Embed Size (px)
Citation preview

U. PIETSCH: Static and Dynamical Valence Charge and Bond Charge Properties 439
phys. stat. sol. (b) 128, 439 (1985)
Subject classification: 4; 13; 22.2
Bektion Physik der Karl-Marx- Universitat Leipzig'), Arbeitsgemeinschaft AIIIBV-Halbleiter
Static and Dynamical Valence Charge and Bond Charge Properties of Zincblende Structure Compounds
BY U. PIETSCH
By means of a simple quantummechanical model of the linear superposition of "two-electron- molecules" both, the valence charge density and the bond charge density of zinc blende structure compounds are constructed. This scheme makes use of the results calculated for elemental semi- conductors. Further it is extended by the self-consistent determination of the effective charge and by an approximation describing the dehybridization of valence orbitals for increasing ionicity. The estimated valence charge densities are in rather good agreement with those published till now from band structure calculations. The model permits us to explain some known empirical properties concerning the ionicity dependence of the bond charge amount, the effective charge, the value 2 e / ~ , defined by the dielectric theory, and the charge dependence of ionic radii. Due to the variation in both, the next neighbow distance and the co-ordination angles the bond charge amount has to vary for a disturbed lattice. The bond charge transfer constants are calculated for thermal displacement and for hydrostatic pressure.
Mit Hilfe eines einfachen quantenmechanischen Modells der linearen Superposition von ,,Zwei- elektronenmolekiilen" werden sowohl die Valenz- (VED) als auch die Bindungsladungsdichte fur Verbindungen mit Zinkblendestruktur simuliert. Das Schema macht von den Ergebnissen Ge- brauch, die bereits fur Elementhalbleiter berechnet wurden. Es wird in der Hinsicht weiter- entwickelt, daR die effektive Ladung der Verbindung selbstkonsistent bestimmt und eine Nahe- rung fur die Dehybridisierung der Valenzorbitale bei zunehmender Ionizitat angegeben wird. Die berechneten VED sind in guter Ubereinstimmung mit bisher bekannten, die im Verlaufe von Bandstrukturberechnungen erhalten wurden. Das Model1 erlaubt eine Erklarung einiger empirisch bekannter Eigenschaften, dazu gehoren die Ionizitatsabhangigkeit des Bindungsladungsbetrages, der effektiven Ladung und der GroRe 2e/e,,, die durch die dielektrische Theorie definiert ist, und die Ladungsabhangigkeit der ionischen Radien. Die Veranderung der nachsten Nachbarabstande und der Bindungswinkel muB zu einer Veranderung des Bindungsladungsbetrages fuhren. Die Bin- dungsladungstransferkonstanten werden fur thermisch induzierte Atomauslenkungen und fur den Ball des hydrostatischen Druckes berechnet.
1. Introduction Many physical properties of the solid state may be explained by the valence electron density (VED) of the crystal. For covalent bonded material its maximum is essentially localized near the middle of the next neighbour distance. Because of the quantum- mechanical overlap of valence orbitals that charge density accumulation is higher than the simple superposition of spherical atomic densities. The concrete VEI) of semiconductors turns out to be the result of the nonlinear screening of ionic potentials by the valence electrons.
Therefore, the evaluation of the exact VED requires self-consistent calculation methods. Their extension led to the use of empirical and semiempirical models for the interpretation of special properties of semiconductors. Today most structural charac-
1) LinnBstr. 5, DDR-7010 Leipzig, GDR.

440 U. PIETSCH
teristics of elemental semiconductors have been found by some authors starting from first principles [l], that means the static VED as well as the polarization of the valence charge caused by external perturbations [2, 31. For zincblende structure compounds (ZBSC) the situation is more difficult because of the consideration of an ionic contribu- tion in addition to the covalent bond. About ten years ago many of their structural properties were scaled in terms of the Phillips ionicity parameter. The ionicity has been introduced by the dielectric theory as a possibility to describe the semicovalent bond in terms of covalent and ionic energy contributions [4]. It profits from the empir- ical fact that the energy properties of ZBSC can be simply scaled by accepting the parameters of elemental semiconductors and using ionic shares. This sophistication was employed first by Cohen and Bergstresser [5] for the reconstruction of the energy band structure of ZBSC by means of the empirical pseudopotential method. Further- more Walter and Cohen [S] succeeded in the calculation of the VED of some ZBSC choosing the same concept. In the following years better VED plots were published benefiting from better pseudopotentials [8, 101 and extended screening concepts
Self-consistently calculated structure properties have been published for three years. Especially Kunc and Martin [12] have been engaged in the computation of the effective charge and the phonon properties of GaAs. Baur et al. [13] provided a better knowledge of the VED of ZBSC. As shown the ionic charge and the bond charge amount vary nonlinearly with increasing difference in the nuclear charges between the sub- lattices in the row Si-Alp-MgS. The bond charge (BC) amount was found to decrease with increasing ionicity. The consideration of the BC and the effective charge are most essential for the interpretation of structural properties. This was done by Rustagi and Weber [14] with regard to the BC-BC and BC-ion interaction for the calculation of the phonon dispersion. The phase transition due to hydrostatic pressure has been investigated by Froyen and Cohen [15] using the local density approximation. Com- monly the VED may be determined by X-ray diffraction methods. A t first the VED of some ZBSC have been published by Sirota et al. [16] using powder diffraction tech- niques. Eecently Gomm [17] has been published VED plots of some AI1’BV semicon- ductors. But this results are not constructive because of the wrong extinction correc- tion. In general the evaluation of the right VED is most complicated with methods of total Fourier transformation of all measurable X-ray structure amplitudes of a crystal. Many of them strongly depend on the core scattering strength and are rather influenc- ed by various experimental corrections than by the distribution of valence electrons. Therefore some special structure amplitudes have been selected being most sensitive to the covalent bond. These reflections are named “weak” (200,222, ...) [18, 191. Bilderback and Colella [20] employed these procedures with success for GaAs and InSb choosing the model of deformable valence orbitals and Pietsch 1211 by help of a BC model. But they are only applicable to compounds having nearly the same or- dinary number of constituents.
Whereas the usual BC models as well as those of deforinable valence orbitals seem to be sufficient for the description of the static VED the interpretation of dynamical VED properties requires rather quantum-mechanical approaches containing the quite close connection between the spherical atomic VED and the overlap charge. In a recent work the author proposed a simple quantum-mechanical model of the VED [22, 231. The total VED and the BC have been constructed by linear superposition of “two- electron molecules”. Each molecule involves an individual overlap charge. The semi- conductor is described by next-neighbour interaction and additional angle-dependent forces. The parameters of the Gaussian-like model wave functions and the metallic contributions are fitted to the VED properties known of the solid state. However,
[7,9,111.

Static and Dynamical Valence Charge and Bond Charge Properties 441
with our model we cannot expect quite exact numerical results. More self-consistent cbalculations are needed. But we are able to predict tendencies of some properties of the ZBSC family. We offer a model for the creation and interpretation of experiments measuring the VED variation caused by different external perturbations.
This paper deals with the following aspects: Firstly the basic assumptions of our model are presented. Secondly an attempt is made to exhibit some properties of the static VED. Thirdly the VED variation is studied caused by different external per- t iirbations in terms of the BC transfer constant)s [21]. Finally a critical discussion of our results is given.
2. Molecule-Theoretic Model of the VED 2.2 Basic assumptions
The VED of ZRSC should be constructed by linear superposition of “two-electron molecules”. Each molecule is supposed to contain all molecule-theoretic sophistica- tion. I n order to build up the tetrahedral co-ordination of a crystal having eight elec- trons in the primitive lattice cell each “molecule” must possess two electrons and two effective core charges. We choose an ansatz function of the molecule density,
1 eJ1’) = ~ {A IW.I(Y)l2 + 4 IYdR - r)IZ +
1 +st + 2 IIA,A,S.I,Y.lr) WAR - r ) } 9 (1)
containing two centres i and j . R means the next neighbour distance. The total VED of crystal is achieved by
(2) @crystadY) = c @?AT) w . 9 Y‘) . 1,3
The operator D(Y, r ’ ) insists on the tetrahedral co-ordination of “molecules” in the ele- mentary cell with respect to any expected bond. According to the V.B. approximation our density approach gives rise to a weighted resonance structure among the neutral AoBo and the ionic structures A+B- and A-B+. The parameters A2,? allow us to pull out the effective charge of participants as well as the ionicity of the compound. This approach relates to the view accepted till now for the covalent-ionic bond in ZBSC [24]. Our VED of the crystal is characterized by the spherical valence density around the atoms
elon,,J(r) = 4A2,, Iw2 3(r)12 (3) and by the BC. The BC localized between the atoms i and j is determined solely by an overlap charge of a single “molecule”,
involving the overlap integral between next neighbour orbitals, Sij = J vi(r) Y ~ ( R - Y) d3r .
The chosen model valence orbitals for a “molecule”
Yi , j ( r ) z= ~ z ~ j l ” { p IS> + 1 8 ~ 2 ) } ; 2 = 1 2 9
29 physiea (b) l2SjZ

44 2 U. PIETSCH
consider the tetrahedral co-ordination of the crystal as well as the covalent and metal- lic bond contributions. The Nf,j are normalization constants. In order to account for the exact hybridization state among the four hybrid orbitals being localized at the same atom the ( p ) portions of a hybrid orbital k or I ,
lsPz)li,l = Nk::/2{Is) + 1 113) } 9
are fixed by the orthogonalization condition
1 + j l k ~ l cos ekl = o , (9) which depends on the co-ordination angle 8 k l between the orbitals. The consideration of (9) permits to improve our next neighbour model by angle-dependent forces being essential in covalently bonded materials.
The additional spherical part in ( 7 ) deals with the metallic contribution of the chem- ical bond. It has been found to be necessary for elemental semiconductors to describe the diminished BC amount from diamond to u-tin.
The screening constants of the preferred Gaussians,
(10) Is) = N t l / z r z exp (--0172) ,
and the parameter pi have been adapted to elemental semiconductors fulfilling the following conditions :
(i) The VED should be characterized by a single maximum in the middle of the next neighbour distance and
(ii) the measurable amount of the BC-dependent X-ray scattering factors (“for- bidden” reflections) should be reconstructed. Despite of the discussion on the VED behaviour of diamond the first condition is justified by experience. Considering the slope of the model orbitals the second condition allows to provide the BC amount being the BC scattering factor,
a t Q = 0. Because of the numerical simplification at and Zi are no longer distinguished.
2.2 Self-consistent determination of the screening constants
The screening constants have to be determined self-consistently with respect to the effective charge of ions. Slater’s rule provides them for neutral atom [32]. It has to be modified using Gaussians and ought to start from those of elemental semiconductors EESC. For Is) and 113) like orbitals they turn out to be
2, treats the Slater screening parameter with respect to 2 - 1 electrons. 2 means the effective electron number. Using (12) the VED is calculated. The normalization con- stants Ai,j are fixed to be a quarter of the valence electron number of free atoms. The next step is the determination of the position r = R,,, where both wave functions

Static and Dynamical Valence Charge and Bond Charge Properties 443
have the same value. The cross-over point R, is assumed to be the border line between the ions. Therefore, the ionic charge of a constituent is obtained after integration of the charge density inside an "ion",
(14) caused by the different penetration amount of both orbitals. Consequently the screen- ing constants of ions have to be transformed taking into account the actual effective charge. That is attended by a change of the Slater atomic screening parameter. At the n-th step it must be varied with respect to the first adaption
+Zi + A 2 = 4n J r 2 e y E D ( r ) dr ,
Zp) = ZLo) - (0.35(2, + 4AZ("--l))) ,
$zi + Az(fl) = A ( n - 1 ) a'
e* = -4AZ("). (17)
(15) considering tetrahedral co-ordination per atom. By use of (15) the screening constants of ions and the VED must be recalculated until the condition
(16) is fulfilled. The self-consistent calculation of the charge transfer between the ions leads to the effective charge of the anion
2.3 Dehgbridization of valence orbitals
In general the determination of the hybridization state of a semiionic bonded atom is an open question. Only for diamond a full /sp3) hybridization seems to appear. We have found an enhanced dehybridization of valence orbitals for elemental semiconductors with increasing ordinary number. This finding correlates with the increased metallic share of the chemical bond in the same direction confirmed by Mossbauer experi- ments [23,25].
On the other hand, the VED of highly ionic compounds is characterized by close spherical shells 11131. Especially the A1BVI1 compounds confirm the tendency of build- ing closed octet shells which is a problem of the overscreening of the ionic potentials by the redistribution of valence electrons.
Therefore, we propose to handle the spherical part of the bond by two different approaches. First the coefficient p should hold constant up to the zincblende to rock- salt phase transition point. The amount of p is assumed to be the same as thab of the corresponding elemental semiconductor.
As a second approximation we decide for a linearly diminished hybridization state of valence orbitals for increased difference between the ordinary number of constitu- ents AZ,,. The actual pci is calculated by
For a rigid lattice A2 remains constant for all tetrahedrally bonded compounds. The coefficient a is different for each row of Mendeleev's table (pESc - from elemental semi- conductor).
2.4 Bond charge transfer constants
According to our model conditions the overlap integral depends on both the variation in the next neighbour distance ARB and the change in the co-ordination angle AOeC. In contrast to elemental semiconductors the movement of a single cation towards the rigid lattice gives rise to a different BC transfer in comparison with a displacement of 29*
444 u. PIETSCR
an anion. This behaviour has to be considered by the BC expansion
BC = BC, + y l k AR, + y e k ARE + 8 1 k AOlk + 8ek AOE f ... = BC, + y l g AR, + ARE . (19)
The difference between the transfer constants YEk, Blk for a thermal cation (k = A) and an anion (k = B) displacement, respectively, depends on the effective charge and the difference in the spherical parts of the valence orbitals of both constituents. The used variation of the metallic portion under pressure corresponds to that given in [21].
3. Results
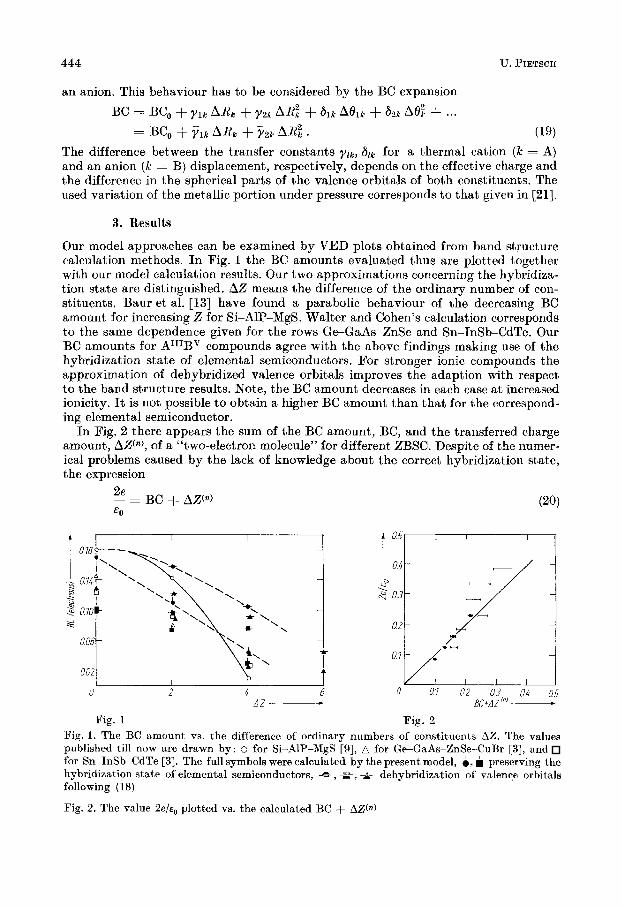
Our model approaches can be examined by VED plots obtained from band structure calculation methods. In Pig. 1 the BC amounts evaluated thug are plotted together with our model calculation results. Our two approximations concerning the hybridiza- tion state are distinguished. AZ means the difference of the ordinary number of con- stituents. Baur et al. [13] have found a parabolic behaviour of the decreasing BC amount for increasing Z for Si-Alp-MgS. Walter and Cohen's calculation corresponds to the same dependence given for the rows Ge-GaAs-ZnSe and Sn-InSb-CdTe. Our BC amounts for AIIIBV compounds agree with the above findings making use of the hybridization state of elemental semiconductors. For stronger ionic compounds the approximation of dehybridized valence orbitals improves the adaption with respect to the band structure results. Note, the BC amount decreases in each case at increased ionicity. It is not possible to obtain a higher BC amount than that for the correspond- ing elemental semiconductor.
In Fig. 2 there appears the sum of the BC amount, BC, and the transferred charge amount, A W ) , of a "two-electron molecule" for different ZBSC. Despite of the numer- ical problems caused by the lack of knowledge about the correct hybridization state, the expression
t I I I I
T t
T I I I
U 2 4 6 42-
Fig. 1 Fig. 2 Fig. 1. The BC amount vs. the difference of ordinary numbers of constituents AZ. The values published till now are drawn by: o for Si-Alp-MgS [g], A for Ge-GaAs-ZnSe-CuBr [3], and for Sn-InSb-CdTe [3]. The full symbols were calculated by the present model, 0 . preserving the hybridization state of elemental semiconductors, =-, *, A- dehybridization of valence orbitals following (1 8)
Fig. 2. The value 2 e / ~ , plotted vs. the calculated BC + AZW)

Static and Dynamical Valence Charge and Bond Charge Properties 445
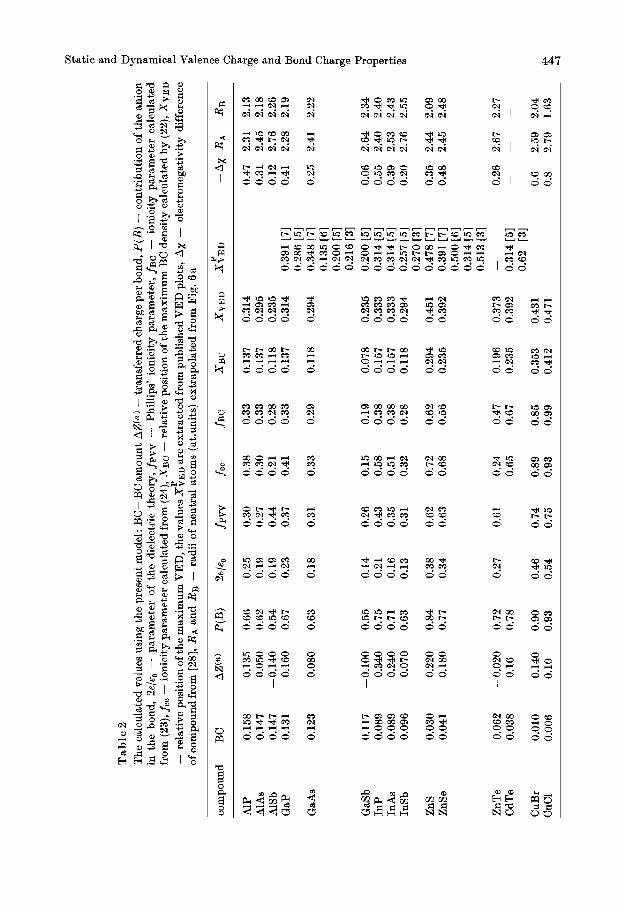
Table 1 The list of the initial parameters of the presented model, at, /&-screening constants, pt metallic portion
compound ‘YA a B PA BB PA PB
AlP AlAs AlSb GaP GaAs GaSb InP InAs InSb ZnS ZnSe ZnTe CdTe CuBr CUCl
0.421 0.396 0.339 0.385 0.368 0.333 0.310 0.294 0.267 0.306 0.299 0.263 0.216 0.214 0.220
0.362 0.340 0.291 0.363 0.347 0.324 0.286 0.272 0.246 0.288 0.282 0.248 0.200 0.212 0.208
0.662 0.547 0.436 0.650 0.540 0.429 0.580 0.501 0.400 0.821 0.633 0.503 0.468 0.773 0.062
0.569 1.8 1.8 0.517 1.8 2.9 0.913 1.8 4.5 0.559 2.9 1.8 0.509 2.9 2.9 0.396 2.9 4.5 0.499 4.5 1.8 0.473 4.5 2.9 0.369 4.5 4.5 0.706 6.2 4.4 0.597 6.2 6.3 0.465 6.2 8.5 0.432 8.5 8.5 0.730 20.0 20.0 0.913 20.0 15.0
seems to be valid up to the phase transition point (E,, means the static dielectric con- stant of the compound).
Table 1 exhibits some material parameters. For ArIrBV compounds the hybridiza- tion state of the corresponding elemental semiconductors has been used. For the other compounds (18) has been employed. The share of the anion in the BC, PBc(B) corre- sponds to the empirical expression given by Hubner [24],
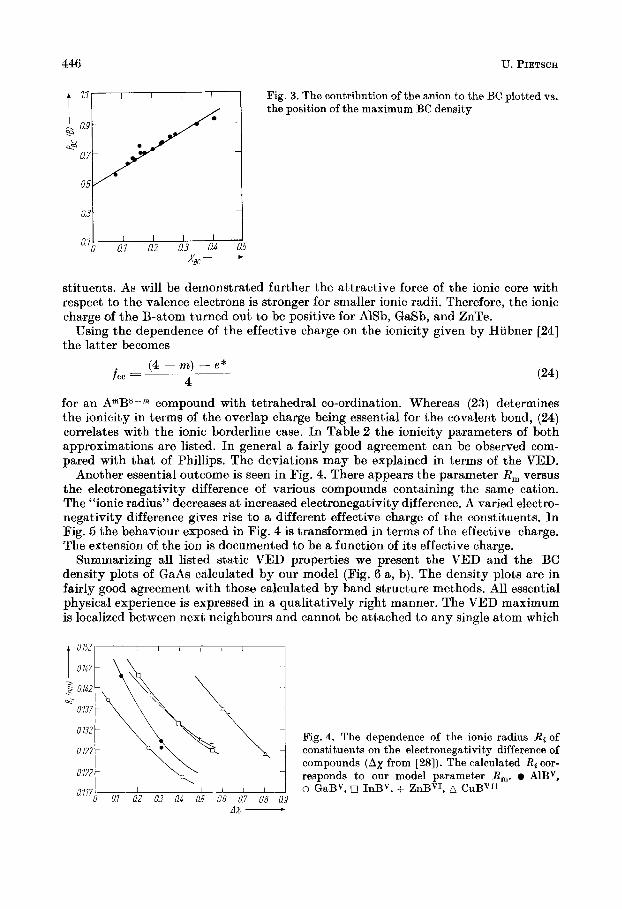
Depending on the Phillips ionicity parameter f p V V the anion contributes more elec- trons to the bond than the cation. Because of the validity of (21) we should test the possibility to define the ionicity by an electron density parameter directly. Therefore, we scale the position of the BC density maximum Rgix measured from the A-atom,
It determines the shift of the BC maximum out of the centre of the next neighbour distance R. In the same manner the relative shift of the VED maximum, XVED, has been obtained applying RVmFD. I n Fig. 3 the share of the anion in the BC is drawn ver- sus XBc. A linear slope is visible. Using (21) and (22) the ionicity parameter may be connected with the parameter XBC by
fBC = AXB‘! . (23) The proportionality coefficient has been fitted to be A = 2.40 f 0.10. Our model pro- vides a fairly linear dependence of Phillips’ ionicity on XVED also. A list of these par- ameters taken from VED plots published till now cannot confirm our experience (Table 2). Therefore, it is better to define the ionicity from XBc.
Another way to determine the ionicity consists in the application of the effective charge given by (17). I ts amount depends on the ratio of the “ionic” radii of the con-
446 U. PIETSCH
U 1.52 I 0132
0127-
0122
Fig. 3. The contribution of the anion to the BC plotted the position of the maximum BC density
I l l I l 1 I
- ~ :.:::\\\: - Fig. constituents 4. The dependence on the electronegativity of the ionic radius difference Rg of of compounds (Ax from [as]). The calculated Ri cor- responds to our model parameter R,. 0 AlBv, o GaBV, 0 InBV, + ZnBVI, A CuBVI1
-
vs.
i
stituents. As will be demonstrated further the attractive force of the ionic core with respect to the valence electrons is stronger for smaller ionic radii. Therefore, the ionic charge of the B-atom turned out to be positive for AlSb, GaSb, and ZnTe.
Using the dependence of the effective charge on the ionicity given by Hubner [24] the latter becomes
(4 - m) - e* 4
f - ec -
for an AmB8--m compound with tetrahedral co-ordination. Whereas (23) determines the ionicity in terms of the overlap charge being essential for the covalent bond, (24) correlates with the ionic borderline case. In Table 2 the ionicity parameters of both approximations are listed. In general a fairly good agreement can be observed com- pared with that of Phillips. The deviations may be explained in terms of the VED.
Another essential outcome is seen in Fig. 4. There appears the parameter R, versus the electronegativity difference of various compounds containing the same cation. The "ionic radius)) decreases a t increased electronegativity difference. A varied electro- negativity difference gives rise to a different effective charge of the constituents. In Fig. 5 the behaviour exposed in Fig. 4 is transformed in terms of the effective charge. The extension of the ion is documented to be a function of its effective charge.
Summarizing all listed static VED properties we present the VED and the BC density plots of GaAs calculated by our model (Fig. 6 a, b). The density plots are in fairly good agreement with those calculated by band structure methods. All essential physical experience is expressed in a qualitatively right manner. The VED maximum is localized between next neighbours and cannot be attached to any single atom which
5 a T
able
2
The
cal
cula
ted
valu
es u
sing
the
pre
sent
mod
el:
RC
-BC
am
ount
AZ(
n) - tr
ansf
erre
d ch
arge
per
bon
d, I
’(B
) - co
ntri
buti
on o
f th
e an
ion
in t
he b
ond,
2e/
e0 - p
aram
eter
of
the
diel
ectr
ic t
heor
y, f
pvv - Ph
illip
s’ i
onic
ity
para
met
er,
fBc - io
nici
ty p
aram
eter
cal
cula
ted
from
(23
), fe
c - io
nici
ty p
aram
eter
cal
cula
ted
from
(24
), X
A~
- re
lati
ve p
ositi
on o
f th
e m
axim
um B
C d
ensi
ty c
alcu
late
d by
(22)
, XV
ED
- r
elat
ive
posi
tion
of t
he m
axim
um V
ED, t
he v
alue
s X
VE
D are
extr
acte
d fr
om p
ublis
hed
VE
D p
lots
, A
x - e
lect
rone
gati
vity
dif
fere
nce
of c
ompo
undf
rom
[28]
, R
A an
d R, - r
adii
of
neut
ral
atom
s (a
t.un
its)
ext
rapo
late
d fr
om F
ig. 6
a
P
Alp
A
lAs
AlS
b GaP
0.15
8 0.
147
0.14
7 0.
131
0.13
5 0.
050
-0.1
40
0.16
0
0.66
0.
25
0.30
0.
38
0.33
0.
62
0.19
0.
27
0.30
0.
33
0.54
0.
19
0.44
0.
21
0.28
0.
67
0.23
0.
37
0.41
0.
33
0.13
7 0.
137
0.11
8 0.
137
0.31
4 0.
295
0.23
5 0.
314
0.47
2.
31
2.13
0.
31
2.45
2.
18
0.12
2.
76
2.26
0.
41
2.28
2.
19
13 a 0.
391
[7]
0.28
6 [5
] 0.
348
[7]
0.13
5 [6
] 0.
200
[5]
0.21
6 [3
] 0.
200
151
0.31
4 [5
] 0.
314
[5]
0.25
7 [5
] 0.
270
[3]
0.47
8 [7
] 0.
391
[7]
0.50
0 [6
] 0.
314
[5]
0.51
3 [3
]
0.31
4 [5
] 0.
62
[3]
-
if a G
aAs
0.12
3 0.
080
0.63
0.
18
0.31
0.
33
0.29
0.
118
0.29
4 0.
25
2.41
2.
22
GaS
b In
P
InA
s In
Sb
0.11
7 0.
089
0.08
9 0.
096
-0.1
00
0.34
0 0.
240
0.07
0
0.55
0.
14
0.26
0.
15
0.19
0.
75
0.21
0.
43
0.58
0.
38
0.71
0.
16
0.35
0.
51
0.38
0.
63
0.13
0.
31
0.32
0.
28
0.07
8 0.
157
0.15
7 0.
118
0.23
5 0.
333
0.33
3 0.
294
0.06
2.
64
2.34
0.
55
2.40
2.
40
0.39
2.
53
2.43
0.
20
2.76
2.
55
ZnS
Z
nSe
0.03
0 0.
041
0.22
0 0.
180
0.84
0.
38
0.62
0.
72
0.62
0.
77
0.34
0.
63
0.68
0.
56
0.29
4 0.
235
0.45
1 0.
392
0.35
2.
44
2.09
0.
48
2.45
2.
48
ZnT
e C
dTe
CuB
r CU
Cl
0.06
2 0.
038
-0.0
20
0.16
0.
72
0.27
0.
61
0.24
0.
47
0.78
0.
65
0.67
0.90
0.
46
0.74
0.
89
0.85
0.
93
0.54
0.
75
0.93
0.
99
0.19
6 0.
235
0.37
3 0.
392
0.26
2.
67
2.27
-
- -
0.35
3 0.
412
0.43
1 0.
471
0.6
2.59
2.
04
0.8
2.79
1.
63
0.01
0 0.
006
0.14
0 0.
10
kP
P
4

448 U. PIETSCH
Fig. 5. The dependence of the ionic radius Ri on the effective charge of constituents e*. x AIIIP, + AIIIP, 0 A’IISb, A AIITe; AIBV, o GaBV, InBV,
-
I
e” - explains the predominantly covalent character of the bond. Further it is somewhat higher compared to band structure calculation results caused by Gaussian-like wave functions. Note, the shift of the VED maximum is clearly higher in comparison with that of the BC. The calculated BC transfer constants are listed in Table 3. In contrast to a thermal displacement the bond angles remain constant for hydrostatic pressure. Because of the different valence electron number a thermal displacement of an anion gives rise to a different transfer constant in comparison with a cation displacement. I n Fig. 7 the BC polarization density of GaAs is shown. The variations correspond to a t.hermal anion displacement of about 0.053 x 10-1O m against the rigid lattice. It is
demonstrated that BC has been transferred from the stretched central bond to the com- pressed ones illustrated on the right-hand side below. The BC density decreases near the cen- tre of the BC maximum. The relative maximum near the anion is caused by the change in the hybridization states of bonding orbitals. For increased ionicity the transfer behaviour is more localized near the sphere of the shifted anion.
a
10 111 plane
Fig. 6. The a) VED of GaAs calculated by the pres- ent model of “two-electron molecules”. The mari- mum VED amounts to 0.167 electron (at units)-3 = = 1.12 x 1030 electrons m-3. The initial parameters are listed in Table 1, contours at 0.033 e (at units)-3; b) BC density of GaAs calculated by the present mod- el. The maximum density amounts to 0.017 electrons (at. units)-3 = 0.12 x 1030 electrons m-3, contours a t 0.0034 electrons (at. units)-3
Static and Dynamical Valence Charge and Bond Charge Properties 449
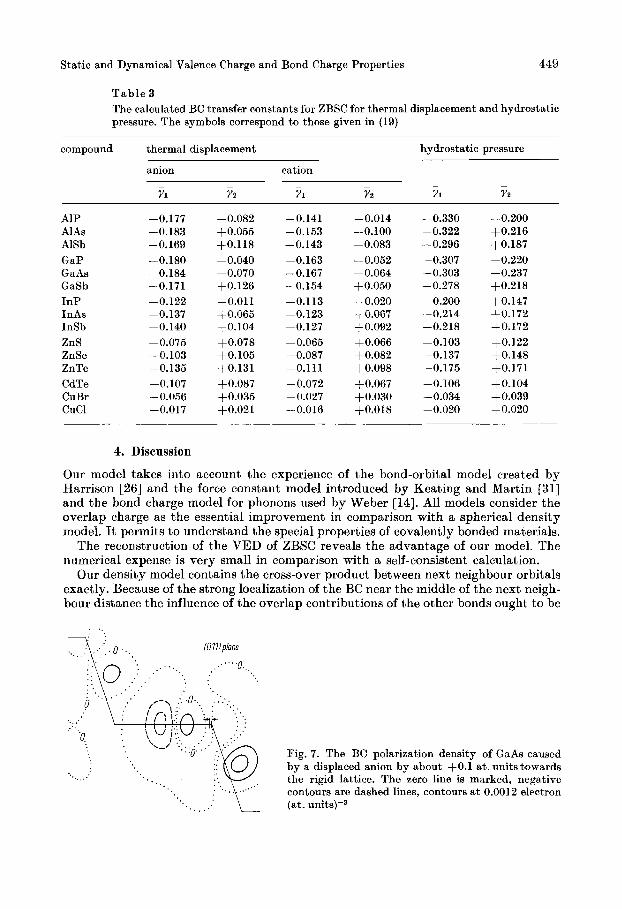
T a b l e 3 The calculated BC transfer constants for ZBSC for thermal displacement and hydrostatic pressure. The symbols correspond t o those given in (19)
compound thermal displacement hydrostatic pressure
anion cation - - - - - - Y1 Y2 YI Yz Y1 Y z
A1 P AlAs AlSb GaP GaAs GaSb InP InAs InSb ZnS ZnSe ZnTe CdTe CuBr CUCl
-0.177 -0.183 -0.169 -0.180 -0.184 -0.171 -0.122 -0.137 -0.140 -0.075 -0.103 -0.135 -0.107 -0.056 -0.017
-0.082 +0.055 +0.118 -0.040 -0.070 +0.126 -0.011 +0.065 +0.104 +0.078 +0.105 +0.131 +0.087 +0.035 $0.021
-0.141
-0.143 -0.153
-0.163 -0.167 -0.154 -0.113 -0.123 -0.127 -0.065 -0.087 -0.111 -b.072 -0.027 -0.016
-0.014 -0.100 -0.083 -0.052 -0.064 +0.050 -0.020 + 0.067 +0.092 +0.066 +0.082 t0.098 +0.067 +0.030 +0.018
-0.330 -0.322 -0.296 -0.307 -0.303 -0.278 -0.200 -0.214 -0.218 -0.103 -0.137 -0.175 -0.106 -0.034 -0.020
-0.200 +0.216 $0.187 -0.220 -0.237 +0.218 +0.147 +0.172 $0.172 t0.122 t0.148 +0.171 $0.104 +0.039 +0.020
4. Discussion
Our model takes into account the experience of the bond-orbital model created by Harrison [26] and the force constant model introduced by Keating and Martin [31] and the bond charge model for phonons used by Weber [l4]. All models consider the overlap charge as the essential improvement in comparison with a spherical density model. It permits to understand the special properties of covalently bonded materials.
The reconstruction of the VED of ZBSC reveals the advantage of our model. The numerical expense is very small in comparison with a self-consistent calculation.
Our density model contains the cross-over product between next neighbour orbitals exactly. Because of the strong localization of the BC near the middle of the next neigh- bour distance the influence of the overlap contributions of the other bonds ought to be
Fig. 7. The BC polarization density of GaAs caused by a displaced anion by about $0.1 at. units towards the rigid lattice. The zero line is marked, negative contours are dashed lines, contours at 0.0012 electron
’.. . . . . . .: (at. units)-3

450 U. PIETSCH
negligible, although the total VED has been built a t each point by linear superposition of molecule densities up to about third next neighbours. A general problem in our model seems to be the knowledge of the dehybridization of valence orbitals with respect t o the ionicity. Theoretical models are absent. It might be solved by using the outcome of a Mossbauer experiment for GaAs:Sn119 given by Antoncik and Lu et al. [25]. Measuring the portion of the Is) like valence orbitals a t the position of the Mossbauer-active atom Sn1l9 the Ip) part of the As atoms was been ob- served to increase but that of the Ga atoms to decrease in comparison with an analo- gous experiment performed on germanium. This result is plausible by the order prin- ciple of Mendeleev’s table. Yet we cannot extract the tendency of closed shells for se- miionic compounds from the above experiment. However, our approximations concern- ing the dehybridization seem to contain physical information enough.
The first essential outcome of our model is the identification of the value 2e/~ , being an important factor in the dielectric theory. It stands for the difference between the screened valence charge of the semiconductor and the positive charge of ionic pseudo- potentials using a linear screening concept. The latter suffices for metals but not for semiconductors. Therefore both the amount 2e/e, and the BC amount decrease in the same way as the metallic contribution in the chemical bond increases. For ZBSC it is necessary to correlate the value 2e/eo to the cross-over product of wave functions and the ionic charge transfer between the constituents. The charge transfer can be consid- ered by the interaction of ionic pseudopotentials and requires a quadratic screening concept. Note, 2e /~ , must not be identified with the overlap charge as often used for ZBSC.
On the other hand, the extracted BC corresponds to the value 2e/s0 using X-ray scattering experiments [21]. This is not surprising because their interpretation is prin- cipally due to free atomic scattering factors.
The identification of our model parameter R, as an “ionic” radius corresponds to usual construction principles. The radii of neutral atoms extrapolated from Fig. 6a confirm the main connection between the extension of valence shells and the turning point of the ionic pseudopotentials given by Heine and Waire [27]. Our result opposes the statement made by Van Vechten and Phillips [28] that “covalent” rigid radii should be valid for all tetrahedrally bonded compounds. Our outcome corresponds to the rule found empirically by Pietsch and Unger 11291 being essential for the interpreta- tion of the lattice parameter variation of doped semiconductors. The calculated trans- fer constants of ZBSC confirm all principles obtained for elemental semiconductors. They decrease with increasing metallic portion of the bond. In addition we have found a decrease in the enhanced ionicity. The ratio rlA/ylB increases from GaAs to CuBr from 1.1 to 2.0 for the thermally induced shift of the ions. Depending on the Ip) part of valence orbitals the transfer constants turn out to be greater for hydrostatic pres- sure (6 = 0) than for thermal displacement. The contribution of the quadratic terms rises with enhanced ionicity. The y 2 k contributes 4% and 107; for GaAs and CuBr, respectively. But with regard to the absolute transfer amount and the experimental possibilities those terms seem to be negligible for practical reasons. However, the experimentally determined transfer constants have been constructed for GaAs and InSb to be yl = -0.185 f 0.005 and -0.165 f 0.005, respectively. For thermal and uniaxial stress induced displacements the co-ordination angles vary. Using the ortho- gonalization condition (9) the Ip) character of the hybrid orbitals has to be changed and the position of the BC must be shifted. Considering (23) it is allowed to correlate that shift in terms of a varied local ionicity. The relative variation of ionicity is greater for compoiinds having a small absolute ionicity of the rigid lattice. Therefore, a uniaxial stress induced displacement of an anion with respect to the [lll] direction by about

Static and Dynamical Valence Charge and Bond Charge Properties 45 1
2% gives rise to a local ionicity variation of about 15% and 25% for GaAs and GaSb, but only 8% and 16% for ZnSe and ZnTe, respectively. Note, the transfer constants are not rather sensitive to the concrete hybridization state. For a thermal displace- ment Fl changes by about 10% only between our two approximations.
Acknowledgements
The author would like to thank Prof. Dr. K. Unger and Dr. L. Pasemann for helpful and critical discussions.
References
[l] M. T. YIN and M. L. COHEN, Phys. Rev. B 26, 5668 (1982). A. ZUNGER and M. L. COHEN, Phys. Rev. B 20,4082 (1979).
[2] A. BALDERESCHI and K. MASCHKE, Proc. XIV. Internat. Conf. Phys. Semiconductors, Edin- burgh 1978.
[3] B. N. HARMON, W. WEBER, and D. R. HAMANN, Phys. Rev. B 25, 1109 (1982). [a] J. C. PHILLIPS, Phys. Rev. 166, 832 (1968).
[5] M. L. COHEN and T. L. BEROSTRESSER, Phys. Rev. 141, 789 (1966). [6] J. P. WALTER and M. L. COHEN, Phys. Rev. B 4, 1877 (1971). [7j R. PICKENHAIN and A. MILCHEV, phys. stat. sol. (b) ii, 571 (1976). [8] J. C. CHELIKOWSKY and M. L. COHEN, Phys. Rev. B 14, 556 (1976). [9] A. BALDERESCHI, K. MASCHKE, A. MILCHEV, R. PICKENHATN, and K. UNGER, phys. stat.
J. A. VAN VECHTEN, Phys. Rev. 182, 891 (1969).
sol. (b) 108, 511 (1981). [lo] C. S. WANG and B. M. KLEIN, Phys. Rev. B 24, 3393 (1981). [ll] L. PASEMANN, Thesis, Freiberg 1978. [12] K. KUNC and R. M. MARTIN, Proc. XVI. Internat. Conf. Semiconductors, Montpellier 1982. [13] J. BAUR, K. MASCHKE, and A. BALDERESCHI, Phys. Rev. B 27, 3720 (1983). [14] K. C. RUSTAGI and W. WEBER, Solid State Commun. 18, 673 (1976). [15] S. FROYEN and M. L. COHEN, Phys. Rev. B 28, 3258 (1983). [16] N. N. SIROTA, Chemical Bond in Semiconductors and Solids, Consultants Bureau, New York
[17] M. GOMM, Thesis, Universitllt Erlangen-Niirnberg 1977. [18] J. J. DEMARCO and R. J. WENS, Phys. Letters 13, 209 (1964). [19] B. DAWSON, Proc. Roy. Soc. A 298, 264 (1967). [20] D. H. BILDERBACK and R. COLELLA, Phys. Rev. B 13, 2479 (1976). [all U. PIETSCH, phys. stat. sol. (b) 103, 93 (1981); 111, K7 (1982). [22] U. PIETSCH, phys. stat. sol. (b) 120, 183 (1983). [23] U. PIETSCH, phys. stat. sol. (b) 126, 595 (1984). [24] K. HUBNER, Bull. SOC. Chim. de France 2, 19 (1975); Ann. Phys. (Germany) 25, 97 (1970). [25j E. ANWNCIK and B. L. Gu, Physica Scripta 26, 835 (1982). [26] W. A. HARRISON, Phys. Rev. B 8, 4487 (1973). [27] V. HEINE and D. WAIRE, Solid State Phys. 24, 249 (1970). [28] J. A. VAN VECHTEN and J. C. PHILLIPS, Phys. Rev. B 2, 2160 (1970). [29] U. PIETSCH and K. UNGER, phys. stat. sol. (a) 80, 165 (1983). [30j J. ST. JOHNS and A. BLOCH, Phys. Rev. Letters 33, 1095 (1974). [31] P. N. KEATING, Phys. Rev. 145, 637 (1966).
R. M. MARTIN, Phys. Rev. B 1, 4005 (1970). [32] J. C. SLATER, Phys. Rev. 36, 57 (1930).
1967.
(Received November 2 , 1984)





























