Embed Size (px)
Citation preview

Journal of Radioanalytical Chemistry, Vol. 23 (1974) 51--62
SOLVENT E X T R A C T I O N OF T E C H N E T I U M ( V I I )
BY 4 - ( 5 - N O N Y L ) P Y R I D I N E A N D ITS S E P A R A T I O N F R O M
U R A N I U M A N D SOME FISSION P R O D U C T S
M. IQBAL, M. EJAZ
Pakistan Institute of Nuclear Science and Technology, Nilore, Rawalpindi (Pakistan)
(Rece ived J a n u a r y I1, 1974)
The extraction of technetium (VII) by 4-(5-nonyl)pyridine has been investigated from different aqueous solutions. Separation from uranium and some fission products has been aeh2eved in nitrate media. From the results of partition experiments, attempts have been made to deduce the nature of the extracted species.
Introduction
Various methods based on solvent extraction are available for the separation of technetium(VII) from chemically similar (manganese and rhenium) and neighbouring (molybdenum and ruthenium) elements 1-6 or from fission products. 6-12 Previous observations lead to the inference that technetium (VII) may be extracted from acid, neutral and alkaline media under appropriate conditions, and that among alI the organic solvents containing oxygen, phosphorus, or nitrogen, anion-exchangers including aromatic amine pyridine and its methyl-substituted derivatives have a high affinity for technetium(VII). It therefore seemed interesting to make extraction separation studies of technetium(VIi) from different aqueous media with an extractant that has not been tested as yet, a high molecular weight aromatic amine, 4-(5-nonyt)pytidine. This extractant may have considerable advantage over pyridine and methyl-substituted pyridines, because its solubility in water is almost negligible.
R e a g e n t s
Experimental
4-(5-nonyl)pyridine (NPy) was obtained from K and K Labs Inc . , Plainview, N. Y. , and was purified by vacuum distillation before use. It is a place yellow oily liquid and has a b.p. of 94 ~ at 0. 8 mm Hg, a refractive index n 20 of 1 485 and a density d 20 of 0. 9208 g / cm 3.
J, Radioanal. Chem. 23 (1974) 4*
5l

M. IQBAL, M. EJAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
Its solubility in water was found to be 1 .2 g/l i tre. Its purity was checked with a Varian Aerograph Chromatography unit, using a 10% earbowax 20M column at 220 ~ which showed a single peak. The detector employed was of alkali f lame ionization type.
Tributyl phosphate (BDH Chemicals Ltd., Poole, England) was purified by washing several times with sodium hydroxide solution and with water, dried over anhydrous sodium sulphate, and redistilled under vacuum.
Nitric, hydrochloric and sulphuric acid solutions were generally prepared from B. D.H. volumetric solution ampoules, and all other chemicals used in this work were of analytical reagent grade.
R a d i o n u c l i d e s
The 6-hr 99mTc was separated from its parent, 66.6-hr 99Mo, by solvent extraction according to the method of F a d d e e v a e t al. 3
99Mo was obtained by the irradiation of MoO 3 in the research reactor of the Pakistan Institute of Nuclear Science and Technology, Nilore, Rawalpindi. The oxide was refluxed with nitric acid (to ensure complete oxidation) and neutralized with sodium hydroxide. 99Mo was freed from its daughter 99mTc and used immediately after purification. In somecases extraction was also carried out with a 99Mo - 99mTc mixture and samples were counted 3 to 4 days after separation. (The concentration of molybdenum in the initial aqueous phases was <10"Smole / litre. )
The 186Re was obtained by neutron irradiation of rhenium.metal. A period of three weeks had elapsed since the tracer was formed, so that all of the 188Re had decayed. The other tracers used in this work were obtained from the Radiochemi( Centre, Amersham, and were pure enough to meet catalog specifications. 233U (obtained from the Radiochemical Centre, Amersham) was purified by solvent extraction 13 before use.
R a d i o c h e m i c a l a s s a y a n d i n s t r u m e n t a t i o n
The equipment used for general alpha-assay was: (1) An argon gs-flow proportional counter, Harwell tyle 3-7/11558, in
conjugation with an EKCO fast scaler, type N 530 F. High voltage was taken from the scaler: and
(2) Nuclear Chicago Corporation alpha-scintillation counter, Model DS-S Serial 1709.
Solid t3-emitting samples were assayed with the aid of an end-window Geiger assembly equipped with a G. E. C. tube, type EHM 2/S. Gamma-ray count rates were determined using a Nuclear Chicago single-channel analyser, model 872.
52 J. RadloanaL Chem. 23 (1974)

M. IQBAL, Ivl. EJAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
coupled with a 3" x 3" thallium-doped sodium iodide weU-type ~/-ray scintil- lation counter.
~, -Spectra were taken using a Nuclear Data ND-4410 Computer System 512/0124 multichannel analyser, model 2560. The detector used with this analyser was a 4" x 3" NaI(T1) crystal.
P r o c e d u r e
Equal volumes of the aqueous phase (cOntaining the tracer) and the organic phase (equilibrated previously with a solution of the same composition as the aqueous phase without tracer) were stirred in a 15 ml graduated centrifuge tube with an electric stirrer for 5 rain at 25 ~ After equilibration the phases were separated by centrifugation. Equal volumes of aliquots from the organic and aqueous phases were pipetted by mieropipetre onto stainless stell or glass planchets in the case of a- and ~-counting. In the case of 7-counting the liquid samples were taken into small 5 ml bottles and counted directly in a ~,-scintillation counter. All data were obtained by both "direct" and "reverse" extractions.
Results and discussion
E x t r a c t i o n o f t e c h n e t i u m ( V I I ) a n d m o l y b d e n u m ( V I ) by 0. 1M N P y / x y l e n e f r o m n i t r i c a c i d s o l u t i o n s
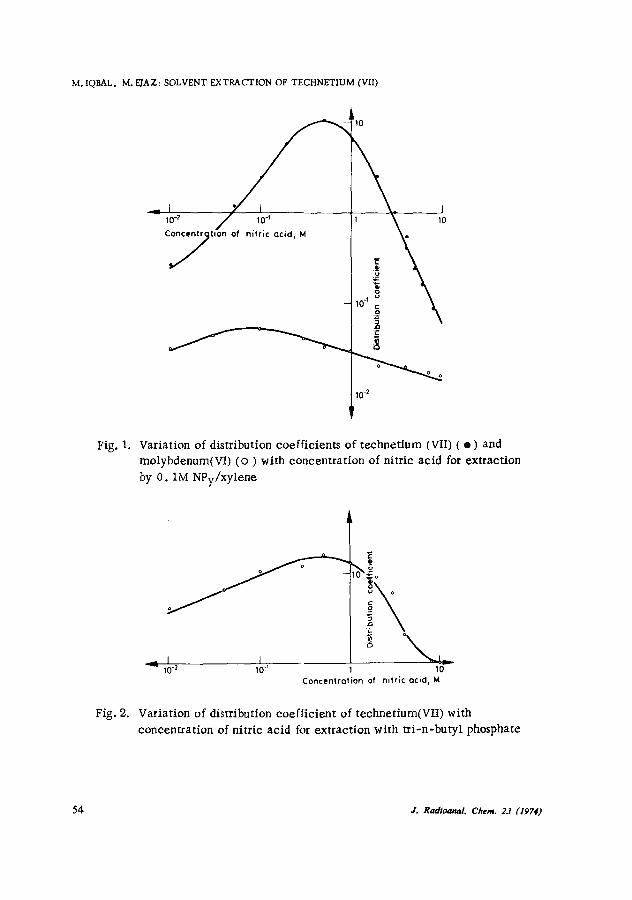
The effects of the nitric acid concentration on the distribution coefficients of technetium (VII) and molybdenum(vi) are shown in Fig. 1. The distribution coefficient for technetium(VIi) increases with increasing acid concentration in the aqueous phase, and attains a maximum at 0. 5M nitric acid before decreasing again at high acidities. The decrease in extraction at high acidities is probably due to the competition of nitric acid, for the available extraetant. Technetium (VII) extraction by NPy parallels the tri-n-butyl phosphate extraction whin in Fig. 2. This indicates that in both systems the extracted technetium(VIi) species is the same. The extracted technetium species may be the monobasic perteehnetate ion. Septivalent metal oxyanions have also been reported to be extracted from dilute acid solutions by amines 4, 14 and weakly basic oxygen-donor extractants. 15-20 The extraction of molybdenum(VI) may also proceed in the form of metal oxyanions. The extraction of oxyanions of molybdenum from acid solutions with organic bases has already been reported. 21 The low extraction of molybdenum(vi) as compared to technetium(VII) may be due to the comparatively low affinity of the reagent for the multibasic molybdate ions.
3". Radloanal. Chem. 23 (1974) 53
M. IQBAL, M. EIAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
10-2 C ~ , ~ t i o n of nitric acid, M
. I
u
_ 1 0 -~
~5
10-2
Fig. 1. Variation of distribution coefficients of technetium (VII) ( �9 ) and molybdenum(VI) ( o ) with concentration of nitric acid for extraction by 0 .1M NPy/xylene
Fig. 2.
10-2 10 -~ 1 0
Concentration of nitric ocid, M
Variation of distribution coefficient of technetium(VII) with concentration of nitric acid for extraction with tri-n-butyl phosphate
54 J. Radloanal. Chem. 23 (1974)

M. IQBAL, M. EIAZ: SOLVENT E X T R A C T I O N OF T E C H N E T I U M (VII)
Fig. 3.
./ i/ - - 10- 2
M o l o r i t y o f / * - n o n g l p y r i d i n e ~ 1,4 / c
! u
o
_ . 10-1
Variation of distribution coefficient of technetium(VII) with concentration of NPy from 0 .5M nitric acid
E x t r a c t i o n f r o m 0. 5M H N O 3 u s i n g d i f f e r e n t c o n c e n t r a t i o n s o f NP
Y With 0 .5M nictric acid, where the highest distribution coefficient was
obtained, the number of NPy molecules in the complex extracted was determined by a graphical method, i. e. from the dope of the line giving the relation of the technetium distribution coefficient to the NPy concentration in logarithmic coordinate (Fig. 3. ). In this case, the numbex of NPy molecules was 2. Hence the complex extracted may be represented as HTcO 4. 2 NPy and the reaction may be expected to proceed as follows:
H + + NO" + aq 3aq 2NC14H23org �9 (C14H23N.. . H . . . NH23C14) + NO3org
+ , - . . + TcO4 (C14H23N.. . H . . . NH23C14 ) NO 3 + TcO 4 ~ (C14H23N.. . H. NH23C14) org aq org
J. Radloanal. Chem. 23 (1974) 55

Concentration of
10-2 10 -1 " 4 I
Fig. 4.
M. IQBAL, M. KIAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
sulphur ic <acid, M
10
D -~
g ~
g
- I
Y Variation of distribution coefficients of technetium(VII) ( �9 ) and molybdenum(VI) (o) with concentration of sulphuric acid for extraction by O. 1M NPy/xylene
E x t r a c t i o n s t u d i e s o f t e c h n e t i u m ( V I I ) by 0. 1M N P y / x y l e n e as a f u n c t i o n o f t h e s o d i u m h y d r o x i d e c o n c e n t r a t i o n o f t h e a q u e o u s p h a s e
Extraction of Te(VII) was studied as a function of the concentration of sodium hydroxide in the aqueous phase. In contrast to the high extraction coefficients obtained for technetium(VII) in the case of pyridine and its methyl-substituted derivatives, 8 no extraction was observed in this case. Since anhydrous salts of septivalent metal oxyanions do not dissolve in dry pyridines, 22 the factor which exerts a decisive influence in the extraction by pyridine bases is apparently the formation and stability of hydrate-solvates. It has been reported that hydrate-
23 solvates are preferentially formed in case of reagents which dissolve more water, and on opposite situation is observed with solvents which dissolve water less well. 24 Thus, it may be assumed that the difference in the extraction behaviour of NPy is possibly due to its inability to form hydrate-solvates.
E x t r a c t i o n o f t e c h n e t i u m ( V I I ) and m o l y b d e n u m ( V I ) by 0 .1M N P y / x y l e n e f r o m s u l p h u r i c a c i d s o l u t i o n s
In Fig. 4 the curves givlng the dependence of the distribution coefficients of Tc(VII) and Mo(VI) on the sulphuric acid concentration show that both of the elements are extracted to some extent from dilute acid solutions. The decrease in
56 Jr. Radloanal. Chem. 23 (1974)

Fig. 5.
M. IQBAL, M. EIAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
Concentration of hgdrochtoric acid, M
10 -2 10 -I 10
10 -1 u g
~ .:-
10-2
!
Variation of distribution coefficients of technetium(VII) ( �9 )and molybdenum(VI) (o) with concentration of hydrochloric acid for extraction by (~. 1M NPy/xylene
extraction of technetium and molybdenum at high acidities may be due to the acid competition for the reagent. The extraction of molybdenum in this case is higher as compared to the corresponding nitrate system. This difference in extraction behaviour may be due to the extraction of sulphate complexes of molybdenum, which have been reported 25 to occur over a wide range of sulphuric acid concentrations.
E x t r a c t i o n o f t e c h n e t i u m ( V I I ) a n d m o l y b d e n u m ( V I ) f r o m h y d r o c h l o r i c a c i d s o l u t i o n s by 0. 1M N P y / x y l e n e
The results presented in Fig. 8 show the distributions of To(VII) and Mo(VI) as a function of the hydrochloric acid concentration of the aqueous phase. The extraction of technetium from weakly acid solutions resembles those for the corresponding nitric and sulphuric acid systems. This indicates that in weakly acid solutions technetium(VII) species are essentially not influenced by the nature of the anion present. At high concentrations of hydrochloric acid anomalous results are ~robably due to some contribution from complexes of reduced technetium i .e . TcCI~-. The extraction of molybdenum(VI) in weak and concentrated acid solutions is probably due to the existence of two types of molybdenum anions. Similar results have been obtained in the case of anion-exchangers. 21, 26
J. Radloanal. Chem. 23 0974) 57

M. IQBAE, M. EJAZ: SOLVENT EXTRACTION OF T E C H N E T I U M (VII)
Fig. 6.
10-2 10-I --el I I
Concen t ra t i on of n i t r i c ac id , M
10
o o
_ 0-2 _
g .2
"~_
i0 -3 ~_ o
f Variation of distribution coefficients of uranium (o) and ruthenium ( �9 ) with concentration of nitric acid for extraction by 0.1M NPy/xylene
In the intermediate region (0.2-2M) low extraction may be due to molybdic cations which have been reported to be formed 25 in this region.
E x t r a c t i o n s t u d i e s o f u r a n i u m and r u t h e n i u m by 0. 1M N P y / x y l e n e f r o m n i t r i c a c i d s o l u t i o n s
The extraction behaviours of uranium and ruthenium were studied as a function of the nitric acid concentration o f the aqueous phase. The extraction isotherms of uranium and ruthenium are presented in Fi,~. 6. The extraction of uranium is similar to that by liquid anion-exchangers. 27 This indicates an identical type of mechanism for file extraction of uranium, i .e. ion pairs of the the type (NPy. H) + [UO2(NO3) 3 ] - are extracted into the organic phase. The dependence of the distribution coefficient on the concentration of NPy from aqueous nitric acid solution of 5M shows a slope of unity (Fig. 7), confirming that one molecule of the reagent is being utilized per molecule of uranyl complex. The extraction of ruthenium is maximum at 1M nitric acid and decreases at high acidities, probably due to the acid competition.
58 J. RadtoanaL Chem. 23 (1974)

M. IQBAL, M. EJAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
Fig. 7.
Moloritg of &- nonglpridine ) M
10- 2 10-~
" ~ 1 ]
/ /o
_ i0 -1
E
t~
10 -2
C5 !
Variation of distribution coefficient of uranium (VI) with concentration of NPy from aM nitric acid
S e p a r a t i o n o f c a r r i e r - f r e e 9 9 T c f r o m i r r a d i a t e d m o l y b d e n u m
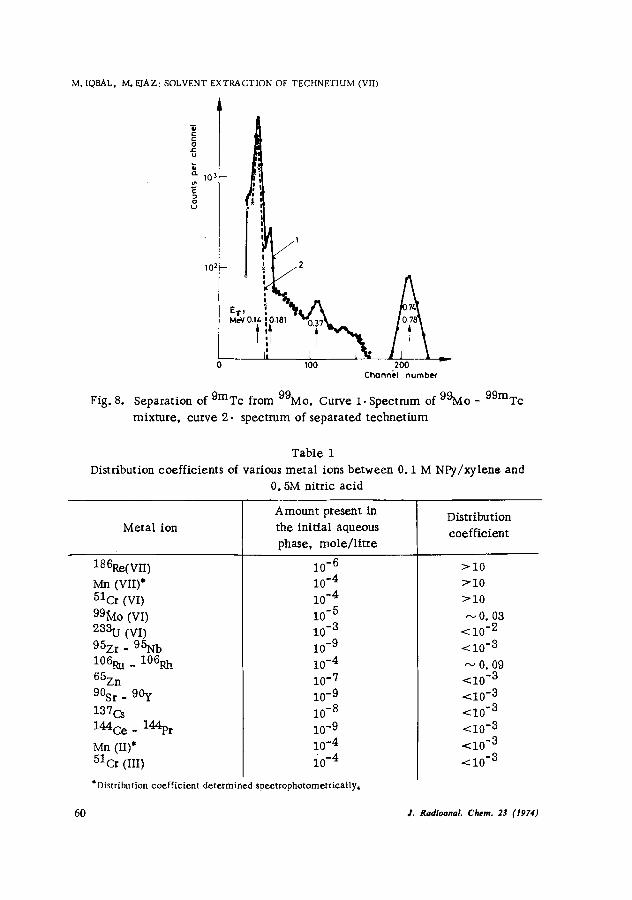
Neutron-irradiated molybdenum oxide was refluxed with nitric acid and neutralized with dilute ammonium hydroxide. The solution was made 0. 5M with respect to nitric acid and equilibrated with an equal volume of 0.1M solution of NPv/xylene for 5 rain, After equilibration the organic phase was separated and s6rubbed with 0.5M nitric acid. The scrubbing step was repeated thrice and finally pure carrier-free technetium(VII) was stripped from the organic phase by using 14M nitric acid. The y-spectrum of the separated 99mTc (Fig. 8) shows that 99Mo and trace activities of niobium and zirconium isotopes, which may be formed from molybdenum by (n, p) and (n, a) reactions, are not extracted by NPy under the conditions studied.
D e c o n t a m i n a t i o n f r o m v a r i o u s e l e m e n t s an d s e p a r a t i o n f r o m m i x e d f i s s i o n p r o d u c t s
To study the selectivity of the extraction separation of technetium with 0.1M NPy/xylene from 0.5M nitric acid, we examined the behaviours of a number of metal ions, including uranium and important fission products. Each element was first studied individually according to the standard procedure described. The data are presented in Table 1.
3". Radioanal. Chem, 23 (1974) 59
M. IQBAL, M. EJAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
Fig. 8,
i: u
a 10 ~
o
10 2 2
ET~ ~
~00 200 Channet number
Separation of 9mTc from 99Mo. Curve i. Spectrum of 99Mo - 99mTc mixture, curve 2. spectrum of separated technetium
Table 1 Distribution coefficients of various metal ions between 0.1 M NPy/xylene and
0.5M nitric acid
Metal ion
186Re(VII) Mn (VII)* 51Cr (VI) 99 fVlo (VI) 233U (VI) 95Zr. 95Nb 106Rn _ 106Rh 65Zn 90Sr. 90y 137Cs 144Ce_ 144pr
Ivln (If)* 51Cr (III)
Amount present in the initial aqueous phase, mole/litre
10-6 10-4 10-4 10-5 10 -3 10-9 10-4 10-7 10-9 10-8 10-9 10-4 1"0-4
*Distribution coefficient determined spectrophotometrically,
Distribution coefficient
>10 ~I0 > i0 ,~0. 03
<10-2 < i0-3 ,',~ O. 09 <10 -3 <i0-3 < i0 -3 <10-3 <10 -3 <10 -3
J. Radloanal. Chem. 23 (1974)
M. IQBAL. M. EJAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
.c E
o c r
10 s
1 0 ~ -
4 10 3 -
10 2 -
1 0 -
i I o.1
Mo - T c
-Nb
\
2
0.2 0.3 0.4 0,5 0.6 0.7 0.8 0.9 l Energg, MeV
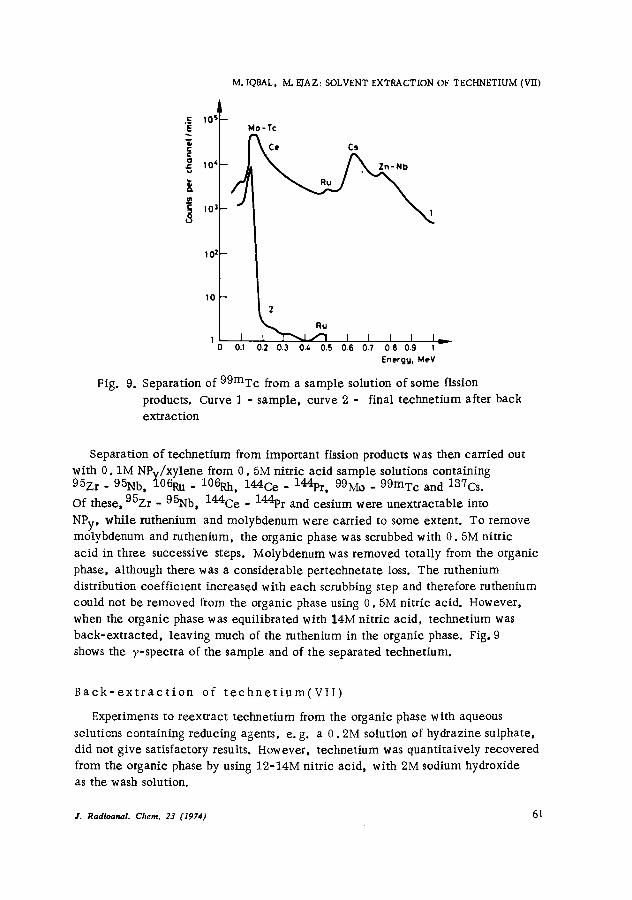
Fig. 9. Separation of 99mTc from a sample solution of some fission products. Curve 1 - sample, curve 2 - final technetium after back extraction
Separation of technetium from important fission products was then carried out with 0.1M NPv/xylene from 0. GM nitric acid sample solutions containing 95Zr - 95Nb, 106Ru - 106Rh, 144Ce - 144pr, 99Mo - 99mTc and 137Cs.
Of these, 9GZr - 95Nb, 144Ce - 144pr and cesium were unextractable into
NPy, while ruthenium and molybdenum were carried to some extent. To remove molybdenum and ruthenium, the organic phase was scrubbed with 0.5M nitric acid in three successive steps. Molybdenum was removed totally from the organic phase, although there was a considerable pertechnetate loss. The ruthenium distribution coefficient increased with each scrubbing step and therefore ruthenium could not be removed from the organic phase using 0.5M nitric acid. However, when the organic phase was equilibrated with 14M nitric acid, technetium was back-extracted, leaving much of the ruthenium in the organic phase. Fig. 9 shows the V-spectra of the sample and of the separated technetium.
B a c k - e x t r a c t i o n o f t e c h n e t i u m ( V I I )
Experiments to reextract technetium from the organic phase with aqueous solutions containing reducing agents, e.g. a 0.2M solution of hydrazine sulphate, did not give satisfactory results. However, technetium was quantitaively recovered from the organic phase by using 12-14M nitric acid, with 2M sodium hydroxide as the wash solution.
J. Radtoanal. Chem. 23 (1974) 6 l

M. IQBAL, M. EIAZ: SOLVENT EXTRACTION OF TECHNETIUM (VII)
References
1. G.E. Boyd , Q . V . L a r s e n , E .E . M o t t a , Rept., AECD-2151, June 1948; Rept., ORNL-2159 and ORNL-2584, 1958.
2. S. T r i b a l a t , J. B e y d o n , Anal. Chim, Acta, 6(1952) 96. 3. M.S. F a d d e e v a , O . N . P a n l o v , V. V. B a k u n i n a , Zh. Neorg. Khim. , 3(1958) 165. 4. J.B. G e r l i t , Proc. Intern. CcmL Peaceful Uses of Atomic Energy, Geneva 1955, 7 (19,56) 145. 5. T . K i b a , A. M i u r a , Y . S u g i u k a , Bull. Chem. Soc. Japan, 36 (1963) 663. 6. E. A n d e r $ , The Radiochemist~y of Technetium, AEC NAS-NS, l~pt.. No. 3021 (1960). 7. G.B.S. S a l a r i a , C.L. R u l f s , P . I . E l v i n g , Anal. Chem. , 35(1963) 983. 8. S . L R i m s h a v , G.F. M a l l i n g , Anal. Chem. , 33(1901) 751. 9. N.H. C a n b e l l , Anal. Chem. , 35(1963)2052.
10. A.A. P o z d n y a k o v , Usp. Khim. , 34(1965) 300. 11. P..J. M e y e r , R.D. O l d h a m , R.P. L a r s e n , Ana l Chem. , 36(1964)1975. 12. G. G o l d s t e i n , J.A. D e a n , Radiochim. Acta, 5(1966) 18. 13. A.S. S o l o v k i n , M . I . K o n a r e v , D . P . A d a l v , ~ . Neorg. Khim., 5(1960) 1861. 14. R. C o l t o n , R . D . P e a c o c k , Q. Rev. Chem. Soc. , 16(1962) 299. 15. J.L B u t h e r , R. M. D i a m o n d , I. Phys. Chem. , 73(1969)675. 16. T.J. C o n o c c h i o b i , M . I . T o c h e r , R . M . D i a m o n d , J . Phys, Chem. , 69(1965) 1106. 17. A.S. K e r t e s , A. B e c k , J. C h e m . Soc. , (1961) 1922. 18. A.V. P e t r o v , A . V . K a r y a k l n , K. V. M a r u n o v a , ZILNeorg. Khim. , 10(1965) 986. 19. J. S p i t s y n , A. V i k t a , A . F . K u z i n , N . N . Z a m o s h n i k o v a , T . S . T o g i l , Dokl.
Akad. Nauk, SSSR, 144 (1962) 1066. 20. A.N. Z e l i n k m a n , L. D r e g a n , Zh. Neorg. Khim. , 12(1967) 261. 21. L.J. A n o k h i n a , N . A . A g r i n s k a y a , V . I . P e t r a s h e n , l~ss, LInorg. Chem. ,
15 (1970) 78. 22. A.A. Z a i t s e v , I . A . L e b e d e v , S . V . P i r o z h k o v , G . N . T a k o v l e v , Rms. J. Inotg.
Chem. , 8 (1963) 1260. 23. P,.N. M a s l o v a , V. V. E o m i n , ZIL Neorg. Khim. , 6(1961) 738. 24. V.V. F o m l n , R. N. M a s l o v a , Zh, Neorg. Khim. , 6(1961) 481. 25. A.K. B a b k o , B . I . N a b i v a n e t s , Zh. Neorg. Khim. , 2(1975) 2085. 26. E . N . G i l b e r t , V . A . P r o n i n , I. M. I v a n o v , S . N. I v a n o v a , A . A ,
V a s i l e v a , P. I. A r t y u k h i n , L. M. G i n d i n , Zh. Neorg. Khim. , 13(1968) 1055. 27. V. M. V d o v e n k o , A . A . L i p o v s k i i , M . G . K u z l n a , Radiokhimiya, 3(1961) 555.
02 J. Radloanal. Chem. 23 (1974)





























