Embed Size (px)
Citation preview

STATI INTERSESSUALI E GENITALI AMBIGUI
LUIGI TARANI Servizio di Genetica Clinica
Dipartimento di Pediatria e Neuropsichiatria Infantile“Sapienza” Università di Roma



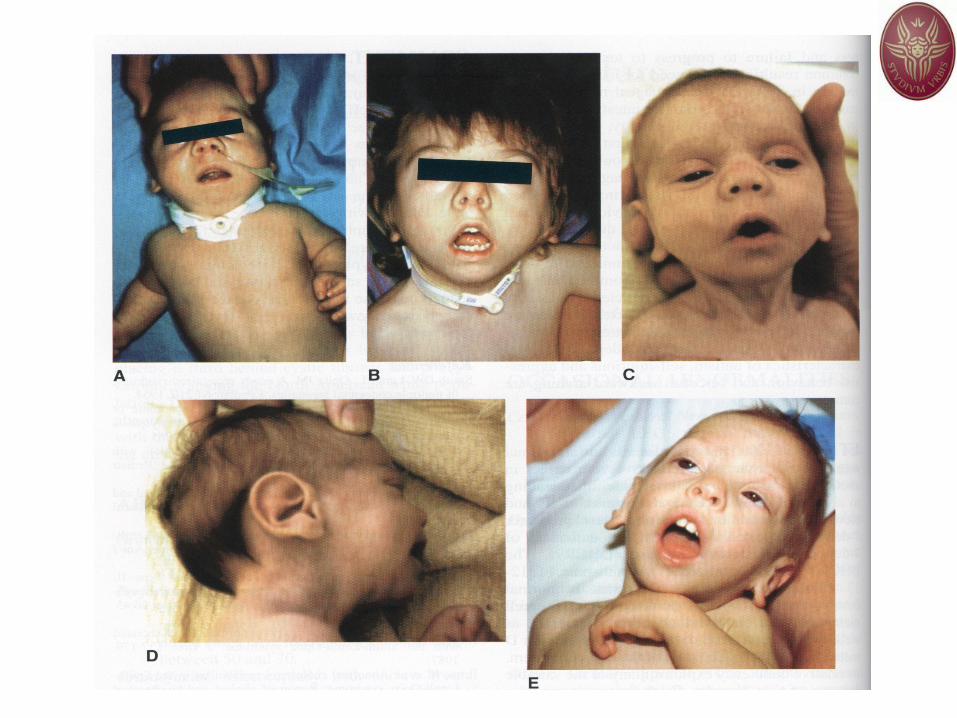
Neonata con SAGC


Genitali ambigui• 1) una disgenesia gonadica o• 2) un’inadeguata azione degli androgeni
determinano lo sviluppo:
O di un maschio ipo-virilizzatoO di una femmina virilizzata

Esame obbiettivo femmina• Il clitoride normale alla nascita ha un
diametro < 6 mm ed una lunghezza < 1 cm (se maggiore: ipertrofico o fallo)
• La fessura vulvare normale: > 5mm• Identificare l’orifizio uretrale• Le grandi labbra alla palpazione devono
essere vuote e non apparire fuse • Rapporto ano-genitale: AF/AC vn 0,37, se
> è segno di virilizzazione

Esame obbiettivo maschioMeato uretrale situato sulla punta del glande,
con cute prepuziale senza schisiIl pene del neonato a termine è lungo >2,5 cm
e largo 0.9-1.3 cm (righello rigido) micropeneSe il pene è ricurvo sospettare ipospadiaI testicoli devo essere palpabili (vol. 1-2 ml)
nello scroto (bifido?) o nel canale inguinale (criptorchidismo?) E.M.S. vn > 11
External Masculinization Score(Ahmed, BJU Intern. 2000)

Diagnosi di ambiguità nel neonato1) Nel neonato che appare maschio:
- micropene - ipospadia- criptorchidismo con scoto bifido
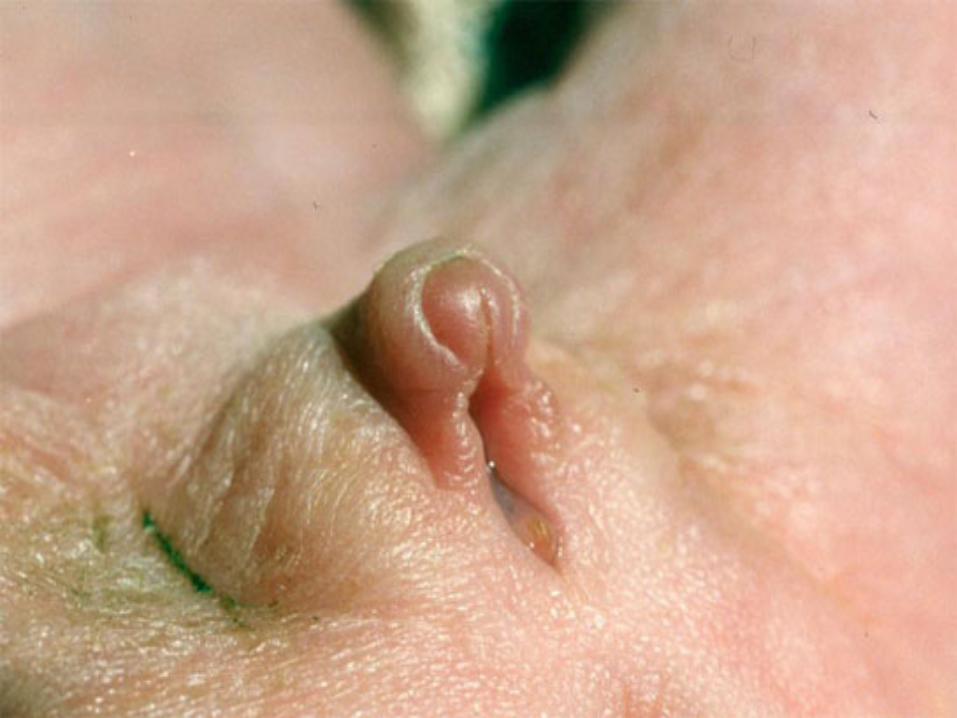
2) Nella neonata che appare femmina- ipertrofia del clitoride - fusione labiale posteriore- vulva corta con meato unico- ernia inguinale con gonade all’interno
3) Ambiguità genitale (Prader), estrofia vescicale 4) Discordanza cariotipo fetale-fenotipo neonato 5) Familiarità (SAGC, FT)

DIFFERENZIAZIONE SESSUALE
• 1) Determinazione del sessoazione dei geni
• 2) Differenziazione primaria:sviluppo delle gonadi
• 3) Differenziazione secondaria:sviluppo dei genitali esterni

Prima delle 8 settimane i.u. Il fenotipo genitale non è riconoscibile
StruttureNeutrali

Cellula GonadicaIndifferenziata(precursore)
Settimana di inizio delladifferenziazione durantelo sviluppo
Struttura Testicolareche si sviluppa
Cellule Germinative:(endoderma saccovitellino)
7 Spermatogonio
Cellule di Supporto:(ectoderma celomatico)
7 C. Sertoli: MIH
Cellule Interstiziali:(mesoderma crestaurogenitale)
8-10 C. Leydig: testosterone
Maschio: la gonade indifferenziata evolve in : Testicolo

DIFFERENZIAZIONE (I FASE) E CRESCITA DEL PENE (II FASE)
8° ------ 12° ----- 40°
Placenta
HCG
Testicolo
Testosterone
Differenziazione sessuale
Ipofisi fetale
LH/FSH
Testicolo
Testosterone
Crescita del pene

Mullerianregression
TIMETABLE OF THE EVENTS OF MALE SEX DIFFERENTIATION
6 8 10 12 14 3624
TestosteroneAMH AMH
Descent of testesTestisform. External genital diff.
Wolffian developmentGermcellmigr.
Trimester First Second Third
Week
AR

Cellula GonadicaIndifferenziata(precursore)
Settimana di inizio delladifferenziazione durantelo Sviluppo
Struttura Ovarica chesi sviluppa
Cellule Germinative:(Endodermiche)
11-12Oogonio – prima divisionemeiotica
Cellule di Supporto:(Ectodermiche)
8-19Cellule della Granulosa: estrogeni
Cellule Interstiziali:(Mesodermiche)
In fasi successive dellosviluppo
Cellule della Teca: androgeni e progesterone
Femmine: la gonade indifferenziata evolve in: ovaio





Determinazione SessualeEVENTO CHIAVE: attivazione genica nelle
cellule somatiche bi-potenti cellule di supporto (PSC pre-Sertoli cells e PGC pre-granulosa cells)
Maschile: UNICA via di attivazione SRY SOX9
Femminile: DUE vie di soppressione di SOX91) antitesticolare: fetale ontogenetica
WNT4 + RSPO1 stabilizza βcatenina inib. SOX9
2) pro-ovarica: fetalepostnataleWNT4 + RSPO1 differ./trofismo ovociti
+ FOXL2 differenziazione/trofismo follicoli(DEFAULT o Z-FACTOR THEORY?)

Modello delle reti di regolazione dei geni nella determinazione del sesso maschile

Modello delle reti di regolazione dei geni nella determinazione del sesso femminile

Geni dello sviluppo sessuale

DIFFERENZIAZIONE SESSUALE
• 1) Determinazione del sessoazione dei geni
• 2) Differenziazione primaria:sviluppo delle gonadi disgenesie
• 3) Differenziazione secondaria:sviluppo dei genitali esterni PEM/F

Relazione genotipo-fenotipo
1) DISGENESIA GONADICA
• Sesso genetico ≠ sesso gonadico= sesso genitale (Femmine XY- Maschi XX)
2) PSEUDO-ERMAFRODITISMO
• Sesso genetico = sesso gonadico ≠ sesso genitale ( FT-SAGC)

MacLaughlin, D. T. et al. N Engl J Med 2004;350:367-378
Functional Abnormalities of the Synthesis and Action of Hormones

Ermafrodito classico(Galleria Borghese. Roma)

Nuova nomenclatura a confronto con la vecchia terminologia (Arch.Dis. Child, 2006; BPRCEM 2010)
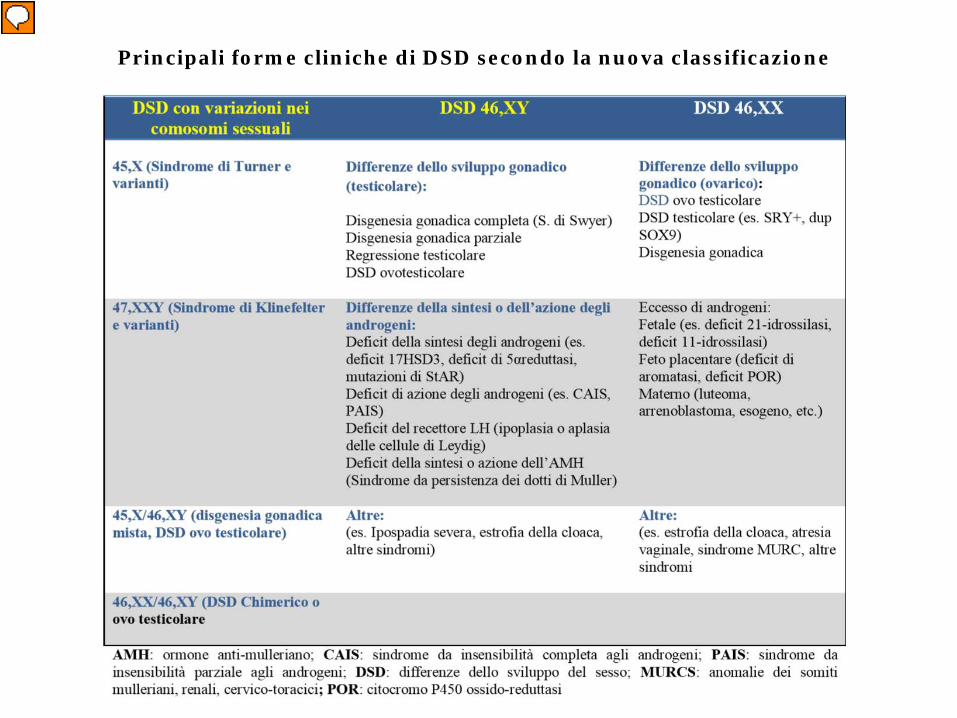
Principali forme cliniche di DSD secondo la nuova classificazione

DSD Cromosomici
1) S. di Turner (45,X) (non ambiguità)2) S di Klinefelter (47,XXY)(non ambiguità)3) Disgenesia Gonadica Mista (45,X/46,XY)4) DSS ovotesticolare (45,X/46,XY o
46,XX/46,XY) o ermafroditismo vero (EV)5) Chimerismo 46,XX/46,XY

DSS 46,XY
A) DISTURBI SVILUPPO TESTICOLARE(Disgenesie gonadiche (DG) e sex reversal)
1 D G completa (femmine XY o S. di Swyer) 2. D G parziale3. Regressione testicolare4. DSS ovotesticolare (EV)

DSD 46,XY
B) DISTURBI ORMONALI (P.E.M.)
1. Difetti della sintesi degli androgeni (T-DHT)2. Difetti dell’azione degli androgeni (FT) 3. Difetto del recettore dell’LH4. Difetti di sintesi ed azione di AMH5. S. di Smith-Lemli-Opitz

MECCANISMO D’AZIONEDEGLI ANDROGENI
TT
T T
DHT
Regolazione gonadotropine
Spermatogenesi
Dotti diWolff
Genitali esterni
Maturazionepuberale
Testicolo
DHT
LH
5αred
CELLULA BERSAGLIO

DIFETTI SINTESI ANDROGENIA) BIOSINTESI TESTOSTERONE- deficit 20-22 desmolasi (+iposurrenalismo)- deficit 3-β-HSD (+iposurrenalismo)- deficit 17-α-idrossilasi (+ipertensione)- deficit 17-20 desmolasi- deficit 17-β-HSD
B) BIOSINTESI DHT- deficit 5-α-reduttasi (test HCG)

RESISTENZA PERIFERICA AGLI ANDROGENI
1) Insensibilità Completa (S. di Morris, femminilizzazione testicolare o FT, CAIS)
2) Insensibilità Parziale (FT parziale, s. di Reifenstein, PAIS)
3) Infertilità maschile (ginecomastia+azoo)4) nei maschi fertili (ginecomastia, ipotrichia)

DSD 46,XX
A) DISTURBI SVILUPPO OVARICO(disgenesie gonadiche e sex reversal)
1. DSS ovotesticolare (EV)2. DSS testicolare (maschi XX)3. Disgenesia gonadica XX

DSD 46,XX
B) DISTURBI ORMONALI (P.E.F.)
1. Eccesso androgeni fetali (SAGC)2. Eccesso androgeni placentari (ARO)3. Eccesso androgeni materni (luteoma)


IPERPLASIA SURRENALICA CONGENITA
• S. Adreno-Genitale Congenita (SAGC, CAH)• 70% delle ambiguità genitali• 21-idrossilasi (1:5000/15.000, 95% SAGC)• - classica (2/3 ipoNa; prevenzione <8set/screen)• - tardiva (st 1:1000; non deficit cortisolo ma
iperandrogenismo; eterozigote composto); • - criptica (+ test ACTH in studi familiari)• 11-idrossilasi (+ipertensione)• 17αidrossilasi, 3βHSD, 20-22desmolasi

SINDROMI CON DSDXY
S. Denis-Drash Assoc. WAGR S. Frasier Displasia camptomelica S. ATR-X S. Smith-Lemli-Opitz
XXAssoc. MURCSMayer-Rokitansky-Kunsters. mani-piedi-genitales. Mc Kusick-Kaufman
S. feto-alcolicaS. FrynsS. Pallister HallSequenza di PotterSequenza
sirenomelicaS. Klippel-TrenaunayS. Opitz (GBBB)S. Proteus

SINDROMI 025.jpg

Sindrome di Pallister Halls. ano-cerebro-digitale
Amartoblastoma ipotalamico (CPHD)Polidattilia postassiale-sindattilia (acro-
meso)Ano imperforato (+ micropene)Cardiopatia, displasia polmonare o renaleMorte precoceGene GLI 3 in 7p13 Autosomica Dominante

S. di Smith-Lemli-OpitzForma neonatale letale e f. lieve (1:30.000)Microcefalia, tempie strette, ptosi
palpebrale, narici antiverse, micrognatia, palatoschisi
Sindattilia 2°-3° dito dei piedi, oligo-polidatt.Anomalie cerebrali RMGenitali ambigui (PEM)Deficit 7-deidrocolesterolo reduttasi (11q13)
con ipocolesterolemia neonatale

DIAGNOSI DSD: Anamnesi
• FAMILIARE: • Insensibilità agli androgeni (FT): X-linked• Difetti Sintesi T e cortisolo (SAGC): AR• Deficit 5-α-reduttasi e Recettore LH: AR
• FARMACOLOGICA (androgeni, alcool)• ORMONALE (infertilità, tumori virilizzanti)

DIAGNOSI DSD: clinica
1) Definire lo stadio di Prader2) Cercare massa palpabile inguino-scotale3) Descrivere il fenotipo generale: idratazione
- IUGR, pigmentazione cutanea, - malformazioni maggiori e dismorfismi
4) BASIC: Bonding, Adrenal, Sex assignment,Imaging, Cytogenetics


Neonato DSD: protocollo SIN1°- 2° giorno: cariotipo, Na-K, glicemia, 17OH-P,
eco addome-pelvi-inguini, PA, Kg3°- 4°g: SRY (qfPCR), T/DHT, LH, FSH, AMH5°- 6°g: genitografia (seno uro-genitale)7°- 8°g: CGH, analisi mutazioni geniche (25%) 9°-10°g : ripetere ormoni, Na, K, Cl Se necessario, successivamente, test HCG o
ACTH, biopsia cute genitale e laparoscopia/tomia per biopsia/rimozione gonadica


La precisione diagnostica favorisce:
• 1) assegnazione del sesso adeguata• 2) indicazione al trattamento
ormonale sostitutivo• 3) terapia dei difetti associati
(surrenali ipo-Na)• 4) prevenzione dei tumori• 5) opzioni sulla fertilità• 6) consulenza valida per tutta la vita

Diagnosi DSD alle varie etàPRENATALE: discordanza cariotipo-fenotipoNEONATALE: genitali ambigui
shock da perdita salinaINFANTILE: ernia inguinale (FT)
androgenizzazione (SAGC incompleta)Tumore di Wilms (WAGR-Denis Drash)
PUBERALE: androgenizzazione (5αRed/17β−HSD, FT parziale, SAGC incompleta)
amenorrea primaria (Swyer/SAGC 17α)telarca o ematuria ciclica maschile (DG/farmaci)
ADULTA: amenorrea, sterilità, tumori (FT)

SCELTA DEL SESSO (1)
• In base a:1) diagnosi2) aspetto e funzionalità dei genitali esterni3) opzioni chirurgiche4) terapia ormonale sostitutiva definitiva5) potenziale fertilità (ovotestis)6) opinione e cultura dei genitori 7) Ri-assegnazione

SCELTA DEL SESSO (2)SVILUPPO PSICO-SESSUALE
• 1) identità sessuale: consapevolezza (3a) in accordo con sesso assegnato (società)
• 2) ruolo sessuale: comportamento (giochi)• 3) orientamento sessuale: attrazione
erotica (etero-, omo-, bi-sessuale)• Adulto DSD: disforia di genere (non
prevedibile) coinvolgerlo nella scelta?

SCELTA DEL SESSO (3)
A) Sicuramente femmina se:• SAGC, FT completa, DG Pura XY e XX,• Difetti sintesi T, resistenza LH
B) Possibilmente maschio se:• FT parziale, DG parziale XY, DG Mista,
C) Sicuramente maschio se: deficit 5-α-reduttasi e 17-β-HSD

Aspetti Medico-legali• Certificato medico all’Ufficiale di Stato
Civile per ottenere la proroga rispetto ai 10 giorni previsti per la Dichiarazione di nascita. Aspettare a dare un nome al neonato (Elia, Andrea…)
• Genitori discordanti su assegnazione giudice tutelare
• Legge 164/82: la riassegnazione deve seguire ad un intervento chirurgico
• 2013 Germania riconosce il “3° sesso”

TERAPIAA) Medica: sostitutiva glico/mineralo-attivi o
sessualiB) Chirurgica: sex, minzione, aspetto adeguati
Separazione uro-genitale (1-2 aa) • Gonadectomia (precoce?)• Vaginoplastica (post-puberale)• Falloplastica (adulto)Precoce: rischio di riassegnazione del sesso Tardiva: rischio di problemi psicologici di identità

Differentiation of internal structures
AndrogensMullerian Inhibiting Substance

DISGENESIE GONADICHE
A) DIST. DETERMINAZIONE SESSUALEReversioni sessuali: Maschi XX
Femmine XY
B) DIST. DIFFERENZIAZIONE GONADE1) Disgenesia gonadica mista2) Ermafroditismo vero

PSEUDOERMAFRODITISMO FEMMINILE
DISTURBI DIFFERENZIAZIONE GENITALE
Iperplasia surrenalica congenita
Deficit aromatasi placentareAssunzione materna di androgeni o
progestinici
Produzione materna di ormoni virilizzanti

PSEUDOERMAFRODITISMO MASCHILE
DISTURBI DIFFERENZIAZIONE GENITALE Resistenza testicolare alle gonadotropine Difetti della biosintesi del testosterone Difetto trasformazione del T in DHT Resistenza periferica agli androgeni Difetti della sintesi, produzione, risposta al
MIF/AMH Assunzione materna di progestinici o
estrogeni

Development of external genitalia: from a common (neutral) structure
Female Male


GENI DELLO SVILUPPO SESSUALE
WT1 11p13SF1 9q33DAX1 Xp21.3SRY Yp11.2SOX9 17q21 DMRT1 9p24.3-25.1 ? 10q26-qter WNT4 1p35
MIF 19p13recMIF 12q13AroP450 15q21.1AZF Yq11.23AR Xq11-125αred1 5p15 5αred2 2p23 CYP21B 6p21-23CYP11B1 8p22


MacLaughlin, D. T. et al. N Engl J Med 2004;350:367-378
Syndromes of Dysgenesis during the Development of the Urogenital Ridge

SF1
WT1
WT1
WT1SRY
SOX9
AMH
SF1
SF1Gonade
indifferenziata
testicolo
ovaio
Cellule dellagranulosa
Cellule della teca
Celluledi Leydig
Cellule del
Sertoli
Regressionedei dottidi Muller
testosterone
SF1
Estrogeni e progesterone
SF1
DAX1
Mesodermaintermedio
?
Lim1 Lhx9Emx2
M33
Wnt4Rspo1
Fgf9
DMRT1,2
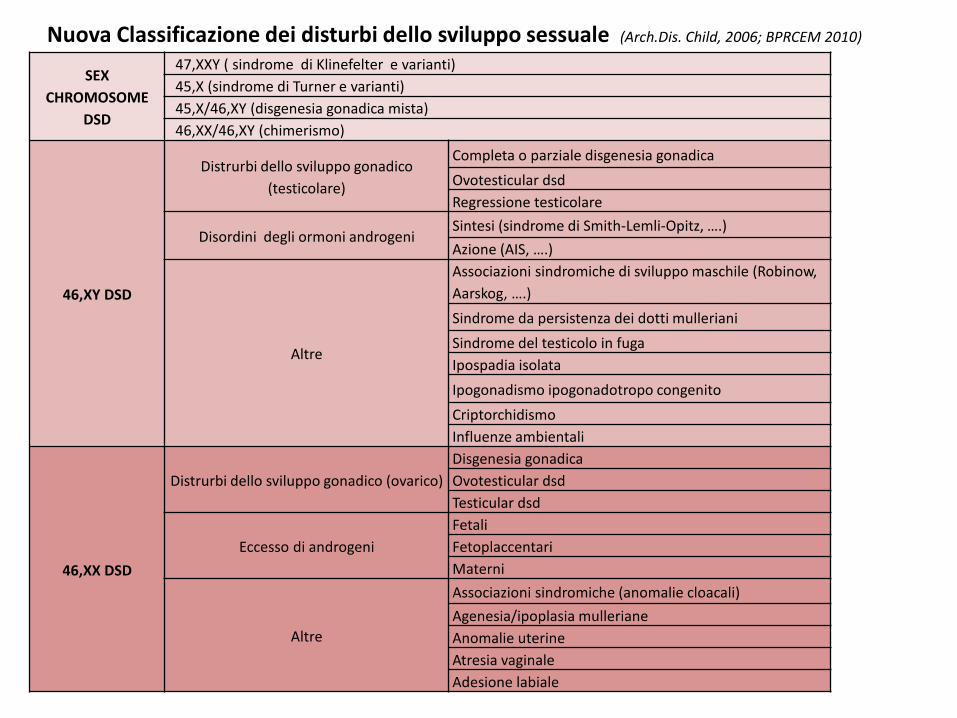
SEX CHROMOSOME
DSD
47,XXY ( sindrome di Klinefelter e varianti)45,X (sindrome di Turner e varianti)45,X/46,XY (disgenesia gonadica mista)46,XX/46,XY (chimerismo)
46,XY DSD
Distrurbi dello sviluppo gonadico (testicolare)
Completa o parziale disgenesia gonadicaOvotesticular dsdRegressione testicolare
Disordini degli ormoni androgeniSintesi (sindrome di Smith-Lemli-Opitz, ….)Azione (AIS, ….)
Altre
Associazioni sindromiche di sviluppo maschile (Robinow, Aarskog, ….)Sindrome da persistenza dei dotti mullerianiSindrome del testicolo in fugaIpospadia isolataIpogonadismo ipogonadotropo congenitoCriptorchidismoInfluenze ambientali
46,XX DSD
Distrurbi dello sviluppo gonadico (ovarico)Disgenesia gonadicaOvotesticular dsdTesticular dsd
Eccesso di androgeniFetaliFetoplaccentariMaterni
Altre
Associazioni sindromiche (anomalie cloacali)Agenesia/ipoplasia mullerianeAnomalie uterineAtresia vaginaleAdesione labiale
Nuova Classificazione dei disturbi dello sviluppo sessuale (Arch.Dis. Child, 2006; BPRCEM 2010)

Diagnosi DSD età successive
• 1) ambiguità non riconosciuta alla nascita• 2) ernia inguinale nella femmina• 3) pubertà ritardata o incompleta• 4) virilizzazione in una femmina• 5) amenorrea primaria• 6) telarca evolutivo in un maschio• 7) ematuria macroscopica ciclica in un
maschio

Diagnosi di ambiguità nel neonato
1) Nel neonato che appare maschio:- micropene e testicoli non palpabili - ipospadia con scoto bifido- ipospadia e criptorchidismo
2) Nella neonata che appare femmina- ipertrofia del clitoride, fusione labiale post.- vulva corta con meato unico- ernia inguinale con gonade all’interno
3) Ambiguità genitale (Prader), estrofia vescicale 4) Discordanza cariotipo fetale-fenotipo neonato 5) Familiarità (SAGC, FT)

Modern Continuum Gender Model
- many configurations are possible - Biological Sex: Male Intersexed Female
Hormones, genitaliasecondary sex characteristics
Gender Expression Masculine Androgynous FeminineDress, posture, roles, identity
Sexual Orientation women mostly both mostly menwomen menAttracted to:
Adapted from Samuel Lurie Asilomar Conference, Oct. 2004; see www.tgtrain.org