical laboratory skills, as well as her willingness to be a collaborator with
me in designing experiments. As we simultaneously came into new roles as a postdoctoral
researcher and graduate student, I have thoroughly enjoyed having Kathleen by my side to grow
as scientists. We also grew together personally, as we felt the terrors and uncertainty of moving
in with a significant other to launching our own passion projects outside of lab. The origin of my
decision to join the Miller Lab starts with an hour long conversation with Mariah Hoye late one
night after class, and I am so glad her persuasion prevailed. I hope an infinitesimal bit of her
work ethic, passion, and undying curiosity for science rubbed off from her bay to mine during
our time together. I express my sincerest gratitude for her patience, willingness to engage in
passionate “discussions” about science, having such a hunk for a husband, and sharing an
impeccable taste for fantasy literature with me. I could not ask for a better coworker and friend to
travel with along our parallel dissertation paths together, and I look forward to seeing the next
steps in life and awards that lay ahead for her.
To friends old and new, thank you for keeping me sane throughout the last 5 years of
this emotional rollercoaster. Our time in The BALSA Group and Washington University
Intramural sports has led to the creation of lasting friendships, no matter how far we reach across
the globe. Although we came from different scientific backgrounds, the shared experience of
pursuing a PhD forms a lasting bond uniting us all, and I appreciate how we all served as
fantastic distractions from science when we are together. I extend a special thanks to my dear
friend Alexandra Russo. You have helped me in more ways than you know, and I am deeply
saddened by the thought of us living apart. Despite its bittersweet nature, I am comforted
knowing we will always be there for each other, no matter what obstacles life throws our way.

xi
To my family, I thank you for your everlasting love and support at every stage of my life.
I cannot say for certain if other families share more moments of laughter and tears, but I would
not have it any other way. To my mother, Pam Self, I thank you for being the foundation of my
life. You can read my mind before I open my mouth, and your love, guidance, support, and
threats to cut me off are essential components which shape the person that I am today. I will
never be able to repay you for everything you have done, but I will certainly try my best to make
you feel as proud as I am of you.
And finally, a million times over, I thank Zoe Gross. Your partnership gives my life
meaning in ways I never thought possible. I love you.
Wade K. Self
Washington University in St. Louis
August 2018

xii
To Ralph, Peggy, David, and all participants in human disease research.
In the face of an intractable disease, your sincerity, positivity, curiosity, and willingness to
sacrifice for the good of mankind is an inspiration to us all.

xiii
“Let us keep looking, in spite of everything. Let us keep
searching. It is indeed the best method of finding, and
perhaps, thanks to our efforts, the verdict we will give
such a patient tomorrow will not be the same we must
give this man today.”
Charcot – 1889
“Isn’t it nice to know a lot? And a little bit, not.”
Little Red Riding Hood – 1986

xiv
Abstract of the Dissertation
Smooth as SILK: Translating Discoveries to New Therapies in Amyotrophic Lateral Sclerosis
by
Wade Kerner Self
Doctor of Philosophy in Biology and Biomedical Sciences
Neurosciences
Washington University in St. Louis, 2018
Professor Timothy Miller, Chair
No effective therapies exist for the treatment of patients suffering from amyotrophic
lateral sclerosis, a devastating neurodegenerative disease characterized by motor neuron loss,
paralysis, and death on average 3 – 5 years after diagnosis. Although multiple promising
candidates have emerged from preclinical experiments in disease models, failure rates of
randomized control trials assessing efficacy in humans are over 95%, demonstrating a need to
improve the translation of preclinical findings to human biology. In this dissertation, I present
original work investigating amyotrophic lateral sclerosis caused by mutations in SOD1
(ALSSOD1
) for the successful translation of SOD1-targeting strategies into meaningful treatment
options for patients. Through a retrospective study of ALSSOD1
natural history, my research
showed that the most common mutation causing ALSSOD1
in North America, Alanine>Valine
(A4V), is rapidly progressing, while different mutations show a wide range of variability in
clinical outcomes. I then describe initial characterization of SOD1 protein half-life in the
cerebrospinal fluid (CSF) of ALSSOD1
patients. Similar to healthy control subjects, I found SOD1

xv
is a long-lived protein in ALS patient CSF, with a half-life on the order of weeks. Moreover,
A4V SOD1 mutant protein showed differential behavior compared to wild type SOD1 in CSF in
one ALS participant, preliminarily suggesting that CSF SOD1 measures may implicate the
pathogenic state of SOD1 protein. Finally, I show that measures of CSF SOD1 protein
production serve as a pharmacodynamics biomarker for Antisense Oligonucleotides (ASOs)
treatment targeting SOD1 mRNA transcripts in the central nervous system (CNS). Lowering of
new SOD1 protein production after ASO treatment preceded lowering of total SOD1 protein
concentration, suggesting measures of new protein production are more sensitive for mRNA-
targeting therapies and are translatable to human trials to assay drug target engagement. Taken
together, these data provide insights that contribute to a further understanding of ALS biology, in
addition to the study design and interpretation of randomized control trials in ALS.

1
Chapter 1: Towards a New Treatment for ALS

2
The average life expectancy in the United States has increased from 68 years in 1950 to
79 years in 2013, and one in four people in the United States are projected to be 65 years or older
by 2060 (Mather et al 2015). These trends present new challenges to human healthcare, including
a higher number of people afflicted with diseases associated with aging. Chief among these are
progressive neurodegenerative diseases, a family of disorders characterized by the loss of
structure and function of neurons in the brain and spinal cord. Neurodegenerative diseases
include Alzheimer’s Disease (AD) and other forms of dementia (Alzheimer 1906), Huntington’s
Disease (HD) (Huntington 1872), Parkinson’s Disease (PD) (Parkinson 1969), and Amyotrophic
Lateral Sclerosis (ALS) (Charcot 1874). Despite the original descriptions of these diseases over
two centuries ago, no current cures exist. If the present therapeutic landscape remains
unchanged, projected costs of neurodegenerative disease patient care will reach over $1 trillion
by 2050 (WHO 2012). Therefore, a dire societal need exists to develop novel therapies to treat
these debilitating diseases.
Many neurodegenerative diseases converge on a similar pathology in the central nervous
system (CNS): the accumulation of insoluble protein aggregates within neuronal populations that
degenerate throughout disease time course. Research into the genetics and pathology that
underlie these diseases revealed multiple proteins that are prone to misfold and aggregate within
neurons (Forman et al 2004). These observations have led to a generally accepted hypothesis that
misfolded proteins impart toxic effects on specific neuronal populations, resulting in cell death.
Therefore, an attractive therapeutic strategy to treat neurodegenerative diseases is to prevent the
accumulation of misfolded proteins within the nervous system to maintain neuronal viability
throughout aging.

3
Despite decades of research that identified potential compounds which ameliorate protein
aggregation pathologies in disease models, clinical trials for neurodegenerative disorders show a
failure rate of over 95% (Mitsumoto et al 2014; Petrov et al 2017), and as high as 99% in
Alzheimer’s Disease (Banik et al 2015; Soejitno et al 2015). Multiple causes may contribute to
these alarming failure rates, including: a lack of understanding of the mechanisms that govern
neurodegeneration, the absence of effective tools to measure biological effects of treatments in
humans, and effective clinical trial design. Moving forward, it is imperative for translational
researchers to consider all facets of preclinical and clinical research in progressive
neurodegenerative diseases to bring more effective or curative therapies to patients. Furthermore,
because neurodegenerative diseases share similar misfolded protein pathologies that converge on
common mechanisms of neuronal death, the development of an effective treatment in one disease
condition may provide valuable insight into the development of therapeutic strategies for
multiple disorders.
Thus, in an effort to better define the biological and clinical manifestations of
neurodegenerative disease, the focus of this dissertation will be ALS, colloquially known as Lou
Gehrig’s Disease, caused by mutations in Superoxide Dismutase 1 (SOD1) (Kiernan et al 2011).
This dissertation will begin with an introduction to the genetics and clinical characteristics of
ALS, the role of mutant SOD1 misfolding in ALS pathobiology and clinical phenotypes, the
development of therapeutic strategies that target SOD1, and previous challenges in translating
scientific discoveries to effective treatments for ALS caused by SOD1 mutations (ALSSOD1
). It
will then discuss the novel research I performed in this dissertation, including a retrospective
cohort study on the most recent understanding of ALS natural history caused by SOD1 mutations
in North America, efforts to understand SOD1 involvement in human ALS through analyzing

4
SOD1 protein turnover within the cerebrospinal fluid (CSF) of ALS patients, and the
development of a new pharmacodynamics biomarker for SOD1-targeting therapies. The
dissertation will conclude with a discussion of the implications of these studies in translating
biomedical research into effective therapies for ALS, as well as future research directions for
both clinical trial design and understanding of human ALS pathobiology.
Amyotrophic Lateral Sclerosis: A relentless degenerative disorder
In 1874, Jean-Marie Charcot first described ALS as a disease of upper and lower motor
neuron degeneration within the brainstem and spinal cord (Charcot 1874). ALS affects ~3
individuals per 100,000 in Europe (Worms 2001) and ~5 individuals per 100,0000 in the United
States (Mehta et al 2016). ALS is the third most common neurodegenerative disorder in the
United States, trailing only Parkinson’s Disease and Alzheimer’s Disease (Kiernan et al 2011).
ALS onset typically begins within the limbs (limb-onset) or facial region (bulbar-onset).
Symptoms will therefore manifest depending on onset location - with brisk reflexes and local
muscle wasting in limb-onset ALS or trouble with speech and swallowing in bulbar-onset
disease (Rowland 2001). Symptoms quickly spread to other regions as additional motor units are
lost, resulting in spasticity, deep tendon reflexes, fasciculation, wasting, and eventual paralysis
(Kiernan et al 2011). ALS is rapidly progressive, where 50% of patients with the disease die
within 30 months from presentation of symptoms and 20% survive between 5 and 10 years after
symptom onset (Talbot 2009). Currently, ALS is diagnosed by an exclusionary process of
eliminating all other possible causes of patient symptoms. The lack of an established biomarker
to accurately diagnose ALS may contribute to poor patient outcomes, as symptoms manifest after
irreversible degeneration of motor neurons has already occurred in the CNS. Upon diagnosis,
only two FDA-approved therapies are available for treatment of ALS: Riluzole (Bensimon et al

5
1994; Lacomblez et al 1996; Miller et al 2012), an inhibitor of glutamate release, and Edaravone
(Abe et al 2017; Kalin et al 2017; Yoshino & Kimura 2006), a free-radical scavenger. These
treatments are not effective for long-term survival of ALS patients, where the median disease
progression in randomized control trials slowed by only ~6 months, and controversy remains
over the reproducibility of results and cost effectiveness of these treatments (Fang et al 2018;
Messori et al 1999). Therefore, a clear need exists to better understand ALS disease pathogenesis
for developing treatments that effectively manage or cure this devastating disease.
Recent advances in genetics have helped to identify potential mechanisms that underlie
disease pathogenesis in familial forms of ALS, which account for ~10% of total cases
(Robberecht & Philips 2013). The first gene implicated in ALS, SOD1 (Rosen et al 1993), was
identified in 1993. For almost 20 years, mutations in SOD1 were the only known cause of
familial ALS. In the last ten years, researchers have seen an explosion in the discovery of new
ALS genes that describe ~60% of familial ALS and 11% of sporadic ALS cases (Renton et al
2014). Of the newly identified genetic causes, the most prevalent are hexanucleotide repeat
expansions found in the first intron of the C9ORF72 gene, accounting for ~40% of familial ALS
in the United States and Europe (Majounie et al 2012; Renton et al 2011), and mutations in
TARDBP and FUS, each accounting for ~5% of familial ALS (Sreedharan et al 2008; Vance et al
2009). These newly discovered genes exhibit a variety of proposed functions, including protein
clearance (UBQLN2 (Deng et al 2011), SQSTM1 (Rubino et al 2012), VCP (Johnson et al 2010),
TBK1 (Cirulli et al 2015)), vesicle trafficking and cytoskeleton dynamics (PFN1 (Wu et al 2012),
OPTN (Maruyama et al 2010), TUBA4A (Smith et al 2014), C9ORF72 (Shi et al 2018),
KIF5A(Nicolas et al 2018)), RNA processing (TDP43 (Ou et al 1995), FUS (Yang et al 1995),

6
MATR3 (Johnson et al 2014), TIA1 (Mackenzie et al 2017)) and reactive oxygen species
metabolism (SOD1 (McCord & Fridovich 1969b)).
ALS is classically described as a motor disorder, but reports suggest that up to 48% of
ALS patients exhibit cognitive abnormalities associated with frontotemporal lobar degeneration,
a progressive dementing condition characterized by neuronal loss in the frontal and anterior
temporal lobes of the brain (Lomen-Hoerth et al 2002; Portet et al 2001). These observations
suggest that ALS and frontotemporal dementia (FTD) fall on a “spectrum” of neurodegenerative
disease states. This claim is supported by genetic evidence, where mutations in multiple genes
cause familial cases of ALS, FTD, and ALS-FTD (Robberecht & Philips 2013). Moreover, the
majority of sporadic and familial ALS and FTD cases share similar pathological features, such as
the presence of protein inclusions containing ubiquitin and p62 in neuronal populations affected
by disease (Neumann et al 2006; Steinacker et al 2008). Although the ALS-FTD spectrum adds
additional complexity to understanding the individual diseases, the similarity of familial genetics
and pathologies suggest a potential unifying mechanism of degeneration underlying these
conditions.
Although ~80% of ALS cases are sporadic with no known genetic cause, 98% of all ALS
cases converge on a shared pathology: cytoplasmic protein inclusions containing TDP-43 in
neurons and glia in the brain and spinal cord (Lomen-Hoerth et al 2002). These observations led
researchers to hypothesize that modeling familial forms of ALS may reveal common
mechanisms that mediate ALS pathogenesis to target for therapeutic intervention. Conversely,
the genetic heterogeneity of ALS may suggest that different ALS subtypes demand unique
treatment strategies. For example, mutations in SOD1 account for ~2% of all ALS cases and,
with the exception of one reported patient (Nakamura et al 2015), present without cognitive

7
deficits or TDP-43-related pathology. Instead, the primary protein pathology observed in motor
neurons is the accumulation of insoluble, ubiquitin-positive aggregates containing mutant SOD1
protein. Despite these discrepancies in pathology between ALSSOD1
and the majority of the ALS
population, almost 25 years of research in understanding the role of SOD1 in ALS has garnered
important insights into mechanisms that govern neurotoxicity associated with protein misfolding,
concurrently leading to the development of novel therapeutic strategies for the treatment of ALS
and other progressive neurodegenerative diseases.
The Role of SOD1 in Amyotrophic Lateral Sclerosis
The SOD1 gene encodes for SOD1, a 153 amino acid, homodimeric, Copper-Zinc
metalloenzyme that catalyzes the conversion of superoxide anion (O2-) into hydrogen peroxide to
regulate the concentration of reactive oxygen species within cells (McCord & Fridovich 1969a).
The SOD1 protein contains eight beta-barrel sheets, connecting loops, two intrachain disulfide
bonds, and two metal-binding domains, with the enzymatic activity of the protein dependent
upon homodimerization (Parge et al 1992). Rosen and colleagues first identified 11 single
amino-acid changes in SOD1 on the basis of genomic DNA sequences in members of 13
different familial ALS families in the United States (Rosen et al 1993). Concurrently, a research
team in Japan identified a Histidine>Arginine amino acid change at residue 46 (H46R) in two
familial ALS families in Japan (Aoki et al 1993). Since these initial discoveries, investigators
have identified over 180 mutations in SOD1 implicated in familial forms of ALS with varying
degrees of penetrance. These mutations are predominantly missense, but also include nonsense
and truncation mutations (for an updated list of all SOD1 mutations, visit the ALS Online
Database). The majority of SOD1 mutations are found in familial ALS, but SOD1 mutations
have also been reported in sporadic ALS (Chio et al 2009b; Chiò et al 2008).

8
Different SOD1 mutations display a broad spectrum of clinical phenotypes that influence
disease onset and disease progression after symptom presentation. Interestingly, in contrast to
other neurodegenerative diseases such as Alzheimer’s Disease where earlier disease onset is
characterized by more aggressive neurodegeneration and rapid disease progression (Lanoiselee et
al 2017), early disease onset in ALSSOD1
is associated with slower disease progression
(Cudkowicz et al 1997; Juneja et al 1997; Rosen et al 1994). However, the relationship between
SOD1 mutation and the heterogeneity of clinical disease characteristics remains poorly
understood. This complexity is seen in family pedigree analyses, where different SOD1
mutations display varying degrees of disease penetrance (Felbecker et al 2010). For example,
description of a large family with ALSSOD1
caused by an I113T mutation shows 50% penetrance
at 60 years, with age of onset ranging from 39 – 67 years (Lopate et al 2010). Given the
heterogeneity in disease penetrance between mutants, one may hypothesize that mutations which
cause fast-progressing forms of disease may exhibit higher penetrance. However, an aggressive
form of ALSSOD1
caused by an I112M mutation in four Mediterranean families had several
obligate carriers in the family pedigree despite an aggressive disease duration of less than 2 years
(Gamez et al 2011). The high variability observed between different mutations and within
families carrying the same SOD1 mutation raises concerns over the validity of these variants as
disease-causing, making the prognosis of ALSSOD1
challenging within the clinic. Therefore, a
better understanding of mutant SOD1 protein behavior in living patients may provide valuable
insight into the prognosis of patients carrying these SOD1 variants.
SOD1 in ALS Pathobiology
SOD1 mutations that cause ALS are found throughout the entire 153 amino acid protein
sequence and, with the exception of the D90A mutation in the Scandanavian population

9
(Andersen et al 1997b), display autosomal dominant inheritance. In line with SOD1 genetics, in
vitro studies have shown that many mutant SOD1 proteins retain enzymatic activity and the
ability to dimerize with both mutant and WT SOD1 (Borchelt et al 1994; Wiedau-Pazos et al
1996). Further, knockout of SOD1 in transgenic mice does not result in global motor neuron
degeneration (Reaume et al 1996; Shefner et al 1999). These data ultimately suggest that SOD1
loss-of-function is not the primary driver of ALSSOD1
.
The common pathology observed in all ALSSOD1
patients is the presence of misfolded
SOD1 protein in ubiquitin-positive, insoluble aggregates within motor neurons (Shibata et al
1996). Accumulating evidence suggests that mutant, misfolded SOD1 acquires a toxic gain-of-
function which leads to motor neuron degeneration. Indeed, the overexpression of a transgene
containing the human SOD1 gene with an ALS-causing G93A point mutation in mice was the
first in vivo model of ALS to show a disease phenotype (Gurney et al 1994). In these transgenic
mice, mutant SOD1 overexpression causes a rapidly progressing degeneration of motor neurons
in the brain and spinal cord, muscle atrophy, and hind-limb paralysis by ~6 months of age. These
animals show the same mutant SOD1 pathology in the brain and spinal cord as human ALS,
where insoluble protein aggregates are observed in the cell body and processes of motor neurons
and glial cell types (Johnston 2000; Shibata et al 1998). These events in transgenic animal
models appear specific to misfolded, mutant SOD1, as over-expression of WT SOD1 protein
does not result in an ALS-like phenotype of motor neuron degeneration (Avraham et al 1988;
Chan et al 1998). Additionally, WT SOD1 overexpression in the presence of mutant SOD1 does
not accelerate the rate of disease in vivo (Bruijn 1998).
Many mechanisms by which misfolded SOD1 causes motor neuron toxicity have been
suggested including impaired protein clearance due to proteasome (Cheroni et al 2009;

10
Matsumoto et al 2005; Urushitani et al 2002) and autophagy (Bandyopadhyay et al 2014;
Rudnick et al 2017; Xie et al 2015) inhibition, mitochondrial dysfunction (Ferri et al 2006;
Israelson et al 2010; Jaarsma et al 2001; Li et al 2010; Liu et al 2004; Pasinelli et al 2004; Vande
Velde et al 2008), endoplasmic reticulum stress (Filezac de L'Etang et al 2015; Lee et al 2016b;
Nagano et al 2015; Saxena et al 2009), and impaired axonal transport (Williamson & Cleveland
1999). Because motor neurons are selectively vulnerable to degeneration in ALS, early efforts
focused on understanding motor neuron-specific cellular mechanisms that contribute to disease.
However, conditional overexpression of mutant SOD1 in neurons yielded conflicting results.
Driving expression of mutant SOD1 with a neuron-specific promoter was not sufficient to
recapitulate the motor neuron degeneration observed with ubiquitous expression of mutant SOD1
in vivo (Pramatarova et al 2001). Other reports showed a mild motor neuron degeneration
phenotype in mice conditionally overexpressing mutant SOD1 in neurons using the Thy1
promoter (Jaarsma et al 2008). However, the Thy1 promoter is also expressed in glial cells,
which confounds the interpretation of degeneration driven by neuron-specific expression of
SOD1 (Kemshead et al 1982). Also, SOD1 pathology is not restricted to motor neurons, as early
characterization of transgenic G85R SOD1 mouse models and human autopsies reported
inclusions of misfolded SOD1 and downregulation of glial glutamate transporters in astrocytes
(Bruijn et al 1997; Kato et al 2000; Lin et al 1998). These observations suggest that cell-
autonomous effects of mutant SOD1 in motor neurons are not solely responsible for ALS
pathology.
Seminal experiments in vivo and in cell culture further revealed the importance of non-
neuronal cell types as contributors to neurotoxicity caused by expression of mutant SOD1.
Chimeric mice expressing WT human SOD1 in glial cells and mutant G37R SOD1 in motor

11
neurons exhibited extended survival compared to mice that ubiquitously express mutant SOD1
(Clement et al 2003). These results were corroborated by genetic experiments where conditional
knockout of mutant SOD1 was performed using a Cre-Lox recombinase system. In this
paradigm, removal of G37R SOD1 in motor neurons resulted in delayed onset of disease without
altering disease progression. However, conditional knockout of G37R SOD1 in either astrocytes
or microglia drastically delayed disease progression (Boillee 2006). Similar removal of mutant
SOD1 from oligodendrocyte progenitors delayed disease onset and progression in transgenic
animal models (Kang et al 2013). Glial cells may also directly contribute to motor neuron
toxicity. Co-culture of astrocytes and oligodendrocytes expressing mutant SOD1 is sufficient to
induce motor neuron cell death. This toxicity is independent on cellular contact, as conditioned
media from glial cell cultures is also sufficient to induce motor neuron toxicity in vitro (Di
Giorgio et al 2008; Di Giorgio et al 2007; Ferraiuolo et al 2016). It is unknown if this toxicity
caused by glia is due to the release of a secreted neurotoxic factor or the result of critical nutrient
depletion for motor neurons by mutant-expressing glial cells. In sum, these data highlight the
critical role of mutant SOD1 expression in glia in mediating motor neuron degeneration in ALS
by non-cell autonomous mechanisms independent of motor neuron function.
One of the proposed mechanisms that drive mechanisms of toxicity in ALS is
inflammation, which involves multiple cell types in both the CNS and periphery. Compromised
blood brain barrier and pericyte dysfunction are early pathological hallmarks of mutant SOD1
transgenic animal models (Zhong et al 2008), indicative of inflammation within the brain and
spinal cord. Outside of the CNS, ALSSOD1
patients present with compromised T-cell function,
resulting in a pro-inflammatory profile that correlates with disease severity and progression
(Beers et al 2017). Within the CNS, mutant SOD1 secreted from neurons can be phagocytosed

12
by surrounding microglia, causing a pro-inflammatory activation of microglia that is ultimately
neurotoxic (Meissner et al 2010; Urushitani et al 2002; Zhao et al 2010). This role of
inflammation in ALS disease progression further demonstrates that non-cell autonomous
mechanisms are not restricted to intracellular activity of mutant SOD1.
The discovery of many non-cell autonomous mechanisms of mutant SOD1 toxicity in
neurons, glia, and peripheral immune cells adds to the complexity of ALS disease pathogenesis
and helps explain the challenges in developing current treatments for patients. However, the
diversity of pathways also provides an opportunity for the development of different therapeutic
strategies to effectively target and ameliorate SOD1-mediated pathology. Furthermore, the wide
array of pathways implicated in ALS also suggest that targeting of SOD1 in all cell types
throughout the entire CNS is important for developing an effective treatment strategy.
An attractive hypothesis relating SOD1 behavior to these toxic pathways, and ultimately
to clinical outcomes, is that SOD1 mutations which cause greater protein destabilization result in
more rapidly progressing forms of disease by catalyzing the conversion of mutant SOD1 into
toxic, misfolded protein species. This hypothesis is supported by experiments using recombinant
SOD1 protein in vitro. Shi and colleagues demonstrated that the failure of dimerization between
mutant and wild type SOD1 protein correlates with a more rapid disease progression (Shi et al
2016). Additional analyses of point mutations at the G93 residue in recombinant SOD1 protein
showed a positive correlation between the propensity for protein aggregation and disease
progression (Pratt et al 2014). In patients, Sato and colleagues tested the hypothesis that unstable
SOD1 mutant protein species would be rapidly turned over and undetectable in anuclear red
blood cells. Indeed, they reported that patients who carried SOD1 mutant protein that was not
detectable in red blood cells exhibited a more rapid disease progression compared to detectable

13
mutant SOD1 protein carriers (Sato et al 2005). Despite the promise of these suggestive
correlations, no current data characterizes the biochemical behavior of mutant and wild type
SOD1 in the CNS of ALS patients, limiting understanding of the biological relevance of these
findings to disease.
Despite extensive descriptions of SOD1 misfolding and aggregation in post mortem brain
and spinal cord tissues of ALS patients, the ability to effectively analyze pathological SOD1
behavior in the CNS of living ALS patients remains elusive. For other neurodegenerative
diseases, researchers developed imaging agents that bind to protein aggregates such as amyloid-β
and microtubule associated protein tau in Alzheimer’s disease to visualize protein misfolding by
positron emission tomography (Klunk et al 2004; Villemagne et al 2018). These imaging agents
are able to bind to β–sheet rich, amyloid aggregate structures and have been instrumental in
characterizing the pathological events that underlie neurodegeneration in AD. By contrast,
misfolded SOD1 forms an amorphous, non-amyloid aggregate, thereby limiting the development
of such agents to identify SOD1 misfolding in ALS (Kerman et al 2010; Mulligan et al 2012).
The primary methods used to tackle this unmet need in ALS are assays that measure
SOD1 protein levels in CSF of patients via enzyme-linked immunosorbent assay (ELISA) or
SOD1 activity assays. However, all studies fail to measure significant differences in CSF SOD1
between ALSSOD1
from controls. Jacobsson and colleagues showed no difference in CSF SOD1
protein activity between ALSSOD1
, sporadic ALS, and control patients (Jacobsson et al 2001).
Further, Zetterstrom and colleagues developed a misfolded SOD1-specific ELISA and tested for
differences in levels of misfolded SOD1 in CSF of 38 neurological controls and 96 ALS patients.
Misfolded SOD1 was present in all samples, but further analysis could not distinguish between
ALS and controls, or ALSSOD1
and sporadic ALS (Zetterstrom et al 2011). In a separate cohort,

14
Winer and colleagues reported in an increase in CSF SOD1 levels in ALSSOD1
compared to
healthy controls. However, CSF SOD1 was also elevated in neurological disease controls,
questioning the specificity of this result to ALS (Winer et al 2013). A key limitation in the
development of SOD1 assays is the challenge of discriminating mutant and WT SOD1 in any
measure. Most single amino acid changes caused by mutations do not drastically change any
bulk biochemical properties of the SOD1 protein such as molecular weight that can be resolved
by methods such as gel electrophoresis. Moreover, single amino acid changes make the
development of novel antibodies that specifically recognize mutant SOD1 protein challenging.
Given these conflicting data, a clear need exists to further investigate SOD1 in human ALS to
better understand its role in both familial and sporadic forms of the disease.
Controversial Role of SOD1 in Sporadic ALS
Because protein misfolding is a hallmark of ALS, and mutant SOD1 protein is prone to
misfolding and aggregation, researchers have investigated the potential for wild-type SOD1
protein to misfold and aggregate. Such data may implicate misfolded SOD1 as a common
disease mechanism in both familial and sporadic forms of ALS in which SOD1 is not mutated. In
vitro perturbations to WT SOD1 such as demetallation and oxidation cause protein misfolding
and denaturation (Rakhit et al 2002; Rodriguez 2002), and these misfolded WT SOD1 species
share similar conformations with mutant SOD1 variants (Durazo et al 2009). Further, co-
expression of mutant SOD1 protein with WT SOD1 protein in cell culture induces WT SOD1
misfolding using a disease-specific epitope antibody (Grad et al 2011). In vivo, crossing mutant
SOD1 transgenic mice with WT SOD1 transgenic mice results in the identification of WT SOD1
in detergent-insoluble spinal cord fractions (Prudencio et al 2010). Additionally, overexpression
of A4V SOD1 in transgenic mice does not show an ALS phenotype unless crossed with human

15
WT SOD1-expressing transgenic mice to co-express both mutant and WT SOD1 protein (Deng
et al 2006). A key limitation of these studies is the use of overexpression paradigms and high
concentrations in vitro, which calls into question the validity of WT SOD1 misfolding in human
ALS pathophysiology.
Despite these observations from mouse models, evidence for misfolded WT SOD1 in
either human ALSSOD1
or sporadic ALS without SOD1 mutations is conflicting. In post-mortem
studies, multiple groups have reported misfolded SOD1 in sporadic ALS spinal cord autopsies
(Bosco et al 2010; Forsberg et al 2010). However, these findings appear to be antibody-specific,
as results could not be recapitulated using different antibodies that recognize misfolded SOD1
protein with overlapping epitopes (Da Cruz et al 2017). Moreover, each of these post-mortem
studies have a relatively small sample sizes to test for misfolded SOD1 positivity in sporadic
ALS samples, limiting the generalizability and interpretation of results.
The limited number of assays that exist to determine the state of misfolded SOD1 protein
in living patients also presents challenges in implicating misfolded SOD1 in sporadic ALS. The
identification of misfolding of wild type SOD1 in sporadic ALS is a key outstanding question for
identifying the relevance of SOD1-targeting therapeutics for ALS outside SOD1 mutant carriers.
Interestingly, lowering of SOD1 in sporadic ALS astrocytes reduces motor neuron toxicity in cell
culture (Haidet-Phillips et al 2011), suggesting that therapeutic targeting of SOD1 may be
beneficial in sporadic ALS. Further data clarifying the role of SOD1 in sporadic ALS may
broaden the utility of SOD1-targeting therapies beyond the ~2% of ALS patients that have
disease caused by SOD1 mutations.

16
Toxicity by Misfolded SOD1: Similarity to other ALS subtypes
Although the presence of misfolded SOD1 pathology outside of ALSSOD1
remains
controversial, the gain-of-toxicity function associated with misfolded proteins may be a common
mechanism that unites many disease subtypes, as other proteins implicated in ALS pathobiology
are prone to misfolding and aggregation. In patients who harbor expansions in the first intron of
C9ORF72 gene, the GGGGCC expansion is unconventionally translated by a non-ATG-related
mechanism to produce dipeptide-repeat proteins that are prone to misfolding and form insoluble
protein aggregates within cells (Ash et al 2013; Mori et al 2013a; Mori et al 2013b; Zu et al
2013). Similar to misfolded SOD1, these dipeptide-repeat proteins are toxic to neurons in vitro
(Donnelly et al 2013; Kwon et al 2014; Mizielinska et al 2014) and in vivo (Chew et al 2015; Liu
et al 2016). Additionally, full-length and C-terminal fragments of TDP-43 found in familial and
sporadic ALS have the ability to induce TDP-43 misfolding and aggregation that is toxic to
neurons (Fang et al 2014; Nonaka et al 2013). Mechanisms of toxicity that have been proposed
for these misfolded protein species are similar to misfolded SOD1(Sareen et al 2013). Therefore,
an understanding of toxicity caused by misfolded SOD1 protein may provide the foundations to
develop therapeutic strategies in other forms of ALS that target different pathological proteins to
test in randomized control clinical trials.
Learning from Past Failures for ALSSOD1
Clinical Trial Design
The average success rate of drug development programs reaching market approval for use
in patients is ~14% (Wong et al 2018). This low probability of success highlights the difficulties
in developing novel therapies for any disease indication. The situation is even more dire in ALS,
where over 50+ randomized control trials have led to two, marginally effective FDA-approved
treatments (Mitsumoto et al 2014). Despite failure rates of over 95% for ALS randomized

17
control trials, previous efforts gleaned valuable lessons to identify key factors that influence
successful clinical trial design. Among these factors include a growing appreciation for the
heterogeneity of clinical disease characteristics in ALS and the need for adequate tools inform on
the biological efficacy of a drug.
Developments in DNA sequencing techniques have provided tremendous insight into the
genetic complexity of disease where, in cases like cancer tumors, subpopulations of cells may
exhibit unique genomic alterations (Fisher et al 2013). Indeed, the growing number of mutations
implicated in ALS suggests that different subtypes of ALS may exist which still result in the
same motor neuron degeneration phenotype. Further, natural history data from all ALS patients
shows median survival time from onset to death ranges from 20 to 48 months, but 10-20% of
ALS patients have survival longer than 12 years (Chio et al 2009a). If we accept the hypothesis
that different rates of disease progression are the result of multiple ALS phenotypes, therapeutic
efficacy of a new treatment may vary between different ALS populations. With the absence of
established prognostic biomarkers for ALS in neurology clinics, the most valuable data available
to help understand clinical outcomes are previous epidemiology studies of natural history in the
ALS population. Therefore, a key component for future trial design is to understand the natural
history of the disease in different genetic, geographic, and topographic variables in ALS
populations.
One proposed solution to account for these differing clinical phenotypes in randomized
control trial design is to stratify participants into fast- and slow-progressing ALS subgroups for
analysis. Indeed, most researchers design ALS randomized control trials to monitor clinical
outcomes after a 12-18 month period. This relatively short time frame demands recruitment of
patients with fast progressing phenotypes to accurately capture changes in survival during the

18
course of a trial. This challenge of trial design is further magnified by the rarity of ALS (~30,000
patients in the United States) (Kiernan et al 2011), limiting the number of patients available for
recruitment into a randomized control trial.
Another proposed solution to address the challenges of disease heterogeneity is the use of
historical controls as an alternative to randomized, placebo-treated participants. For example, a
multicenter, randomized control trial testing the efficacy of lithium carbonate in improving
survival enrolled 107 unique ALS patients and matched these participants with 249 historical
controls pooled from previous trials (Miller et al 2011). For subpopulation stratification or the
use of historical controls to be effective in a prospective trial design, it is imperative to
understand if clinical disease characteristics change over time, as improvements in standard of
care may confound the ability to accurately match or stratify ALS subpopulations for effective
interpretation of treatment efficacy.
An additional glaring failure of previous ALS clinical trials is the lack of tools to
properly determine drug target engagement, ie.) pharmacodynamics biomarkers, in the CNS.
From 2004 – 2014, researchers performed 23 large, multicenter, randomized clinical trials for
disease-modifying ALS drugs with multiple therapeutic targets (Aggarwal et al 2010; Beghi et al
2013; Berry et al 2013; Chiò et al 2010; Cudkowicz et al 2013; Cudkowicz Merit et al 2006;
Dupuis et al 2012; Gordon et al 2007; Graf et al 2005; Kaufmann et al 2009; Lauria et al 2009;
Meininger et al 2006; Meininger et al 2004; Meininger et al 2009; Milane et al 2009; Miller et al
2007; Mosley et al 2007; Pascuzzi et al 2010; Rosenfeld et al 2008; Shefner et al 2004; Sorenson
et al 2008; Verstraete et al 2012). All trials failed to show a difference in the primary clinical
outcomes of survival or change in the ALS Functional Rating Scale (ALSFRS) compared to
placebo. Further, 22 of the 23 trials lacked a pharmacodynamics biomarker to determine if the

19
experimental drug effectively engaged with its intended pathological target. Equally troubling,
recent phase III randomized control trials whose data supported the approval of Edaravone failed
to analyze the drug’s mechanism of action to scavenge free radicals in the CNS (2017; Abe et al
2017). Failure to identify drug target engagement limits the interpretation of all results in these
trials, as investigators confine interpretation of results to assessment of clinical effects. These
analyses leave an understanding of the underlying human biology with the experimental
treatment unknown. Especially in a disease such as ALS with unknown pathogenesis, it is
essential to understand if new treatments have any biological effects on desired targets to modify
hypotheses in the field and focus new drug development efforts. Despite the progress of
technologies to assay biofluid-based markers of neurodegeneration such as neurofilament light-
chain in plasma and CSF (Bacioglu et al 2016; Gaiani et al 2017; Lu et al 2015) or extracellular
domain P75 in urine (Shepheard et al 2017), no methods currently exist to test objective
biomarkers in ALS patients. Therefore, investigators must strive to include pharmacodynamics
biomarkers in trial design to provide more insight into the relationship between improved clinical
outcomes to effective changes in ALS pathobiology.
Therapeutic Strategies for ALSSOD1
Given the accumulating evidence of mutant SOD1 toxicity to motor neurons, an
attractive therapeutic strategy is to eliminate the presence of misfolded SOD1 protein species
within the CNS. The predominant strategies under investigation for targeting misfolded SOD1
protein in neurons include: 1) promoting clearance of misfolded SOD1 protein through the
ubiquitin-proteasome system and autophagy degradation pathways, 2) enhancing the activity of
SOD1 molecular chaperones to prevent protein misfolding, and 3) lowering protein synthesis of
SOD1 within cells. Despite studies demonstrating improved survival in preclinical disease

20
models using these therapeutic strategies, key limitations inhibited translation of these
approaches to successful clinical trials in human ALS patients.
Cells clear mutant SOD1 through the ubiquitin-proteasome system (UPS) and autophagy
pathways (Kabuta et al 2006). These pathways provide investigators with multiple targets to test
the hypothesis that enhanced mutant SOD1 protein clearance will lead to improved disease
outcomes. Indeed, researchers identified multiple E3 ligases that specifically interact with mutant
SOD1 to promote protein clearance within motor neurons. Overexpression of the E3 ligases
Dorfin and NEDL1 led to reduced motor neuron toxicity by mutant SOD1 in vitro and increased
overall survival in transgenic animal models (Miyazaki et al 2004; Niwa et al 2002; Sone et al
2010). Additionally, treatment with small molecules that increase proteasome activity (Lee et al
2010) and mTOR-dependent autophagy (Castillo et al 2013; Gomes et al 2010) enhance the
clearance of misfolded proteins and reduce neuronal toxicity in cell culture.
However, conflicting studies cast doubt on the feasibility of targeting the UPS and
autophagy pathways in vivo. In the case of autophagy, treatment with rapamycin led to an
exacerbation of motor neuron degeneration in SOD1G93A
transgenic mice (Zhang et al 2011).
Similarly, treatment with another small molecule, trehalose, to enhance autophagy in SOD1G85R
transgenic mice saw improvements in overall survival but did not alter disease onset. These
results suggest authophagy enhancement is not sufficient to ameliorate all motor neuron toxicity
caused by mutant SOD1. Moreover, the importance of ubiquitin in synaptic maintenance (Wilson
et al 2002) suggests that enhancing proteasome activity may result in detrimental effects to
neurons. Therefore, despite an understanding of the role that these pathways play in the clearance
of mutant protein from cells, the complexities of these molecular pathways in vivo have hindered
the development of an effective therapeutic strategy targeting protein clearance mechanisms.

21
Similar challenges are seen in the development of strategies to prevent protein misfolding
by enhancing the unfolded protein response (UPR) and molecular chaperones associated with the
heat shock proteins (Lindquist 1986). Researchers hypothesized that these target pathways would
enhance the stability of mutant SOD1 and prevent toxic protein misfolding and aggregation for
efficient protein clearance. Researchers observed initial promise when SOD1G93A
transgenic
mice treated with arimoclomol, a co-inducer of heat shock protein expression through the
activation of heat shock factor-1, resulted in increased survival (Kieran et al 2004), leading to a
randomized control trial to test arimoclomol in ALS patients (Benatar et al 2018). Also, Nagy
and colleagues demonstrated that transgenic overexpression of the chaperone HSP110 improves
survival in SOD1G85R
transgenic mice without altering SOD1 mRNA or protein levels (Nagy et
al 2016). However, activation of other heat shock proteins do not exhibit a similar response, as
overexpression of HSP70 in SOD1G93A
transgenic mice is not sufficient to reduce mutant SOD1
protein toxicity (Liu et al 2005). Moreover, inhibition of HSP90 by a small molecule showed
opposite effects in lowering misfolded protein toxicity in vitro and in vivo (Cha et al 2014),
providing insight into the tight regulation of the HSP protein family in vivo. Therefore, these
conflicting data highlight that although HSP chaperones and regulators of cellular stress response
pathways are an attractive target for therapeutic intervention, complications of specificity,
sustained activation, and compensatory mechanisms of different chaperones result in the absence
of effective drug candidates for treatment of ALS.
The complications associated with targeting the maintenance of SOD1 led researchers to
test a therapeutic intervention further upstream of proteostasis: the production of new SOD1
protein. Because most SOD1 mutations are caused by a single missense mutation, current
technologies are limited in their ability to specifically target the mutant allele. However, the

22
observation that genetic deletion of the SOD1 gene is generally well-tolerated in vivo (Reaume et
al 1996; Shefner et al 1999) suggests that therapeutics lowering global SOD1 synthesis is
feasible. However, identification of small molecule therapeutics that lower SOD1 transcription
remains elusive, as Wright and colleagues failed to identify any small molecules that alter SOD1
promoter activity from a screen of 1,040 FDA-approved compounds (Wright et al 2010).
Further downstream from transcriptional control, another attractive strategy includes
RNAi-based SOD1 gene silencing. Two parallel studies initially demonstrated the therapeutic
efficacy of RNAi-based approaches in vivo by transducing SOD1G93A
transgenic mice with
lentiviral vectors expressing short-hairpin RNA (shRNA) targeting SOD1 mRNA transcripts.
Ralph and colleagues demonstrated that intramuscular lentiviral injection of SOD1 shRNA
delayed disease onset by 100% and increased survival by more than 80% (Ralph et al 2005b).
Concurrently, Raoul and colleagues showed increases in overall survival of 20% compared to
untreated animals after intraspinal lentivirus injection (Raoul et al 2005). In both instances,
improvements in survival were the result of delaying motor neuron degeneration in the CNS, as
Miller and colleagues showed that restricted SOD1 mRNA silencing in muscle is not sufficient
to improve animal survival (Miller et al 2006). Further advancements in the delivery of SOD1
shRNA by adenoviral vector AAV9 showed improvements in delayed disease onset and overall
survival through peripheral administration (Foust et al 2008) and injection into the motor cortex
(Thomsen et al 2014) of SOD1 transgenic models.
Despite the promise of RNAi-based approaches in preclinical models of ALSSOD1
,
significant traction fails to exist for translation to clinical trials. Lack of progress for a DNA-
based therapeutic strategy is primarily driven by continued safety concerns with viral packaging
in which there is no ability to “turn off” shRNA expression. Additionally, the need for shRNA to

23
be processed by endogenous transcriptional machinery such as Dicer and Argonaute proteins
presents risks of toxicity through competition with endogenous mRNAs for proper RNA
homeostasis. Indeed, Grimm and colleagues showed that high levels of shRNA within liver cells
result in oversaturation of micro-RNA processing machinery, leading to animal fatality (Grimm
et al 2006). Due to these concerns, an opportunity exists for an alternative approach that utilizes
RNAi independent of viral-based approaches that uses alternative cellular machinery. One such
treatment that has gained significant traction is the use of antisense oligonucleotides.
Antisense Oligonucleotides for the Treatment of ALSSOD1
Antisense oligonucleotides (ASOs) are short, single-stranded sequences of DNA
composed of a phosphate backbone and ribose sugar rings (Schoch & Miller 2017). ASOs bind
by standard Watson-Crick base pairing (Watson & Crick 1953) to specific mRNA in the nucleus
and cytoplasm of cells to exert gene modulating effects. The sequence design and chemical
modifications to the phosphate backbone and sugar groups result in multiple potential
mechanisms of action for ASOs, including mRNA transcript degradation by recruitment of
RNase enzymes (DeVos & Miller 2013a), splicing modifications (Schoch et al 2016), stalling of
translation (Bennett & Swayze 2010), or targeting microRNAs (Janssen et al 2013; Koval et al
2013). ASOs present multiple advantages over shRNA technology for RNAi-based therapies.
First, ASOs are readily taken up by tissues, most likely through interaction with high- and low-
binding plasma proteins, internalized into lysosomal or endosomal compartments, and trafficked
within cells (Geary et al 2015). Further, chemical modifications increase resistance of ASOs to
nuclease degradation and eliminate toll-like receptor responses (Henry et al 2000), resulting in
superior pharmacokinetic properties of ASOs without the need for viral packaging. Although
these highly negatively charged DNA-like molecules do not cross the blood brain barrier,

24
empirical data in non-human primates demonstrates that intrathecal delivery of ASOs results in
broad distribution within the CNS (DeVos et al 2017; Kordasiewicz et al 2012; Smith et al
2006). Researchers performed the first clinical trials using ASOs in acute myelogenous
leukemia (Bayever et al 1993) over 25 years ago, and the first market approval for an antisense
therapeutic for the treatment of cytomegalovirus retinitis in patients with immunodeficiency
occurred in 1998 (Marwick). These previous successes highlight the feasibility of using ASOs
for the treatment of human disease. Indeed, the recent approval of ASO treatments for two
neuromuscular diseases: Duchenne muscular dystrophy (Mendell et al 2016) and spinal muscular
atrophy (Finkel et al 2017) further supports the prospects of translating ASO treatments for
neurodegenerative diseases into medical practice.
As with previous studies using shRNA, ALSSOD1
is an ideal candidate on which to test an
ASO therapy. Using ASOs that contain a phosphorothioate DNA backbone designed to recruit
RNase H effectively degrade SOD1 mRNA transcripts (Wu et al 2004). Smith and colleagues
first demonstrated the broad distribution of these SOD1-lowering ASOs in the CNS after
infusion into cerebrospinal fluid (CSF) in rats and rhesus monkeys. ASOs targeting human
SOD1 mRNA resulted in global lowering of SOD1 mRNA in the brain and spinal cord and
improved survival of SOD1G93A
transgenic rats relative to saline-treated animals by ~10% (Smith
et al 2006). Further optimization of SOD1 ASOs for sequence specificity and tolerability led to
increases in overall survival of ~40% with a single, intrathecal bolus injection of SOD1 ASO
compared to artificial CSF-treated animals (McCampbell et al 2018). Excitingly, SOD1 ASOs
administered via intrathecal injection were shown to be safe in a human Phase I clinical trial
(Miller et al 2013). This pioneering work in the use of ASOs for ALSSOD1
has led to the
development of ASOs for the treatment of other neurodegenerative diseases such as Alzheimer’s

25
Disease (DeVos et al 2017), Huntington’s Disease (Kordasiewicz et al 2012), and Parkinson’s
Disease (Zhao et al 2017b) that are being pursued in human randomized control clinical trials.
Despite these promising data, key challenges in clinical trial execution remain for successful
translation of SOD1 ASOs as effective therapeutics for SOD1ALS
.
Considerations for ASO Clinical Trial Design in ALSSOD1
Because of the unresolved role of SOD1 outside of ALSSOD1
, current ASO clinical trials
limit patient population recruitment to those patients with known SOD1 mutations. This
population only accounts for ~2% of all ALS cases, drastically lowering the number of subjects
available. As previously discussed, the ALSSOD1
population displays heterogeneity in clinical
phenotypes dependent upon each mutation. The mean disease duration of ALSSOD1
is 3.9 ± 5.5
years (Cudkowicz et al 1997; Juneja et al 1997), ranging from rapidly progressing disease with a
mean disease duration of ~1 year such as A4V (Rosen et al 1994), to slowly progressing disease
where patients live 20+ years after diagnosis such as I104F (Abe 1997) and I151T (Kostrzewa et
al 1996). A4V is a mutation of particular interest, as it accounts for ~50% of North American
ALSSOD1
with little variability in disease progression (Juneja et al 1997). With a high percentage
of rapidly progressing patients and a rare disease population, an SOD1 ASO clinical trial may
benefit from stratification and the use of historical controls. However, multicenter ALSSOD1
natural history studies past the year 2000 are largely absent, as most current research focuses on
characterization of newly identified SOD1 mutations. Thus, recent changes to ALSSOD1
prognosis
are not well understood. It is possible that advancements in ALS standard of care have improved
ALSSOD1
patient outcomes. Supporting this claim, a retrospective analysis of ALS patients cared
for from 1984 – 1999 and 1999 – 2004 suggests improvement to contemporary patient standard
of care in the 21st century (Czaplinski et al 2006b). Therefore, it is important to understand if

26
revised disease management results in changes to ALSSOD1
epidemiology since initial
characterization of SOD1 mutations in the late 1990s for the optimization of current SOD1 ASO
clinical trial design.
In contrast with previous trials, targeting SOD1 by ASO treatment provides an exciting
opportunity to incorporate pharmacodynamics biomarkers into outcomes measures. Lowering of
SOD1 mRNA and protein in brain and spinal cord leads to subsequent lowering of CSF SOD1
protein levels after ASO treatment in ALS rodent and non-human primate models (McCampbell
et al 2018; Winer et al 2013). As mRNA is difficult to isolate from CSF, it is encouraging to
demonstrate the feasibility of more easily available, protein-based ASO pharmacodynamics
biomarkers. Moreover, these data provided the rationale to evaluate the effects of ASO treatment
on SOD1 protein in CSF as a secondary objective in an ongoing SOD1 ASO Phase I trial
(NCT02623699). However, in a rapidly progressing disease such as ALS, it is imperative to
assess treatment efficacy as quickly as possible to maximize patient outcomes. To that end,
SOD1 protein measurement in CSF may not be the fastest assessment of ASO
pharmacodynamics. Protein concentration is dependent on both the production of new protein,
but also on the clearance of existing protein as determined by protein half-life. SOD1 is a long-
lived protein in the human CNS, with a previously calculated half-life of ~25 days in CSF (Crisp
et al 2015) . Therefore, further potential exists to develop pharmacodynamics biomarkers that
more directly and quickly reflect the mechanism of ASO action to lower target mRNA levels and
inhibit the synthesis of new protein.
Summary of Dissertation
The work in subsequent chapters represents new insights into the translation of
preclinical drug candidates toward new therapies for ALSSOD1
. Chapter 2 describes the largest

27
effort in the last 20 years to study the natural history of ALSSOD1
in North America. In this
retrospective cohort study analyzing 175 ALSSOD1
patients cared for past the year 2000, I found
that mean age of onset and median survival of ALS caused by SOD1 mutations is consistent
across previous natural history studies. Additionally, I demonstrated that the A4V point mutation
remains an aggressive form of disease with a significantly shorter median survival after symptom
onset with lower variability compared to non-A4V mutation carriers. Given that the A4V
mutation is the most common variant in North America, my power analysis suggests that the
design of a randomized control trial with SOD1 A4V carriers to test therapeutic interventions in
ALSSOD1
is possible.
Chapter 3 describes my efforts to characterize SOD1 protein turnover in the CSF of ALS
participants using stable isotope labeling kinetics (SILK) analysis (Bateman et al 2006). We
describe preliminary results to characterize SOD1 protein behavior in the CSF of both ALSSOD1
and sporadic ALS patients. Similar to normal controls, SOD1 is a long-lived protein in the CSF
of ALS participants, with a protein half-life of ~20-25 days. Excitingly, I also show that mutant
SOD1 protein exhibits different behavior than WT SOD1 in the CSF of SOD1 mutant carriers, as
a participant harboring an A4V SOD1 mutation exhibits changes in CSF protein behavior
between mutant and wild-type protein. Interestingly, two participants with different SOD1
mutations (E100K, A89V) show similar CSF SOD1 protein behavior compared to WT SOD1.
These data suggest that, similar to the differences in ALSSOD1
clinical phenotypes by mutation,
different SOD1 mutant proteins may show differential biochemical properties in human CSF.
Chapter 4 highlights the development of a protein-based pharmacodynamics biomarker
for ASO treatment in the CNS. Using a stable isotope labeling method to measure incorporation
of 13
C6-leucine into newly produced proteins after ASO treatment, I show significant lowering of

28
target protein synthesis in CNS tissues and CSF after ASO treatment in transgenic animal
models. Excitingly, we observed lowering of target protein synthesis at an earlier time point than
protein concentration-based measures in tissue and CSF, suggesting that new protein production
may be a more sensitive pharmacodynamics biomarker than protein concentration in clinical
trials. The final chapter will then discuss these results and consider future directions for research
into developing effective treatments for ALS.

29
Chapter 2: Defining SOD1 ALS Natural History to Guide
Therapeutic Clinical Trial Design

30
ABSTRACT
Understanding the natural history of familial amyotrophic lateral sclerosis (ALS) caused
by SOD1 mutations (ALSSOD1
) will provide key information for optimizing clinical trials in this
patient population. In this study, we sought to establish an updated natural history of ALSSOD1
in
a retrospective cohort study from 15 medical centers in North America. We evaluated records
from one hundred seventy-five ALS patients with genetically confirmed SOD1 mutations, cared
for after the year 2000. Age of onset, survival, ALS function rating scale (ALS-FRS) scores, and
respiratory function were analyzed as primary measures. Patients with the A4V (Ala-Val) SOD1
mutation (SOD1A4V
), the largest mutation population in North America with an aggressive
disease progression, were distinguished from other SOD1 mutation patients (SOD1Non-A4V
) for
analysis. Mean age of disease onset was 49.7 ± 12.3 years (mean ± standard deviation) for all
SOD1 patients, with no statistical significance between SOD1A4V
and SOD1Non-A4V
(p=0.72,
Kruskal-Wallis). Total SOD1 patient median survival was 2.7 years. Mean disease duration for
all SOD1 was 4.6 ± 6.0 years and 1.4 ± 0.7 years for SOD1A4V
. SOD1A4V
survival probability
(median survival 1.2 years) was significantly decreased compared to SOD1Non-A4V
(median
survival 6.8 years) (p<0.0001, log-rank). A statistically significant increase in ALS-FRS decline
in SOD1A4V
compared to SOD1Non-A4V
subjects (p=0.02) was observed, as well as a statistically
significant increase in ALS-FVC decline in SOD1A4V
compared to SOD1Non-A4V
(p=0.02).
Similar to previous investigations, SOD1A4V
remains an aggressive, but relatively homogeneous
form of ALS. These SOD1-specific ALS natural history data will be important for the design
and implementation of clinical trials in the ALSSOD1
patient population.

31
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease affecting
both upper and lower motor neurons, resulting in severe weakness and fatal respiratory failure
within 2-5 years. The annual incidence of ALS is 1.7 in 100,000 people (Sorenson et al 2002),
yet the only FDA-approved treatment, riluzole, has minimal effects on disease course(Miller et al
2009).
Inherited or familial ALS (FALS) accounts for roughly 10% of all ALS cases(Byrne et al
2011). The second most common cause of FALS is the gene encoding copper zinc superoxide
dismutase 1 (SOD1), which accounts for 10-20% of FALS and mostly follow an autosomal
dominant inheritance pattern(Rosen et al 1994), although an aspartic acid to alanine (D90A)
point mutation found in the Scandanavian population displays recessive inheritance(Andersen
2001). Multiple efforts (ClinicalTrials.gov NCT00706147)(Gros-Louis et al 2010; Lange et al
2013; Miller et al 2005; Miller et al 2013; Patel et al 2014; Ralph et al 2005a; Raoul et al 2005;
Ray et al 2005; Riboldi et al 2014; Smith et al 2006; Winer et al 2013) have focused on targeted
therapeutic approaches for SOD1-related ALS (ALSSOD1
).
To test the efficacy of SOD1-focused therapeutics, an ALSSOD1
natural history dataset is
required to determine the patient sample size needed to observe the desired effect. Considering
the rare nature of SOD1 mutations, these historical control data may also be used in future
clinical trials provided that updated data show no change in disease measures. Given the
heterogeneity among different SOD1 mutations(Cudkowicz et al 1997), it is important to
clinically stratify mutations in SOD1 according to disease duration and rate of progression. The
A4V (SOD1A4V
) is known for an exceptionally aggressive disease course and relatively low

32
inter-patient clinical variability, and represents about 50% of ALSSOD1
in the United
States(Cudkowicz et al 1997; Rosen et al 1993).
We report the findings from a multicenter retrospective chart review of genetically-
confirmed ALSSOD1
cases cared for after the year 2000 and discuss implications for future
ALSSOD1
clinical trials.

33
METHODS
Data Acquisition
We corresponded with 37 sites within the Northeast ALS Consortium Network (NEALS),
an international, independent, non-profit group of research sites who collaboratively conduct
ALS clinical research. Sites performed a retrospective chart review of their respective ALSSOD1
patients cared for from the year 2000 to present. Data were sent to the coordinating center at the
Washington University in St. Louis and entered into a central database. Information for all
patients was de-identified, and data collection and processing were approved by the local IRB
committees. The following 17 centers sent charts as part of this study: Washington University
School of Medicine, Northwestern University Feinberg School of Medicine, University of
Washington Medical Center, Neurology Clinical Research Institute at Massachusetts General
Hospital, Mayo Clinic Florida, Emory University School of Medicine, Johns Hopkins
University, Wake Forest School of Medicine, Perelman School of Medicine at the University of
Pennsylvania, Methodist Neurological Institute at The Methodist Hospital, Neurosciences
Institute at Albany Medical Center, University of Utah, Sunnybrook Health Sciences Centre,
University of California Irvine, University of Michigan, University of Kansas Medical Center,
and Cedars-Sinai Medical Center.
Data collection and analysis
All sites received a standardized form with the following items requested: gender, genetic
mutation, month/year of birth, month/year of symptom onset, month/year of death, month/year of
permanent ventilation (if applicable), ALS-Functional Rating Scale (ALS-FRS) at time points
where available, and forced vital capacity (FVC) at time points where available. Symptom onset
was defined as the first reported loss of function, such as limb weakness or dysarthria.

34
Genetic testing was performed by individual institutions at the time of diagnosis, and
sites were asked to only report the specific SOD1 mutation found. Data were collected and
analyzed from individual patients or families of index cases with confirmed SOD1 mutations
only; mutations recorded as “self-reported” were not included in the analysis. All mutations are
reported in this study using the traditional numbering nomenclature, i.e. not counting the first
(ATG) codon. When time of onset could not be narrowed down to more than a range of several
months, e.g. “fall 2011”, the onset was determined to be the midpoint of that range, e.g. “October
2011.” Patients still alive at the time of data collection were reported as such, and their last
recorded FVC or ALS-FRS, whichever came later, was used for survival analyses. ALS-FRS is
understood to be the revised scale (ALSFRS-R), a 12-item questionnaire with the highest,
normal score being 48(Cedarbaum et al 1999). When both sitting and lying FVC readings were
available, sitting values were used. Additional clinical features, such as site of onset and
presence of non-motor symptoms and signs, were not analyzed in this study.
Statistical Analysis
SAS Version 9.3 (Cary, NC) was used to perform all statistical analyses. Descriptive
statistics were provided for gender, age, and disease duration by mutation group (A4V and Non-
A4V) and total patients, respectively. Additionally, mean, standard deviation of age, and disease
duration were calculated for each interested mutation. The primary study endpoints included
ALS-FRS and FVC while secondary endpoint was overall survival (OS). OS was defined as the
date of symptom onset to death from any cause or last follow-up. Kaplan-Meier (KM) curves for
OS were generated that provide unadjusted survival estimates for total sample and between
mutation groups (Non-A4V vs. A4V), respectively. Difference between mutations groups were
determined by log-rank testing.

35
The study sample includes ALS-FRS and FVC measurements for multiple visits. To
account for correlations among repeated measures from the same patient, the longitudinal data
was analyzed using a generalized estimating equation (GEE) model with log link function to
examine the change in ALS-FRS and FVC measurements over the first three time points
recorded for a patient. The autoregressive of first order as working correlation structure was used
and the patients with missing values at any assessment number were excluded from GEE
analysis. The GEE model includes the group indicator, time points, the interaction term between
group and time points. The p-values of the interaction term from type 3 analysis in the GEE
model were estimated to assess whether the measurements across all time points between A4V
and Non-A4V groups were significantly different. The least square mean and standard error of
the mean were provided.

36
RESULTS
Descriptive Features
Applicable data were collected from 15 medical centers. A total of 175 subjects with
confirmed SOD1 mutations were analyzed. Of subjects collected, 63 had SOD1A4V
mutations
(36.4%), the most common mutation observed in this cohort. 112 subjects had SOD1Non-A4V
mutations (63.6%). There was a slight male dominance in this cohort with a male-female ratio of
1.30:1 in all SOD1 subjects, but there was no significant difference in the male-female ratio
between SOD1A4V
(1.33:1) and SOD1Non-A4V
(1.29:1) subjects (p = 0.9089, Chi-Squared testing)
(Table 2.1).
A total of 36 distinct, missense SOD1 mutations were found in this cohort. The most
common SOD1Non-A4V
mutation was I113T (Ile-Thr) in 32 subjects (18.3%), followed by G41D
(Gly-Asp) (11 subjects; 6.4%), and E40K (Glu-Lys) (10 subjects; 5.7%). The frequencies and
clinical characteristics of all subjects included in this study are shown in Table 2.2.

37
SD = Standard Deviation
a. χ
2 test for categorical variable; Kruskal-Wallis test for continuous variable; Log-rank test for overall
survival.
Total SOD1 SOD1A4V
SOD1Non-A4V
p valuea
No. of subjects (%) 175 (100) 63 (36.4) 112 (63.6)
Male-Female Ratio 1.30 : 1 1.33 : 1 1.29 : 1 0.91
Mean age at onset
(yr) ± SD (n)
49.7 ± 12.3 (162) 50.0 ± 12.4 (57) 49.5 ± 12.3 (105) 0.72
Mean disease duration
(yr) ± SD (n)
4.6 ± 6.0
(134)
1.4 ± 0.7
(51)
6.6 ± 7.0
(83)
<0.0001
Median survival (yr) 2.7 1.2 6.8 <0.0001
Table 2.1 Clinical Characteristics of SOD1A4V
versus SOD1Non-A4V
mutation populations in
familial ALS

38
Mutationa
Exon Codon Substitution No. of
Patients (%)
Mean Age at Onset
(yr) ± SD (n)
Mean Durationb
(yr) ± SD (n)
Median
Duration
(yr)
1 4 Ala-Val 63 (36.6) 49.9 ± 12.3 (57) 1.4 ± 0.7 (51) 1.2
4 113 Ile-Thr 32 (18.3) 54.2 ± 10.7 (29) 5.3 ± 4.8 (22) 3.3
2 41 Gly-Asp 11 (6.4) 34.2 ± 7.8 (8) 23.5 ± 14 (4) 21
4 100 Glu-Lys 10 (5.7) 39.6 ± 5.9 (10) 8.9 ± 5.2 (8) 9.1
4 100 Glu-Gly 5 (2.9) 42.6 ± 9.1 (5) 9 ± 6.2 (4) 7.2
4 89 Ala-Val 4 (2.3) 56 ± 10.4 (4) 8 + 1.6 (2) 8
1 4 Ala-Thr 3 (1.7) 50 ± 10.1 (3) 0.8 ± 0.05 (2) 0.8
4 90 Asp-Ala 3 (1.7) 57.7 ± 9.6 (3) 9.7 ± 5.4 (2) 9.7
4 93 Gly-Ala 3 (1.7) 49 ± 12.5 (3) 2.2 ± 0.6 (2) 2.2
5 137 Thr-Ala 3 (1.7) 38.3 ± 9.2 (3) 9.6 ± 7 (3) 6.4
1 14 Val-Gly 3 (1.7) 50.3 ± 21.0 (3) 2.6 ± 1 (2) 2.6
1 6 Cys-Ser 2 (1.2) 54 ± 16.9 (2) 4.1 ± 0.5 (2) 4.1
5 133c Glu-Ala 2 (1.2) 62 ± 2.8 (2) 0.9 ± 0.6 (2) 0.9
2 49 Glu-Lys 2 (1.2) 59 ± 19.8 (2) 6.8 ± 1.9 (2) 6.8
5 141 Gly- --- 2 (1.2) 42.5 ± 2.1 (2) 1.1 ± 0.7 (2) 1.1

39
Mutationa
Exon Codon Substitution No. of
Patients (%)
Mean Age at Onset
(yr) ± SD (n)
Mean Durationb
(yr) ± SD (n)
Median
Duration
(yr)
2 37 Gly-Arg 2 (1.2) 26.5 ± 6.4 (2) 7.8
2 41 Gly-Ala 2 (1.2) 44.5 ± 10.6 (2) 17.3 ± 0 (2) 17.3
4 85 Gly-Arg 2 (1.2) 56.5 ± 9.2 (2) 2.2 ± 0.3 (2) 2.2
5 139 Asn-Lys 2 (1.2) 51 ± 2.8 (2) 1.5 ± 0.7 (2) 18.5
5 148 Val-Gly 2 (1.2) 49.5 ± 0.7 (2) 0.7 ± 0.4 (2) 0.7
1 4c Ala-His 1 (0.6) 50
5 125c Asp-Ala 1 (0.6) 63 1.1
3 76 Asp-Tyr 1 (0.6) 75 3.7
4 108c Gly-Arg 1 (0.6) 67 1.7
1 10c Gly-Ala 1 (0.6) 59 4.8
5 141 Gly-Ala 1 (0.6) 43
2 37 Gly-Val 1 (0.6) 36 4.6
2 41 Gly-Ser 1 (0.6) 67 0.4
4 93 Gly-Ser 1 (0.6) 45
2 43 His-Arg 1 (0.6) 56 0.3
2 46 His-Arg 1 (0.6) 47 23.4

40
Mutationa
Exon Codon Substitution No. of
Patients (%)
Mean Age at Onset
(yr) ± SD (n)
Mean Durationb
(yr) ± SD (n)
Median
Duration
(yr)
5 144 Leu-Phe 1 (0.6) 66 6.2
5 144 Leu-Ser 1 (0.6) 56 7.8
1 14 Val-Met 1 (0.6) 54 4.1
4 87 Val-Met 1 (0.6) 39 8.8
5 134 Thr-Cys 1(0.6) 55
N/A 1 (0.6)
Total 175 d (100%) 162
d 134
d
aThe mutations are ordered by decreasing number of affected patients. Nomenclature corresponds to
amino acid sequence excluding ATG start codon.
bCensored data are included in determination of mean or median duration
cMutations that are not listed in the ALSoD Database as of 5/5/15
dTotal number of subjects with available data
Table 2.2 Disease characteristics by mutation type in SOD1 familial ALS

41
Age of Disease Onset
Age of onset data was provided for 162 (92.5%) total subjects, with data for 57 SOD1A4V
and 105 SOD1Non-A4V
patients available for analysis. The mean age of onset for all subjects was
49.7 ± 12.3 years (mean ± standard deviation). The mean age of onset varied across different
mutations, with G37R (Gly-Arg) and D133A (Asp-Ala) mutations associated with the youngest
and oldest mean ages at onset, respectively (Figure 2.1A). There was no significant difference in
mean age of onset between SOD1A4V
(50.0 ± 12.4 years) and SOD1Non-A4V
(49.5 ± 12.3 years)
subjects (p = 0.72, Kruskal-Wallis) (Table 2.1).
Disease Duration
Disease duration information was available for 134 (76.6%) of total subjects, including
51 SOD1A4V
and 83 SOD1Non-A4V
patients. The mean disease duration was 4.6 ± 6.0 years for all
SOD1 subjects, 1.4 ± 0.7 years for SOD1A4V
subjects, and 6.6 ± 7.0 years for SOD1 Non-A4V
(Table 2.1). Disease duration varied between mutation type, ranging from rapidly progressing
mutations such as A4T (Ala-Thr) (0.8 ± 0.1 years) to slow progressing mutants such as G41D
(Gly-Asp) (23.5 ± 14.0 years) (Figure 2.1B).
Kaplan-Meier survival analysis was performed comparing the survival probabilities from
time of disease onset to death between SOD1A4V
and SOD1Non-A4V
(Figure 2.2). Of 120 subjects
analyzed, 35 were SOD1A4V
and 85 were SOD1Non-A4V
. The median survival for all subjects was
2.7 years, 1.2 years for SOD1A4V
, and 6.8 for SOD1non-A4V
. SOD1A4V
median survival was
significantly shorter than SOD1Non-A4V
by log-rank test (p <0.0001). No SOD1A4V
subjects
survived to four years (Figure 2.2B).

42
(A.) The mean age at onset and (B.) mean disease duration were reported for each mutation
population with a minimum n = 2. There was no statistical significance between all SOD1
patients (n =162), SOD1Non-A4V
patients (n = 105), SOD1A4V
patients (n = 57) (p = 0.7217,
Kruskal-Wallis). (A.) Average age at onset varied amongst mutations, with the SOD1G37R
and
SOD1D133A
mutations associated, respectively, with the youngest and oldest mean ages at onset
among mutations with more than 1 patient. (B.) Average disease duration varied with different
mutations, with SOD1A4T
and SOD1G41D
mutations associated, respectively, with the shortest and
longest disease durations. SOD1A4V
disease duration (n =51) is significantly shorter than
SOD1Non-A4V
disease duration (n = 83) (p < 0.0001, Kruskall-Wallis). Error bars represent
standard deviation.
Figure 2.1 Mean age at onset and disease duration for ALSSOD1

43
Figure 2.2 Decreased survival probability from time of disease onset for SOD1A4V
compared to SOD1Non-A4V
familial ALS
(A.) Plot for survival probability of all SOD1 mutation patients. 112 subjects were analyzed. The
median survival for all SOD1 patients is 2.7 years. The survival probabilities for SOD1 patients
and the corresponding 95% confidence intervals were 0.82 (0.73, 0.88), 0.51 (0.41, 0.60), and
0.43 (0.33, 0.53) at years 1, 3, and 5, respectively, after disease onset. (B.) Plot for survival
probability for SOD1A4V
vs. SOD1Non-A4V
mutation patients. Analysis consisted of 33 SOD1A4V
and 79 SOD1Non-A4V
. The presence of SOD1A4V
mutations was associated with shorter survival
(p < 0.0001, log-rank). The survival probability for SOD1A4V
patients and the corresponding
95% confidence intervals were 0.60 (0.41, 0.75), 0.04 (0.00, 0.15), and 0.00 (n/a, n/a) at years 1,
3 and 5, respectively, after disease onset. No SOD1A4V
patients survived to year 4. The survival
probability of SOD1Non-A4V
patients was 0.91 (0.82, 0.96), 0.70 (0.59, 0.79), and 0.60 (0.48, 0.71)
at 1, 3 and 5 years after disease onset.

44
FRS and FVC
Only three data points were considered for FRS and FVC. 30 SOD1A4V
and 75 SOD1Non-
A4V subjects were used for ALS-FRS and FVC analysis. The least mean and standard error of
ALS-FRS for SOD1A4V
were 37.8 ± 1.4, 30.0 ± 2.0, and 22.3 ± 3.7; while those of SOD1Non-A4V
were 35.9 ± 0.9, 32.9 ± 1.1, and 29.9 ± 1.3 at time points 1, 2, and 3, respectively. A statistically
significant difference was seen in ALS-FRS across three time points in SOD1A4V
compared to
SOD1Non-A4V
subjects (p = 0.02, Figure 2.3A). Additionally, 37 SOD1A4V
and 81 SOD1Non-A4V
subjects were used for FVC analysis. Dates of permanent ventilation were rarely available (less
than 5% of total subjects), most likely due to death without receiving tracheal ventilation, and so
were not included in final analysis. Similarly, a statistically significant increase in ALS-FVC
decline was observed in SOD1A4V
compared to SOD1Non-A4V
(p = 0.02, Figure 2.3B). The least
mean and standard error of FVC for SOD1A4V
were 86.2 ± 4.1, 58.8 ± 5.1, and 50.7 ± 9.8; while
those of SOD1Non-A4V
were 73.2 ± 2.8, 62.2 ± 3.2, and 54.7 ± 4.0 at time points 1, 2, and 3,
respectively.

45
(A.) Least squares mean of ALS-FRS over multiple assessment periods. SOD1A4V
patients
decline from mean ALS-FRS 37.6 (35.1, 40.6) (95% CI) at assessment point 1, 29.9 (26.2, 34.3)
at assessment point 2, to 22.3 (16.1, 31.0) at assessment point 3. SOD1Non-A4V
patients decline
from mean ALS-FRS 35.9 (34.1, 37.8) at assessment point 1, 35.9 (34.1, 37.8) at assessment
point 2, to 29.9 (27.4, 32.2) at assessment point 3.The SOD1A4V
FRS decline is significantly
increased compared to SOD1Non-A4V
(p = 0.0168). (B.) Least squares mean of FVC over multiple
assessment periods. SOD1A4V
patients decline from mean ALS-FRS 86.2 (78.6, 94.5) (95% CI)
at assessment point 1, 58.8 (49.6, 69.8) at assessment point 2, to 50.8 (34.8, 74.0) at assessment
point 3. SOD1Non-A4V
patients decline from mean FVC 74.7 (69.5, 80.4) at assessment point 1,
63.4 (57.5, 69.8) at assessment point 2, to 55.6 (48.4, 64.0) at assessment point 3.The SOD1A4V
FVC decline is significantly increased compared to SOD1Non-A4V
(p = 0.0168). Error bars
represent standard error of the mean (SEM).
Figure 2.3 SOD1A4V
ALS patients exhibit greater decline in ALS-FRS and FVC rates compared
to SOD1Non-A4V
patients

46
DISCUSSION
We collected data from 15 North American centers, studying 175 subjects with
confirmed SOD1 mutations, cared for from the year 2000 to present. The number of patients
available for this study represent the largest effort to study ALSSOD1
natural history in almost 20
years. Combining data from multiple studies(Andersen et al 1997a; Andersen et al 2003;
Cudkowicz et al 1998; Cudkowicz et al 1997; Gellera et al 2001; Juneja et al 1997; Rosen et al
1994; Sato et al 2005; Stewart et al 2006) suggests mean disease duration for all SOD1 is 3.9 ±
5.5 years and SOD1A4V
is 1.4 ± 0.9 years. Three of these studies (Cudkowicz et al 1997; Juneja
et al 1997; Rosen et al 1994) represent 92% of the 199 reported patients and use data collected
more than 20 years ago and published in the 1990’s. Our calculated disease duration for all
SOD1 of 4.6 ± 6.0 and SOD1A4V
of 1.4 ± 0.7 years indicates no significant change in the natural
history of the disease since these historical data were collected. Our results provide updated
survival data that could serve in the design and interpretation of future clinical trials specific for
ALSSOD1
patients.
A total of 36 missense SOD1 mutations were captured, with the four most common
(SOD1A4V
, SOD1I113T
, SOD1G41D
, and SOD1E100K
) accounting for two-thirds of all subjects. Our
data show a smaller percentage (36.4%) of SOD1A4V
among all ALSSOD1
subjects than
previously reported (50%). This may reflect the recent expansion in genetic testing and
improvement of ALS care, leading to increased detection of SOD1 mutations with less
aggressive or typical presentations. Interestingly, male predominance was present, although
mild, in ALSSOD1
subjects as a whole (M:F of 1.3). These findings may reflect potential bias in
the dataset, as they are in contrast to a recent literature review (McCombe & Henderson 2010).
However, ALSSOD1
cohorts published earlier report similar male predominance (Cudkowicz et al

47
1997). Age of onset for all SOD1 patients (49.7 ± 12.3 years) was not statistically different from
SOD1A4V
age of onset (50.0 ± 12.4 years), which might be unexpected due to the aggressive
nature of SOD1A4V
. However, these findings are consistent with previous reports (Cudkowicz et
al 1997) in which onset is similar between SOD1A4V
and all SOD1 patients, while progression is
more rapid in the SOD1A4V
population.
Our results are limited by the design of this study as retrospective. As with all
retrospective studies, inherent biases exist based upon the availability of data collected. Of the
original 37 NEALS medical centers which we corresponded, 17 participated in this study. No
obvious variables such as geographic location or size of center were linked to the 20 medical
centers that did not respond. Correspondingly, data included in the study have come from clinical
centers spanning wide geographical regions and likely capture a significant portion of the
ALSSOD1
population in North America. However, assumptions must be made when compiling
survival data from multiple centers, including consistency of standard of care. Also, given that
our data were drawn from large ALS referral centers, survival and progression rate numbers may
be different than outcomes in the community.
Natural history data is optimally collected prospectively, and in fact a portion of our data
from 2013-2014 was collected as such. Efforts to collect prospective natural history data on
ALSSOD1
, and FALS in general, should be considered in the future and will be better able to
capture real-time clinical information, such as site of onset and presence of extra-motor findings,
in addition to more closely documenting paraclinical endpoints such as ALS-FRS, FVC and
time-to-permanent-ventilation. Despite these limitations, the rarity of ALSSOD1
coupled with the
urgent need for enabling new clinical trials helps justify the use of a retrospective study design.

48
In clinical trials with a rare disease population, such as ALSSOD1
, historical controls may
be implemented to assess outcomes. As recruitment of patients to a trial in a rare disease
population is challenging, the use of historical controls would be valuable to design an
adequately-powered trail. However, it is important to validate if historical data has changed over
time. In contrast to our results focusing on ALSSOD1
, previous natural history studies (Czaplinski
et al 2006b; Qureshi et al 2009) of the ALS population suggest an increase in survival and
conclude this may reflect improvement in respiratory, nutritional and supportive clinical care.
These findings may suggest that, although overall ALS survival has improved over time,
ALSSOD1
may represent a unique ALS subpopulation. Although our data suggests the ALSSOD1
has not changed over time, the use of historical controls in clinical trials is challenging, and key
limitations exist in our data for direct implementation as historical controls. To properly match
patients in trials with historical controls, additional data such as standard of care (riluzole use,
non-invasive ventilation), cognitive status, and site of disease onset must be collected, leading to
enhanced comparisons with a future cohort. Future prospective studies will focus on these
missing data in future prospective natural history studies in ALSSOD1
or other ALS disease
populations. Nevertheless, the data reported regarding updated survival statistics for this
population are one important consideration in the use of historical controls.
The first reported cause of FALS was a mutation in SOD1, and it is now widely accepted
that the pathogenesis of mutations in SOD1 is related to a gain of toxic function of the SOD1
protein. Therefore, global reduction of toxic SOD1 in ALS has been recognized as a possible
therapeutic intervention. Our work with SOD1 antisense oligonucleotides (Crisp et al 2015;
Miller et al 2005; Miller et al 2013; Smith et al 2006; Winer et al 2013) and others
(ClinicalTrials.gov NCT00706147, (Gros-Louis et al 2010; Lange et al 2013; Patel et al 2014;

49
Ralph et al 2005a; Raoul et al 2005; Ray et al 2005; Riboldi et al 2014) have focused on targeted
SOD1-lowering therapeutics approaches for ALSSOD1
. Given the heterogeneity of disease
progression in different mutation carriers, one may envision the design of a trial that stratifies
results from patients with rapidly progressing disease as defined by unchanged natural history
data. For example, our analysis of the SOD1A4V
patient population in this study suggests that
SOD1A4V
disease characteristics are homogeneous, and natural history has remained unchanged
over time with a median survival of 1.2 years. As SOD1A4V
is the most common mutation found
in North America, recruitment for a SOD1A4V
-focused trial for therapeutic efficacy may be
feasible. Assuming an accrual interval of 1 year and additional follow-up interval of 2 years, 52
SOD1A4V
subjects per group (new treatment and placebo) are needed to achieve at least 80%
power at a 0.05 significance level to detect the statistical difference when the median survival in
the treatment group is 2.4 years using two-sided log-rank test, where hazard ratio of new
treatment to control is 0.5. In contrast, our findings suggest that the median survival in all SOD1
subjects is 2.7 years, and thus 88 SOD1 subjects per group (new treatment and placebo) are
needed when the median survival probability in the treatment group is 5.4 years, where hazard
ratio of new treatment to control is 0.5. Although the effect sizes discussed here are large, we
believe such a result is feasible, as SOD1 antisense oligonucleotides directly inhibit the cause of
motor neuron toxicity and death in ALSSOD1
. The prior Phase I trial using SOD1 antisense
oligonucleotides (Miller et al 2013) in this same population recruited 21 unique patients,
suggesting that such a trial is feasible.
Overall, the data presented here will help guide clinical trial design and may serve as a
historical control database for ALSSOD1
interventional trials, with the hope of demonstrating that
one of the upcoming therapies extends survival.

50
ACKNOWLEDGEMENTS
Funding provided by Seattle VA Medical Center, Department of Veterans Affairs
research funds (T.D.B.) for collection of SOD1 data at University of Washington; Amyotrophic
Lateral Sclerosis Association (E.R.) for collection of SOD1 clinical information at MGH;
Harvard NeuroDiscovery Center (E.R.) for collection of SOD1 clinical information at
Massachusetts General Hospital; the Dr. Anne B. Young Neuroscience Translational Medicine
Fellowship (Massachusetts General Hospital Neurology and Biogen Idec) (E.R.) for collection of
SOD1 clinical information at Massachusetts General Hospital; Muscular Dystrophy Association
(T.M.M.) for design and conduct of the study, collection of clinical information, data
management and analysis, interpretation of the data, and preparation and review of the
manuscript; NIH/NINDS R25NS065743 (E.R.) for collection of SOD1 clinical information at
Massachusetts General Hospital, R01NS078398 (T.M.M.) for salary support to T.M.M. during
the conduct of the study, U01NS084970 (T.M.M.) for salary support to T.M.M. during the
conduct of the study.

51
Chapter 3: Characterizing SOD1 Protein Kinetics in the
Cerebrospinal Fluid of Individuals with ALS

52
INTRODUCTION
Autosomal dominant mutations in SOD1 were the first reported genetic cause of ALS
(Rosen et al 1993). Accumulating evidence suggests that a gain of toxicity function associated
with the pathological misfolding of mutant SOD1 protein results in the death of motor neurons
(Ilieva et al 2009). In vitro characterization of mutant, recombinant SOD1 supports this
hypothesis, as data suggest that the altered stability of mutant SOD1 proteins correlates with
faster disease progression (Pratt et al 2014; Shi et al 2016). However, the ability to effectively
analyze pathological SOD1 behavior in the central nervous system (CNS) of living, human
patients remains elusive, limiting the ability to confirm the biological relevance of in vitro
observations to ALS prognosis.
A key biochemical characteristic of mutant SOD1 protein cell culture (Borchelt et al
1994) and in vivo (Farr et al 2011) is accelerated turnover caused by protein instability compared
to wild-type (WT) SOD1. Our group also demonstrated that mutant G93A SOD1 protein exhibits
faster protein turnover, indicated by a shortened protein half-life, compared with WT SOD1
protein in the CNS of transgenic rat models. Moreover, misfolded G93A SOD1 protein displayed
a shorter half-life than the total G93A SOD1 protein pool. These observed effects in the brain
and spinal cord were mirrored in the CSF of these animals, suggesting that measures of protein
turnover within CSF are a sufficient proxy for CNS protein biology (Crisp et al 2015). Therefore,
differences in mutant and WT SOD1 half-life in CSF may provide insight into the identification
of misfolded SOD1 within the CNS of ALS patients for the first time.

53
Outside of ALSSOD1
, the role of SOD1 in sporadic ALS is controversial. In post-mortem
studies, multiple groups reported misfolded SOD1 in sporadic ALS spinal cord autopsies (Bosco
et al 2010; Forsberg et al 2010). However, others are unable to reproduce these results (Da Cruz
et al 2017). These findings have direct therapeutic implications, as SOD1-targeting therapies are
currently under development for the treatment of ALSSOD1
(Benatar et al 2018; Miller et al 2013),
but only ~2% of the ALS population carry SOD1 mutations. Further data that supports a
biological role for pathological, misfolded SOD1 in sporadic ALS would be an important finding
to broaden the utility of targeting SOD1 for therapeutic intervention in the 90% of ALS patients
with no genetic causes (Kiernan et al 2011).
We previously calculated the half-life of CSF SOD1 in normal, health human subjects as
~25 days using stable isotope labeling kinetics (SILK) analysis via tandem liquid
chromatography-mass spectrometry (LC-MS/MS) (Crisp et al 2015). In these studies, we
administered 13
C6-Leucine via the drinking water of participants fed a low leucine diet. However,
feedback from healthy control participants suggested dissatisfaction with this method of labeling.
Considering ALS patients already exhibit a compromised quality of life, it is imperative to
design clinical studies that ease the burden on the participant population, while still capturing
accurate measures protein turnover.
Here, we performed a preliminary analysis of an ongoing, multicenter study
(NCT03449212) to determine SOD1 protein half-life in ALS patients. In this study, we wish to
test the hypothesis that CSF SOD1 kinetics is altered in ALS by comparing: 1. Total SOD1
protein turnover between ALS participants and healthy controls, and 2. Mutant versus WT SOD1
protein turnover within ALS participants with SOD1 mutations. To improve participant
satisfaction, we employed a modified protocol to deliver 13
C6-Leucine via intravenous infusion

54
to patients over a 16 hour labeling period, and compared results to our previous 10 day oral
labeling paradigm data. In addition to understanding ALS pathobiology, calculating CSF SOD1
half-life will also be a key parameter to model pharmacokinetic-pharmacodynamics (PK-PD)
relationships of experimental SOD1-targeting therapies currently in clinical trials for the
treatment of ALSSOD1
.
METHODS
Human Subjects
This study involving human subjects was approved by the Washington University Human
Studies Committee and the Clinical Research Unit (CRU) Advisory Committee (an Institute of
Clinical and Translational Sciences (ICTS) Resource Unit) and the Institutional Review Board of
the Partners Human Research Committee at Massachusetts General Hospital. Written informed
consent was obtained from all participants prior to inclusion in the study.
The study population consists of subjects with: known SOD1 positive ALS status, SOD1
positive carriers without ALS, known C9ORF72 positive ALS status, C9ORF72 positive carriers
without ALS, sporadic ALS, and non-ALS (normal controls). Inclusion criteria includes (with
exceptions for each subgroup): males or females aged 18 years or older, positive for SOD1
mutation, positive for C9ORF72 repeat expansion, diagnosis of definite, probable, or possible
ALS in accordance with El Escorial Criteria (ALS only) (Westeneng et al 2018), able to hold
position and breathe comfortably for duration of lumbar puncture procedure. Exclusion criteria
for all groups include: invasive ventilator dependence, medical inability to undergo lumbar
puncture, any active dermatologic disease, any connective tissue disease, any known or
suspected abnormal CSF pressure or intracranial/intraspinal tumors, use of anticoagulant
medication that cannot be safely withheld until coagulation parameters have normalized prior to

55
lumbar puncture and for up to a week following the lumbar puncture, blood dyscrasia, abnormal
bleeding diathesis, the use of dialysis for renal failure, safety lab values greater than 2X the
upper limit of normal, allergy to lidocaine, pregnancy, or any contradiction for lumbar puncture.
13C6-Leucine Administration and Sample Collection
Participants were admitted to the Clinical Research Unit (Center for Applied Research
Sciences, Washington University), received a low-leucine diet (Washington University Research
Kitchen), and administered an intravenous infusion of 10 g 13
C6-leucine (Cambridge Isotope
Laboratories CLM-2262, Cambridge, MA) at a rate between 4 – 10 mg/kg/hr dependent on the
participant’s weight for 16 hours. Venous blood draws were collected at designated time points
during the infusion period. CSF, venous blood, and urine were collected over five follow up
lumbar punctures (LPs) from day 8 after infusion through day 120.
Participants from Massachusetts General Hospital and select participants from
Washington University received 10 g of 13
C6-Leucine via the oral labeling method described
previously(Crisp et al 2015). Briefly, participants consumed 13
C6-leucine after dissolving 330 mg
13C6-leucine in 120 mL of tap water + Kool-Aid® flavored powder three times per day (total
daily dose = 990 mg) for 10 days. Overnight fasting blood was collected at day 1, 4, 8, and 10 of
the 13
C6-leucine labeling period. CSF via lumbar puncture and venous samples were collected at
four designated time points between 14-85 days after 13
C6-leucine labeling was initiated.
Blood was centrifuged at 1000xg for 10 minutes at room temperature, and 500 uL plasma
was aliquoted into low-binding 1.7 mL tubes (Axygen, MCT-175-L-C) and frozen on dry ice.
CSF (20-25 mL) was centrifuged at 1000xg for 10 minutes at 4°C, and 1 mL aliquots were

56
frozen on dry ice. Urine (10 mL) was divided into 1 mL aliquots and frozen on dry ice. All
samples were stored at -80°C before use.
Isolation and mass spectrometric analysis of CSF SOD1, free 13
C6-Leucine in CSF and Plasma,
and 13
C6-Leucine labeled total protein
SOD1 was immunoprecipitated from 1 mL of CSF using 50 uL of M-270 Epoxy
Dynabeads (Invitrogen, Carlsbad, CA) coupled to anti-SOD1 (mouse monoclonal; Millipore
Sigma S2147, Burlington, MA) supplemented with 0.1% Tween-20 and protease inhibitors for 2
hours at room temperature. Before immunoprecipitation, 100 ng of N15-labeled recombinant
SOD1 (ProSpec-Tany Technogene, Rehovot, Israel) was added to each sample. In SOD1 mutant
carriers, an additional 50 ng of mutant recombinant SOD1 was added to each sample. Protein
samples were washed with 25 mM ammonium bicarbonate, reduced with DL-Dithiothreitol in
100 mM triethylammonium bicarbonate for 30 minutes at 37°C, alkylated with iodoacetamide
for 30 minutes at RT in the dark, and washed two times with 25 mM ammonium bicarbonate.
SOD1 was eluted from magnetic beads using 50 uL of formic acid. The formic acid eluent was
transferred to a new tube and lyophilized via speed vacuum (Labconco, CentriVap), washed with
100 uL methanol, and lyophilized again via speed vacuum. Dry samples were resuspended in 20
uL of 50 mM triethylammounium bicarbonate, pH 8.8.
Proteolytic digestion was performed on resuspended samples with either 600 ng of
Endoproteinase GluC (Millipore Sigma 11047817001) at 25°C for 16 hours, or a combination
digest with 250 ng LysC (Millipore Sigma 11047825001) at 37°C for 6 hours followed by 250
ng Trypsin (Millipore Sigma 11418475001) at 37°C for 12 hours. Samples were desalted and
washed with TopTipC18 10-200 uL Tip-Columns (Glygen TT2C18.96, Columbia, MD),

57
lyophilized via speed vacuum, and resuspended in 25 uL of 2% acetonitrile/0.1% formic acid
prior to liquid-chromatography-triple quadrupole-mass spectrometry analysis (Xevo, Waters).
The 13
C6-Leucine/12
C6-Leucine tracer-to-tracee ratio (TTR) of SOD1 peptides was
quantified by comparing the area under the curves for each peptide in the presence or absence of
13C6-Leucine. SOD1 TTR measurements were calibrated using a
13C6-leucine labeled SOD1
standard curve derived from HEK cell lysates, as described previously (Crisp et al 2015). SOD1
protein concentration was quantified by comparing the area under the curve of CSF SOD1
peptide to the recombinant, N15-labeled peptide (N14-SOD1:N15-SOD1), and calibrated to a
recombinant SOD1 protein standard curve. See Supplementary Tables 1 and 2 for a summary of
peptides analyzed.
Plasma free, plasma protein-bound, and CSF protein-bound 13
C6-leucine was determined
by precipitation of proteins with 10% tricholoroacetic acid overnight at 4°C, followed by
capillary chromatography-negative chemical ionization-quadrupole-mass spectrometry (Agilent
6890N Gas Chromatograph and Agilent 5973N Mass Selective Detector) as described previously
(Potter et al 2013).
Compartmental Modeling
Modeling was conducted using SAAM II (The Epsilon Group, Charlottesville, NC).
Kinetic data for plasma free 13
C6-leucine, CSF total protein, plasma total protein, and CSF SOD1
species for each subject were incorporated into a compartmental model that uses the shape of the
plasma leucine enrichment time course as a “front end” to describe the enrichment time course
shapes for the product proteins (Supplemental Figure 1). Each protein (CSF SOD1, CSF total
protein, and plasma total protein) is assumed to be at steady state over the ~120 day time course.

58
The total rate of production of each protein was thus assumed to be constant over time, but the
rate at which isotopically labeled protein is synthesized was dependent on the mole fraction of
isotopically labeled plasma leucine monitored over the ~120 day period. The model describes the
turnover of each protein as unlabeled protein is initially replaced with labeled protein, and then
labeled protein is cleared and replaced with unlabeled protein. It was necessary to assume that
each protein was kinetically heterogeneous, consisting of a mixture of faster and slower turning
over components, in order to optimally fit the shapes of each protein enrichment time course. In
addition, the slower component of SOD1 was optimally described as a minimal 2-compartment
delay chain, i.e. two compartments in series. All CSF SOD1 peptides analyzed were used as
individual replicates at each time point for modeling. All TTR values were converted to mole-
fraction label (MFL) for modeling using the following equation:
Modeling results are reported as fractional turnover rates (FTR, pools/d; the fraction of the pool
turning over per day) and an estimate of the error (reported as a coefficient of variation) for each.
Protein half-life was then calculated by the following equation:
Statistical Analysis
All quantitative results are expressed as mean ± standard deviation. Due to low sample
sizes within each subgroup examined (n = 2-3), no formal statistical tests were performed.

59
RESULTS
Participant Demographics
To date, 12 individual participants initiated the protocol (Table 3.1). Of these, 1
participant withdrew from the study due to complications associated with the lumbar puncture
procedure, resulting in a current study completion rate of over 90% (11/12). The mean
participant age was 54.5 ± 9.5 years, and the majority of participants were male (75%, 9/12) and
Caucasian (92%, 11/12). Of the 11 participants analyzed, 1 received the 10 day oral labeling
protocol of 13
C6-Leucine, while the remaining received a 16 hour IV infusion of 13
C6-Leucine.
Protein Kinetics in Participants via 16 hour, 13
C6-Leucine intravenous infusion
We employed a labeling protocol where we administered 10g of 13
C6-Leucine via IV
infusion over a 16-h time period, with subsequent blood draws up to 24 hours from label
administration. An example of the tracer kinetics observed for participants with this protocol is
seen in Figure 3.1. Over the five time points between 7 – 120 days when we collected CSF,
maximum 13
C6-Leucine enrichment in CSF total protein and SOD1 approaches ~1% and decays
over time (Figure 3.1B). Calculations of CSF SOD1 half-life in normal, healthy controls by
compartmental modeling confirmed CSF SOD1 is a long-lived protein, with half-life on the
order of weeks. This is similar to our previous calculations with the 10 day oral labeling method
(Figure 3.1B, Table 3.2). We also calculated the half-life of CSF total protein on order of
weeks, which is similar to previous measures of the predominant protein found in human CSF-
human serum albumin (half-life of 15-20 days) (Iwao et al 2006). Taken together, these data
validate the utility of our 16 h- IV infusion protocol to assay CSF protein kinetics.

60
Participant Disease Type Disease Stage Labeling
Method Age Race Gender
Control 1
16 hour IV 60 Caucasian Female
Control 2
16 hour IV 51 Caucasian Male
Control 3
16 hour IV 43 African
American Female
ALS 1 SOD1 (A89V) Symptomatic 10 day oral 48 Caucasian Male
ALS 2 SOD1 (E100K) Symptomatic 16 hour IV 49 Caucasian Male
ALS 3 SOD1 (A4V) Asymptomatic 16 hour IV 52 Caucasian Male
ALS 4* SOD1 (A4V) Asymptomatic 10 day oral 37 Caucasian Male
ALS 5 Sporadic Symptomatic 16 hour IV 64 Caucasian Male
ALS 6 Sporadic Symptomatic 16 hour IV 62 Caucasian Male
ALS 7 Sporadic Symptomatic 16 hour IV 56 Caucasian Male
ALS 8 C9ORF72 Symptomatic 16 hour IV 69 Caucasian Female
ALS 9 C9ORF72 Asymptomatic 16 hour IV 63 Caucasian Male
* Participant withdrew from study after first lumbar puncture
Table 3.1 Participant Demographics

61
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0
0 .0
T im e (d a y s )
Mo
le F
ra
cti
on
La
be
l (M
FL
)
0 .10
0 .20
0 .30
0 .40
0 .50
P la s m a F re e L e u c in e
P la s m a T o ta l P ro te in
1 2 0
T im e (d a y s )
Mo
le F
ra
cti
on
La
be
l (M
FL
)
7 14 31 63
0 .005
0 .010
0 .015
0 .030
0 .045 C S F S O D 1 : t1 /2 = 1 7 .1 d a y s
C S F T o ta l P ro te in : t1 /2 = 1 6 .4 d a y s
P la s m a F re e L e u c in e
P la s m a T o ta l P ro te in : t1 /2 = 4 .1 d a y s
1
A .
B .
Figure 3.1. Representative Kinetic Curve of a participant labeled with 10 g 13
C6-Leucine
via 16 hour intravenous infusion.
(A.) 13
C6-Leucine plasma enrichment during the first 24 hours of procedure. Peak 13
C6-Leucine
enrichment in plasma free leucine is observed at end of 16 hour labeling period near 40% of total
leucine pool, with rapid clearance of 13
C6-leucine maximum incorporation into plasma protein
observed within the first 8 hours of the chase period. (B.) 13
C6-Leucine enrichment in CSF and
plasma proteins over time. Representative protein half-life values (t1/2) calculated from
compartmental modeling corresponds to participant Control 1.

62
Total CSF SOD1 Behavior in ALS
To analyze total CSF SOD1 protein behavior in ALS, we categorized participants into
four groups: SOD1 mutation carriers, sporadic ALS, C9ORF72 repeat expansion carriers, and
healthy controls (Table 3.2). In addition to ALSSOD1
and sporadic ALS groups, analyzing CSF
SOD1 protein in the C9ORF72 group serves as a negative control to test for specificity of altered
SOD1 behavior, as SOD1 pathology is not implicated in the C9ORF72 disease population.
Finally, the healthy control group serves as a control to identify differences in SOD1 protein
behavior due to disease pathology. At this time, the sample sizes of these individual subgroup are
not sufficient to determine any statistically significant differences between the groups (n = 2 – 3
per group).
Normal control subjects had an average CSF SOD1 protein concentration of 164.7 11.2
ng/mL, with an average CSF SOD1 protein half-life of 20.6 8.5 days. The ALS groups (SOD1,
sporadic, and C9ORF72) appear to show elevated CSF SOD1 concentration compared with
normal controls, ranging from 191.0 6.5 ng/mL (C9ORF72) to 270.5 62.5 ng/mL (SOD1).
Moreover, average CSF SOD1 half-life in ALS groups ranged between 25.0 7.1 (sporadic) to –
29.3 20.2 days (SOD1). Due to the variable nature and small sample size in these groups, no
we cannot draw definitive conclusions that suggest there are clear changes in in total CSF SOD1
protein turnover in ALS versus controls.
.
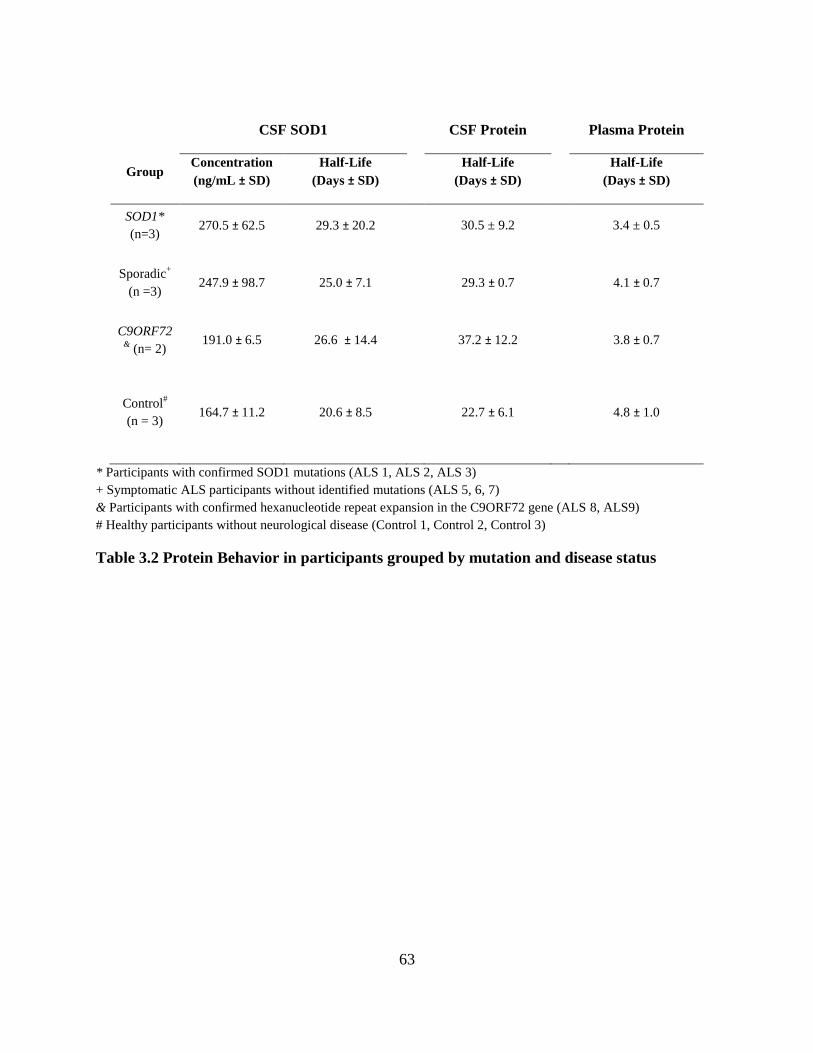
63
CSF SOD1 CSF Protein Plasma Protein
Group Concentration
(ng/mL ± SD)
Half-Life
(Days ± SD)
Half-Life
(Days ± SD)
Half-Life
(Days ± SD)
SOD1*
(n=3) 270.5 ± 62.5 29.3 ± 20.2 30.5 ± 9.2 3.4 ± 0.5
Sporadic+
(n =3) 247.9 ± 98.7 25.0 ± 7.1 29.3 ± 0.7 4.1 ± 0.7
C9ORF72& (n= 2)
191.0 ± 6.5 26.6 ± 14.4 37.2 ± 12.2 3.8 ± 0.7
Control#
(n = 3) 164.7 ± 11.2 20.6 ± 8.5 22.7 ± 6.1 4.8 ± 1.0
* Participants with confirmed SOD1 mutations (ALS 1, ALS 2, ALS 3)
+ Symptomatic ALS participants without identified mutations (ALS 5, 6, 7)
& Participants with confirmed hexanucleotide repeat expansion in the C9ORF72 gene (ALS 8, ALS9)
# Healthy participants without neurological disease (Control 1, Control 2, Control 3)
Table 3.2 Protein Behavior in participants grouped by mutation and disease status

64
Mutant versus Wild-Type CSF SOD1 in SOD1 Mutation Carriers
Previous efforts to characterize CSF SOD1 protein concentration and turnover analyzed
the entire SOD1 protein pool. However, three participants had genetically confirmed point
mutations in the SOD1 gene with a familial history of disease (Table 3.1). These individuals
carry one copy of the mutant SOD1 gene, and one copy of WT SOD1, and an advantage of our
mass spectrometry-based SILK analysis is the ability to distinguish these two proteins within
CSF. Thus, we are able to test hypotheses related to differential behavior in both forms of the
protein with regards to SOD1 protein concentration and half-life. Two participants were
previously diagnosed with ALS and were similar in age (48 and 49 years old). Interestingly,
natural history data suggests the median disease duration in these two mutant SOD1 carriers is
similar (A89V median survival: 8 years, E100K median survival: 9.1 years, (Bali et al 2017)).
An additional participant carried an A4V mutation, the most common mutation in the North
American population, with rapidly aggressive disease progression (median survival: 1.2 years
(Bali et al 2017)).
Average WT CSF SOD1 protein concentration ranged from 96.4 ng/mL to 214.4 ng/mL
in participants (Table 3.3). In symptomatic participants carrying A89V and E100K mutations,
the concentration of mutant CSF SOD1 protein appeared elevated relative to WT CSF SOD1,
while the half-life of mutant and WT CSF SOD1 were similar within each individual patient.
Contrary to these findings, the asymptomatic participant carrying an A4V mutation showed a ~9-
fold decrease in mutant CSF SOD1 concentration. Due to limitations in instrument sensitivity,
SOD1 A4V half-life could not be calculated. These discrepancies appear to be independent from
global changes in protein metabolism, as the plasma total protein half-life is similar between all

65
patients (Table 3.3). Thus, these data suggest that different SOD1 mutations may exhibit
differential protein behavior within human CSF.

66
CSF WT SOD1 CSF Mutant SOD1
Participant Mutation
Concentration
(ng/mL)
Half-Life
(Days)
Concentration
(ng/mL)
Half-Life
(Days)
ALS 1 A89V 137.7 26.3 205 25.7
ALS 2 E100K 96.4 31.6 135.4 28.8
ALS 3 A4V 214 25.3 23.6 -
Table 3.3 CSF SOD1 protein in participants with SOD1 mutations

67
DISCUSSION
These data provide the first insight into SOD1 protein kinetics in the central nervous system
of patients with ALS. Although limited in overall interpretation due to small sample size, this
preliminary analysis provides a foundation for future data interpretation when the remaining 15
currently enrolled and future participants complete this study. To our knowledge, this study is
also the first examination of mutant versus WT protein kinetics in an autosomal dominant
neurological disorder. These findings are valuable to understand disease pathobiology in human
ALS. Further, these data also provide crucial values for parameters in randomized control
clinical trial design to test new for this devastating disease.
Our previous efforts to calculate SOD1 protein half-life in human CSF employed an oral
labeling method with 13
C6-Leucine (Crisp et al 2015). However, the 16-hour infusion protocol
described here provides multiple advantages over an oral labeling paradigm for tracer-based
kinetic analysis. From an analytical perspective, our IV protocol allows for a higher sampling of
plasma leucine time points during the 13
C6-Leucine labeling period, as previous measurements of
13C6-Leucine input into plasma were sampled with 2-3 blood draws over a 10-day period. This
higher sampling frequency limits the variance in tracer input modeling, improving the quality of
fit of the model to data. Our kinetic analysis calculated CSF total protein half-life on the order of
20-30 days, which is relatively similar to the previously half-life of human serum albumin, the
predominant protein found in Human CSF, between 15-20 days (Iwao et al 2006). Importantly,
the 16 hour IV protocol is less burdensome for patients compared to adhering to a strict diet and
self-administered labeling paradigm over a 10 day labeling period. Therefore, controlled
administration of 13
C6-Leucine with IV infusion may limits variability in study compliance and

68
improves the overall participant experience in an already invasive clinical study associated with
multiple lumbar punctures.
Calculations of SOD1 protein half-life are directly applicable to current clinical trials in ALS,
as multiple experimental therapeutics target SOD1 by inhibiting protein synthesis (Lange et al ;
Miller et al 2013) or promoting protein degradation (Benatar et al 2018). For example, Antisense
Oligonucleotides that target SOD1 mRNA for degradation rely on measures of CSF SOD1
protein concentration as a biomarker to assay target engagement of ASO with SOD1 mRNA in
the central nervous system of patients (Winer et al 2013). However, decreases in CSF SOD1
protein concentration also rely on clearance of SOD1 protein that is unaffected by ASO
treatment, and this rate of protein clearance is determined by protein half-life. Therefore, our
calculations of CSF SOD1 half-life as ~25 days here and in previous work (Crisp et al 2015)
establish a critical parameter to understand the relationship between SOD1 ASO treatment and
maximum lowering of CSF SOD1 protein concentration (Figure 3.2).
Our preliminary results show similar values of total SOD1 protein half-life between
participants with SOD1 mutations and controls. However, comparison of mutant versus WT
SOD1 protein in an A4V SOD1 participant suggests that A4V SOD1 protein exhibits altered
protein behavior compared to WT SOD1. An attractive hypothesis to explain this phenomenon is
that SOD1 half-life correlates to the overall stability of the protein, suggesting that alterations in
protein half-life indicate protein misfolding. Indeed, these results have been observed with
different SOD1 mutants in cell culture (Borchelt et al 1994), and measures of mutant SOD1
protein stability in patient red blood cells appear to correlate with rate of disease progression
(Sato et al 2005). Interestingly, the half-life of E100K and A89V SOD1 in two participants
showed similar measures of protein half-life to WT SOD1, suggesting mutant-specific

69
differences in SOD1 kinetics between ALS participants. However, the small sample size of this
experiment, combined with an inability to confirm the misfolded state of CSF SOD1 protein
leaves any interpretation of these results at this stage to be speculative. Our mass spectrometry-
based approach has multiple advantage for future studies, as we are able to interrogate multiple
forms of SOD1 protein within a sample. In addition to distinguishing mutant versus WT SOD1
protein, future investigation should also search for posttranslational modifications of CSF SOD1
that have been implicated in forms of pathological SOD1, such as deamidation (Schmitt & Agar
2017), SUMOylation (Niikura et al 2014), and disulfide reduction (Bosco et al 2010; Nagano et
al 2015; Tiwari & Hayward 2003).
Similar efforts to analyzed CSF SOD1 half-life can be employed to investigate the potential
role of SOD1 in sporadic ALS. Such data will contribute to resolving the controversial
identification of misfolded, WT SOD1 in post-mortem tissue sections (Bosco et al 2010; Da
Cruz et al 2017; Kerman et al 2010) or by CSF ELISA (Zetterstrom et al 2011). An important
consideration in developing experiments to identify changes in CSF SOD1 protein behavior is to
ensure that alterations in CSF SOD1 protein concentration or half-life are specific to sporadic
ALS. For example, Zetterstrom and colleagues failed to distinguish differences in misfolded
SOD1 between ALS patients and normal controls, questioning the validity of their misfolded
SOD1 ELISA. To help control for changes observed in CSF SOD1 protein in sporadic ALS,
future studies will also measure CSF SOD1 concentration and half-life in other neurological
disease states, such as Alzheimer’s Disease and Parkinson’s Disease. These participants will help
to control for any effects observed due to overall neurodegeneration. An additional consideration
is the overall variance in CNS protein metabolism individual subjects. Thus, incorporation of
other CSF protein half-life measures such as microtubule-associated protein tau (Sato et al 2018),

70
0 5 0 1 0 0 1 5 0 2 0 0
0
5 0
1 0 0
T im e (d a y s )
SO
D1
Pro
tein
(%
of
init
ial)
S O D 1 P ro te in : t1 /2 = 2 5 d a y s
S O D 1 A S O C o n c e n tra tio n
T im e to m a x im u m in h ib it io n
1 2 3 d a y s
Figure 3.2 Theoretical modeling of CSF SOD1 protein levels after ASO treatment.
Indirect response model used to simulate SOD1 protein levels after 4 doses of SOD1 antisense
oligonucleotides using a 1 compartment drug model with inhibition of input response model:
Where Kin = 1, Kout = SOD1 half-life (25 days), IC50 = 10, and Cp is the SOD1 ASO
concentration. SOD1 protein concentrations were normalized to % of initial value. Drug (SOD1
Antisense Oligonucleotide) tissue concentrations simulated after 4 bolus doses administered on
Days 0, 28, 56, and 84. Assumed drug t1/2 = 30 days.

71
apolipoprotein E (Wildsmith et al 2012), and alpha synuclein (Fanara et al 2012) will provide
valuable information to distinguish the specificity of SOD1 changes in protein turnover for ALS
participants. These data will provide valuable information for the field to determine the utility of
SOD1-targeting therapeutics in the remaining 98% of ALS patients without SOD1 mutations.
In sum, our work characterizing CSF SOD1 half-life will lead to a better understanding of
ALS biology, while also helping to optimize clinical trial design advance SOD1-targeted
therapies through the therapeutic pipeline for ALS.
ACKNOWLEDGEMENTS
We thank Jennifer Jockel-Balsarotti, Debbie King, Jesse Markway, Amber Malcolm,
Robert Bucelli, Arun Varadhachary, and members of the CRU at Washington University School
of Medicine for their efforts in participant recruitment, lumbar puncture and blood draw
procedures, and clinical management of this study. We also thank Drs. James Bollinger and
Kwasi Mawuenyega of Dr. Randall Bateman’s lab, Drs. Cheryl Frankfater, John Turk, and Bruce
Patterson for continued collaboration in our LC-MS/MS, GC-MS, and kinetic modeling analyses,
respectively. Research reported in this publication was supported by the Washington University
Institute of Clinical and Translational Sciences grant UL1TR002345, sub-award TL1TR002344
from the National Center for Advancing Translational Sciences (NCATS), and the National
Institute for Neurological Disorders and Stroke grant R-01-NS098716-01 (TM Miller, PI) from
the National Institutes of Health.
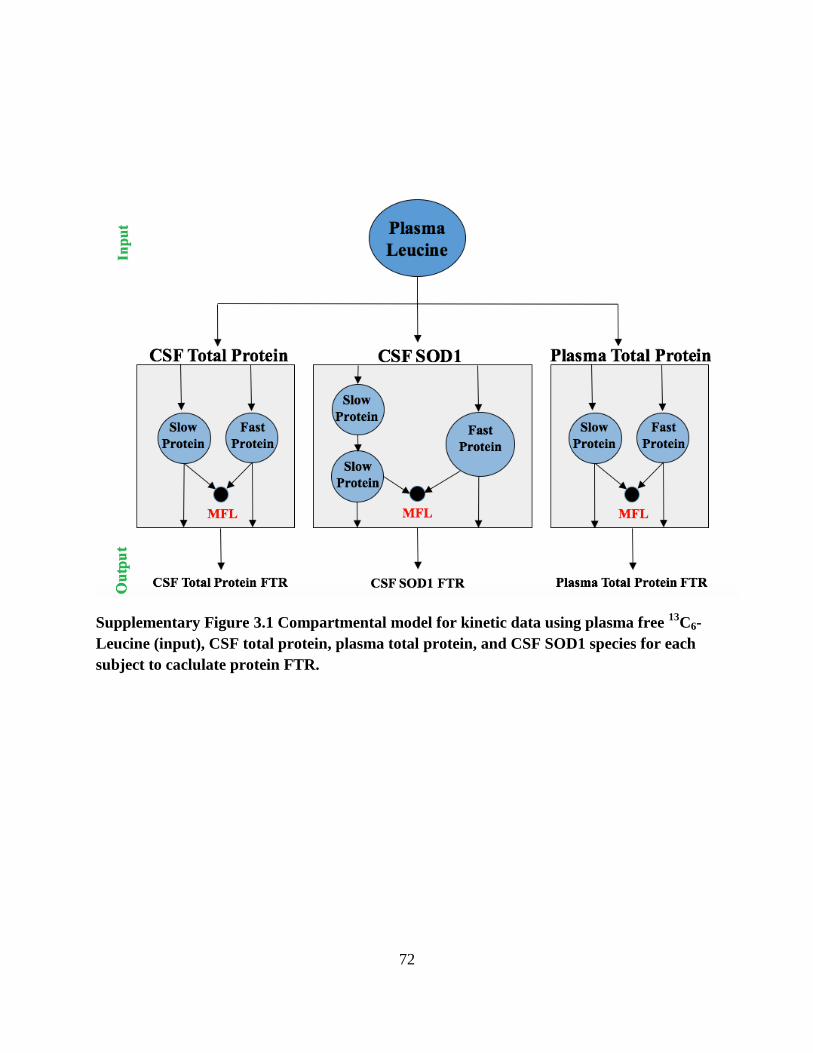
72
Supplementary Figure 3.1 Compartmental model for kinetic data using plasma free 13
C6-
Leucine (input), CSF total protein, plasma total protein, and CSF SOD1 species for each
subject to caclulate protein FTR.

73
Peptide Sequence Charge Label Precursors
Ions Product Ion
Cone
Voltage
(V)
Collision
Energy (V)
24SNGPVKVWGSIKGLTE
39 +++ WT 557.9737 547.3086 35 19
707.4087 35 14
735.9194 35 14
804.4462 35 14
13C6-Leu 559.9804 553.3287 35 19
710.4187 35 14
738.9294 35 14
810.4663 35 14
N15 564.6206 553.2908 35 19
715.3849 35 14
744.3942 35 14
813.4195 35 14
40GLHGFHVHE
48 ++ WT 516.7541 308.1717 35 13
649.3205 35 18
725.3365 35 18
748.3889 35 18
13C6-Leu 519.7642 314.1918 35 13
655.3406 35 18
725.3365 35 18
754.409 35 18
N15 524.2319 313.1569 35 13
659.2909 35 18
735.3069 35 18
759.3563 35 18
79RHVGDLGNVTADKDGVADVSIE
100 +++ WT 756.3803 425.2199 35 26
474.7541 35 26
565.2841 35 31
910.9479 35 21
13C6-Leu 758.387 428.23 35 26
477.7642 35 26
565.2841 35 31
913.958 35 21
N15 766.0183 432.1992 35 26
482.2319 35 26
575.2545 35 31
923.4109 35 21
101DSVISLSGDHC[+57.0]IIGRTLVVHE
121 +++ WT 769.7302 302.1347 35 26

74
Peptide Sequence Charge Label Precursors
Ions Product Ion
Cone
Voltage
(V)
Collision
Energy (V)
846.9279 35 21
946.986 35 21
1003.528 35 21
13C6-Leu 771.7369 846.9279 35 21
849.938 35 21
949.9961 35 21
1006.5381 35 21
N15 779.0359 305.1258 35 26
857.8953 35 21
958.9504 35 21
1015.991 35 21
122KADDLGKGGNEE
133 ++ WT 616.7913 315.1663 35 22
430.1932 35 22
690.3053 35 22
803.3894 35 22
13C6-Leu 619.8014 315.1663 35 22
430.1932 35 22
690.3053 35 22
809.4095 35 22
N15 624.2691 319.1544 35 22
624.2691 435.1784 35 22
624.2691 699.2786 35 22
624.2691 813.3597 35 22
134STKTGNAGSRLAC[+57.0]GVIGIAQ
153 +++ WT 654.3459 681.3331 35 22
730.8674 35 22
815.9201 35 17
872.4622 35 17
13C6-Leu 656.3527 684.3432 35 22
733.8774 35 22
818.9302 35 17
875.4722 35 17
N15 662.9869 690.805 35 22
740.8377 35 22
826.8875 35 17
883.9281 35 17
Supplementary Table 3.1 Transition ions used for SOD1 tandem liquid chromatography-
mass spectrometry analysis using GluC digestion protocol.

75
Peptide Sequence Charge Label Precursors
Ions
Product
Ion
Cone Voltage
(V)
Collision Energy
(V)
4AVC[+57.0]VLK
9 (WT) ++ WT 345.2044 260.1969 35 16
359.2653 35 16
519.2959 35 10
13C6-Leu 348.2144 266.217 35 16
365.2854 35 16
525.3161 35 10
N15 348.694 263.188 35 16
363.2534 35 16
524.2811 35 10
79
HVGDLGNVTADK91
++ WT 613.3122 333.1769 35 22
434.2245 35 22
533.293 35 22
647.3359 35 22
817.4414 35 22
989.4948 35 22
13C6-Leu 616.3223 333.1769 35 22
434.2245 35 22
533.293 35 22
647.3359 35 22
823.4615 35 22
995.5099 35 22
N15 621.2885 337.165 35 22
439.2097 35 22
539.2752 35 22
655.3122 35 22
827.4188 35 22
1001.452 35 22
116TLVVHEK
122 ++ WT 413.2541 276.1554 35 14
306.1792 35 14
413.2143 35 14
512.2827 35 14
611.3511 35 14
13C6-Leu 416.2551 276.1554 35 14
306.1792 35 14
413.2143 35 14
512.2827 35 14
611.3511 35 14
N15 418.2303 279.1465 35 14

76
Peptide Sequence Charge Label Precursors
Ions
Product
Ion
Cone Voltage
(V)
Collision Energy
(V)
310.1673 35 14
419.1965 35 14
519.262 35 14
619.3274 35 14
4VVC[+57.0]VLK
9 ++ A4V 359.22 260.1969 35 18
519.2959 35 14
618.3643 35 14
13C6-Leu 362.2301 266.217 35 18
525.3161 35 14
624.3845 35 14
N15 362.7096 263.188 35 18
524.2811 35 14
624.3466 35 14
79HVGDLGNVTVDK
91 ++ A89V 627.3279 361.2082 32 22
462.2558 32 22
561.3243 32 22
675.3672 32 22
845.4727 32 22
1017.5211 32 22
13C6-Leu 630.3379 361.2082 32 22
462.2558 32 22
561.3243 32 22
675.3672 32 22
851.4928 32 22
1023.5412 32 22
N15 635.3041 365.1963 32 22
467.241 32 22
567.3065 32 22
683.3435 32 22
855.4431 32 22
1029.4855 32 22
101DSVISLSGDHC[+57.0]IIGR
115 +++ E100K 543.6069 507.7429 28 14
607.801 28 14
664.343 28 16
713.8772 28 14
13C6-Leu 545.6136 507.7429 28 14
610.811 28 14
667.3531 28 16
716.8873 28 14
N15 550.2538 514.7222 28 14

77
Peptide Sequence Charge Label Precursors
Ions
Product
Ion
Cone Voltage
(V)
Collision Energy
(V)
615.7772 28 16
672.8178 28 14
722.8505 28 14
Supplementary Table 3.2 Transition ions used for SOD1 tandem liquid chromatography-
mass spectrometry analysis using LysC/Trypsin digestion protocol for mutant peptide
detection

78
Chapter 4: Protein Production is an Early Biomarker for RNA-
Targeting Therapies

79
ABSTRACT
Clinical trials for progressive neurodegenerative disorders such as Alzheimer’s Disease
and Amyotrophic Lateral Sclerosis have been hindered due to the absence of effective
pharmacodynamics markers to assay target engagement. We tested whether measurements of
new protein production would be a viable pharmacodynamics tool for RNA-targeted therapies.
To test this hypothesis, transgenic animal models expressing human proteins implicated in
neurodegenerative disorders -- microtubule-associated protein tau (hTau) or superoxide
dismutase-1 (hSOD1) -- were treated with antisense oligonucleotides (ASOs) delivered to the
central nervous system to target these human mRNA transcripts. Simultaneously, animals were
administered 13
C6-leucine via drinking water to measure new protein synthesis after ASO
treatment. Measures of new protein synthesis and protein concentration were assayed at
designated time points after ASO treatment using targeted proteomics. ASO treatment lowered
hTau mRNA and protein production (measured by 13
C6-leucine-labeled hTau protein) earlier
than total hTau protein concentration in transgenic mouse cortex. In the CSF of hSOD1
transgenic rats, ASO treatment lowered newly generated hSOD1 protein driven by decreases in
newly synthesized hSOD1 protein, not overall protein concentration 30 days after treatment. At
later time points, decreases in newly generated protein driven by overall lowering of protein
concentration, not the rate of newly synthesized protein. Taken together, measures of newly
generated protein show earlier pharmacodynamics changes for RNA-lowering therapeutics
compared with total protein concentration. However, a therapeutic window exists to measure
new protein production before pharmacodynamics effects reach steady state. Measuring new
protein production in CSF may be a promising early pharmacodynamics marker for RNA-
targeted therapeutics.

80
INTRODUCTION
The number of persons aged 65 and older globally is projected to triple by the year 2050
(WHO 2012), increasing the number of people living with adult-onset, progressive
neurodegenerative diseases in the population. There is a dire need to develop novel therapeutics
for these conditions, as current FDA-approved drugs do little more than manage symptoms, and
no treatments exist to reverse disease pathology. Despite promise of new therapies in preclinical
models over the past 30 years, these compounds have failed in Amyotrophic Lateral Sclerosis
(ALS) and Alzheimer’s Disease (AD) clinical trials at rates greater than 95 and 99%,
respectively (Cummings et al 2014; Petrov et al 2017). One important way to improve the
current treatment landscape is to focus on the development of novel methods to determine
whether a therapeutic has engaged the intended target in human clinical trials.
A central hypothesis in neurodegenerative disease is that the accumulation of misfolded
proteins over time imparts selective toxicity to specific neuronal populations, causing
progressive neurodegeneration and manifestation of symptoms. This pathological misfolding of
proteins in AD (Braak & Braak 1995), ALS (Neumann et al 2006; Shibata et al 1996),
Parkinson’s Disease (PD) (Bethlem & Den Hartog Jager 1960), and Huntington’s Disease (HD)
(DiFiglia et al 1997) suggests that an approach to inhibit the production of toxic proteins within
the cell may be an effective therapy. One such approach includes lowering levels of RNA to
decrease protein translation using molecules such as antisense oligonucleotides (ASOs) or small
interfering RNA (siRNA). ASOs are short, DNA-like molecules that can be designed to
selectively bind mRNA transcripts and cause the catalytic destruction of particular mRNAs
(DeVos & Miller 2013a). ASOs that lower mRNA levels and thus decrease translation of
proteins prone to misfolding show great promise in preclinical models of tauopathy (DeVos et al

81
2017), ALS (Becker et al 2017; Donnelly et al 2013; Smith et al 2006), and HD (McBride et al
2011), and are relatively safe in humans (Miller et al 2013). These promising data have led to the
launch of multiple clinical trials for ASOs in neurodegenerative diseases (NCT03225846,
NCT02623699, NCT02519036, NCT03186989).
For RNA-targeted therapeutics, it is crucial to establish measurements that validate
compound engagement with its desired biological target, mRNA (Mitsumoto et al 2014).
However, it is challenging to isolate mRNA from cerebrospinal fluid (CSF), the primary biofluid
available to assay target engagement in the human central nervous system (CNS). Because
mRNA translation leads to the production of new protein, protein-based biomarkers in CSF
represent an attractive avenue to pursue target engagement for RNA-targeted therapeutics. In this
study, we used nonradioactive, stable isotope labeling with an essential amino acid (Bateman et
al 2007; Bateman et al 2006) to identify inhibition of new protein production after ASO
treatment. Using two preclinical models of ASO targets in clinical trials for neurodegenerative
disease, we tested the hypotheses that stable isotope labeling with 13
C6-Leucine will: 1) identify
pharmacodynamics at an earlier time point than protein concentration-based measures and 2)
show measurable effects in CSF to support translation of this assay approach from animal
models to human CNS. These results would support the use of new protein synthesis
measurements in clinical trials for RNA-lowering therapies in neurodegenerative diseases.

82
METHODS
Transgenic Animals
Transgenic mice expressing human tau (hTau) (Andorfer et al 2003) (Jackson
Laboratories, Bar Harbor, ME) were maintained through in-house breeding on a C57/Bl6
background. Transgenic rats expressing human superoxide dismutase-1 (hSOD1WT
) (Chan et al
1998) were provided by Pak Chan (Stanford University) and maintained through in-house
breeding on a Sprague-Dawley background. Genotyping was performed by PCR amplification of
tail DNA (Supplementary Table 1). All breeding and experimental protocols were approved by
the Institutional Animal Care and Use Committee at Washington University and conducted
according to the NIH Guide for Care and Use of Laboratory Animals.
Antisense Oligonucleotides (ASOs)
ASOs that recruit RNase-H to degrade target mRNA and an inactive, control ASO with
no sequence specificity were provided by Ionis Pharmaceuticals (DeVos et al 2017; McCampbell
et al 2018) (Table 4.1). All ASOs were diluted in artificial CSF before use.
Surgical Procedures
All animals were anesthetized by 2-5% inhalant isoflurane, received constant isoflurane
flow during the procedure, and were given an injection of bupivacaine prior to incision.
Following the surgical procedure, animals received carprofen and were placed on a 37°C heating
pad for recovery.
For intracerebroventricular injections, 60-day-old hTau transgenic mice received 300 µg
of ASO at -1.0mm M/L, -0.3mm A/P, -3.0mm D/V from Bregma (DeVos & Miller 2013b).

83
ASO Sequence/Chemistry
hTau ASO CCoGoToTTTCTTACCAoCoCT
hSOD1 ASO TToAoATGTTTATCAoGoGAT
Inactive ASO CCoToAoTAGGACTATCCAoGoGoAA
Table 4.1. Antisense Oligonucleotides
Oligonucleotides containing phosphothioate backbone modifications: unmodified phosphodiester
linkage (red o), 2’-O-methoxyethylribose, (orange) and constrained ethyl (blue) groups. All
cytosine residues are 5’methylcytosine.

84
Similarly, 120-day-old hSOD1 transgenic rats received 1000 µg of ASO injected at -1.4mm
M/L, -0.4mm A/P, -3.5mm D/V from Bregma. For intrathecal ASO injection, 120-day-old
hSOD1 transgenic rats received 1000 µg of ASO between the L3 and L5 vertebrae (Mazur et al
2017). Both male and female transgenic animals were used in these studies, and researchers were
blinded to the identity of treatment throughout the duration of the experiment.
13C6-Leucine Oral Labeling and Tissue Collection
Methods for oral labeling in rodents with 13
C6-Leucine were described previously using
unlabeled L-leucine or labeled, 13
C6-L-Leucine (Cambridge Isotope Laboratories, Cambridge,
MA) dissolved in water at 5 mg/mL (Crisp et al 2015). Labeling was consistent in all
experiments between groups (Supplementary Figure 1).
At indicated time points after surgery, animals were anesthetized with isoflurane and
perfused with cold phosphate buffered saline (PBS) containing 0.03% heparin. Before perfusion,
CSF was extracted from rat cisterna magna via a 23x¾” winged infusion butterfly needle
(Terumo, Somerset, NJ). Blood was collected from mechanically ruptured vena cavae at
beginning of perfusion, and the brain and spinal cord were harvested after perfusion. All samples
were flash frozen in liquid nitrogen and stored at -80 °C before use.
mRNA Isolation and Analysis
mRNA isolation from flash-frozen tissues was performed using the RNeasy® Mini Kit
(Qiagen, Venlo, Netherlands). The EXPRESS One-Step Superscript qRT-PCR Universal Kit
(Invitrogen, Carlsbad, CA) was used for reverse transcription and amplification of SOD1 and
MAPT mRNA transcripts. Comparative analysis by the ΔΔCt method, with GAPDH mRNA
transcripts as an internal control, was performed with the ABI PRISM 7500 Fast Real-Time
System (Applied Biosystems, Waltham, MA) (Supplementary Table 1).

85
N15 Recombinant Protein Standards
N15-labeled, recombinant hTau protein (isoform 2N4R 411) was a generous gift from
Drs. Isabelle Huvent and Guy Lippens (Université des Sciences et Technologie de Lille 1,
France).
Recombinant hSOD1 was produced in Rosetta 2 E Coli (Novagen) using an hSOD1
cDNA construct with an N-terminal GST tag subcloned into a pGEX4T-1 backbone. Bacterial
cultures were inoculated overnight in 15N-Celtone Complete Medium (Cambridge Isotope
Laboratories, Inc) with shaking at 37°C and induced with 1mM IPTG for hSOD1 production.
Protein was isolated by incubation with 600 µL of glutathione sepharose 4B beads (GE
Healthcare) for 30 minutes, 1 unit of thrombin (Thrombin Cleavage Capture Kit, Millipore) for
20 hours, and 30 µL of streptavidin agarose for 30 minutes, performed at room temperature.
Samples were spun at 1,000xg for 5 minutes, and supernatant containing N15-labeled, hSOD1
recombinant protein was collected.
Tissue Homogenization, Protein Isolation, and Mass Spectrometric Analysis
hTau transgenic mouse brain tissue was homogenized in PBS plus protease inhibitors
(P8340; Sigma-Aldrich, St. Louis, MO). Protein concentration of brain lysate was determined by
Pierce BCA assay (ThermoFisher Scientific, Waltham, MA). Total hTau was
immunoprecipitated from 100 µg of tissue lysate using 30 µL of CNBr-activated Sepharose
beads (GE Healthcare, Chicago, IL) crosslinked to anti-hTau antibody (Tau 1 monoclonal,
produced by Nicholas M. Kanaan, Michigan State University) at 3 mg antibody/g of beads in 1
mL PBS plus 1% NP-40, 5 mM guanidine, and protease inhibitors for 1.5 hours at room
temperature. Before immunoprecipitation, 5 ng of N15-lableled hTau was added to each sample.
hTau was digested using 400 ng of sequencing-grade endoproteinase Trypsin (Promega,

86
Madison, WI) for 16 hours at 37°C. Samples were lyophilized and reconstituted in 25 uL of 2%
acetonitrile/0.1% formic acid solution prior to liquid chromatography- quadrupole-orbitrap mass
spectrometry analysis (OrbiTrap Fusion, ThermoFisher).
hSOD1 was isolated from transgenic rat tissue or CSF as described previously (Crisp et al
2015). Before immunoprecipitation, 200 ng of N15-labeled hSOD1 was added to each brain or
spinal cord sample, and 50 ng of N15 SOD1 was added to CSF samples. Isolated hSOD1 was
digested with 600 ng of sequencing-grade endoproteinase GluC in 25 µL of 50 mM NaHCO3
buffer (pH 8.8) for 17 hours at room temperature. After solid phase extraction, samples were
lyophilized and resuspended in 25 µL 2% acetonitrile/0.1% formic acid solution prior to liquid
chromatography-triple quadrupole mass spectrometry analysis (Xevo TQ-S, Waters).
Newly synthesized proteins were quantified by analyzing the incorporation of 13
C6-
Leucine into target proteins, indicated by the tracer-to-tracee ratio (TTR), by comparing the area
under the chromatogram curve for SOD1 or tau peptides containing 13
C6-Leucine or 12
C6-
Leucine. Relative protein concentration was quantified by measuring the ratio between the area
under the curve of protein immunoprecipitated from animal tissue and N15-labeled recombinant
protein (Supplementary Tables 4.2 and 4.3). To account for non-steady state conditions due to
mRNA lowering in this experiments, newly generated protein was also analyzed by multiplying
the relative protein concentration by protein TTR at each time point. For figure presentation, data
for hTau is represented by the TPSLPTPPTR peptide and data for hSOD1 is represented by the
GLHGFHVHE peptide.
Plasma-free 13
C6-Leucine abundance was analyzed from TCA precipitation of 100 µL rat
or mouse plasma via capillary gas chromatography-negative chemical ionization-quadrupole
mass spectrometry (Agilent) as described previously (Crisp et al 2015).

87
Statistical Analysis
All statistical analyses were performed using Graphpad Prism 7.0 (GraphPad Software).
All hypothesis tests performed were two-sided assuming a normal distribution of data. P < 0.05
was determined significant. Based on previous results (DeVos et al 2017; McCampbell et al
2018), a cutoff of 50% target mRNA reduction relative to the mean mRNA levels in inactive
ASO-treated animals was used as inclusion criteria for subsequent analysis. In CSF analysis,
samples were excluded if blood contamination was visibly present or if the volume of CSF
collected was less than 75 uL. For relative quantification, samples were normalized to a single
sample in each experiment from the inactive ASO treatment group. Quantitative results are
expressed as mean difference and 95% confidence interval.

88
RESULTS
Lowering New hTau Production Precedes Protein Concentration Changes after ASO Treatment
We performed ICV injection of an ASO targeting human tau mRNA (DeVos et al 2017)
in transgenic mice expressing the human tau transgene (hTau mice) to understand lowering of
tau mRNA and total protein over time. To analyze newly generated hTau protein, we
administered 13
C6-Leucine in the drinking water at designated time points (Figure 4.1A). We
organized the timing of ASO treatment, 13
C6-Leucine administration, and euthanasia to observe
consistent, maximum label incorporation of proteins within the CNS at each time point assayed .
hTau ASO treatment lowered hTau mRNA in the cerebral cortex over time (Figure
4.1B). As early as two days post-injection, hTau ASO treatment lowered hTau mRNA levels by
~50% relative to inactive ASO, with further change of hTau mRNA by ~72% by 23 days post-
injection. hTau ASO treatment also decreased hTau protein levels over time (Figure 4.1C), with
significant effects of ~32% observed at 23 days. The discrepancy between overall mRNA
lowering and protein lowering is consistent with tau as a long-lived protein in the CNS (Sato et
al 2018).
Using percent of 13
C6-Leucine incorporation (TTR) as a measure of newly synthesized
protein, hTau ASO treatment lowered of newly synthesized hTau protein over time (Figure
4.1D). We observed lowering of new hTau protein production at 13 days after ASO treatment by
~71%. Combined, these data suggest that newly generated hTau protein is a reliable measure of
hTau ASO pharmacodynamics in central nervous system tissue, with newly generated hTau
showing an earlier (13 vs. 23 days) and greater magnitude of effect (71% vs. 32%) compared to
hTau protein concentrations (Figure 4.1E).

89
(A.) Experimental Design. hTau transgenic mice received 300 μg of either hTau ASO or inactive
ASO via intracerebroventricular injection and were labeled with13
C6-Leucine (13
C6-Leu) at
designated time points. (B.) hTau mRNA levels in temporal cortex by qPCR. Compared to
inactive ASO control, hTau ASO treatment lowered hTau mRNA over time (2-Way ANOVA F3,24
= 10.04, p = 0.0145). hTau mRNA levels are significantly lowered as early as two days after
ASO treatment by 50% (95% CI: 34.8%, 66.9%), and remain lowered as late as 23 days post
treatment by 72% (55.5%, 87.2%). (C.) hTau protein levels relative to N15-labeled recombinant
standard in temporal cortex by LC-MS. hTau ASO treatment lowers hSOD1 protein over time
Figure 4.1 Lowered hTau protein synthesis precedes protein lowering after ASO
treatment in hTau transgenic mice

90
(2-Way ANOVA F3,24 = 3.374, p = 0.0349). This effect is observed at 23 days post ASO treatment
by 32% (0.05%, 64.0%). (D.) Newly synthesized 13
C6-Leu-labeled hTau in temporal cortex by
LC-MS. Values are measured as the tracer to trace ratio (TTR) of 13
C6-Leu-labeled:12
C6-Leu-
unlabeled hTau. Lowering of 13
C6-Leu-labeled hTau is observed over time (2-Way ANOVA,
F3,24 = 5.748, p = 0.0041), and as early as 13 days post ASO treatment by 71% (95% CI: 29.4%,
123%). (E.) Newly generated hTau protein measured by multiplying hTau protein concentration
and TTR to account for non-steady state conditions. **** p < 0.0001, *** p < 0.001, ** p < 0.01.
n = 4 per treatment group per time point. Error bars in figures represent standard error of the
mean (SEM).

91
Lowering of 13
C6-Leucine-labeled hSOD1 Occurs in CSF after ASO Treatment
hSOD1WT
rats were treated with an ICV injection of 1000 µg hSOD1 ASO in the lateral
ventricle and administered 13
C6-Leucine in drinking water (Figure 4.2A). Based upon previous
measurements of hSOD1 half-life in these rats (Crisp et al 2013), and that lowering of CSF
SOD1 after ASO treatment in the G93A SOD1 transgenic rat model was observed at 42 days
post ASO treatment (Winer et al 2013), we euthanized animals 30 days after treatment to assay
hSOD1 total protein levels and new hSOD1 protein production to look for early ASO
pharmacodynamics. hSOD1 ASO treatment lowered hSOD1 mRNA globally within the CNS
(Figure 4.2B), leading to overall hSOD1 protein lowering by ~62% in cortex and ~40% in
lumbar spinal cord (Figure 4.2C). Similarly, we observed a decrease in newly synthesized
hSOD1 protein by ~34% in cortex and ~50% in lumbar spinal cord (Figure 4.2D). At this time
point, lowering of both hSOD1 protein concentration and newly synthesized hSOD1 protein
drive overall decreases in newly generated hSOD1 protein (Figure 4.2E).
In CSF, hSOD1 treatment failed to lower hSOD1 protein concentration (Figure 4.2F).
However, SOD1 ASO treatment decreased newly synthesized 13
C6-Leucine-labeled hSOD1 in
CSF by ~30% relative to the inactive ASO treatment group (Figure 2G). This effect of lowered
13C6-Leucine-labeled hSOD1 is specific to CSF, as we did not measure any differences in
peripheral hSOD1 protein in plasma (Supplemental Figure 4.1). To test SOD1 protein
concentration at a later time point, we treated a separate cohort of animals without 13
C6-Leucine
labeling using the same ASO treatment paradigm and analyzed CSF hSOD1 concentration at 50
days post-surgery (Supplemental Figure 4.5). Although we observed lowering in hSOD1
mRNA throughout the CNS 50 days post-ASO treatment (Supplemental Figure 4.5A), we did
not observe decreases in CSF hSOD1 protein concentration (Supplemental Figure 4.5B),

92
suggesting an even longer incubation time may be required to observe protein lowering in CSF.
Taken together, these data suggest changes in newly synthesized, 13
C6-Leucine-labeled target
proteins drives early pharmacodynamics biomarker effects in the CSF after ASO treatment
(Figure 4.2H).

93
Figure 4.2 Lowered 13
C6-Leucine-labeled hSOD1 observed in cerebrospinal fluid after ASO
treatment in hSOD1 transgenic rats

94
(A.) Experimental Design. hSOD1 transgenic rats received 1000 μg of either hSOD1 ASO or
inactive ASO via intracerebroventricular injection at designated time points. (B.) hSOD1 mRNA
levels in the CNS by qPCR. hSOD1 mRNA levels are lowered globally in the CNS, with mRNA
lowering of ~70% in cortex and lumbar spinal cord and ~55% in cerebellum. (C.) hSOD1 protein
levels relative to N15-labeled recombinant standard in CNS cortex by LC-MS. Lowering of
hSOD1 protein is observed in both cortex at ~61% (95% CI: 41.9%, 81.3%) and lumbar spinal
cord at ~40% (24.6%, 54.3%). (D.) 13
C6-Leucine (13
C6-Leu) labeled hSOD1 in CNS by LC-MS.
Values are measured as the tracer to trace ratio (TTR) of 13
C6-Leu-labeled:12
C6-Leu-unlabeled
hSOD1. Significant lowering of 13
C6-Leu-labeled hSOD1 is observed in both cortex at ~34%
(15.5%, 51.9%) and lumbar spinal cord at ~50% (33.3%, 66.1%). (E.) Newly generated hSOD1
protein measured by multiplying hSOD1 protein concentration and TTR in tissues. (F.) hSOD1
protein levels relative to N15-labeled recombinant standard in CSF by LC-MS. No changes in
CSF hSOD1 protein levels were observed (Student’s T-Test t = 0.448, p = 0.67). (G.) 13
C6-Leu
labeled hSOD1 in CSF by LC-MS. Significant lowering of 13
C6-Leu-labeled hSOD1 by ~30%
(95% CI: 12.0%, 48.4%) observed relative to inactive ASO control (Student’s T Test t = 3.923, p
= 0.0057). (H.) Newly generated hSOD1 protein measured by multiplying hSOD1 protein
concentration and TTR in CSF. One sample in inactive ASO group was excluded in CSF
analysis due to blood contamination during CSF collection. **** p < 0.0001, *** p < 0.001, ** p
< 0.01. Error bars in figures represent SEM.

95
A Therapeutic Window Exists for Newly Synthesized Protein Pharmacodynamics
To enhance the translational relevance of our findings, we performed similar experiments
in hSOD1 rats using intrathecal ASO injection, the method of drug delivery used in current
clinical trials. We analyzed CNS tissue 10, 30, and 60 days after ASO treatment compared to
inactive ASO control to better understand longitudinal changes in hSOD1 protein (Figure 4.3A).
Contrary to hSOD1 ASO delivery by ICV injection, we observed differential effects of hSOD1
mRNA lowering in CNS tissues after intrathecal ASO injection. We observed a significant
hSOD1 mRNA decrease in the lumbar spinal cord over time, with an average hSOD1 mRNA
lowering at 60 days by ~66%. However, hSOD1 treatment did not significantly lower hSOD1
mRNA in the cortex or cerebellum over time (Figure 4.3B). Because it is likely that SOD1
mRNA will need to be lowered globally in the CNS to affect SOD1 CSF levels, we focused on
protein measurements in the spinal cord, where we confidently documented mRNA lowering.
To further understand the relationship between protein lowering and lowering of newly
synthesized hSOD1 after ASO treatment, we analyzed hSOD1 protein in lumbar spinal cord.
hSOD1 ASO treatment decreased hSOD1 protein levels across all time points measured, and
treatment effect was greatest at day 60, with 50% lowering of hSOD1 protein (Figure 4.3C).
hSOD1 ASO treatment significantly lowered newly synthesized hSOD1 compared to inactive
ASO-treated animals (Figure 4.3D). Interestingly, ASO treatment decreased newly synthesized
hSOD1 at 10 and 30 days by 40 and 45%, but there was no difference in 13
C6-Leucine-labeled
hSOD1 between groups at day 60 due to lowering of total hSOD1 protein concentration.
Therefore, at later time points, decreases in newly generated hSOD1 protein by ASO treatment
are driven by overall lowering of protein concentration (Figure 4.3E).

96
Figure 4.3 A therapeutic window exists to observe lowered protein synthesis
pharmacodynamics in the central nervous system

97
(A.) Experimental Design. hSOD1 transgenic rats received 1000 ug of either hSOD1 ASO or
inactive ASO at designated time points via intrathecal injection. (B.) hSOD1 mRNA levels in the
CNS by qPCR. hSOD1 mRNA levels are significantly lowered in spinal cord after hSOD1 ASO
treatment (2 Way ANOVA F2,39 = 10.91, p = 0.002), with a maximum lowering of ~66% (95%
CI: 54.6%, 78.2%) 60 days post-treatment. No significant hSOD1 mRNA lowering was
observed in frontal cortex (2 Way ANOVA F2,40 = 1.48, p = 0.24) or cerebellum. (C.) hSOD1
protein levels relative to N15-labeled recombinant standard in lumbar spinal cord by LC-MS.
Lowering of hSOD1 protein is observed at over time (2 Way ANOVA F2,36 = 10.04, p = 0.0004),
with a maximum lowering by ~50% (95% CI: 42.5%, 79.8%) at Day 60. (D.) 13
C6-Leu labeled
hSOD1 in CNS by LC-MS. Values are measured as the tracer to trace ratio (TTR) of 13
C6-Leu-
labeled:12
C6-Leu-unlabeled hSOD1. Significant lowering of newly synthesized hSOD1 observed
after hSOD1 ASO Treatment (2 Way ANOVA F2,36 = 6.269, p = 0.0046). Lowering of 13
C6-
Leu-labeled hSOD1 is observed 10 and 30 days after ASO treatment by ~40-45% (30%, 88%).
60 days post ASO treatment, there was no difference in 13
C6-Leu-labeled hSOD1 between groups
(adjusted p value = 0.9976). (E.) Newly generated hSOD1 protein measured by multiplying
hSOD1 protein concentration and TTR in tissues. **** p < 0.0001, *** p < 0.001, * p < 0.05.
hSOD1 ASO: n = 5 - 10 per time point, Inactive ASO: n = 5 – 7 per time point. Error bars in
figures represent SEM.

98
DISCUSSION
Our data show that, following treatment with a mRNA lowering therapeutic, the lowering
of newly synthesized protein occurs before lowering of total protein concentration in tissue.
Further, our findings indicate that labeled, newly generated protein measures direct effects of
protein translation in the source tissues, which are not seen in CSF protein concentrations at early
time points. Given the direct measurement of protein translation, labeling protein production has
higher sensitivity than protein concentration-based measures for mRNA-lowering therapies.
These studies suggest that identifying newly generated protein after ASO treatment is an
effective protein-based biomarker for assaying ASO target engagement (Figure 4.4).
Previous work supports the use of CSF protein concentration as a promising
pharmacodynamics biomarker for ASO therapy (Gendron et al 2017; Winer et al 2013), and CSF
SOD1 concentration is a secondary outcome measurement in an ongoing Phase I clinical trial
(NCT0262399). However, a key limitation in the use of CSF protein concentration as a measure
of ASO efficacy is the indirect mechanism informed by this assay. ASOs inhibit the production
of new protein, and measures of protein concentration also rely on transport to CSF and the
clearance of existing protein which may be unaffected by ASO treatment. Especially in a rapidly
progressing neurodegenerative disease such as ALS (Bali et al 2017), it is important to develop
pharmacodynamics tools that identify direct mechanisms of action at early time points that
reflect the actual drug effect in the tissue of interest.
Our study design is translatable to humans, as stable isotope labeling is safe and effective
in measuring the kinetics of proteins implicated in neurodegenerative disease within human CSF
(Bateman et al 2007; Bateman et al 2006; Bertram & Tanzi ; Crisp et al 2015; Sato et al 2018).

99
(A.) Normal protein turnover at steady state synthesis includes production (top arrow) and
clearance (bottom arrow) rates in the CNS. (B.) Early after treatment, lowering in new protein
synthesis is observed due to lowering of target mRNA. (C.) In addition to lowering of new
protein synthesis, proteins present before 13
C6-labeling are cleared at the rate of turnover or half-
life. (D.) mRNA lowering reaches steady state, resulting in a consistent lowered protein synthesis
rate. Protein clearance remains constant and faster than production, leading to a new lowered
steady state protein concentration detected at a later time and less magnitude.
Figure 4.4 A model of the interplay between new protein production and protein
concentration pharmacodynamics in RNA-lowering treatment strategies

100
However, these experiments were performed over a 36-hour time period, as the half-life of
amyloid beta peptide is ~9 hours in CSF (Bateman et al 2006). To our knowledge, the results
presented here are the first observations measuring new protein production of long-lived protein
targets in the CNS after therapeutic intervention. Our approach can be directly applied to
multiple RNA-directed strategies, such as siRNAs (Pfister et al 2009) and ASOs (Huynh et al
2017; Zhao et al 2017a), that target long-lived proteins in neurodegeneration for clinical trials.
Besides its lowered protein production readout, stable isotope labeling could be a powerful tool
to examine increased protein production for therapeutic strategies such as gene therapy
(Tuszynski et al 2015) and splicing ASO treatments (Finkel et al 2017; Schoch et al 2016).
Moreover, an understanding of protein kinetics using stable isotope labeling can also inform on
assays for therapies that target protein turnover, such as those approaches that activate protein
clearance mechanisms (Lee et al 2010) or promote proper protein folding (Das et al 2015).
Another advantage of our mass spectrometry-based assay is the ability to specifically
isolate mutant from wild type protein in patient populations. Many familial forms of
neurodegenerative diseases are caused by autosomal dominant mutations [31] and, thus, exhibit
both mutant and normal protein forms. However, it is currently unknown if mutant proteins
display altered protein kinetics compared to their wild type counterparts in humans. Future
studies in symptomatic, mutation-carrying patients are needed to characterize the half-life of
these target proteins in human CSF. Protein half-life measures will be crucial to determine the
timing required to observe pharmacodynamics effects in humans when designing randomized-
control clinical trials.
One limitation in this study is the inability to identify target protein lowering in CSF after
SOD1 ASO treatment. Without this effect, the relationship between lowered protein production

101
and total protein concentration cannot be determined in the biological fluid important for human
clinical trials. Previous data suggests this limitation may be due to the drug delivery paradigm in
these experiments. The current study used a single bolus injection of ASO in rodent models
while previous studies that observed total target protein lowering in CSF after ASO treatment in
non-human primate models were performed with multiple intrathecal ASO injections (DeVos et
al 2017; McCampbell et al 2018). Additionally, both transgenic animal models used here
express wild type forms of the target protein, where no pathological misfolding of either hSOD1
or hTau is observed (Andorfer et al 2003; Chan et al 1998). Therefore, future studies should
examine how new protein production changes in the presence of misfolded proteins within the
CNS.
In sum, the approach described here provides a framework for the development of assays
that isolate the direct mechanism of action of target engagement in neurodegenerative disease
therapies, with the hope that such data will lead to a higher success in clinical trial outcomes and
approval of more therapies for patients.
ACKNOWLEDGEMENTS
We thank Drs. Cheryl Frankfater and John Turk of the Biomedical Mass Spectrometry
Core at Washington University in St. Louis for assistance with gas chromatography-mass
spectrometry analysis. Research reported in this publication was supported by the Washington
University Institute of Clinical and Translational Sciences grant UL1TR002345, sub-award
TL1TR002344 from the National Center for Advancing Translational Sciences (NCATS), the
National Institute for Neurological Disorders and Stroke grant U01NS084870, grant R-01-
NS098716-01 (TM Miller, PI) and R-01-NS095773 (RJ Bateman, PI) from the National
Institutes of Health, as well as the MetLife Foundation Award (RJ Bateman, PI) (NIH). The

102
content is solely the responsibility of the authors and does not necessarily represent the official
view of NIH.

103
Primer Sequence
MAPT Genotyping Forward
5’-ACTTTGAACCAG GATGGCTGAGCCC-3’
MAPT Genotyping Reverse 5’-CTGTGCATGGCTGTCCACTAACCTT -3’
SOD1 Genotyping Forward 5’-TTGTTCAGAAAACTCTCTCCAACTTTGCACT-3’
SOD1 Genotyping Reverse 5’- GGGTTTTAACGTTTAGGGGCTACTCTACTG -3’
MAPT qPCR Forward 5’-AGAAGCAGGCATTGGAGAC-3’
MAPT qPCR Reverse 5’-TCTTCGTTTTACCATCAGCC-3’
MAPT qPCR Probe 5’-/56-FAM/ACGGGACTGGAAGCGATGACAAAA/3IABkFQ/-3’
SOD1 qPCR Forward 5’-TGCATCATTGGCCGCA-3’
SOD1 qPCR Reverse 5’-TTTCTTCATTTCCACCTTTGCC-3’
SOD1 qPCR Probe 5'- /56FAM/ACTGGTGGTCCATGAAAAAGCAGATGACTT/36-TAMTph/-3'
GAPDH qPCR Forward 5’-TGCCCCCATGTTGTGATG -3’
GAPDH qPCR Reverse 5’-TGTGGTCATGAGCCC TTCC -3’
GAPDH qPCR Probe 5’- /56-FAM/AATGCATCCTGCACCACCAACTGCTT/3IABkFQ/-3’
Supplementary Table 4.1 Primers and probes used for genotyping and qPCR analysis of
hTau (MAPT), hSOD1 (SOD1), and mouse GAPDH (GAPDH) DNA

104
Sequence Label Precursors Product Collision Energy (V)
24SNGPVKVWGSIKGLTE
39 557.9737 547.3086 19
557.9737 707.4087 14
557.9737 735.9194 14
557.9737 804.4462 14
13
C6-Leu 559.9804 553.3287 19
559.9804 710.4187 14
559.9804 738.9294 14
559.9804 810.4663 14
N15 564.6206 553.2908 19
564.6206 715.3849 14
564.6206 744.3942 14
564.6206 813.4195 14 40
GLHGFHVHE48
516.754 308.1717 13
516.754 649.32 18
516.754 725.336 18
516.754 748.388 18
13
C6-Leu 519.764 314.1918 13
519.764 655.3406 18
519.764 725.3365 18
519.764 754.4090 18
N15 524.2319 313.1569 13
524.2319 659.2909 18
524.2319 735.3069 18
524.2319 759.3563 18
78RHVGDLGNVTADKDGVADVSIE
99 756.3083 425.2199 26
756.3083 474.7541 26
756.3083 565.2841 31
756.3083 910.9479 21
13
C6-Leu 758.3870 428.2300 26
758.3870 477.7642 26
758.3870 565.2841 31
758.3870 913.9850 21
N15 766.0183 432.1992 26
766.0183 482.2319 26
766.0183 575.2545 31
766.0183 923.4109 21
Supplementary Table 4.2 Transition ions used for hSOD1 tandem LC-MS/MS.
GLHGFHVHE peptide measurements were used as representative data in Figures.

105
Sequence Label Precursors Product Collision Energy (V)
212TPSLPTPPTR
221 533.7982 668.3726 20
13
C6-Leu 536.8083 668.3726 20
N15 540.2789 677.3459 20
243LQTAPVPMPDLK
254 655.3629 658.3729 15
13
C6-Leu 658.3729 896.4910 15
260IGSTENLK
267 431.2374 691.3621 15
13
C6-Leu 434.2475 697.3822 15
Supplementary Table 4.3 Transition ions used for hTau tandem LC-MS/MS. TPSLPTPPTR
peptide measurements were used as representative data in Figures.

106
Plasma 13
C6-Leucine values were analyzed in all samples analyzed and compared between
groups. (A.) hTau ICV ASO plasma 13
C6-Leucine at Day 23 post surgery, as seen in Figure 1. No
differences were observed (Student’s T-test, p =0.4496). (B.) hSOD1 ICV ASO plasma 13
C6-
Leucine at Day 30. No differences were observed (Student’s-T test) (C.) hSOD1 IT ASO plasma 13
C6-Leucine at all time points assessed. No differences were observed (2-Way ANOVA). Error
bars in figures represent SEM.
Supplementary Figure 4.1 No differences in 13
C6-Leucine oral labeling between
treatment groups in all experiments performed.

107
Linear regression analysis comparing peptides shows strong correlations exist between all three
peptide measurements. All p values < 0.0001 for each analysis.
Supplementary Figure 4 Correlation between all hTau peptides measured

108
Linear regression analysis comparing peptides shows strong correlations exist between all three
peptide measurements. All p values <0.0001 for each analysis.
Supplementary Figure 4.3 Correlation between all hSOD1 peptides measured.

109
(A.) hSOD1 protein levels relative to N15-labeled recombinant standard by LC-MS in plasma.
No changes in plasma hSOD1 protein levels were observed (Student’s T-test). (B.) 13
C6-Leu
labeled hSOD1 in plasma by LC-MS. No changes in plasma 13
C6-Leu labeled hSOD1 was
observed (Student’s T-Test). Error bars in figures represent SEM.
Supplementary Figure 4.4 No pharmacodynamics observed in the periphery after 1000 ug
hSOD1 ASO intracerebroventricular injection.

110
Supplementary Figure 5 No protein concentration pharmacodynamics observed in CSF
after 1000 ug hSOD1 ASO intracerebroventricular injection at 50 days post-treatment.
(A.) hSOD1 mRNA levels in the CNS by qPCR. hSOD1 mRNA levels are significantly lowered
in spinal cord and frontal cortex by ~70% and 50%, respectively, after ASO treatment relative to
inactive ASO controls (Student’s T-test). (B.) hSOD1 protein levels relative to N15-labeled
recombinant standard by LC-MS. No changes in CSF hSOD1 protein levels were observed
(Student’s T-test). n = 5 for each treatment group, with one CSF sample excluded from each
group due to blood contamination. **** p < 0.0001. Error bars in figures represent SEM.

111
Chapter 5: Summary and Future Directions

112
Advances in elucidating the genetic causes of ALS and generating models of disease
have rapidly enhanced our understanding of ALS pathophysiology in recent years, leading to the
development of multiple drug candidates for this devastating condition. However, multiple
challenges still exist for successful translation of preclinical discoveries to effective therapies in
the clinic. In this dissertation, I presented novel findings that further the understanding of human
ALS epidemiology and CSF SOD1 biology. Further, these findings generate crucial parameters
that can be directly implemented into randomized control clinical trial design for ALS patients.
ALSSOD1
clinical outcomes remain unchanged over time
In Chapter 2, I detailed findings from a retrospective cohort study to examine ALSSOD1
across 15 medical centers in the United States and Canada. Multiple studies in the late 1990’s
and early 2000’s characterized the natural history of the ALSSOD1
population, but our efforts to
analyze 175 subjects represent the largest patient population in almost 20 years. With the advent
of multidisciplinary care for the ALS patient population, previous studies reported an
improvement in ALS natural history, as defined by slower disease progression, from the end of
the 19th
century to the beginning of the 20th
century (Czaplinski et al 2006b; Qureshi et al 2009).
Therefore, it is important to determine if similar trends exist specifically in the ALSSOD1
population, as such trends will improve our understanding of clinical outcomes and influence
randomized control trial design for new potential treatments in this disease population.
Our calculated mean disease duration for all ALSSOD1
is similar to previous studies
(Table 2.1). Of particular interest in ALSSOD1
epidemiology is the A4V point mutation that
accounts for ~50% of the disease population in North America. Indeed, the highest percentage of
patients in our analysis carried the A4V mutation, and our calculated mean disease duration of
1.4 ± 0.7 years is unchanged compared to previous studies (Table 2.2). These data have

113
important implications, as the contrast to the trend of slower disease progression in the entire
ALS population over time suggests ALSSOD1
may be a unique subpopulation of disease.
As multiple therapeutic candidates are emerging to target SOD1 in ALS (Benatar et al
2018; Miller et al 2013), our natural history data have important implications for randomized
control trial design. Previous ALS trial enrollment is low and highly variable between study
sites, presenting a need to design more efficient clinical trials (Bedlack et al 2008). From our
natural history data, multiple opportunities exist for such considerations in the ALSSOD1
population. The increased prevalence of A4V mutations in the SOD1 population suggests
stratification of trial data to this specific mutation population. Such a strategy may be feasible
due to the limited variability in survival of SOD1A4V
patients, as our survival analysis showed no
SOD1A4V
patients living past 3.5 years (Figure 2.2). Indeed, our power calculations showed that
only 52 SOD1A4V
subjects per treatment group are needed to show hazard ratio of 0.5 the
experimental and control treatment groups. In contrast, 88 subjects per treatment group from the
entire SOD1 population are needed to detect the same statistical difference in median survival.
Moreover, unchanged natural history of ALSSOD1
may provide support for the use of
historical controls in early phase (I/II) trials (Donofrio & Bedlack), as seen in previous studies
(Miller et al 2011). The use of historical controls addresses many of the challenges associated
with previous clinical trial design, including reduced cost, improved efficacy, and appeal to
patients (Gordon 2009). Czaplinski and colleagues argued that historical controls are particularly
useful for experimental compounds that have received FDA approval for other indications, as
data for safety in humans already exists (Czaplinski et al 2006a). However, multiple limitations
exist in the use of historical controls such as matching bias, and the lack of key clinical
information such as comorbidities, current medication use, site of disease onset, and others.

114
Therefore, in future ALS natural history studies, it is important to collect data in a prospective
manner, capturing all clinically relevant data in the ALSSOD1
population. These data will be
crucial for not only improving clinical trial design in ALS, but further understanding the factors
that contribute to ALS pathobiology to better understand patient prognosis.
Understanding drivers of ALSSOD1
heterogeneity
My results also show that the remaining, non-A4V ALSSOD1
population exhibits highly
variable measures in mean age of disease onset (49.5 ± 12.3 years) and mean disease duration
(6.6 ± 7.0 years) (Table 2.1). We identified 36 missense variants in our cohort (Figure 2.1), and
150+ variants have been identified as pathogenic in ALS, yet these variants exhibit a varying
degree of disease penetrance (Felbecker et al 2010; Lopate et al 2010). Taken together, these
observations question the validity of all SOD1 variants as pathogenic. Further efforts must be
made in understanding this variability in the global ALSSOD1
population, as the majority of data
have focused on the North American disease population. Interestingly, Rechtman and colleagues
reported gender and ethnic differences in ALS incidence and age at diagnosis in the United
States (Rechtman et al 2015). Particular geographic areas of interest to update ALSSOD1
natural
history include Europe (Gamez et al 2006), Canada (Eisen et al 2008), and Asia (Chen et al
2015). Therefore, a combination of natural history data and more rigorous genome sequencing
data with advanced techniques (MacArthur et al 2014) may provide valuable insight into
understanding the pathogenicity of each SOD1 variant as a contributor to disease.
A missing component in many natural history studies, including our data, is an analysis
considering comorbidities and environmental factors as contributors to disease outcomes.
Environmental and lifestyle factors such as chemical toxins (Su et al 2016), smoking (Alonso et

115
al 2010; Armon 2009), military service (Weisskopf et al 2015; Weisskopf et al 2005), and
physical activity (Beghi et al 2010; Pupillo et al 2014) have all been implicated as risk factors of
ALS. These environmental factors may contribute to the heterogeneity of outcomes observed
between and within different SOD1 ALS variants, highlighting one of the key limitations in
retrospective cohort analyses. A further understanding of the interplay between genetics and
environmental contributions to ALS disease pathogenesis will be invaluable for the application
of prognostic models to individual patient management (Westeneng et al 2018). Further,
understanding of environmental factor contribution may help in formulating inclusion criteria for
randomized control trials, limiting the variability in primary and secondary outcome measures to
improve the statistical power of Phase II/III efficacy studies.
CSF SOD1 is a long-lived protein in ALS
In Chapter 3, I showed the first efforts to determine CSF SOD1 protein concentration and
kinetics in ALS patients using stable isotope labeling of essential amino acids combined with
tandem liquid chromatography-mass spectrometry. In these participants, 13
C6-Leucine-labeled
SOD1 persists as long as 120 days after the end of the 16 hour labeling period, further validating
previous observations (Crisp et al 2015) that SOD1 is a long-lived protein within the CNS of
humans (Figure 3.1). Additionally, CSF SOD1 protein concentration appeared to be increased in
ALS participants compared to controls (Table 3.2), similar to previous results measured by CSF
SOD1 ELISA (Winer et al 2013). Variability and limited sample size inhibited conclusive
differences in CSF SOD1 half-life between groups, with a calculated average of ~20 – 25 days
(Table 3.2). CSF SOD1 half-life is a crucial parameter for randomized control trial design for
experimental therapies in ALSSOD1
, as CSF SOD1 protein concentration is a secondary outcome
measure in current trials that inhibit the synthesis of new SOD1 protein (NCT01083667,

116
NCT02623699). Effects observed by these experimental treatments are influenced by the
intrinsic synthesis and degradation rates of SOD1 in the CNS at steady state, as defined by the
protein’s half-life. This relationship is clearly illustrated through PK-PD modeling, as we
demonstrated how a calculated CSF SOD1 half-life of 25 days will result in an incubation time
of ~120 days to observe changes in CSF SOD1 protein concentration after SOD1 ASO treatment
(Figure 3.2). These kinetic parameters are also valuable for improving participant experience in
clinical trials, as an understanding of the PK-PD relationship of experimental treatments may
reduce the number of invasive procedures such as lumbar punctures that must be performed in
order to establish therapeutic efficacy of drug target engagement.
Analyzing CSF SOD1 protein behavior to elucidate contributions to ALS pathobiology
Besides considerations for clinical trial design, an understanding of SOD1 kinetics may
provide valuable insight into the pathogenic status of SOD1 in ALS. Previous efforts to identify
misfolded SOD1 in biofluids of living ALS patients, even those with SOD1 mutations, have
remained elusive. Additionally, the role of misfolded SOD1 in sporadic ALS remains
controversial. Similar to observations of alterations in protein turnover in human
neurodegenerative disease, determination of CSF SOD1 protein turnover may provide an
opportunity to interrogate the stability of SOD1 protein within the CNS of ALS patients.
Moreover, if the degree of misfolded, aggregated protein in the brain and spinal cord of ALS
patients relates to protein stability, an attractive hypothesis is that potential changes in SOD1
protein kinetics may better inform on the prognosis of ALS patients. Currently, we are
underpowered to test this hypothesis, as to-date, we only have three participants (each harboring
different SOD1 mutations). However, my analysis in these SOD1 mutant carriers is the first to
dissociate wild type from mutant protein kinetics in the cerebrospinal fluid of patients with

117
mutations that cause an autosomal dominant form of a neurodegenerative disease. With the
recruitment of more participants, we hope to further investigate questions related to the
potentially misfolded state of mutant SOD1 protein including: the ability to identify misfolded
SOD1 in CSF, the role of SOD1 dimerization affecting protein stability in vivo, and how SOD1
stability may differentiate between pathogenic and benign variants as contributors to ALS
pathobiology.
Accumulating evidence in vitro and in vivo suggests that we may see altered CSF SOD1
protein kinetics in ALS, indicative of SOD1 protein misfolding in the CNS. In cell culture, the
half-life of soluble, mutant SOD1 is accelerated compared to WT protein (Borchelt et al 1994).
Similar behavior of mutant SOD1 is observed in vivo, where the half-life of both G93A SOD1
(Crisp et al 2015) and G85R SOD1 (Farr et al 2011) are accelerated compared to WT hSOD1 in
transgenic animal models. Moreover, isolation of misfolded G93A SOD1 by
immunoprecipitation with an antibody that specifically recognizes misfolded hSOD1 revealed
that misfolded SOD1 shows faster protein kinetics than the entire G93A SOD1 pool (Crisp et al
2015). Faster protein turnover is also observed in other models of neurological disorders, as
mutations in glial fibrillary acidic protein which cause similarly decrease the half-life of mutant
protein compared to wild type in animal models of Alexander’s Disease (Moody et al 2017).
Further, Sato and colleagues used the presence of mutant SOD1 protein in red blood cells as a
marker for altered protein turnover, where fast SOD1 turnover by erythrocytes suggests very
misfolded proteins will not be present in these cells (Sato et al 2005). Because SOD1 is a
ubiquitously-expressed protein, isolation from blood limits the ability to specifically understand
SOD1 protein behavior in the CNS. However, we previously observed that CSF SOD1 half-life
is similar to SOD1 from the brain and spinal cord (Crisp et al 2015), suggesting that our stable

118
isotope labeling method to measure CSF protein half-life will be informative of SOD1
biochemistry within the human CNS.
Identification of misfolded SOD1 protein in humans by protein turnover will be of great
value to developing ALS biomarkers, as the amorphous structure of SOD1 aggregates
(Matsumoto et al 2006; Matsumoto et al 2005) impedes the development of imaging tracers to
visualize protein misfolding events in human CNS tissues. For example, efforts to develop
positron emission tomography radiotracers that bind amyloid-β (Klunk et al 2004) and tau
(Johnson et al 2016) protein aggregates in Alzheimer’s Disease have proven successful due to
the β-sheet rich, amyloid structures formed by these proteins. Therefore, to confirm that
alterations in SOD1 protein kinetics describes misfolded SOD1 protein, future work must
continue to develop other biochemical assays that specifically identify misfolded, pathogenic
SOD1. A newly emerging biochemical property of misfolded proteins that cause
neurodegenerative diseases is the amplification of a protein aggregate by templated
conformational changes, ie.) “seeding”, similar to prion diseases (Kaufman & Diamond 2013;
Prusiner 1982). Multiple assays have been developed to identify pathogenic protein seeds for
proteins such as tau (Holmes et al 2013), alpha synuclein (Groveman et al 2018), and huntingtin
(Tan et al 2015) in both CNS tissue and CSF of AD, PD, and HD patients. Despite the non-
amyloid structure of SOD1 aggregates, multiple research groups demonstrated the ability of
recombinant and cellular/tissue derived mutant SOD1 to seed intracellular SOD1 and propagate
extracellularly (Ayers et al 2014; Chia et al 2010; Munch et al 2011). Therefore, the
development of an SOD1 seeding assay may overcome previous limitations of antibody capture-
based methods to identify misfolded SOD1 species, while helping to establish the relationship
between CSF SOD1 turnover and protein misfolding.

119
Future studies must also aim to elucidate the biochemical composition of SOD1 in human
CSF to understand the relationship of a CSF biomarker to protein misfolding in the CNS.
Dimerization is a unique feature of SOD1 structure compared to other proteins that contribute to
neurodegeneration, and has been studied extensively to understand mutant SOD1 toxicity.
Thermodynamic analyses of recombinant SOD1 protein (Khare & Dokholyan 2006; Lindberg et
al) and imaging of SOD1 aggregation in live cells (Kim et al 2014) suggest that dimer
destabilization is the first step in the production of SOD1 monomers that are capable of
misfolding and forming protein aggregates. These observations are further supported by our
natural history data, as A4 SOD1 mutants (A4V, A4T), and C-terminal missense and truncation
mutants (V148G, G141X) are found at the SOD1 dimerization interface (Keerthana &
Kolandaivel 2015) and show fast mean disease durations of less than 2 years. Moreover, the
strongest correlation of SOD1 biochemical parameters in vitro and ALS clinical phenotypes is
rate of disease progression and aggregation propensity of mutant protein (Pratt et al 2014;
Prudencio et al 2009; Wang et al 2008).
An added layer of complexity presents itself when considering ALSSOD1
, as three potential
SOD1 dimer pairs are possible: WT-WT, WT-Mutant, and Mutant-Mutant. Both mutant
homodimers and WT-mutant heterodimers are implicated in motor neuron toxicity. In a subset of
transgenic models, multiple reports suggest that crossing mutant SOD1-expressing transgenic
mice with WT hSOD1-expressing mice accelerates disease phenotypes (Deng et al 2006; Wang
et al 2009). Similarly, Witan and colleagues created artificial heterodimer and homodimer
constructs of mutant and WT SOD1 and showed WT-mutant heterodimers induce a more toxic
phenotype than mutant-mutant homodimers in C. Elegans (Witan et al 2008). The process of
heterodimerization also appears to correlate with SOD1ALS
disease progression, as Shi and

120
colleagues demonstrated faster disease progression in SOD1 mutants that have a more favorable
Gibbs free energy association of heterodimerization between mutant and WT recombinant SOD1
protein (Shi et al 2016).
Despite these extensive characterizations in vitro or overexpression paradigms in vivo, there
are relatively few analyses examining SOD1 monomer and dimer state in humans that validate
the physiological relevance of these laboratory findings to human ALS. The mass spectrometry-
based methods I developed to quantify protein concentration and turnover of mutant and WT
SOD1 protein in human CSF provide the foundation to investigate these questions in living ALS
participants. Further technical developments of “Top-Down” analyses to examine intact protein
(Padula et al 2017) should thus aim to establish a relationship between CSF SOD1 biochemical
characteristics and ALS clinical phenotypes. Future experiments using induced pluripotent stem
cell-derived motor neuron should also examine how these observations in human CSF relate to
motor neuron toxicity. By using iPSC’s derived from SOD1 mutant carriers, one can also create
isogenic WT SOD1 or mutant SOD1-expressing lines and compare the three cell lines to test
hypotheses describing the role of mutant SOD1 protein stability, dimer stability, and the
presence of WT SOD1 modulating motor neuron toxicity (Imamura et al 2017; Wainger et al
2014).
Multiple studies have suggested a relationship between mutant SOD1 stability and rate of
disease progression after symptom onset, but no work has reported a correlation with age of
disease onset. One possible hypothesis that explains these results is that changes in proteostasis
associated with aging drive the rate of disease onset, as protein degradation rates decrease over
time within cells (McShane et al 2016). Previous work supports this hypothesis in other
neurodegenerative diseases, as Patterson and colleagues demonstrated a highly significant

121
correlation between age and slow amyloid-beta turnover rates, suggesting that slowing of
amyloid-beta turnover increases the likelihood of protein misfolding that leads to deposition
(Patterson et al 2015). Similarly, single cell analysis of motor neurons in culture suggests that
slower turnover of proteins implicated in ALS correlates to a cell’s susceptibility to degeneration
(Barmada et al 2014), and aging is one of the strongest risk factors for ALS (Niccoli et al 2017).
SOD1 is degraded by the ubiquitin proteasome system and macroautophagy within cells (Kabuta
et al 2006), two pathways that slow over time (Dhondt et al 2017), suggesting that age-dependent
effects of SOD1 turnover in the CNS and CSF may be observed in ALSSOD1
. Families that carry
the A4V SOD1 mutation are an ideal population to investigate the association between aging,
SOD1 turnover, and clinical phenotypes, as the mutation exhibits a high degree of disease
penetrance, with a rapid disease progression after symptom onset with little variability between
patients. To determine if these changes in SOD1 turnover relate to motor neuron degeneration
during disease time course, additional analyses should examine changes in CSF SOD1 kinetics
with prognostic biomarkers associated with neuronal death, such as neurofilament light chain in
CSF or plasma (Bacioglu et al 2016; Gaiani et al 2017; Lu et al 2015) or p75 extracellular
domain fragments found in patient urine (Shepheard et al 2017).
Future investigation of the ALSSOD1
population will be critical to establish any
relationship between SOD1 half-life measures and disease pathobiology, and will determine the
feasibility of identifying misfolded WT SOD1 by kinetics-based measures in ALS patients that
do not have SOD1 mutations. Without a basic understanding of mutant SOD1 protein behavior,
any investigation into kinetics-based approaches to test the hypothesis of misfolded SOD1 in
sporadic ALS without SOD1 mutations will be difficult to interpret. Although the identification
of SOD1 involvement in sporadic ALS would have a significant impact on treatment strategies

122
for the entire ALS population, further investigation in ALSSOD1
may confirm that pathogenic
mechanisms associated with SOD1 mutations are specific to this subpopulation. If the
interpretation of changes in CSF SOD1 kinetics is ultimately specific to ALSSOD1
, future efforts
should aim to use similar approaches on other proteins implicated in disease to expand the scope
of the relationship between protein turnover and ALS pathogenesis.
Of particular interest is TDP-43, the protein found in insoluble aggregates of motor
neurons in the remaining 98% of ALS patients that do not carry SOD1 mutations (Neumann et al
2006). Stable isotope labeling with 13
C6-Leucine to measure TDP-43 protein kinetics in CSF of
ALS patients is theoretically feasible, and the recent identification of TDP-43 in exosomes
derived from CSF suggests that a CNS-specific pool of TDP-43 can be analyzed to measure
protein turnover (Feneberg et al 2018; Feneberg et al 2014). Similar experiments described
previously for CSF SOD1 can be directly translated to studies of TDP-43 in ALS, as the two
proteins share similar characteristics of misfolding such as the involvement of post-translational
modifications (Fujishiro et al 2009; Igaz et al 2008), and prion-like propagation (Nonaka et al
2013). Aggregates of TDP-43 and C-terminal fragments of TDP-43 also appear to form amyloid
structures (Bigio et al ; Garnier et al 2017), opening the possibility for future development of an
imaging agent to observe TDP-43 aggregates in living ALS patients. Combining our SILK
analysis with imaging would be invaluable to understand the relationship between CSF protein
biomarkers and disease progression in the brain and spinal cord of ALS patients, as CSF protein
biomarkers may provide a great cost effectiveness to implement in in the clinic compared to high
costs associated with PET imaging scans (Mitka). In sum, future investigations of CSF protein
kinetics should aim to identify a mechanistic link between protein homeostasis and
neurodegeneration to better understand disease biology. Such findings would be crucial to help

123
understand the variability in prognosis and clinical phenotypes in ALS, with the hopes of leading
to improved patient outcomes. Further, as new therapeutic candidates emerge that correct TDP-
43 pathology (Becker et al 2017), an understanding of TDP-43 turnover in the CNS will be
needed.
New Protein Production is a viable biomarker for RNA-lowering therapies
Understanding of protein turnover has direct application for experimental therapies under
investigation for the treatment of ALS. In Chapter 4, I described how measurements of new
protein production using 13
C6-Leucine labeling serves as a pharmacodynamics assay to
determine target engagement after therapeutic treatment. The primary focus of
pharmacodynamics measures for ASOs in neurodegenerative diseases is target protein
concentration in cerebrospinal fluid, confirmed by results in preclinical disease models that
suggest changes in CNS protein levels are reflected in CSF (DeVos et al 2017; McCampbell et al
2018). Excitingly, we observed changes in newly produced protein at earlier time points than
protein concentration in both the CNS and CSF after ASO treatment (Figures 4.1, 4.2),
suggesting that measures of newly produced protein are more sensitive measures of target
engagement for treatments that inhibit protein synthesis. As stable isotope labeling is safe in
humans (Crisp et al 2015; Ovod et al 2017; Sato et al 2018), measures of new protein production
to determine drug target engagement are directly translatable to randomized control trials with
drugs that inhibit SOD1 synthesis (Thomsen et al 2014) or promote protein clearance (Kieran et
al 2004). As SOD1 ASO treatment is currently in clinical trials and using CSF SOD1 protein
concentration as a secondary outcome (NCT02623699), our measures of new protein production
can also be implemented into future studies. As most previous multicenter randomized control
trials in ALS have failed to analyze any pharmacodynamics biomarkers (Mitsumoto et al 2014),

124
our results open a new opportunity to use protein kinetics to better understand the effects of
experimental drugs on human ALS biology.
Protein Turnover as a measure of pharmacodynamics for SOD1-targeting therapies
My research specifically focused on new protein production using stable isotope labeling
with 13
C6-Leucine, but measures of protein turnover and half-life also have potential utility as
pharmacodynamics biomarkers. For example, an emerging class of drugs targets the proper
folding and maturation of SOD1 to prevent mutant SOD1 toxicity. In a chaperone-mediated
strategy, Das and colleagues developed a small molecule that inhibits protein phosphatase 1 to
increase the availability of chaperones to bind to misfolded SOD1. Treatment with this molecule
lowered misfolded SOD1 protein levels without changing total SOD1 protein concentration in
G93A SOD1 transgenic mice, preventing motor neuron loss and rescuing motor deficits (Das et
al 2015). Nagy and colleagues showed similar results through genetic overexpression of HSP110
in mutant SOD1 transgenic animals (Nagy et al 2016). In an alternative approach, Capper and
colleagues recently described a cysteine-reactive small molecule that promotes dimerization of
SOD1 protein, restoring the monomer-dimer equilibrium of A4V SOD1 to wild-type in cell
culture (Capper et al 2018). These data suggest that measures of protein concentration will not
serve as a successful pharmacodynamics biomarker. However, stabilizing mutant SOD1 protein
may lead to alterations in protein turnover, suggesting that half-life measurements of mutant
SOD1 protein after treatment may be a viable biomarker. These hypotheses can be directly tested
in vivo using our current labeling methods in preclinical models and humans, but also have the
utility to be used as an assay to screen compounds that target proteostasis in neuronal cell culture
(Sato et al 2018; Zhang et al 2014). To ultimately serve as a pharmacodynamics biomarker, it is
imperative to determine the effect that such treatments will have on CSF SOD1. Such findings

125
have utility to both identify better measures of drug mechanism of action, but also our
understanding of the biology underlying SOD1 release from cells into CSF.
Understanding the mechanisms of CNS protein deposition into the CSF
Despite significant progress in our understanding of the correlations between CNS and
CSF protein behavior, the mechanisms that govern SOD1 protein release into the extracellular
space remain poorly understood. Traditionally thought to be a passive process associated with
motor neuron degeneration, emerging evidence suggests that human cells secrete misfolded
proteins through an active process (Fontaine et al 2016; Lee et al 2016a). Multiple groups
reported unconventional secretion of mutant SOD1 in lower-order organisms such as yeast
(Cruz-Garcia et al 2017) and human neurons through interaction with neurosecretory vesicles
(Urushitani et al 2006). An additional factor implicated in extracellular release of proteins is
neuronal activity, as increased neuronal activity leads to elevated levels of both amyloid beta
(Cirrito et al 2003) and tau (Yamada et al 2014). Evidence suggests mutant SOD1-expressing
neurons exhibit a hyperexcitability phenotype, so neuronal activity in the brain and spinal cord
may influence extracellular release of SOD1 (Wainger et al 2014). Although previous data has
primarily focused on neuronal release of misfolded proteins, it is also unknown how each cell
type of the brain contributes to the pool of SOD1 found in CSF. Experiments in both transgenic
animal models and motor neuron cell cultures can contribute to characterizing changes in SOD1
turnover and extracellular release over time and through disease time course. Excitingly, new
drug candidates that target proper SOD1 folding and maturation provide an opportunity to
provide a more complete picture on the role of SOD1 protein turnover affecting the behavior of
CSF SOD1 protein by measures of protein concentration and half-life in vivo.

126
Similarly, no studies have examined the clearance of either mutant or WT SOD1 protein
from CSF. The identification of paravascular and dural lymphatic vascular systems that drive
interstitial fluid and CSF flow (Aspelund et al 2015; Iliff et al 2012) present exciting
opportunities to determine how the clearance of SOD1 protein influences measures of CSF
SOD1 protein half-life. Within both vascular systems, the clearance of small fluorescent tracers
and soluble amyloid beta peptide from CSF required specific receptors such as Aquaporin 4 and
Vascular Endothelial Growth Factor Receptor. The molecular weight of SOD1 dimers (32 kDA)
is similar to the size of fluorescent tracers used in these studies, suggesting experiments
examining tagged SOD1 clearance from CSF are feasible to test hypotheses related to similar
ligand-receptor relationships for the clearance of SOD1 protein in animal model systems.
Therefore, future endeavors into understanding the mechanisms that drive SOD1 entrance and
clearance from CSF will further advance our understanding of CSF protein biology and its utility
as a biomarker in disease states.
Concluding Remarks
The original report of ASOs targeting SOD1 in ALS was released over 10 years ago
(Smith et al 2006), pioneering the use of RNA-lowering therapies for diseases of the CNS. The
recent approval of ASOs that modulate splicing in juvenile-onset spinal muscular atrophy (Finkel
et al 2017) pave the way for potential of RNA-lowering therapies for progressive
neurodegenerative diseases to be realized in the near future. However, multiple challenges will
persist as these promising therapeutic candidates advance through the clinic, and the SOD1 ASO
randomized control trials will define the ability of the neurology field to learn from past failures
in clinical trial design. In this dissertation, I provided new insights into ALSSOD1
epidemiology,
CSF SOD1 protein behavior, and pharmacodynamics biomarker development to hopefully lead

127
to successful approval of ASO treatment in ALSSOD1
. These challenges, among others, will
continue to present themselves in the development of RNA-targeted therapies for other types of
ALS, Alzheimer’s Disease, Parkinson’s Disease, Huntington’s Disease, Charcot-Marie Tooth
Disease (Zhao et al 2018), as well as future disorders with yet to be defined targets. In sum, I
hope the work described in this dissertation provides the foundation for future investigators in
disease research, where experiments in the laboratory and clinic combine to reveal breakthroughs
that ultimately lead to improvements in our understanding of human biology to combat and one
day cure these devastating neurological diseases.

128
References
Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre,
randomised, double-blind, placebo-controlled trial. The Lancet Neurology 12:339-45
2017. Open-label 24-week extension study of edaravone (MCI-186) in amyotrophic lateral
sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 18:55-63
Abe K. 1997. Clinical and molecular analysis of neurodegenerative diseases. Tohoku J Exp Med
181:389-409
Abe K, Aoki M, Tsuji T, Itoyama Y. 2017. Safety and efficacy of edaravone in well defined
patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-
controlled trial. Lancet Neurol 16:505-12
Aggarwal SP, Zinman L, Simpson E, McKinley J, Jackson KE, et al. 2010. Safety and efficacy
of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a
randomised, double-blind, placebo-controlled trial. The Lancet Neurology 9:481-8
Alonso A, Logroscino G, Jick SS, Hernan MA. 2010. Association of smoking with amyotrophic
lateral sclerosis risk and survival in men and women: a prospective study. BMC Neurol
10:1471-2377
Alzheimer A. 1906. Über einen eigenartigen schweren Erkrankungsprozeβ der Hirnrincle.
Neurol Central 25
Andersen PM. 2001. Genetics of sporadic ALS. Amyotroph Lateral Scler Other Motor Neuron
Disord 2:S37-41
Andersen PM, Nilsson P, Keranen ML, Forsgren L, Hagglund J, et al. 1997a. Phenotypic
heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase
mutations in Scandinavia. Brain 120 ( Pt 10):1723-37
Andersen PM, Nilsson P, Keranen ML, Forsgren L, Hagglund J, et al. 1997b. Phenotypic
heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase
mutations in Scandinavia. Brain 120:1723-37
Andersen PM, Sims KB, Xin WW, Kiely R, O'Neill G, et al. 2003. Sixteen novel mutations in
the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of
discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord
4:62-73
Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, et al. 2003. Hyperphosphorylation
and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem
86:582-90
Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, et al. 1993. Mild ALS in Japan
associated with novel SOD mutation. Nat Genet 5:323-4
Armon C. 2009. Smoking may be considered an established risk factor for sporadic ALS.
Neurology 73:1693-8
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, et al. 2013. Unconventional translation
of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to
c9FTD/ALS. Neuron 77:639-46

129
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, et al. 2015. A dural lymphatic
vascular system that drains brain interstitial fluid and macromolecules. The Journal of
Experimental Medicine 212:991-9
Avraham KB, Schickler M, Sapoznikov D, Yarom R, Groner Y. 1988. Down's syndrome:
abnormal neuromuscular junction in tongue of transgenic mice with elevated levels of
human Cu/Zn-superoxide dismutase. Cell 54:823-9
Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, et al. 2014. Experimental
transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol 128:791-803
Bacioglu M, Maia LF, Preische O, Schelle J, Apel A, et al. 2016. Neurofilament Light Chain in
Blood and CSF as Marker of Disease Progression in Mouse Models and in
Neurodegenerative Diseases. Neuron 91:56-66
Bali T, Self W, Liu J, Siddique T, Wang LH, et al. 2017. Defining SOD1 ALS natural history to
guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry 88:99-105
Bandyopadhyay U, Nagy M, Fenton WA, Horwich AL. 2014. Absence of lipofuscin in motor
neurons of SOD1-linked ALS mice. Proc Natl Acad Sci U S A 111:11055-60
Banik A, Brown RE, Bamburg J, Lahiri DK, Khurana D, et al. 2015. Translation of Pre-Clinical
Studies into Successful Clinical Trials for Alzheimer's Disease: What are the Roadblocks
and How Can They Be Overcome? J Alzheimers Dis 47:815-43
Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, et al. 2014. Autophagy induction enhances
TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 10:677-85
Bateman RJ, Munsell LY, Chen X, Holtzman DM, Yarasheski KE. 2007. Stable isotope labeling
tandem mass spectrometry (SILT) to quantify protein production and clearance rates. J
Am Soc Mass Spectrom 18:997-1006
Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. 2006. Human
amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo.
Nat Med 12:856-61
Bayever E, Iversen PL, Bishop MR, Sharp JG, Tewary HK, et al. 1993. Systemic administration
of a phosphorothioate oligonucleotide with a sequence complementary to p53 for acute
myelogenous leukemia and myelodysplastic syndrome: initial results of a phase I trial.
Antisense Res Dev 3:383-90
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, et al. 2017. Therapeutic reduction of ataxin-
2 extends lifespan and reduces pathology in TDP-43 mice. Nature advance online
publication
Bedlack RS, Pastula DM, Welsh E, Pulley D, Cudkowicz ME. 2008. Scrutinizing enrollment in
ALS clinical trials: room for improvement? Amyotroph Lateral Scler 9:257-65
Beers DR, Zhao W, Wang J, Zhang X, Wen S, et al. 2017. ALS patients' regulatory T
lymphocytes are dysfunctional, and correlate with disease progression rate and severity.
JCI Insight 2:89530
Beghi E, Logroscino G, Chio A, Hardiman O, Millul A, et al. 2010. Amyotrophic lateral
sclerosis, physical exercise, trauma and sports: results of a population-based pilot case-
control study. Amyotroph Lateral Scler 11:289-92
Beghi E, Pupillo E, Bonito V, Buzzi P, Caponnetto C, et al. 2013. Randomized double-blind
placebo-controlled trial of acetyl-L-carnitine for ALS. Amyotrophic Lateral Sclerosis and
Frontotemporal Degeneration 14:397-405

130
Benatar M, Wuu J, Andersen PM, Atassi N, David W, et al. 2018. Randomized, double-blind,
placebo-controlled trial of arimoclomol in rapidly progressive <em>SOD1</em> ALS.
Neurology
Bennett CF, Swayze EE. 2010. RNA Targeting Therapeutics: Molecular Mechanisms of
Antisense Oligonucleotides as a Therapeutic Platform. Annual Review of Pharmacology
and Toxicology 50:259-93
Bensimon G, Lacomblez L, Meininger V. 1994. A controlled trial of riluzole in amyotrophic
lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 330:585-91
Berry JD, Shefner JM, Conwit R, Schoenfeld D, Keroack M, et al. 2013. Design and Initial
Results of a Multi-Phase Randomized Trial of Ceftriaxone in Amyotrophic Lateral
Sclerosis. PLoS One 8:e61177
Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease: J Clin Invest.
2005 Jun 1;115(6):1449-57. doi:10.1172/JCI24761.
Bethlem J, Den Hartog Jager WA. 1960. The incidence and characteristics of Lewy bodies in
idiopathic paralysis agitans (Parkinson's disease). J Neurol Neurosurg Psychiatry 23:74-
80
Bigio EH, Wu JY, Deng HX, Bit-Ivan EN, Mao Q, et al. Inclusions in frontotemporal lobar
degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis
(ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid:
Acta Neuropathol. 2013 Mar;125(3):463-5. doi: 10.1007/s00401-013-1089-6. Epub 2013
Feb 3.
Boillee S. 2006. Onset and Progression in Inherited ALS Determined by Motor Neurons and
Microglia. Science 312:1389-92
Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, et al. 1994. Superoxide dismutase 1 with
mutations linked to familial amyotrophic lateral sclerosis possesses significant activity.
Proc Natl Acad Sci U S A 91:8292-6
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, et al. 2010. Wild-type and mutant
SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat
Neurosci 13:1396-403
Braak H, Braak E. 1995. Staging of Alzheimer's disease-related neurofibrillary changes.
Neurobiol Aging 16:271-8
Bruijn LI. 1998. Aggregation and Motor Neuron Toxicity of an ALS-Linked SOD1 Mutant
Independent from Wild-Type SOD1. Science 281:1851-4
Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, et al. 1997. ALS-linked SOD1
mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease
with SOD1-containing inclusions. Neuron 18:327-38
Byrne S, Walsh C, Lynch C, Bede P, Elamin M, et al. 2011. Rate of familial amyotrophic lateral
sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 82:623-
7
Capper MJ, Wright GSA, Barbieri L, Luchinat E, Mercatelli E, et al. 2018. The cysteine-reactive
small molecule ebselen facilitates effective SOD1 maturation. Nat Commun 9:018-04114
Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, et al. 2013. Trehalose delays the
progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons.
Autophagy 9:1308-20

131
Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, et al. 1999. The ALSFRS-R: a revised
ALS functional rating scale that incorporates assessments of respiratory function. BDNF
ALS Study Group (Phase III). J Neurol Sci 169:13-21
Cha JR, St Louis KJ, Tradewell ML, Gentil BJ, Minotti S, et al. 2014. A novel small molecule
HSP90 inhibitor, NXD30001, differentially induces heat shock proteins in nervous tissue
in culture and in vivo. Cell Stress Chaperones 19:421-35
Chan PH, Kawase M, Murakami K, Chen SF, Li Y, et al. 1998. Overexpression of SOD1 in
transgenic rats protects vulnerable neurons against ischemic damage after global cerebral
ischemia and reperfusion. J Neurosci 18:8292-9
Charcot J. 1874. De la sclérose latérale amyotrophique. Prog Med 2:341-453
Chen L, Zhang B, Chen R, Tang L, Liu R, et al. 2015. Natural history and clinical features of
sporadic amyotrophic lateral sclerosis in China. J Neurol Neurosurg Psychiatry 86:1075-
81
Cheroni C, Marino M, Tortarolo M, Veglianese P, De Biasi S, et al. 2009. Functional alterations
of the ubiquitin-proteasome system in motor neurons of a mouse model of familial
amyotrophic lateral sclerosis. Hum Mol Genet 18:82-96
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, et al. 2015. C9ORF72 repeat
expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits.
Science 14
Chia R, Tattum MH, Jones S, Collinge J, Fisher EM, Jackson GS. 2010. Superoxide dismutase 1
and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death
mechanism in amyotrophic lateral sclerosis. PLoS One 5:e10627
Chiò A, Borghero G, Calvo A, Capasso M, Caponnetto C, et al. 2010. Lithium carbonate in
amyotrophic lateral sclerosis. Neurology 75:619
Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, et al. 2009a. Prognostic factors in
ALS: A critical review. Amyotroph Lateral Scler 10:310-23
Chio A, Mora G, Calvo A, Mazzini L, Bottacchi E, Mutani R. 2009b. Epidemiology of ALS in
Italy: a 10-year prospective population-based study. Neurology 72:725-31
Chiò A, Traynor BJ, Lombardo F, Fimognari M, Calvo A, et al. 2008. Prevalence of
<em>SOD1</em> mutations in the Italian ALS population. Neurology 70:533-7
Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, et al. 2003. In vivo assessment of
brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-
beta metabolism and half-life. J Neurosci 23:8844-53
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, et al. 2015. Exome sequencing in
amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436-41
Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, et al. 2003. Wild-type
nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science
302:113-7
Crisp MJ, Beckett J, Coates JR, Miller TM. 2013. Canine degenerative myelopathy: biochemical
characterization of superoxide dismutase 1 in the first naturally occurring non-human
amyotrophic lateral sclerosis model. Exp Neurol 248:1-9
Crisp MJ, Mawuenyega KG, Patterson BW, Reddy NC, Chott R, et al. 2015. In vivo kinetic
approach reveals slow SOD1 turnover in the CNS. J Clin Invest 125:2772-80
Cruz-Garcia D, Brouwers N, Duran JM, Mora G, Curwin AJ, Malhotra V. 2017. A diacidic motif
determines unconventional secretion of wild-type and ALS-linked mutant SOD1. J Cell
Biol 9:201704056

132
Cudkowicz ME, McKenna-Yasek D, Chen C, Hedley-Whyte ET, Brown RH, Jr. 1998. Limited
corticospinal tract involvement in amyotrophic lateral sclerosis subjects with the A4V
mutation in the copper/zinc superoxide dismutase gene. Ann Neurol 43:703-10
Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, et al. 1997. Epidemiology of
mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol 41:210-
21
Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, et al. 2013.
Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis
(EMPOWER): a randomised, double-blind, phase 3 trial. The Lancet Neurology 12:1059-
67
Cudkowicz Merit E, Shefner Jeremy M, Schoenfeld David A, Zhang H, Andreasson Katrin I, et
al. 2006. Trial of celecoxib in amyotrophic lateral sclerosis. Annals of Neurology 60:22-
31
Cummings JL, Morstorf T, Zhong K. 2014. Alzheimer’s disease drug-development pipeline: few
candidates, frequent failures. Alzheimer's Research & Therapy 6:37
Czaplinski A, Haverkamp LJ, Yen AA, Simpson EP, Lai EC, Appel SH. 2006a. The value of
database controls in pilot or futility studies in ALS. Neurology 67:1827-32
Czaplinski A, Yen AA, Simpson EP, Appel SH. 2006b. Slower disease progression and
prolonged survival in contemporary patients with amyotrophic lateral sclerosis: is the
natural history of amyotrophic lateral sclerosis changing? Arch Neurol 63:1139-43
Da Cruz S, Bui A, Saberi S, Lee SK, Stauffer J, et al. 2017. Misfolded SOD1 is not a primary
component of sporadic ALS. Acta Neuropathol 134:97-111
Das I, Krzyzosiak A, Schneider K, Wrabetz L, D'Antonio M, et al. 2015. Preventing proteostasis
diseases by selective inhibition of a phosphatase regulatory subunit. Science 348:239-42
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, et al. 2011. Mutations in UBQLN2
cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature
477:211-5
Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, et al. 2006. Conversion to the amyotrophic lateral
sclerosis phenotype is associated with intermolecular linked insoluble aggregates of
SOD1 in mitochondria. Proc Natl Acad Sci U S A 103:7142-7
DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, et al. 2017. Tau reduction
prevents neuronal loss and reverses pathological tau deposition and seeding in mice with
tauopathy. Sci Transl Med 9
DeVos SL, Miller TM. 2013a. Antisense oligonucleotides: treating neurodegeneration at the
level of RNA. Neurotherapeutics 10:486-97
DeVos SL, Miller TM. 2013b. Direct intraventricular delivery of drugs to the rodent central
nervous system. J Vis Exp 12:50326
Dhondt I, Petyuk VA, Bauer S, Brewer HM, Smith RD, et al. 2017. Changes of Protein Turnover
in Aging Caenorhabditis elegans. Mol Cell Proteomics 16:1621-33
Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. 2008. Human embryonic stem cell-
derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-
causing mutation. Cell Stem Cell 3:637-48
Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. 2007. Non-cell autonomous effect
of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci
10:608-14

133
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, et al. 1997. Aggregation of huntingtin in
neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990-3
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, et al. 2013. RNA toxicity from the
ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80:415-28
Donofrio PD, Bedlack R. Historical controls in ALS trials: a high seas rescue?: Neurology.
2011 Sep 6;77(10):936-7. doi: 10.1212/WNL.0b013e31822cfcb6. Epub 2011 Aug 3.
Dupuis L, Dengler R, Heneka MT, Meyer T, Zierz S, et al. 2012. A Randomized, Double Blind,
Placebo-Controlled Trial of Pioglitazone in Combination with Riluzole in Amyotrophic
Lateral Sclerosis. PLoS One 7:e37885
Durazo A, Shaw BF, Chattopadhyay M, Faull KF, Nersissian AM, et al. 2009. Metal-free
superoxide dismutase-1 and three different amyotrophic lateral sclerosis variants share a
similar partially unfolded beta-barrel at physiological temperature. J Biol Chem
284:34382-9
Eisen A, Mezei MM, Stewart HG, Fabros M, Gibson G, Andersen PM. 2008. SOD1 gene
mutations in ALS patients from British Columbia, Canada: clinical features,
neurophysiology and ethical issues in management. Amyotroph Lateral Scler 9:108-19
Fanara P, Wong PY, Husted KH, Liu S, Liu VM, et al. 2012. Cerebrospinal fluid-based kinetic
biomarkers of axonal transport in monitoring neurodegeneration. J Clin Invest 122:3159-
69
Fang T, Al Khleifat A, Meurgey JH, Jones A, Leigh PN, et al. 2018. Stage at which riluzole
treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective
analysis of data from a dose-ranging study. Lancet Neurol 7:30054-1
Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, et al. 2014. Full-length TDP-43 forms toxic
amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat
Commun 5
Farr GW, Ying Z, Fenton WA, Horwich AL. 2011. Hydrogen-deuterium exchange in vivo to
measure turnover of an ALS-associated mutant SOD1 protein in spinal cord of mice.
Protein Sci 20:1692-6
Felbecker A, Camu W, Valdmanis PN, Sperfeld AD, Waibel S, et al. 2010. Four familial ALS
pedigrees discordant for two SOD1 mutations: are all SOD1 mutations pathogenic? J
Neurol Neurosurg Psychiatry 81:572-7
Feneberg E, Gray E, Ansorge O, Talbot K, Turner MR. 2018. Towards a TDP-43-Based
Biomarker for ALS and FTLD. Mol Neurobiol 19:018-0947
Feneberg E, Steinacker P, Lehnert S, Schneider A, Walther P, et al. 2014. Limited role of free
TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph Lateral Scler
Frontotemporal Degener 15:351-6
Ferraiuolo L, Meyer K, Sherwood TW, Vick J, Likhite S, et al. 2016. Oligodendrocytes
contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc Natl
Acad Sci U S A 113:E6496-E505
Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, et al. 2006. Familial ALS-superoxide
dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad
Sci U S A 103:13860-5
Filezac de L'Etang A, Maharjan N, Cordeiro Brana M, Ruegsegger C, Rehmann R, et al. 2015.
Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER
stress in ALS. Nat Neurosci 5

134
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, et al. 2017. Nusinersen versus Sham
Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 377:1723-32
Fisher R, Pusztai L, Swanton C. 2013. Cancer heterogeneity: implications for targeted
therapeutics. British Journal of Cancer 108:479-85
Fontaine SN, Zheng D, Sabbagh JJ, Martin MD, Chaput D, et al. 2016. DnaJ/Hsc70 chaperone
complexes control the extracellular release of neurodegenerative-associated proteins.
EMBO J 35:1537-49
Forman MS, Trojanowski JQ, Lee VM. 2004. Neurodegenerative diseases: a decade of
discoveries paves the way for therapeutic breakthroughs. Nat Med 10:1055-63
Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, et al. 2010. Novel
antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS
One 5:0011552
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. 2008. Intravascular
AAV9 preferentially targets neonatal neurons and adult astrocytes. Nature Biotechnology
27:59-65
Fujishiro H, Uchikado H, Arai T, Hasegawa M, Akiyama H, et al. 2009. Accumulation of
phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta
Neuropathol 117:151-8
Gaiani A, Martinelli I, Bello L, Querin G, Puthenparampil M, et al. 2017. Diagnostic and
Prognostic Biomarkers in Amyotrophic Lateral Sclerosis: Neurofilament Light Chain
Levels in Definite Subtypes of Disease. JAMA Neurol 6
Gamez J, Caponnetto C, Ferrera L, Syriani E, Marini V, et al. 2011. I112M SOD1 mutation
causes ALS with rapid progression and reduced penetrance in four Mediterranean
families. Amyotroph Lateral Scler 12:70-5
Gamez J, Corbera-Bellalta M, Nogales G, Raguer N, Garcia-Arumi E, et al. 2006. Mutational
analysis of the Cu/Zn superoxide dismutase gene in a Catalan ALS population: should all
sporadic ALS cases also be screened for SOD1? J Neurol Sci 247:21-8
Garnier C, Devred F, Byrne D, Puppo R, Roman AY, et al. 2017. Zinc binding to RNA
recognition motif of TDP-43 induces the formation of amyloid-like aggregates. Sci Rep
7:017-07215
Geary RS, Norris D, Yu R, Bennett CF. 2015. Pharmacokinetics, biodistribution and cell uptake
of antisense oligonucleotides. Adv Drug Deliv Rev 87:46-51
Gellera C, Castellotti B, Riggio MC, Silani V, Morandi L, et al. 2001. Superoxide dismutase
gene mutations in Italian patients with familial and sporadic amyotrophic lateral
sclerosis: identification of three novel missense mutations. Neuromuscul Disord 11:404-
10
Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang Y-J, et al. 2017. Poly(GP) proteins are a
useful pharmacodynamic marker for <em>C9ORF72</em>-associated amyotrophic
lateral sclerosis. Science Translational Medicine 9
Gomes C, Escrevente C, Costa J. 2010. Mutant superoxide dismutase 1 overexpression in NSC-
34 cells: effect of trehalose on aggregation, TDP-43 localization and levels of co-
expressed glycoproteins. Neurosci Lett 475:145-9
Gordon PH. 2009. A placebo arm is not always necessary in clinical trials of amyotrophic lateral
sclerosis. Muscle Nerve 39:858-60

135
Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, et al. 2007. Efficacy of
minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial.
The Lancet Neurology 6:1045-53
Grad LI, Guest WC, Yanai A, Pokrishevsky E, O'Neill MA, et al. 2011. Intermolecular
transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S
A 108:16398-403
Graf M, Ecker D, Horowski R, Kramer B, Riederer P, et al. 2005. High dose vitamin E therapy
in amyotrophic lateral sclerosis as add-on therapy to riluzole: results of a placebo-
controlled double-blind study. Journal of Neural Transmission 112:649-60
Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, et al. 2006. Fatality in mice due to
oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441:537-41
Gros-Louis F, Soucy G, Lariviere R, Julien JP. 2010. Intracerebroventricular infusion of
monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1
mutant delays mortality in a mouse model of ALS. J Neurochem 113:1188-99
Groveman BR, Orru CD, Hughson AG, Raymond LD, Zanusso G, et al. 2018. Rapid and ultra-
sensitive quantitation of disease-associated alpha-synuclein seeds in brain and
cerebrospinal fluid by alphaSyn RT-QuIC. Acta Neuropathol Commun 6:018-0508
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, et al. 1994. Motor neuron
degeneration in mice that express a human Cu,Zn superoxide dismutase mutation.
Science 264:1772-5
Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, et al. 2011. Astrocytes from
familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824-8
Henry S, Stecker K, Brooks D, Monteith D, Conklin B, Bennett CF. 2000. Chemically modified
oligonucleotides exhibit decreased immune stimulation in mice. J Pharmacol Exp Ther
292:468-79
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, et al. 2013. Heparan sulfate proteoglycans
mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad
Sci U S A 110:29
Huntington G. 1872. On chorea. Medical and Surgical Reporter of Philadelphia 26:317-21
Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, et al. 2017. Age-Dependent Effects
of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis.
Neuron 96:1013-23
Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, et al. 2008. Enrichment of C-terminal
fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in
spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J
Pathol 173:182-94
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, et al. 2012. A Paravascular Pathway Facilitates
CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes,
Including Amyloid β. Science Translational Medicine 4:147ra11-ra11
Imamura K, Izumi Y, Watanabe A, Tsukita K, Woltjen K, et al. 2017. The Src/c-Abl pathway is
a potential therapeutic target in amyotrophic lateral sclerosis. Science Translational
Medicine 9
Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, et al. 2010. Misfolded Mutant SOD1
Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron
67:575-87

136
Iwao Y, Anraku M, Yamasaki K, Kragh-Hansen U, Kawai K, et al. 2006. Oxidation of Arg-410
promotes the elimination of human serum albumin. Biochim Biophys Acta 4:743-9
Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. 2001. CuZn
superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic
mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol
102:293-305
Jaarsma D, Teuling E, Haasdijk ED, De Zeeuw CI, Hoogenraad CC. 2008. Neuron-Specific
Expression of Mutant Superoxide Dismutase Is Sufficient to Induce Amyotrophic Lateral
Sclerosis in Transgenic Mice. Journal of Neuroscience 28:2075-88
Jacobsson J, Jonsson PA, Andersen PM, Forsgren L, Marklund SL. 2001. Superoxide dismutase
in CSF from amyotrophic lateral sclerosis patients with and without CuZn-superoxide
dismutase mutations. Brain 124:1461-6
Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, et al. 2013. Treatment of
HCV infection by targeting microRNA. N Engl J Med 368:1685-94
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, et al. 2010. Exome
sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857-64
Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, et al. 2014. Mutations in the Matrin 3 gene
cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664-6
Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, et al. 2016. Tau positron emission
tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79:110-9
Johnston JA. 2000. Formation of high molecular weight complexes of mutant Cu,Zn-superoxide
dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proceedings of the
National Academy of Sciences 97:12571-6
Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. 1997. Prognosis in familial
amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and
ala4val mutations in Cu,Zn superoxide dismutase. Neurology 48:55-7
Kabuta T, Suzuki Y, Wada K. 2006. Degradation of amyotrophic lateral sclerosis-linked mutant
Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol
Chem 281:30524-33
Kalin A, Medina-Paraiso E, Ishizaki K, Kim A, Zhang Y, et al. 2017. A safety analysis of
edaravone (MCI-186) during the first six cycles (24 weeks) of amyotrophic lateral
sclerosis (ALS) therapy from the double-blind period in three randomized, placebo-
controlled studies. Amyotroph Lateral Scler Frontotemporal Degener 18:71-9
Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, et al. 2013. Degeneration and impaired
regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat
Neurosci 16:571-9
Kato S, Takikawa M, Nakashima K, Hirano A, Cleveland DW, et al. 2000. New consensus
research on neuropathological aspects of familial amyotrophic lateral sclerosis with
superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons
and astrocytes. Amyotroph Lateral Scler Other Motor Neuron Disord 1:163-84
Kaufman SK, Diamond MI. 2013. Prion-like propagation of protein aggregation and related
therapeutic strategies. Neurotherapeutics 10:371-82
Kaufmann P, Thompson John LP, Levy G, Buchsbaum R, Shefner J, et al. 2009. Phase II trial of
CoQ10 for ALS finds insufficient evidence to justify phase III. Annals of Neurology
66:235-44

137
Keerthana SP, Kolandaivel P. 2015. Interaction between dimer interface residues of native and
mutated SOD1 protein: a theoretical study. JBIC Journal of Biological Inorganic
Chemistry 20:509-22
Kemshead JT, Ritter MA, Cotmore SF, Greaves MF. 1982. Human Thy-1: expression on the cell
surface of neuronal and glial cells. Brain Res 236:451-61
Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, et al. 2010. Amyotrophic lateral sclerosis is a
non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial
form. Acta Neuropathol 119:335-44
Khare SD, Dokholyan NV. 2006. Common dynamical signatures of familial amyotrophic lateral
sclerosis-associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc Natl
Acad Sci U S A 103:3147-52
Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. 2004.
Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease
progression in ALS mice. Nat Med 10:402-5
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, et al. 2011. Amyotrophic lateral
sclerosis. Lancet 377:942-55
Kim J, Lee H, Lee JH, Kwon DY, Genovesio A, et al. 2014. Dimerization, oligomerization, and
aggregation of human Amyotrophic lateral sclerosis Cu/Zn-superoxide dismutase 1
mutant forms in live cells. Journal of Biological Chemistry
Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, et al. 2004. Imaging brain amyloid in
Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 55:306-19
Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, et al. 2012. Sustained
therapeutic reversal of Huntington's disease by transient repression of huntingtin
synthesis. Neuron 74:1031-44
Kostrzewa M, Damian MS, Muller U. 1996. Superoxide dismutase 1: identification of a novel
mutation in a case of familial amyotrophic lateral sclerosis. Hum Genet 98:48-50
Koval ED, Shaner C, Zhang P, du Maine X, Fischer K, et al. 2013. Method for widespread
microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet
22:4127-35
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, et al. 2014. Poly-dipeptides encoded by the
C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345:1139-
45
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. 1996. Dose-ranging study of
riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study
Group II. Lancet 347:1425-31
Lange DJ, Andersen PM, Remanan R, Marklund S, Benjamin D. 2013. Pyrimethamine decreases
levels of SOD1 in leukocytes and cerebrospinal fluid of ALS patients: a phase I pilot
study. Amyotroph Lateral Scler Frontotemporal Degener 14:199-204
Lange DJ, Shahbazi M, Silani V, Ludolph AC, Weishaupt JH, et al. Pyrimethamine Significantly
Lowers CSF/SOD1 in ALS Patients With SOD1 Mutations. Annals of Neurology:n/a-n/a
Lanoiselee HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, et al. 2017. APP, PSEN1,
and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of
familial and sporadic cases. PLoS Med 14
Lauria G, Campanella A, Filippini G, Martini A, Penza P, et al. 2009. Erythropoietin in
amyotrophic lateral sclerosis: A pilot, randomized, double-blind, placebo-controlled
study of safety and tolerability. Amyotrophic Lateral Sclerosis 10:410-5

138
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, et al. 2010. Enhancement of proteasome activity by
a small-molecule inhibitor of USP14. Nature 467:179-84
Lee JG, Takahama S, Zhang G, Tomarev SI, Ye Y. 2016a. Unconventional secretion of
misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells.
Nat Cell Biol 18:765-76
Lee S, Shang Y, Redmond SA, Urisman A, Tang AA, et al. 2016b. Activation of HIPK2
Promotes ER Stress-Mediated Neurodegeneration in Amyotrophic Lateral Sclerosis.
Neuron 91:41-55
Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, et al. 2010. ALS-linked mutant superoxide
dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein
import. Proc Natl Acad Sci U S A 107:21146-51
Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, et al. 1998. Aberrant RNA processing
in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in
amyotrophic lateral sclerosis. Neuron 20:589-602
Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase:
Disulfide reduction prevents dimerization and produces marginally stable monomers:
Proc Natl Acad Sci U S A. 2004 Nov 9;101 34 4 29 426 15 276 99 56 281 97 277 99 332
100 278 127 100 8 261 262 443 32 334 19 92 3 334 265 377 94 31 317 77 101 6 101 100
8 23 288 7 277 312 10 104 279 43 9 7 89 325 93(45):15893-
8:1383:62:529:884:91:12791:9010:523:1851:12571:15932:16607:601:7021:5984:73:361
7:933:16215:1744:313:4322:515:53:10869:R81:1077:21612:215:6084:765:639:334:511:
99:5976:9256:101:2872:413:1145:985:873:461:581:15499:4899:818:903:6109:367:1161
5. Epub 2004 Nov 2 doi:10.1073/pnas.0403979101.
Lindquist S. 1986. The heat-shock response. Annu Rev Biochem 55:1151-91
Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, et al. 2004. Toxicity of Familial ALS-Linked
SOD1 Mutants from Selective Recruitment to Spinal Mitochondria. Neuron 43:5-17
Liu J, Shinobu LA, Ward CM, Young D, Cleveland DW. 2005. Elevation of the Hsp70
chaperone does not effect toxicity in mouse models of familial amyotrophic lateral
sclerosis. Journal of Neurochemistry 93:875-82
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, et al. 2016. C9orf72 BAC Mouse Model with
Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 90:521-34
Lomen-Hoerth C, Anderson T, Miller B. 2002. The overlap of amyotrophic lateral sclerosis and
frontotemporal dementia. Neurology 59:1077-9
Lopate G, Baloh RH, Al-Lozi MT, Miller TM, Fernandes Filho JA, et al. 2010. Familial ALS
with extreme phenotypic variability due to the I113T SOD1 mutation. Amyotroph Lateral
Scler 11:232-6
Lu CH, Macdonald-Wallis C, Gray E, Pearce N, Petzold A, et al. 2015. Neurofilament light
chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology
1:0000000000001642
MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, et al. 2014. Guidelines for
investigating causality of sequence variants in human disease. Nature 508:469-76
Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, et al. 2017. TIA1 Mutations in
Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation
and Alter Stress Granule Dynamics. Neuron 95:808-16

139
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, et al. 2012. Frequency of the C9orf72
hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and
frontotemporal dementia: a cross-sectional study. Lancet Neurol 11:323-30
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, et al. 2010. Mutations of optineurin in
amyotrophic lateral sclerosis. Nature 465:223-6
Marwick C. First "antisense" drug will treat CMV retinitis: JAMA. 1998 Sep 9;280(10):871.
Mather M, Jacobsen L, Pollard KM. 2015. Aging in the United States. Population Reference
Burea 70
Matsumoto G, Kim S, Morimoto RI. 2006. Huntingtin and mutant SOD1 form aggregate
structures with distinct molecular properties in human cells. J Biol Chem 281:4477-85
Matsumoto G, Stojanovic A, Holmberg CI, Kim S, Morimoto RI. 2005. Structural properties and
neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase
1 aggregates. J Cell Biol 171:75-85
Mazur C, Fitzsimmons B, Kamme F, Nichols B, Powers B, Wancewicz E. 2017. Development of
a simple, rapid, and robust intrathecal catheterization method in the rat. J Neurosci
Methods 280:36-46
McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, et al. 2011. Preclinical Safety of
RNAi-Mediated HTT Suppression in the Rhesus Macaque as a Potential Therapy for
Huntington's Disease. Molecular Therapy 19:2152-62
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, et al. 2018. New SOD1
Antisense Oligonucelotides markedly extend survival and reverse muscle denervation in
SOD1 ALS rodent models. In Press.
McCombe PA, Henderson RD. 2010. Effects of gender in amyotrophic lateral sclerosis. Gend
Med 7:557-70
McCord JM, Fridovich I. 1969a. Superoxide dismutase. An enzymic function for erythrocuprein
(hemocuprein). J Biol Chem 244:6049-55
McCord JM, Fridovich I. 1969b. The utility of superoxide dismutase in studying free radical
reactions. I. Radicals generated by the interaction of sulfite, dimethyl sulfoxide, and
oxygen. J Biol Chem 244:6056-63
McShane E, Sin C, Zauber H, Wells JN, Donnelly N, et al. 2016. Kinetic Analysis of Protein
Stability Reveals Age-Dependent Degradation. Cell 167:803-15
Mehta P, Kaye W, Bryan L, Larson T, Copeland T, et al. 2016. Prevalence of Amyotrophic
Lateral Sclerosis - United States, 2012-2013. MMWR Surveill Summ 65:1-12
Meininger V, Asselain B, Leigh PN, Ludolph A, Lacomblez L, Guillet P. 2006. Reply from the
authors [6]. Neurology 66:1786-7
Meininger V, Bensimon G, Bradley WG, Brooks BR, Douillet P, et al. 2004. Efficacy and safety
of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials. Amyotrophic
Lateral Sclerosis and Other Motor Neuron Disorders 5:107-17
Meininger V, Drory VE, Leigh PN, Ludolph A, Robberecht W, Silani V. 2009. Glatiramer
acetate has no impact on disease progression in ALS at 40 mg/day: A double- blind,
randomized, multicentre, placebo-controlled trial. Amyotrophic Lateral Sclerosis 10:378-
83
Meissner F, Molawi K, Zychlinsky A. 2010. From the Cover: Mutant superoxide dismutase 1-
induced IL-1 accelerates ALS pathogenesis. Proceedings of the National Academy of
Sciences 107:13046-50

140
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, et al. 2016. Longitudinal effect of
eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann
Neurol 79:257-71
Messori A, Trippoli S, Becagli P, Zaccara G. 1999. Cost effectiveness of riluzole in amyotrophic
lateral sclerosis. Italian Cooperative Group for the Study of Meta-Analysis and the
Osservatorio SIFO sui Farmaci. Pharmacoeconomics 16:153-63
Milane A, Tortolano L, Fernandez C, Bensimon G, Meininger V, Farinotti R. 2009. Brain and
plasma riluzole pharmacokinetics: effect of minocycline combination. J Pharm Pharm
Sci 12:209-17
Miller R, Bradley W, Cudkowicz M, Hubble J, Meininger V, et al. 2007. Phase II/III randomized
trial of TCH346 in patients with ALS. Neurology 69:776-84
Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, et al. 2009. Practice parameter
update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and
respiratory therapies (an evidence-based review): report of the Quality Standards
Subcommittee of the American Academy of Neurology. Neurology 73:1218-26
Miller RG, Mitchell JD, Moore DH. 2012. Riluzole for amyotrophic lateral sclerosis
(ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 14
Miller RG, Moore DH, Forshew DA, Katz JS, Barohn RJ, et al. 2011. Phase II screening trial of
lithium carbonate in amyotrophic lateral sclerosis: examining a more efficient trial
design. Neurology 77:973-9
Miller TM, Kaspar BK, Kops GJ, Yamanaka K, Christian LJ, et al. 2005. Virus-delivered small
RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann Neurol 57:773-6
Miller TM, Kim SH, Yamanaka K, Hester M, Umapathi P, et al. 2006. Gene transfer
demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in
familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 103:19546-51
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, et al. 2013. An antisense
oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial
amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol
12:435-42
Mitka M. PET imaging for Alzheimer disease: are its benefits worth the cost?: JAMA. 2013 Mar
20;309(11):1099-100. doi: 10.1001/jama.2013.2101.
Mitsumoto H, Brooks BR, Silani V. 2014. Clinical trials in amyotrophic lateral sclerosis: why so
many negative trials and how can trials be improved? The Lancet Neurology 13:1127-38
Miyazaki K, Fujita T, Ozaki T, Kato C, Kurose Y, et al. 2004. NEDL1, a novel ubiquitin-protein
isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J Biol Chem
279:11327-35
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, et al. 2014. C9orf72 repeat
expansions cause neurodegeneration in Drosophila through arginine-rich proteins.
Science 345:1192-4
Moody LR, Barrett-Wilt GA, Sussman MR, Messing A. 2017. Glial fibrillary acidic protein
exhibits altered turnover kinetics in a mouse model of Alexander disease. Journal of
Biological Chemistry 292:5814-24
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, et al. 2013a. Bidirectional transcripts of
the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide
repeat proteins. Acta Neuropathol 126:881-93

141
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, et al. 2013b. The C9orf72 GGGGCC
repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science
339:1335-8
Mosley RL, Gordon PH, Hasiak CM, Van Wetering FJ, Mitsumoto H, Gendelman HE. 2007.
Glatiramer acetate immunization induces specific antibody and cytokine responses in
ALS patients. Amyotrophic Lateral Sclerosis 8:235-42
Mulligan VK, Kerman A, Laister RC, Sharda PR, Arslan PE, Chakrabartty A. 2012. Early steps
in oxidation-induced SOD1 misfolding: implications for non-amyloid protein aggregation
in familial ALS. J Mol Biol 421:631-52
Munch C, O'Brien J, Bertolotti A. 2011. Prion-like propagation of mutant superoxide dismutase-
1 misfolding in neuronal cells. Proc Natl Acad Sci U S A 108:3548-53
Nagano S, Takahashi Y, Yamamoto K, Masutani H, Fujiwara N, et al. 2015. A cysteine residue
affects the conformational state and neuronal toxicity of mutant SOD1 in mice -
Relevance to the pathogenesis of ALS. Hum Mol Genet 11
Nagy M, Fenton WA, Li D, Furtak K, Horwich AL. 2016. Extended survival of misfolded G85R
SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc Natl Acad
Sci U S A 113:5424-8
Nakamura M, Bieniek KF, Lin WL, Graff-Radford NR, Murray ME, et al. 2015. A truncating
SOD1 mutation, p.Gly141X, is associated with clinical and pathologic heterogeneity,
including frontotemporal lobar degeneration. Acta Neuropathol 28:28
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, et al. 2006. Ubiquitinated
TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science
314:130-3
Niccoli T, Partridge L, Isaacs AM. 2017. Ageing as a risk factor for ALS/FTD. Hum Mol Genet
26:R105-R13
Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, et al. 2018. Genome-wide Analyses
Identify KIF5A as a Novel ALS Gene. Neuron 97:1268-83
Niikura T, Kita Y, Abe Y. 2014. SUMO3 modification accelerates the aggregation of ALS-
linked SOD1 mutants. PLoS One 9
Niwa J, Ishigaki S, Hishikawa N, Yamamoto M, Doyu M, et al. 2002. Dorfin ubiquitylates
mutant SOD1 and prevents mutant SOD1-mediated neurotoxicity. J Biol Chem
277:36793-8
Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, et al. 2013. Prion-like
properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4:124-34
Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. 1995. Cloning and characterization
of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1
TAR DNA sequence motifs. J Virol 69:3584-96
Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, et al. 2017. Amyloid beta
concentrations and stable isotope labeling kinetics of human plasma specific to central
nervous system amyloidosis. Alzheimers Dement 11:32518-9
Padula MP, Berry IJ, O′Rourke MB, Raymond BBA, Santos J, Djordjevic SP. 2017. A
Comprehensive Guide for Performing Sample Preparation and Top-Down Protein
Analysis. Proteomes 5:11
Parge HE, Hallewell RA, Tainer JA. 1992. Atomic structures of wild-type and thermostable
mutant recombinant human Cu,Zn superoxide dismutase. Proc Natl Acad Sci U S A
89:6109-13

142
Parkinson J. 1969. An essay on the shaking palsy. Archives of Neurology 20:441-5
Pascuzzi RM, Shefner J, Chappell AS, Bjerke JS, Tamura R, et al. 2010. A phase II trial of
talampanel in subjects with amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis
11:266-71
Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, et al. 2004. Amyotrophic lateral
sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord
mitochondria. Neuron 43:19-30
Patel P, Kriz J, Gravel M, Soucy G, Bareil C, et al. 2014. Adeno-associated virus-mediated
delivery of a recombinant single-chain antibody against misfolded superoxide dismutase
for treatment of amyotrophic lateral sclerosis. Mol Ther 22:498-510
Patterson BW, Elbert DL, Mawuenyega KG, Kasten T, Ovod V, et al. 2015. Age and amyloid
effects on human central nervous system amyloid-beta kinetics. Ann Neurol 78:439-53
Petrov D, Mansfield C, Moussy A, Hermine O. 2017. ALS Clinical Trials Review: 20 Years of
Failure. Are We Any Closer to Registering a New Treatment? Front Aging Neurosci 9
Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, et al. 2009. Five siRNAs targeting three
SNPs in Huntingtin may provide therapy for three-quarters of Huntington’s disease
patients. Current biology : CB 19:774-8
Portet F, Cadilhac C, Touchon J, Camu W. 2001. Cognitive impairment in motor neuron disease
with bulbar onset. Amyotroph Lateral Scler Other Motor Neuron Disord 2:23-9
Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA. 2001. Neuron-specific
expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor
impairment. J Neurosci 21:3369-74
Pratt AJ, Shin DS, Merz GE, Rambo RP, Lancaster WA, et al. 2014. Aggregation propensities of
superoxide dismutase G93 hotspot mutants mirror ALS clinical phenotypes. Proc Natl
Acad Sci U S A 111:14
Prudencio M, Durazo A, Whitelegge JP, Borchelt DR. 2010. An examination of wild-type SOD1
in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum Mol
Genet 19:4774-89
Prudencio M, Hart PJ, Borchelt DR, Andersen PM. 2009. Variation in aggregation propensities
among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet
18:3217-26
Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136-44
Pupillo E, Messina P, Giussani G, Logroscino G, Zoccolella S, et al. 2014. Physical activity and
amyotrophic lateral sclerosis: a European population-based case-control study. Ann
Neurol 75:708-16
Qureshi M, Schoenfeld DA, Paliwal Y, Shui A, Cudkowicz ME. 2009. The natural history of
ALS is changing: improved survival. Amyotroph Lateral Scler 10:324-31
Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, et al. 2002. Oxidation-induced
misfolding and aggregation of superoxide dismutase and its implications for amyotrophic
lateral sclerosis. J Biol Chem 277:47551-6
Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, et al. 2005a. Silencing mutant SOD1
using RNAi protects against neurodegeneration and extends survival in an ALS model.
Nat Med 11:429-33
Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, et al. 2005b. Silencing mutant SOD1
using RNAi protects against neurodegeneration and extends survival in an ALS model.
Nature Medicine 11:429-33

143
Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, et al. 2005. Lentiviral-mediated
silencing of SOD1 through RNA interference retards disease onset and progression in a
mouse model of ALS. Nat Med 11:423-8
Ray SS, Nowak RJ, Brown RH, Jr., Lansbury PT, Jr. 2005. Small-molecule-mediated
stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase
mutants against unfolding and aggregation. Proc Natl Acad Sci U S A 102:3639-44
Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, et al. 1996. Motor neurons in
Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell
death after axonal injury. Nat Genet 13:43-7
Rechtman L, Jordan H, Wagner L, Horton DK, Kaye W. 2015. Racial and ethnic differences
among amyotrophic lateral sclerosis cases in the United States. Amyotroph Lateral Scler
Frontotemporal Degener 16:65-71
Renton AE, Chio A, Traynor BJ. 2014. State of play in amyotrophic lateral sclerosis genetics.
Nat Neurosci 17:17-23
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, et al. 2011. A hexanucleotide
repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD.
Neuron 72:257-68
Riboldi G, Zanetta C, Ranieri M, Nizzardo M, Simone C, et al. 2014. Antisense oligonucleotide
therapy for the treatment of C9ORF72 ALS/FTD diseases. Mol Neurobiol 50:721-32
Robberecht W, Philips T. 2013. The changing scene of amyotrophic lateral sclerosis. Nat Rev
Neurosci 14:248-64
Rodriguez JA. 2002. Familial Amyotrophic Lateral Sclerosis-associated Mutations Decrease the
Thermal Stability of Distinctly Metallated Species of Human Copper/Zinc Superoxide
Dismutase. Journal of Biological Chemistry 277:15932-7
Rosen DR, Bowling AC, Patterson D, Usdin TB, Sapp P, et al. 1994. A frequent ala 4 to val
superoxide dismutase-1 mutation is associated with a rapidly progressive familial
amyotrophic lateral sclerosis. Hum Mol Genet 3:981-7
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, et al. 1993. Mutations in Cu/Zn
superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis.
Nature 362:59-62
Rosenfeld J, King RM, Jackson CE, Bedlack RS, Barohn RJ, et al. 2008. Creatine monohydrate
in ALS: Effects on strength, fatigue, respiratory status and ALSFRS. Amyotrophic
Lateral Sclerosis 9:266-72
Rowland LP. 2001. How amyotrophic lateral sclerosis got its name: the clinical-pathologic
genius of Jean-Martin Charcot. Arch Neurol 58:512-5
Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, et al. 2012. SQSTM1 mutations in
frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79:1556-
62
Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, et al. 2017. Distinct roles for motor
neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proceedings of
the National Academy of Sciences
Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, et al. 2013. Targeting RNA foci in
iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci
Transl Med 5:3007529
Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, et al. 2018. Tau Kinetics
in Neurons and the Human Central Nervous System. Neuron 98:861-4

144
Sato T, Nakanishi T, Yamamoto Y, Andersen PM, Ogawa Y, et al. 2005. Rapid disease
progression correlates with instability of mutant SOD1 in familial ALS. Neurology
65:1954-7
Saxena S, Cabuy E, Caroni P. 2009. A role for motoneuron subtype–selective ER stress in
disease manifestations of FALS mice. Nature Neuroscience 12:627-36
Schmitt ND, Agar JN. 2017. Parsing disease-relevant protein modifications from epiphenomena:
perspective on the structural basis of SOD1-mediated ALS. J Mass Spectrom 52:480-91
Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, et al. 2016. Increased 4R-Tau Induces
Pathological Changes in a Human-Tau Mouse Model. Neuron 90:941-7
Schoch KM, Miller TM. 2017. Antisense Oligonucleotides: Translation from Mouse Models to
Human Neurodegenerative Diseases. Neuron 94:1056-70
Shefner JM, Cudkowicz ME, Schoenfeld D, Conrad T, Taft J, et al. 2004. A clinical trial of
creatine in ALS. Neurology 63:1656
Shefner JM, Reaume AG, Flood DG, Scott RW, Kowall NW, et al. 1999. Mice lacking cytosolic
copper/zinc superoxide dismutase display a distinctive motor axonopathy. Neurology
53:1239-46
Shepheard SR, Wuu J, Cardoso M, Wiklendt L, Dinning PG, et al. 2017. Urinary p75ECD: A
prognostic, disease progression, and pharmacodynamic biomarker in ALS. Neurology
22:0000000000003741
Shi Y, Acerson MJ, Abdolvahabi A, Mowery RA, Shaw BF. 2016. Gibbs Energy of Superoxide
Dismutase Heterodimerization Accounts for Variable Survival in Amyotrophic Lateral
Sclerosis. J Am Chem Soc 138:5351-62
Shi Y, Lin S, Staats KA, Li Y, Chang WH, et al. 2018. Haploinsufficiency leads to
neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 5
Shibata N, Hirano A, Kobayashi M, Dal Canto MC, Gurney ME, et al. 1998. Presence of Cu/Zn
superoxide dismutase (SOD) immunoreactivity in neuronal hyaline inclusions in spinal
cords from mice carrying a transgene for Gly93Ala mutant human Cu/Zn SOD. Acta
Neuropathol 95:136-42
Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, et al. 1996. Intense superoxide
dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial
amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp
Neurol 55:481-90
Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, et al. 2014. Exome-wide rare variant analysis
identifies TUBA4A mutations associated with familial ALS. Neuron 84:324-31
Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, et al. 2006. Antisense
oligonucleotide therapy for neurodegenerative disease. J Clin Invest 116:2290-6
Soejitno A, Tjan A, Purwata TE. 2015. Alzheimer's Disease: Lessons Learned from
Amyloidocentric Clinical Trials. CNS Drugs 29:487-502
Sone J, Niwa J, Kawai K, Ishigaki S, Yamada S, et al. 2010. Dorfin ameliorates phenotypes in a
transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci Res 88:123-35
Sorenson EJ, Stalker AP, Kurland LT, Windebank AJ. 2002. Amyotrophic lateral sclerosis in
Olmsted County, Minnesota, 1925 to 1998. Neurology 59:280-2
Sorenson EJ, Windbank AJ, Mandrekar JN, Bamlet WR, Appel SH, et al. 2008. Subcutaneous
IGF-1 is not beneficial in 2-year ALS trial. Neurology 71:1770-5
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, et al. 2008. TDP-43 mutations in familial
and sporadic amyotrophic lateral sclerosis. Science 319:1668-72

145
Steinacker P, Hendrich C, Sperfeld AD, Jesse S, von Arnim CA, et al. 2008. TDP-43 in
cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic
lateral sclerosis. Arch Neurol 65:1481-7
Stewart HG, Andersen PM, Eisen A, Weber M. 2006. Corticomotoneuronal dysfunction in ALS
patients with different SOD1 mutations. Clin Neurophysiol 117:1850-61
Su FC, Goutman SA, Chernyak S, Mukherjee B, Callaghan BC, et al. 2016. Association of
Environmental Toxins With Amyotrophic Lateral Sclerosis. JAMA Neurol 73:803-11
Talbot K. 2009. Motor neuron disease: the bare essentials. Pract Neurol 9:303-9
Tan Z, Dai W, van Erp TG, Overman J, Demuro A, et al. 2015. Huntington's disease
cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol Psychiatry 20:1286-93
Thomsen GM, Gowing G, Latter J, Chen M, Vit JP, et al. 2014. Delayed disease onset and
extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after
suppression of mutant SOD1 in the motor cortex. J Neurosci 34:15587-600
Tiwari A, Hayward LJ. 2003. Familial amyotrophic lateral sclerosis mutants of copper/zinc
superoxide dismutase are susceptible to disulfide reduction. J Biol Chem 278:5984-92
Tuszynski MH, Yang JH, Barba D, U HS, Bakay RA, et al. 2015. Nerve Growth Factor Gene
Therapy: Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol
72:1139-47
Urushitani M, Kurisu J, Tsukita K, Takahashi R. 2002. Proteasomal inhibition by misfolded
mutant superoxide dismutase 1 induces selective motor neuron death in familial
amyotrophic lateral sclerosis. J Neurochem 83:1030-42
Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. 2006. Chromogranin-
mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral
sclerosis. Nat Neurosci 9:108-18
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, et al. 2009. Mutations in FUS, an
RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science
323:1208-11
Vande Velde C, Miller TM, Cashman NR, Cleveland DW. 2008. Selective association of
misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria.
Proceedings of the National Academy of Sciences 105:4022-7
Verstraete E, Veldink JH, Huisman MHB, Draak T, Uijtendaal EV, et al. 2012. Lithium lacks
effect on survival in amyotrophic lateral sclerosis: a phase IIb randomised sequential
trial. Journal of Neurology, Neurosurgery & Psychiatry 83:557-64
Villemagne VL, Dore V, Burnham SC, Masters CL, Rowe CC. 2018. Imaging tau and amyloid-
beta proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol 16:9
Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, et al. 2014. Intrinsic membrane
hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep
7:1-11
Wang L, Deng H-X, Grisotti G, Zhai H, Siddique T, Roos RP. 2009. Wild-type SOD1
overexpression accelerates disease onset of a G85R SOD1 mouse. Human Molecular
Genetics 18:1642-51
Wang Q, Johnson JL, Agar NY, Agar JN. 2008. Protein aggregation and protein instability
govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol 6:0060170
Watson JD, Crick FH. 1953. Molecular structure of nucleic acids; a structure for deoxyribose
nucleic acid. Nature 171:737-8

146
Weisskopf MG, Cudkowicz ME, Johnson N. 2015. Military Service and Amyotrophic Lateral
Sclerosis in a Population-based Cohort. Epidemiology 26:831-8
Weisskopf MG, O'Reilly EJ, McCullough ML, Calle EE, Thun MJ, et al. 2005. Prospective
study of military service and mortality from ALS. Neurology 64:32-7
Westeneng H-J, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, et al. 2018. Prognosis for
patients with amyotrophic lateral sclerosis: development and validation of a personalised
prediction model. The Lancet Neurology 17:423-33
WHO. 2012. Dementia: A public health priority World Health Organization
Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, et al. 1996. Altered reactivity of
superoxide dismutase in familial amyotrophic lateral sclerosis. Science 271:515-8
Wildsmith KR, Basak JM, Patterson BW, Pyatkivskyy Y, Kim J, et al. 2012. In vivo human
apolipoprotein E isoform fractional turnover rates in the CNS. PLoS One 7:4
Williamson TL, Cleveland DW. 1999. Slowing of axonal transport is a very early event in the
toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2:50-6
Wilson SM, Bhattacharyya B, Rachel RA, Coppola V, Tessarollo L, et al. 2002. Synaptic defects
in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease.
Nat Genet 32:420-5
Winer L, Srinivasan D, Chun S, Lacomis D, Jaffa M, et al. 2013. SOD1 in cerebral spinal fluid
as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol
70:201-7
Witan H, Kern A, Koziollek-Drechsler I, Wade R, Behl C, Clement AM. 2008. Heterodimer
formation of wild-type and amyotrophic lateral sclerosis-causing mutant Cu/Zn-
superoxide dismutase induces toxicity independent of protein aggregation. Hum Mol
Genet 17:1373-85
Wong CH, Siah KW, Lo AW. 2018. Estimation of clinical trial success rates and related
parameters. Biostatistics:kxx069-kxx
Worms PM. 2001. The epidemiology of motor neuron diseases: a review of recent studies. J
Neurol Sci 191:3-9
Wright PD, Huang M, Weiss A, Matthews J, Wightman N, et al. 2010. Screening for inhibitors
of the SOD1 gene promoter: pyrimethamine does not reduce SOD1 levels in cell and
animal models. Neurosci Lett 482:188-92
Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, et al. 2012. Mutations in the profilin 1 gene
cause familial amyotrophic lateral sclerosis. Nature 488:499-503
Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. 2004. Determination of the Role of the
Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. Journal of
Biological Chemistry 279:17181-9
Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH. 2015. Endolysosomal Deficits Augment
Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron
87:355-70
Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, et al. 2014. Neuronal activity regulates
extracellular tau in vivo. The Journal of Experimental Medicine 211:387-93
Yang X, Nagasaki K, Egawa S, Maruyama K, Futami H, et al. 1995. FUS/TLS-CHOP chimeric
transcripts in liposarcoma tissues. Jpn J Clin Oncol 25:234-9
Yoshino H, Kimura A. 2006. Investigation of the therapeutic effects of edaravone, a free radical
scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph Lateral Scler
7:241-5

147
Zetterstrom P, Andersen PM, Brannstrom T, Marklund SL. 2011. Misfolded superoxide
dismutase-1 in CSF from amyotrophic lateral sclerosis patients. J Neurochem 117:91-9
Zhang G, Deinhardt K, Neubert TA. 2014. Stable isotope labeling by amino acids in cultured
primary neurons. Methods Mol Biol:1142-4_5
Zhang X, Li L, Chen S, Yang D, Wang Y, et al. 2011. Rapamycin treatment augments motor
neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis.
Autophagy 7:412-25
Zhao HT, Damle S, Ikeda-Lee K, Kuntz S, Li J, et al. 2018. PMP22 antisense oligonucleotides
reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J Clin Invest
128:359-68
Zhao HT, John N, Delic V, Ikeda-Lee K, Kim A, et al. 2017a. LRRK2 Antisense
Oligonucleotides Ameliorate alpha-Synuclein Inclusion Formation in a Parkinson's
Disease Mouse Model. Mol Ther Nucleic Acids 8:508-19
Zhao HT, John N, Delic V, Ikeda-Lee K, Kim A, et al. 2017b. LRRK2 Antisense
Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease
Mouse Model. Molecular Therapy. Nucleic Acids 8:508-19
Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, et al. 2010. Extracellular mutant SOD1
induces microglial-mediated motoneuron injury. Glia 58:231-43
Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, et al. 2008. ALS-causing SOD1 mutants
generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11:420-2
Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, et al. 2013. RAN proteins and RNA foci
from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proceedings
of the National Academy of Sciences 110:E4968-E77

148
Curriculum Vitae Wade K. Self
Washington University in St. Louis
Email: [email protected]
Education
2013- Current Washington University in St. Louis, St. Louis, MO
Doctoral Candidate (Expected August 2018)
Division of Biology and Biomedical Sciences
Graduate Program: Neuroscience
2009-2013 Case Western Reserve University, Cleveland, OH
Bachelor of Science in Engineering (May 2013)
GPA: 3.83
Major: Biomedical Engineering, Tissue Engineering Track
Research Experience
2013 - Current Doctoral Candidate
Washington University in St. Louis School of Medicine
Department of Neurology
Principal Investigator: Timothy Miller, M.D, Ph.D.
Dissertation: Understanding superoxide dismutase 1 turnover to improve
patient outcomes in amyotrophic lateral sclerosis
2012- 2013 Research Assistant
Case Western Reserve University, Department of Pathology
Principal Investigator: Xiongwei Zhu, Ph.D.
Project Description: Determining the aberrant localization of SREBP2 in
neurons of post mortem Alzheimer’s Disease hippocampal brain sections
2012 Summer Undergraduate Research Fellow
Mayo Clinic, Department of Neuroscience
Principal Investigator: Guojun Bu, Ph.D.
Project Description: Screening small molecule modulators of APOE
expression in astrocytes for the treatment of Alzheimer’s Disease
2010 - 2012 Undergraduate Research Assistant
Case Western Reserve University, Department of Biomedical Engineering,
Supervisor: Jeffrey Capadona, Ph.D.
Project Description: Preventing neuronal loss after intracortical
microelectrode implantation for electrical functional stimulation

149
Selected Grants and Academic Awards
2018 Burroughs Wellcome Travel Fund, Association of Clinical and Translational
Science National Meeting, Washington D.C.
2017 3rd
Place Outstanding Poster Award, Association of Clinical and Translational
Science National Meeting, Washington D.C.
2017 Outstanding Citizenship Award, Clinical Research Training Center, Washington
University School of Medicine
2016 Outstanding Continuing Mentor, Young Scientist Program, Washington
University in St. Louis
2016 - 2018 TL1 Predoctoral Clinical Research Fellow, Washington University in St. Louis
Institute of Clinical and Translational Sciences
2014 Graduate Research Fellowship Program Honorable Mention, National Science
Foundation
2013 B.S.E. Biomedical Engineering magna cum laude, Case Western Reserve
University
2013 Biomedical Engineering Scholarship Award, Department of Biomedical
Engineering, Case Western Reserve University
2013 Philip K. “Nip” Heim Award, Department of Athletics, Case Western Reserve
University
2012 Alpha Eta Mu Beta Biomedical Engineering Honors Society
2012 Tau Beta Pi Engineering Honors Society
2012 Louis Spencer Dupre Scholarship, Mayo Clinic
2012 Summer Undergraduate Research Fellowship, Mayo Clinic
2012 Cristina A. Camardo Memorial Award, Department of Biomedical Engineering,
Case Western Reserve University
2011 Case Alumni Association Undergraduate Research Grant Award,
Case Western Reserve University
2009-2013 Trustee’s Scholarship, Case Western Reserve University
Teaching Experience
2017 Teaching Fellow
Course: When I’m 64: Transforming your Future
Washington University in St. Louis
Professors: Brain Carpenter, Ph.D., Nancy Marie-Howell, Ph.D., Susan Stark,
Ph.D.
2015 Tutor
Course: Cellular Neurobiology
Washington University School of Medicine

150
Professor: Paul Taghert, Ph.D.
2015 Teaching Assistant
Course: Neural Sciences
Washington University School of Medicine
Professor: David Van Essen, Ph.D.
2012 Teaching Assistant
Course: EBME 370- Introduction to Biomedical Engineering
Case Western Reserve University
Professor: Steven Eppell, Ph.D.
Students Mentored
2017 – 2018 Ajit Perla, High School Student, St. Louis County, MO
2015 - 2018 Jacob Alex, Undergraduate Student, Washington University in St. Louis
2013 - 2016 Mykeal Franklin, High School Student, St. Louis MO
Peer- Reviewed Publications
1. Self W.K., Schoch K.M., Alex J., Barthelemy N., Bollinger J.G., Sato C., Cole T.,
Kordasiewicz H.B., Bateman R.J., Miller T.M. (2018) Protein production is an early
biomarker for RNA-targeting therapies. Under Review
2. Bali T.*, Self W.K.*, Siddique T., Wang LH., Ratti E., Boylan KB., Glass JD., Maragakis
N.J., Caress J.B., Scherer S.S., Appel S.H., Wymer J.P., Gibson S., Zinman L., Mozaffar
T., Jockel-Balsarotti J., Allred M., Liu J., Fisher E.R., Lopate G., Pestronk A., Cudkowicz
M.E., Miller T.M. (2016) Defining SOD1 ALS natural history to guide therapeutic clinical
trial design. Journal of Neurology, Neurosurgery, and Psychiatry. 10.1136/jnnp-2016-
313521.
3. Crisp M.J., Mawuenyega K.G., Patterson B.W., Reddy N.C., Chott R., Self W.K., Weihl
C.C., Jockel-Balsarotti J., Varadachary A., Bucceli R., Yarasheski K.E., Bateman R.J.,
Miller T.M. (2015). Slow CNS SOD1 turnover rate in rats and humans quantified using a
novel in vivo stable isotope protein labeling approach. Journal of Clinical Investigation.
125(7):2772-80
4. Potter K.A., Buck A.C, Self W.K., Callanan M.E., Sunil S., Capadona J.R. (2013). The
effect of reseveratrol on neurodegeneration and blood brain barrier stability surrounding
intracortical microelectrodes. Biomaterials. 34(29), 7001-15.
5. Potter K.A., Buck A.C, Self W.K., Capadona J.R. (2012). Stab injury and device
implantation within the brain results in inversely multiphasic neuroinflammatory and
neurodegenerative responses. Journal of Neural Engineering. 9(4):046020.

151
* Both authors contributed equally to the publication
Book Chapters
1. Self W.K., Miller T.M. (2016) Antisense Oligonucleotides for Amyotrophic Lateral
Sclerosis. Translational Neuroscience – Fundamental Approaches for Neurological
Disorders. IBSN: 978-1-4899-7654-3
Selected Abstracts
1. Self, WK, Schoch KM, Bollinger JG, Mauwenyega KG, Cole T, Kordasiewicz H, Bennett
CF., Bateman RJ, Miller TM. Protein production as an early pharmacodynamics
biomarker for RNA-targeting therapy in neurodegenerative diseases. AAT-AD/PD Joint
Meeting, March 2018. (Podium)
2. Self, WK., Bollinger JG., Mawuenyega KG., Patterson B., Baker-Nigh A., Jockel-
Balsarotti J., Turk J., Bateman RJ., Miller, TM. Determining SOD1 half-life for clinical
trial design. Association for Clinical and Translational Science, Washington D.C., April
2017. (Poster)
3. Self, WK., Mauwenyega KG., Wegener AJ., Kirmess, K., Cole T., Kordiewicz H.,
Yarasheski KE., Bennett CF., Bateman RJ., Miller TM. Stable Isotope Labeling of Amino
Acids Identifies Pharmacodynamics of next generation antisense oligonucleotides
targeting human SOD1 transcripts in an ALS animal. CHSL Neurodegeneration, Cold
Spring Harbor, NY, November 2016. (Poster)
4. Bali T.*, Self W.K.*, Siddique T., Wang LH., Ratti E., Boylan KB., Glass JD., Maragakis
N.J., Caress J.B., Scherer S.S., Appel S.H., Wymer J.P., Gibson S., Zinman L., Mozaffar
T., Jockel-Balsarotti J., Allred M., Liu J., Fisher E.R., Lopate G., Pestronk A., Cudkowicz
M.E., Miller T.M. Survival and disease progression in SOD1 Familial ALS in North
America. American Academy of Neurology, Vancouver, BC, April 2016. (Poster)
5. Self, WK., Mauwenyega KG., Wegener AJ., Kirmess, K., Cole T., Kordiewicz H.,
Yarasheski KE., Bateman RJ., Miller TM. A novel pharmacodynamics biomarker for
antisense oligonucleotide-mediated protein reduction in ALS. Translational Science 2016,
Association for Clinical and Translational Science, Washington D.C, April 2016. (Poster)
6.
Self W.K., Wren M.C., Bu G. Identification of Compounds that Modulate Expression of
Apolipoprotein E in Astrocytes.Mayo Clinic SURF Seminar, Jacksonville, FL, July 2012.
(Podium)
7. Ravikumar M., Self. W.K., Jain S., Selkirk S.M., Capadona J.R. Understanding
Inflammation-mediated Neurodegeneration. Department of Veterans Affairs Research

152
Week, Cleveland, OH, May 2011. (Poster)
Memberships
2016 – Present American Academy of Neurology
2016 – Present Association for Clinical and Translational Science
2015 – Present Society for Neuroscience
Professional Experience
2017 – Present Analyst, Life Sciences Fund, Cultivation Capital Venture Firm, St. Louis, MO
2016 – Present President and Co-Founder, Pocket PhDs, Inc. St. Louis, MO
2015 – 2016 Idea Supporter, The BALSA Foundation. St. Louis, MO
2015 – 2017 Director of Operations, Project Manager, The BALSA Group, St. Louis MO





























