Embed Size (px)
Citation preview

STRATEGIES TO IMPROVE THE AGING, BARRIER AND MECHANICAL PROPERTIES OF CHITOSAN,
WHEY AND WHEAT GLUTEN PROTEIN FILMS
Idoia Olabarrieta
AKADEMISK AVHANDLING
Som med tillstånd av Kungliga Tekniska Högskolan framlägges till offentlig granskning för avläggande av teknisk doktorsexamen fredagen den 27 maj 2005, kl 10.00 I sal K2, Teknikringen 28, KTH, Stockholm. Avhandlingen försvaras på engelska.

STRATEGIES TO IMPROVE THE AGING, BARRIER AND MECHANICAL PROPERTIES OF CHITOSAN, WHEY AND
WHEAT GLUTEN PROTEIN FILMS
ABSTRACT
Chitosan, Whey Protein Isolate (WPI) and vital wheat gluten (WG) are three biomaterials that have quite promising properties for packaging purposes. They have good film forming properties and good gas barrier properties in dry conditions. Moreover, because they are produced from industrial waste of food processing, they offer an ecological advantage over polymers made from petroleum. However, their physicochemical characteristics still must be improved for them to be of commercial interest for the food packaging industry. The purpose of this work was to study different strategies aiming to improve the water resistance and aging properties of these polymers, which are some of the key disadvantages of these materials.
The produced solution cast chitosan and WPI films were characterised with scanning electron microscopy (SEM), density measurements and thermogravimetry. The water vapour transmission rate was determined at a relative humidity of 11%. In the first part, mechanical properties of solid films and seals were assessed by tensile testing. WG film’s tensile properties and oxygen and water vapour permeabilities were measured as a function of aging time. The changes in the protein structure were determined by infrared spectroscopy and size-exclusion high-performance liquid chromatography and the film structure was revealed by optical and scanning electron microscopy. Gluten-clay nanocomposites were characterised by tensile testing, X-ray diffraction and transmission electron microscopy.
The incorporation of a hydrophobic biodegradable polymer, poly (ε-caprolactone), PCL, in both chitosan and whey protein, yielded a significant decrease in water vapour transmission rate. It was observed that a certain amount of the PCL particles were ellipsoidal in chitosan and fibrous in WPI. The obtained data also indicated that the particle shape had an important influence in the water vapour transmission rate.
In the second part, the aging properties of WG films, plasticized with glycerol and cast from water/ethanol solutions with pH=4 or pH=11 were investigated. WG films made from alkaline solutions were mechanically more time-stable than the acidic ones, the latter being initially very ductile but turning brittle towards the end of the aging period. The protein solubility measurements indicated that the protein structure of the acidic films was initially significantly less aggregated than the in basic films. During aging the acidic films lost more mass than the basic films through slow evaporation of volatiles (water/ethanol) and through migration of glycerol to the paper support. The oxygen permeability was also lower for the basic films.
In the last part, the properties of new and aged glycerol-plasticized WG films at acidic and basic conditions containing ≤4.5 wt% natural or quaternary-ammonium-salt-modified montmorillonite were studied. Films of WG with montmorillonite were possible to produce by solution casting. The aging rate of acidic and basic films was unaffected by the incorporation of clay. However, the large reduction in water vapour permeability for most systems suggested that the clay sheets were evenly distributed within the films. The film prepared from basic solution and containing natural clay was almost completely exfoliated as revealed by transmission electron microscopy and X-ray diffraction. The best water vapour barrier properties were obtained by using modified clay.
Keywords: biodegradable polymers, chitosan, whey protein, wheat gluten, poly(ε-caprolactone), montmorillonite, food packaging, permeability, mechanical properties, aging, pH, solubility, migration, solution casting.

STRATEGIES TO IMPROVE THE AGING, BARRIER AND MECHANICAL PROPERTIES OF CHITOSAN, WHEY AND
WHEAT GLUTEN PROTEIN FILMS
SAMMANFATTNING
Kitosan, WPI (Whey Protein Isolate / vassleproteinisolat) och vitalt vetegluten (vital wheat gluten, WG) är tre biomaterial med ganska lovande förpackningsegenskaper. De har goda egenskaper för filmformning och de är utmärkta gasbarriärer i torrt tillstånd. Dessutom tillverkas de av biprodukter från livsmedelsindustrin, vilket ger en ekologisk fördel jämfört med petroliumbaserade plaster. Deras fysikokemiska karakteristika måste emellertid förbättras innan de blir kommersiellt intressanta som livsmedelsförpackningar. Syftet med det här arbetet var att studera olika strategier för att förbättra egenskaperna avseende resistans mot vattenånga och åldring, vilka är de huvudsakliga svagheterna hos de här polymererna.
De tillverkade filmerna av kitosan och WPI karakteriserades med hjälp av svepelektronmikroskop (SEM), densitetsmätningar och termogravimetri. Transmissionsgraden för vattenånga bestämdes vid en relativ fukthalt av 11%. I den första delen fastställdes mekaniska egenskaper för filmer och förseglingar med dragprov. Veteglutenfilmernas mekaniska egenskaper och syre- respektive vattenångpermeabilitet uppmättes som en funktion av åldringstiden. Förändringarna i proteinstrukturen fastställdes med infraröd spektroskopi och vätskekromatografi och filmstrukturen studerades med optisk- och svepelektronmikroskopi. Gluten-lera nanokompositer karakteriserades med dragprovning, röntgendiffraktionsmätning och transmissionselektronmikroskopi.
Tillsatsen av en hydrofob bionedbrytbar polymer, poly (ε-kaprolakton), PCL, i både kitosan och vassle, medförde en märkbar minskning av transmissionsgraden för vattenånga. Experimenten klargjorde att en viss mängd av PCL-partiklarna var ellipsoidformade i kitosan och hade en mer avlång struktur i vassle. Erhållna data visade också att partikelformen hade en betydande påverkan på transmissionsgraden för vattenånga.
I den andra delen undersöktes åldringsegenskaperna för veteglutenfilmer, med glycerol som mjukgörare, gjutna från vatten/etanollösningar med pH=4 eller pH=11. Veteglutenfilmer från alkaliska lösningar var mekaniskt mer tidsstabila än de från sura, de senare var initialt mycket elastiska men blev mot slutet av åldringsperioden spröda. Mätningar av proteinlösligheten indikerade att proteinstrukturen för de sura filmerna initialt var signifikant mindre aggregerade än de basiska filmerna. Under åldring förlorade de sura filmerna mer massa än de basiska filmerna genom långsam evaporering av flyktiga ämnen (vatten/etanol) och genom migration av glycerol till pappersunderlaget. Syrepermeabiliteten var också lägre för de basiska filmerna.
I den sista delen studerades egenskaperna hos nya och åldrade glycerol-mjukgjorda veteglutenfilmer, vid basiska och sura förhållanden, innehållande ≤4.5 vikt% naturliga eller kvartenär ammoniiumsalt-modifierad montmorillonit. Det var möjligt att tillverka veteglutenfilmer med montmorillonit genom gjutning. Åldringshastigheten för sura och basiska filmer var opåverkad av inblandingen av lera. Den stora reduktionen, med avseende på vattenångpermeabilitet för de flesta systemen påvisade emellertid att ”lerflaken” var jämt fördelade i filmerna. Filmen som preparerades från basisk lösning och innehöll naturlig lera var nästan fullständigt exfolierad såsom visats med transmissionselektronmikroskopi och röntgendiffraktionsstudier. De bästa vattenångbarriäregenskaperna erhölls med modifierad lera.
Nyckelord: bionedbrytbara polymerer, kitosan, vassleprotein, vetegluten, poly(ε-kaprolakton), montmorillonit, livsmedelsförpackningar, permeabilitet, mekaniska egenskaper, åldring, pH, löslighet, migration, filmgjutning

LIST OF PUBLICATIONS This thesis is a summary of the following publications: I. ‘Transport properties of chitosan and whey blended with poly(ε-caprolactone)
assessed by standard permeability measurements and microcalorimetry’. I. Olabarrieta, D. Forsström, U. W. Gedde, M. S. Hedenqvist. Polymer, Vol.42, 4401-4408 (2001).
II. ‘Mechanical and physical properties of chitosan and whey blended with poly(ε-
caprolactone). I. Olabarrieta, A. Jansson, U. W. Gedde, M. S. Hedenqvist. International Journal of Polymeric Materials, 51: 275-289, (2002).
III. ‘Aging properties of films of plasticized vital wheat gluten cast from acidic and
basic solutions’. I. Olabarrieta, M. Gällstedt, J.R. Sarasua, E. Johansson and M. S. Hedenqvist. Submitted to Biomacromolecules.
IV. ‘Properties of new and aged montmorillonite-wheat gluten composite films’.
I. Olabarrieta, M. Gällstedt, I. Ispizua, J. R. Sarasua, M. S. Hedenqvist. To be submitted to Journal of Agricultural and Food Chemistry.

I
TABLE OF CONTENTS
1 INTRODUCTION ...............................................................................................1 1.1 BACKGROUND.............................................................................................................. 1 1.2 PURPOSE AND SCOPE OF THE STUDY .................................................................... 2 1.3 EDIBLE AND BIODEGRADABLE MATERIALS IN FOOD PACKAGING.............. 2
1.3.1 Biodegradable polymers.............................................................................................. 3 1.3.2 Biopolymer films ........................................................................................................ 4
1.4 BARRIER PROPERTIES - PERMEABILITY................................................................ 5 1.4.1 Oxygen permeability ................................................................................................... 6 1.4.2 Water vapour permeability.......................................................................................... 7
1.5 MECHANICAL PROPERTIES....................................................................................... 8 1.6 CHITOSAN...................................................................................................................... 9
1.6.1 Chitosan films ........................................................................................................... 10 1.7 WHEY PROTEIN .......................................................................................................... 11
1.7.1 Whey Protein Isolate films........................................................................................ 12 1.8 WHEAT GLUTEN......................................................................................................... 12
1.8.1 Wheat gluten films .................................................................................................... 14 1.9 PLASTICIZERS IN PROTEIN FILMS......................................................................... 15 1.10 DRAWBACKS OF BIOPOLYMER FILMS................................................................. 16
1.10.1 Water sensitivity........................................................................................................ 16 1.10.2 Aging of biodegradable polymers ............................................................................. 16
1.11 STRATEGIES TO ENHANCE FILM PROPERTIES AND LIFE TIME.................... 17 1.12 HYBRID COMPOSITE STRUCTURES....................................................................... 19
1.12.1 Poly(ε-caprolactone) composites .............................................................................. 19 1.12.2 Nanoclay nanocomposites......................................................................................... 20
2 EXPERIMENTAL.............................................................................................23 2.1 MATERIALS ................................................................................................................. 23 2.2 FILM FORMATION...................................................................................................... 23
2.2.1 WPI films .................................................................................................................. 23 2.2.2 Chitosan films ........................................................................................................... 23 2.2.3 PCL/Chitosan films ................................................................................................... 24 2.2.4 PCL/WPI films.......................................................................................................... 24 2.2.5 Gluten films............................................................................................................... 24 2.2.6 Gluten/MMT films .................................................................................................... 25
2.3 METHODS and INSTRUMENTS................................................................................. 25 2.3.1 Permeability .............................................................................................................. 25 2.3.2 Microcalorimetry....................................................................................................... 26 2.3.3 Tensile test ................................................................................................................ 27 2.3.4 Content of volatile mass............................................................................................ 27 2.3.5 Microscopy................................................................................................................ 27 2.3.6 Water uptake ............................................................................................................. 28 2.3.7 Spectroscopy ............................................................................................................. 28 2.3.8 Protein solubility ....................................................................................................... 28 2.3.9 Characterization of Polymer-Clay Nanocomposites ................................................. 29 2.3.10 Packaging related tests .............................................................................................. 29

II
3 RESULTS AND DISCUSSION........................................................................31 3.1 BIOPOLYMER/PCL BLENDS ..................................................................................... 31
3.1.1 Oxygen permeability ................................................................................................. 31 3.1.2 Water vapour permeability........................................................................................ 32 3.1.3 Mechanical properties ............................................................................................... 37 3.1.4 Other packaging related properties............................................................................ 40
3.2 AGING OF WHEAT GLUTEN FILMS........................................................................ 41 3.2.1 General film characteristics....................................................................................... 41 3.2.2 Mechanical properties of unaged and aged WG films .............................................. 42 3.2.3 Permeability .............................................................................................................. 45 3.2.4 Protein solubility of unaged and aged WG films ...................................................... 46 3.2.5 Mass loss ................................................................................................................... 48
3.3 GLUTEN /MONTMORILLONITE FILMS .................................................................. 50 3.3.1 Mechanical properties ............................................................................................... 50 3.3.2 Permeability .............................................................................................................. 51 3.3.3 X-ray and TEM ......................................................................................................... 53
4 CONCLUSIONS................................................................................................55
5 ACKNOWLEDGEMENTS ..............................................................................57
6 REFERENCES ..................................................................................................59

Introduction
1
1 INTRODUCTION
1.1 BACKGROUND
Synthetic polymers have become very important since their development and huge boost under the World War II. This gave cause to the first important commercial modern plastic appearances in the 1940s. Packaging is one application area that was revolutionised by oil-based polymers such as polyethylene, polypropylene, polystyrene, poly(ethylene terephtalate) and poly (vinyl-chloride). These polymers quickly found wide acceptance, in different packaging applications, due to their attractive properties such as flexibility, toughness, low weight, processability and low price [1]. Almost all foods, whether fresh or processed, are enclosed in some form of packaging, from processing and manufacturing through handling and storage all the way to the consumer [2]. Packaging protects food from the outside environment. The use of commodity plastics such as polyolefins provides many conveniences including light weight and desirable physical and mechanical properties with a favourable cost performance for the food industry and the consumer. However, the growing dependence on synthetic polymers has raised a number of environmental concerns. Most plastic materials are not biodegradable and are derived from non-renewable resources. The property of durability which makes these materials so useful also ensures their persistence in the environment and complicates their disposal which is becoming an ever growing problem for many of the plastics now in use. The three main strategies available for the management of plastic waste are: incineration, recycling and landfill [3]. The limited availability of landfills, the pollution from incineration and the cost and limitations of recycling, together with more restrictive customer demands and government legislation about plastics and waste materials drives the plastics industry to search for other solutions. Packaging materials for foodstuff, like any other short-term storage packaging material, have been under scrutiny for many years as heavy contributors to the plastic waste-management problems. Interest to maintain or enhance food quality while reducing packaging waste has encouraged the exploration of new packaging materials, such as edible and biodegradable films from renewable resources. During the last two decades the interest in edible and biodegradable materials has increased due to their environmental benefits [4-9]. The use of these materials, due to their biodegradable nature, could at least to some extent solve the waste problem. In this case, both renewable biomass feedstock (crops) and agro-industrial waste emerge as the key alternatives to produce new biodegradable materials. Indeed, agro-industrial waste offers a great promise as feedstock because of the low price, but also because its conversion contributes to solve the disposal problem by turning waste into useful products [10].

Introduction
2
1.2 PURPOSE AND SCOPE OF THE STUDY The present thesis focuses on the potential of three biopolymers to be used in food packaging. The purpose of the study is based in finding strategies to improve the properties of whey and wheat gluten (proteins) and chitosan (polysaccharide) films. This study is a contribution to the ongoing research with aim to improve the properties of these potential food packaging materials.
The scope of the thesis can be divided in three different parts:
• The first part deals with the improvement of water vapour permeability of whey and chitosan by mixing them with a hydrophobic biodegradable polymer, poly(ε-caprolactone), PCL. By blending these polymers and testing the packaging related properties, as well as water and gas permeability, the potential of these blends as food packaging films is analysed.
• The second part is focused on the aging problems of the wheat gluten protein. That is, the influence of different parameters in the long term properties of this material.
• The third part consists in the blending of wheat gluten with nanoclays to study the nanoparticles’ impact on the mechanical properties and the permeability of the films.
The final aim is to find combinations and methods for these biopolymers that make these renewable materials a commercially interesting alternative to materials commonly used today. 1.3 EDIBLE AND BIODEGRADABLE MATERIALS IN FOOD
PACKAGING Certain newly discovered characteristics of natural biopolymers could make them the choice for different types of wrappings and films [11-13]. Indeed, many researchers believe that biodegradable packaging has a bright future. Growing environmental awareness and consumer power coupled with the inexorable rise in pre-packaged disposable meals means that food manufacturers and packagers are increasingly being targeted to improve their environmental performances. The use of natural polymers dates back to the time of the ancient Egyptians. But nowadays, with the exception of cellulose, due to the big dominancy of synthetic plastics, biopolymers have been slow to reach commercial maturity. That is because they have generally had higher costs and less optimal physical properties than conventional plastics. In addition, there have not been sufficient incentives for downstream processors to incorporate the biodegradable materials into their products. Things are changing, however. New large-scale production systems are bringing down the costs of biodegradable polymers, and sophisticated polymerization and blending techniques are improving the material’s mechanical properties. In addition, food and beverage producers, seeking goodwill from an increasingly environmentally conscious public, have begun to employ biodegradable plastics for a variety of

Introduction
3
packaging applications. In some cases, local and national laws are also encouraging the use of degradable materials.
1.3.1 Biodegradable polymers
Unlike most other plastics, biodegradable polymers can be broken down in the environment. A polymer is referred to as biodegradable when after the degradation-process it is fully reduced to natural products (such as carbon dioxide, water, methane or biomass) by micro-organisms (bacteria or fungi) and/or enzymes [14, 15]. Materials used to make biodegradable plastics fall into two broad categories [6]:
• Natural polymers (biopolymers): made from renewable or natural resources
• Fossil or non-renewable: made synthetically from oil
A schematic diagram of the types of biobased polymers or biopolymers is showed in Figure 1. Biobased polymers may be divided into three main categories based on their origin and production [13]:
• Category 1: Polymers directly extracted/removed from biomass. Examples are polysaccharides such as starch and cellulose and proteins like casein and gluten.
• Category 2: Polymers produced by classical chemical synthesis using renewable biobased monomers. A good example is polylactic acid, a biopolyester polymerised from lactic acid monomers. The monomers themselves may be produced via fermentation of carbohydrate feedstock.
• Category 3: Polymers produced by microorganisms or genetically modified bacteria. To date, this group of biobased polymers consists mainly of the polyhydroxyalkonoates, but developments with bacterial cellulose are in progress.
Figure 1: Schematic presentation of types of biobased polymers and origin of the three
materials used in this work.

Introduction
4
In general, compared to conventional plastics derived from mineral oil, biobased polymers have more diverse chemistry and architecture of the side chains giving the material scientist unique possibilities to tailor the properties of the final package [13]. The basic materials researched to use as edible and biodegradable films and coatings in food packaging are the ones directly extracted from biomass (category 1). The most commonly available, are extracted from marine and agricultural animals and plants and they are based in: proteins, polysaccharides and lipids. The polysaccharides are natural polymers. They either act as energy-rich food stores in plants (starch) and animals (glycogen), or have structural roles in the plant cell wall (cellulose, pectin) or the tough outer skeleton of insects and similar creatures (chitin) [16]. To date, the principal polysaccharides of interest for material production have been cellulose, starch, gums, and chitosan [13]. Proteins, contrary to homopolymers or copolymers in which one or two monomers are repeated, are heteropolymers comprised of more than 20 different amino acids, each with specific sequences and structures. The side chains of these amino acids are highly suitable for chemical modification which is helpful to the material engineer when tailoring the required properties of the packaging material [17]. Due to this molecular diversity, proteins have considerable potential for the formation of linkages that differ with respect to their position, nature, and/or energy. Proteins can be divided into proteins from plant origin (e.g. gluten, soy, pea and potato) and proteins from animal origin (e.g. casein, whey, collagen, and keratin) [16]. All these biopolymers are by nature hydrophilic and somewhat crystalline, factors causing problems, especially in relation to packaging of moist products. The excess of disulphide cross-linking and thermal degradation of these polymers are also two main problems in their processing and performance. On the other hand, these polymers produce materials with excellent gas barriers [18]. Three interesting materials in this category are whey, gluten and chitosan, (see Figure 1) which are presented in this report. The interest in these materials and the development of their applications in food packaging have increased due to large surpluses of the raw materials, which are produced in large amounts as by-products of agro-industrial processes. Even if they are used in several applications there is still a great disposal of these by-products. 1.3.2 Biopolymer films
The purpose with developing biopolymer films is to offer commercially interesting replacement alternatives to existing synthetic, non biodegradable products, e.g. in applications for food packaging. Current research in the area cover modified biopolymers and blends. But these approaches still require further research and development before they become competitive to the commonly used synthetic polymers [3].

Introduction
5
Advantages of edible and biodegradable films compared to other polymeric materials are [4]:
• They contribute to the reduction of environmental pollution, since they are produced from renewable and biodegradable ingredients.
• Some biopolymers are edible, so these kinds of films could be consumed with the product. This could be important since it represents an environmentally ideal package (no dispose).
• The films may be used in multi-layer packaging, e.g. with cellulosic material resulting in a totally biodegradable package.
• The films can enhance the organoleptic properties of packaged foods, provided that various components (flavouring, colouring, and sweeteners) are incorporated to them.
• The films can supplement the nutritional value of foods. This is particularly true for protein films.
• The films can function as carriers for antimicrobial and antioxidant agents.
• The films can regulate transfer of moisture, oxygen, carbon dioxide, lipid, and aroma and flavour compounds in food systems to increase product shelf life and improve quality.
The most common materials used to form edible films and coatings are polysaccharides, proteins and lipid compounds. Many of these materials have good film-forming properties. Films formed from polysaccharides are hydrophilic and provide efficient barriers to oils and lipids, but their moisture barrier properties are poor. Protein based films are highly interesting, as they confer more potential functional properties. Many lipid compounds such as animal and vegetable fats have been used to make edible films and coatings. Lipid films have excellent moisture barrier properties but they can cause textural and organoleptical problems due to their oxidation and their waxy taste [5]. Properties of biopolymers and biodegradable polymer films depend on the raw material that they are based on, the additives used and on the (chemical) modifications during production. Water vapour- and oxygen permeability and tensile test are the most frequently used methods to determinate film characteristics for food packaging films [4]. 1.4 BARRIER PROPERTIES - PERMEABILITY A key characteristic of glass and metals as packaging materials is their high barrier properties to gases and vapours. While polymers can provide an attractive balance of properties such as flexibility, toughness, lightweight, formability and printability, they do allow the transport of gases and vapours to some extent. The selection of a barrier polymer for a particular application typically involves tradeoffs between permeation, mechanical and aesthetic properties as well as economic and recycling considerations. Quality and shelf life are reduced when food, through interaction with the outside environment, gains or loses moisture or aroma, takes up oxygen (leading to oxidative rancidity), or becomes contaminated with micro-organisms [12]. There is an ongoing

Introduction
6
interest in optimising property sets of barrier polymers to provide an efficient and economical method for packaging and for extending the shelf life of packaged foods and beverages [19].
Barrier properties are determined by the steady-state rate of mass transport through the films [20, 21]. The permeability coefficient, P, can be defined by:
( ) ( )( ) ( ) ( )filmindroppressuretimearea
thicknessfilmpermeantofvolumeP⋅⋅
⋅=
The permeability coefficient is not only a function of the chemical structure of the polymer, it also depends on many physical factors such as density, crystallinity, orientation, cross-linking, plasticizers, moisture sensitivity and temperature. Thus, film properties should be compared at as near identical testing conditions as possible as the conditions at which the test or analysis is carried out affect the results. Oxygen Transmission Rate (OTR) and Water Vapour Transmission Rate (WVTR) are two of the most important parameters that affect food. The challenge in use of the barrier properties of proteins is to select biopolymer, plasticizer and film-formation conditions that optimize the desired barrier properties, while achieving other desirable properties as film flexibility and strength. 1.4.1 Oxygen permeability
The transfer of oxygen from the environment to food has an important effect on food quality and shelf life. Oxygen causes food deterioration such as lipid and vitamin oxidation, leading to sensory and nutrient changes [2]. Due to the large amount of hydrogen bonds biopolymer films are hydrophilic, which makes them excellent barriers to non-polar substances, such as oxygen and some aroma compounds [22]. This hydrophilicity makes their gas barrier properties very much dependent on the humidity conditions for the measurements. That is why the gas permeability of these materials may increase manifold when humidity increases. Modification of polymer structure combined with optimised selection of plasticizer may produce, at low to intermediate RH, biodegradable films with oxygen barrier properties that are as good as those of poly(vinylidene chloride) (PVDC) and ethylene vinyl alcohol copolymer (EVOH) films (see Figure 2). Gas permeability measurements of wheat gluten films, generally conducted at fixed temperature and dry conditions have shown high oxygen barrier properties [23-25]. Many biopolymer film research studies are focused in oxygen and carbon dioxide permeability since these two gases influence the rates of oxidation and respiration in the enclosed food, as fruits and vegetables [26-29]. In most cases a biopolymer showing a good O2 permeation, will also be a good barrier to CO2, as well as many organic vapours and odours [30]. The development of edible films and coatings with selective gas permeability is potentially of great interest for controlling respiratory exchange, achieving a modified atmosphere effect, modified atmosphere packaging (MAP) [25, 28, 31].
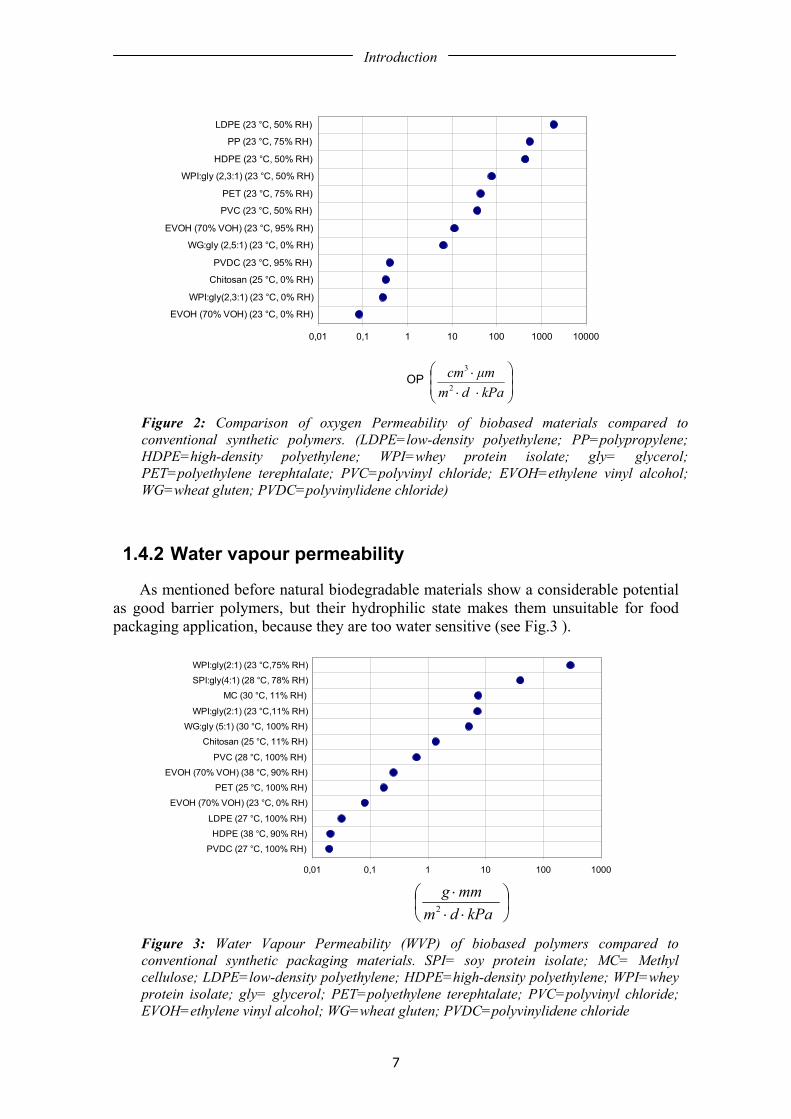
Introduction
7
OP ⎟⎟⎠
⎞⎜⎜⎝
⎛
⋅⋅⋅
kPadmµmcm
2
3
Figure 2: Comparison of oxygen Permeability of biobased materials compared to conventional synthetic polymers. (LDPE=low-density polyethylene; PP=polypropylene; HDPE=high-density polyethylene; WPI=whey protein isolate; gly= glycerol; PET=polyethylene terephtalate; PVC=polyvinyl chloride; EVOH=ethylene vinyl alcohol; WG=wheat gluten; PVDC=polyvinylidene chloride)
1.4.2 Water vapour permeability
As mentioned before natural biodegradable materials show a considerable potential as good barrier polymers, but their hydrophilic state makes them unsuitable for food packaging application, because they are too water sensitive (see Fig.3 ).
WVP ⎟⎠⎞
⎜⎝⎛
⋅⋅⋅
kPadmmmg
2
Figure 3: Water Vapour Permeability (WVP) of biobased polymers compared to conventional synthetic packaging materials. SPI= soy protein isolate; MC= Methyl cellulose; LDPE=low-density polyethylene; HDPE=high-density polyethylene; WPI=whey protein isolate; gly= glycerol; PET=polyethylene terephtalate; PVC=polyvinyl chloride; EVOH=ethylene vinyl alcohol; WG=wheat gluten; PVDC=polyvinylidene chloride
0,01 0,1 1 10 100 1000 10000
LDPE (23 °C, 50% RH)
PP (23 °C, 75% RH)
HDPE (23 °C, 50% RH)
WPI:gly (2,3:1) (23 °C, 50% RH)
PET (23 °C, 75% RH)
PVC (23 °C, 50% RH)
EVOH (70% VOH) (23 °C, 95% RH)
WG:gly (2,5:1) (23 °C, 0% RH)
PVDC (23 °C, 95% RH)
Chitosan (25 °C, 0% RH)
WPI:gly(2,3:1) (23 °C, 0% RH)
EVOH (70% VOH) (23 °C, 0% RH)
0,01 0,1 1 10 100 1000
WPI:gly(2:1) (23 °C,75% RH)
SPI:gly(4:1) (28 °C, 78% RH)
MC (30 °C, 11% RH)
WPI:gly(2:1) (23 °C,11% RH)
WG:gly (5:1) (30 °C, 100% RH)
Chitosan (25 °C, 11% RH)
PVC (28 °C, 100% RH)
EVOH (70% VOH) (38 °C, 90% RH)
PET (25 °C, 100% RH)
EVOH (70% VOH) (23 °C, 0% RH)
LDPE (27 °C, 100% RH)
HDPE (38 °C, 90% RH)
PVDC (27 °C, 100% RH)
⎟⎠⎞
⎜⎝⎛
⋅⋅⋅
kPadmmmg
2

Introduction
8
Normally protein films have quite high water vapour permeability compared to waxes, which are often used as moisture barrier coatings on fruits, vegetables, confections and drugs and low density polyethylene (LDPE) packaging film, which commonly is used to protect food and drugs from moisture [32]. The poor resistance of WG films to water vapour is due to the hydrophilic nature of the protein and to the substantial amount of hydrophilic plasticizer added to impart adequate film flexibility. Understanding the way of transport of water vapour through the film is important for improving the moisture barrier properties of WG films. Water vapour transport through polymer films proceeds through [33]:
(i) absorption of water vapour on to the polymer surface (ii) solution of water vapour into the polymer matrix (iii) diffusion of water vapour through the polymer (iv) desorption of water vapour from the other surface of the polymer
Permeability is the rate of transport of a molecule through a polymer as a result of the combined effects of diffusion and solubility. The permeability coefficient (P) is the product of the diffusion (D) and solubility (S) coefficients:
P (mol/m⋅s⋅Pa) = D (m2/s) x S (mol/m3⋅Pa)
The diffusion coefficient indicates how fast a permeant will move within the polymer, while the solubility coefficient gives the amount of the permeant taken (absorbed) by the polymer from a contacting phase [34]. Ideally, when no interaction occurs between a polymer film and the permeating water vapour, P is independent of the apparent equilibrium water vapour pressure corresponding to the water activity (aw) of the film. Water vapour permeabilities of hydrophobic films, such as polyethylene, are independent of the water vapour pressure. However, protein-based films, similar to other hydrophilic films, exhibit water vapour pressure-dependent permeability [35]. Hydrophilic biopolymer materials, such as protein films, deviate from this ideal behaviour due to interactions of permeating water molecules with polar groups in the film structure [22, 36, 37]. Water vapour permeability measurements of WG films have been reported [23, 24, 38, 39]. 1.5 MECHANICAL PROPERTIES Mechanical properties are as important to edible and biodegradable films as barrier properties are. Having adequate mechanical strength and being free of minor defects ensure the integrity of a film. Interactions between hydrocolloids and small molecules, including water, plasticizers, lipids, and other additives dispersed in the space of the matrix, contribute to the mechanical behaviour of films. Quantitative information on the mechanical parameters of edible films is also essential for the packaging design process [4]. Among the many mechanical properties of plastic materials, tensile properties are the most frequently considered, evaluated, and used throughout the industry. Tensile testing provides these useful data: yield strength, fracture strength (ultimate tensile strength), modulus of elasticity (Young's modulus), and elongation at yield and

Introduction
9
break[40]. Maximum tensile strength is the maximum tensile stress that a film can sustain. Strain is the maximum change in length of the test specimen before breaking. Elastic modulus is the fundamental measure of film stiffness [41]. The test conditions will be important since the mechanical properties of hydrophilic films are also affected by the interactions with the surrounding environment. The stress-strain behaviour of these polymers is strongly dependent on temperature and relative humidity. Films conditioned in the environment of 50 %RH are weaker than films equilibrated with less RH because of the plasticizing effect of the water molecules [4]. As temperature increases, Young’s modulus and the tensile yield strength as well as maximum tensile strength generally decrease. However, the elongation at yield and break tend to increase. Additives and plasticizers also affect the mechanical properties. As the plasticizer content increases, tensile strength decreases and elongation increases [4, 41, 42]. 1.6 CHITOSAN Chitosan is a derivative of chitin, which is the second most abundant polymer on earth after cellulose [43]. Chitin is a naturally occurring polysaccharide found in the major structural component of the exoskeleton of invertebrates (crab, shrimps, krill, squid, etc) and the cell walls of e.g. many fungi. The natural occurrence of chitin is widespread, but the only practical and considerable source is the exoskeleton of commercially harvested shellfish [44]. The biodegradation of chitin is very slow in crustacean shell waste. Accumulation of large quantities of discards from processing of crustaceans has become a major concern in the food processing industry [45]. The seafood industry produces large amounts of chitin and most of it is wasted. Japan and USA are the main producers while India, China, Pakistan, Norway and Island, among others, are now manufacturing chitin as a by-product from their growing seafood industries. Even though chitin is the second most abundant natural polymer after cellulose, the production volumes are relatively much lower than cellulose [46]. The main reasons are the high costs for the extraction of chitin and differences between batches depending on the source due to the difficulties in preparing uniformly reproducible charges in bulk quantities from various marine organisms [47]. Chitosan is commercially available from a number of suppliers in various grades of purity, molecular weight and degree of deacetylation [48]. It was reported that the degree of deacetylation is one of the more important chemical characteristics [49], which could influence the performance of chitosan in many of its applications [50-52]. In addition, the degree of deacetylation, which determines the content of free amino groups in the polysaccharides [48], can be employed to differentiate between chitin and chitosan. For instance, chitin with a degree of deacetylation of 75% or above is generally known as chitosan [53]. The process of deacetylation involves the removal of acetyl groups from the molecular chain of chitin, leaving behind a complete amino group (-NH2). Chitosan versatility depends mainly on these high degree chemical reactive amino groups.

Introduction
10
The structure of chitin and chitosan are shown in Figure 4. Their chemical structures are very similar to that of cellulose. Chitin is composed of repeating units of 1,4 linked 2 deoxy-2-acetoamido-β-D-glucose. Chitosan consists of repeating units of (2 amino-2deoxy-D-glucose). So the difference is that instead of the -OH group bounded at C-2 in each glucose unit of cellulose, or acetylated amino group (-NHCOCH3) in chitin there is an amino group (-NH2) in chitosan [50, 54, 55]. The predomination of partially deacetylized groups makes this polymer soluble in dilute organic acids, providing clear, homogenous, and viscous solutions or gels with a consistent structure, thus being ideal for food coatings [47, 56]. The chemically active groups in chitosan structure are the free amino groups and the hydroxyl groups, with both being susceptible to modifications [56].
Figure 4: Molecular structures of chitin, chitosan and cellulose. [57]
Chitin and chitosan biopolymers possess some distinct properties that give them extraordinary potential in a broad spectrum of commercial, consumer and pharmaceutical products [54]:
• Anti-microbial, which makes chitin and chitosan potent bacteria and virus fighters.
• Highly positively charged, which makes chitosan an effective binder to negatively charged metals, biochemical, macromolecules and cells.
• Biodegradable, which makes chitin and chitosan ideal for decreasing the environmental impact of packaging and other products.
• Biocompatible, non-toxic naturally occurred polymer, which is biodegradable to normal body constituents.
Several papers have been written about the film properties of chitosan, and most work on chitin and chitosan is performed in Japan [31, 43, 47]. Chitin and the derivatives, in particular chitosan, are currently used only in small quantities as coagulants for water treatment, additives for shampoos and cosmetics, medical materials such as artificial skin and sutures, textiles and dietary fibres [43, 46, 51]. 1.6.1 Chitosan films
Chitosan films are clear, tough, and flexible with good oxygen barrier properties [58, 59]. Chitosan based coatings can protect foods from fungal decay and modify the atmospheres of fresh fruits and vegetables [12].

Introduction
11
Figure 5:Molecular structure of chitosan.
Like cellulose, both chitin and chitosan are crystalline due to their linear linkages. The high content of hydrogen bonds and the crystallinity give these films low oxygen permeability. The properties of chitosan makes it very interesting for applications as a packaging material and for edible coatings [55, 60-62]. Makino and Hirata [31] have shown that a biodegradable laminate consisting of chitosan-cellulose and polycaprolactone can be used in modified atmosphere packaging of fresh produce. However, many problems still have to be solved before chitosan can be of commercial interest, replacing e.g. poly(ethylene-co-vinyl alcohol). Processing of chitosan is a relatively unexplored field, compared to the more conventional materials and the majority of the applications are still in a developing stage [63]. 1.7 WHEY PROTEIN Liquid whey, a by-product from cheese manufacturing, is produced in large quantities and its annual production is continuously rising. Even if it has generally been disposed of as animal feed or used in infant formulas, sport food, clinical nutrition products etc. much of this whey is not utilised; creating serious waste disposal problems [64]. Typically milk contains around 87% water and 13% solids. Liquid whey is the thin, watery portion of milk that is obtained by coagulation and removal of the curd (casein) during cheese production. Whey protein accounts for approximately 5% of total solids of the milk (see Figure 6) [65]. The main components of liquid whey are whey proteins, lactose, water, fats and inorganic minerals [2, 66, 67]. Whey protein products can be classified according to their composition. In particular, they are divided according to their protein content. Usually the whey is purified by membrane filtration to concentrate the protein, a procedure that is very costly. Whey protein concentrate (WPC) contains 25–80% protein. Whey protein isolate (WPI) is nearly all protein (>90%) [2]. The wide range of protein concentration in these products depends on the membrane used and the pre-treatment prior to membrane separation. The whey protein used in this study (WPI) is refined from liquid whey and purified. Purification with respect to the whey protein is performed to enhance its film forming properties. Whey protein contains five principal proteins, which are α-lactalbumins, β-lactoglobulins, bovine serum albumin, immunoglobulins, and proteose-peptones. The major protein fraction is β-lactoglobulin [65, 68].

Introduction
12
Figure 6: Bovine milk fractions
1.7.1 Whey Protein Isolate films
The WPI films are excellent barriers against oxygen [41, 69] and since the uptake of oxygen in food causes rancidity they would be suitable for biodegradable food packaging in multi-component foods. The films could function as controlled-release carriers for antioxidants and antimicrobial agents, as well as supplementing the nutritional value of foods. Consequently, the interest in the study of edible and biodegradable films made from milk proteins has been outlined in the literature [69-77]. Since β-lactoglobulin is the dominating protein in whey its properties tend to dominate as well [63]. Because of the globular nature of these proteins, production of films requires pH/heat-denaturation to open the globular structure, break existing disulphide bonds and form new intermolecular disulphide bonds. These proteins undergo temperature denaturation reactions at temperatures above 73°C [78]. Native whey proteins are globular proteins containing most of the hydrophobic and sulfhydryl groups hidden in the interior of the molecule. Formation of whey protein films has mainly involved heat denaturation of whey proteins in aqueous solutions. Heating modifies the three-dimensional structure of the protein, exposing internal -SH and hydrophobic groups, which promote intermolecular disulfide bonding and hydrophobic interactions upon drying [77, 79]. 1.8 WHEAT GLUTEN Gluten is the main storage protein in wheat and corn. Wheat gluten (WG) is the cohesive and elastic mass that is leftover after starch is washed away from wheat flour dough. Commercially, it is an industrial by-product of wheat starch production via wet milling. Dry wheat flour comprises 9–13% protein and 75–80% starch [80]. The fractionated wheat starch is removed for e.g. fermentation to ethanol and sweeteners. The production of WG has increased in the last decade due to the increased usage of ethanol for fuel, and will probably increase in the near future [81].
Introduction
13
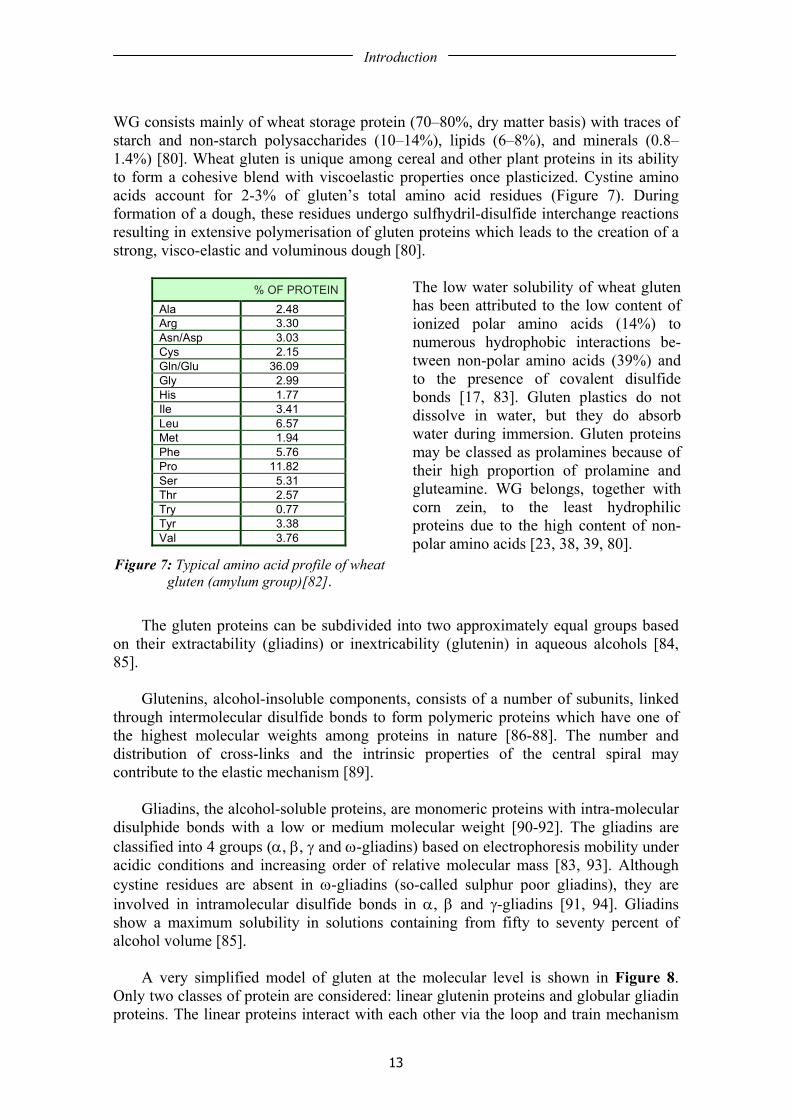
WG consists mainly of wheat storage protein (70–80%, dry matter basis) with traces of starch and non-starch polysaccharides (10–14%), lipids (6–8%), and minerals (0.8–1.4%) [80]. Wheat gluten is unique among cereal and other plant proteins in its ability to form a cohesive blend with viscoelastic properties once plasticized. Cystine amino acids account for 2-3% of gluten’s total amino acid residues (Figure 7). During formation of a dough, these residues undergo sulfhydril-disulfide interchange reactions resulting in extensive polymerisation of gluten proteins which leads to the creation of a strong, visco-elastic and voluminous dough [80].
% OF PROTEINAla 2.48 Arg 3.30 Asn/Asp 3.03 Cys 2.15 Gln/Glu 36.09 Gly 2.99 His 1.77 Ile 3.41 Leu 6.57 Met 1.94 Phe 5.76 Pro 11.82 Ser 5.31 Thr 2.57 Try 0.77 Tyr 3.38 Val 3.76
Figure 7: Typical amino acid profile of wheat gluten (amylum group)[82].
The low water solubility of wheat gluten has been attributed to the low content of ionized polar amino acids (14%) to numerous hydrophobic interactions be-tween non-polar amino acids (39%) and to the presence of covalent disulfide bonds [17, 83]. Gluten plastics do not dissolve in water, but they do absorb water during immersion. Gluten proteins may be classed as prolamines because of their high proportion of prolamine and gluteamine. WG belongs, together with corn zein, to the least hydrophilic proteins due to the high content of non-polar amino acids [23, 38, 39, 80].
The gluten proteins can be subdivided into two approximately equal groups based on their extractability (gliadins) or inextricability (glutenin) in aqueous alcohols [84, 85]. Glutenins, alcohol-insoluble components, consists of a number of subunits, linked through intermolecular disulfide bonds to form polymeric proteins which have one of the highest molecular weights among proteins in nature [86-88]. The number and distribution of cross-links and the intrinsic properties of the central spiral may contribute to the elastic mechanism [89]. Gliadins, the alcohol-soluble proteins, are monomeric proteins with intra-molecular disulphide bonds with a low or medium molecular weight [90-92]. The gliadins are classified into 4 groups (α, β, γ and ω-gliadins) based on electrophoresis mobility under acidic conditions and increasing order of relative molecular mass [83, 93]. Although cystine residues are absent in ω-gliadins (so-called sulphur poor gliadins), they are involved in intramolecular disulfide bonds in α, β and γ-gliadins [91, 94]. Gliadins show a maximum solubility in solutions containing from fifty to seventy percent of alcohol volume [85]. A very simplified model of gluten at the molecular level is shown in Figure 8. Only two classes of protein are considered: linear glutenin proteins and globular gliadin proteins. The linear proteins interact with each other via the loop and train mechanism

Introduction
14
and by disulphide bonding. In the diagram, for simplicity, disulphide bonds are not shown. As a first approximation, the chains are imagined to interact with the globular proteins by non-bonding forces such as Van der Waals interactions. The gliadin proteins fit in the spaces of the glutenin network acting as a plasticizer and giving to the compound the viscoelastic properties [89]. It is believed that glutenins contribute to the elastic properties of dough, whereas gliadins contribute to the viscous properties [95-97].
Figure 8: A very simplified model of molecular structure of gluten. HMW subunits (glutenin) are approximated by linear polymers, interchain disulphide links are not shown. Other polymers (such as gliadins) are approximated by spheres [89].
1.8.1 Wheat gluten films
WG proteins can be utilized to make films with novel functional properties, such as selective gas barrier properties and rubber-like mechanical properties. WG-based materials are homogeneous, transparent, mechanically strong, and relatively water insoluble. They are biodegradable and a priori biocompatible, apart from some WG-specific characteristics such as allergenicity. They are also edible when food-grade additives are used, and the presence of impurities is avoided. Several studies have been done on gluten films obtained by casting technique or thermal film forming processes [24, 37, 39, 63, 79, 80, 95, 98-101]. WG proteins are insoluble in water and require a complex solvent system with basic or acidic conditions in the presence of alcohol. Generally, changing the pH of the medium disrupts hydrogen and ionic interactions, while ethanol disrupts hydrophobic interactions [80]. The isoelectric point for WG was estimated to 7,5 [102]. Gennadios et al. [37] showed that WG films did not form in the protein’s isoelectric region (pH 7-8), most likely impeded by intermolecular protein repulsive forces. Also WG dispersion at pH 5-6 is very poor, resulting in films of uneven thickness and containing big particles of coagulated protein particles, which were unsuitable for property evaluation. However, adjusting the solution to acidic or basic conditions (above and below this pH) will favour the protein solubility and the film forming process [103-105]. It is well-known that heat treatment affects protein conformation and improves some film properties depending on heating conditions [106]. Temperature has an aggregating effect on wheat gluten inducing the formation of covalent bonds between proteins. Rising the temperature can denaturate globular proteins. This means that gliadin proteins when denaturated lose the native conformation and they open, exposing previously unexposed intramolecular disulfide bonds which now can be reduced and form other new intermolecular disulfide bonds [69].

Introduction
15
The drying step is also a critical part of the film forming process which can affect the final film properties [106]. During drying, all volatile disruptive agents are progressively eliminated. Solvent removal increases the concentration of WG proteins. Consequently, active sites for bond formation become free and close enough to each other to create new interactions. New hydrogen bonds, hydrophobic interactions and disulfide bonds contribute to formation of a good oxygen barrier three-dimensional network [5, 17, 22, 23, 27, 80, 107]. There are many parameters which must be controlled when making WG films. Functional properties of gluten films are highly dependent on WG concentration in solution, pH, additives, solvent polarity, temperature, and drying rate [37, 38]. 1.9 PLASTICIZERS IN PROTEIN FILMS Proteins by themselves, including whey and wheat gluten, form brittle films, due to extensive interactions between protein chains through hydrogen bonding, electrostatic forces, hydrophobic bonding, and/or disulfide cross-linking [27]. This brittleness makes these films useless in packaging film applications. The usual approach to enhance film forming properties is to add a plasticizer. Plasticizers are hydrophilic low molecular weight liquids that reduce protein chain-to-chain interaction. Gluten protein plasticization is a complex phenomenon that can be primarily explained by the ability of the hydrophilic plasticizer molecules to share hydrogen bonds with the protein network [108]. The result is an increase of the free volume and the mobility of polymer chains, lower glass transition temperature and more flexible and soft films. The most commonly used plasticizers are poly-functional alcohols, such as polyethylene-glycol, glycerol and sorbitol [72, 73, 77, 109-111]. In this study glycerol was used. Glycerol is a high boiling point plasticizer. It is water soluble, polar, non-volatile, and protein miscible. It is also harmless as plasticizer for films in contact with foodstuff, and is frequently used as sweetener in foodstuff [41]. These properties make glycerol a suitable plasticizer to use with a compatible water-soluble polymer [75]. The increase in film flexibility is also accompanied by an increase in film permeability, which depends on the type and amount of plasticizer [41, 42]. This increase in permeability is undesirable for food quality, so there is a need to optimise the use of plasticizer to offer the right balance of desired mechanical and barrier properties for the desired application [27, 36, 42, 69, 104, 111, 112]. Water is the most ubiquitous and uncontrollable plasticizer for hydrophilic biopolymer material films, thus the surrounding relative humidity has a large effect on the film properties [100]. The plasticizing and/or swelling effect of water on hydrophilic (polar) polymers also result in increased permeability values [25, 41, 77].

Introduction
16
1.10 DRAWBACKS OF BIOPOLYMER FILMS During this introduction the potential of biodegradable films for applications in food packaging have been pointed out. Nevertheless, despite the benefits that these polymers offer there are still significant obstacles that must be overcome, such as retailer and consumer scepticism, material costs, the added costs of switching technologies. Moreover, biopolymer film properties still have some restrictions and drawbacks which limit their use in large-scale applications. Apart from not yet competitive mechanical properties, biopolymers films are still difficult to process in comparison with synthetic polymers. Some of the main drawbacks and limitations of biopolymer films are listed below.
1.10.1 Water sensitivity
As pointed out earlier the major challenge for the material manufacturer is the by nature hydrophilic behaviour of many biobased polymers, as many food applications demand materials that are resistant to moist conditions. Most of the biobased materials are not soluble or are difficult to dissolve in water but they show large water up-take (swelling) and high water permeability. Moreover, films made by biological materials do change their mechanical and barrier properties in high moisture conditions, which is also a great disadvantage. This water sensitivity can develop a more spontaneous, rapid, non controllable degradation under the influence of bacteria (the disadvantage of biodegradability).
1.10.2 Aging of biodegradable polymers
One of the challenges for the successful use of biodegradable polymer products is to achieve controlled lifetime. Products must remain stable and function properly during storage and intended use, but after that they should biodegrade efficiently. Only by appropriately controlling water activity, pH, nutrients, temperature, oxygen levels and time can package integrity and microbial stability be assured. Thus, biodegradable polymer films may be safely stored in dry environments and used with dry food products over a relatively long period of time, whereas acceptable time of storage in moist environments or time of use with moist foods would be limited [18]. Biodegradable polymers suffer a change in their properties during time, aging, which make them not suitable for commercial applications. The aging of a biodegradable film can be due to physical or chemical reactions in the polymer matrix. The most common aging processes that films use to go through are:
• Physical aging: Migration of additives from the matrix.
This is a physical process that the films suffer when plasticizers migrate to the surface. Generally, migration of these low molecular compounds leads to stiffer and less extendable polymers, which may decrease the protective function of the packaging and consequently reduce the shelf life of the packaged food. Plasticizer molecular weight, concentration and the hydrophobic or hydrophilic character influence the migration. Water, as one of the most powerful plasticizer for these types of films, also migrates with time from the matrix to the surface and evaporates leaving a more brittle film [100].

Introduction
17
• Chemical aging: Oxidation Some studies have shown that formation of disulfide bonds by thiol oxidation occurred during wheat gluten film storage, even in conditions (temperature and relative humidity) under which the mobility of molecules is reduced. Therefore, mechanical properties of the films might be changed with film aging, depending on the rate of thiol oxidation during film drying and storage [90, 113]. The sulfhydril in the cystein amino acid is responsible for the formation of disulphide cross links during oxidation [63]. This is a process which is accelerated when heating the film solution to make films, and it is of major importance for achieving good mechanical properties of the final product [114]. During time of storage, oxidation of not reacted thiol groups and a reorganisation of the intra-molecular disulfide bonds to intermolecular disulfide bonds via thiol-disulfide exchange reactions can occur. This will induce an increase of protein aggregation and a brittleness of the film structure [88].
1.11 STRATEGIES TO ENHANCE FILM PROPERTIES AND
LIFE TIME The attempts to improve the properties of biopolymer films are made in several ways by different researchers. Most of them apply their modifications as ‘pre-treatments’, meaning that the changes occur in the film-forming solution. Some other attempts are performed using ‘post-treatments’ (applied on the film) [101]. Here the most commonly used methods are summarized: Plasticizer
• In response to the problems caused by the use of a low molecular weight plasticizer as glycerol (increase in diffusion of gas and water vapour through the film and migration of the plasticizer), it is possible to replace glycerol with a higher molecular weight compound that has hydrophobic substituents [108, 109]. This may help to decrease WVP and plasticizer migration and hence improve the fracture strain of gluten films [111]. Substances studied as possible replacements to glycerol are amphiphilic substances, including fatty acids (lauric, stearic and oleic acids) [109], octanoic and palmitic acids, dibutyl phthalate and tartrate [115]. Trying to reduce the plasticizer migration, many other studies have been done comparing the effects of various hydrophilic plasticizers (differing in their chain length) on the mechanical properties of protein films [41, 108, 116].
Cross-linking
• Another alternative to improve protein film properties is to modify the polymer network through cross-linking of the polymer chains. The presence of reactive side groups in proteins makes it possible to cross-link the polypeptide chains through chemical, enzymatic, or physical treatments in order to enhance functionality of derived protein films. In several studies proteins have been cross-linked with aldehydes. Some different aldehydes have been used for this

Introduction
18
type of reaction, particularly formaldehyde [117], glutaraldehyde and glyoxal [101, 118-120]. However, although highly reactive, these components have a major disadvantage: their toxicity [37]. This must be taken into account when synthesizing biopolymer materials.
• Enzynmatic cross-linking with transgluminase has been performed with whey protein [75, 121] and wheat gluten [105], with subsequent analyses of the mechanical properties, water vapour transferability, solubility, and hydrolyzability of the resulting films.
• Gamma irradiation as pre-treatment on the film-forming solution has also been applied. Cross-linking induced using gamma irradiation was found to be an effective method for the improvement of both barrier and mechanical properties of some edible films and coatings [122-125]. Another weaker form of electromagnetic radiation is ultraviolet (UV) radiation [101]. This irradiation method has been used by Rhim et al [125] to cross-link protein films.
• Thermal treatment of the solution. Properties of several proteins have been substantially modified by controlled thermal treatments [22, 36, 38, 79, 106, 114]. These studies have shown that heating at elevated temperatures increase the film strength and water resistant properties due to the formation of intermolecular disulfide bonds necessary to form intact film. [23, 36, 41, 77, 113, 126].
Blends
• One extensively used method to enhance the water vapour barrier properties of films have been the incorporation of hydrophobic compounds such as lipids into the film-forming solution [60, 71, 74, 127, 128]. The incorporation of lipid compounds (waxes and fatty acids) helps to limit the moisture migration since the lipids help lessen water vapour transmission and the proteins or polysaccharides give the films strength and thus help improve structural integrity [129]. Various investigations have examined and confirmed the effectiveness of composite edible films with fatty acids (stearic or palmitic acids), paraffin or bee wax, fatty acid esters and carnouba wax [130, 131]. Blends with lipids were still brittle so the addition of a plasticizer was needed [111]. Morillon et al [132] and McHugh et al [128] have published extensive reviews of the factors involved in the water permeability of edible films based on lipid incorporation.
• The still unfavourable properties of biopolymer films prevent any direct use on
the market. However, the use of these films in multilayer packaging seems to be a good alternative. By laminating biopolymer films between other synthetic films their lack of structural integrity and characteristic functionality can be solved [31].
• Preparation of composite films through combined use of compatible
polysaccharides and proteins to improve properties of biopolymers is another way of trying to improve film mechanical properties [133-138].

Introduction
19
• Natural fibre reinforced composites are an attractive research area because natural fibres are eco-friendly, sustainable, inexpensive, have low density, in combination with acceptable mechanical properties and biodegradability. Additionally, these fibres have excellent thermal and sonic insulation properties. Natural fibres from grass, hemp, and ramie have been reported as reinforcements of polymer films [139].
• A fairly new group of composites has emerged, called nanocomposites, in
which the reinforcing material has dimensions of less than or equal to one nanometer. Some examples of nano reinforcements currently coming forward are nano clays, cellulose nanowhiskers, ultrafine titanium dioxide and carbon nanotubes [136].
In this study two different strategies were followed to try to improve the materials properties. First a composite structure with a high crystalline biodegradable material (PCL) was made. Through the second one a nanocomposites clay-gluten system was tried. 1.12 HYBRID COMPOSITE STRUCTURES
1.12.1 Poly(ε-caprolactone) composites
Blending is an important method in polymer manufacturing and has received increasing attention because of the strong economic incentives arising form the use of polymer blends [140]. These blends may combine the advantages of both components and have better properties than either component [141]. Poly(ε-caprolactone), (PCL) is recognised as one of the few synthetic polymers which are biodegradable. PCL is a linear aliphatic polyester, which is approximately 50% crystalline [142]. Some of the reasons for choosing this material for blending with chitosan and whey are: • It is highly compatible with a wide range of polymers [143]. • It is crystalline and hydrophobic, which could help to reduce the water vapour
permeability of chitosan and whey films. • It is completely biodegradable into non-toxic substances so the mixture of this
polymer with chitosan and whey biopolymers would still be biodegradable [142]. • It has good mechanical properties.

Introduction
20
Some properties of PCL are listed in the following table:
Table 1: Physical properties of PCL [144].
Poly(ε-caprolactone)
Density ρ (g/cm3) 1.11 – 1.146
Tensile strength σ (Mpa) 20.7 – 42
Tensile modulus E (Gpa) 0.21 – 0.44
Ultimate strain ε (%) 300 – 1000
Glass transition temperature Tg (°C) -60 – -65
Melting point Tm (°C) 58 – 65
The interesting properties of this polyester have made many researches to use it in blends with natural polymers such as protein [133, 137, 141], cellulose [145], starch [146, 147] and chitin [148]. 1.12.2 Nanoclay nanocomposites
Polymers filled with low amounts of layered silicate, dispersed at nanoscale level, are most promising materials characterized by a combination of chemical, physical and mechanical properties that cannot be obtained with macro- or microscopic dispersions of inorganic fillers [149, 150]. Due to the nanometer-size particles obtained by dispersion, these nanocomposites exhibit distinctly improved mechanical, thermal, optical and physicochemical properties when compared with the pure polymer or conventional (microscale) composites as firstly demonstrated by Toyota researchers, Kojima and co-workers [151] for nylon-clay nanocomposites. The surface of the clay-particles is of major importance. The compatibility between the clay nanoparticle and the matrix decides the ability for dispersion and exfoliation (together with the molecular mobility of the matrix), that in turn decides the final properties of the film. The blocking effect, which also is a function of the total amount of free volume in the penetrating matrix, is important for the final gas transmission properties. Bad compatibility results in a higher free volume in the phase boundary between the montmorillonite particle and the biopolymer film, and poor exfoliation and distribution. If the oxygen diffuse in the high-permeable phase boundary and the montmorillonites are poorly exfoliated, then it will give no improvement [130, 150, 152].

Introduction
21
Morphological descriptors such as intercalation and exfoliation are commonly used to describe the state of aggregation of the individual sheets of clay in the polymer matrix (see Figure 9) [153]. Conventional composites or microcomposites are defined when clay acts as a conventional filler. Intercalated nanocomposites refer to the state where the gallery regions between the aluminosilicate sheets are swollen by polymer chains while retaining the regular stacking arrangement. Delaminated or exfoliated nanocomposites represent the situation when 1 nm-thick layers are completely delaminated and dispersed in the matrix, forming a monolithic structure on the microscale. The latter configuration is of particular interest because it maximises the polymer-clay interactions, making the entire surface of the layers available for the polymer. This should lead to most dramatic changes in mechanical and physical properties [154]. Montmorillonite is one of the most used clays to produce nanocomposites. The layer thickness in momtmorillonite is around 1nm. These layers are organized in stacks with periodic Van der Waals gaps in between them called galleries or interlayers. The compatibility between clay mineral and the organic polymer may be substantially enhanced by replacing the charge balancing interlayer cations with e.g. alkyl ammonium ions. The modified clay then, has lower surface energy and is more compatible with organic polymers. These polymers can be able to intercalate easier within the galleries to exfoliate the nanoclay layers [150].
Figure 9: Scheme of different types of composite arising from the intercalation of layered silicates and polymers: (a) phase separated microcomposite; (b) intercalated nanocomposite;
(c) exfoliated nanocomposite. [150]

Introduction
22
Several strategies have been considered to prepare polymer-layered silicate nanocomposites whereof three are the most used ones: In-situ polymerisation [151, 155-159], melt intercalation [160-162] and solution method [163, 164]. In this study the latter method was used to form gluten-clay nanocomposites. Here the organoclay and the polymer are dissolved in a polar organic solvent. Layered silicates can be easily dispersed due to the weak forces that stack the layers together. The polymer is then adsorbed onto the laminated sheets and when the solvent is evaporated an ordered multilayer structure is formed (see Figure 10).
Figure 10: Schematic sketch of solvent based synthesis. Drawn after [149]

Experimental
23
2 EXPERIMENTAL
2.1 MATERIALS MD Foods Ingredients (Denmark) supplied WPI, LACPRODAN DI-9224. Chitosan (deacetylation degree of 84.7%) of molar mass Mw ≈ 400 000 g·mol-1 was provided as platelets by Fluka Biochemika (Sweden). The vital wheat gluten (WG) powder (84.8 wt% WG proteins, 8.1 wt% wheat starch, 5 wt% water, 1.34 wt% fat and 0.76 wt% ash) was kindly supplied by Reppe AB, Lidköping, Sweden. PCL grade TONE© P-300 (Mw = 10000 g mol-1 and Mw/Mu = 1.7) was supplied as granules from Union Carbide USA. Two types of commercial nanoclays were used: Cloisite®Na+ and Cloisite®10A, obtained from Southern Clay Products Inc. (Texas, USA). Cloisite®Na+ is a natural sodium-rich montmorillonite. Cloisite®10A is a natural montmorillonite modified with a quaternary ammonium salt. Chloroform (tri-chloro-methane stabilised with 1% ethanol) was purchased from Lab Scan (Sweden). The de-ionised water was prepared in a Miele Aqua Purificator G7749. Acetic acid (glacial 100%) was purchased from MERCK (Germany). Teflon© coated aluminium foil, Bytac© Type AF-21, was supplied by Norton Performance Plastics Corporation. Glycerol with a purity of 99.5% was supplied by Karlshamns Tefac AB, Karlshamn, Sweden. The ethanol (95%) was obtained from Kemetyl AB, Sweden. Sodium hydroxide was supplied by Sigma. Aldrich Chemie GmbH, Germany. Iodine, resublimed GR for analysis, was supplied by Merck KGaA, Germany. 2.2 FILM FORMATION All chitosan, whey, wheat gluten and composite films were produced by solution casting method (see Figure 11). 2.2.1 WPI films
Whey films were produced by first stirring an aqueous solution of 12%(w/w) whey and 6%(w/w) glycerol at room temperature. The solution was kept at 73°C for 20 min in order to denaturalise the protein. The solution was decanted into petri dishes covered by Teflon coated aluminium (20g in each) and subsequently dried for 48h at room temperature and 30% relative humidity.
Figure 11: Film casting technique
2.2.2 Chitosan films
Chitosan, though insoluble in water, does dissolve in aqueous organic acids to give viscous solutions at a pH below 6.5. So acetic acid was used to protonise the chitosan molecule (-NH2 -NH3
+) in order to make the polymer soluble in water and make it
Solution
Teflon coated aluminium
FILM CASTING

Experimental
24
possible to cast and dry the solution into transparent polymer films. First 1% (w/w) chitosan and 1% acetic acid were dissolved in water during high-speed stirring. Stirring was continued for approximately 30 min, until all the chitosan was dissolved. To ensure homogeneity the solution was mixed in a blender for 4 min. The solution was then kept untouched for 2h before it was decanted into petri dishes covered by Teflon© coated aluminium. Approximately 20 g of solution was poured into each petri dish. The films were dried for 48 h at room temperature and 30% relative humidity. 2.2.3 PCL/Chitosan films
An aqueous solution containing 1%(w/w) chitosan and 1% acetic acid and a chloroform solution containing 10%(w/w) PCL were each decanted into separate glass tubes. Both solutions were mixed with a magnetic stirrer for 20 minutes. The chitosan solution was then mixed in a blender for 4 min. Then both solutions were put together and mixed for another 4 min. The new solution was kept untouched for 30 min. Then it was dried in a vacuum oven for 10 min before it was decanted into petri dishes covered with Teflon© coated aluminium foil. Films were subsequently obtained after the solution had dried for 24h at 23°C and 30%RH. Blends were prepared with 5, 10 and 15% by weight of PCL with respect to the chitosan added. 2.2.4 PCL/WPI films
The WPI and the PCL solutions were made separately as described above. The solutions were separately stirred until homogeneous solutions were obtained and they were then mixed together in a blender for 4 min. The new solution was cooled to room temperature. Subsequently a layer of foam was removed from the surface and then the solution was heated to 60°C for 20 min. After being cooled to room temperature, the solution was dried in a vacuum oven for 10 min before it was decanted into petri dishes covered with Teflon© coated aluminium at 23°C and 30%RH. Blends were prepared with 5, 10 and 15% by weight of PCL with respect to the WPI added. 2.2.5 Gluten films
The films were produced by first stirring 30 g WG powder and 9.9 g glycerol in 135 g ethanol. After homogeneity was attained, 90 g of deionised water was added slowly. Solutions with pH=4 or pH=11 was obtained by adding, respectively, acetic acid or sodium hydroxide. The solutions were thereafter heated to 75°C during 20 min (2.5 °C/min) and kept at that temperature for 10 minutes under gentle stirring. 300-400 µm-thick films were obtained by pouring the solution into petri dishes and allowing it to dry for 2 days at 23°C and 50%RH. The petri dishes were coated with a release agent layer of polytetrafluoroethylene supported by an aluminium foil (Bytac Type AF-21; Norton Performance Plastics Corp., Wayne, NJ). Films were conditioned in a climate chamber at 23°C and 50% RH for at least 48 h and then placed with the same side facing up on a porous paper support (Blotting Paper 1600, 220 diam. from VWR International).

Experimental
25
2.2.6 Gluten/MMT films
The gluten/MMT films were produced following the method shown in Figure 12. 18 g of WG powder and 6 g of glycerol were stirred in 90 g of ethanol until a homogeneous solution was obtained. Then 60 g deionised water was added slowly. Consequently the mass ratio of water and ethanol was 1:1.5. Solutions with pH=4 and pH=11 were obtained by adding, respectively, acetic acid and sodium hydroxide. The solutions were thereafter heated to 75°C during 20 min (2.5 °C/min). Ethanol solutions with 0.75, 1.5, 3 and 4.5 wt.% natural and modified montmorillonite were prepared. These were ultrasonicated for three minutes, in an Elma Transsonic T420 apparatus. Subsequently the solutions were poured into the 70°C gluten solution and then stirred for 10 minutes. 300-400 µm-thick films were obtained by pouring the solutions into petri dishes and after drying for two days at 23°C and 50%RH. The petri dishes were coated with a release agent layer of polytetrafluoroethylene supported by an aluminium foil (Bytac Type AF-21; Norton Performance Plastics Corp., Wayne, NJ). Films were conditioned in an environmental chamber at 23°C and 50% RH for at least 48 h prior to testing.
Figure 12: Flow chart presenting the different steps of the solution approach.[154]
2.3 METHODS AND INSTRUMENTS 2.3.1 Permeability
Oxygen permeability test
The oxygen permeability of the films, at 23°C, was determined, according to ASTM D 3985-95, by using a Mocon OX-TRAN TWIN that was equipped with a coulometric oxygen sensor. The specimens, tightly sandwiched between two pieces of aluminium foil (see Figure 13) had a circular active area of 5 cm2. These films were mounted in isolated diffusion cells and subsequently purged with nitrogen gas (2% hydrogen) to obtain the background oxygen leakage. After background correction, the specimen was exposed to flowing oxygen gas (99.95%) at atmospheric pressure.
Figure 13: Polymer sample glued together in between two alumin-ium foils

Experimental
26
Measurements were interrupted when steady-state was attained. The oxygen transmission rate was normalized with respect to the oxygen pressure and the film thickness to yield the oxygen permeability (OP). Water vapour permeability test The water vapour transmission rate (WVTR) of each sample was measured using a Mocon Permatran-W Twin (Minneapolis, MN) according to ASTM F 1249-90. Specimens were tightly sandwiched between two pieces of aluminium foil leaving a 5 cm2 exposure area for the WVTR measurements (see Figure 13). Water vapour permeabilities were evaluated for the films by applying a small RH gradient (11%) across film samples. As explained by Gennadios et al (1990) [23], this low RH was used in order to avoid swelling and non-Fickian behaviour of the films. Before the measurements, the specimens were conditioned in isolated diffusion cells with one side in contact with an atmosphere of a saturated LiCl-solution (giving 11% RH) and the other side facing dry nitrogen gas. The WVTR was normalized with respect to the water vapour pressure and the film thickness to yield the water vapour permeability (WVP). 2.3.2 Microcalorimetry
A microcalorimeter system (2277 Thermal Activity Monitoring, Thermometric AB, Sweden) of the heat conduction type equipped with a 4 ml stainless steel RH perfusion ampoule, referred to as TAM, was used for the measurements. The RH perfusion ampoule was designed to humidify a gas to a specified relative humidity by mixing a flow of dry gas with a flow of gas of 100% relative humidity. A closed and empty 4-ml stainless steel ampoule was used on the reference side. The film sample was tightly fastened onto the upper part of the steel ampoule by a stainless steel ring that was anchored using a screw. In the present case water was added to the lower compartment (inside the steel ampoule). The measurement principle is based on the assumption that there is an equilibrium exchange of water molecules between the liquid, the lower compartment, the film and the upper compartment at steady-state. It is also assumed that the heat of condensation at the lower film surface is the same as the heat of evaporation at the upper film surface and that the heat of solution and desolution of water in the film are included in these terms. The calorimetric response at steady-state measured by the microcalorimeter is the heat flow due to evaporation of water from the liquid. The water vapour transmission rate at steady-state through the film, Qo, is given by Eq. 1:
pHtMP
Qv
e
∆⋅Α⋅∆⋅⋅
=ο
Eq. 1: The water vapour transmission rate at steady-state
Where Pe is the heat flow rate, M is the molar mass of water and t is the film thickness. A is the exposed surface area of the film (A = 0.2827 cm2) and ∆p is the water vapour pressure difference across the film. The heat of water evaporation from the liquid, ∆Hv, was measured for pure water by first putting 0.4 g of water into a steel ampoule with an inner diameter of 6 mm, which in turn was inserted into the perfusion ampoule. Before insertion, the water contained in the steel ampoule was weighed. After

Experimental
27
approximately 15 h, the perfusion ampoule was withdrawn and weighed and the heat of evaporation calculated. In order to achieve a relative humidity of 11% in the lower compartment, a solution saturated with LiCl crystals was used instead of pure water. The heat of evaporation of water in this case was obtained by a procedure similar to that for pure water. 2.3.3 Tensile test
The stress-strain properties of chitosan and whey films were obtained at 23°C (50% RH) according to SCAN-P 38:80 (34) using an Alwetron TCT 10 tensile tester. Three samples with a length of 50 mm and a width of 15 mm were tested for each blend. The thickness of each specimen was taken as the average of five readings. The strain rate was 100 mm/min and. The clamped length of the specimen was 40mm. The WG films were tensile tested at 50% RH and 23°C, using a ZwickZ010 tensile tester controlled by testXpert 7.1® computer program supplied from Zwick GmbH & Co, Germany. Dumpbell shaped specimens were punched out from the films with a length and width of the narrow section of 16.0±1.0 mm and 4.0±0.1 mm, respectively (ISO 37:1994(E)). The measurements were performed as described in ASTM 882-01 with a crosshead speed of 100 mm/min and a clamp distance of 40 mm. 10 replicates of each sample were measured. The tensile strength was defined as the maximum stress in the nominal stress-strain curve. 2.3.4 Content of volatile mass
The loss of volatile mass of gluten films was measured as described in ASTM D 644-94. Prior to testing the samples were conditioned in a room at 50%RH and 23C. The test pieces were weighed and then dried for 24 h at 105°C in a Nûve FN400 oven (LabRum Klimat AB, Sweden). The specimens were subsequently cooled in desiccators at 0% RH and 23°C and then weighed to determine the loss of volatile mass. Three replicates from each sample were measured. 2.3.5 Microscopy
Scanning Electron Microscopy Scanning electron microscopy was performed on fractured cross-sections and surface of gold/palladium (60%/40%) sputtered chitosan, whey and gluten composite specimens, using a JEOL JSM-5400 scanning electron microscope. Optical Microscopy Starch turns deep blue (almost black) in the presence of a small amount of iodine vapour. This reaction serves as a test to check the presence of starch in the surface of the gluten films. Iodine powder was dispersed in the bottom of a hermetically closed recipient. The WG films were hanged inside the recipient in order to expose the entire surface to the sublimed iodine. The samples were kept in iodine vapour for 24 hours.

Experimental
28
The structure of the surfaces was analysed with a Leitz Ortholux POL BK II optical microscope. 2.3.6 Water uptake
Small whey and chitosan test specimens were cut and put on aluminium foils. The specimens were dried in a vacuum oven for two days and then weighed. After that they were stored in a chamber at 11%RH and 23°C for two days and then they were weighed again. The water uptake was obtained as the percentage of gained weight. 2.3.7 Spectroscopy
UV/VIS spectroscopy
A Hewlett-Packard 8415A Diode Array spectrometer was used to measure the light absorption in whey and chitosan films. IR spectroscopy
Fourier transform infrared spectra (12 scans) of gluten films were recorded on a Perkin Elmer Spectrum 2000 FTIR spectrometer equipped with a normal single reflection ATR accessory (Gloden Gate) from Specac Ltd. (Kent, England). 2.3.8 Protein solubility
A biochemical control parameter of the changes in the gluten polymer network is the percentage of protein, which is insoluble in non-ionic detergents such as sodium dodecyl sulphate (SDS). The percentage of SDS-insoluble protein is a well established quantity, which reflects the covalent cross-linking degree of the protein network [165, 166] Amount and size distribution of proteins extractable with sodium dodecyl sulfate (SDS) and SDS+sonication of cast films were obtained using size-exclusion high performance liquid chromatography (SE-HPLC), together with a three-step extraction procedure according to Gällstedt et al [63]. Proteins soluble in dilute SDS were extracted in the first step and additional proteins were extracted with repeated sonications [63]. The new films were analyzed approximately 6 days after being prepared. Aged films were at least 120 days old. In the first step, 16.5 mg of each film were suspended in 1.5 ml 0.5% SDS-phosphate buffer (pH 6.9) and vortexed for 10 s. The suspension was then stirred for 5 min at 2000 rpm and centrifuged for 30 min at 10000 rpm to obtain the supernatant protein. In the second step, the pellet was re-suspended in the SDS buffer and sonicated in an ultrasonic disintegrator (Soniprep 150, Tamro, Mölndal, Sweden) for 30 s, amplitude 5, fitted with a 3 mm exponential microtip. The samples were then centrifuged (30 min, 10000 rpm) to obtain a supernatant of proteins. For the third step, the pellet was again re-suspended in the SDS buffer and sonicated as above for 30+60+60s. The extracts were filtered through 0.45 mm filters (Millipore, Durapore Membrane Filters) before the SE-HPLC operation. SE-HPLC analyses were performed on a Waters HPLC system using a BIOSEP SEC-4000 Phenomenex column. Separation was

Experimental
29
obtained after 30 min by loading 20 µl of sample into an eluant of 50% (v/v) acetonitrile and water containing 0.1% (v/v) trifluoroacetic acid at a flow rate of 0.2 ml min-1. Proteins were detected by UV absorption at 210 nm. The amount of extracted proteins after each of the extraction steps were normalized the protein solubility of unprocessed WG powder exposed to the third extraction step [63]. The SE-HPLC chromatograms were divided into polymeric and monomeric fractions (Johansson et al 2001) and the protein polymer/monomer ratio was calculated [63]. 2.3.9 Characterization of Polymer-Clay Nanocomposites
X-ray Diffraction (XRD) X-ray diffractograms of films were obtained in a Siemens D5000 diffractometer with Cu radiation (50 kV, 40 mA). The scanning speed and the step size were 0.15°/min and 0.02°. The specimen rotation speed was 15 rpm. Transmission Electron Microscopy (TEM) Specimen preparation constitutes a major difficulty in transmission electron microscopy analysis. TEM specimens need to be sufficient thin in order to optimise the so-called mass thickness contrast. One way to prepare TEM specimen from polymer samples is ultramicrotomy. Samples were embedded in epoxy and curated at 60 degrees during 24 hours. Then, using an RMC ultramicrotome, 50 nm thick sections were obtained at cryogenic temperature (-80 or -120°C) (Figure 14). After heating to room temperature the sections were removed from the knife using a grid with a water droplet.
Figure 14: Ultramicrotomy - the sample is moved across a knife-edge (diamond). [154]
The grids were then transferred to the transmission electron microscope (TEM) Philips Tecnai 10 operating at 80 kV for observation of the clay-polymer structure. 2.3.10 Packaging related tests
Printability A red ethanol-based dye was applied as a single straight line on the surface of the films. After intervals of 2s and 30s, a dried poly(methyl methacrylate) plate was drawn over the dye line to smear the line out. The distance of the smearing was measured. The

Experimental
30
shape and amount of dye left on the dye line after this ‘draw-down’ procedure were analysed qualitatively. Seal strength Measurements Films (thickness = 0.1-0.3mm) were welded in a Multivac A300 during 10 consecutive 0.2 s heat pulses. The seal strength of the welded films was determined in an Alwetron TCT10 tensile tester at 23±2°C (50% relative humidity) and 100 mm/min strain rate. The clamped length of the specimen was 40 mm. Transparency The transparency of the films was assessed by measuring the distance (L) between the specimen (polymer film) and a letter, at which the letter became invisible when viewed through the specimen. The letter had a thickness of 300 µm and was printed with black ink on white paper. The distance between the polymer film and the observer was 40 cm. Flexural Stiffness The stiffness of the blends was obtained by the standard test according to SCAN-P 29:95 using a Lorentzen and Wettre stiffness tester. The force to bend the specimen through an angle of 7.5°C was monitored. The specimen had the dimensions 55mm x 40 mm (clamp width) and the distance between the clamp and the point where the force was applied was 10mm. The samples were conditioned at 23°C and 50% RH for 3 h before the measurements. The stiffness index (Nm2/g) was obtained from the force (F) and the density (w) of the specimen: Si = F/w3. The stiffness index was calculated as the average of 3-4 specimens. Folding Endurance Test The folding test was performed according to SCAN-P 17:77 using a Lorentzen and Wettre folding endurance tester. Specimens preconditioned at 23°C for 24h at 50%RH (chitosan blends) and 25%RH (WPI blends) were subjected to cyclic folding through 180° until fracture occurred. Specimens were fixed in vibrating clamps at one end and the vibrational amplitude caused imposed folding of the specimen. The 10-logarithm of the number of cycles until fracture (log(Nf)) was calculated and averaged over 5 samples.

Results and discussion
31
3 RESULTS AND DISCUSSION
3.1 BIOPOLYMER/PCL BLENDS In this section the results of the study of chitosan and whey protein films blended with polycaprolactone are discussed. Attempts to find a procedure to blend PCL with chitosan and whey are reported and mechanical and barrier properties of these films are discussed. 3.1.1 Oxygen permeability
The oxygen permeability was measured in pure WPI and chitosan films and in some blends of chitosan. Table 2: Comparison between oxygen permeability values in this study and earlier studies.
Film Conditions OP (cc µm/m2 day kPa) Ref.
Chitosan 25°C, 0%RH 0.325 ± 0.14 this study
Chitosan PCL5% 25°C, 0%RH 0.10 ± 0.05 this study
Chitosan PCL10% 25°C, 0%RH 0.04 ± 0.04 this study
Chitosan PCL15% 25°C, 0%RH 0.27 ± 0.11 this study
WPI 25°C, 0%RH 0.28 ± 0.15 this study
Chitosan unspecified 0.708 [167] Chitosan (plasticized) 25°C, 0%RH 0.46±0.33 [55]
Chitosan (unplasticised)
unspecified 3.3±0.1 [60]
WPI: glycerol (5.7:1) 23°C, 50%RH 18.5 [41] WPI: glycerol (2.3:1) 23°C, 50%RH 76.1 [41] LDPE 23°C, 50%RH 1865 [30] HDPE 23°C, 50%RH 427 [30] EVOH (70%VOH) 23°C, 0%RH 0.1 [30] EVOH (70%VOH) 23°C, 95%RH 12 [30] PVDC based films 23°C, 50%RH 0.4-5 [30] It was proved that chitosan and whey (at low relative humidity) had a lower oxygen permeability than that of poly(vinylidiene chloride), and similar to that of dry ethylene vinyl alcohol copolymers, which are commercial polymer films commonly used as high oxygen barriers. Film permeabilities were much lower than values reported by Muzzarelli in 1974 (0.708 cc µm/m2 day kPa) [167] and Wong in 1992 (3.3 cc µm/m2 day kPa) [60], but the conditions in these studies were not specified. The values were in reasonable agreement

Results and discussion
32
with the data obtained by Butler [55], where the values are higher but of the same magnitude. In this case it should be noted that the chitosan was plasticized, which could be an explanation to the higher permeability to oxygen. PCL lowered the values of oxygen permeability even if there was no relation found between the percentage of PCL and the permeability decrease. Thus, it was found that the gas barrier properties of these materials were very good in dry conditions. The oxygen barrier properties of these edible polymer films indicate good potential as food packaging supplements. 3.1.2 Water vapour permeability
Pure chitosan and WPI water vapour permeability values were compared and found in agreement with former research studies. (To compare the data a unit conversion was done. This took into account that PH2O [11%RH, 24.8°C] = 0.00346 atm. Table 3: Comparison between WVP-values for pure chitosan and WPI films in this study and earlier studies.
Chitosan films
Composition WVP
(g/m day atm) Conditions Reference
Chitosan 0.135 24.8° C, 0-11 %RH this study Chitosan plasticized 0.18-0.34 25° C, 11-50%RH [55] Chitosan 0.315 23° C, 0-50%RH [60] WPI: glycerol (2:1) 0.723 24.8° C 0-11%RH this study WPI: glycerol (15:1) 4.59 26.3°C 0-88%RH [71] WPI: glycerol (2.3:1) 9.33 25°C 0-77%RH [48] WPI: glycerol (1.6:1) 12.13 25° C 0-65.1%RH [77] The values confirmed the influence of the plasticizer, temperature and relative humidity.
When adding PCL to the films a substantial decrease of the water vapour permeability was obtained, as shown in Table 4.
Table 4: Water vapour permeability for blends with different percentage of PCL and at 11%RH and 24,8 °C.
Permeability (g.µm/m2.day) PCL (%)
Chitosan films WPI films
0 469.44 2513.58 5 160.82 2065.86
10 110.40 1774.76 15 46.70 813.46

Results and discussion
33
Chitosan blends with a volume fraction of 18.3 vol% (15 wt%) of PCL particles exhibited a 90% decrease in water vapour permeability compared with the pure chitosan films. WPI blends with 16.8 vol% (15 wt%) PCL showed a 70% decrease in water vapour permeability compared to pure WPI. Maxwell derived a relationship for predicting the electrical conductivity of a system of low conducting spheres in a high-conducting matrix [168]. The assumption in this derivation was that the content of spheres is low enough to ensure that no direct contact exist between neighbouring spheres. Considering the physical similarity between solute permeability and electrical conductivity, it was possible to apply the Maxwell equation to this study. Maxwell‘s equation was used to predict data that describe the impedance of a heterogeneous system of dispersed low permeable spheres (PCL) in a permeable matrix.
( )( )⎥
⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢
⎣
⎡
−−−+
−+=
m
mm
QQυ
υ
11213
1
Eq. 2: Maxwell’s equation
Here q is the ratio of the water vapour transmission rate of PCL to that of chitosan/WPI. The water transmission rate of PCL is 0.069 (g.µm/m2.day) at 100% RH, hence very small compared to the values of chitosan and WPI. Thus, for this study q can be neglected. νm is the relative amount of permeable matrix (chitosan or WPI) in the blend. SEM pictures (Figure 15 and Figure 16) showed that a great amount of the PCL particles have ellipsoidal shape in chitosan and long shape in WPI films instead of spherical shape as supposed in the Maxwell equation. The PCL particles were small (in the order of 1-10 µm) indicating good miscibility (compatibility) between the two components.
Figure 15: Chitosan SEM micrograph showing the morphology of the cross section surface of specimen containing 15-wt% PCL particles in the chitosan matrix
Figure 16: Whey SEM micrograph showing the morphology of the surface of specimen containing 5-wt% PCL particles in the WPI matrix

Results and discussion
34
As showed in Figure 19 and Figure 21 the resulting Maxwell curves don’t fit the data obtained in this study.
Ellipsoidal particles, especially if they are oriented parallel to the film surface, are more effective than spherical particles in decreasing the permeability. This is because the solute diffusion path (tortuosity) is increasing with the width-to-thickness ratio of the particles.
Figure 17: Tortuosity of the permeant particle across the film with hydrophobic PCL particles.
Figure 18: Schematic diagram of the PCL particle
Thus, the model of Fricke (see Eq. 3) was applied to the same system, because in this model the low conducting particles can also have ellipsoidal shapes [169]. Again, the similarity between electrical conductivity and solute permeability was taken into account, making it possible to use the model here. It was considered that the PCL particles were the low-conducting ellipsoids and that the chitosan and the WPI particles were the conducting or permeable matrix:
Eq. 3: Frickes equation
The parameter k1 and k2 are described as:
and
where w is the thickness and t is the width of the less permeable particles (PCL). C∞,d and C∞,m are the water saturation concentrations in the dispersed and matrix components. In this case the ratio of water saturation concentration in Eq. 3 is close to zero and it simplifies to:
w
t
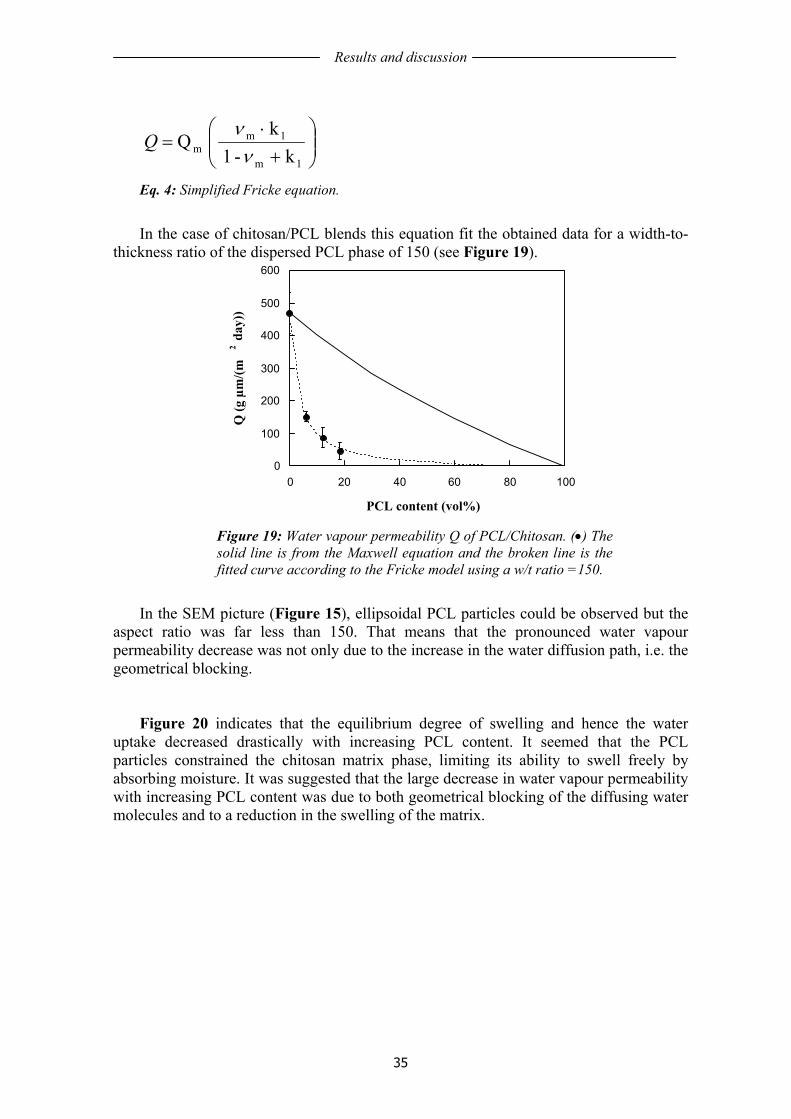
Results and discussion
35
⎟⎟⎠
⎞⎜⎜⎝
⎛+
⋅=
1m
1mm k -1
k Q
νν
Q
Eq. 4: Simplified Fricke equation.
In the case of chitosan/PCL blends this equation fit the obtained data for a width-to-thickness ratio of the dispersed PCL phase of 150 (see Figure 19).
0
100
200
300
400
500
600
0 20 40 60 80 100
Q (g
µm
/(m2 d
ay))
PCL content (vol%) Figure 19: Water vapour permeability Q of PCL/Chitosan. (•) The solid line is from the Maxwell equation and the broken line is the fitted curve according to the Fricke model using a w/t ratio =150.
In the SEM picture (Figure 15), ellipsoidal PCL particles could be observed but the aspect ratio was far less than 150. That means that the pronounced water vapour permeability decrease was not only due to the increase in the water diffusion path, i.e. the geometrical blocking. Figure 20 indicates that the equilibrium degree of swelling and hence the water uptake decreased drastically with increasing PCL content. It seemed that the PCL particles constrained the chitosan matrix phase, limiting its ability to swell freely by absorbing moisture. It was suggested that the large decrease in water vapour permeability with increasing PCL content was due to both geometrical blocking of the diffusing water molecules and to a reduction in the swelling of the matrix.
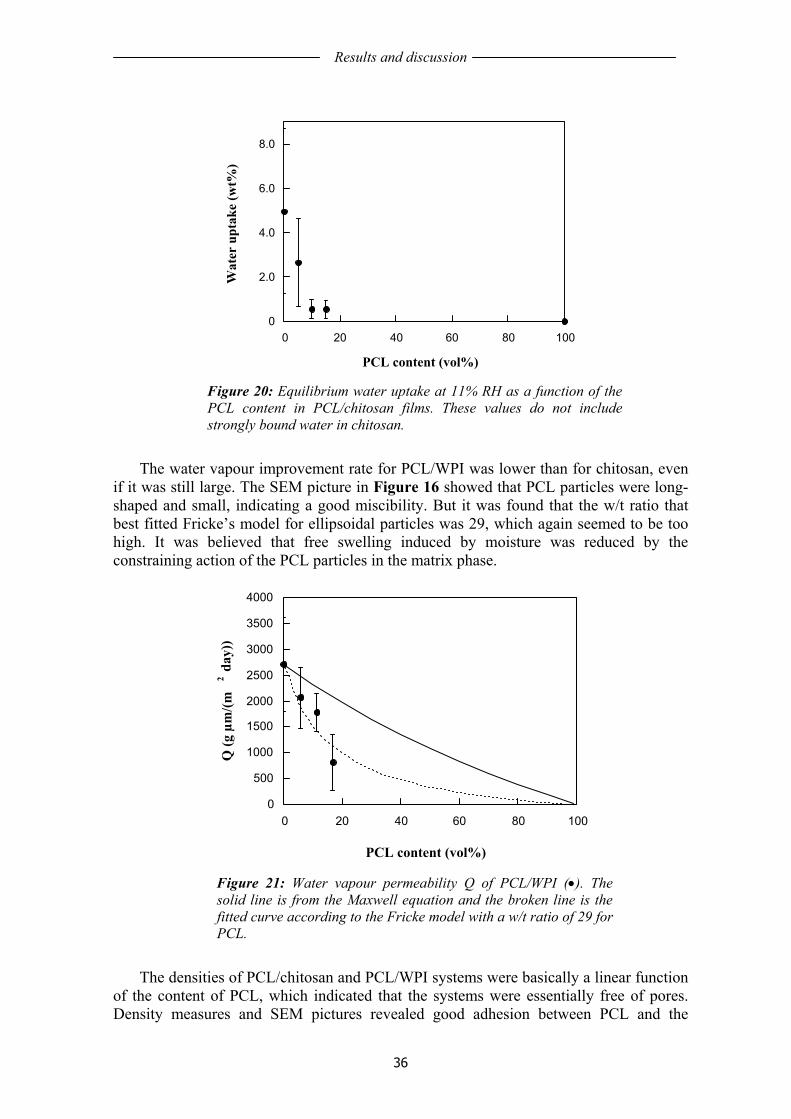
Results and discussion
36
0
2.0
4.0
6.0
8.0
0 20 40 60 80 100
Wat
er u
ptak
e (w
t%)
PCL content (vol%) Figure 20: Equilibrium water uptake at 11% RH as a function of the PCL content in PCL/chitosan films. These values do not include strongly bound water in chitosan.
The water vapour improvement rate for PCL/WPI was lower than for chitosan, even if it was still large. The SEM picture in Figure 16 showed that PCL particles were long-shaped and small, indicating a good miscibility. But it was found that the w/t ratio that best fitted Fricke’s model for ellipsoidal particles was 29, which again seemed to be too high. It was believed that free swelling induced by moisture was reduced by the constraining action of the PCL particles in the matrix phase.
0
500
1000
1500
2000
2500
3000
3500
4000
0 20 40 60 80 100
Q (g
µm
/(m2 d
ay))
PCL content (vol%) Figure 21: Water vapour permeability Q of PCL/WPI (•). The solid line is from the Maxwell equation and the broken line is the fitted curve according to the Fricke model with a w/t ratio of 29 for PCL.
The densities of PCL/chitosan and PCL/WPI systems were basically a linear function of the content of PCL, which indicated that the systems were essentially free of pores. Density measures and SEM pictures revealed good adhesion between PCL and the

Results and discussion
37
matrices. Blends with a volume fraction of 18.3% PCL particles lowered the water vapour transmission rate of chitosan by 90%. 16.8% vol% PCL particles lowered the water vapour permeability of WPI by 70%. 3.1.3 Mechanical properties
The tensile test that was performed on pure materials lead to the following results: Table 5: Mechanical properties of pure chitosan and WPI films.
Yield stress (MPa)
Yield strain (%)
Young’s modulus (MPa)
Fracture stress (MPa)
Fracture strain (%)
Chitosan 59.703 3.6 1657.6 67.4 12.5
WPI 1.196 4 31.271 2.165 100 WPI data seemed to be in agreement with earlier studies [42, 64, 74], even though the values of this study were a bit lower, which can be due to different drying or testing conditions. When comparing the two materials it can be said that WPI is tougher but weaker than chitosan. In PCL/chitosan blends, the fracture stress decreased linearly with increasing PCL content, as shown in Figure 22. This was also confirmed by the strain values that decreased only mildly with increasing PCL content (Figure 23).
0
10
20
30
40
50
60
70
80
0 20 40 60 80 100
Nom
inal
stre
ss (M
Pa)
PCL content (vol%) Figure 22: Yield stress (•) and fracture stress (o) for PCL/ chitosan blends. The line is drawn to show the linearity.
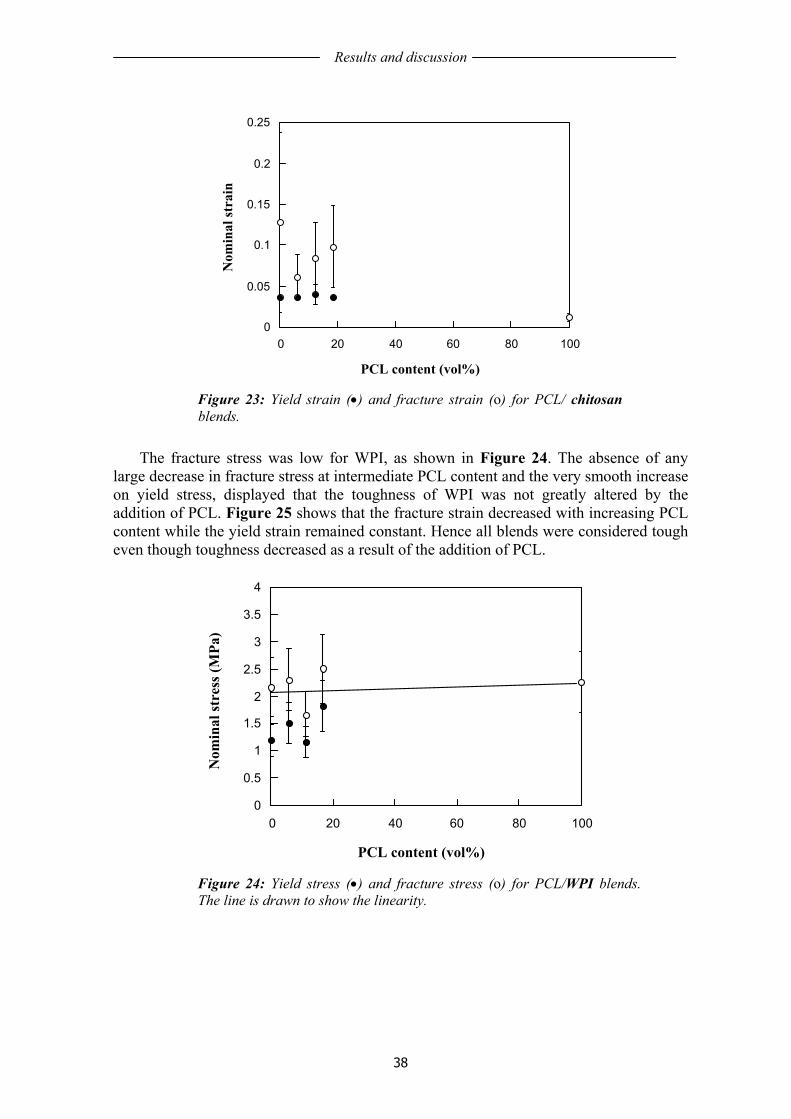
Results and discussion
38
0
0.05
0.1
0.15
0.2
0.25
0 20 40 60 80 100
Nom
inal
stra
in
PCL content (vol%) Figure 23: Yield strain (•) and fracture strain (o) for PCL/ chitosan blends.
The fracture stress was low for WPI, as shown in Figure 24. The absence of any large decrease in fracture stress at intermediate PCL content and the very smooth increase on yield stress, displayed that the toughness of WPI was not greatly altered by the addition of PCL. Figure 25 shows that the fracture strain decreased with increasing PCL content while the yield strain remained constant. Hence all blends were considered tough even though toughness decreased as a result of the addition of PCL.
0
0.5
1
1.5
2
2.5
3
3.5
4
0 20 40 60 80 100
PCL content (vol%)
Nom
inal
stre
ss (M
Pa)
Figure 24: Yield stress (•) and fracture stress (o) for PCL/WPI blends. The line is drawn to show the linearity.
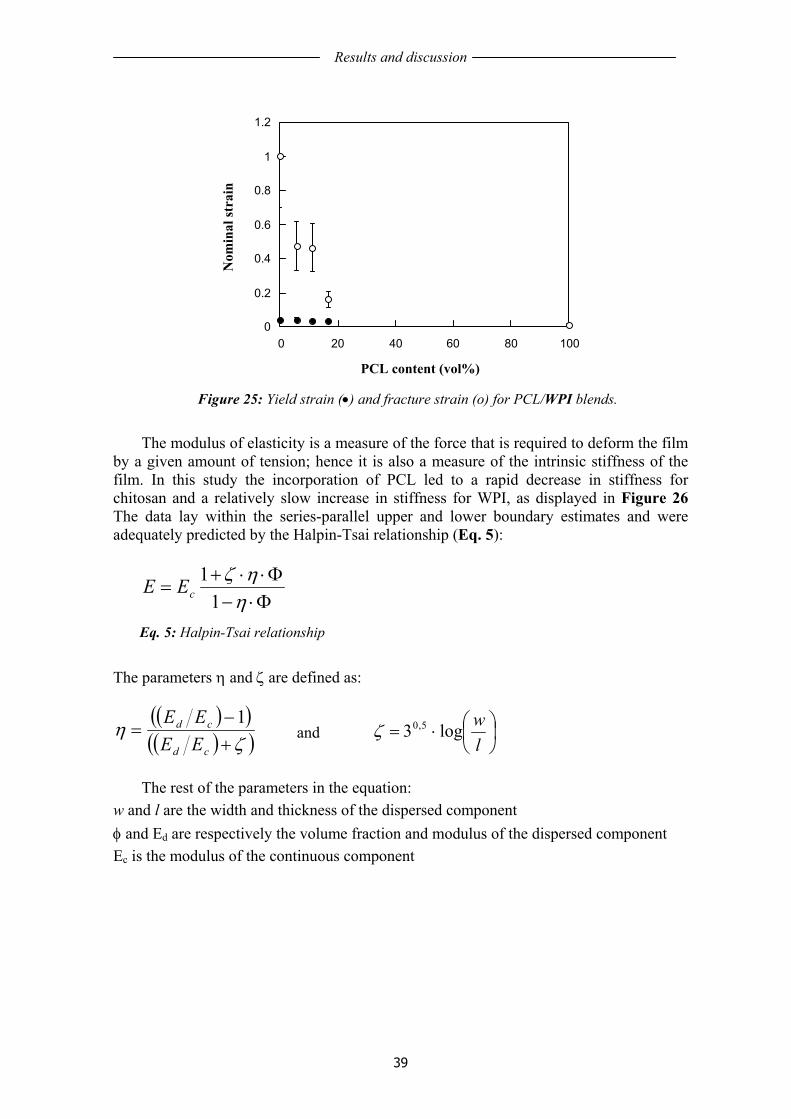
Results and discussion
39
0
0.2
0.4
0.6
0.8
1
1.2
0 20 40 60 80 100
Nom
inal
stra
in
PCL content (vol%) Figure 25: Yield strain (•) and fracture strain (o) for PCL/WPI blends.
The modulus of elasticity is a measure of the force that is required to deform the film by a given amount of tension; hence it is also a measure of the intrinsic stiffness of the film. In this study the incorporation of PCL led to a rapid decrease in stiffness for chitosan and a relatively slow increase in stiffness for WPI, as displayed in Figure 26 The data lay within the series-parallel upper and lower boundary estimates and were adequately predicted by the Halpin-Tsai relationship (Eq. 5):
Φ⋅−Φ⋅⋅+
=η
ηζ1
1cEE
Eq. 5: Halpin-Tsai relationship
The parameters η and ζ are defined as:
( )( )( )( )ζ
η+−
=cd
cd
EEEE 1
and ⎟
⎠⎞
⎜⎝⎛⋅=
lwlog3 5,0ζ
The rest of the parameters in the equation: w and l are the width and thickness of the dispersed component φ and Ed are respectively the volume fraction and modulus of the dispersed component Ec is the modulus of the continuous component

Results and discussion
40
0
500
1000
1500
2000
0
50
100
150
200
0 20 40 60 80 100
You
ng's
mod
ulus
(MPa
)
PCL content (vol%)
Young's m
odulus (MPa)
Figure 26: Modulus for PCL/chitosan blends (•) and PCL/WPI blends (o). The solid line represents best fits of the Halpin Tsai-model for a width-to-thickness ratio of 1.5 (PCL/ chitosan) and 50 (PCL/WPI). The dashed lines are upper and lower boundary estimates.
The best fit of the PCL/chitosan data yielded an average width-to-thickness ratio of 1.5 for the dispersed PCL phase. This is in agreement with the SEM picture (Figure 15). The width-to-thickness ratio of the PCL domains in PCL/WPI was found to be approximately 50. This value seems to be rather high, but it may be explained by the fact that the PCL domains are long (Figure 16). The Halpin-Tsai model predicts a value of unity for ζ, clearly inconsistent with the moduli data. Lewis and Nielsen modified the Halpin-Tsai model and using their model it was possible to obtain a perfect fit for PCL/WPI as shown in Figure 26. The modified model yielded a fibre length-to-diameter ratio of 4-6. 3.1.4 Other packaging related properties
The sealability of a packaging is one of the most important properties when considering its use on wrapping or bag making equipment and, of course, the integrity of the seal is also important to the ultimate package. It was found that it was impossible to seal chitosan and its PCL blends with standard heat sealing equipment. Thus, sealing chitosan requires alternative techniques. WPI and the WPI blends were readily sealable, but the seals were brittle. The fractures, when the tensile tests were performed, occurred at the seal lines. The seal strength of the PCL/WPI blends decreased with increasing PCL content. For packaging materials it is also essential to know the bending stiffness of the material, this is done by measuring the stiffness index. The stiffness can be considered as the film’s resistance to distortion and, in particular, to bending. Young’s modulus is one measure of the inherent stiffness of a film. The stiffness index appeared to be much higher for chitosan than for WPI, in agreement with the Young’s modulus values that were obtained. The addition of PCL to chitosan reduced the bending stiffness, as was expected considering the tensile modulus data.

Results and discussion
41
The resistance to repeated flexure or creasing is important for the use of the films. In essence, the resistance to flexing was measured by repeated folding of the films backwards and forward, at a given pace. The number of cycles until failure was recorded as the flex or folding resistance. Interestingly, the resistance to repeated folding was lower for WPI than for chitosan, whereas the strain data (Figure 23 and Figure 25) showed that WPI was the tougher material. It seems that the fatigue properties of WPI, which were measured indirectly through the folding test, were inferior to those of chitosan. Even if the addition of PCL reduced the bending stiffness of chitosan, the log(Nf) in the folding test was not lowered, which is promising for future packaging applications. The effects of adding PCL to WPI were different. In agreement with data for modulus and fracture strain presented earlier, the bending stiffness increased and log(Nf) decreased with the addition of PCL. Regarding the printing properties, it was proved that a red dye on pure chitosan and on pure WPI yielded a solid thick line. The red dye was absorbed more rapidly in WPI than in chitosan and the general quality of the print was better on WPI. The impact of PCL on the printability of WPI was negligible. These results therefore suggested that PCL was not present on the surface of the WPI blends. Transparency is an often-desired property for food or medical packaging materials. The optical clarity of chitosan was higher than that of WPI. WPI contained globular particles and a ‘wavy’ surface that decreased the optical clarity. The optical clarity of the chitosan blends was not affected below 5% PCL. Above 5%, the transparency decreased and the blends became more opaque. However, transparency was acceptable even for films with 15 wt% PCL. The uniform colour suggested a homogeneous dispersion of the PCL phase. As regards WPI the transparency also decreased gradually with increasing PCL content and here the samples turned from beige to white. The homogeneous colour suggested that the PCL particles were uniformly distributed in the blends. UV-VIS spectroscopy revealed that light transmission was high; between 220 nm and 1100 nm for pure chitosan; and between 320 and 1100 nm for pure WPI. 3.2 AGING OF WHEAT GLUTEN FILMS Biopolymers, including WG films, suffer from aging. In order to limit aging, it is important to reveal and understand the mechanisms and reasons for the time-dependent physical and chemical changes. This study aims towards the understanding of the mechanisms responsible for the aging of films of plasticized vital/commercial wheat gluten cast from water-ethanol solutions at pH=4 and pH=11. The physicochemical, mechanical and permeation properties were obtained as a function of aging time. 3.2.1 General film characteristics
The film and film forming properties are strongly dependent on the pH of the dispersion and is normally inferior close to the isoelectric point, which for WG is on the order of 7.5 [102]. In accordance to Gennadios et al.[37] WG homogeneous films are obtained at pH 2-4 and pH 9-13 whereas films are of poor quality at pH 5-6 and do not form at all at pH 7-8.

Results and discussion
42
The choice of casting pH=4 and pH=11 WG solutions was based on the idea of having acidic and basic solutions with approximately the same net intermolecular electrostatic repulsion, i.e. the same pH distance from the isoelectric point.
The films prepared from basic solution (hereafter referred to as b-films) appeared more homogeneous and smooth than the films prepared from acidic solutions (a-films). SEM revealed a undulating a-film upper surface and an uneven film thickness (Figure 27 and Figure 28). The lower surface, i.e. the surface that faced the Teflon-coated petri dish during casting, was always smooth for both films. IR spectroscopy on dehydrated samples showed that the a-film surface was richer in glycerol than the corresponding b-film, as indicated by a more pronounced 850 cm-1 peak, associated with the C-C-O stretch or CH2 twist of glycerol [170], in the former IR spectrum. IR, which covers a depth of a few microns, also indicated that there was more glycerol on the lower side than the upper side of the cast films. Most probably the gravitational force led to that a limited amount of glycerol migrated downwards through the solidifying film during the casting process. Both the a- and b-films contained particles at the surface but also in the bulk. Iodine staining indicated that these were starch granules.
Figure 27: SEM micrograph of the upper faces of a- film
Figure 28: SEM micrograph of upper face of b-film
3.2.2 Mechanical properties of unaged and aged WG films
The unaged b-film had a higher modulus, tensile strength and lower elongation at break than the unaged a-film (see Figure 29-Figure 32). This was in accordance with the results of Gennadios et al. [37] and Kayserilioglu et al.[79], who reported stronger films at alkaline conditions. According to Morel et al. 2000, [90] at high pH disulfide bonds are cleaved and the released thiol groups would become available for formation of new disulfide linkages, which gives strength to the film. The unaged a-films were very ductile. However, the ductility dropped and the stiffness and strength increased monotonically with time and the film became brittle (see Figure 29-Figure 32). In contrast to this, the ductility and stiffness of the b-film changed only marginally with time. The stiffness of the a-film increased 32 times during 120 days
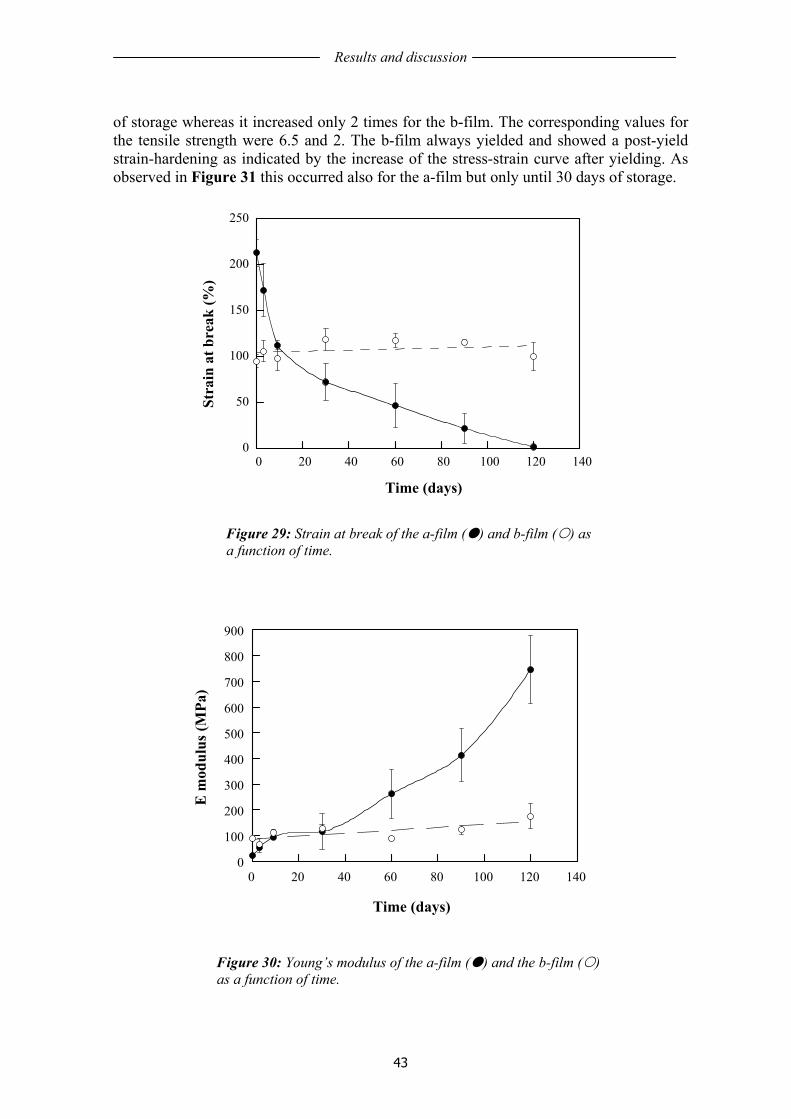
Results and discussion
43
of storage whereas it increased only 2 times for the b-film. The corresponding values for the tensile strength were 6.5 and 2. The b-film always yielded and showed a post-yield strain-hardening as indicated by the increase of the stress-strain curve after yielding. As observed in Figure 31 this occurred also for the a-film but only until 30 days of storage.
0
50
100
150
200
250
0 20 40 60 80 100 120 140
Stra
in a
t bre
ak (%
)
Time (days)
Figure 29: Strain at break of the a-film ( ) and b-film ( ) as a function of time.
0
100
200
300
400
500
600
700
800
900
0 20 40 60 80 100 120 140
E m
odul
us (M
Pa)
Time (days)
Figure 30: Young’s modulus of the a-film ( ) and the b-film ( ) as a function of time.
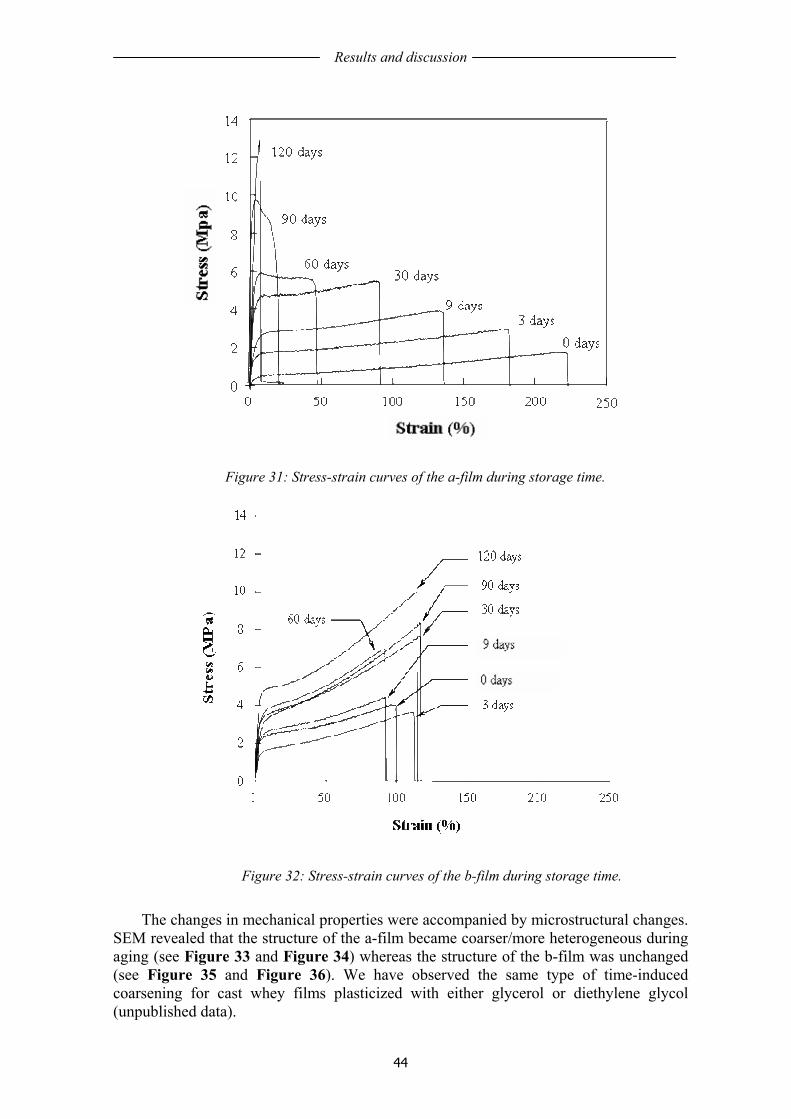
Results and discussion
44
Figure 31: Stress-strain curves of the a-film during storage time.
Figure 32: Stress-strain curves of the b-film during storage time.
The changes in mechanical properties were accompanied by microstructural changes. SEM revealed that the structure of the a-film became coarser/more heterogeneous during aging (see Figure 33 and Figure 34) whereas the structure of the b-film was unchanged (see Figure 35 and Figure 36). We have observed the same type of time-induced coarsening for cast whey films plasticized with either glycerol or diethylene glycol (unpublished data).

Results and discussion
45
Figure 33: SEM micrograph of cross-section of unaged tensile fractures a-film
Figure 34: SEM micrograph of cross-section of aged tensile fractures a-film
Figure 35: SEM micrograph of cross-section of unaged tensile fractures b-film
Figure 36: SEM micrograph of cross-section of aged tensile fractures b-film
3.2.3 Permeability
The permeability of water and oxygen provide additional information on the differences in the structure of the different films. It is expected that if the film looses plasticizing molecules during aging that the oxygen permeability would decrease.
Table 6: Oxygen permeability of a-films and b-films
Oxygen permeability (cm3 mm/(m2 day atm)) Days of storage a-film b-film
0 1.5 (0.1) 0.5 (0.2)
90 2.5 (1.0) 0.9 (0.6) This was however not the case. The OP increased with aging (see Table 6). This suggested that the loss and agglomeration of plasticizer yielded a less dense and more

Results and discussion
46
permeable structure. The OP of the a-film was approximately 3 times larger than that of the b-film, indicating that the more heterogeneous a-film structure was also more permeable. In addition the scatter in OP data (standard deviation, Table 6) increased with aging and was largest for the a-film, reflecting its large heterogeneity. Even though the undulating surface of the a-film led to an overestimation of the thickness, and consequently an overestimation of the OP, simply because the calliper might not have reached the bottom of every valley, it could not explain the large OP differences between the a- and b-films. Also, the water vapour permeability increased with aging. However the differences between the a- and b-films were small. The more heterogeneous a-film probably contained a larger amount of voids/capillaries between starch, glycerol and protein particles than the b-film.
Table 7: Water vapour permeability of a-films and b-films
Water vapor permeability (g mm/(m2 day atm)) Days of storage a-film b-film
17 240 (28) 310 (140)
60 920 (500) 1150 (530)
120 690 (520) 670 (300)
The small difference in WVP is probably due to that WVP is less sensitive to the presence of voids than OP [171]. This is explained by that the high surface energy of water leads to that it adheres to the surfaces of capillaries, cavities and voids, whereas oxygen is filling the same. Consequently the concentration of oxygen (and also the WVP) increases more rapidly than the concentration of water with increasing amounts of capillaries and voids. 3.2.4 Protein solubility of unaged and aged WG films
The SE-HPLC data revealed that the protein solubility of the unaged a-film was considerably higher than the corresponding b-film (see Figure 37). The protein solubility of the a-film was high already after the SDS-treatment, which dissolved protein molecules by breaking mainly secondary bonds. However, the subsequent sonication, which is considered to break primarily disulphide bonds, increased the protein solubility significantly. Interestingly, the sonication had a larger impact on the a-film than on the b-film. Figure 38 showed that a significant part of the dissolved matter of the a-film was large proteins. In fact, after the last sonication extraction, more than one third of the dissolved proteins were “polymeric” proteins. Only a small part of the dissolved b-film proteins were large proteins. Since it is known that basic conditions promote protein denaturation by reducing the number of intermolecular and intramolecular S-S bonds [35], and that the conformation of the protein molecule is more stable in acidic conditions [172], it is likely that the b-film experienced a larger degree of denaturation during film formation than the a-film. In addition, other aminoacid crosslinking reactions, not involving thiol groups, e.g. the formation of lysinoalanine, occurs more readily at high pH [79]. Following the denaturation, the b-film proteins would then show a more extensive aggregation and polymerisation than those in the a-film. This would explain the
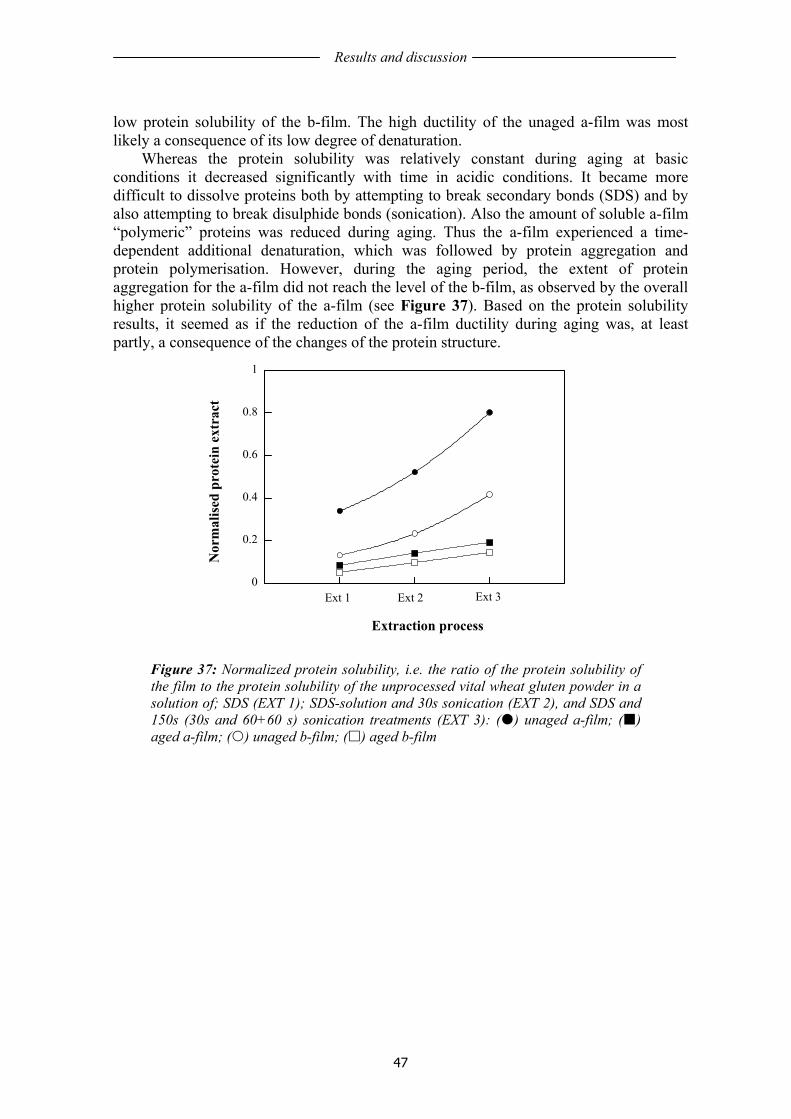
Results and discussion
47
low protein solubility of the b-film. The high ductility of the unaged a-film was most likely a consequence of its low degree of denaturation. Whereas the protein solubility was relatively constant during aging at basic conditions it decreased significantly with time in acidic conditions. It became more difficult to dissolve proteins both by attempting to break secondary bonds (SDS) and by also attempting to break disulphide bonds (sonication). Also the amount of soluble a-film “polymeric” proteins was reduced during aging. Thus the a-film experienced a time-dependent additional denaturation, which was followed by protein aggregation and protein polymerisation. However, during the aging period, the extent of protein aggregation for the a-film did not reach the level of the b-film, as observed by the overall higher protein solubility of the a-film (see Figure 37). Based on the protein solubility results, it seemed as if the reduction of the a-film ductility during aging was, at least partly, a consequence of the changes of the protein structure.
0
0.2
0.4
0.6
0.8
1
Nor
mal
ised
pro
tein
ext
ract
Extraction process
Ext 1 Ext 2 Ext 3
Figure 37: Normalized protein solubility, i.e. the ratio of the protein solubility of the film to the protein solubility of the unprocessed vital wheat gluten powder in a solution of; SDS (EXT 1); SDS-solution and 30s sonication (EXT 2), and SDS and 150s (30s and 60+60 s) sonication treatments (EXT 3): ( ) unaged a-film; ( ) aged a-film; ( ) unaged b-film; ( ) aged b-film
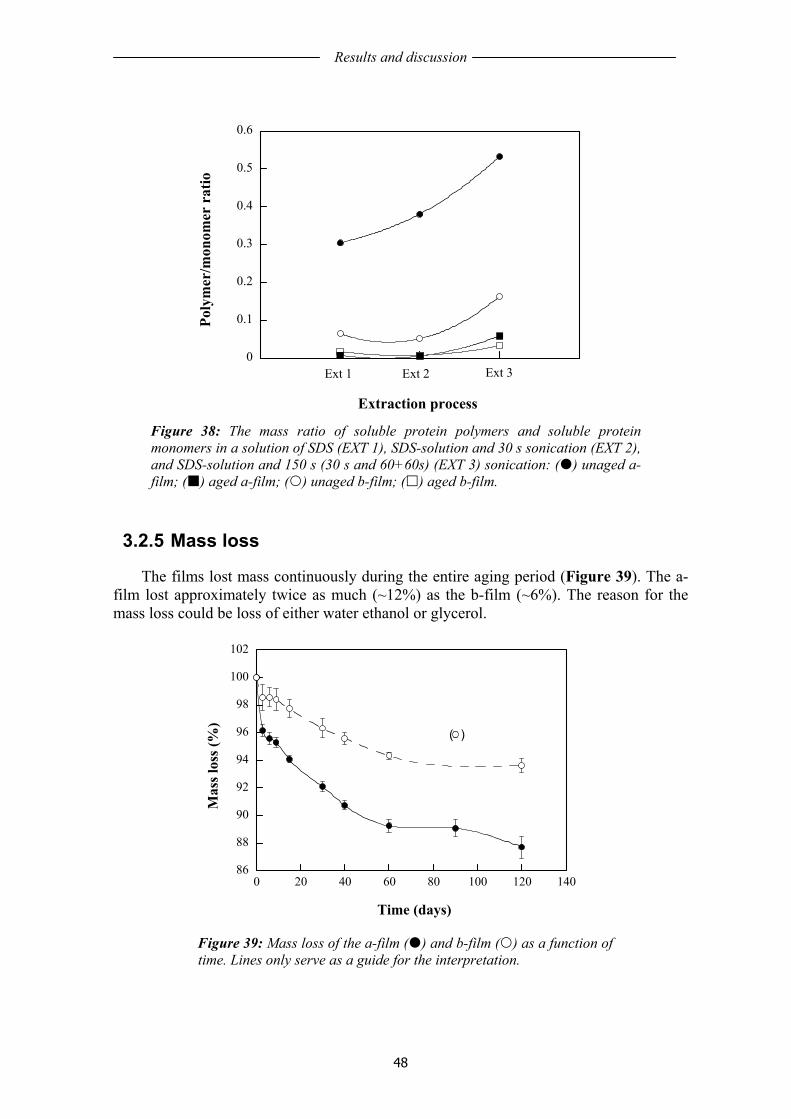
Results and discussion
48
0
0.1
0.2
0.3
0.4
0.5
0.6
Poly
mer
/mon
omer
rat
io
Extraction process
Ext 1 Ext 2 Ext 3
Figure 38: The mass ratio of soluble protein polymers and soluble protein monomers in a solution of SDS (EXT 1), SDS-solution and 30 s sonication (EXT 2), and SDS-solution and 150 s (30 s and 60+60s) (EXT 3) sonication: ( ) unaged a-film; ( ) aged a-film; ( ) unaged b-film; ( ) aged b-film.
3.2.5 Mass loss
The films lost mass continuously during the entire aging period (Figure 39). The a-film lost approximately twice as much (~12%) as the b-film (~6%). The reason for the mass loss could be loss of either water ethanol or glycerol.
86
88
90
92
94
96
98
100
102
0 20 40 60 80 100 120 140
Time (days)
( )
Mas
s los
s (%
)
Figure 39: Mass loss of the a-film ( ) and b-film ( ) as a function of time. Lines only serve as a guide for the interpretation.
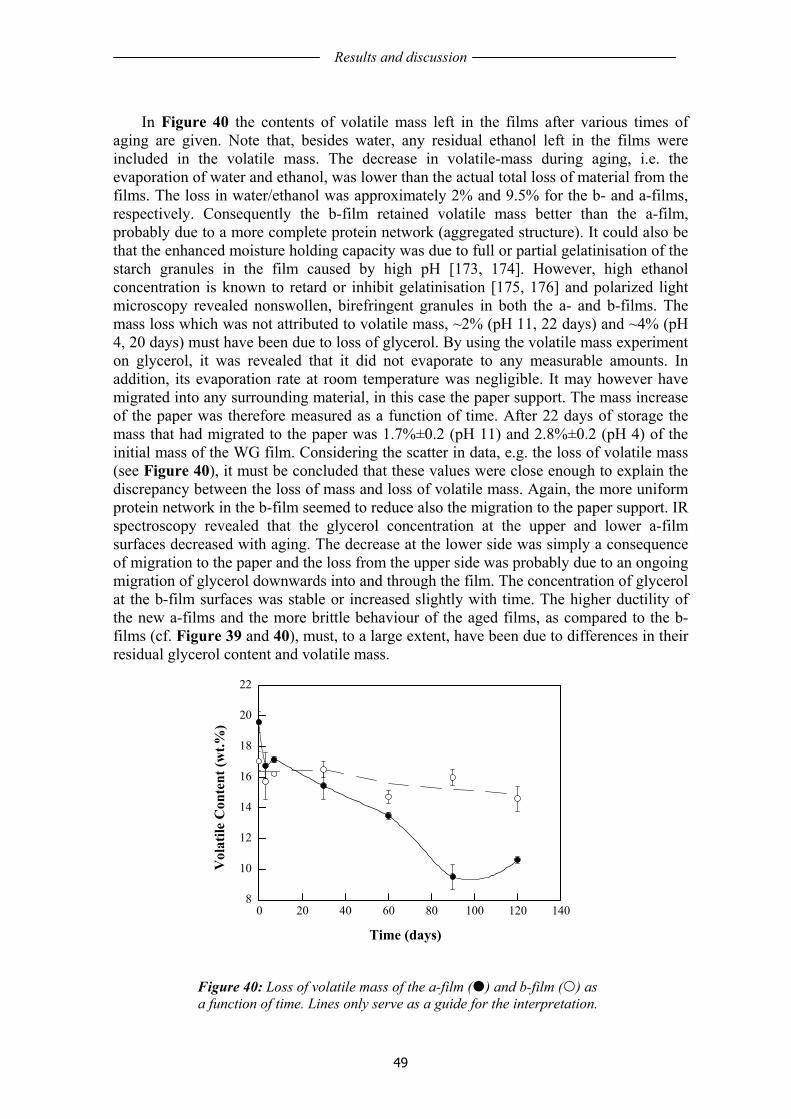
Results and discussion
49
In Figure 40 the contents of volatile mass left in the films after various times of aging are given. Note that, besides water, any residual ethanol left in the films were included in the volatile mass. The decrease in volatile-mass during aging, i.e. the evaporation of water and ethanol, was lower than the actual total loss of material from the films. The loss in water/ethanol was approximately 2% and 9.5% for the b- and a-films, respectively. Consequently the b-film retained volatile mass better than the a-film, probably due to a more complete protein network (aggregated structure). It could also be that the enhanced moisture holding capacity was due to full or partial gelatinisation of the starch granules in the film caused by high pH [173, 174]. However, high ethanol concentration is known to retard or inhibit gelatinisation [175, 176] and polarized light microscopy revealed nonswollen, birefringent granules in both the a- and b-films. The mass loss which was not attributed to volatile mass, ~2% (pH 11, 22 days) and ~4% (pH 4, 20 days) must have been due to loss of glycerol. By using the volatile mass experiment on glycerol, it was revealed that it did not evaporate to any measurable amounts. In addition, its evaporation rate at room temperature was negligible. It may however have migrated into any surrounding material, in this case the paper support. The mass increase of the paper was therefore measured as a function of time. After 22 days of storage the mass that had migrated to the paper was 1.7%±0.2 (pH 11) and 2.8%±0.2 (pH 4) of the initial mass of the WG film. Considering the scatter in data, e.g. the loss of volatile mass (see Figure 40), it must be concluded that these values were close enough to explain the discrepancy between the loss of mass and loss of volatile mass. Again, the more uniform protein network in the b-film seemed to reduce also the migration to the paper support. IR spectroscopy revealed that the glycerol concentration at the upper and lower a-film surfaces decreased with aging. The decrease at the lower side was simply a consequence of migration to the paper and the loss from the upper side was probably due to an ongoing migration of glycerol downwards into and through the film. The concentration of glycerol at the b-film surfaces was stable or increased slightly with time. The higher ductility of the new a-films and the more brittle behaviour of the aged films, as compared to the b-films (cf. Figure 39 and 40), must, to a large extent, have been due to differences in their residual glycerol content and volatile mass.
8
10
12
14
16
18
20
22
0 20 40 60 80 100 120 140
Vol
atile
Con
tent
(wt.%
)
Time (days)
Figure 40: Loss of volatile mass of the a-film ( ) and b-film ( ) as a function of time. Lines only serve as a guide for the interpretation.
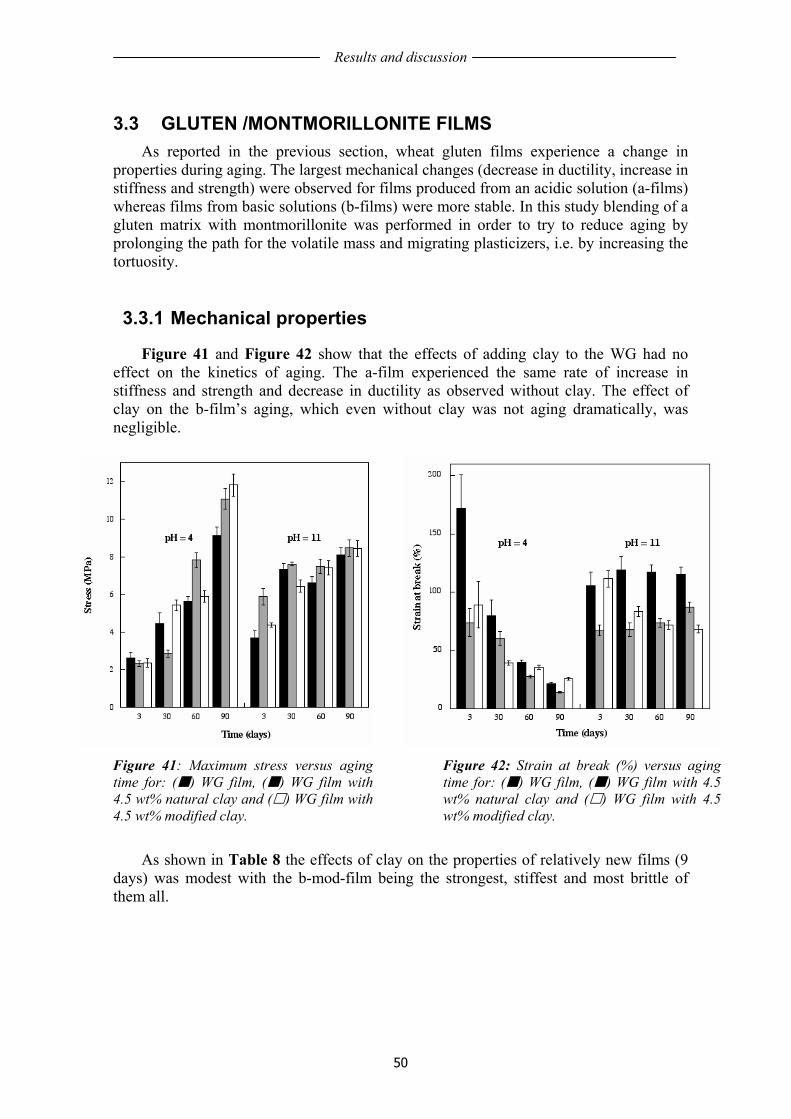
Results and discussion
50
3.3 GLUTEN /MONTMORILLONITE FILMS As reported in the previous section, wheat gluten films experience a change in properties during aging. The largest mechanical changes (decrease in ductility, increase in stiffness and strength) were observed for films produced from an acidic solution (a-films) whereas films from basic solutions (b-films) were more stable. In this study blending of a gluten matrix with montmorillonite was performed in order to try to reduce aging by prolonging the path for the volatile mass and migrating plasticizers, i.e. by increasing the tortuosity.
3.3.1 Mechanical properties
Figure 41 and Figure 42 show that the effects of adding clay to the WG had no effect on the kinetics of aging. The a-film experienced the same rate of increase in stiffness and strength and decrease in ductility as observed without clay. The effect of clay on the b-film’s aging, which even without clay was not aging dramatically, was negligible.
Figure 41: Maximum stress versus aging time for: ( ) WG film, ( ) WG film with 4.5 wt% natural clay and ( ) WG film with 4.5 wt% modified clay.
Figure 42: Strain at break (%) versus aging time for: ( ) WG film, ( ) WG film with 4.5 wt% natural clay and ( ) WG film with 4.5 wt% modified clay.
As shown in Table 8 the effects of clay on the properties of relatively new films (9 days) was modest with the b-mod-film being the strongest, stiffest and most brittle of them all.

Results and discussion
51
Table 8: Effects of clay added to WG films. Values within parenthesis are standard deviations.
9 days Clay
Content (%)
Maximum strength (MPa)
Fracture strain (%)
Young’s Modulus (MPa)
a 0 3.52 (0.09) 112.26 (9.08) 94.53 (8.05)
a-nat 4.5 2.33 (0.15) 75.5 (14.01) 64.97 (14.60)
a-mod 4.5 2.35 (0.18) 67.45 (7.63) 92.27 (10.72)
b 0 5.41 (0.97) 97.84 (12.77) 111.07 (15.01)
b-mod 4.5 7.13 (0.48) 62.82 (0.99) 148.51 (8.86)
b-nat 4.5 5.02 (0.24) 134.55 (8.77) 81.14 (8.37)
The modest change was due to the small amount of clay in the films (4.5wt%=~1-2 vol%). The choice of the content of clay was based on the idea of allowing the individual clay layers to orient independently and thereby to allow for a more complete exfoliation. 3.3.2 Permeability
In order to see if the small content of clay chosen was the reason why there were no effects on the aging kinetics, water permeation data was obtained. Figure 6 shows that the small amount of clay greatly effected the water barrier properties. The permeability of the composite film was modelled by first considering the sheets as oblate randomly oriented exfoliated spheroids uniformly distributed within the matrix [169, 177]. The permeability of the clay-WG film (Pc) is:
Eq. 6. Pc =Pm vm
τ
where Pm is the permeability of the polymer matrix and vm is the volume fraction of glycerol/protein in the composite. τ is the tortuosity factor which increases with increasing blocking efficiency of the filler. Provided that the montmorillonite layers were completely delaminated and that the reduction in permeability was a function solely of the content of filler and its width (w) and thickness (l), the tortuosity factor could be estimated using the following relationship:
Eq. 7. X =1− vm
τ −1
where X is obtained from
Eq. 8.
3XX0.6160.785
1lw
+−−
=
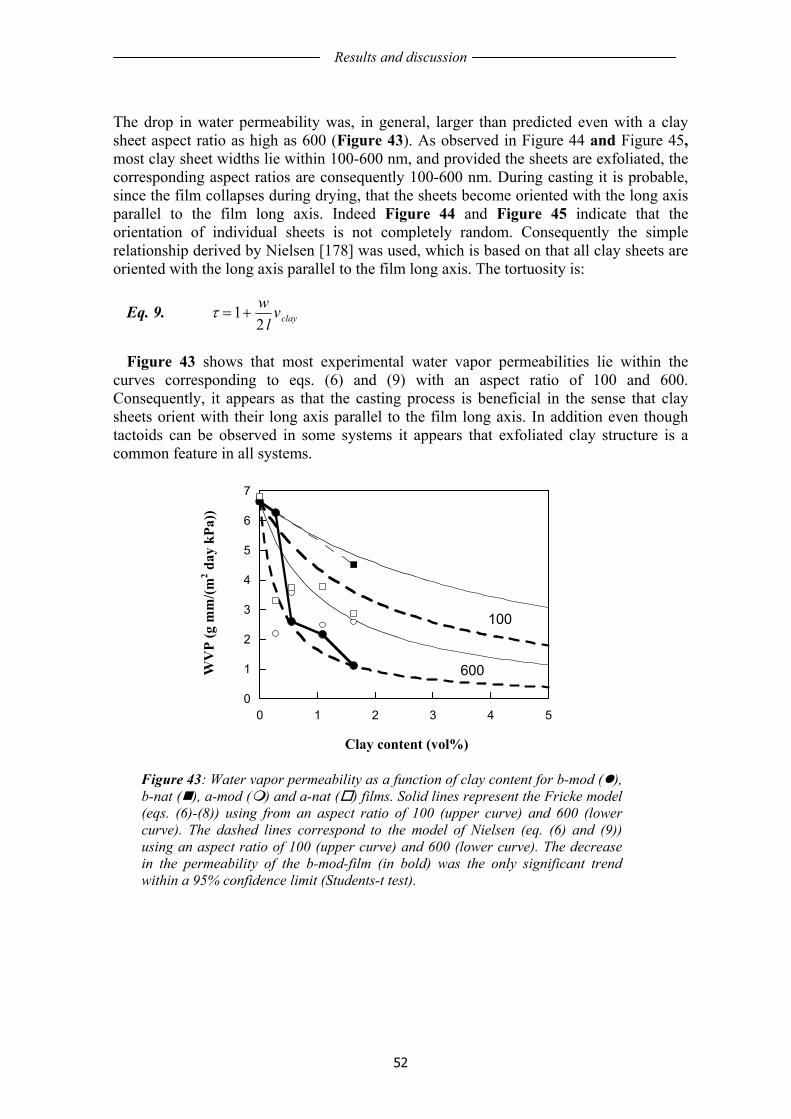
Results and discussion
52
The drop in water permeability was, in general, larger than predicted even with a clay sheet aspect ratio as high as 600 (Figure 43). As observed in Figure 44 and Figure 45, most clay sheet widths lie within 100-600 nm, and provided the sheets are exfoliated, the corresponding aspect ratios are consequently 100-600 nm. During casting it is probable, since the film collapses during drying, that the sheets become oriented with the long axis parallel to the film long axis. Indeed Figure 44 and Figure 45 indicate that the orientation of individual sheets is not completely random. Consequently the simple relationship derived by Nielsen [178] was used, which is based on that all clay sheets are oriented with the long axis parallel to the film long axis. The tortuosity is:
Eq. 9. τ =1+w2 l
vclay
Figure 43 shows that most experimental water vapor permeabilities lie within the curves corresponding to eqs. (6) and (9) with an aspect ratio of 100 and 600. Consequently, it appears as that the casting process is beneficial in the sense that clay sheets orient with their long axis parallel to the film long axis. In addition even though tactoids can be observed in some systems it appears that exfoliated clay structure is a common feature in all systems.
0
1
2
3
4
5
6
7
0 1 2 3 4 5
Clay content (vol%)
WV
P (g
mm
/(m2 d
ay k
Pa))
600
100
Figure 43: Water vapor permeability as a function of clay content for b-mod ( ), b-nat ( ), a-mod ( ) and a-nat ( ) films. Solid lines represent the Fricke model (eqs. (6)-(8)) using from an aspect ratio of 100 (upper curve) and 600 (lower curve). The dashed lines correspond to the model of Nielsen (eq. (6) and (9)) using an aspect ratio of 100 (upper curve) and 600 (lower curve). The decrease in the permeability of the b-mod-film (in bold) was the only significant trend within a 95% confidence limit (Students-t test).
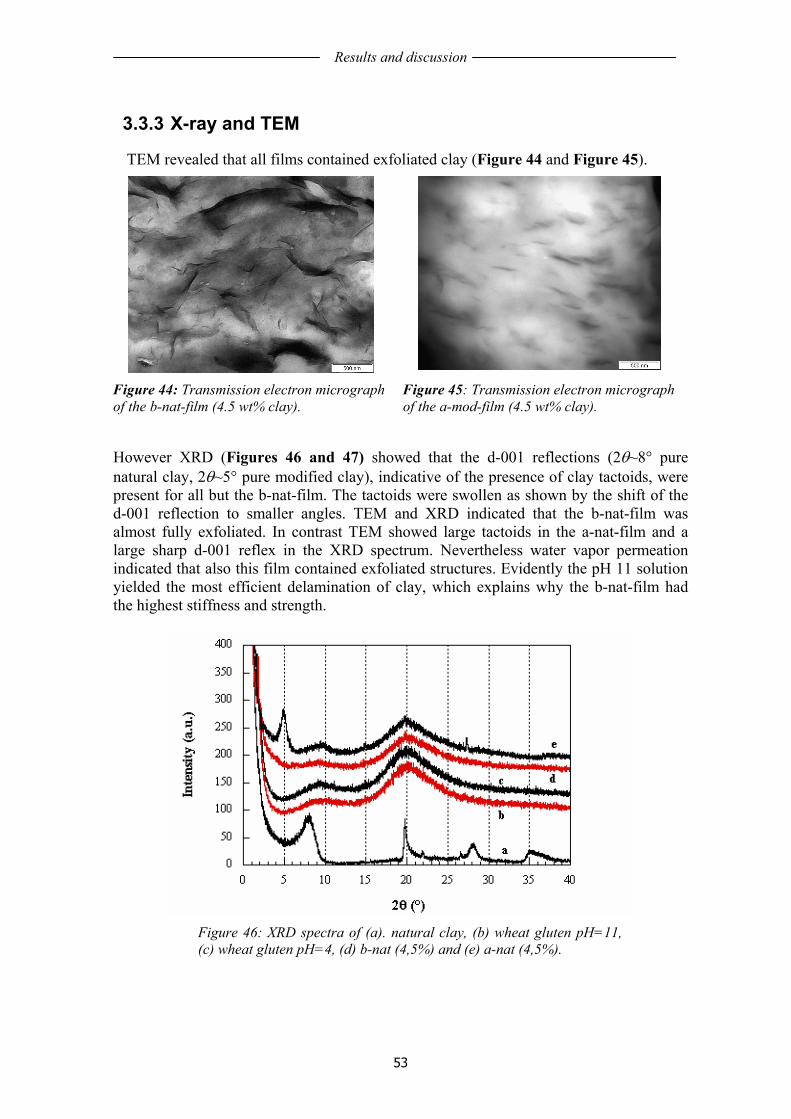
Results and discussion
53
3.3.3 X-ray and TEM
TEM revealed that all films contained exfoliated clay (Figure 44 and Figure 45).
Figure 44: Transmission electron micrograph of the b-nat-film (4.5 wt% clay).
Figure 45: Transmission electron micrograph of the a-mod-film (4.5 wt% clay).
However XRD (Figures 46 and 47) showed that the d-001 reflections (2θ~8° pure natural clay, 2θ~5° pure modified clay), indicative of the presence of clay tactoids, were present for all but the b-nat-film. The tactoids were swollen as shown by the shift of the d-001 reflection to smaller angles. TEM and XRD indicated that the b-nat-film was almost fully exfoliated. In contrast TEM showed large tactoids in the a-nat-film and a large sharp d-001 reflex in the XRD spectrum. Nevertheless water vapor permeation indicated that also this film contained exfoliated structures. Evidently the pH 11 solution yielded the most efficient delamination of clay, which explains why the b-nat-film had the highest stiffness and strength.
Figure 46: XRD spectra of (a). natural clay, (b) wheat gluten pH=11, (c) wheat gluten pH=4, (d) b-nat (4,5%) and (e) a-nat (4,5%).
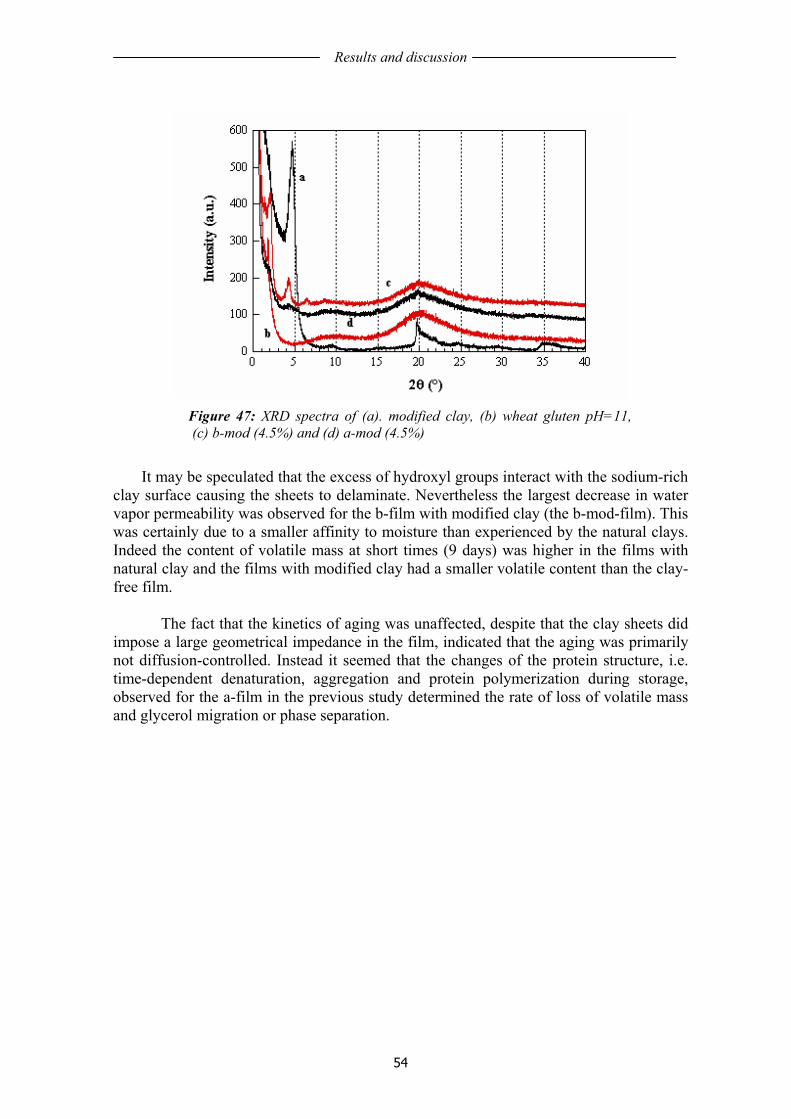
Results and discussion
54
Figure 47: XRD spectra of (a). modified clay, (b) wheat gluten pH=11, (c) b-mod (4.5%) and (d) a-mod (4.5%)
It may be speculated that the excess of hydroxyl groups interact with the sodium-rich clay surface causing the sheets to delaminate. Nevertheless the largest decrease in water vapor permeability was observed for the b-film with modified clay (the b-mod-film). This was certainly due to a smaller affinity to moisture than experienced by the natural clays. Indeed the content of volatile mass at short times (9 days) was higher in the films with natural clay and the films with modified clay had a smaller volatile content than the clay-free film.
The fact that the kinetics of aging was unaffected, despite that the clay sheets did
impose a large geometrical impedance in the film, indicated that the aging was primarily not diffusion-controlled. Instead it seemed that the changes of the protein structure, i.e. time-dependent denaturation, aggregation and protein polymerization during storage, observed for the a-film in the previous study determined the rate of loss of volatile mass and glycerol migration or phase separation.

Conclusions
55
4 CONCLUSIONS
Films of chitosan or whey blended with PCL
• Poly (ε-caprolactone) (PCL) was ‘compatible’ with both whey and chitosan and it was possible to produce films of these polymer blends, using casting technique.
• The addition of PCL to whey protein isolate (WPI) and chitosan reduced the water vapour and oxygen permeabilities. Chitosan showed lower water vapour permeability than WPI. The composites were, however, too water sensitive to be used in direct contact with water.
• Chitosan films showed higher strength and stiffness, but lower elongation at break, than WPI films. The addition of PCL had different effects on the mechanical properties of the two materials. PCL reduced the Young’s modulus in chitosan but increased it in WPI.
• Chitosan films were found to be difficult to heat sealable. The WPI films were sealable, but they had a low sealing strength.
• Chitosan films had a high folding endurance, which was not altered by the addition of PCL. High folding endurance indicated good fatigue properties for this material, which could be promising for future packaging applications. Surprisingly WPI, even if it had higher fracture strain than chitosan, had much lower resistance to folding.
Aging of wheat gluten films
• The aging properties of the wheat gluten cast films depended strongly on the pH of the solution. Gluten films made with acidic solutions experienced a significant increase in fracture stress and Young’s modulus, while alkaline films remained more stable during the storage time. The homogeneity, thickness and surface smoothness were higher for the alkaline films. The increased film homogeneity of the alkaline films yielded enhanced oxygen barrier properties.
• The gluten films made from acidic solution experienced a larger loss of volatiles
and migration of glycerol to the paper that supported the films during the storage. The time-induced decrease of fracture strain of the acidic film was due to the loss of plasticizing molecules (water, ethanol and glycerol), which, in turn, was due to a lower degree of protein aggregation/polymerisation as compared to the basic film. The more aggregated protein in the alkaline films seemed to be more effective in retaining the plasticizing molecules within the film.

Conclusions
56
Composite films of wheat gluten and montmorillonite
• It was possible to produce films of wheat gluten with montmorillonite clay by solution casting technique. The water vapour permeabilities indicated that the clay particles effectively acted as geometrical obstacles in the wheat gluten matrix.
• The clay did not affect the aging rate of the films made from acidic and basic solution.
• The montmorillonite clay showed a higher degree of exfoliation in films cast from alkaline solution than from acidic solution. The water vapour permeability was lower for films with the less hygroscopic modified montmorillonite.
This study shows the potential of these biopolymers as blending materials for producing films by casting method. This can serve as a base for further research, which should be directed towards maintaining the oxygen barrier properties while improving water sensitivity, water vapour barrier and mechanical properties.

Acknowledgements
57
5 ACKNOWLEDGEMENTS
First I would like to express my very sincere gratitude to my supervisor, Associate Professor Mikael Hedenqvist, for his support, his encouragement, his help and his sense of humour. I also thank Professor Ulf W. Gedde for his kindness and for accepting me in his group.
Prof. Ann-Christine Albertsson, Prof. Anders Hult, Prof. Bengt Stengberg and Prof. Sigbritt Karlsson are thanked for their leadership and for making this department scientifically stimulating. Associate Prof. Mats Johansson and Prof. Eva Malmström are thanked for always being helpful and so nice and the administrative personnel of the department, especially Maggan, Maria and Viktoria for their help with all administrative matters and their kindness. I also thank Inger for her valuable assistance in every kind of matters and Ove for being an authentic help with all the technical problems I had.
I thank people from the Engineering Faculty in Bilbao (University of Basque Country) for their kindness, especially Prof. Jose Ramon Sarasua, for his help, encouragement and interesting talks about this work.
Västsvenska Lantmännens Research Foundation (VL-stiftelsen) is acknowledged for financial support. Peter Baeling, Martin Svensson and Kurt Villwock at Lantmännen are thanked for all the valuable discussions during ‘gluten meetings’.
I am indebted to Mikael Gällstedt for helping me so much, always finding time to answer and discuss my questions about “gluten et al.”, for his guidance, optimism, laugh and late chats. I wish also thank all my former colleges at Packforsk, now part of STFI-Packforsk, for their kindness and support in both work and personal matters.
I am grateful to Mari Jose Suarez, Inma Angulo and my very good friend Laura Dominguez (hejsan!) at the Research Institute Gaiker, in Basque Country, for their encouragement at the beginning of my polymer career and for being the origin of this long journey.
I would like to show my gratitude to all the people who made the work at the Polymer department a great experience for me. I would like to mention: the ‘Spanish group’ for giving me the support and courage I needed in every moment, especially Marie for her optimisms, generosity, kindness and the good moments we spent together (and more to come); Kamyar (R&R at Beckers) and Eugenia (Photoshop Queen and promising artist) for being great friends and a second family to me; Ana for the nice times in the gym and in the dancing courses, remember that we could have been a promising artistic ‘magdans’ couple (it’s never too late); Natalia (honeeeeeeey!) for her funny craziness, for taking care of me and being a beautiful person and Fran for having the guts to take over Kamyar’s place as the only male in our Friday’s girl lunches. I will always remember the good times with the rest of Polymer’s girl power as Clara S., Linda F., Maria E, Annika L., Viktoria B. among others. Past and present members of Ulf’s Group: Mikael Krook., Gunnar K., Janis R., Anders B., Bereket N., Alessandro M., Richard O., David N., Henrik U., Roland K, Bruska A., Stefanie R., Jinglan, Carol, and Sung-Woo; the fika-rum people; former room mates as Margaretha, Johan, Anders, John, Jonas (sorry for leaving the MOCON nightmare to you) and to all of those of you I have not forgotten to mention but I didn’t have the space to do it, thank you for making the department a nice and enjoyable place to work.

Acknowledgements
58
My friends outside the department are also thanked, especially Natalia H. for being so sweet, unconditional friend and my moral support here in Stockholm. My friends in Bilbao are thanked for being there, no matter the distance, for never remember the word biopolymer and for making me enjoy the trips home.
My warmest gratitude goes to my family for their support and their love, and for always being ready to help me, maite zaituztet! Also warm thanks to Karin and Rolf and the rest of the family Weström for taking so well care of me and for being to me a real family.
This work is dedicated to Pär for his love and dedication, for being the best that have ever happened to me.

References
59
6 REFERENCES
1. Goddard, R., Packaging Materials. 1990: Pira. 2. Anker, M., Edible and Biodegradable Whey Protein Films as Barriers in Foods and
Food Packaging, in Department of Food Science. 2000, Chalmers University of Technology: Göteborg.
3. Huang, S.J., Polymer Waste Management - biodegradation, incineration and recycling, in Selected Papers Presented at the International Workshop on Controlled Life Cycle of Polymeric Materials, Stockholm, 1994. Journal Macromolecular Science - Pure Applied Chemistry, 1995. 32(4): p. 593-597.
4. Anker, M., Edible and biodegradable films and coatings for food packaging. a literature review. 1996, Chalmers University of Technology: Göteborg.
5. Guilbert, S., B. Cuq, and N. Gontard, Recent innovations in edible and/or biodegradable packaging materials. Food Additives and Contaminants, 1997. 14(6): p. 741-751.
6. Flieger, M., et al., Biodegradable plastics from renewable sources. Folia Microbiologica (Prague, Czech Republic), 2003. 48(1): p. 27-44.
7. Scott, G., 'Green' Polymers. Polymer Degradation and Stability, 2000. 68: p. 1-7. 8. Trznadel, M., Biodegradable polymer materials. International Polymer Science and
Technology, 1995. 22(12): p. 58-65. 9. Swifft, G., Opportunities for Environmentally Degradable Polymers, in Selected
Papers Presented in The International Workshop on Controlled Life-Cycle of Polymeric Materials, Stockholm, 1994. Journal Macromolecular Science - Pure Applied Chemistry, 1995. 32(4): p. 641 - 651.
10. Demicheli, M., Biodegradable plastics from renewable sources, in IPTS Report, Vol 10., http://www.jrc.es, Editor. 1996.
11. Petersen, K., et al., Potential of biobased materials for food packaging. Trends in Food Science & Technology, 1999. 10(2): p. 52-68.
12. Krochta, M. and C.D. Mulder-Johnston, Edible & Biodegradable Polymer Films. 1997, Department of Food Science and Technology, University of California-Davis.
13. Weber, C.J., et al., Biobased packaging materials for the food industry. 2000, KVL. 14. Albertsson, A.C. and S. Karlsson, Chemistry and biochemistry of polymer
biodegradation, in Chemistry and Technology of Biodegradable Polymers, E. G.J.L. Griffin, Editor. 1994, Blackie Academic & Professional: London. p. 7-17.
15. Karlsson, S. and A.-C. Albertsson, Biodegradable Polymers and Environmental Interaction. Polymer Engineering and Science, 1998. 38(8): p. 1251-1253.
16. Nelson, L.N. and M.M. Cox, Lehninger Principles of Biochemistry. 3rd ed. 2000, New York: Worth Publishers.
17. Cuq, B., N. Gontard, and S. Guilbert, Proteins as agricultural polymers for packaging production. Cereal Chemistry, 1998. 75(1): p. 1-9.
18. Krochta, J.M. and C. DeMulder Johnston, Edible and Biodegradable Polymer Films: challenges and Opportunities. Food Technology, 1997. 51: p. 61-74.
19. Freeman, B.D., S.N. Dhoot, and M.E. Stewart, Barrier Polymers, in Encyclopedia of Polymer Science and Technology. 2002, John Wiley & Sons, Inc.

References
60
20. Robertson, G.L., Food Packaging: Principles and Practice. 1993, Marcel Dekker. 21. Pauly, S., Permeability and Diffusion Data, in Polymer Handbook, J. Brandrup,
E.H. Immergut, and E.A. Grulke, Editors. 1999, John Wiley & Sons, Inc. 22. Miller, K.S. and J.M. Krochta, Oxygen and Aroma Barrier Properties of Edible
Films: A Review. Trends in Food Science & Technology, 1997. 8: p. 228-237. 23. Gennadios, A. and C.L. Weller, Edible films and coatings from wheat and corn
proteins. Food Technology (Chicago, IL, United States), 1990. 44(10): p. 63-9. 24. Aydt, T.P., C.L. Weller, and R.F. Testin, Mechanical and barrier properties of
edible corn and wheat protein films. Transactions of the ASAE, 1991. 34(1): p. 207-11.
25. Gontard, N., et al., Influence of Relative Humidity and Film Composition on Oxygen and Carbon Dioxide Permeabilities of Edible Films. Journal of Agricultural and Food Chemistry, 1996. 44(4): p. 1064-9.
26. Pavlath, A.E., D.S.W. Wong, and T.F. Kumosinski, New coatings for cut fruits and vegetables. CHEMTECH, 1993(2): p. 36-40.
27. Gennadios, A., Protein-based Films and Coatings, ed. A. Gennadios. 2002: CRC Press LLC, USA.
28. Farber, J.N., et al., Microbiological safety of controlled and modified atmosphere packaging of fresh and fresh-cut produce. Comprehensive Reviews in Food Science and Food Safety, 2003. 2.
29. Emmambux, M.N., S. Iannace, and M. Stading. Cereal biopolymer films, coatings and other industrial products. in Workshop on the proteins od srrghum and millets: enhancing nutritional and functional properties for Africa. 2003. Pretoria, South Africa: P.S. Belton nad J.R.N Taylor.
30. Salame, M., Barrier Polymers, in The Wiley Encyclopedia of Packaging Technology, M. Bakker, Editor. 1986, John Wiley & Sons. p. 48-54.
31. Makino, Y. and T. Hirata, Modified atmosphere packaging of fresh produce with a biodegradable laminate of chitosan-cellulose and polycaprolactone. Postharvest Biology and Technology, 1997. 19: p. 247-254.
32. Krochta, J.M., Proteins as Raw Materials for Films and Coatings: Definitions, Current Status and Opportunities., in Protein-based films and coatings, A. Gennadios, Editor. 2002.
33. Debeaufort, F., A. Voilley, and P. Meares, Water vapor permeability and diffusivity through methylcellulose edible films. Journal of Membrane Science, 1994. 91(1-2): p. 125-33.
34. Bastioli, C., Biodegradable materials, in Willey Encyclopedia of Packaging Technology, A.L.B.K.S. Marsh, Editor. 1997, John Willey % Sons.
35. Roy, S., et al., Water vapor transport parameters of a cast wheat gluten film. Industrial Crops and Products, 2000. 11: p. 43-50.
36. Ali, Y., V.M. Ghorpade, and M.A. Hanna, Properties of thermally-treated wheat gluten films. Industrial Crops and Products, 1997. 6: p. 177-184.
37. Gennadios, A., et al., Effect of pH on Properties of Wheat Gluten and Soy Protein Isolate Films. Journal of Agricultural and Food Chemistry, 1993. 41: p. 1835-1839.

References
61
38. Gontard, N., S. Guilbert, and J.-L. Cuq, Edible Wheat Gluten Films: Influence of the Main Process VAriables on Film Properties using Response Surface Methodology. Journal of Food Science, 1992. 57(1): p. 190-199.
39. Herald, T.J., et al., Degradable wheat gluten films: preparation, properties and applications. Journal of Food Science, 1995. 60(5): p. 1147-50.
40. Nielsen, L.E. and R.F. Landel, Mechanical Properties of Polymers and Composites. Second Edition ed, ed. L.L. Faulkner. 1994, New York: Marcel Dekker, Inc.
41. McHugh, T.H. and J.M. Krochta, Sorbitol -vs Glycerol-Plasticized Whey Protein Edible Films: Integrated Oxygen Permeability and Tensile Property Evaluation. Journal of Agricultural and Food Chemistry, 1994. 42(4): p. 841-845.
42. Sothornvit, R. and J.M. Krochta, Oxygen Permeability and Mechanical Properties of Films from Hydrolyzed Whey Protein. J. Agric. Food Chem, 2000. 48(9): p. 3913-3916.
43. Kurita, K., Chemistry and application of chitin and chitosan. Polymer Degradation and Stability, 1997. 59: p. 117-120.
44. Hirano, S., Production and application of chitin and chitosan in japan, in Chitin and Chitosan, A. Skjak-Braek, and Sanford, Editor. 1989, Elsevier Science Publishing Co.: London. p. 37-43.
45. Shahidi, F. and J. Synowiecki, Isolation and Characterization of Nutrients and Value-Added Products from Snow Crab (Chinoecetes Opilio) and Shrimp (Pandalus Borealis) Processing Discards. J. Agric. Food Chem., 1991. 39: p. 1527-1532.
46. Shahidi, F., J.K.V. Arachchi, and Y.-J. Jeon, Food Applications of chitin and chitosan. Trends in Food Science & Technology, 1999. 10: p. 37-51.
47. Peter, M.G., Applications and Environmental aspects of chitin and chitosan. Journal of macromolecular Science- Pure Apllied Chemistry, 1995. 32(4): p. 629-640.
48. Khan, T.A., K.K. Peh, and H.S. Ch'ng, Reporting degree of deacetylation values of chitosan: the influence of analytical methods. Journal of Pharmacy & Pharmaceutical Sciences [online computer file], 2002. 5(3): p. 205-212.
49. Li, Q., et al., Applications and properties of chitosan. Journal of Bioactive and Compatible Polymers, 1992. 7(4): p. 370-97.
50. Lower, E.S., Polymer from the Sea: Chitin & Chitosan. Manufacturing Chemist., 1984: p. 47.
51. Knorr, D., Recovery and Utilization of Chitin and Chitosan in Food Processing Waste Management. 1991. Food Technology: p. 114-122.
52. Khan, T.A., K.K. Peh, and H.S. Ch'ng, Mechanical, bioadhesive strength and biological evaluations of chitosan films for wound dressing. Journal of Pharmacy & Pharmaceutical Sciences [online computer file], 2000. 3(3): p. 303-311.
53. Li, J., J.F. Revol, and R.H. Marchessault, Effect of degree of deacetylation of chitin on the properties of chitin crystallites. Journal of Applied Polymer Science, 1997. 65(2): p. 373-380.
54. Luyen, D.v. and V. Rossbach, Chitin and chitosan; potential fiber raw materials. Technical textiles, 1992. 35: p. 19-20.
55. Butler, B.L., et al., Mechanical and Barrier Properties of Edible Chitosan Films as affected by Composition and Storage. Journal of Food Science, 1996. 61(5): p. 953-961.

References
62
56. Darder, M., Biopolymer-Clay Nanocomposites Based on Chitosan Intercalated in Montmorillonite. Chem. Mater., 2003. 15: p. 3774-3780.
57. Han, S.-M., Chitin and chitosan, via http://user.chollian.net, Seoul National University of Technology.
58. Kittur, F.S., K.R. Kumar, and R.N. Tharanathan, Functional packaging properties of chitosan films. Z. Lebensm Unters Forsch A, 1998. 206: p. 44-47.
59. Hoagland, P.D. and N. Parris, Chitosan/Pectin Laminated Films. Journal of Agricultural Food Chemistry, 1996. 44(7): p. 1915-1919.
60. Wong, D.W.S., et al., Chitosan-lipid films: microstructure and surface energy. Journal of Agricultural and Food Chemistry, 1992. 40(4): p. 540-4.
61. Shepherd, R., S. Reader, and A. Falshaw, Chitosan functional properties. Glycoconjugate journal, 1997. 14: p. 535-542.
62. Krasavtsev, V., et al., Study and selection of chitosan characteristics for packaging materials and preservation of fish products. Advances in Chitin Science, 2002. 5: p. 543-546.
63. Gällstedt, M., Films and Composites Based on Chitosan, Wheat Gluten or Whey Proteins-Their packaging related mechanical and barrier properties, in Fibre and Polymer Technology. 2004, Technical Royal School, KTH: Stockholm.
64. Anker, M., M. Stading, and A.M. Hermansson, Effects of pH and Gel State on the Mechanical Properties, Moisture Coontents, and Glass Transition Temperatures of Whey Protein Films. J. Agric. Food Chem., 1999. 47(5): p. 1878-1886.
65. Swaisgood, H.E., Characteristics of Milk, in Food Chemistry, O.R. Fennema, Editor. 1996, Marcel Dekker, Inc.
66. Foegeding, E.A., et al., Advances in modifying and understanding whey protein functionality. Trends in Food Science & Technology, 2002. 13: p. 151-159.
67. Lindmark-Mansson, H., R. Fonden, and H.-E. Pettersson, Composition of Swedish dairy milk. International Dairy Journal, 2003. 13(6): p. 409-425.
68. Mulvihill, D.M., Functional milk protein products. Special Publication - Royal Society of Chemistry, 1994. 150(Biochemistry of Milk Products): p. 94-113.
69. Maté, J.I. and J.M. Krochta, Comparison of Oxygen and Water Vapor Permeabilities of Whey Protein Isolate and beta-Lactoglobulin Edible Films. J. Agric. Food Chem., 1996. 44(10): p. 3001-3004.
70. Gallstedt, M., et al., Transport and tensile properties of compression-molded wheat gluten films. Biomacromolecules, 2004. 5(5): p. 2020-8.
71. Shellhammer, T.H. and J.M. Krochta, Whey Protein Emulsion Film Performance as Affected by Lipid Type and Amount. Journal of Food Science, 1997. 62(2): p. 390-394.
72. Sothornvit, R. and J.M. Krochta, Plasticizer Effect on Oxygen Permeability of b-Lactoglobulin Films. Journal of Agricultural and Food Chemistry, 2000. 48(12): p. 6298-6302.
73. Anker, M., M. Stading, and A.-M. Hermansson, Mechanical Properties, Water Vapor Permeability, and Moisture Contents of b-Lactoglobulin and Whey Protein Films Using Multivariate Analysis. Journal of Agricultural and Food Chemistry, 1998. 46(5): p. 1820-1829.

References
63
74. Fairley, P., et al., Mechanical Properties and Water Vapor Permeability of Edible Films from Whey Protein Isolate and Sodium Dodecyl Sulfate. J.Agric. Food Chem,, 1996. 44(2): p. 438-443.
75. Mahmoud, R. and P.A. Savello, Mechanical Properties of and Water Vapor Transferability Through Whey Protein Films. Journal of Dairy Science, 1992. 75(4): p. 942-946.
76. Pérez-Gago, M.B., P. Nadaud, and J.M. Krochta, Water Vapor Permeability, Solubility, and Tensile Properties of Heat-denaturated versus Native Whey Protein Films. Journal of Food Science, 1999. 64(6): p. 1034-1037.
77. McHugh, T.H., J.-F. Aujard, and J.M. Krochta, Plasticized Whey Protein Edible Films: Water Vapor Permeability Properties. Journal of Food Science, 1994. 59(2): p. 416-419.
78. Perez-Gago, M.B. and J.M. Krochta, Formation and Properties of Whey Protein Films and Coatings, in Protein based films and Coatings, A. Gennadios, Editor. 2002.
79. Kayserilioglu, B.S., et al., Mechanical and Biochemical Characterization of Wheat Gluten Films as a Function of pH and Co-solvent. Starch/Stärke, 2001. 53: p. 381-386.
80. Guilbert, S., et al., Formation and Properties of Wheat Gluten Films and Coatings, in Protein based films and Coatings, A. Gennadios, Editor. 2002.
81. EUR-Lex, T.C.o.t.E.U.d., DIRECTIVE 2003/30/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 8 May 2003 on the promotion of the use of biofuels or other renewable fuels for transport. Official Journal of the European Union, 2003.
82. Tate&Lyle, A.G., Wheat protein, in www.amylumgroup.com. 83. Jones, R.W., N.W. Taylor, and F.R. Senti, Electrophoresis and fractionation of
wheat gluten. Archives of Biochemistry and Biophysics, 1959. 84(2): p. 363-376. 84. Singh, H. and F. MacRitchie, Application of Polymer Science to Properties of
Gluten. Journal of Cereal Science, 2001. 33(3): p. 231-243. 85. Martin, W.M., Electrokinetic properties of proteins. I. Isoelectric point and
solubility of wheat proteins in aqueous solutions of ethanol. Journal of Physical Chemistry, 1931. 35: p. 2065-90.
86. Wahlund K. -G., et al., Size Characterisation of Wheat Proteins, Particularly Glutenin, by Asymmetrical Flow Field-Flow Fractionation,. Journal of Cereal Science, 1996. 23(2): p. 113-119.
87. Egorov, T.A., et al., Characterisation of high Mr wheat glutenin polymers by agarose gel eletrophoresis and dynamic light scattering. FEBS Letters, 1998. 434(1-2): p. 215-217.
88. Lindsay, M.P. and J.H. Skerritt, The glutenin macropolymer of wheat flour doughs: structure-function perspectives. Trends in Food Science & Technology, 1999. 10(8): p. 247-253.
89. Belton, P.S., On the elasticity of Wheat Gluten. Journal of Cereal Science, 1999. 29: p. 103-107.
90. Morel, M.H., et al., Protein Insolubilization and Thiol Oxidation in Sulfite-Treated Whea Gluten Films during Aging at Various Temperatures and Relative Humidities. Journal Agricultural Food Chemistry, 2000. 48: p. 186-192.

References
64
91. Shewry, P.R. and A.S. Tatham, Disulfide bonds in wheat gluten proteins. Journal of Cereal Science, 1997. 25(3): p. 207-227.
92. Singh, H. and F. MacRitchie, Use of sonication to probe wheat gluten structure. Cereal Chemistry, 2001. 78(5): p. 526-529.
93. Woychik, J.H., J.A. Boundy, and R.J. Dimler, Starch gel electrophoresis of wheat gluten proteins with concentrated urea. Archives of Biochemistry and Biophysics, 1961. 94(3): p. 477-482.
94. Hernandez-Munoz, P., et al., Gliadins polymerized with cysteine: Effects on the physical and water barrier properties of derived films. Biomacromolecules, 2004. 5(4): p. 1503-1510.
95. Hernandez-Munoz, P., R. Villalobos, and A. Chiralt, Effect of thermal treatments on functional properties of edible films made from wheat gluten fractions. Food Hydrocolloids, 2004. 18(4): p. 647-654.
96. Takeda, K., Y. Matsumura, and M. Shimizu, Emulsifying and Surface Properties of Wheat Gluten undes Acidic Conditions. Food Chemistry and Toxicology, 2001. 66(3): p. 393-399.
97. Blanch, E.W., et al., New Insight into the Solution Structures of Wheat Gluten Proteins from Raman Optical Activity. Biochemistry, 2003. 42(19): p. 5665-5673.
98. Anker, C.A., G.A. Foster, Jr., and M.A. Loader, Preparing gluten-containing films and coatings, in U.S. 1972, (General Mills, Inc.). Us. p. 3 pp.
99. Ghezelbash, M., Plasticizers in wheat gluten and whey protein isolate packaging films, in Department of Fibre and Polymer Technology. 2002, Royal Institute of Technology, KTH: Stockholm. p. 47.
100. Gontard, N. and S. Ring, Edible Wheat Gluten Film: Influence of Water Content on Glass Transition Temperature. Journal of Agricultural and Food Chemistry, 1996. 44(11): p. 3474-3478.
101. Micard, V., et al., Properties of Chemically and Physically Treated Whet Gluten Films. Journal Agricultural Food Chemistry, 2000. 48: p. 2948-2953.
102. Wu, Y.V. and R.J. Dimler, Hydrogen-Ion Equilibria of Wheat Gluten. Archives of biochemistry and biophysics FIELD Publication Date:1963 Aug, 1963. 102: p. 230-7. FIELD Reference Number: FIELD Journal Code:0372430 FIELD Call Number:.
103. Lens, J.-P., et al., Influence of processing and storage conditions on the mechanical and barrier properties of films cast from aquous wheat gluten dispersions. Industrial Crops and Products, 2003. 17: p. 119-130.
104. Gontard, N., S. Guilbert, and J.L. Cuq, Water and glycerol as plasticizers affect mechanical and water vapor barrier properties of an edible wheat gluten film. Journal of Food Science, 1993. 58(1): p. 206-211.
105. Larre, C., et al., Properties of deamidated gluten films enzymatically cross-linked. Journal of Agricultural and Food Chemistry, 2000. 48(11): p. 5444-5449.
106. Kayserilioglu, B.S., et al., Drying Temperature and Relative Humidity Effects on Wheat Gluten Film Properties. Journal Agricultural Food Chemistry, 2003. 51: p. 964-968.
107. Paz, H.M. and N. Gontard, Oxygen and Carbon Dioxide Permeability of Wheat Gluten Film: Effect of Relative Humidity and Temperature. Journal of Agricultural and Food Chemistry, 1997. 45(10): p. 4101-4105.

References
65
108. Mangavel, C., et al., Molecular Determinants of the Influence of Hydrophilic Plasticizers on the Mechanical Properties of Cast Wheat Gluten Films. Journal of Agricultural and Food Chemistry, 2003. 51(5): p. 1447-1452.
109. Pommet, M., et al., Study of wheat gluten plasticization with fatty acids. Polymer, 2003. 44: p. 115-122.
110. Pouplin, M., A. Redl, and N. Gontard, Glass Transition of Wheat Gluten Plasticized with Water, Glycerol, or Sorbitol. Journal of Agricultural and Food Chemistry, 1999. 47(2): p. 538-543.
111. Irissin-Mangata, J., et al., New plasticizers for wheat gluten films. European Polymer Journal, 2001. 37(8): p. 1533-1541.
112. Hernandez-Munoz, P. and R.J. Hernandez. Effect of Storage and Hydrophilic Plasticizers on Functional Properties of Wheat Gluten Glutenin Fraction Films. in Worldpak2002, 13th Proc IAPRI Conf. 2002.
113. Micard, V., et al., Thermal properties of raw and processed wheat gluten in relation with protein aggregation. Polymer, 2000. 42(2): p. 477-485.
114. Roy, S., et al., Physical and molecular properties of wheat gluten films cast from heated film-forming solutions. Journal of Food Science, 1999. 64(1): p. 57-60.
115. Di Gioia, L. and S. Guilbert, Corn Protein-Based Thermoplastic Resins: Effect of Some Polar and Amphiphilic Plasticizers. Journal of Agricultural and Food Chemistry, 1999. 47(3): p. 1254-1261.
116. Anker, M., M. Stading, and A.-M. Hermansson, Aging of Whey Protein Films and the Effect on Mechanical and Barrier Properties. Journal of Agricultural and Food Chemistry, 2001. 49(2): p. 989-995.
117. Hernandez-Munoz, P., et al., Formaldehyde cross-linking of gliadin films: Effects on mechanical and water barrier properties. Biomacromolecules, 2004. 5(2): p. 415-421.
118. Galietta, G., et al., Mechanical and Thermomechanical Properties of Films Based on Whey Proteins as Affected by Plasticizer and Crosslinking Agents. J. Dairy Sci., 1998. 81(12): p. 3123-3130.
119. Orliac, O., et al., Effects of additives on the mechanical properties, hydrophobicity and water uptake of thermo-moulded films produced from sunflower protein isolate. Polymer, 2002. 43(20): p. 5417-5425.
120. Rhim, J.W., et al., Solubility, tensile, and color properties of modified soy protein isolate films. Journal of agricultural and food chemistry, 2000. 48(10): p. 4937-41.
121. Mahmoud, R. and P.A. Savello, Solubility and hydrolyzability of films produced by transgluminase catalytic crosslinking of whey protein. J. Dairy Sci., 1993. 76: p. 29-35.
122. Ciesla, K., et al., Gamma irradiation influence on physical properties of milk proteins. Radiation Physics and Chemistry, 2004. 71(1-2): p. 95-99.
123. Sabato, S.F., et al., Mechanical and barrier properties of cross-linked soy and whey protein based films. Journal of Agricultural and Food Chemistry, 2001. 49(3): p. 1397-1403.
124. Lacroix, M., et al., Use of g-irradiation to produce films from whey, casein and soya proteins: structure and functional characteristics. Radiation Physics and Chemistry, 2002. 63(3-6): p. 827-832.

References
66
125. Rhim, J.W., et al., Properties of ultraviolet irradiated protein films. Lebensmittel-Wissenschaft und -Technologie, 1999. 32(3): p. 129-133.
126. Pommet, M., et al., Aggregation and degradation of plasticized wheat gluten during thermo-mechanical treatments, as monitored by rheological and biochemical changes. Polymer, 2004.
127. Quezada Gallo, J.A., et al., Lipid hydrophobicity, physical state and distribution effects on the properties of emulsion-based edible films. Journal of Membrane Science, 2000. 180(1): p. 37-46.
128. McHugh, T.H., Protein-lipid interactions in edible films and coatings. Nahrung, 2000. 44(3): p. 148-151.
129. Gontard, N., et al., Edible composite films of wheat gluten and lipids: water vapor permeability and other physical properties. International Journal of Food Science and Technology, 1994. 29(1): p. 39-50.
130. Carrado, K.A., Synthetic organo- and polymer-clays: preparation, characterization, and materials applications. Applied Clay Science, 2000. 17: p. 1-23.
131. Bertan, L.C., et al., Effect of fatty acids and 'Brazilian elemi' on composite films based on gelatin. Food Hydrocolloids, 2005. 19(1): p. 73-82.
132. Morillon, V., et al., Factors Affecting the Moisture Permeability of Lipid-Based Edible Films: A Review. Critical Reviews in Food Science and Nutrition, 2002. 42(1): p. 67-89.
133. Corradini, E., et al., Mechanical, thermal and morphological properties of poly(e-caprolactone)/zein blends. Polymers for Advanced Technologies, 2004. 15(6): p. 340-345.
134. Averous, L. and N. Boquillon, Biocomposites based on plasticized starch: thermal and mechanical behaviours. Carbohydrate Polymers, 2004. 56(2): p. 111-122.
135. Xu, Y.X., et al., Chitosan-starch composite film: preparation and characterization. Industrial Crops and Products, 2005. 21(2): p. 185-192.
136. Fischer, S., Natural polymers reinforced by inorganic nano-particles. 2005, TNO Industrial Technology: Eindhoven, The Netherlands.
137. John, J., J. Tang, and M. Bhattacharya, Processing of biodegradable blends of wheat gluten and modified polycaprolactone. Polymer, 1998. 39(13): p. 2883-2895.
138. Lim, S.W., et al., Structure and properties of biodegradable gluten/aliphatic polyester blends. Euopean Polymer Journal, 1999. 35: p. 1875-1881.
139. Espert, A., Strategies for improving mechanical properties of polypropylene/cellulose composites, in Fibre and Polymer Technology. 2005, The Royal Institute of Technology: Stockholm.
140. Olabisi, O., L.M. Roberson, and M.T. Show, Polymer-polymer Miscibility. 1979, New York: Academic Press.
141. Zhong, Z. and X.S. Sun, Properties of soy protein isolate/polycaprolactone blends compatibilized by methylene diphenyl diisocyanate. Polymer, 2001. 42(16): p. 6961-6969.
142. Goldberg, D., A review of the biodegradability and utility of poly(caprolactone). Journal of Environmental Polymer Degradation, 1995. 3(2): p. 61-7.

References
67
143. Goldberg, D., et al., Polycaprolactone: a biodegradable additive for olefinic and other polymers. Book Pap. - Int. Nonwoven Fabr. Conf., 1990: p. 55-66.
144. Velde, K.V.d. and P. Kiekens, Biopolymers: overview of several properties and consequences on their applications. Polymer Testing, 2002. 21: p. 433-442.
145. Krook, M., et al., Barrier and mechanical properties of pulp fiber/polymer laminates and blends. Polymer Engineering and Science, 2000. 40(1): p. 143-156.
146. Averous, L., et al., Properties of thermoplastic blends: starch-polycaprolactone. Polymer, 2000. 41(11): p. 4157-4167.
147. Avella, M., et al., Preparation of biodegradable polyesters/high-amylose-starch composites by reactive blending and their characterization. Journal of Applied Polymer Science, 2002. 83(7): p. 1432-1442.
148. Chen, B., K. Sun, and T. Ren, Mechanical and viscoelastic properties of chitin fiber reinforced poly(e-caprolactone). European Polymer Journal, 2005. 41(3): p. 453-457.
149. Zanetti, M., S. Lomakin, and G. Camino, Polymer layered silicate nanocomposites. Macromolecular Materials and Engineering, 2000. 279: p. 1-9.
150. Alexandre, M. and P. Dubois, Polymer-layered silicate nanocomposites: preparation, properties and uses of a new class of materials. Materials Science & Engineering, R: Reports, 2000. R28(1-2): p. 1-63.
151. Okada, A., et al., US Patent, in Polymer Reprints. 1988. p. 447. 152. Giannelis, E.P., R. Krishnamoorti, and E. Manias, Polymer-silicate
nanocomposites: model systems for confined polymers and polymer brushes. Advances in Polymer Science, 1999. 138(Polymers in Confined Environments): p. 107-147.
153. Bharadwaj, R.K., et al., Structure-property relationships in cross-linked polyester-clay nanocomposites. Polymer, 2002. 43(13): p. 3699-3705.
154. Kornmann, X., Synthesis and characterization of thermoset-clay nanocomposites, in Division of Polymer Engineering. 2001, Luleå University of Technology: Luleå.
155. Kornmann, X., H. Lindberg, and L.A. Berglund, Synthesis of epoxy-clay nanocomposites. Influence of the nature of the curing agent on structure. Polymer, 2001. 42(10): p. 4493-4499.
156. Kornmann, X., H. Lindberg, and L.A. Berglund, Synthesis of epoxy-clay nanocomposites: influence of the nature of the clay on structure. Polymer, 2000. 42(4): p. 1303-1310.
157. Kornmann, X., et al., Nanocomposites based on montmorillonite and unsaturated polyester. Polymer Engineering and Science, 1998. 38(8): p. 1351-1358.
158. Usuki, A., et al., Synthesis of nylon 6-clay hybrid. Journal of Materials Research, 1993. 8: p. 1179.
159. Kawasumi, M., et al., Process for producing composite material. 1989, Toyota Motor Co. Japan. p. 1-12.
160. Vaia, R.A., H. Ishii, and E.P. Giannelis, Synthesis and properties of two-dimensional nanostructures by direct intercalation of polymer melts in layered silicates. Chemistry of Materials, 1993. 5(12): p. 1694-6.

References
68
161. Vaia, R.A. and E.P. Giannelis, Polymer Melt Intercalation in Organically-Modified Layered Silicates: Model Predictions and Experiment. Biomacromolecules, 1997. 30: p. 8000-8009.
162. Cho, W.-J. and D.R. Paul, Nylon 6 nanocomposites by melt compounding. Polymer, 2001. 42: p. 1083-1094.
163. Jeon, H.G., et al., Morphology of polymer/silicate nanocomposites. High-density polyethylene and a nitrile copolymer. Polymer Bulletin (Berlin), 1998. 41(1): p. 107-113.
164. Sohn, J.I., et al., Viscoelasticity and relaxation characteristics of polystyrene/clay nanocomposite. Journal of Materials Science, 2003. 38(9): p. 1849-1852.
165. Redl, A., et al., Rheological Properties of gluten plasticized with glycerol: dependence on temperature, glycerol content and mixing conditions. Rheol. Acta, 1999. 38: p. 311-320.
166. Domenek, S., et al., Biodegradability of wheat gluten based bioplastics. Chemosphere, 2004. 54(4): p. 551-559.
167. Muzzarelli, R.A.A., Natural Chelating Polymers; Alginic Acid, Chitin, and Chitosan. 1974. 254 pp.
168. Hedenqvist, M. and U.W. Gedde, Diffusion of small-molecule penetrants in semicrystalline polymers. Progress in Polymer Science, 1996. 21(2): p. 299-333.
169. Fricke, H., A Mathematical Treatment of the Electric Conductivity and Capacity of Disperse Systems I. THE ELECTRIC CONDUCTIVITY OF A SUSPENSION OF HOMOGENEOUS SPHEROIDS. Physical Review, 1924. 24: p. 575-587.
170. Socrates, G., Infrared and Raman Characteristics Group Frequencies. 3rd. Ed. ed. 2001: Wiley.
171. Backman, A., J. Lange, and M.S. Hedenqvist, Transport properties of uniaxially oriented aliphatic polyketone. Journal of Polymer Science, Part B: Polymer Physics, 2004. 42: p. 947-955.
172. Wu, Y.V. and R.J. Dimler, Hydrogen-Ion Equilibria of Wheat Gluten in Guanidine-Hydrochloride Solutions. Archives of biochemistry and biophysics, 1964. 108: p. 490-6.
173. Mangels, C.E. and C.H. Baileys, Relation of concentration to action of gelatinizing agents on starch. Journal American Chemical Society, 1993. 55: p. 1981-1988.
174. Oosten, B.J., Interactions between starch and electrolytes. Starch/Stärke, 1990. 42: p. 327-330.
175. Eastman, J.E., Cold-water-soluble granular starch for gelled food compositions. 1984: U.S.A.
176. Chen, J. and J. Jane, Preparation of granular cold-water-soluble starches prepared by alcoholic-alkaline treatment. Cereal Chemistry, 1994. 71: p. 618-622.
177. Krook, M., Packaging Related Properties of Polymer Nanocomposites and Pulp Fiber/Polymer Blends and Laminates, in Fibre and Polymer Technology. 2004, Institute of Royal Technology, KTH: Stockholm.
178. Nielsen, L.E., Models for the permeability of filled polymer systems. Journal of Macromolecular Science, Chemistry, 1967. A1(5): p. 929-942.





























