Embed Size (px)
Citation preview

Solid State Physics
Study Support
Prof. RNDr.Pavel Koštial,CSc.
Ostrava 2015
VYSOKÁ ŠKOLA BÁŇSKÁ – TECHNICKÁ UNIVERZITA OSTRAVA
FAKULTA METALURGIE A MATERIÁLOVÉHO INŽENÝRSTVÍ

1
Title:Solid State Physics
Code:
Author: Prof. RNDr. Pavel Koštial,CSc.
Edition: first, 2015
Number of pages: 76
Academic materials for the Advanced Engineering Materials study programme at the
Faculty of Metallurgy and Materials Engineering.
Proofreading has not been performed.
Execution: VŠB - Technical University of Ostrava

2
STUDY INSTRUCTIONS
This text serves as the basic orientation material for the study of the subject Physics of
Solids for the second stage of higher education. It is an overview of the concepts of the
subject of study, but it is not a classic textbook. The material is also varied with a series of
images that should make them more legible. For a deeper understanding is needed, however,
to resort to classical textbooks .
The subject is included in the master's study and study support is divided into parts,
chapters, which correspond logically divided the study drug but not equally extensive.
Estimated time to study chapters can vary widely, so are large chapters further divided into
numbered subsections and answers them structure described below. The material is suitable
for students with a focus on material engineering, diagnostics of materials, chemistry, as well
as other interested parties of technical fields.
At the outset, this chapter is given the time needed to study the substance. Time is only
indicative.. Further such goals are to be achieved student after studying this chapter and
specific knowledge. Followed by the interpretation of the study material, the introduction of
new concepts, explanations, all of which is accompanied by images and tablesi.At the end of
each chapter are repeated the main concepts.. To verify fully mastered the material chapter, a
student at the and of every chapeter have of several theoretical questions.
Communication
Prof. RNDr.Pavel Koštial, CSc..
email: [email protected]
tel. +420597324498

3
CONTENT
1. Tensors 4
1.1 Transformation of coordinates 4-13
1.2 Properties of tensors 13-17
1.3 Stress tensor 17-20
1.4 Strain tensor 20-23
1.5 Elastic constants tensor 23-25
1.6 Compressibility 25
1.7 Impact of lattice symmetry on tensor representation 26-30
2.The basics of statistical physics
and quantum mechanics 32
2.1 Statistical interpretation of a physical systems
description with many particles 32-36
2.2 The basics of wave particle description 37-39
2.3 Quantum statistics 39-43
2.4 Schrodinger equation 43-44
2.5 The phace space 45-46
3. Viscous and viscous-elastic behaviourof materials 48
3.1 Charasteristics of viscous and
viscous elastic materials 49-63
3.2 Models of viscous elastics materials 63-70
3.3 Payne effect 70-74

4
1 Tensors
Study time 5 hours
Objectives
you will be familiar with the tensor description of the anisotropic properties of
materials
you will learn about the basis of tensors number in applications on materials
you will learn about calculations of specific physical quantities in anisotropic
materials
you will understand the relationship between the structure of material and its
mathematical description
LLecture
1.1.Transformation of coordinates
The term mass point, whose position in space is determined by the position
vector r , is one of the fundamental concepts of classical mechanics. The first
derivation of the position vector by time determines the velocity, and the second
derivation the mass point acceleration.
In the case of an N system of mass points the number of coordinates
determining the position of such a system is equal to 3N . If the mass points are
independent, then the number of degrees of freedom is also 3N .
From the elementary course on physics we know that the mass point
position is always determined with regard to a selected reference system. This
system allows us to describe the mass point position in general N with

5
independent variables qi, (i=1, 2, ..N), which characterise the system position with
N degrees of freedom, and we call them generalized coordinates.
Their first derivation by time we call generalized velocity and the second
derivation generalized acceleration..
The full determination of such a mechanical system requires to know N
number of generalized coordinates and N number of generalized velocities.
A good example of such generalized coordinates can be illustrated by the
polar coordinates r and , suitable e.g. to describe a mass point movement on a
circle.
Movement of the mechanical system is determined by kinetic equations,
such as the differential relations between coordinates and velocities. Motion
equations, expressed in generalized coordinates, have also generalized forms.
From the elementary physics course we know that we can distinguish scalar
physical quantities (also called zero-order tensors), which are fully
determined by entering numbers (scalar values), which are independent of
coordinate system selection. This can be described mathematically in a three
dimensional space using the scalar function of coordinates f(x 1 , x 2 , x 3 ), which is
invariant against the coordinate axes transformations, and so it holds that f(x 1 , x 2
, x 3 ) = f(x 1 ', x 2 x 3 ', '). These include physical quantities such as mass, density,
temperature, and more.
In contrast to them, there are also vector quantities (also called tensors of the
first order). If we identify a suitable coordinate system, which is dependant on
transformation rules (see relations in 1.13, 1.14), then the vector is fully
determined by entering its components´ values in the direction of these coordinate
axes.
Thus, we understand the vector also as an ordered triple system (in a
general n-th system) of the scalar values - vector coordinates in the selected
coordinate system. Among vector physical quantities belong acceleration, force,
etc.

6
The vector is also the triple of the scalar function partial derivative f(x 1 , x 2
, x 3), which we call the scalar function gradient because
'
i
k
k'
i x
x.
x
f
x
f
. 1.1
Gradient components in the new system are expressed through the components f
/ xk , in the old system using coefficients xk / xi’.
As to the correlation, we can view the scalar quantity as a mathematical
object, enabling the vector to be multiplied by a number. This claim will be
understood better, if we take as an example Ohms law in its generalized form. The
relationship between current density vector i and the vector of the electric field
strength E is given by a well-known relationship
i = E. 1.2
For the components of the current density vector the following relations
apply
332211 Ei,Ei,Ei 1.3
and the electrical conductivity is in this case (in given physical conditions) a
scalar quantity. From the mathematical point of view, therefore, it is truly a
multiplication of the electric field intensity vector by a number.
However, the situation is different if the material is anisotropic. Then an
electricity intensity vector is not collinear with the current density vector, such as
in the previous case, and components of the current density vector are identified
as linear combinations of the electrical intensity vector components in the form
i E E E
i E E E
i E E E
1 11 1 12 2 13 3
2 21 1 22 2 23 3
3 31 1 32 2 33 3
,
,
.
1.4

7
It is clear that the electrical conductivity is no longer a scalar quantity, but
each of the values ij has its specific physical importance.
If the electric field is applied in the direction of the axis x 1 , then the vector
of the electric field intensity components are
E = (E 1 , 0.0) 1.5
and the current density vector will include
131312121111 Ei;Ei;Ei . 1.6
Importance of the current density vector components can be seen in Figure 1.1.
Figure 1.1. Importance of the current density vector components
Thus, the electrical conductivity in the case of an anisotropic crystalline material
is generally determined by nine independent components, which form the tensor
of the second order (named after the number of components indices), which can
be written in the form of a matrix
11 12 13
21 22 23
31 32 33
. 1.7

8
The second order tensor may be seen as a mathematical object
(operator), which clearly defines the relation between the two vector
quantities.
Later, we will demonstrate that the fourth order tensor is determined by the
functional relationship between two tensors of the second order.
Equation 1.3, which illustrates the dependence of the current density vector on the
electric field intensity vector, can also be written in the reduced form
32,1,i,32,1,j,Ei3
1j
jiji
1.8
or
,3,2,1j,iEi jiji 1.9
while we have used the so-called Einstein summation convention, under which
we always add up following the index, which is repeated twice. In this case, it is
the index j, which we call the summational index . Index i is called the free
index.
Many times, when solving physical tasks, we are confronted with the
problem that it is necessary to pass from one coordinate system (related, for
example, to the crystallographic orientation) to the coordinate system of work, in
which the task is solved (e.g. the dynamics of the elastic waves propagation in the
crystal, in a specified direction, and in a given plane). We will try to explain some
parts of these interesting problems here.
Before we proceed to study the issue of transformations, let us define one
general rule, which must be met, regardless of what type of transformation we are
dealing with.

9
In the transition (transformation) from one coordinate system to
another, only the form of the transformed quantity notation changes, but not
the transformed quantity itself .
To keep this rule, it is necessary to define the transformation rules, which
will be based on Figure 1. 2.
For transforming coordinate axes we will assume that both systems have a
common origin. Relations between the "old" and "new" axes are determined by
a table (matrix)
"the old axis" 1.10
"the new axis"
333231
232221
131211
aaa
aaa
aaa
The first index in the matrix refers to the 'new' axes and the second one to the
"old" axes.

10
Figure 1.2. Transformation of the coordinate axes
Note .
From transformation theory we know that the so-called relations orthogonality
must be met, from which the first can be written in the form (the sum of
squares for direction cosine equals to one)
.1aaa
,1aaa
,1aaa
2
33
2
32
2
31
2
23
2
22
2
21
2
13
2
12
2
11
In the reduced form these equations can be expressed as 1aa jkik for i=j.
The second condition (orthogonality ) is met if the equation
.0aaaaaa
,0aaaaaa
,0aaaaaa
231322122111
133312321131
332332223121
Reduced notation of these equations will be 0aa jkik for ji .

11
Let us turn to the concrete transformations. First, we will look at the
transformation of the vector. Let us take vector p with components p1, p2, p3. the
components of the vector p in the new coordinate axes (broken line) will be given
as the projections of components p1, p2, p3 (taken as vectors in the direction of
unbroken axes, see Figure 1.3) through direction cosine in the form
( ) ( ) ( )'133
'
122
'
111
'
1 xxcosp+xxcosp+xxcosp=p 1.11
or
313212111
'
1 papapap . 1.12
Using the Einstein summation convention, 1.11 will transferred to
jij
'
i pap . 1.13
For the inverse transformation we have similarly
,
jiji pap . 1.14
If we compare both transformation relations, we can see that the order of
summation indices differs in 1.14 and 1.13. In the case of direct transformation
the summation indices are mutually "closer" than in the case of the inverse
one.
Based on previous interpretation, we can easily make sure that the following
transformation matrices are correct.
Symmetry with the beginning of the coordinate axes is determined by the
matrix

12
100
010
001
SS . 1.15
From the plane of symmetry perpendicular to the axis x 3 the following
applies
100
010
001
3XM . 1.16
The coordinate system rotations by the angle 2 / 3 about the diagonal dice
is determined by matrix
001
100
010
32;111 . 1.17
For the torsion of an angle around axes x 1 and x 2 these transformation
matrices should have the form
cossin0
sincos0
001
;x1 1.18
and
cos0sin
010
sin0cos
;x 2.

13
Figure 1.3. Transformation of the vector
1.2. Properties of tensors
In the previous section we have dealt with the issues of zero-order tensors
(scalars) and the first order (vectors). In this section we will deal with tensors of
higher-orders and the mathematical apparatus, which describes them. First, we
will examine transformation relations applicable to tensors of higher-orders. The
transformation of a tensor (let us see it as a mathematical object enabling the
functional relationship between two vectors) will be explained on a general
example, using the vectors of effect u and the causes p. We will proceed
following the scheme
u’ u p p’. 1.19
Then we gradually get
kik
'
i uau . 1.20
If we use k as a free index and l as a summation index, then, in accordance with
I.13, we get
.pT=u lklk 1.21
By analogy, we replace the summation index l for the free now, and we have
'
jjll pap 1.22

14
or
.paTapTauau '
jjlkliklklikkik
'
i 1.23
After arrangement we get the relation
'
j
'
ij
'
i pTu , 1.24
where
kljlik
'
ij TaaT , 1.25
which we call the transformational relation of the second order tensor.
Let us stress that in equation I.25 there are i, j free indices and k,l summation
indices .
It is necessary to highlight one fact, and that is the difference between the
transformation matrix and the tensor components Tkl , which represent a
physical quantity. In both cases, in their mathematical notation technically they
are the same mathematical objects, containing nine members (if we consider the
second-order tensor). But it is the only similarity.
The matrix of transformation coefficients (aij) is not a physical
quantity, but the file of components T kl forming the tensor T kl is.
Based on this claim, it is possible to form the following definition of the tensor.
If in any coordinate system of nine T kl coefficients (describing some
physical quantity) linearly binds the vectors components p and u , then in the
transition from one system to the other the [ Tkl ] tensor are transformed in
accordance with the equation 1.25, and these components form the tensor of the
second order.
In general, we can say that the tensor of the r order is the operator, which consists
of a set of 3 r constants, having r indices, which are transformed according to the formula
....lmn..
'
...ijk.. AA ..nkjmil .
Let us explain now the geometric interpretation of the second-order tensor.
For this purpose we will analyse the quadric equation in the form

15
1xxS jiij , 1.26.
where S ij will be considered as a component of the tensor Sij .
If we divide the equation 1.26 into components, we will get
.1xSxxSxxSxxS
xSxxSxxSxxSxS
2
333233213313223
2
222122131132112
2
111
1.27
Note:
By a symmetrical (antisymetrical) minus sign applies ) tensor we
understand a tensor, whose components meet the equation Sij = Sji.
If we assume that this tensor is symmetrical, we get
.1xxS2xxS2xxS2xSxSxS 211213313223
2
333
2
222
2
111 1.28
If we compare this relation with the transformational relation for tensor 2.7, we
see that the quadric, as well as the symmetric tensor of the second order, are
subject to the same transformational relation.
Characteristic area, determined by the quadric 1.27, is the
characteristic area of the symmetrical tensor Sij .
It is important to note that the quadric has main axes, which are
perpendicular to each other.
The quadric equation can be adjusted for a simplified shape (if main quadric axes
are identical to the selected coordinate system)
.1xSxSxS 2
33
2
22
2
11 1.29
If we compare the quadric equation to the traditional equation of the quadratic
surface
1c
x
b
x
a
x2
2
3
2
2
2
2
2
1 , 1.30

16
we are getting for the main half axes the values
,S
1,
S
1,
S
1
321
1.31
where S i are diagonal components of the tensor Sij .
If all the components of the Si are positive, then the characteristic surface is an
ellipsoid. If there are two coefficients positive and one negative, the area is a uni-
axial hyperboloid. If there are two coefficients negative and one positive, the
characteristic area is two axial hyperboloids (see Figure 1.4).
Figure 1.4. Ellipsoid (a), uni-axial hyperboloid (b), two axial
hyperboloids(c)
.0S ijij 1.32
i j is Kronecker's symbol. Three roots define the main axes of the characteristic
surface and it can be demonstrated that all the three main axes are mutually
orthogonal, and that the following ’= S1, ’’= S2, ’’’= S3 applies.
Quantities are also called eigenvalues of tensor Sij .
The sum of the eigenvalues of tensor Sij is its invariant.

17
Vectors determined by the equation 1.33 are called the eigenvalues of tensor
Sij . Eigenvalues of the tensor Sij are real values and the directions
determined by these values are mutually orthogonal.
From a practical point of view, we often need to determine the value of the
tensor variable in a given direction. For this problem solving, depending on the
coordinate system we recognize two cases, which again we will demonstrate
using the electrical conductivity of a tensor.
1.Let the direction cosines 3,21 ll,l determine the selected direction of
electrical conductivity. If the electric field is oriented in this direction, then we
can express it using the form El,El,ElE 321 . Then for a current density applies
El,El,Eli 332211 . The component of the current density projected in the E
direction will be ElElEli 3
2
32
2
21
2
1 . Then the electrical conductivity in
direction li will be 3
2
32
2
21
2
1 lll .
2. Let the coordinate system be oriented arbitrarily toward the main axes of
the electrical conductivity tensor.
If we have direction cosines li with an arbitrary coordinate system, so it is
ii l.EE . The component of the current density i in the direction E will be E/Ei
or written as tensor E
Ei ii . For the electrical conductivity we get
2
ijij
2
ii
E
EE
E
Ei , or jiij ll .
1.3. Stress tensor
We have understood these forces tacitly as the "point effect". In fact, real
forces operate within a certain area, and thus we no longer talk about the power,
but about the mechanical stress that causes distortion. In the basic course of
physics you have already analysed these terms, but for an isotropic body.
It was stated that the "mechanical stress is homogeneous throughout the
whole body", "all its particles are statistically balanced", "volume powers and

18
volume moments are negligible". Now we will broaden our knowledge with a
comprehensive view of the issue.
We will follow the Figure 1.5 with the unit cube.
Figure 1.5. Distribution of stress in a homogeneously tensed solid matter.
From this picture it is clear that the components of tensor distortions [Tij]
are positive in the case of stresses causing dilatation to a solid body, and negative,
if these tensions cause any contraction. It is also clear that the diagonal
components of the stress tensor T 11, T 22, T 33 represent normal stress components,
and components T ij for i j are tangential components.
But let us return to the definition of the mechanical stress concept. We will use
Figure I. 6.
On the surface of S2, which normally is parallel to the x2 axis, the stresses T12,
T22, T32 operate , where e.g. T12 is defined by the relation
2
1
0S12
S
FlimT2
1.33
or generally
k
i
0Sik
S
FlimTk
. 1.34

19
Figure 1.6. Definition of mechanical stress. Element of the surface is
perpendicular to the coordinate axis (a). Arbitrarily oriented element
of the area (b).
T ik represents the i –th component of a force applied to the surface
perpendicular to the axis xk . Perhaps its environment, based on the law of action
and reaction, will act with an equally large, but reversed force.
In conclusion, here are some special cases of the stress tensor. For uniaxial
tension the stress tensor is in the form
T11 0 0
0 0 0
0 0 0
. 1.35
For the two axes stress there will apply
T
T
11
22
0 0
0 0
0 0 0
. 1.36
"Pure" shear stress will be expressed in the form

20
0 0
0 0
0 0 0
12
12
T
T
. 1.37
Hydrostatic pressure p shall be expressed by the matrix
p
p
p
0 0
0 0
0 0
. 1.38
1.4. Strain tensor
Strain tensor describes the "response" of a solid body to the stress
operation. In this section, we will analyse also other processes in connection
with the changes of the body dimensions, such as e.g. thermal expansion. We
shall start with the already known Figure 1.7 , which describes the deformation of
a "unilaterally fixed" bar oriented in the direction of the F force.
If we take on this rod a length piece with the M and N end points, which
Figure 1. 7. Deformation of this "unilaterally fixed" rod under the influence
of the F force
have the coordinates x and x + x, then after the deformation influenced by F
force we will get

21
the new coordinates of the mentioned point in the form of x + u(x) and x + x +
u(x +x), where u(x) is the connected function of coordinates.
Relative elongation of the MN element is then defined in the vicinity of the
M point (i.e. for x 0) by the first derivative as per x in the form
dx
du
x
ulime
0x
. 1.39
Let us stress that distortion is a dimensionless value . From the above
mentioned explanation it is clear that the influence of the F force should cause
only the linear deformation (the motion of atoms in the direction of the force
operation), if we do not consider changing the diameter of the bar used.
But the reality is more complicated and here is no physical reason for the
real material to act in such a way. The movement of atoms is dependent inter alia
also on the crystallographic system and, therefore, also on the type of chemical
bonds operating among atoms.
Strain tensor can be expressed using the relation
j
k
i
k
i
j
j
iij
x
u
x
u
x
u
x
u
2
1S , 1.40
Nine constituents Sij form the second-order tensor [Sij], which we call the strain
tensor.
Most of them are small deformations and the following applies
i
j
i
iij
x
u
x
u
2
1S , 1.41
While we have neglected the second-order expression
j
k
i
k
x
u
x
u
1.42
This approximation then leads to the so-called linear elastic theory, but
basically this deformation is non-linear.

22
Let us look at the importance of the individual components of the strain
tensor.
It is clear that e11 represents the elongation per unit of length, shown on the
x1axis. Quantity e 21 represents the counterclockwise rotation.
By analogy, we would also get the physical sense of the e 12 component, which
represents the rotation of the same element in a clockwise direction.
Any second-order tensor can be factored out into symmetric and
antisymmetric parts ijijij Se , where the tensor
)ee(2
1S jiijij 1.43
represents the deformation and the tensor
)ee(2
1jiijij 1.44
its rotation.
In case of three-dimensional deformations we receive the strain tensor in the form
3332233113
3223222112
3113211211
ij
e)ee(2
1)ee(
2
1
)ee(2
1e)ee(
2
1
)ee(2
1)ee(
2
1e
S . 1.45
Diagonal components represent elongation of a solid body. In any coordinate
system the relative change in volume dV
dVdV ' is an invariant and there
applies
332211 SSS . 1.46
Non-diagonal components after their deformation in relation to their
original position form an angle / 2 - 2 S12. The change of the angle between

23
the two mutually perpendicular sections dxi, dxj is due to shear strain Sij, and thus
equal to -2S ij .
So far we have dealt with changes of the samples length due to physical
stresses. Changes of the samples length, however, can also have different causes,
and this can be e.g. thermal expansion.
For small temperature changes we can consider the deformation to be
homogeneous, and for the strain tensor we can write
TS ijij . 1.47
ij are called coefficients of expansion. Let us note that both [ ij ] and [ Sij ]
are second-order tensors.
1.5. Elastic constants tensor
In these course tasks we will expect these distortions to be small (flexible)
and they can be described by the first-order element in the Taylor series, as it
comes from the relationship
T S TT
SS
T
S SS S
ij kl ijij
kl Skl
kl
ij
kl mn SklSmn
kl mn
0
1
2
0
2
00
....
1.48
As it applies T ij (0) = 0, we can write (after neglecting higher members in the
Taylor series)
klijklij ScT , 1.49
where

24
cT
Sijkl
ij
kl Skl
0
. 1.50
Expression 1.50 is a modulus of elasticity tensor and the expression 1.49
we call the generalized Hook´s law . The tensor of elastic constants is the fourth-
order tensor and it has 3 4 =
81 components. Stress and strain tensors are
symmetrical, and therefore the values of elastic constants do not change when
switching the first two or the other two indexes, and so it is
c c c cijkl jikl ijkl ijlk ; . 1.51
Relationship 1.51 reduces the number of independent elastic constants to 36.
Further reduction, as we shall see later, will be caused by the symmetry of
crystals.
In respect to the above stated fact that the order of indices (e.g. i, j ) is
commutable, there are only six pairs of combinations, numbered from one to six,
as follows:
(11) 1, (22) 2, (33) 3, (23) = (32) 4, (31) = (13) 5, (12) = (21) 6.
1.52
With the unevenly pairs of indices the "renumbering" can be determined in a way
that we add figure 3 to the missing number of the triple group 1,2,3.
If we would like to express distortion as a stress function, then the
generalized Hook´s law has the form
,TsS klijklij 1.53
where the set of values sijkl, the so-called coefficients of compliance, is again the
fourth-order tensor. For the mathematical expression of the elastic constants
tensor [cijkl] and the compliance tensor [sijkl] in the form of matrix it applies that
s= (c)-1
or s c = , where is the Kronecker symbol, which we can
also interpret as a matrix, in which the both tensors s, c are mutually inverse.

25
Note.
In technical practices we often do not use the elastic constants (or
compliance) introduced above, but the Young's modulus (E), the shear module
(G), and the Poisson coefficient ( ) . Relations between the elastic constants (or
compliance) and the above mentioned modules for the isotropic material are as
follows.
.cc2
1cG
,12
EG,
G
1
E
12ss2,
Es,
E
1s
121144
12111211
1. 6. Compressibility
By compressibility we understand a decrease in volume, due to versatile
unit hydrostatic pressure. In relation 1.54 we will replace Tkl by the relationTkl = -
pkl, and thus we get
ijkkklijklij pspsS . 1.54
We will identify the change in volume by the relation = Sii = - psiikk, from
which for this compressibility we get - / p = siikk = k, and this represents the
new invariant type of tensor.
For the cubic crystallographic system or for isotropic solid matter we can
express the compressibility using 3 (s11 + 2s12).
In practice we often introduce this volume module compression as the
reversed value of compression k in the form K=1/k =1/3(s11+2s12)= E / 3 (1 -
2).

26
1. 7. Impact of lattice symmetry on tensor representation
The periodicity of lattice reduces the number of possible elements of
symmetry and creates relationships between some of them, and so there are only
seven groups of symmetries, each of which determines one crystallographic
system.
There are two basic types of elementary symmetric operations. It is a direct
and inverse symmetry.
Elementary direct symmetries are related to the axis of symmetry, with the axis
of symmetry An of the n order, where n is a whole number if by rotating the
crystal around this axis by angle 2 / n this crystal gets into the starting position,
in which it was before rotating.
Inverse symmetry is bound to the centre of symmetry and the inverse axis.
The centre of symmetry C corresponds to an inverse transformation of the
crystal with respect to a point.
The inverse axis of the n order, denoted as An
i re-transforms the crystal
from the initial position by turning it using the 2 / n angle and the subsequent
inversion with regard to the point. Thus the equation An
i = AnC = C An
applies.
The plane of symmetry is a special case of inversion axis of the second
order, or the mirror plane M perpendicular to the axis at the point of inversion,
and thus A 2
i = M.
Equally, the centre of symmetry is equivalent to the inverse axis of the first
order, and so it is C = A1i .
As we have already mentioned above, the periodicity of the crystal structure,
except for reducing the types of symmetry, also leads to the relationships of
equivalence between the individual symmetries .
As a result of crystals symmetry only certain types of symmetrical
operations may exist and only the axis of a certain order symmetry. Overall,

27
there are thirty-two classes of crystals point symmetry. For the three
dimensional lattice there are fourteen types of so-called Bravaiselementary
lattice types. These fourteen types of lattices are grouped in seven
crystallographic systems, according to the seven types of elementary cells.
Principles applicable to the so-called point symmetry of the crystal lattices can be
summed up into the following statements.
1. Each straight line parallel to the axis of symmetry of the n order, which
passes through a node of lattice, is the axis of symmetry of the same order.
2. Each axis of symmetry passing through the node is identical with the
lattice order.
3. Each node of the lattice is the centre of symmetry.
4. The axis of symmetry of the order n 2 creates n of the perpendicular
axes of the second order.
In the next section we will focus on symmetrical operations, which are
permitted in respect to the lattice periodicity, and which determine the seven
crystallographic systems.
For greater clarity there are the individual symmetrical operations and geometric
parameters of the individual crystallographic systems stated in Figure 1.8 .
At the very beginning of our chapter on the impact of symmetry on the
tensor representation of physical quantities, we will deal with second-order
symmetrical tensors, which describe physical properties which have central
symmetry. These tensors have six independent components, if they relate to any
coordinate system.
If, however, the crystal has a certain symmetry, the number of independent
components is declining. The symmetry of the characteristic area of the tensor is
identical to the symmetry of the given physical property described by the tensor.
In the context of physical properties symmetry, and the point symmetry of
the crystal it is useful to come closer to the so-called Neuman´s principle that
says:

28
Elements of physical quantity symmetry, which characterises the
crystal, involve elements of point group crystal symmetry.
Physical quantity may therefore have a certain symmetry, which is manifested
independently of the symmetry in a given crystal. In accordance with Neuman´s
principle, however, the physical property must also have all the elementary
symmetries, which have a crystal. For example, the flexibility of a hexagonal
crystal is not only centrally symmetrical, but also has elements of symmetry of
this crystalline system.
If we introduce transformation matrix l , we get
...pqr..
'
..ijk.. SS ......... r
k
q
j
p
i lll , 1.55
Therefore, the invariance of the physical properties in relation to
selected symmetrical operations leads to the transformation equation
...pqr.....ijk.. SS ......... krjqip lll 1.56
Let us go, however, to some specific cases. The centre of symmetry is described
using matrix 1.15 or in a short entry ij
j
i , whereij is the Kronecker symbol.
Then ...ijk.....ijk.. SS ......... kkjjii lll ,
or for the tensor of n order it is valid S S...ijk..
n
...ijk.. 1
If n is odd, then (- 1) n =
-1, and therefore all tensor components are zero.
This result can also be interpreted, so that centrally symmetrical crystals cannot
have physical properties described by the tensors of unpaired orders. This applies,
for example, to piezoelectric and pyroelectric phenomenon, which are described
precisely by tensors of the third order. If n is paired, there are no changes caused
by the centre of symmetry.
At the very beginning of our chapter on the impact of symmetry on the
tensor representation of physical quantities, we will deal with second-order
symmetrical tensors, which describe physical properties which have a central

29
symmetry. These tensors have six independent components, if they relate to any
coordinate system.
If, however, the crystal has a certain symmetry, the number of independent
components is declining. The symmetry of the characteristic area of a tensor is
identical to the symmetry of the given physical property described by the tensor.
In the context of physical properties symmetry, and the point symmetry of
the crystal it is useful to come closer to the so-called Neuman´s principle that
says:
Elements of physical quantity symmetry, which characterises the
crystal, involve elements of point group crystal symmetry.
The physical quantity may therefore have a certain symmetry, which is
manifested independently of the symmetry in a given crystal. In accordance with
Neuman´s principle, however, the physical property must also have all the
elementary symmetries, which have a crystal. For example, the flexibility of a
hexagonal crystal is not only centrally symmetrical, but also has the elements of
symmetry of this crystalline system.
If we introduce transformation matrix l, we get
...pqr..
'
..ijk.. SS ......... r
k
q
j
p
i lll , 1.57
Therefore, the invariance of the physical properties in relation to selected
symmetrical operations leads to the transformation equation
...pqr.....ijk.. SS ......... krjqip lll 1.58
Let us go, however, to some specific cases. The centre of symmetry is described
by the relationship ij
j
i , where ij is the Kronecker symbol. Then, or more
precisely for the tensor of the n order there is valid S S...ijk..
n
...ijk.. 1
If n is odd, then (- 1) n =
-1, and therefore all tensor components are zero.
This result can also be interpreted, so that centrally symmetrical crystals
cannot have physical properties described by tensors of unpaired orders.
This applies, for example, to piezoelectric and pyroelectric phenomenon, which

30
are described precisely by tensors of the third order. If n is paired, there are no
changes caused by the centre of symmetry. Relevant symmetrical operations in
crystalographic systems is presented in Figure 1.8.
Figure 1.8. Crystallographic systems and their relevant symmetrical operations
Summary of terms
Transformation of coordinates and tensors, properties of the second-order tensors,
stress tensors, distortions and Hook´s law in general form, compressibility, the impact of
lattice symmetry on the the number of independent components of the tensor.
Questions to the topic
1. How to transform the second-order tensor?
2. What relationship determines the stress tensor constituents?
3. Explain Hook´s law in its general form.
4. How can we calculate the compressibility of anisotropic material?

31
5. What is the impact of symmetry on the number of independent components of the second-
order tensor?
6. Explain the term invariant tensor, and what it determines?

32
2. The basics of statistical physics and quantum mechanics
Study time 5 hours
Objectives
you will be familiar with the statistical description of large groups of
particles.
you will get acquainted with the basics of the wave description of
particles.
you will understand the difference between the micro and macro world.
you will understand the importance of introducing the concept of phase
space.
LLecture
2.2 1. Statistical interpretation of a physical systems description with
many particles
In this section we will formulate the basic relations of a multi-
particle system description and monitor their progress, depending on the
changes of various physical parameters. Why is there a need for the
introduction of another mathematical approach, as we were used to
traditional Newton´s physics? The answer to this question is presented
in Table 2.1.
From the numbers above we can see that the multi-particle systems
descriptions, such as e.g. gases, using the differential equations of
Newton´s mechanics, would be extremely difficult, but in particular
scientifically difficult to solve.
Therefore, for the description of multi-particle systems the methods of
mathematical statistics are used. Mathematical probability is defined as
the ratio of expected phenomena to all 1n possible phenomena n ,
which can be expressed by the relation

33
n
nP 1 < 1 2.1.
and this ratio is always less than one.
In the physics of many-particle systems, we are talking about a statistics
file , which is composed of a large number of identical sub-populations
(even infinitely large). Every part of the subsets system acts as the entire
file. An acceptable status of the system is the one that is contrary to its
physical properties.
For example those states, when SiO 2 has a crystalline structure are
unacceptable if we had at the beginning the glass form of a material. At
low temperatures it is physically impossible to achieve the
transformation of glass to crystal.
If there is a g of eligible population, then the file has g of subsets.
With a large number of repeated random phenomena the
coincidence has the character of necessity. For a small number of
phenomena the term of probability loses its sense.
Table 2.1. Dimension characteristics of molecules world
Number of molecules in the volume of one
kilomol in gas
2610.025,6
Number of molecules in the volume of 1m3 2510.668,2
Velocity of molecules s/m1010 32
Diameter of molecules m10 10
The transport mean free path of molecules m10 10
Amount of collisions in 1 second 109 1010
Let us focus further on the probability trend when tossing more
coins. Every coin has a reverse side ( R ) and obverse ( O ) side and

34
when tossing a set of coins the different ratio of R:O appears. To
describe such a process we use combinatorics. If every coin is an
individual element of the coins file, and we number them from 1 to 10,
and we take into consideration only one side of them, we can build out
of these ten numbered coins a total of 3,628,800 arranged series of
different sequences of numbered coins, as
36288001......8.9.10!10!n .
After each toss of these coins we get the different ratio of R and O in
the series, but with the identical ratio and in various combinations. If we
consider variations of both sides of the coins in the series with ten coins
( R= 0-10, L=10 - 0), according to the rules of combinatorics, the
number of all variations is 1024210 .
For example, for four coins we receive 1624 variations. Hence, we
can make 16 patterns with four coins, in which there are different
variations in sides R,O. The probability of phenomenon, in which when
tossing ten coins we will get all R or only O is 10
10
22
1
, and for
that case we need to toss them at least 1,024 times.
For a number of equal and repeating cases the following relationship
applies
!nn!n
!nn
!n!n
!n
11
21
21
, 2.2.
where 21 nnn is the number of expected phenomena.
For 21 nn we get relationship
!n!n
!ng
11
, 2.3.
which was called by Planck thermodynamic probability, and it is also
called the statistical weight, or the degree of system degeneration ( the
number of equal conditions) .
Probability that we will toss Ln1 and Rn2 will be given by the

35
relation
nn
21
2
!L!R
!LR
2
!n!n
!n
P
, where
21
n
21 nnn,10242,Ln,Rn .
Let us now further investigate the interpretation of thermodynamic
probability. Imagine the two identifiable (marked, but identical)
molecules in an enclosed space with a filter, which divides the space
into two halves. Possible distribution of molecules in each part (
statistical conditions or also microstates ) is in the Table 2.2.
If we apply this example of the above mentioned relations, we will
get the total number of microstates of 422 , but only three
thermodynamic (also called macrostates) states, or the state of gas
determined by the group of molecules, regardless of which individual
molecule creates this state of gas. Equal distribution of molecules on
both sides of the filter appears twice ( 2!1!1
!2
!n!n
!ng
11
) and the
total probability P of this state is thus 50 percent.
Lets´ investigate the further development of such a twin-
microstate system with an increasing number of molecules. For four
molecules we get 1624 states, while the even distribution will
appear in six cases, or 6!2!2
!4
!n!n
!ng
11
.
The probability of the spontaneous compression of molecules in one
part of this system decreases with an increasing number of molecules. If
it was 25 percent for two molecules, in case of four molecules it will be
only 6.25 percent, and with ten molecules the figure will be 410.8
percent.
What is more interesting, the thermodynamic probability g of the
state of equilibrium (distribution), on the other hand, will grow quickly.
With ten molecules these states reach 252, with 100 molecules it is even
2810.15,11 , and with a thousand molecules we get 29910.7,2 . Based on
these considerations it is clear that the most probable distribution of a

36
large number of identical particles is the one which will ensure an equal
distribution of molecules. Of course, other states are also possible (they
can occur e.g. during their fluctuation within the system), but their
probability is very low.
Table 2.2. Number of microstates and their probability in a two-particle system
Microstate I Microstate II Number of
states
P [%]
1 25
1 25
1 25
1 25
We had that probability defined previously by the relation q
niP
. The mean value of any physical quantity A can be determined using
the relation
in.iAq
iP.iAAn
i
n
i
11
1. 2.4
We know that the probability of a particle to be in the state with energy
E is proportional to the Boltzmann distribution
kT
Eexp i , if there is no
replacement of particles present.
n
i
Z
i
n
i
i
kT
E
kT
Eexp.iA
A
1
1
, 2.5.
Z- statistical sum.

37
2. 2. The basics of wave-particle description
As we already know from the basic course on physics, under certain
conditions particles behave as waves, which can be easily demonstrated
on Bragg´s scattering of X-rays in a crystal lattice, as shown in Figures
2.1 and 2.2.
Figure 2. 1. Bragg´s scattering of x-rays in a crystal lattice
Figure 2. 2. Bragg´s condition
Based on these experiments Louis de Broglie formulated the
following relations between the wavelength of an electron and its
momentum.
mv
h
p
h . 2.6

38
Wave functions themselves does not provide any relevant physical
information, as it is comprehensive.
Physical signification, however, has the product of real and imaginary
components 22222* B+A=Bi-A=ψψ , the so-called probability
density , which represents a real number. This product gives the probability
that a particle will occur at a given time and at a given space.
On the basis of such a wave function interpretation then W.
Heisenberg formulated the so-called uncertainties relation, which does
not to simultaneously allow the same accuracy to measure a coordinate
and momentum because of the principle.
ΔxΔp≥h/ 2π. 2.7
Let us now describe the n particles system using the wave
functions. Such a "total" wave function can be expressed as a product of
functions of the individual parts in the form
( ) ( ) ( ) ( ).nψ.......2ψ1ψ=N:::::.2.1ψ 2.8
If there is one particle in state a and the second in state b, the probability
density should not change and there must apply
( ) ( )22
1,2ψ=2,1ψ. 2.9
Wave functions ( )1,2ψ after replacement of particles may be either
symmetrical, or antisymmetric, and so one of the two following relations
applies
( ) ( )1,2ψ=2,1ψ ,
( ) ( )1,2ψ=2,1ψ- . 2.10
If there is particle 1 in state a and particle 2 in state b, or particle 1 in
state b and particle 2 in state a, this equation can be applied
( ) ( )
( ) ( ).1ψ2ψ=ψ
,2ψ1ψ=ψ
ba2
ba1
2.11

39
Both functions properly describe the system, and so the resulting
function of the whole system is given by their linear combination
( ) ( ) ( ) ( )( )1ψ2ψ+2ψ1ψ2
1=ψ babas
,
( ) ( ) ( ) ( )( )1ψ2ψ-2ψ1ψ2
1=ψ babaa
. 2.12
Antisymmetric features describe those particles with a half-integer
spin, so-called Fermions ( electrons, protons, neutrons..)
Particles with an integer spin, such as photons, are described using
symmetrical functions, and they are also referred to as Bosons,
2. 3. Quantum statistics
As we already know from the previous parts of this textbook,
which was dealing with the quantum-mechanical principles of the micro
world, an electron can be in only one energy state at the same time,
with two different orientations of spins (the Pauli principle). Particles
operating under this principle are also called fermions (and include, e.g.
also 3 He - 2 protons, 1 neutron). Let us return to the model used in
Chapter 4, and we will assume that the system (with only one electron
possessing energy , or a free-vacant, and its energy is equal to zero) is
in the thermal and diffusion contact with a reservoir, as seen in Figure
2.3.
Figure 2. 3. Possible states of a fermion according to the Pauli
1. R
e
s
e
r
v
o
i
r
vacant
energy
=0
2. R
e
s
e
r
v
o
i
r
1
electro
n
energy

40
principle
Large amount 9 (not only there is an exchange of energy taking
place within the system, but also an exchange of particles), we can write
for fermions the following
Z = 1 + exp(-/), 2.13
because kTkTkTkT
0
e1eeeZ
. For the mean value of the
system occupancy we get the expression
kT
kT
e.1
e.n
, 2.14.
or we define the function
( )
1+e
1=εn=)ε(f
kT
με
, 2.15
which we call Fermi - Dirac distribution.
In the subject of solid physics the chemical potential at absolute
zero is called Fermi energy or even the Fermi level.
If at any temperature the value the energy of a system is equal to the
chemical potential, then the medium-sized occupancy of the system is
equal to one-half, which is based on the relationship
.5,011
1
1kT/exp
1f
Development of the Fermi-
Dirac partition function is shown in Figure 2. 4.

41
0 0,2 0,4 0,6 0,8 1,0 1,2 1,4
0,2
1,0
0,8
0,6
0,4
1f( )=
Figure 2. 4. Fermi-Dirac distribution 5,0 μττ
Note:
It should be noted that the occupation of the levels in the Fermi-
Dirac partition function is identical to the probability of its
occupancy.
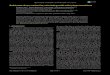
Temperature dependence of the Fermi-Dirac function for
K50000k
T FF
ε is shown in Figure 2. 5.
In addition to fermions there is also another class of particles with
an integer spin (e.g. photons, 4 He, 2 protons, 2 neutrons), which are
subject to other statistical rules as fermions. To these particles the Pauli
principle does not apply, and so the system, which is in thermal and
diffusion contact with the reservoir can be filled with any number of
particles, and for a large amount this expression is valid
x1
1=x=e=
kT
εnμnexp=)kT,μ(Z
0=n
n
0=n
n
kT
εμ
0=n
∑∑∑∞∞∞
, 2.16.
where x = exp((-)/kT). The sum of geometric series for x 1 converts
a for x 1 diverges. For the mean number of particles in the system a
42
similar procedure as the one for fermions is applied and we gradually
get
987654321
0,8
0,6
0,4
0,2
1,0
1,2
0
500 K5000 K
2,5.10 4K
5.10 4K
10.10 4K
Figure 2. 5. Temperature dependence of Fermi-Dirac functions for
K50000k
T FF
ε .
.1x
1
1x
1
1
x1
x
Z
Z.dx
d.x
x
xdx
d.x
x
x.n
n1
0n
n
0n
n
0n
n
0n
n
2.17
Expression
1e
1n
kT
2.18
we call Bose-Einstein distribution for bosons, which is shown in Figure
2. 6.
Note.
Unlike the Fermi-Dirac distribution the probability of the level

43
occupancy is not in the bosons statistics identical to its occupancy, as it
was in the case with fermions. The less the energy, the greater the
number of particles.
210-1-20
Boseho -Einsteinoverozdelenie
Fermiho -Diracoverozdelenie
n (
1
2
3
4
Figure 2. 6. Bose-Einstein and Fermi-Dirac distribution.
In Figure 2.6 you can also see that for the 'great power' both
distributions valid for the world of quanta within limits are getting
near each other (the unit for the denominator ceases to play an
important role) and pass into the "traditional" Maxwell-Boltzmann
distribution in (if we contemplate the pre-exponential member).
.
2. 4. Schrödinger equation
Let's write an equation for a wave travelling in the direction of the axis x in
the form
Ψ=Aexp[-iω(t-x/v)]. 2.19
As it applies
E=hν
and 2.20
λ=h/p

44
we can rewrite the equation 2.19 as
Ψ=Aexp[-i/ħ(Et-xp)], 2.21
which is valid for a free particle .
In order to monitor the dynamics of phenomena, which means
changes in coordinates, or time, we have to calculate the relevant
derivations.
2
2
2
2 Ψp-=
x∂
Ψ∂
2.22.
ΨiE-=
t∂
Ψ∂
For the total energy of particles the following relation applies
V+m2
p=E
2
, 2.23
or after adjustment
ΨV+m2
Ψp=ΨE
2
. 2.24
Further applies
t
Ψ
i-=ΨEΨ
iE-=
t
Ψ
∂
∂→
∂
∂
2.25
Ψp=x
Ψ- 2
2
2
2
∂
∂
where we come to the time-dependent form of Schrödinger equation of
0=ΨEm2
+x
Ψ→ΨV-
x
Ψ
m2=
t
Ψ
i 22
2
2
22
∂
∂
∂
∂
∂
∂, 2.26
45
2. 5. The phase space
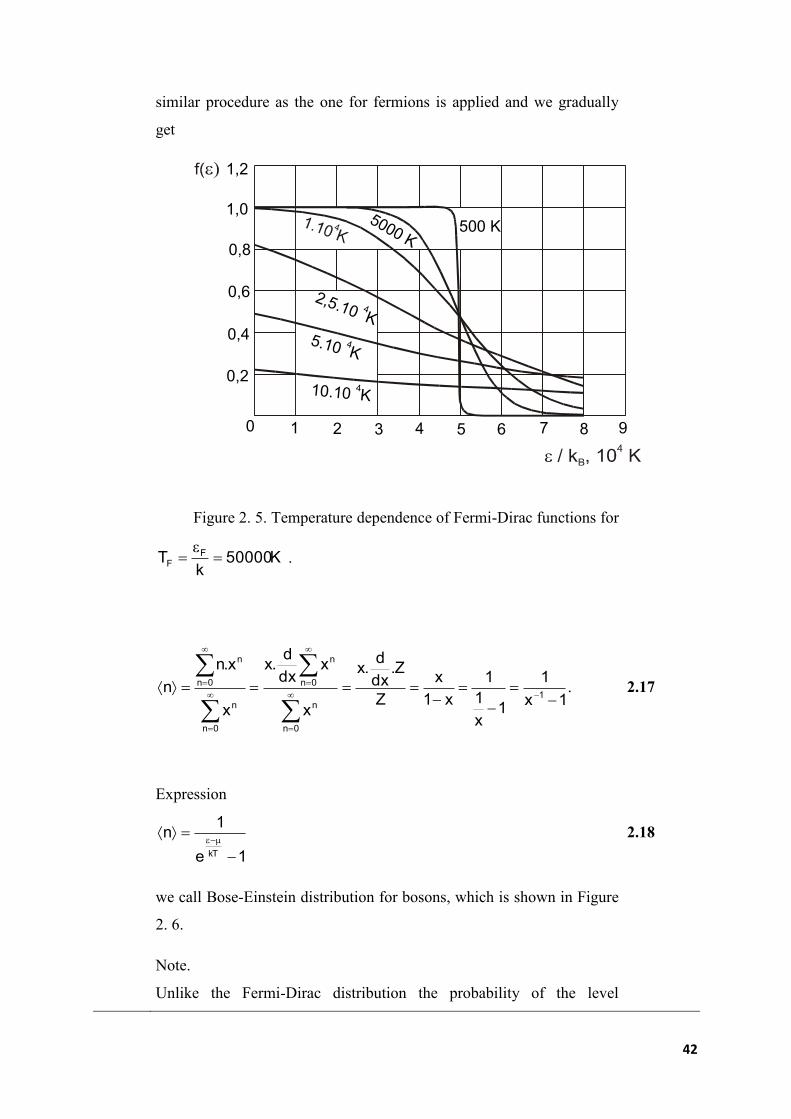
In traditional mechanics, the immediate status of a particle is
clearly described by the value of coordinates and pulse, which, as we
know, are in principle also measurable with the same accuracy. The laws
of Newton´s mechanics with regard to their deterministic nature enable
us to identify the behaviour of particles at the time. Entering
instantaneous values of generalized coordinates q and generalized pulse
p is equivalent to the point within the two-dimensional space p vs. q,
which we call phase space (see Figure 2.7).
Figure 2. 7. Definition of phase space
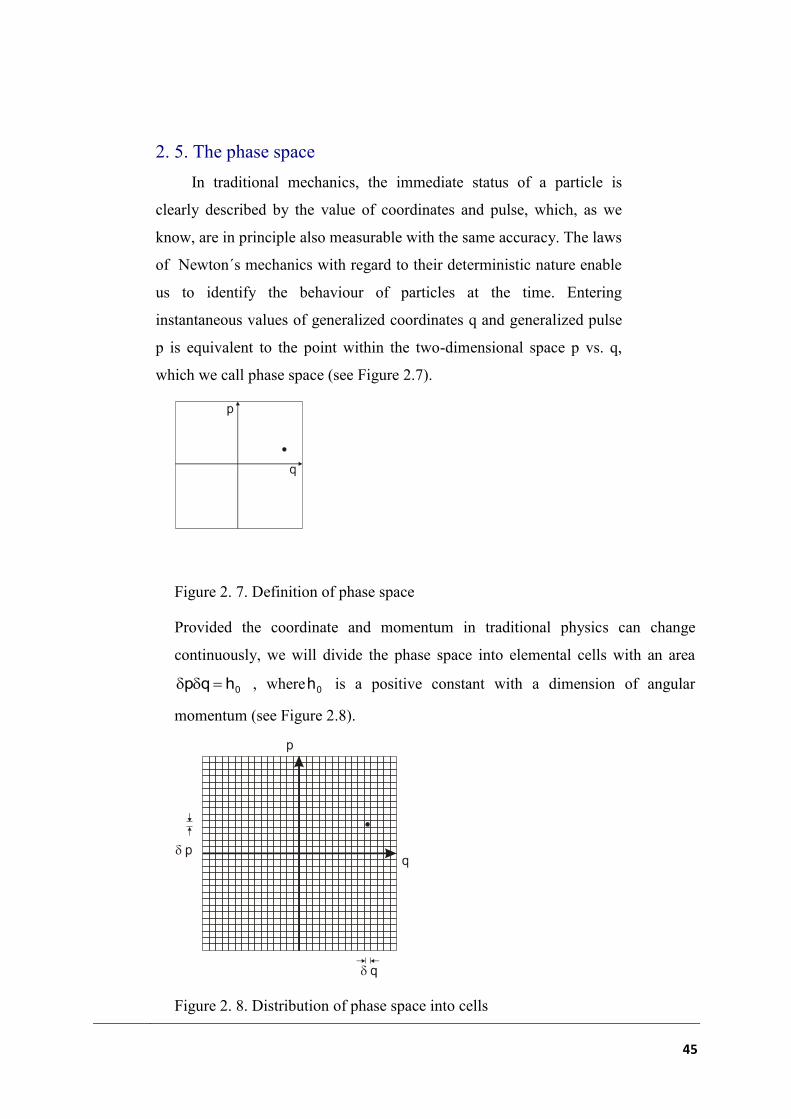
Provided the coordinate and momentum in traditional physics can change
continuously, we will divide the phase space into elemental cells with an area
0hqp , where 0h is a positive constant with a dimension of angular
momentum (see Figure 2.8).
Figure 2. 8. Distribution of phase space into cells
46
In order for the status particles to be described it is sufficient to
prove that the coordinate is in the range of phase space qq,q and
the momentum is from the interval pp,p . Now, we need to answer
the question what size must constant 0h be? In traditional physics its
value may be infinitely small, and our task is reduced to answer the
question, what is the "serial " number of a cell in the phase space, in
which the coordinate and momentum of a particle are situated?
As we know from quantum mechanics, the volume of a phase
space cell is clearly determined by the Heisenberg uncertainty principle.
In conclusion here are practical technical applications of the phase
space.
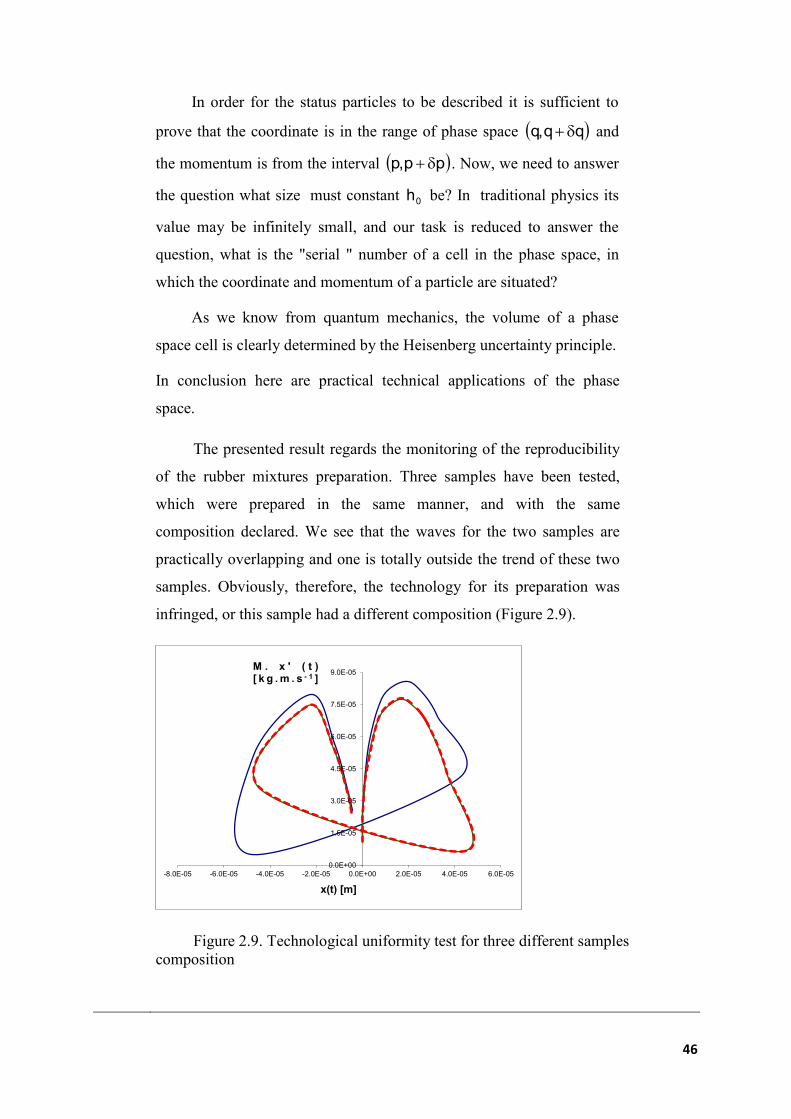
The presented result regards the monitoring of the reproducibility
of the rubber mixtures preparation. Three samples have been tested,
which were prepared in the same manner, and with the same
composition declared. We see that the waves for the two samples are
practically overlapping and one is totally outside the trend of these two
samples. Obviously, therefore, the technology for its preparation was
infringed, or this sample had a different composition (Figure 2.9).
Figure 2.9. Technological uniformity test for three different samples
composition
0.0E+00
1.5E-05
3.0E-05
4.5E-05
6.0E-05
7.5E-05
9.0E-05
-8.0E-05 -6.0E-05 -4.0E-05 -2.0E-05 0.0E+00 2.0E-05 4.0E-05 6.0E-05
M . x ' ( t ) [ k g . m . s - 1 ]
x(t) [m]

47
Summary of terms
Statistical description multi-particle systems, mean values of physical quantities, wave particle
description, probability density, Fermi-Dirac distribution function, Bose-Einstein distribution
function, Schrödinger equation, the phase space.
Questions to the topic
1. How is the probability of a phenomenon defined?
2.What is the thermodynamic probability?
3. How do we calculate the mean value of a physical quantity?
4. Characterize Fermi-Dirac statistics.
5. Characterize Bose-Einstein statistics.
6. Explain the Schrödinger equation.
7.What are symmetrical and antisymmetric wave functions?
8. Explain the term the wave function of particles.
9. When can the electron be seen as a traditional particle?
10. Explain the Heisenberg uncertainty principle.
11. Explain the concept of the phase space.

48
3. Viscous and viscous-elastic behaviour of materials
Time for learning: 5 hours
you will be familiar with viscous and viscous-elastic concepts.
you will learn about these environments models.
you will get acquainted with the characteristics of real materials.
you will learn to understand microstructural aspects of materials flow.
you will understand the processes accompanying material flow.
Lecture
3. 1. Characteristics of viscous and viscous-elastic environments
The term viscosity is most frequently associated with liquid and gaseous
states. Viscosity is a physical phenomenon caused by Van der Waals´ forces
acting among particles in liquid or gas during their motions. If this movement is
only of a "shear" nature, then, as we already know from the basic course on
physics, Newton´s viscous law applies as
dy
dvTyx , 3.1.
where T is stress and η is the dynamic viscosity . We call them Newton´s fluids.
Real fluids, however, are different from Newton´s ones. Also water, as a liquid
with a relatively low viscosity, responds "flexibly", if we apply to it e.g. enormous
stresses in short pulses. This phenomenon is well known e.g from "spoiled
jumps" into water, when we hit our abdomen (the flexible response of water is
felt sufficiently in that very moment). Development over time of a mechanically
loaded viscous environment thus also depends on the duration of load action.
We know from our experience that on asphalt roads so-called "tracks"
appear, caused by heavy lorries and vehicles, especially during the summer time.

49
Certainly, you can recall the prints of female high-heel shoes, the so-called
needles, in asphalt during the summer months. We also know that if we heat
honey, it becomes more liquid, and we can find even more similar examples.
There is, however, one fact, namely that material viscosity is, along with the
length of exposure and the size of attached mechanical tension (a so-called
thixotropic substance) also a function of temperature.
In our interpretation of this phenomenon we will draw on the so-called the
hole model of fluids. In this model, it is assumed that fluid is composed of
molecules and of vacant places, so-called holes, which may appear in a liquid, e.g.
after the evaporation of some molecules. The jump of a molecule into a new, more
energy-advantageous position is only possible if the neighbouring place is free,
and thus there is a hole. The concentration of such holes (the ratio of vacant places
n to the total sum of the nodes N of this structural network) will be
RT/HeN
n , 3.2.
Where ΔH is enthalpy, which is needed to create a hole (and it is approximately
equal to 2/5.ΔH cal. ), R is the gas constant and T is the absolute temperature.
Molecules oscillate around equilibria positions, and 2ν-times per second attack the
surrounding potential barriers. Then for the frequency of possible jumps of
molecules we get
BA
RT/QRT/H HHQ;e2eN
n2
1
. 3.3
The first of enthalpies represents the activation enthalpy, necessary for the
creation of holes, and the second represents the height of barriers, which the
molecule must overcome in self-diffusion. If we identify the coordination
number of the z molecule (the number of nearest neighbours is z), then for the
frequency of jumps it is zτ
1=ν .
If the diffusion process is assisted by the external force F, then the
molecules will be moving in the direction of this force and the mean speed of

50
molecules will be proportional to this force, the coefficient of proportion is called
the mobility of molecules.
Fqv . 3.4
If between the liquid layers, which are mutually slipping, the distance δ is equal
to the atomic distance, then the following relationship applies
δq
1=η 3.5
and for dynamic viscosity coefficient we get (without stating further steps, which
are of a mathematical character)
RT/Q
33e
νδ2
zkT=
δ
τzkT=η , 3.6
where k is the Boltzmann constant. We can see that the dynamic viscosity
coefficient decreases with increasing temperature, as we have discussed
above. The mentioned model is of course a qualitative one, and for different types
of materials and temperature ranges of other relations are also used.
Now we extend our knowledge of the physical interpretation of viscous flow
to polymers and rubbers. It is well known that molecular "jumping over" takes
place mainly in amorphous areas, while for crystallite they occur in their
interfaces or degraded areas. It may also result in a partial turn of all crystallites.
The kinetics of flow is described using two different processes.
Let us analyse the first process. Molecules jumping over in the direction of
flow lead to an arrangement with less entropy without changing their internal
energy; this process is called entropic elasticity. The process is shown in Figure
3.1a. It can also cause filling states with higher energy, which is conditioned by
the existence of nodal points, and this is so-called energy flexibility, and it
corresponds to the situation displayed in Figure 3.1b.
In the later case, molecules or their parts distract from their surroundings
and the nodal points shall cease to exist. The distribution of this potential energy
does not change on average, as there are new potential barriers being built, which
have the same energy distribution as the original one. This flow is called
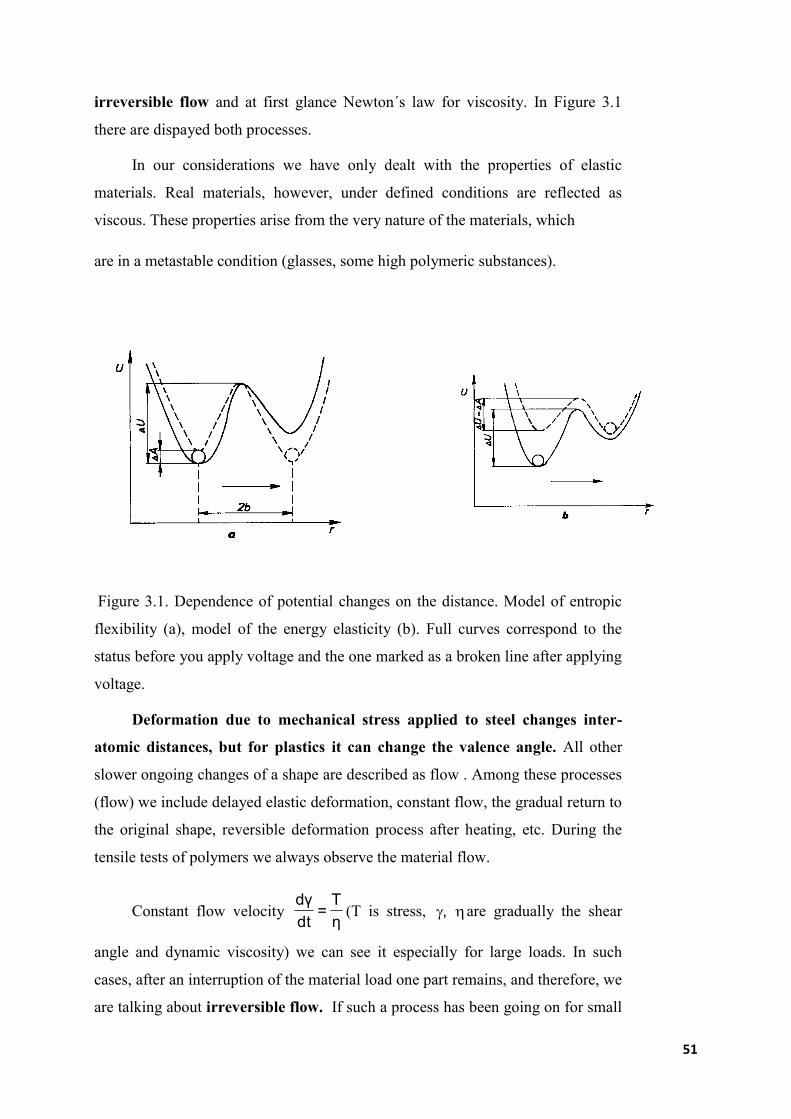
51
irreversible flow and at first glance Newton´s law for viscosity. In Figure 3.1
there are dispayed both processes.
In our considerations we have only dealt with the properties of elastic
materials. Real materials, however, under defined conditions are reflected as
viscous. These properties arise from the very nature of the materials, which
are in a metastable condition (glasses, some high polymeric substances).
Figure 3.1. Dependence of potential changes on the distance. Model of entropic
flexibility (a), model of the energy elasticity (b). Full curves correspond to the
status before you apply voltage and the one marked as a broken line after applying
voltage.
Deformation due to mechanical stress applied to steel changes inter-
atomic distances, but for plastics it can change the valence angle. All other
slower ongoing changes of a shape are described as flow . Among these processes
(flow) we include delayed elastic deformation, constant flow, the gradual return to
the original shape, reversible deformation process after heating, etc. During the
tensile tests of polymers we always observe the material flow.
Constant flow velocity η
T=
dt
γd(T is stress, , are gradually the shear
angle and dynamic viscosity) we can see it especially for large loads. In such
cases, after an interruption of the material load one part remains, and therefore, we
are talking about irreversible flow. If such a process has been going on for small

52
stresses, we can say that this material does not have supple strength. Supple
strength was first seen in crystals, and this deformation is usually called plastic.
For illustration in Figure 3.2 there is shown the rate of change for the original
right angle γ of the prism, on which tangential stress is applied, as a function of
the attached mechanical stress for various types of materials.
Deformation of vulcanised natural rubber is almost immediate, and it is
permanent only for a tiny part of this material. After that load interruption
material returns to its original state and hysteresis is small (the hysteretic curve
has a small area), as we can see in Figure 3.3.
The measured elastic deformation of rubber is composed of two parts. This
deformation leads both to a reduction in entropy due to the increase in the
orderliness of this system, and this process will also be reflected in a fall of
specific heat capacity of this material. Module E 1 flowing from this decrease in
entropy is directly proportional to its absolute temperature. In non-ideal
vulcanized rubber one part of deformation work is bound to internal energy,
resulting in a change of inter-atomic distances, but in particular the changes of
valence angles and the corresponding change of module E 0 . For the overall
module then we get10 E
1
E
1
E
1 , or
T
tatankonš
E
1
E
1
0
.
3. 2. Newton´s fluid (a), non-Newton fluid (b), substance with supple
strength (c)

53
3.3. Hysteretic curve. Mechanical stress versus deformation
As can be seen from Figure 3.4 with increasing temperature the module
increases (curve a), which is the result of the entropic nature of the flexibility
rubber, but for plastics it decreases (curve b), as the share of contributions of
reversible, and time-dependent deformation increases.
Change of the module between points A and B depends on the transition to
the ideal flexible behaviour of the material under the glass transition temperature.
Rubber substance (a), viscous elastic substances (b).
Figure 3. 4. Dependence of module E on temperature.
The module of rubber is significantly lower than those of the other
substances. The Elastic deformation of rubber in many cases exceeds the initial

54
length of material. It is therefore not possible in one diagram to display the stress
and deformation of rubber, plastic and steel.
Such a comparison is only possible using a logarithmic scale, as seen in
Figure 3. 5.
Let us now describe polymers in terms of their structures and physical
properties. As with all substances, also for polymers we can define three basic
states: gas, liquid and solid. Despite the general similarities, in polymers there are
certain specifics properties, which distinguish them from low molecular
substances.
.
3. 5. Tensile waves in a logarithmic scale for rubber (a), viscous elastic
substance (b), steel (c)
In general the gaseous state is characterised by an intensive flow of particles,
where the distances between molecules are great. As the interaction of particles,
within the meaning of attractive force, decreases with the sixth square distance,
for molecules in gaseous state the mutual attractive forces in view of the distance
between molecules can be practically disregarded.

55
In the liquid state the thermal movement of molecules is still very intense, but in
contrast to the gas state, the distances between molecules are considerably smaller
and the molecules strongly affect each other.
In the solid state, the intensity of heat movement is reduced so much that it
is not sufficient to break up the molecular contacts. Molecules will take a
permanent, defined position within the space, and only perform vibratory
movements with a frequency of 10 13-14
Hz . The distances between molecules do
not differ too much from the liquid state, which can be easily proven by
comparing the density of substances in all three states. While the difference in
density between gas and liquid reaches several orders, the difference between the
density of liquid and solid substances is small. It can be said that the distance
between the particles are approximately on the same level of molecular sizes.
From differences in molecules mobility also some significant differences in
properties result.
For the liquid state the intensive translational movement of molecules along
with rotary and vibration movement is typical, in the solid state all molecules are
occupying more or less constant positions, and translational movement is reduced
to a minimum. While liquid easily changes its shape with minimum force, e.g.
using its own weight (gravity force), for the deformation of a solid body we
usually need to apply a relatively large force.
When discussing arrangement in a solid state, it should be noted that there
are two possible arrangements. The first one is random arrangement, similar to
those in a liquid state, with the significantly lower thermal mobility of particles.
The second one is a crystalline arrangement, where molecules are arranged
regularly showing a clear symmetry along spatial axes. It can be seen that the
solid materials can exist in different structural phases, which have different way of
molecule arrangement, but it is not always possible to separate these stages from
one another. In this case we can distinguish between two definitions of phases,
according to the structural or thermodynamic point of view, and in the second
case, the phases are different as to the thermodynamic parameters, and they are
separated in clearly distinguishable interfaces and they are separable.

56
Typical examples of a material containing various structural phases is the
coexistence of a crystalline state, with a regular arrangement of particles into
crystalline lattices and an overcooled liquid state, in which the mobility of
molecules is low, but their arrangement in the space is random, without signs of
symmetry. In the event of accidental deposit in the area we are talking about the
so-called amorphous state (from the Greek "morphe = shape", "amorphous =
formless" ). Phases of this material are chemically identical, but they significantly
differ in their arrangement. As an example of phases defined from the
thermodynamic point of view we can mention a composite composed of two
different materials, such as soft clay suspension in water.
Below the melting temperature of water the whole system is in solid state,
with two thermodynamically different components, separated by a strictly defined
interface and mutually detachable. For polymers we can introduce as an example
the composite of a plastic and inorganic filler.
When we discuss the phase conditions, in which the different substances are
under different conditions, we do not consider a gaseous state for polymers,
because of the molecular cohesion given the length of chains, and thus the
resulting number of contacts is much higher than the strength of the covalent
bindings. That is why, before there could appear a drift of individual
macromolecules to their gaseous state, there will be the destruction of the chain
caused by thermal degradation. The liquid and solid state, the nature of phase
transitions is more complex than for the low-molecular substances. For this
discussion it is favourable, if we do not envisage the mobility of entire
macromolecules, but these will be divided into the parts of a chain - segments, in
typical cases e.g. 12 to 60 carbons in the main chain. At low temperatures, the
equilibrium position of segments is constant and movement is limited to vibration
or rotary-vibratory oscillations about equilibrium positions. A polymer behaves in
the same way as a low-molecular substance in a solid state. When applying the
deformation, the entire system observes Hooke´s law, Young's elastic modulus is
high, and the material is fragile. As the behaviour of a polymer is similar to glass,
this state is called the vitreous status. Vitreous status is observed at low
temperatures up to the so-called temperature of vitreous transition. In the area of
vitreous transition temperature T g the qualitative change of segments movement

57
appears, which in the area above T g changes to rotation. A chain may take a large
number of different conformational shapes, material has a lower modulus of
elasticity and it behaves as a highly elastic body. We are talking about the so-
called a highly elastic, or rubber state. This state is typical for the linear
polymers and except for polymers it is not familiar in any other materials.
By further increasing temperature the rotary movement of segments
becomes more intense, to finally allow the first realignments of segments and later
of the whole macromolecules. After reaching the so-called creep temperature a
polymer is in a viscous-plastic state, when it comes to non-reversible flow. Above
this temperature, polymers are heat-plastically formable.
These considerations are also valid for the non-arranged amorphous solid
phase. Many polymers, however, create a crystalline phase, and this almost
without exception coexists with the amorphous phase in the form of so-called
semicrystallic materials. The crystalline phase is acting in the same way as the
solid one, and above the so-called melting temperature, when the crystallites
crash, it is transformed into the liquid phase, the viscous-plastic one. From the
temperature point of view, at which there are significant transformations taking
place, we differentiate between the temperatures
Vitreous transition - T g
melting - T m
creep - Tf
while Tg < Tm ~ Tf..
Change of the state, the transformation of an amorphous substance into the
crystalline and vice versa, or the transformation of one crystalline system to
another is termed as a phase, or a state transition. Next to each other several stages
may exist, which are in thermodynamic equilibrium, separated by a clearly
identifiable interface and one phase area of finite dimensions. At the interface the
internal energy is changing, and so do the specific heat and the thermal expansion
coefficient. Because of the nature of this transformation we are talking about the
phase transitions of the first or the second order.

58
Phase transitions are determined from dependencies f internal energy
changes on the rise in temperature. Experimentally, the easiest way to determine
them is to use their thermal dependence on the specific volume. In the phase
transition of the 1st order on both dependencies the jump change is evident. A
typical phase transition of the 1st order is melting, or a reversed action -
solidification at a point melting/solidification point. At this point for example, for
melting we supply heat into the system, i.e. internal energy grows, but the
temperature does not change. Only after the entire crystalline share is melted, the
temperature, now of a liquid state, starts to rise again. A similar picture can be
seen if we watch the change of a specific volume with temperature, where at the
melting point there is a significant change of this parameter without any change in
temperature.
At the phase transition of the 2nd order we are seeing for these
dependencies a break instead of a jump. Dependence e.g. of the volume on
temperature is linear, when direction line dV / dT is a coefficient of thermal
expansion. During the phase transition, this direction line will change, but its
dependency will still be continuous, uninterrupted. In fact, in most cases of
polymers there is a bigger or a smaller bending instead of a break occurring,
within the temperature interval of several degrees (typically 5 - 15 o
C). Then the
temperature of vitreous transition can be set using linear extrapolation as the
intersection of two straight lines.
The way the melt gets back into the solid state, depends on the structure of a
polymer chain and its speed of cooling.
If the matter has non-symmetrical molecules, there cannot be a regular
arrangement, and material does not crystallize. Under all circumstances an
amorphous solid phase will form. On the other hand, even at the maximum
regularity of the polymer structure, the entire volume of the overcooled melt will
virtually never crystallize. After reduction in temperature to a temperature of
crystallization, the polymeric segments begin to organise themselves into the
crystalline lattice. Segments that are already built into a crystalline lattice have
well defined positions and their translational movement will stop. At the same
time, the mobility of neighbouring segments decreases, which therefore crystallize

59
more slowly, or even have no chance to organize themselves into any regular
shape and create an amorphous phase, i.e. a frozen solid, but non-crystalline
structure.
From this point of view, the superposition of crystallization speed and the
mobility of segments with a decreasing temperature of crystallisation is
interesting. The lower the crystallisation temperature, the greater the driving force,
which forces segments to organize themselves into some crystalline lattice.
Simultaneously, the viscosity of the overcooled melt grows and the mobility of
segments decreases. Therefore, the dependence of the crystallization speed from T
m is to maximum, when during small supercooling the driving force of
crystallization is small, while at high supercooling the driving force is great, but
the mobility of segments, allowing their arrangement, is low.
Finally, in the T g area the translational movement of segments virtually
stops and a part of the material, which did not manage to crystallize, is being
transformed into a vitreous condition. It is clear that at a sufficiently high speed of
crystallization it is theoretically possible to obtain each substance in the form of
amorphous glass.
Another consequence of this process is that, as long as the cooling is not
infinitely slow, there are always areas in the material which are not in
thermodynamic balance. This leads to the so-called secondary crystallization
phenomenon, when the temperature increases to the area (far below the melting
temperature) the mobility of segments may increase so that the additional
crystallization will be possible.
As it was said above, vitreous transition temperature Tg is defined as a
temperature at which a bend or a break appears, depending on the specific volume
on temperature. In the same temperature field the break of temperature
dependencies of thermodynamic functions is occurs, such as enthalpy and
entropy. The first derivation of the basic thermodynamic functions, the
temperature coefficient of volume dV/dT, and the temperature coefficient of
enthalpy dH/dT = Cp (heat capacity) changes in a leap. Also, leap changes are
occurring for temperature coefficients in transport properties, such as viscosity,

60
gas diffusion and tension relaxation, and modulus of elasticity rises by several
orders. Absorption of mechanical and electrical energy reaches the maximum.
To determine the temperature of the vitreous transition there are several methods
that are using the changes of the properties in the Tg. T g can be determined for
example dilatometrically, monitoring the heat expansion. Another commonly used
method is calorimetry, particularly differential scanning calorimetry DSC . One
important method is also the dynamic-mechanical analysis, which in a gradual
increase of temperature directly detects the release of certain movements, while
the most significant response is to be observed with the release of movements in
the main chain segments, which correspond to temperature T g .
The methods for the determination of T g are related one important
parameter, the so-called free volume. The concept of the free volume can be
explained using the following reflection. In a solid body molecules are not quite
closely stacked, among them there are free vacancies. These relate to the freezing,
or to the limitation of movement in the real time and in a certain condition.
Increasing temperature leads to thermal volume expansion. Yet, in the volume
itself a molecule does not change with temperature and practically the only
changes take place in space between molecules and, at the same time, the mobility
of molecules increases. According to the theory of Cohen and Turnbulla, in the
liquid there is a systematic redistribution in the size of vacancies, when a
molecule can skip only into an area of at least a minimum volume. Leap
frequency is not determined by energy factors, but by the factors of probabilities,
while an important determinant is the critical mass. Probability that a certain
molecule jumps to another location is given by the probability that there is a large
enough vacancy in its vicinity.
Volume thermal expansivity is caused by an increase in free volume.
Probability can be expressed as variable f, the share of occupied and vacant
space. The dependence of f on temperature is linear
f = f (T - T), 3.7

61
where T and T is the system temperature and the reference temperature and f is
the thermal expansion temperature. Based on these assumptions the relationships
for the diffusion coefficient D can be derived in the form
D = D exp (-1 / f (T - T) 3.8
or other quantities, like viscosity η
η = η exp (-1 / f (T - T) 3.9
An alternative to the probability theory is the energy theory. According to
these the jump of a fluid particle is only possible if the particle has a certain
surplus energy, which will allow it to overcome the energy barrier. Temperature
dependencies are the two parameters and have the shape of the Arrhenus equation
D = D e-Ea/RT
, 3.10
where Ea can be regarded as an activation energy, or the temperature coefficient
of the process. In doing so, it should be noted that the overall energy system
depends on temperature, but the energy of each individual particle (or moving
segment in case of a polymer) is not identical with the other particles and that it is
not constant. Particles (segments) are mutually clashing and giving their energy to
one another, while some particle may randomly take energy sufficient to break the
energy barrier constructed surrounding particles. When this occurs, a reordering -
a translation movement - takes place.
At the temperature T = 0 K the diffusion coefficient drops to zero and
viscosity gains infinite value.
In 1921 Vogel, Fulcher and Tamman derived during their study of inorganic
glasses viscosity one important empirical equation, allowing a direct calculation
of viscosity melt η for any temperature T, if there is experimentally determined
constant B and its viscosity at the reference temperature T
log = log + B / (T - T) 3.11
By comparing these relations we can see that

62
f = log e / B. 3.12
The second key semi-empiric equation proposed by Williams - Landel -
Ferry in 1953, when the reference temperature selected Tg and g was the
viscosity at Tg. This equation is of the form
log / g = -c1g (T - Tg) / c2g + T - Tg 3.13
and it is so important that it is known under the definition: WLF equation.
According to he WLF equation, the η viscosity at a temperature T can be
calculated on the basis of the known viscosity g at a temperature T g , where c 1g
and c 2g are empirical parameters.
WLF equation may be derived from the Vogel equation, so that the basic
parameters will be represented by the coordinates of a reference point g a Tg.
After substitution
c1g = B / Tg - T 3.14
and
c2g = Tg - T 3.15
we get the WLF equation. In this case, the constants c 1g and c 2g are almost truly
independent of temperature, if we use T g as the reference temperature. Universal
(average) values of constants are c 1g = 17.4 and c 2g = 52 K .
And on the basis of previous relations it can be expressed
f = 1/2,3 c1gc2g,
3.16
fg = 1/2,3c1g
When applying universal values of constants c 1g and 2g ,we get the universal
values of f = 4.8 x 10 -4
K and f g = 0.025 . At the same time we observe a good
compliance f of the universal with experimental values measured for many

63
polymers. Also, from the definition for f g it implies that at T g the free volume is
2.5% of the total volume of body, i.e. 97.5 % are occupied by matters of
molecules. When temperature rises by 100 o
C above T g, the parameter fg will
reach a value of 0.075 g , i.e. , the free volume will be increased to 7.5 %.
This result can be interpreted also in a way that under T g the free volume is
virtually constant. In this case, the measured thermal expansion of a material in a
vitreous condition will be only the measure of the expansion of molecules itself.
Based on the considerations of Simha and Boyer, the volume of molecules Vm can
be obtained at absolute zero by extrapolation from the liquid.
As it was said above, T g is formally the transition of the 2nd order . From
the thermodynamic standpoint, however, this claim is not quite accurate, because
the measurements of thermodynamic quantities in the environment of T g are not
at equilibrium, but the time factor (the rate of temperature change) plays a role
here, as well as the history of a sample (the method for preparation). Therefore, T
g cannot be considered as a thermodynamically defined transition. The result is the
fact that the experimental determination of T g for a specific polymer depends on
the method used, and to some extent on the conditions of the experiment.
Therefore, within the professional literature the mentioned values T g differ,
sometimes by more than dozens of o
C .
Thermodynamic theory, which was presented by Gibbs and Di Marzio, says
that the equilibrium transition actually exists and it is situated in the surroundings
of Vogel temperatureT. In practice, however, these measurements are
impracticable, because it would require infinitely slow changes in temperature. A
relatively reliable picture can be obtained using a grid-model, with which we can
calculate a conformational energy system and conformational entropy. With
decreasing temperature, the number of viable macro-conformations reduces, while
of course the energy-poorer conformation prevail. At the limit temperature of
absolute zero in a perfectly frozen condition all macro-conformations should be
trans, the number of possible statuses would be 1 and conformational entropy
zero. Real systems freeze several tens of degrees above this temperature, where
the number of conformational transitions is small and transitions rare.

64
3.2. Models of viscous elastic materials
Let us now follow the issue of viscous environment phenomenologically.
For elastic environment, as we already know, Hook´s law applies in its symbolic
form
σ=Eε . 3.17
Newton´s law of viscous creep can be written as
σ = η ε’, 3.18
where ε’ represents the strain rate.
Equation 3.18 does not describe distortions causing changes in volume during a
deformation of the environment, but it only describes shear deformation.
Nevertheless, in the dissemination of the elastic waves in viscous environments,
under certain conditions deformations of volume and the consequent contribution
to viscosity environment arise, also called bulk viscosity η'.
The real environment, as we have said, has viscous and elastic properties.
Measured time dependence of the shear deformation, as shown in Figure 3.6a, we
can therefore divide into three components, namely elastic, viscous, and relaxation
(Figure 3.6b, c, d).
Let us suppose that the stress is composed of the 'flexible stress" σ E and
from "viscous stress" σ η
'
ηE 'ηε+εE=σ+σ=σ 3.19
This equation is also called Kelvin´s equation.
If, at the time t=0 we apply the rectangular pulse of the mechanical tension T0 to
such an environment, then the environment will be described by relations
( )E
η=τ;e-1
E
σ=ε τ/t0
. 3.20

65
Figure 3. 6. Experimental time-dependent shear deformation (a). Distribution
of deformation into flexible (b), viscous (c), and relaxation component (d)
For a sufficiently long time in the environment there is created a time-
independent constant deformation E
σ=ε
0
∞ and the environment acts as non-
viscous . Reaction of the environment to the applied stress is probably not
immediate, but it progressively achieves the maximum value. Similarly, after
an interruption of the stress, this deformation exponentially decreases to zero,
while the time of relaxation is determined by the relationshipE
η=τ . This type
of deformation is called the delayed deformation.
Another approach to describe a viscous elastic environment was used by
Maxwell, who anticipated that within the environment two types of
deformations occur, elastic and viscous.
ε=εE+εη . 3.21
After derivation 3.6 by time and the substitution from relations 3.2 and 3.3 we
get
η
σ+
E
σ=ε
'
'. 3.22
From this equation it is clear that this deformation is composed of a
deformation proportional to the stress and viscous creep.
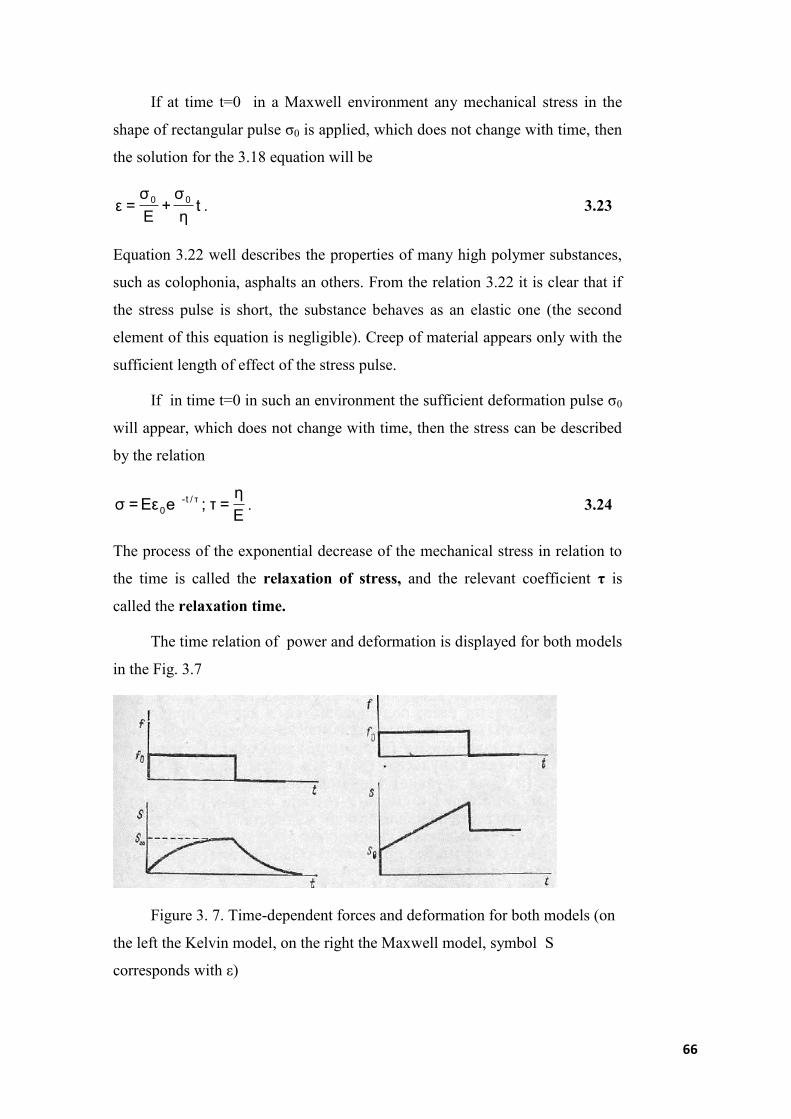
66
If at time t=0 in a Maxwell environment any mechanical stress in the
shape of rectangular pulse σ0 is applied, which does not change with time, then
the solution for the 3.18 equation will be
tη
σ+
E
σ=ε
00. 3.23
Equation 3.22 well describes the properties of many high polymer substances,
such as colophonia, asphalts an others. From the relation 3.22 it is clear that if
the stress pulse is short, the substance behaves as an elastic one (the second
element of this equation is negligible). Creep of material appears only with the
sufficient length of effect of the stress pulse.
If in time t=0 in such an environment the sufficient deformation pulse σ0
will appear, which does not change with time, then the stress can be described
by the relation
E
η=τ;eεE=σ τ/t-
0 . 3.24
The process of the exponential decrease of the mechanical stress in relation to
the time is called the relaxation of stress, and the relevant coefficient τ is
called the relaxation time.
The time relation of power and deformation is displayed for both models
in the Fig. 3.7
Figure 3. 7. Time-dependent forces and deformation for both models (on
the left the Kelvin model, on the right the Maxwell model, symbol S
corresponds with ε)

67
Now let us briefly mention the mechanical models for the above
environment, which allow the modelling of real materials. The flexible
environment is in these models represented by a spring and the viscous one by a
cylinder moving
in a piston in a viscous liquid (figure 3.8 a, b). These are the fundamental
elements.
Figure 3. 8. The flexible environment model (a), the viscous environment model
(b)
Kelvin´s environment can be modelled using fundamental elements and
connecting them parallely, on condition that their connection element AB is
stationary and the CD moves on rails, which prevents this system´s deformation
(see Figure 3.9). Then the increase of both basic elements will be the same and the
total force acting on the model will be equal to the sum of forces applied to both
elements. In such a way both elastic and viscous stresses in Kelvin´s environment
are comprised.
From this model we can see that the immediate deformation of such a model
would require infinitely great strength, which is not possible. With the gradual
distortion caused by constant stress we gradually reach a constant value of
deformation.

68
Figure 3.9. Model of Kelvin´s environment
Maxwell´s environment can be modelled using the series connections of
basic elements, as seen in Figure 3. 10. In this case the same force acts on both
elements and their extensions are composite
Figure 3. 10. Model of the Maxwell´s environment
Real viscous elastic environments are behaving in a more complex way. One of
the models is the so-called Knezer´s model shown in Figures 3. 11.
In Figure 3.12 the flow curve of a rubber mixture is shown, which is qualitatively
very similar to Knezerś model.

69
On the basis of acquired knowledge we can now define a comprehensive
Young's modulus, which is described schematically in Figure 3.13, and from
which we get that the viscous elastic properties of a viscous model are
described by the imaginary part of this module and by the elastic real part
of the module.
Figure 3. 11. Knezer´s model (left diagram, right time course of the force and
deformation)
.
Figure 3. 12. The flow curve of the rubber mixture

70
Figure 3.13. Complex Young's modulus
Module and the viscosity are thus frequency-dependent, which is typical
for the viscous elastic materials.
The operator of the effective module will have in Maxwell´s environment the
shape
dt
dτ+1
dt
dη
=E . 3.25
For the harmonic signal we get
i1
iE .
If we divide the expression for the effective module into its real and imaginary
parts, we get
2222
22
1i
1EE
. 3.26
Quantity
E1
EERe
22
22
3.27
is called the dynamic module.
221EIm
1 3.28
is called the dynamic viscosity.
From relations 3.26 and 3.27 we can see that at low frequencies (ωτ<< 1)
Maxwell´s environment acts as a viscous liquid, because its dynamic module is
very small and its dynamic viscosity is frequency-independent. With the
growth of its frequency the dynamic viscosity begins to fall, the dynamic
module is growing, and for very high frequencies (ωτ>>1) Maxwell´s
environment acts like a solid. This process is called the relaxation of viscosity
.

71
In the case of Kelvin´s environment the dynamic module and its dynamic
viscosity are frequency-independent.
3.3 Payne effect
Adding some filler to a polymeric matrix significantly affects the behaviour
of individual dynamic characteristics depending on increasing the instance of the
dynamic deformation, with regard to the quality of interactions established
between the particles in the fillers themselves, and between the particles in a filler
and the polymer matrix. The quality of these interactions can be assessed by
measuring the dynamic-mechanic properties of the prepared composites at various
amplitudes of the dynamic deformation.
The values of the elastic module (E') for not filled vulcanizates do not change
significantly with a raising level of dynamic distortion; in the case of stuffed
vulcanizates, however, there is a decrease in the module, and this effect is more
pronounced for vulcanizates with a higher content of filler. This phenomenon is
called the Payne effect.
In the case of a leaking module (E' ), like in the case of the elastic module,
the increase of its values with the increasing degree of polymer filling in a whole
range of dynamic distortions is observed. Depending on the increase in
deformation, however, there are no monotonous value decreases observed, as in
the case of an elastic module, but the maximum at a certain deformation,
characteristic for a given pair of the polymer-filler. It is assumed that the
development of the loss module is controlled by extinction and the recovery
of the filler particles networks. Consequently, the loss module is dependent on
the speed of extinction and recovery of the network. The speed of extinction and
recovery of the network is based on the dynamic deformation phenomenon. When
the distortion is large, the disappearance of a network is significant, and its
recovery practically does not occur. If the deformation is sufficiently large for the
network to recover at a given frequency, the impact of the network on the loss
module will be eliminated.

72
Similarly, if a deformation is small (or the network is sufficiently strong) to
trigger the extinction of a network, the loss module is not dependent on the given
deformation, but it will mainly depend on the contribution of the polymeric matrix
during the cyclic dynamic deformations of the composite.
A change of each of the modules (elastic and loss) depending on the size of
dynamic distortion is based on other mechanisms. While an elastic module
depends to a large extent on the presence of a physical network (its gradual
disappearance in with the increasing dynamic deformation) consisting of
filler particles in a polymeric matrix, the loss-making module is affected by
the repeated process of dissolution and the recovery of its structure during
the cyclical stress.
The polymer-filler interaction causes the absorption of the polymer chains to
the surface of the filler, thus limiting the mobility of the polymer segments. The
result is the creation of a polymeric layer on the filler surface, where the
viscosity and the module of polymer will increase. A very high module in
proximity to the surface of the filler particles within the polymer shell with an
increasing distance from the surface filler will fall, until at a certain distance it
will be the same as the module for the polymer matrix. If there are two or more
particles of the filler or units which are close together enough, they create an
agglomerate of a mechanism of mutual breaches of the polymer shells in filler
particles. The network, set up using such a mechanism, will be less rigid than the
network, set up using a direct contact between the aggregates. This type of
network can begin to fade already at relatively small deformations.
In the case of a network created by the direct contact of aggregates the
process of repeated extinction and subsequent recovery of a network causes
higher loss energy. This shows that at high temperatures the internal friction
between the aggregates is the dominant mechanism. When reducing
temperature, the polymer gets into the glass transition area, where the
polymer mix has the major proportion of the losses of the included
mechanical energy.
If the network consists of the mutually breached shells of particles, the
mechanism of contribution to the energy losses is different than the one for the

73
network described above. At higher temperatures the polymer matrix is in a
rubber condition, but the polymer adsorbed on the filler surface is in the
phase of transition. This leads us to the fact that the shell can absorb more
energy. As the temperature increases, the thickness of the shell decreases, which
increases the mobility of chains, which leads to a lower hysteresis. Reversed re-
agglomeration of the filler during cyclic dynamic deformation reaches a higher
degree.
The most significant impact on the dynamic properties changes of
polymers is represented by the morphology of the filler particle, namely the
size of particles or their specific surface, and particles structure. With an
increasing specific surface of the soot at the constant replacement, a sharper
decrease in the elastic module is observed, with an increasing degree of the
dynamic deformation. An increase in the Payne effect indicates the great ability of
small particles (aggregates) in soot to agglomerate in a polymeric matrix.
From these considerations it is clear that the measurements of the
Payne effect may contribute significantly to the characterisation of
interactions between the components of the filled vulcanizate in the dynamic
conditions.
We will monitor the Payne effect on the developments of the dynamic
characteristics of polymers with a different content of fillers, according to the
degree of the dynamic deformation.
The addition of filler into the polymeric matrices in general affects the
increase of elastic module values, which is caused by the nature of a filler,
which behaves as a Hook´s element. By comparing the values of modules for a
given temperature, frequency, the amplitude of distortion, and the selection of a
pair polymer matrix - we can see that the values of modules grow with the degree
of filling .
In Figures 3.14 and 3.15 there are displayed the dependences of modules of
E and G for the tread mixture. It is visible that even during the static
measurements of elastic modules the Payne effect is demonstrated by a decrease
in the values for both modules.

74
Figure 3.14. Dependence of Young´s modulus on the perpendicular
operating pressure
Figure 3 15. Dependence of the shearing module on perpendicularly
operating pressure
Summary of terms
Transformation of coordinates and tensors, properties of the second-order tensors,
stress tensors, distortions and Hook´s law in general form, compressibility, the impact of
lattice symmetry on the the number of independent components of the tensor.
1.Define Newton's liquid.
2. Qualitatively describe the material flow from a microscopic point of view.

75
3. What is entropic flexibility?
4. Describe the glass stage conditions.
5. What equations describe the glass transition in polymers?
6. Explain Kelvin´s model.
7. Explain Maxwell´s model.
8. Explain Knezer´s model.
9. Explain the fundamentals of the complex Young´s model.
10. Explain Payne phenomenon.
Questions to the topic
1. How to transform the second-order tensor?
2. What relationship determines the stress tensor constituents?
3. Explain Hook´s law in its general form.
4. How can we calculate the compressibility of anisotropic material?
5. What is the impact of symmetry on the number of independent components of the second-
order tensor?
6. Explain the term invariant tensor, and what it determines?

76