Embed Size (px)
Citation preview

Structural asymmetry in a conserved signaling systemthat regulates division, replication, and virulence ofan intracellular pathogenJonathan W. Willetta,b, Julien Herroua,b, Ariane Briegelc, Grant Rotskoffa, and Sean Crossona,b,d,1
aDepartment of Biochemistry and Molecular Biology, University of Chicago, Chicago, IL 60637; bHoward Taylor Ricketts Laboratory, University of Chicago,Argonne National Laboratory, Argonne, IL 60439; cDivision of Biology and Biological Engineering, California Institute of Technology, Pasadena, CA 91125;and dThe Committee on Microbiology, University of Chicago, Chicago, IL 60637
Edited by Graham C. Walker, Massachusetts Institute of Technology, Cambridge, MA, and approved June 2, 2015 (received for review February 13, 2015)
We have functionally and structurally defined an essential proteinphosphorelay that regulates expression of genes required forgrowth, division, and intracellular survival of the global zoonoticpathogen Brucella abortus. Our study delineates phosphoryl trans-fer through this molecular pathway, which initiates from the sensorkinase CckA and proceeds through the ChpT phosphotransferase totwo regulatory substrates: CtrA and CpdR. Genetic perturbation ofthis system results in defects in cell growth and division site selec-tion, and a specific viability deficit inside human phagocytic cells.Thus, proper control of B. abortus division site polarity is necessaryfor survival in the intracellular niche. We further define the struc-tural foundations of signaling from the central phosphotransferase,ChpT, to its response regulator substrate, CtrA, and provide evi-dence that there are at least two modes of interaction betweenChpT and CtrA, only one of which is competent to catalyze phos-phoryltransfer. The structure and dynamics of the active site on eachside of the ChpT homodimer are distinct, supporting a model inwhich quaternary structure of the 2:2 ChpT–CtrA complex enforcesan asymmetric mechanism of phosphoryl transfer between ChpTand CtrA. Our study provides mechanistic understanding, from thecellular to the atomic scale, of a conserved transcriptional regulatorysystem that controls the cellular and infection biology of B. abortus.More generally, our results provide insight into the structural basisof two-component signal transduction, which is broadly conservedin bacteria, plants, and fungi.
Brucella abortus | two-component system | cell cycle | ChpT | CtrA
Brucellosis, caused by Brucella spp., is among the most com-mon zoonotic diseases worldwide (1). These intracellular
pathogens are estimated to cause at least 500,000 new humaninfections each year and, in areas of Africa, Asia, and SouthAmerica, inflict significant agricultural losses due to decreasedlivestock production (2, 3). Survival of Brucella within mam-mals is linked to their ability to infect and survive insideprofessional phagocytic cells (2). If left untreated in humanhosts, Brucella eventually spread to multiple tissue types,which can lead to a range of debilitating chronic sequelaeincluding reticuloendothelial, cardiovascular, gastrointestinal,and neurological damage.Brucella are members of the α-proteobacteria, a diverse class of
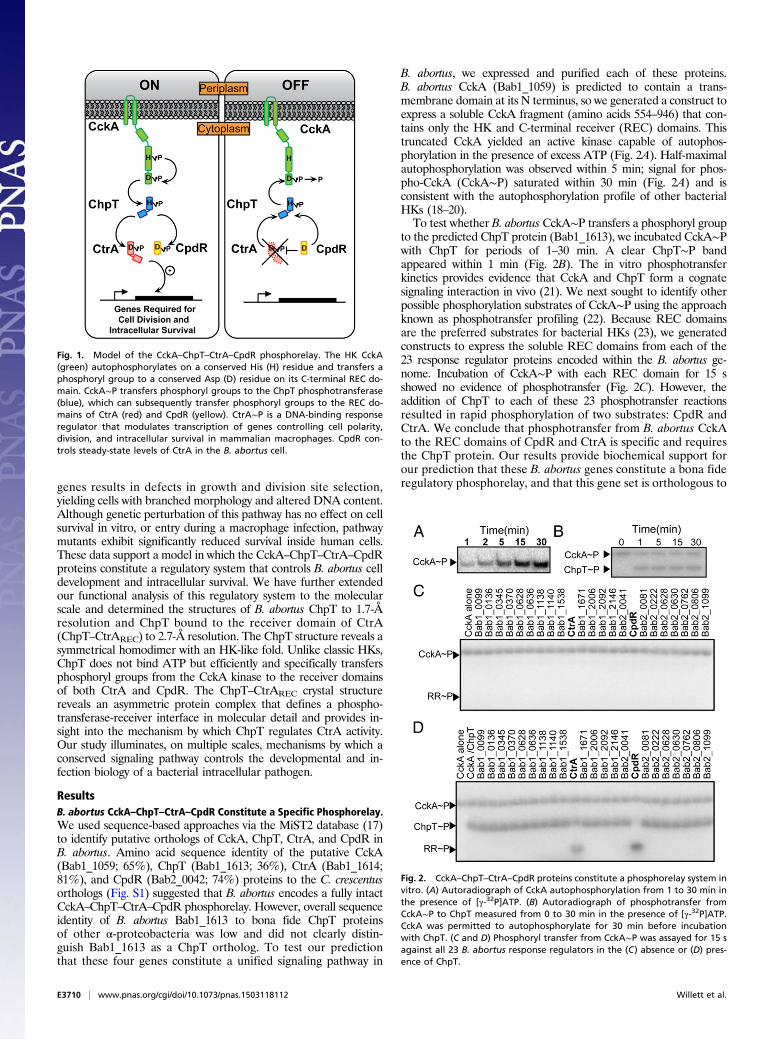
Gram-negative species adapted for growth across a range of en-vironmental conditions including plant surfaces and roots, aquaticand soil ecosystems, and the interior of mammalian cells (4, 5).Among the central regulatory systems controlling the α-proteo-bacterial cell cycle is a multistep phosphorelay composed of fourproteins: (i) the hybrid sensor histidine kinase (HK) CckA, (ii) thehistidine phosphotransferase ChpT, (iii) the DNA-binding re-sponse regulator CtrA, and (iv) the phospho-receiver proteinCpdR (Fig. 1). Our current understanding of this conserved reg-ulatory system is based largely on studies of the related aquaticbacterium Caulobacter crescentus (6, 7). In Caulobacter, auto-phosphorylated CckA transfers phosphoryl groups to a conserved
histidine on ChpT. ChpT∼P subsequently transfers phosphorylgroups to either CpdR or CtrA (8). CtrA is a regulator of cell cycleand developmental gene transcription; its activity is controlled byphosphorylation and by proteolysis. Specifically, CtrA is active as atranscription factor when phosphorylated (CtrA∼P); CtrA proteinis stabilized in the cell by CpdR∼P and is proteolyzed when CpdRis in its unphosphorylated form (9). Thus, ChpT regulates CtrA-dependent transcription both by phosphorylating CtrA and con-trolling CtrA protein stability via CpdR.The genes encoding the CckA–ChpT–CtrA–CpdR regulatory
system have been identified in several α-proteobacteria. However,there is notable diversity in the transcriptional output of this sys-tem across the clade (10–13), which likely reflects the breadth ofniches inhabited by these species (14). The function of this systemin Brucella abortus, which is capable of infecting, growing, andreplicating inside mammalian cells, is poorly understood, althoughprevious studies of Brucella CtrA have revealed a possible role inthe control of cell division (15). It is further known that geneticperturbation of the DNA methyltransferase CcrM, which is di-rectly regulated by CtrA, results in a virulence defect (16).In this study, we define the full set of genes encoding the
B. abortus CckA–ChpT–CtrA–CpdR phosphorelay and charac-terize molecular and structural requirements of signaling throughthis conserved pathway. These four proteins comprise an essentialphosphorelay that controls B. abortus cell growth, division, andinfection biology. Expression of conditional mutant alleles of these
Significance
Brucella abortus is an intracellular bacterial pathogen that in-flicts a significant health burden on both humans and theirlivestock on a global scale. We demonstrate that an essentialregulatory system controls the growth and morphology ofB. abortus, and that this system is required for survival insidemammalian host cells. Using experimental and computationaltools of structural biology, we further define how the proteincomponents of this regulatory pathway interact at the atomicscale. Our results provide evidence for multiple, asymmetricmodes of binding between essential pathway proteins thatcontrol transcription. The multimodal molecular interactions weobserve provide evidence for new layers of allosteric control ofthis conserved gene regulatory system.
Author contributions: J.W.W. and S.C. designed research; J.W.W., J.H., A.B., and G.R. per-formed research; J.W.W., J.H., A.B., G.R., and S.C. contributed new reagents/analytic tools;J.W.W., J.H., A.B., G.R., and S.C. analyzed data; and J.W.W. and S.C. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors have beendeposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4QPK and 4QPJ).1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1503118112/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1503118112 PNAS | Published online June 29, 2015 | E3709–E3718
MICRO
BIOLO
GY
PNASPL
US
genes results in defects in growth and division site selection,yielding cells with branched morphology and altered DNA content.Although genetic perturbation of this pathway has no effect on cellsurvival in vitro, or entry during a macrophage infection, pathwaymutants exhibit significantly reduced survival inside human cells.These data support a model in which the CckA–ChpT–CtrA–CpdRproteins constitute a regulatory system that controls B. abortus celldevelopment and intracellular survival. We have further extendedour functional analysis of this regulatory system to the molecularscale and determined the structures of B. abortus ChpT to 1.7-Åresolution and ChpT bound to the receiver domain of CtrA(ChpT–CtrAREC) to 2.7-Å resolution. The ChpT structure reveals asymmetrical homodimer with an HK-like fold. Unlike classic HKs,ChpT does not bind ATP but efficiently and specifically transfersphosphoryl groups from the CckA kinase to the receiver domainsof both CtrA and CpdR. The ChpT–CtrAREC crystal structurereveals an asymmetric protein complex that defines a phospho-transferase-receiver interface in molecular detail and provides in-sight into the mechanism by which ChpT regulates CtrA activity.Our study illuminates, on multiple scales, mechanisms by which aconserved signaling pathway controls the developmental and in-fection biology of a bacterial intracellular pathogen.
ResultsB. abortus CckA–ChpT–CtrA–CpdR Constitute a Specific Phosphorelay.We used sequence-based approaches via the MiST2 database (17)to identify putative orthologs of CckA, ChpT, CtrA, and CpdR inB. abortus. Amino acid sequence identity of the putative CckA(Bab1_1059; 65%), ChpT (Bab1_1613; 36%), CtrA (Bab1_1614;81%), and CpdR (Bab2_0042; 74%) proteins to the C. crescentusorthologs (Fig. S1) suggested that B. abortus encodes a fully intactCckA–ChpT–CtrA–CpdR phosphorelay. However, overall sequenceidentity of B. abortus Bab1_1613 to bona fide ChpT proteinsof other α-proteobacteria was low and did not clearly distin-guish Bab1_1613 as a ChpT ortholog. To test our predictionthat these four genes constitute a unified signaling pathway in
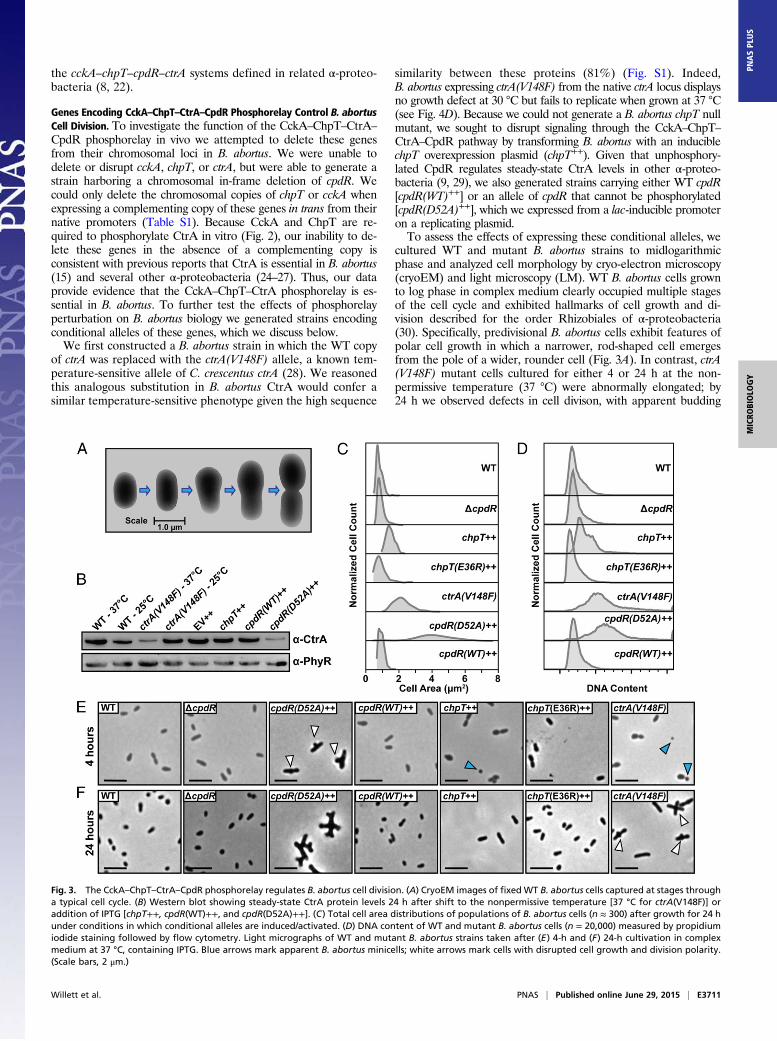
B. abortus, we expressed and purified each of these proteins.B. abortus CckA (Bab1_1059) is predicted to contain a trans-membrane domain at its N terminus, so we generated a construct toexpress a soluble CckA fragment (amino acids 554–946) that con-tains only the HK and C-terminal receiver (REC) domains. Thistruncated CckA yielded an active kinase capable of autophos-phorylation in the presence of excess ATP (Fig. 2A). Half-maximalautophosphorylation was observed within 5 min; signal for phos-pho-CckA (CckA∼P) saturated within 30 min (Fig. 2A) and isconsistent with the autophosphorylation profile of other bacterialHKs (18–20).To test whether B. abortus CckA∼P transfers a phosphoryl group
to the predicted ChpT protein (Bab1_1613), we incubated CckA∼Pwith ChpT for periods of 1–30 min. A clear ChpT∼P bandappeared within 1 min (Fig. 2B). The in vitro phosphotransferkinetics provides evidence that CckA and ChpT form a cognatesignaling interaction in vivo (21). We next sought to identify otherpossible phosphorylation substrates of CckA∼P using the approachknown as phosphotransfer profiling (22). Because REC domainsare the preferred substrates for bacterial HKs (23), we generatedconstructs to express the soluble REC domains from each of the23 response regulator proteins encoded within the B. abortus ge-nome. Incubation of CckA∼P with each REC domain for 15 sshowed no evidence of phosphotransfer (Fig. 2C). However, theaddition of ChpT to each of these 23 phosphotransfer reactionsresulted in rapid phosphorylation of two substrates: CpdR andCtrA. We conclude that phosphotransfer from B. abortus CckAto the REC domains of CpdR and CtrA is specific and requiresthe ChpT protein. Our results provide biochemical support forour prediction that these B. abortus genes constitute a bona fideregulatory phosphorelay, and that this gene set is orthologous to
Fig. 1. Model of the CckA–ChpT–CtrA–CpdR phosphorelay. The HK CckA(green) autophosphorylates on a conserved His (H) residue and transfers aphosphoryl group to a conserved Asp (D) residue on its C-terminal REC do-main. CckA∼P transfers phosphoryl groups to the ChpT phosphotransferase(blue), which can subsequently transfer phosphoryl groups to the REC do-mains of CtrA (red) and CpdR (yellow). CtrA∼P is a DNA-binding responseregulator that modulates transcription of genes controlling cell polarity,division, and intracellular survival in mammalian macrophages. CpdR con-trols steady-state levels of CtrA in the B. abortus cell.
Fig. 2. CckA–ChpT–CtrA–CpdR proteins constitute a phosphorelay system invitro. (A) Autoradiograph of CckA autophosphorylation from 1 to 30 min inthe presence of [γ-32P]ATP. (B) Autoradiograph of phosphotransfer fromCckA∼P to ChpT measured from 0 to 30 min in the presence of [γ-32P]ATP.CckA was permitted to autophosphorylate for 30 min before incubationwith ChpT. (C and D) Phosphoryl transfer from CckA∼P was assayed for 15 sagainst all 23 B. abortus response regulators in the (C) absence or (D) pres-ence of ChpT.
E3710 | www.pnas.org/cgi/doi/10.1073/pnas.1503118112 Willett et al.
the cckA–chpT–cpdR–ctrA systems defined in related α-proteo-bacteria (8, 22).
Genes Encoding CckA–ChpT–CtrA–CpdR Phosphorelay Control B. abortusCell Division. To investigate the function of the CckA–ChpT–CtrA–CpdR phosphorelay in vivo we attempted to delete these genesfrom their chromosomal loci in B. abortus. We were unable todelete or disrupt cckA, chpT, or ctrA, but were able to generate astrain harboring a chromosomal in-frame deletion of cpdR. Wecould only delete the chromosomal copies of chpT or cckA whenexpressing a complementing copy of these genes in trans from theirnative promoters (Table S1). Because CckA and ChpT are re-quired to phosphorylate CtrA in vitro (Fig. 2), our inability to de-lete these genes in the absence of a complementing copy isconsistent with previous reports that CtrA is essential in B. abortus(15) and several other α-proteobacteria (24–27). Thus, our dataprovide evidence that the CckA–ChpT–CtrA phosphorelay is es-sential in B. abortus. To further test the effects of phosphorelayperturbation on B. abortus biology we generated strains encodingconditional alleles of these genes, which we discuss below.We first constructed a B. abortus strain in which the WT copy
of ctrA was replaced with the ctrA(V148F) allele, a known tem-perature-sensitive allele of C. crescentus ctrA (28). We reasonedthis analogous substitution in B. abortus CtrA would confer asimilar temperature-sensitive phenotype given the high sequence
similarity between these proteins (81%) (Fig. S1). Indeed,B. abortus expressing ctrA(V148F) from the native ctrA locus displaysno growth defect at 30 °C but fails to replicate when grown at 37 °C(see Fig. 4D). Because we could not generate a B. abortus chpT nullmutant, we sought to disrupt signaling through the CckA–ChpT–CtrA–CpdR pathway by transforming B. abortus with an induciblechpT overexpression plasmid (chpT++). Given that unphosphory-lated CpdR regulates steady-state CtrA levels in other α-proteo-bacteria (9, 29), we also generated strains carrying either WT cpdR[cpdR(WT)++] or an allele of cpdR that cannot be phosphorylated[cpdR(D52A)++], which we expressed from a lac-inducible promoteron a replicating plasmid.To assess the effects of expressing these conditional alleles, we
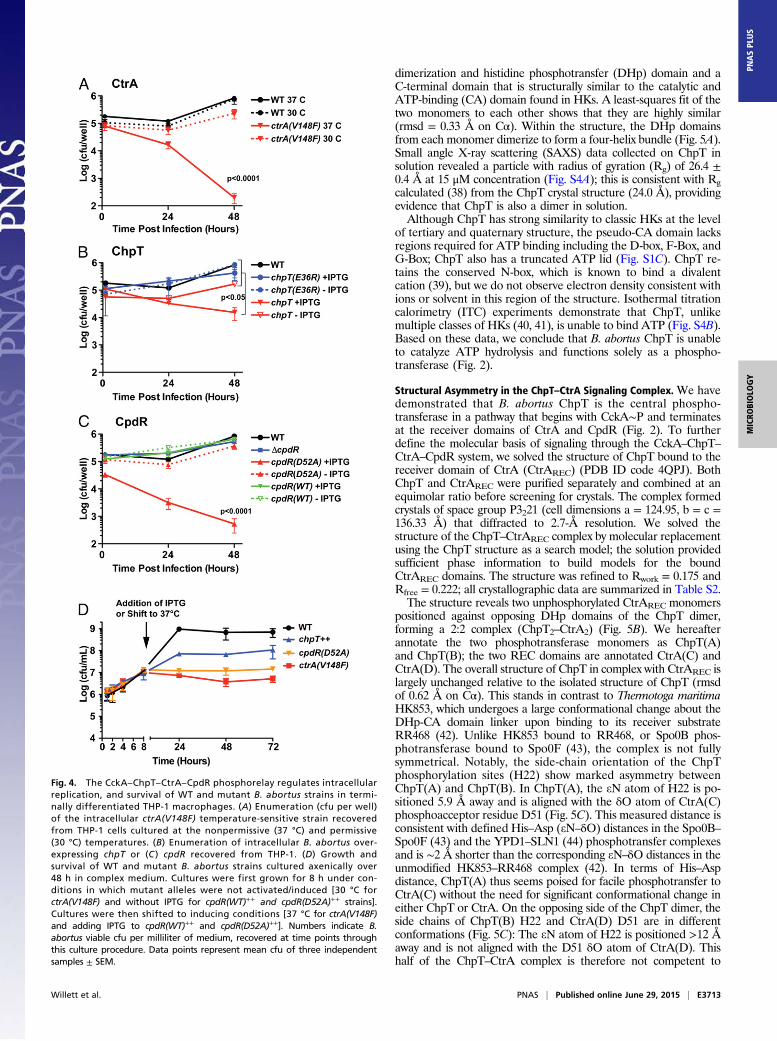
cultured WT and mutant B. abortus strains to midlogarithmicphase and analyzed cell morphology by cryo-electron microscopy(cryoEM) and light microscopy (LM). WT B. abortus cells grownto log phase in complex medium clearly occupied multiple stagesof the cell cycle and exhibited hallmarks of cell growth and di-vision described for the order Rhizobiales of α-proteobacteria(30). Specifically, predivisional B. abortus cells exhibit features ofpolar cell growth in which a narrower, rod-shaped cell emergesfrom the pole of a wider, rounder cell (Fig. 3A). In contrast, ctrA(V148F) mutant cells cultured for either 4 or 24 h at the non-permissive temperature (37 °C) were abnormally elongated; by24 h we observed defects in cell divison, with apparent budding
Fig. 3. The CckA–ChpT–CtrA–CpdR phosphorelay regulates B. abortus cell division. (A) CryoEM images of fixed WT B. abortus cells captured at stages througha typical cell cycle. (B) Western blot showing steady-state CtrA protein levels 24 h after shift to the nonpermissive temperature [37 °C for ctrA(V148F)] oraddition of IPTG [chpT++, cpdR(WT)++, and cpdR(D52A)++]. (C) Total cell area distributions of populations of B. abortus cells (n ≈ 300) after growth for 24 hunder conditions in which conditional alleles are induced/activated. (D) DNA content of WT and mutant B. abortus cells (n = 20,000) measured by propidiumiodide staining followed by flow cytometry. Light micrographs of WT and mutant B. abortus strains taken after (E) 4-h and (F) 24-h cultivation in complexmedium at 37 °C, containing IPTG. Blue arrows mark apparent B. abortus minicells; white arrows mark cells with disrupted cell growth and division polarity.(Scale bars, 2 μm.)
Willett et al. PNAS | Published online June 29, 2015 | E3711
MICRO
BIOLO
GY
PNASPL
US

from the midcell position (Fig. 3 E and F and Fig. S2A). CtrA(V148F) protein is still present in the cell after growth at 37 °C for24 h, although at slightly decreased levels (Fig. 3B). Induction ofchpT expression (chpT++) by the addition of isopropyl β-D-1-thi-ogalactopyranoside (IPTG) for 4 and 24 h increased the pro-portion of elongated cells; we further observed the presence ofminicells in these cultures (Fig. 3 E and F and Fig. S2B).Finally, we measured the effects of cpdR allele expression on
B. abortus cell morphology and on steady-state CtrA levels. Be-cause unphosphorylated CpdR activates proteolysis of CtrA inC. crescentus (29), we tested whether unphosphorylated CpdR af-fects steady-state levels of CtrA in B. abortus. After inducing cpdR(WT)++ or cpdR(D52A)++ expression with IPTG for 24 h, we ob-served a significant reduction in CtrA protein in the strain ex-pressing cpdR(D52A)++, which is missing the site of aspartylphosphorylation. Expression of cpdR(WT)++ had no effect onsteady-state CtrA levels assessed by Western blot (Fig. 3B). Thisresult is consistent with a model in which unphosphorylated CpdRdestabilizes CtrA in B. abortus. We further analyzed cpdR(WT)++
and cpdR(D52A)++ overexpression strains by cryoEM and LM andobserved large elongated cells with apparent budding at the midcellafter 4 h of cpdR(D52A)++ induction (Fig. 3E); after 24 h of in-duction we observed large B. abortus cells that were highly branched,which is consistent with a defect in proper cell division (Fig. 3Fand Fig. S2). Overexpression of cpdR(WT)++ or deletion of cpdR(ΔcpdR) did not result in a gross defect in cell morphology (Fig. 3).CryoEM and LM images of a small number of cells provide
evidence that perturbation of the CckA–ChpT–CtrA–CpdRpathway affects cell growth and division. We next imaged a largenumber of fixed WT and mutant B. abortus cells, extracted the2D cell contours, and analyzed these cell contours to determine2D cell area (31). This analysis permitted us to more thoroughlyquantify variation in cell area across all strains. After 24 h ofgrowth at 37 °C in complex medium (adding IPTG where ap-plicable) the mean cell area of WT B. abortus was 0.67 ± 0.19 μm2.However, mean cell areas of ctrA(V148F) (2.1 ± 0.57 μm2), chpT++
(1.1 ± 0.26 μm2), and cpdR(D52A)++ (3.9 ± 1.4 μm2) were signif-icantly increased (one-way ANOVA; Dunnett’s post test; P <0.0001) compared with WT. The ΔcpdR (0.75 ± 0.22 μm2) andcpdR(WT)++ (0.79 ± 0.19 μm2) strains exhibited less increase in cellarea relative to WT but were still significantly larger than WT (P <0.001). We further tested whether the DNA content of these mu-tant strains differed from WT B. abortus by staining DNA withpropidium iodide and analyzing DNA content using flow cytom-etry. The ctrA(V148F), chpT++, and cpdR(D52A)++ strains all havehighly increased DNA content relative to WT (Fig. 3D), consistentwith the gross cell division defects of these mutants.To assess the impact that CckA–ChpT–CtrA–CpdR pathway
perturbation has on CtrA-dependent transcription, we quantifiedtranscript levels of the CtrA-regulated gene ccrM (15), by quanti-tative reverse-transcription PCR (qRT-PCR). After 4 h of growthat the nonpermissive temperature or addition of IPTG, ctrA(V148F), chpT++, and cpdR(D52A)++ strains have significantly de-creased ccrM transcript abundance compared with WT B. abortusor an empty vector control (Fig. S3). We conclude that thesegenetic perturbations reduce CtrA-dependent transcription inB. abortus. Overall, our data support a model in which phosphor-elay through the CckA–ChpT–CtrA–CpdR pathway regulatesB. abortus processes that determine cell growth, cell cycle, and celldivision via control of CtrA-dependent transcription.
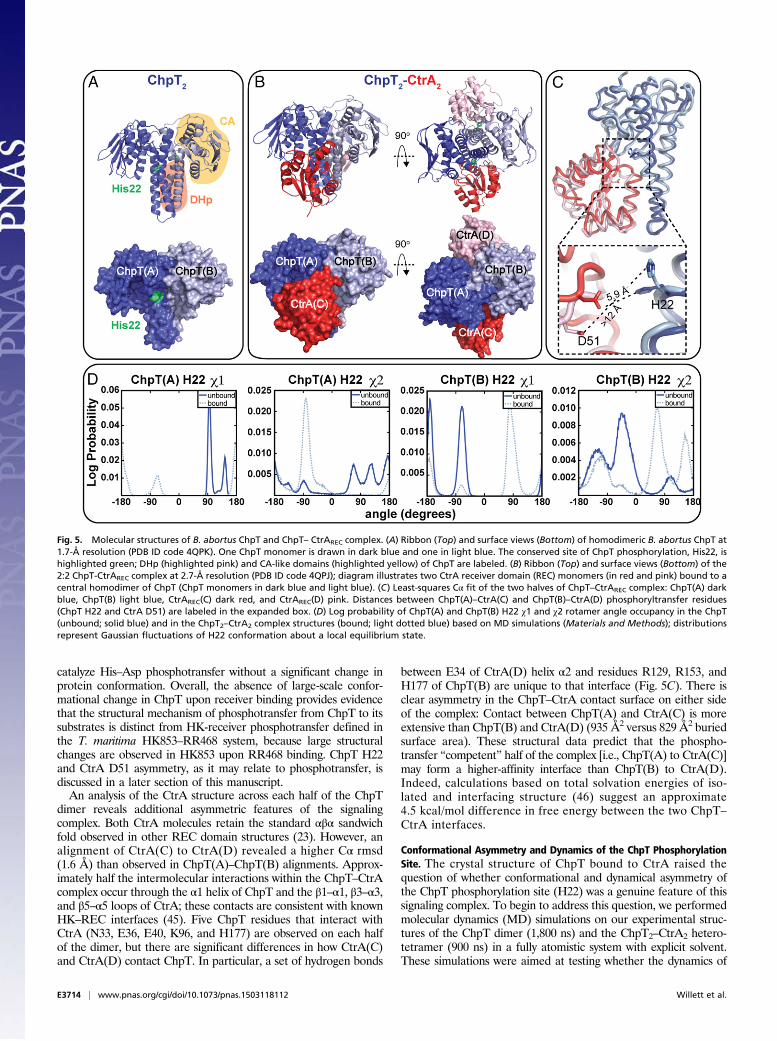
The CckA–ChpT–CtrA–CpdR Pathway Is Required for Intracellular Survivalof B. abortus in Human Macrophages. A natural niche of B. abortus isthe interior of mammalian cells. We assessed the effect CckA–ChpT–CtrA–CpdR pathway perturbation has on entry, replication,and survival inside terminally differentiated THP-1 macrophages.Before infection, overnight cultures ofWT and conditional B. abortusmutant strains were grown under noninducing conditions in rich
medium. Conditional alleles were then activated/induced by cul-turing for 4 h at 37 °C with IPTG (where indicated) before infectingmacrophages. WT and mutant B. abortus cells were added to THP-1 cells at a multiplicity of infection (MOI) of 100 cfu per macro-phage. The initial 1 h post infection (hpi) time point in theseexperiments generally reflects B. abortus entry. No statisticallysignificant differences were observed at this time point, indicatingthere is no defect in macrophage entry of any mutant strain (Fig. 4A–C). In contrast, there is a significant reduction in intracellularB. abortus replication and/or survival in the ctrA(V148F), chpT++,and cpdR(D52A)++ strains relative to WT and uninduced controlsat 48 hpi. The number of ctrA(V148F) cells isolated from THP-1(at 37 °C) is ∼1 log lower at 24 hpi and 3.5 logs lower than WTB. abortus at 48 hpi (Fig. 4A). ctrA(V148F) infections performed atthe permissive temperature (30 °C) revealed no significant decreasein cfu at 48 hpi compared with WT cultured under equivalentconditions. Overexpression of chpT++ resulted in a 1.0 log decreasein the number of cells recovered from THP-1 compared withuninduced and empty vector controls at 48 hpi (Fig. 4B). The mostsevere intracellular defect was observed in the strain overexpressingcpdR(D52A)++: We observed a 1.5 log decrease in cells recoveredfrom THP-1 at 24 hpi and a 3.5 log reduction of recovered cells at48 hpi relative to the uninduced control. There was no difference inrecovery of B. abortus expressing cpdR(WT)++ or the ΔcpdR in-frame deletion strain relative to WT (Fig. 4).We next tested whether mutant attenuation observed in mac-
rophages is due to a general loss of B. abortus viability, or whetherCckA–ChpT–CtrA–CpdR pathway perturbation results in a rep-lication/survival defect that is specific to the intracellular niche.We first cultured B. abortus ctrA(V148F), chpT++, and cpdR(D52A)++ conditional mutants under noninducing conditions inliquid growth medium for 8 h, enumerating bacteria over thisgrowth period. At 8 h (or 1 × 107 cfu/mL) we shifted to the re-strictive temperature (37 °C) and induced expression of conditionalalleles with IPTG; we chose to shift cells at this point because thiswas the approximate density of cells used for macrophage in-fections. We enumerated cfu of these induced/activated B. abortusmutants at similar time points assayed in our THP-1 macrophageinfection experiments (ref. 24, 48 h). Upon induction/activation, thectrA(V148F) and cpdR(D52A)++ mutants were completely inhibitedin replication whereas the chpT++ mutant replicated, but at a lowerrate than WT. In contrast to what we observed in macrophages,neither the ctrA(V148F), chpT++, nor cpdR(D52A)++ strains lostviability under these conditions (over 48 h) (Fig. 4D). We concludethe reduced numbers of ctrA(V148F), chpT++, and cpdR(D52A)++
cells recovered from THP-1 reflects a specific requirement forthe CckA–ChpT–CtrA–CpdR pathway for survival inside macro-phages. We further conclude that this signaling pathway does notgenerally control features of the cell required for B. abortus entryinto mammalian macrophages.
ChpT: A Histidine Phosphotransferase with an HK-Like Structure.Having established the importance of the CckA–ChpT–CtrA–CpdR system in B. abortus cellular and infection biology, we nextsought to characterize the structural basis of phosphotransferthrough this conserved pathway. To this end, we purified, crystal-lized, and solved the structure of the ChpT phosphotransferase(PDB ID code 4QPK). ChpT formed tetragonal crystals of spacegroup P43 (a = b = 70.84, c = 87.01 Å) that diffracted to 1.7-Åresolution; we phased the ChpT crystal structure by molecular re-placement using a model based on structures of a C. crescentushomolog (32, 33). B. abortus ChpT was refined to Rwork = 0.164and Rfree = 0.188. Crystallographic data and refinement statisticsare summarized in Table S2.The crystallographic asymmetric unit contains two ChpT mole-
cules, organized as a symmetric dimer. The structure is similar toHKs (34–36) and other histidine phosphotransfer (Hpt) proteins(32, 33, 37). Each ChpT monomer is composed of an N-terminal
E3712 | www.pnas.org/cgi/doi/10.1073/pnas.1503118112 Willett et al.
dimerization and histidine phosphotransfer (DHp) domain and aC-terminal domain that is structurally similar to the catalytic andATP-binding (CA) domain found in HKs. A least-squares fit of thetwo monomers to each other shows that they are highly similar(rmsd = 0.33 Å on Cα). Within the structure, the DHp domainsfrom each monomer dimerize to form a four-helix bundle (Fig. 5A).Small angle X-ray scattering (SAXS) data collected on ChpT insolution revealed a particle with radius of gyration (Rg) of 26.4 ±0.4 Å at 15 μM concentration (Fig. S4A); this is consistent with Rgcalculated (38) from the ChpT crystal structure (24.0 Å), providingevidence that ChpT is also a dimer in solution.Although ChpT has strong similarity to classic HKs at the level
of tertiary and quaternary structure, the pseudo-CA domain lacksregions required for ATP binding including the D-box, F-Box, andG-Box; ChpT also has a truncated ATP lid (Fig. S1C). ChpT re-tains the conserved N-box, which is known to bind a divalentcation (39), but we do not observe electron density consistent withions or solvent in this region of the structure. Isothermal titrationcalorimetry (ITC) experiments demonstrate that ChpT, unlikemultiple classes of HKs (40, 41), is unable to bind ATP (Fig. S4B).Based on these data, we conclude that B. abortus ChpT is unableto catalyze ATP hydrolysis and functions solely as a phospho-transferase (Fig. 2).
Structural Asymmetry in the ChpT–CtrA Signaling Complex. We havedemonstrated that B. abortus ChpT is the central phospho-transferase in a pathway that begins with CckA∼P and terminatesat the receiver domains of CtrA and CpdR (Fig. 2). To furtherdefine the molecular basis of signaling through the CckA–ChpT–CtrA–CpdR system, we solved the structure of ChpT bound to thereceiver domain of CtrA (CtrAREC) (PDB ID code 4QPJ). BothChpT and CtrAREC were purified separately and combined at anequimolar ratio before screening for crystals. The complex formedcrystals of space group P3221 (cell dimensions a = 124.95, b = c =136.33 Å) that diffracted to 2.7-Å resolution. We solved thestructure of the ChpT–CtrAREC complex by molecular replacementusing the ChpT structure as a search model; the solution providedsufficient phase information to build models for the boundCtrAREC domains. The structure was refined to Rwork = 0.175 andRfree = 0.222; all crystallographic data are summarized in Table S2.The structure reveals two unphosphorylated CtrAREC monomers
positioned against opposing DHp domains of the ChpT dimer,forming a 2:2 complex (ChpT2–CtrA2) (Fig. 5B). We hereafterannotate the two phosphotransferase monomers as ChpT(A)and ChpT(B); the two REC domains are annotated CtrA(C) andCtrA(D). The overall structure of ChpT in complex with CtrAREC islargely unchanged relative to the isolated structure of ChpT (rmsdof 0.62 Å on Cα). This stands in contrast to Thermotoga maritimaHK853, which undergoes a large conformational change about theDHp-CA domain linker upon binding to its receiver substrateRR468 (42). Unlike HK853 bound to RR468, or Spo0B phos-photransferase bound to Spo0F (43), the complex is not fullysymmetrical. Notably, the side-chain orientation of the ChpTphosphorylation sites (H22) show marked asymmetry betweenChpT(A) and ChpT(B). In ChpT(A), the eN atom of H22 is po-sitioned 5.9 Å away and is aligned with the δO atom of CtrA(C)phosphoacceptor residue D51 (Fig. 5C). This measured distance isconsistent with defined His–Asp (eN–δO) distances in the Spo0B–Spo0F (43) and the YPD1–SLN1 (44) phosphotransfer complexesand is ∼2 Å shorter than the corresponding eN–δO distances in theunmodified HK853–RR468 complex (42). In terms of His–Aspdistance, ChpT(A) thus seems poised for facile phosphotransfer toCtrA(C) without the need for significant conformational change ineither ChpT or CtrA. On the opposing side of the ChpT dimer, theside chains of ChpT(B) H22 and CtrA(D) D51 are in differentconformations (Fig. 5C): The eN atom of H22 is positioned >12 Åaway and is not aligned with the D51 δO atom of CtrA(D). Thishalf of the ChpT–CtrA complex is therefore not competent to
Fig. 4. The CckA–ChpT–CtrA–CpdR phosphorelay regulates intracellularreplication, and survival of WT and mutant B. abortus strains in termi-nally differentiated THP-1 macrophages. (A) Enumeration (cfu per well)of the intracellular ctrA(V148F) temperature-sensitive strain recoveredfrom THP-1 cells cultured at the nonpermissive (37 °C) and permissive(30 °C) temperatures. (B) Enumeration of intracellular B. abortus over-expressing chpT or (C ) cpdR recovered from THP-1. (D) Growth andsurvival of WT and mutant B. abortus strains cultured axenically over48 h in complex medium. Cultures were first grown for 8 h under con-ditions in which mutant alleles were not activated/induced [30 °C forctrA(V148F) and without IPTG for cpdR(WT)++ and cpdR(D52A)++ strains].Cultures were then shifted to inducing conditions [37 °C for ctrA(V148F)and adding IPTG to cpdR(WT)++ and cpdR(D52A)++]. Numbers indicate B.abortus viable cfu per milliliter of medium, recovered at time points throughthis culture procedure. Data points represent mean cfu of three independentsamples ± SEM.
Willett et al. PNAS | Published online June 29, 2015 | E3713
MICRO
BIOLO
GY
PNASPL
US
catalyze His–Asp phosphotransfer without a significant change inprotein conformation. Overall, the absence of large-scale confor-mational change in ChpT upon receiver binding provides evidencethat the structural mechanism of phosphotransfer from ChpT to itssubstrates is distinct from HK-receiver phosphotransfer defined inthe T. maritima HK853–RR468 system, because large structuralchanges are observed in HK853 upon RR468 binding. ChpT H22and CtrA D51 asymmetry, as it may relate to phosphotransfer, isdiscussed in a later section of this manuscript.An analysis of the CtrA structure across each half of the ChpT
dimer reveals additional asymmetric features of the signalingcomplex. Both CtrA molecules retain the standard αβα sandwichfold observed in other REC domain structures (23). However, analignment of CtrA(C) to CtrA(D) revealed a higher Cα rmsd(1.6 Å) than observed in ChpT(A)–ChpT(B) alignments. Approx-imately half the intermolecular interactions within the ChpT–CtrAcomplex occur through the α1 helix of ChpT and the β1–α1, β3–α3,and β5–α5 loops of CtrA; these contacts are consistent with knownHK–REC interfaces (45). Five ChpT residues that interact withCtrA (N33, E36, E40, K96, and H177) are observed on each halfof the dimer, but there are significant differences in how CtrA(C)and CtrA(D) contact ChpT. In particular, a set of hydrogen bonds
between E34 of CtrA(D) helix α2 and residues R129, R153, andH177 of ChpT(B) are unique to that interface (Fig. 5C). There isclear asymmetry in the ChpT–CtrA contact surface on either sideof the complex: Contact between ChpT(A) and CtrA(C) is moreextensive than ChpT(B) and CtrA(D) (935 Å2 versus 829 Å2 buriedsurface area). These structural data predict that the phospho-transfer “competent” half of the complex [i.e., ChpT(A) to CtrA(C)]may form a higher-affinity interface than ChpT(B) to CtrA(D).Indeed, calculations based on total solvation energies of iso-lated and interfacing structure (46) suggest an approximate4.5 kcal/mol difference in free energy between the two ChpT–CtrA interfaces.
Conformational Asymmetry and Dynamics of the ChpT PhosphorylationSite. The crystal structure of ChpT bound to CtrA raised thequestion of whether conformational and dynamical asymmetry ofthe ChpT phosphorylation site (H22) was a genuine feature of thissignaling complex. To begin to address this question, we performedmolecular dynamics (MD) simulations on our experimental struc-tures of the ChpT dimer (1,800 ns) and the ChpT2–CtrA2 hetero-tetramer (900 ns) in a fully atomistic system with explicit solvent.These simulations were aimed at testing whether the dynamics of
Fig. 5. Molecular structures of B. abortus ChpT and ChpT– CtrAREC complex. (A) Ribbon (Top) and surface views (Bottom) of homodimeric B. abortus ChpT at1.7-Å resolution (PDB ID code 4QPK). One ChpT monomer is drawn in dark blue and one in light blue. The conserved site of ChpT phosphorylation, His22, ishighlighted green; DHp (highlighted pink) and CA-like domains (highlighted yellow) of ChpT are labeled. (B) Ribbon (Top) and surface views (Bottom) of the2:2 ChpT-CtrAREC complex at 2.7-Å resolution (PDB ID code 4QPJ); diagram illustrates two CtrA receiver domain (REC) monomers (in red and pink) bound to acentral homodimer of ChpT (ChpT monomers in dark blue and light blue). (C) Least-squares Cα fit of the two halves of ChpT–CtrAREC complex: ChpT(A) darkblue, ChpT(B) light blue, CtrAREC(C) dark red, and CtrAREC(D) pink. Distances between ChpT(A)–CtrA(C) and ChpT(B)–CtrA(D) phosphoryltransfer residues(ChpT H22 and CtrA D51) are labeled in the expanded box. (D) Log probability of ChpT(A) and ChpT(B) H22 χ1 and χ2 rotamer angle occupancy in the ChpT(unbound; solid blue) and in the ChpT2–CtrA2 complex structures (bound; light dotted blue) based on MD simulations (Materials and Methods); distributionsrepresent Gaussian fluctuations of H22 conformation about a local equilibrium state.
E3714 | www.pnas.org/cgi/doi/10.1073/pnas.1503118112 Willett et al.
ChpT H22 in the isolated ChpT structure and in the ChpT–CtrAcomplex are inherently asymmetric across the twofold ChpTdimer axis.As described above, our crystal structure of ChpT–CtrA
revealed two distinct conformations of the ChpT H22. MD sim-ulations show that H22 stably occupies multiple χ1/χ2 rotamerangles on chains A and B, both when bound and unbound to CtrA(Fig. 5D). When not bound to CtrA, ChpT(A) H22 evinces sub-stantial heterogeneity: Five stable rotameric states are apparent.The rotamer conformations occupied by H22 differ in each ChpTmonomer both when bound and not bound to CtrA. These resultsprovide evidence that the significant asymmetry we observe inconformations of ChpT H22 in our complex crystal structure is notan artifact of crystallization, but rather reflects true asymmetry inprotein conformational dynamics at this site. We conclude thatsmall differences in side-chain and backbone structure, likelyenforced by ChpT–ChpT and ChpT–CtrA interaction at thequaternary level, affect the energy landscape of H22 conforma-tional transitions. This in turn affects the primary rotamer con-formations that H22 samples on each side of the ChpT dimer.
Structural and Functional Analysis of ChpT Phosphoryltransfer. Theexperimental crystal structures of B. abortus ChpT and the 2:2ChpT–CtrAREC complex provide a foundation for understandingthe molecular basis of phosphoryltransfer through the CckA–
ChpT–CtrA–CpdR system. Using these data, we constructedpoint mutations in components of the pathway to test predictionsof our structural models in vitro. We initially tested whetherChpT residues observed to interact with CtrA in our crystalstructure are required for the initial phosphotransfer step in thispathway (i.e., CckA to ChpT). In our experimental structure,three residues within the DHp domain of ChpT (N33, E36, andE40) and three residues within the CA domain (K96, R153, andH177) interact extensively with CtrA in the asymmetric ChpT2–
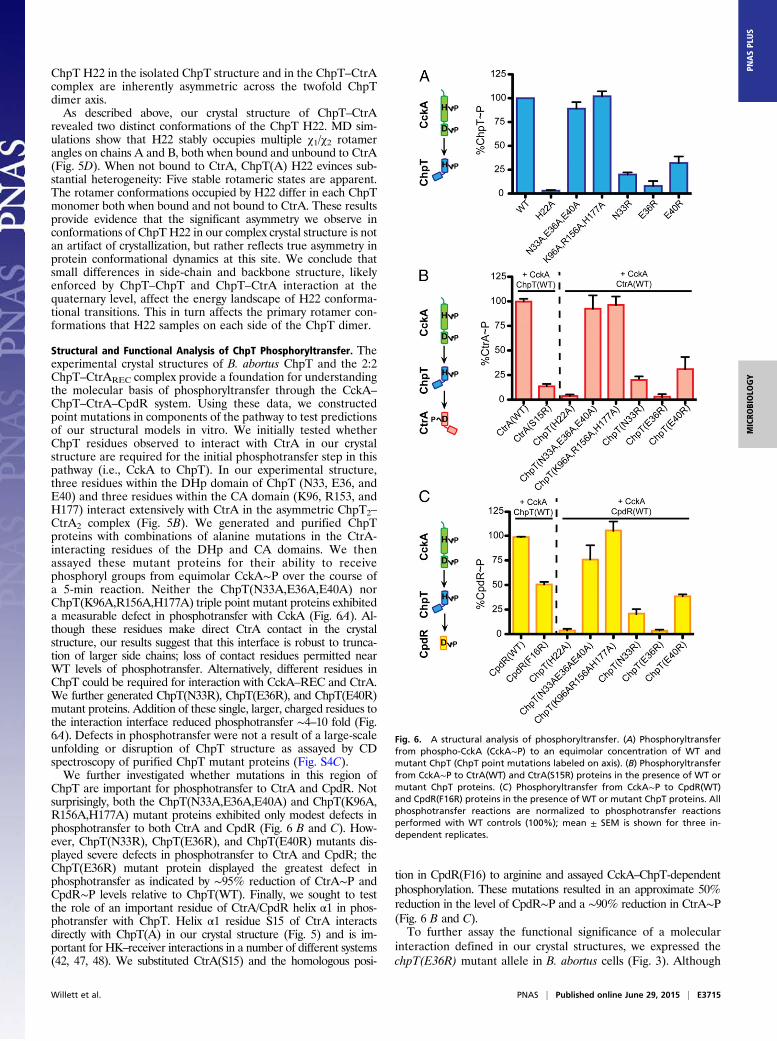
CtrA2 complex (Fig. 5B). We generated and purified ChpTproteins with combinations of alanine mutations in the CtrA-interacting residues of the DHp and CA domains. We thenassayed these mutant proteins for their ability to receivephosphoryl groups from equimolar CckA∼P over the course ofa 5-min reaction. Neither the ChpT(N33A,E36A,E40A) norChpT(K96A,R156A,H177A) triple point mutant proteins exhibiteda measurable defect in phosphotransfer with CckA (Fig. 6A). Al-though these residues make direct CtrA contact in the crystalstructure, our results suggest that this interface is robust to trunca-tion of larger side chains; loss of contact residues permitted nearWT levels of phosphotransfer. Alternatively, different residues inChpT could be required for interaction with CckA–REC and CtrA.We further generated ChpT(N33R), ChpT(E36R), and ChpT(E40R)mutant proteins. Addition of these single, larger, charged residues tothe interaction interface reduced phosphotransfer ∼4–10 fold (Fig.6A). Defects in phosphotransfer were not a result of a large-scaleunfolding or disruption of ChpT structure as assayed by CDspectroscopy of purified ChpT mutant proteins (Fig. S4C).We further investigated whether mutations in this region of
ChpT are important for phosphotransfer to CtrA and CpdR. Notsurprisingly, both the ChpT(N33A,E36A,E40A) and ChpT(K96A,R156A,H177A) mutant proteins exhibited only modest defects inphosphotransfer to both CtrA and CpdR (Fig. 6 B and C). How-ever, ChpT(N33R), ChpT(E36R), and ChpT(E40R) mutants dis-played severe defects in phosphotransfer to CtrA and CpdR; theChpT(E36R) mutant protein displayed the greatest defect inphosphotransfer as indicated by ∼95% reduction of CtrA∼P andCpdR∼P levels relative to ChpT(WT). Finally, we sought to testthe role of an important residue of CtrA/CpdR helix α1 in phos-photransfer with ChpT. Helix α1 residue S15 of CtrA interactsdirectly with ChpT(A) in our crystal structure (Fig. 5) and is im-portant for HK–receiver interactions in a number of different systems(42, 47, 48). We substituted CtrA(S15) and the homologous posi-
tion in CpdR(F16) to arginine and assayed CckA–ChpT-dependentphosphorylation. These mutations resulted in an approximate 50%reduction in the level of CpdR∼P and a ∼90% reduction in CtrA∼P(Fig. 6 B and C).To further assay the functional significance of a molecular
interaction defined in our crystal structures, we expressed thechpT(E36R) mutant allele in B. abortus cells (Fig. 3). Although
Fig. 6. A structural analysis of phosphoryltransfer. (A) Phosphoryltransferfrom phospho-CckA (CckA∼P) to an equimolar concentration of WT andmutant ChpT (ChpT point mutations labeled on axis). (B) Phosphoryltransferfrom CckA∼P to CtrA(WT) and CtrA(S15R) proteins in the presence of WT ormutant ChpT proteins. (C) Phosphoryltransfer from CckA∼P to CpdR(WT)and CpdR(F16R) proteins in the presence of WT or mutant ChpT proteins. Allphosphotransfer reactions are normalized to phosphotransfer reactionsperformed with WT controls (100%); mean ± SEM is shown for three in-dependent replicates.
Willett et al. PNAS | Published online June 29, 2015 | E3715
MICRO
BIOLO
GY
PNASPL
US

overexpression of WT chpT (chpT++) results in a defect in cellmorphology, DNA content, and intracellular survival, over-expression of chpT(E36R) does not have a statistically significanteffect on any of these phenotypes (Figs. 3 and 4). This result isconsistent with the inability of chpT(E36R) to facilitate phosphor-yltransfer in vitro (Fig. 6). As a control, we confirmed that WTChpT and the ChpT(E36R) alleles were expressed at similar levelsin the B. abortus cell (Fig. S2D).
DiscussionProtein phosphorelays regulate many important processes inbacteria, including asymmetric cell division, sporulation, andmulticellular development (49, 50). The conserved CckA–ChpT–CtrA–CpdR phosphorelay controls transcription of a diverse setof genes involved in multiple aspects of α-proteobacterial cellu-lar biology (8, 10, 14). To date, the identities and functions of thegenes encoding this pathway in the intracellular pathogenB. abortus had remained largely undefined. We have identifiedand biochemically reconstituted the complete B. abortus CckA–
ChpT–CtrA–CpdR system in vitro and provide evidence that itcomprises an essential and specific phosphorelay that regulatesreplication, cell division, and survival in the intracellular niche.ChpT is central in this pathway and shuttles phosphoryl groupsbetween the HK CckA and two receiver substrates, CtrA andCpdR. CtrA is a classical DNA-binding response regulator,whereas CpdR is a single domain receiver that regulates steady-state CtrA levels in the cell (Fig. 3). Our data support a model inwhich the regulatory topology of this pathway is conserved be-tween B. abortus and C. crescentus, two α-proteobacterial speciesthat inhabit widely different environmental niches and havedistinct cellular features.We have discovered that B. abortus mutants defective in sig-
naling through this pathway have no deficiency in entry into ahuman macrophage cell line but exhibit reduced intracellularsurvival over a 48-h timescale (Fig. 4). This survival defect isspecific to the intracellular niche because expression of condi-tional ctrA(V148F), chpT++, and cpdR(D52A)++ alleles thatstrongly perturb the phosphorelay does not result in cell death inaxenic culture (Fig. 4D). This result raises the question of whatspecific genes are under transcriptional control of the CckA–
ChpT–CtrA–CpdR system, and which of these genes are re-quired for survival of B. abortus inside mammalian cells. A pre-vious study has identified a CtrA binding site in front of the celldivision protein ftsE (15), which is consistent with observed celldivision defects. However, there are certainly other direct targets ofthis essential response regulator, many of which are likely to beimportant for the intracellular lifestyle of B. abortus. Like thePhoPQ two-component system of Salmonella, this phosphorelaymay control cell surface remodeling during infection, which isknown to be important for intracellular survival (51).The transcriptional output of conserved phosphorelays like
the CckA–ChpT–CtrA–CpdR system (14) or more simple, ar-chetypal two-component systems like FixL–FixJ (52) are oftentailored to the unique physiologies of the species in which theyare encoded. We note that consensus CtrA binding sites, basedon published B. abortus CtrA targets (15), are present in front ofseveral known B. abortus virulence factors including superoxidedismutase (sodC) and flagellar regulatory gene ftcR (53, 54).Defining genes in the Brucella CtrA regulon that uniquely con-trol its function as a facultative intracellular pathogen is an im-portant area of future investigation.The structure of ChpT is related to classic HKs and is distinct
from phosphotransferase proteins like Ypd1, which lack a CA-like domain (44). It is likely that ChpT arose through HK geneduplication and subsequently evolved to function solely as aphosphotransferase. Indeed, the ChpT CA domain does not bindATP, supporting a model in which ChpT structure and functionhave diverged from an ancestral HK (32). This raises the question
of what function ChpT–CA plays if it does not bind nucleotide. Itis possible that the ChpT–CA domain is conserved because it hasan important structural role in binding to receiver domain sub-strates. Certainly, the crystal structure of ChpT bound to CtrARECreveals several direct interactions between CtrAREC and ChpT–CA (Fig. 5 and Fig. S5 B, C, and D) that are related to those instructurally defined HK–REC systems (42). The ChpT–CA do-main may simply enhance stability of ChpT–REC complexes andfacilitate efficient phosphotransfer. Alternatively, ChpT–CA mayconfer additional regulatory capacity on the system. For example,the binding of different nucleotide forms by the CA domain canmodulate both kinase and phosphatase activities of HKs (18, 55,56). Although ChpT does not bind ATP, there may be other smallmolecule ligands that can serve to regulate interaction of ChpTwith its receiver substrates. The clear presence of a glycerolmolecule in the electron density maps of the ChpT–CA bindingpocket (Fig. S5A) demonstrates that CA has the capacity to ac-commodate small molecules and suggests that small-moleculebinding to ChpT–CA could allow for the integration of additionalsignals into this important pathway.The experimental structure of the ChpT–CtrA heterotetramer
revealed three residues in the DHp-like domain (N33, E36, andE40) and three residues in the CA-like domain of ChpT (K96,R153, and H177) that interact with CtrA on each half of the ChpThomodimer (Fig. 5B). These ChpT residues correspond to theso-called specificity residues that govern specific HK-to-responseregulator (RR) phosphoryltransfer (48, 57). Our structure thusprovides evidence that the ChpT phosphotransferase interacts withits cognate substrates in a manner that is structurally similar toclassic HK–RR interactions. Alanine mutagenesis showed that theChpT–RR structural interface is robust to multiple substitutionsunder the tested in vitro conditions. This result is consistentwith mutagenesis studies of the kinases CrdS from Myxococcusxanthus and EnvZ from Escherichia coli, in which substitution ofselect CrdS and EnvZ specificity residues had minimal impact onphosphoryltransfer to their cognate RRs in vitro (58, 59). However,it may be the case that these substitutions affect phosphoryltransferspecificity in vitro or signaling in B. abortus cells.A notable structural feature reported in this study is asym-
metry in the positions of the ChpT phosphorylation site (H22)and the CtrA phosphoryl receiver residue (D51) (Fig. 5 C andD). H22 occupies identical rotamer conformations in the struc-ture of ChpT alone but two distinct rotamer conformations oneach side of the ChpT homodimer in the 2:2 ChpT–CtrA com-plex. On one side of the complex, H22 is in line with D51 ofCtrA, which is well-positioned to receive a phosphoryl group. Onthe opposite side of the complex, both H22 and D51 adopt dif-ferent rotamer conformations and would not be able to undergophosphotransfer without a large change in H22 and D51 side-chain conformation. The half of the complex that is positioned toundergo phosphotransfer has a larger ChpT–CtrA contact sur-face than the half that is incompatible with phosphotransfer.These structural data suggest that the ChpT dimer has at leasttwo distinct interaction modes with CtrA substrate: one that iscompetent to catalyze phosphotransfer and one that buries lessChpT–REC surface area and is structurally incompetent forphosphotransfer. Although the asymmetry of ChpT–CtrA in-teraction observed in our structure may be a function of em-bedment in the crystal lattice, long-time-scale MD simulationspresented herein support a model in which the histidine phos-phorylation sites on each half of the ChpT dimer have distinctstructural and dynamical properties, particularly in the context ofthe 2:2 ChpT–CtrA complex (Fig. 5 B–D and Fig. S5). Thus, atany instant only one side of the ChpT–CtrA complex may becompetent to undergo a His–Asp phosphotransfer reaction. Ourstructures and simulations do not directly address the effectsof adding phosphoryl groups to H22 or D51. The affects of
E3716 | www.pnas.org/cgi/doi/10.1073/pnas.1503118112 Willett et al.

phosphoryl addition to H22 or D51 on protein structure anddynamics are a topic of ongoing investigation.We note that structural asymmetry has been reported for other
HKs. Thus, the structural and dynamical asymmetry we observeacross the central twofold axis of the 2:2 ChpT–CtrA heterodimermay reflect a general (but not necessarily universal) feature of two-component phosphorelays. Indeed, it is established that the auto-phosphorylation equilibria of each subunit of dimeric E. coli NtrBdiffer by nearly two orders of magnitude (60). The structures ofHKs HK853 (34), DesK (61), YF1 (62), VicK (36), and CpxA (35)adopt different tertiary structures in the frame of the central HKdimer axis. Structures of VicK, YF1, and CpxA in particular showhighly asymmetric positioning of the CA domain: One CA isproximal to the site of histidine phosphorylation and the second CAdomain is distal. Moreover, recent work on T. maritima HK853provides structural evidence for asymmetry in the histidine auto-kinase reaction (63), even though the ATP-binding CA domainsoccupy symmetrical positions in the unphosphorylated structure(34). Although ChpT is not a bona fide HK, our study providesevidence that this protein has retained some structural featurescommon to other two-component His–Asp phosphorelay systemsand may thus serve as an excellent model to investigate the struc-tural and dynamical mechanisms that underpin function of thesecomplex regulatory systems.
Materials and MethodsBacterial Culture and Strain Construction. All strains and primers used in thisstudy are detailed in Tables S3 and S4. E. coli strains were constructed usingroutine cloning techniques. All B. abortus strains were maintained andconstructed using previously described techniques (64) under biosafety level3 (BSL3) conditions per Centers for Disease Control and Prevention (CDC)rules and regulations governing the use of select agents. Further details onstrain generation and maintenance are provided in Supporting Information.
In Vitro Kinase and Phosphotransfer Assays. Genes encoding proteins to beoverexpressed were cloned into pET28a (Novagen), expressed in RosettaE. coli (DE3), and purified using an N-terminal His6-tag. All kinase andphosphotransfer reactions were performed following previously publishedmethods (20, 21, 65). For complete protocols see Supporting Information.
Crystallization of ChpT and the ChpT–CtrA Complex. All crystallization condi-tions used the hanging drop vapor diffusion technique. The structure of ChpTalone was obtained by mixing CckAsoluble (20 mg·mL−1 final) and ChpT(4.7 mg·mL −1 final) and seeding using a horse hair in the following crystalliza-
tion buffer: 0.2 M ammonium sulfate, 0.1 M sodium acetate pH 5.5, and10% (wt/vol) PEG 2000 MME. The ChpT2–CtrA2 complex was obtained by mixingChpT (8.4 mg·mL −1) and CtrA (5.3 mg·mL −1) after concentration using the fol-lowing crystallization buffer: 0.1 M Hepes pH 7.0 and 8% (wt/vol) PEG 8000.After mixing 1.5 μL of protein solution and 1.5 μL of crystallization buffer against500 μL of crystallization buffer in the well, all crystals grew for 7 d and weremounted after soaking for 1 min in crystallization buffer supplemented with25% (vol/vol) glycerol.
Crystallographic Data Collection, Processing, Phasing, and Refinement. Alldiffraction data were collected on beamline 21-ID-F (Life Sciences Collabo-rative Access Team, Advanced Photon Source) and reduced using the HKL2000 software suite. All structures were solved by molecular replacement inPHENIX (66) using models that were initially based off C. crescentus ChpT(33). Model building and refinement was conducted using Coot and PHENIX,accessed through the SBGrid consortium (67). Coordinates of the B. abortusChpT (PDB ID code 4QPK) and the 2:2 ChpT–CtrA complex (PDB ID code4QPJ) have been deposited in the Protein Data Bank.
Biophysical Analyses of ChpT Structure. Analyses of ChpT by CD spectroscopy,ITC, and SAXS were performed using previously published methods (59, 68, 69)and are described in detail in Supporting Information. SAXS data on ChpT werecollected at the Advanced Photon Source beamline 18-ID (BioCAT).
Molecular Dynamics Simulations. Fully atomistic models of ChpT, CtrA, and theChpT–CtrA complex were constructed based on the crystallographic datapresented in this study. The simulations were run for 1,851, 1,000, and 886 ns,respectively. Further details are provided in Supporting Information.
Imaging and Analysis of Brucella Cells. Before analysis by LM, flow cytometry,and CryoEM, B. abortus cells were fixed following established BSL3 B. abortusprotocols at the University of Chicago Ricketts Laboratory. All infection assaysusing differentiated THP-1 macrophages used an MOI of 100. All resultspresented are the mean ± SEM. Details of sample preparation and analysisare outlined in Supporting Information.
ACKNOWLEDGMENTS. We thank Aretha Fiebig and members of the S.C.laboratory for discussions and guidance during the preparation of thismanuscript and Elena Solomaha and Ryan Duggan for technical assistance.This project has been funded in whole or in part with federal funds fromNIH–National Institute of Allergy and Infectious Diseases Grants U19 AI107792and R01 AI107159 (to S.C.). J.W.W. is supported by NIH Ruth KirschsteinPostdoctoral Fellowship F32 GM109661. Funding for LS-CAT Sector 21 wasprovided by the Michigan Economic Development Corporation and theMichigan Technology Tri-Corridor Grant 085P1000817. Small angle X-rayscattering at Advanced Photon Source–BioCAT is supported by NIH GrantP41 GM103622.
1. Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV (2006) The new globalmap of human brucellosis. Lancet Infect Dis 6(2):91–99.
2. Atluri VL, Xavier MN, de Jong MF, den Hartigh AB, Tsolis RM (2011) Interactions of thehuman pathogenic Brucella species with their hosts. Annu Rev Microbiol 65:523–541.
3. Dean AS, et al. (2012) Clinical manifestations of human brucellosis: A systematic re-view and meta-analysis. PLoS Negl Trop Dis 6:e1929.
4. Batut J, Andersson SG, O’Callaghan D (2004) The evolution of chronic infectionstrategies in the alpha-proteobacteria. Nat Rev Microbiol 2(12):933–945.
5. Philippot L, et al. (2010) The ecological coherence of high bacterial taxonomic ranks.Nat Rev Microbiol 8(7):523–529.
6. McAdams HH, Shapiro L (2009) System-level design of bacterial cell cycle control. FEBSLett 583(24):3984–3991.
7. Tsokos CG, Laub MT (2012) Polarity and cell fate asymmetry in Caulobacter crescentus.Curr Opin Microbiol 15(6):744–750.
8. Biondi EG, et al. (2006) Regulation of the bacterial cell cycle by an integrated geneticcircuit. Nature 444(7121):899–904.
9. Iniesta AA, McGrath PT, Reisenauer A, McAdams HH, Shapiro L (2006) A phospho-signaling pathway controls the localization and activity of a protease complex criticalfor bacterial cell cycle progression. Proc Natl Acad Sci USA 103(29):10935–10940.
10. Laub MT, Chen SL, Shapiro L, McAdams HH (2002) Genes directly controlled by CtrA,a master regulator of the Caulobacter cell cycle. Proc Natl Acad Sci USA 99(7):4632–4637.
11. Leung MM, Brimacombe CA, Beatty JT (2013) Transcriptional regulation of the Rho-dobacter capsulatus response regulator CtrA. Microbiology 159(Pt 1):96–106.
12. Wang H, et al. (2014) The CtrA phosphorelay integrates differentiation and com-munication in the marine alphaproteobacterium Dinoroseobacter shibae. BMC Ge-nomics 15:130.
13. Zan J, Heindl JE, Liu Y, Fuqua C, Hill RT (2013) The CckA-ChpT-CtrA phosphorelaysystem is regulated by quorum sensing and controls flagellar motility in the marinesponge symbiont Ruegeria sp. KLH11. PLoS ONE 8(6):e66346.
14. De Nisco NJ, Abo RP, Wu CM, Penterman J, Walker GC (2014) Global analysis of cell
cycle gene expression of the legume symbiont Sinorhizobium meliloti. Proc Natl Acad
Sci USA 111(9):3217–3224.15. Bellefontaine AF, et al. (2002) Plasticity of a transcriptional regulation network
among alpha-proteobacteria is supported by the identification of CtrA targets in
Brucella abortus. Mol Microbiol 43(4):945–960.16. Robertson GT, et al. (2000) The Brucella abortus CcrM DNA methyltransferase is es-
sential for viability, and its overexpression attenuates intracellular replication in
murine macrophages. J Bacteriol 182(12):3482–3489.17. Ulrich LE, Zhulin IB (2010) The MiST2 database: A comprehensive genomics resource
on microbial signal transduction. Nucleic Acids Res 38(Database issue):D401–D407.18. Gutu AD, Wayne KJ, Sham LT, Winkler ME (2010) Kinetic characterization of the
WalRKSpn (VicRK) two-component system of Streptococcus pneumoniae: De-
pendence of WalKSpn (VicK) phosphatase activity on its PAS domain. J Bacteriol
192(9):2346–2358.19. Willett JW, Kirby JR (2011) CrdS and CrdA comprise a two-component system that is
cooperatively regulated by the Che3 chemosensory system in Myxococcus xanthus.
MBio 2(4):e00110–e00111.20. Willett JW, et al. (2013) Specificity residues determine binding affinity for two-com-
ponent signal transduction systems. MBio 4(6):e00420–e13.21. Skerker JM, Prasol MS, Perchuk BS, Biondi EG, Laub MT (2005) Two-component signal
transduction pathways regulating growth and cell cycle progression in a bacterium: A
system-level analysis. PLoS Biol 3(10):e334.22. Laub MT, Biondi EG, Skerker JM (2007) Phosphotransfer profiling: Systematic map-
ping of two-component signal transduction pathways and phosphorelays. Methods
Enzymol 423:531–548.23. Stock AM, Robinson VL, Goudreau PN (2000) Two-component signal transduction.
Annu Rev Biochem 69:183–215.
Willett et al. PNAS | Published online June 29, 2015 | E3717
MICRO
BIOLO
GY
PNASPL
US

24. Barnett MJ, Hung DY, Reisenauer A, Shapiro L, Long SR (2001) A homolog of the CtrAcell cycle regulator is present and essential in Sinorhizobium meliloti. J Bacteriol183(10):3204–3210.
25. Brilli M, et al. (2010) The diversity and evolution of cell cycle regulation in alpha-proteobacteria: A comparative genomic analysis. BMC Syst Biol 4:52.
26. Kim J, Heindl JE, Fuqua C (2013) Coordination of division and development influencescomplex multicellular behavior in Agrobacterium tumefaciens. PLoS ONE 8(2):e56682.
27. Quon KC, Marczynski GT, Shapiro L (1996) Cell cycle control by an essential bacterialtwo-component signal transduction protein. Cell 84(1):83–93.
28. Jacobs C, Domian IJ, Maddock JR, Shapiro L (1999) Cell cycle-dependent polar local-ization of an essential bacterial histidine kinase that controls DNA replication and celldivision. Cell 97(1):111–120.
29. Smith SC, et al. (2014) Cell cycle-dependent adaptor complex for ClpXP-mediatedproteolysis directly integrates phosphorylation and second messenger signals. ProcNatl Acad Sci USA 111(39):14229–14234.
30. Brown PJB, et al. (2012) Polar growth in the Alphaproteobacterial order Rhizobiales.Proc Natl Acad Sci USA 109(5):1697–1701.
31. Pincus Z, Theriot JA (2007) Comparison of quantitative methods for cell-shape anal-ysis. J Microsc 227(Pt 2):140–156.
32. Blair JA, et al. (2013) Branched signal wiring of an essential bacterial cell-cyclephosphotransfer protein. Structure 21(9):1590–1601.
33. Fioravanti A, et al. (2012) Structural insights into ChpT, an essential dimeric histidinephosphotransferase regulating the cell cycle in Caulobacter crescentus. Acta Crys-tallogr Sect F Struct Biol Cryst Commun 68(Pt 9):1025–1029.
34. Marina A, Waldburger CD, HendricksonWA (2005) Structure of the entire cytoplasmicportion of a sensor histidine-kinase protein. EMBO J 24(24):4247–4259.
35. Mechaly AE, Sassoon N, Betton JM, Alzari PM (2014) Segmental helical motions anddynamical asymmetry modulate histidine kinase autophosphorylation. PLoS Biol12(1):e1001776.
36. Wang C, et al. (2013) Mechanistic insights revealed by the crystal structure of a his-tidine kinase with signal transducer and sensor domains. PLoS Biol 11(2):e1001493.
37. Varughese KI, Madhusudan, Zhou XZ, Whiteley JM, Hoch JA (1998) Formation of anovel four-helix bundle and molecular recognition sites by dimerization of a responseregulator phosphotransferase. Mol Cell 2(4):485–493.
38. Svergun D, Barberato C, Koch MHJ (1995) CRYSOL - A program to evaluate x-raysolution scattering of biological macromolecules from atomic coordinates. J ApplCryst 28:768–773.
39. Dutta R, Yoshida T, Inouye M (2000) The critical role of the conserved Thr247 residuein the functioning of the osmosensor EnvZ, a histidine Kinase/Phosphatase, in Es-cherichia coli. J Biol Chem 275(49):38645–38653.
40. Alexandre MT, et al. (2010) Electronic and protein structural dynamics of a photo-sensory histidine kinase. Biochemistry 49(23):4752–4759.
41. Bilwes AM, Quezada CM, Croal LR, Crane BR, Simon MI (2001) Nucleotide binding bythe histidine kinase CheA. Nat Struct Biol 8(4):353–360.
42. Casino P, Rubio V, Marina A (2009) Structural insight into partner specificity andphosphoryl transfer in two-component signal transduction. Cell 139(2):325–336.
43. Varughese KI, Tsigelny I, Zhao H (2006) The crystal structure of beryllofluoride Spo0Fin complex with the phosphotransferase Spo0B represents a phosphotransfer pre-transition state. J Bacteriol 188(13):4970–4977.
44. Xu Q, Porter SW, West AH (2003) The yeast YPD1/SLN1 complex: Insights into mo-lecular recognition in two-component signaling systems. Structure 11(12):1569–1581.
45. Capra EJ, Laub MT (2012) Evolution of two-component signal transduction systems.Annu Rev Microbiol 66:325–347.
46. Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystallinestate. J Mol Biol 372(3):774–797.
47. Laub MT, Goulian M (2007) Specificity in two-component signal transduction path-ways. Annu Rev Genet 41:121–145.
48. Skerker JM, et al. (2008) Rewiring the specificity of two-component signal trans-duction systems. Cell 133(6):1043–1054.
49. Kroos L (2007) The Bacillus and Myxococcus developmental networks and theirtranscriptional regulators. Annu Rev Genet 41:13–39.
50. Shapiro L, McAdams HH, Losick R (2009) Why and how bacteria localize proteins.Science 326(5957):1225–1228.
51. Guo L, et al. (1997) Regulation of lipid A modifications by Salmonella typhimuriumvirulence genes phoP-phoQ. Science 276(5310):250–253.
52. Crosson S, McGrath PT, Stephens C, McAdams HH, Shapiro L (2005) Conserved mod-ular design of an oxygen sensory/signaling network with species-specific output. ProcNatl Acad Sci USA 102(22):8018–8023.
53. Gee JM, et al. (2005) The Brucella abortus Cu,Zn superoxide dismutase is required foroptimal resistance to oxidative killing by murine macrophages and wild-type viru-lence in experimentally infected mice. Infect Immun 73(5):2873–2880.
54. Léonard S, et al. (2007) FtcR is a new master regulator of the flagellar system ofBrucella melitensis 16M with homologs in Rhizobiaceae. J Bacteriol 189(1):131–141.
55. Igo MM, Ninfa AJ, Stock JB, Silhavy TJ (1989) Phosphorylation and dephosphorylationof a bacterial transcriptional activator by a transmembrane receptor. Genes Dev 3(11):1725–1734.
56. Jiang P, Atkinson MR, Srisawat C, Sun Q, Ninfa AJ (2000) Functional dissection of thedimerization and enzymatic activities of Escherichia coli nitrogen regulator II andtheir regulation by the PII protein. Biochemistry 39(44):13433–13449.
57. Podgornaia AI, Laub MT (2013) Determinants of specificity in two-component signaltransduction. Curr Opin Microbiol 16(2):156–162.
58. Capra EJ, et al. (2010) Systematic dissection and trajectory-scanning mutagenesis ofthe molecular interface that ensures specificity of two-component signaling path-ways. PLoS Genet 6(11):e1001220.
59. Willett JW, Kirby JR (2012) Genetic and biochemical dissection of a HisKA domainidentifies residues required exclusively for kinase and phosphatase activities. PLoSGenet 8(11):e1003084.
60. Jiang P, Peliska JA, Ninfa AJ (2000) Asymmetry in the autophosphorylation of thetwo-component regulatory system transmitter protein nitrogen regulator II of Es-cherichia coli. Biochemistry 39(17):5057–5065.
61. Albanesi D, et al. (2009) Structural plasticity and catalysis regulation of a thermo-sensor histidine kinase. Proc Natl Acad Sci USA 106(38):16185–16190.
62. Diensthuber RP, Bommer M, Gleichmann T, Möglich A (2013) Full-length structure ofa sensor histidine kinase pinpoints coaxial coiled coils as signal transducers andmodulators. Structure 21(7):1127–1136.
63. Casino P, Miguel-Romero L, Marina A (2014) Visualizing autophosphorylation in his-tidine kinases. Nat Commun 5:3258.
64. Kim HS, Caswell CC, Foreman R, Roop RM, 2nd, Crosson S (2013) The Brucella abortusgeneral stress response system regulates chronic mammalian infection and is con-trolled by phosphorylation and proteolysis. J Biol Chem 288(19):13906–13916.
65. Kim HS, Willett JW, Jain-Gupta N, Fiebig A, Crosson S (2014) The Brucella abortusvirulence regulator, LovhK, is a sensor kinase in the general stress response signallingpathway. Mol Microbiol 94(4):913–925.
66. Adams PD, et al. (2010) PHENIX: A comprehensive Python-based system for macro-molecular structure solution. Acta Crystallogr D Biol Crystallogr 66(Pt 2):213–221.
67. Morin A, et al. (2013) Collaboration gets the most out of software. eLife 2:e01456.68. Herrou J, Crosson S (2013) myo-inositol and D-ribose ligand discrimination in an ABC
periplasmic binding protein. J Bacteriol 195(10):2379–2388.69. Herrou J, Rotskoff G, Luo Y, Roux B, Crosson S (2012) Structural basis of a protein
partner switch that regulates the general stress response of α-proteobacteria. ProcNatl Acad Sci USA 109(21):E1415–E1423.
70. Khan SR, Gaines J, Roop RM, 2nd, Farrand SK (2008) Broad-host-range expressionvectors with tightly regulated promoters and their use to examine the influence ofTraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol74(16):5053–5062.
71. Pronk S, et al. (2013) GROMACS 4.5: A high-throughput and highly parallel opensource molecular simulation toolkit. Bioinformatics 29(7):845–854.
72. Lindorff-Larsen K, et al. (2010) Improved side-chain torsion potentials for the Amberff99SB protein force field. Proteins 78(8):1950–1958.
73. Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: A linear constraint solverfor molecular simulations. J Comput Chem 18:1463–1472.
74. Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: A newmolecular dynamics method. J Appl Phys 52:7182–7190.
75. Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N·log(N) method forEwald sums in large systems. J Chem Phys 98:10089–10092.
76. Iancu CV, et al. (2006) Electron cryotomography sample preparation using the Vi-trobot. Nat Protoc 1(6):2813–2819.
77. Tivol WF, Briegel A, Jensen GJ (2008) An improved cryogen for plunge freezing.Microsc Microanal 14(5):375–379.
78. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of imageanalysis. Nat Methods 9(7):671–675.
E3718 | www.pnas.org/cgi/doi/10.1073/pnas.1503118112 Willett et al.

Supporting InformationWillett et al. 10.1073/pnas.1503118112SI Materials and MethodsBacterial Culture and Strain Construction. All strains used in thisstudy are detailed in Table S3. E. coli strains used for cloning weregrown in LB (37 °C) supplemented with 50 μg·mL−1 kanamycin(Kan) or 100 μg·mL−1 ampicillin when required. All strains wereconstructed using standard cloning techniques. Primers are listed inTable S4. Site-directed mutagenesis was performed using the PCR-sewing method. All constructs were verified by DNA sequencing.All B. abortus strains were grown on either Schaedler agar
(Difco) supplemented with 5% defibrinated bovine blood (SBA)or Brucella broth (Difco) and grown at 37 °C supplemented with5% CO2 [with the exception of the ctrA(V148F) temperature-sensitive strain, which was grown at 30 °C]. When needed, formaintenance of plasmids, B. abortus strains were supplementedwith 50 μg·mL−1 Kan to maintain selection. All studies on liveB. abortus strains were performed at the University of ChicagoHoward T. Ricketts Laboratory under BSL3 conditions per CDCrules and regulations.Overexpression of chpT and cpdR(WT) and cpdR(D52A) was
performed by cloning into the replicating, IPTG-inducible vectorpSRK (70). Expression was induced by the addition of 2 mM IPTG.
Purification of His6-Tagged Proteins Expressed in E. coli. Genesencoding proteins to be overexpressed were cloned into pET28a(Novagen) and expressed in Rosetta E. coli(DE3). All proteinsused in the kinase and phosphotransfer assays were purifiedusing the following method: For each strain, a 10-mL overnightculture was used to inoculate a 250-mL culture of Terrific Brothsupplemented with Kan50 and grown (37 °C, 220 rpm) untilOD600 reached 0.7–0.8. Cultures were then induced with 1 mMIPTG and grown for 4–6 h at 20 °C, before being pelleted bycentrifugation in a Sorvall F10S-6 × 500y rotor at 6,000 × g for10 min. Cell pellets were stored at −20 °C until purification. Forpurification, cell pellets were thawed and suspended in 10 mL oflysis buffer [25 mM Tris pH 7.6, 125 mM NaCl, 10 mM imidazole,1% (vol/vol) Triton X-100, 5 μg·mL−1 DNaseI, 50 μg·mL−1 PMSF,and 3 mg·mL−1 lysozyme]. Samples were incubated for an hourand pelleted to remove insoluble cell debris. Lysates were addedto a column containing 4 mL of Chelating Sepharose FF (GE)charged with Ni2+ and equilibrated with column buffer (25 mMTris pH 7.6, 125 mM NaCl, and 10 mM imidazole). CckA andREC domain proteins were dialyzed overnight at 4 °C againststorage buffer (25 mM Tris pH 7.6, 125 mM NaCl, and 50%glycerol) and stored at −20 °C. After purification ChpT proteinswere dialyzed against 25 mM Tris pH 7.6 and 125 mM NaCl andaliquots were snap-frozen in liquid nitrogen and stored at −80 °C.For protein crystallography 50 mL of overnight E. coli cultures
were added to 2 L of LB, grown with shaking (37 °C, 220 rpm)until OD600 reached 0.6, and induced with 1 mM IPTG. After 4 h,cells were pelleted as above and stored at −20 °C until purifica-tion. For purification, cells were thawed and resuspended in50 mL buffer A (25 mM Tris pH 7.6 and 125 mMNaCl) and lysedby three passages through a French pressure cell. Cells wereclarified by a 20,000 × g spin and lysates were loaded onto acolumn loaded with 10 mL chelating resin listed above, equili-brated in buffer A. Resin was washed five times with three columnvolumes of column buffer and eluted with elution buffer (25 mMTris pH 7.6, 125 mM NaCl, and 500 mM Imidazole). All proteinswere dialyzed against 2 L of dialysis buffer (25 mM Tris pH 7.6and 125 mM NaCl), concentrated using 3-kDa (molecular weightcut-off, MWCO) centrifugal filters (Amicon; Millipore). All pro-teins were assayed for purity by 14% SDS/PAGE.
In Vitro Kinase and Phosphotransfer Assays. All kinase and phos-photransfer reactions were carried out in 1× kinase buffer (10×kinase buffer, 250 mM Tris pH 7.6, 500 mM KCl, 10 mM MgCl2,10 mM MnCl2, 10 mM CaCl2, and 10 mM DTT) (59). Reactionswere incubated at room temperature and were initiated by ad-dition of an ATP mix (25 μM ATP and 0.3 μM [γ-32P]ATP). Atthe indicated times, aliquots were removed and stopped by ad-dition to an equal volume of SDS-loading buffer. In phospho-transfer reactions, CckA was allowed to autophosphorylate for30 min before addition to ChpT and or CpdR/CtrA. All proteinswere used at a final concentration of 5 μM. Samples were re-solved by 15% SDS/PAGE. The dye front, which contains un-incorporated ATP, was removed and gels were subsequentlyexposed for 2–4 h on a phosphor screen before visualizationusing a FX Pro Plus Molecular Imager (Bio-Rad).Phosphotransfer profiling experiments were performed fol-
lowing previously published methods (20, 21). Briefly, CckA wasallowed to autophosphorylate for 30 min and incubated witheither ChpT or buffer for 5 min before proceeding with phos-photransfer reactions. CckA or CckA–ChpT solutions were in-cubated with each REC domain for 15 s before reactions werestopped by addition of an equal volume of SDS/PAGE buffer.The final concentration of proteins in these profiling experi-ments was 2.5 μM CckA, 6.6 μM ChpT, and 12.5 μM of eachREC domain. AcP labeling of REC domains was performed aspreviously described (20, 59). Reactions were resolved by run-ning on a Precast Criterion 18% SDS/PAGE gel (Bio-Rad) andvisualized as above.
Crystallization of ChpT and the ChpT–CtrA Complex. Proteins werepurified following the methods described above. All crystalliza-tion conditions used the hanging drop vapor diffusion technique.The final concentration of proteins used for screening for theChpT–CtrA complex were 8.4 mg·mL−1 ChpT and 5.3 mg·mL−1
CtrA. The structure of ChpT alone was obtained by mixingCckAsoluble (20 mg·mL −1 final) and ChpT (4.7 mg·mL−1 final) insolution after purification and concentration. Initial screeningwas performed in a 96-well plate format using a Mosquito robot(TTP Labtech), which mixed 0.1 μL of protein solution and0.1 μL of the crystallization solution and subsequently equili-brated this drop against 75 μL of crystallization buffer. Com-mercial crystallization kits were used for initial crystallizationscreening (Nextel Suites; Qiagen). After manual refinement in24-well plates (Hampton Research) the best crystals for ChpT–CtrA were obtained in the following crystallization buffer at19 °C: 0.1 M Hepes pH 7.0 and 8% (wt/vol) PEG 8000. The bestcrystals for ChpT alone were obtained after seeding using a horsehair in the following crystallization buffer: 0.2 M ammonium sul-fate, 0.1 M sodium acetate pH 5.5, and 10% (wt/vol) PEG 2000MME. For each crystallization condition, manual drops were setup by adding 1.5 μL of protein solution and 1.5 μL of crystalli-zation buffer against 500 μL of crystallization buffer in the well.Crystals grew for 7 d and were mounted after soaking for 1 min incrystallization buffer supplemented with 25% (vol/vol) glycerol.
Crystallographic Data Collection, Processing, Phasing, and Refinement.All diffraction data were collected on beamline 21-ID-F (LifeSciences Collaborative Access Team, Advanced Photon Source)using a MAR Mosaic 225 detector. Datasets were obtained fromfrozen, loop-mounted single crystals at 100 K using an oscillationrange of 1° and reduced using the HKL 2000 software suite.
Willett et al. www.pnas.org/cgi/content/short/1503118112 1 of 11

All structures were solved using by molecular replacement inPHENIX (66) using models that were initially based off thestructure of C. crescentus ChpT (33). All model building and re-finements were conducted using Coot and PHENIX, which aremaintained by the SBGrid consortium (67). Coordinates of theB. abortus ChpT (PDB ID code 4QPK) and the 2:2 ChpT–CtrAcomplex (PDB ID code 4QPJ) have been deposited in the ProteinData Bank.
CD Spectroscopy. Protein samples for CD spectroscopy were pu-rified as described above, dialyzed overnight in CD buffer (25 mMNa phosphate pH 7.6 and 50 mM NaCl) and diluted to a finalconcentration of 25 μM in CD buffer. Samples were analyzed usinga 1 mM cuvette on an Aviv 202 CD spectrometer. Data shown arethe average of three separate spectral scans.
ChpT–ATP Binding Measurements Using ITC. Proteins for ITC anal-ysis were purified as detailed above and dialyzed overnight againstITC buffer (25 mMTris pH 7.6, 125 mMNaCl, and 1 mMMgCl2)and concentrated using Amicon Ultra 3k MWCO spin columns.Samples were subsequently purified further using a HiPrep 26/60Sephacryl S-200 (GE) gel-filtration column equilibrated in ITCbuffer and again dialyzed overnight against ITC buffer.All binding measurements were performed using aMicroCal200
iTC (GE). All solutions and samples were extensively degassed.The cell of the MicroCal200 iTC was filled with ChpT at con-centrations of 200 μM (monomer). ChpT protein concentrationswere determined using the calculated extinction coefficient ofChpT. The syringe was filled with 1 mM ATP (Sigma-Aldrich)resuspended in ITC buffer. Titrations were initially performed at25 °C with constant stirring. To confirm that the absence of ob-served binding between ChpT and ATP was not due to high-affinitybinding reactions with ΔH values near 0, we performed sepa-rate ITC experiments at 16 °C (where there was also no ap-parent binding).
MD Simulations. Fully atomistic models of ChpT, CtrA, and theChpT–CtrA complex were constructed based on the crystallo-graphic data presented in this study. Missing side chains wereadded to the models using a least-squares fit to the backboneatoms when necessary. All simulations were run with the Gro-macs MD package, version 4.6.5 (71). Each structure was sol-vated in TIP3 water with Na+ and Cl− counter ions to achieve aneutral periodic cell at 150 mM concentration. The Amber99sb-ILDN forcefield was used (72). The systems contained 95,606,20,830, and 157,620 atoms, respectively. Each system wasenergy-minimized using a gradient descent method to removehigh-energy contacts between the protein and the solvent andrelax any steric clashes. All bonds were replaced by constraintsusing the LINCS algorithm (73). To equilibrate the systems, 10-nssimulations in the NPT ensemble were performed at 300 K, usingthe Parrinello–Rahman barostat at a pressure of 1 bar (74).Samples were collected from simulations of the NVT ensemble,which was maintained by a Nosé–Hoover thermostat with a ref-erence temperature of 300 K. The simulations were run for 1,851,1,000, and 886 ns, respectively. A 5-fs time step was used in allproduction simulations. Electrostatics were evaluated every fourtime steps using the particle mesh Ewald method (75).
SAXS. SAXS data on ChpT were collected at the AdvancedPhoton Source beamline 18-ID (BioCAT, Argonne NationalLaboratory). Purified ChpT was suspended in 25 mM Tris pH 7.6and 125 mM NaCl at a concentration of 15 μM. Data werecollected using an Aviex CCD detector and analyzed in IgorPro.
LM and CryoEM of B. abortus. Brucella cells were grown in rich mediafor the indicated time and fixed with 4% (vol/vol) paraformaldehydein PBS for 30 min. Samples were washed two times in PBS and
suspended in EM imaging buffer (20 mM Tris pH 7.6, 50 mMglucose, and 10 mM EDTA) and samples were confirmed tocontain no viable cells, following established BSL3 B. abortusprotocols at the University of Chicago Ricketts Laboratory. Phase-contrast images of cells were collected using a Leica DM 5000Bmicroscope with an HCX PL APO 63×/1.4 N.A. Ph3 objective.Images were acquired with a mounted Orca-ER digital camera(Hamamatsu) controlled by the Image-Pro software suite (MediaCybernetics).From visualization by cryoEM fixed cells were mixed with
BSA-treated 10 nm colloidal gold solution to avoid aggregation(76). Three to four microliters of a cell culture–gold solutionmixture were applied to glow discharged copper Quantifoilgrids (Quantifoil Micro Tools). The grids were blotted eithermanually or automatically using the Vitrobot (FEI) and plungedinto a liquid ethane/propane mixture (76, 77). The grids werekept in liquid nitrogen until they were loaded into a Gatan 626cryoholder and examined on an FEI T12 electron microscope at120 keV.
Analysis of Brucella Cell Shape and DNA Content. Cells analyzed byLM and flow cytometery were first grown to the indicated time inrich media, fixed, and removed fromBSL3 containment as detailedabove. Phase-contrast images were obtained as detailed as aboveand uniformly thresholded using ImageJ (78). Cell contours wereextracted and cell area analyzed using the open source Celltoolapplication (31). The following numbers of cells were analyzed forcell shape analysis: WT (n = 611), ΔcpdR (n = 1,395), ctrA(V148F)(n = 216), chpT++ (n = 140), chpT(E36R)++ (n = 261),cpdR(WT)++ (n = 321), and cpdR(D52A)++ (n = 113).Flow cytometery to measure DNA content was performed on a
BDFACSAria II cytometer. Propidium iodide was added (10 μg/mLfinal) to 1 mL of fixed cells before analysis by flow cytometery. Datafor 20,000 individual cells were collected and analyzed using theFlowJo software suite (FlowJo Enterprise).
THP1 Macrophage Infection Assays. Human monocytic THP-1 cells(ATCC TIB-202) were maintained by culture in RPMI-1640supplemented with 10% (vol/vol) heat-inactivated FBS and 2 mML-glutamine. For monocytic differentiation of THP-1 cells, 1 × 105
cells in RPMI supplemented with phorbol ester 12-O-tetradeca-noylphorbol-13-acetate (40 μg·mL−1) were added to a each well of96-well plate, and cells were grown for 24–48 h before proceed-ing with infections. All THP-1 cells were grown at 37 °C with 5%(vol/vol) atmospheric CO2.For infections, B. abortus cells were resuspended in warmed
RPMI and added at an MOI of 100 cfu per macrophage cell. Tosynchronize infections after the addition of Brucella cells, plateswere centrifuged at 200 × g for 5 min. After a 30-min incubation,media was removed and replaced with RPMI supplemented with100 μg·mL−1 gentamycin to kill extracellular bacteria. Afterward,samples were incubated for 1 h, after which monolayers werewashed with PBS and THP-1 cells were lysed by addition of0.01% Triton X-100. Serial dilutions were prepared and viablecells were determined by plating on TSA plates. For longer timepoints, the RPMI was removed and replaced with RPMI sup-plemented with 50 μg·mL−1 gentamycin. Additional sampleswere lysed as above and serial dilutions plated. All THP-1 assayswere performed in triplicate using independent B. abortus cul-tures. The results presented illustrate the mean ± the SEM. ForTHP-1 cell infections at the ctrA(V148F) permissive growthtemperature of 30 °C, THP-1 cells were initially prepared asabove, but after infections cells were grown at 30 °C with 5%(vol/vol) atmospheric CO2.
Western Blot Analysis of CtrA Levels. For Western blot analysis,overnight cultures of B. abortus were diluted to OD600 of 0.1 andcultured for 4 h after shift to the nonpermissive growth condition
Willett et al. www.pnas.org/cgi/content/short/1503118112 2 of 11

or addition of IPTG as indicated. Five milliliters of cultures werespun down and resuspended in 4× SDS-loading buffer treatedwith DNaseI and 15 μL was resolved by SDS/PAGE. PrimaryCtrA polyclonal antiserum was a gift from Peter Chien, Universityof Massachusetts, Amherst, MA, and Brucella α-PhyR polyclonalantiserum was used as a loading control. α-Rabbit antibody wasused as a secondary, which was conjugated to Alexa 488 (LifeTechnologies).
RNA Isolation and qRT-PCR. RNA was extracted following a pre-viously published method (65) after 4 h of growth at the non-permissive temperature or IPTG addition in rich medium (startingat an OD600 = 0.1). cDNA was generated using the SuperScript IIIcDNA kit (Invitrogen) and qPCR was carried out using a SYBRGreen PCR kit (Life Technologies). Relative quantification of theccrM transcript was calculated using the 2−ΔΔCt method, usingrplK as the reference gene.
Willett et al. www.pnas.org/cgi/content/short/1503118112 3 of 11

cckAREC
fli
Bab1_1059 Bab1_1060 Bab1_1061Bab1_1058
KINASEPAS4PAS8PAS9
TM1 TM2
CckA
chpT ctrA
Bab1_1613 Bab1_1614 Bab1_1615
ChpT
CtrA
DUF2328
REC HTH
cpdR
Bab2_0042Bab2_0041 Bab2_0043
CpdR REC36% 81%
62%
48%
0 100 200 300 400 500 600 700 800Residue Number
A
C
D
B
Bab1_1612
Bab1_1611
ChpTBa 1 MSLPVTLSALDLGALLCSRICHDIISPVGAINNGLELLEEG-------GADEDAMALIKSSARNASARLQFARIA---FGAAGSAGVQID---TGChpTCc 1 ------VQGPDFAAMLAARLCHDFISPASAIVSGLDLLEDP----SAQDMRDDAMNLIASSARKLADLLQFTRVAFGA----SASAENFD---SRHK853Tm 1 -----------MKTEFIANISHELRTPLTAIKAYAETIYNSLGELDLSTLKEFLEVIIDQSNHLENLLNELLDFSRLER---KSLQINREKVDLCEnvZEc 1 -----VKQLADDRTLLMAGVSHDLRTPLTRIRLATEMMSEQD-----GYLAESINKDIEECNA---IIEQFIDYLRT------GQEMPMEMADLN
ChpTBa 83 DAQNVATEYFRNEK----P-EFTW-EGARVLLP--- KNKVKLLLNMLLIGNGA----IPRGGSLAVRLEGSDTDPRFVITVKGRMLRVPPKFLEChpTCc 79 ELEKLAQGVFAHVR----PTLDW-QIEPQAMN---- KPSSRAVLNIAQIAASA----LPAGGVATVKGVAADGRFSIIADAKGPRARLRPEVLAHK853Tm 82 DLVESAVNAIKEFASSHNVNVLFESNVPCPVEAYIDPTRIRQVLLNLLNNGVKYSKKDAPDK-YVKVILDEKDGGVLIIVEDNGIGIPDHAKDRIEnvZEc 77 AVLGEVIAAESGYEREIETALY-----PGSIEVKMHPLSIKRAVANMVVNAARYGNG------WIKVSSGTEPNRAWFQVEDDGPGIAPEQRKHL
α1
β2 β3 β4 β5
β1
β6 β7
α2
α3
ATP-lid
α4 α5
α6
ChpTBa 164 LHSGAAPEE-------PIDAHSVQPYYTLLLAEEAGMKISIHATAEDIVFSAE---------------ChpTCc 160 GLKGEPLAE-------GLGGPWVQAAYLNALVRAAGGQIAVEIGEDRASIAAWVPA------------HK853Tm 176 FEQFYRVDSSLTYEVPGTGLGLAITKEIVEL--HGGRIWVESEVGKGSRFFVWIPKDRAGEDNRQDN-EnvZEc 161 FQPFVRG----DSARTISGTGLGLAIVQRIVDNHNGMLELGTSERGGLSIRAWLPVPVTRAQGTTKEG
--
CtrA-REC 1 --MRVLLIEDDSAIAQSIELMLKSESFNVYTTDLGEEGIDLGKLY--DYDIILLDLNLPDMSGYEVLRTLRLSK--VKTPILILSGMAGIEDKVRCpdR 1 -MKRILLAEDDNDMRRFLVKALEKAGYHVTHFDNGASAYERLQEE--PFSLLLTDIVMPEMDGIELARRATEID-----PDLKI-FITGFAAVALCckA-REC 1 GSATVLLVEDEDAVRMGGVRALQSRGYTVHEAASGVEALEIMEELGGEVDIVVSDVVMPEMDGPTLLRELRKTH-----PDIKFIFVSGYAEDAFRR468 1 MSKKVLLVDDSAVLRKIVSFNLKKEGYEVIEAENGQIALEKLSEF--TPDLIVLDIMMPVMDGFTVLKKLQEKEEWKRIPVIVLTAKGGEEDESL
CtrA-REC 90 GLGFGAD---DYMTKPFHKDELIARIHAIVRR--CpdR 87 NPDSDAPRDAKVLSKPFHLRDLVNEIEKMLIAA-CckA-REC 91 ARNLPADAKFGFLPKPFSLKQLATTVKEMLEKQDRR468 94 ALSLGAR---KVMRKPFSPSQFIEEVKHLLNE--
β1
β5
α1 β2 β3 β4
α5
α2 α3 α4
Tm
Tm
DHp CA
N-Box D-Box
G2-BoxF-Box
Fig. S1. (A) Genome context of cckA (green), chpT (blue), ctrA (red), and cpdR (yellow) in B. abortus strain 2308. Locus numbers are indicated above eachgene. Percent amino acid sequence identity to C. crescentus orthologs is indicated below each gene. (B) Domain organization of B. abortus CckA, ChpT, CtrA,and CpdR proteins. All proteins are drawn to scale. (C) C. crescentus and B. abortus ChpT phosphotransferase proteins aligned with the sequences of HKs E. coliEnvZ and T. maritima HK853. Residues involved in protein dimerization (blue circles) and interaction with CtrA(C) (small red arrow) and CtrA(D) (small red star)are indicated. The conserved histidine phosphorylation site is highlighted red. (D) Alignment of B. abortus CtrA, CpdR, CckA, and T. maritima RR468 RECdomains. Site of aspartyl phosphorylation is highlighted red. Residues that interact with ChpT(A) (small blue arrow) and ChpT(B) (small blue star) are indicated.All secondary structure is indicated above the sequence (spiral, helix; horizontal arrow, beta strand).
Willett et al. www.pnas.org/cgi/content/short/1503118112 4 of 11
A B C
D
chpT++ctrA(V148F)
α-HA
HA
-Chp
T-W
T
HA
-Chp
T-E3
6R
WT
B. a
bort
us
cpdR(D52A)++
Fig. S2. Cryoelectron micrographs of B. abortus (A) ctrA(V148F), (B) chpT++, and (C) cpdR(D52A)++ overexpression strains. (D) Western blot of HA-tagged ChpTand ChpT(E36R) proteins from B. abortus cell lysate resolved by SDS/PAGE. WT B. abortus cell lysate was run and blotted as a negative control.
Willett et al. www.pnas.org/cgi/content/short/1503118112 5 of 11
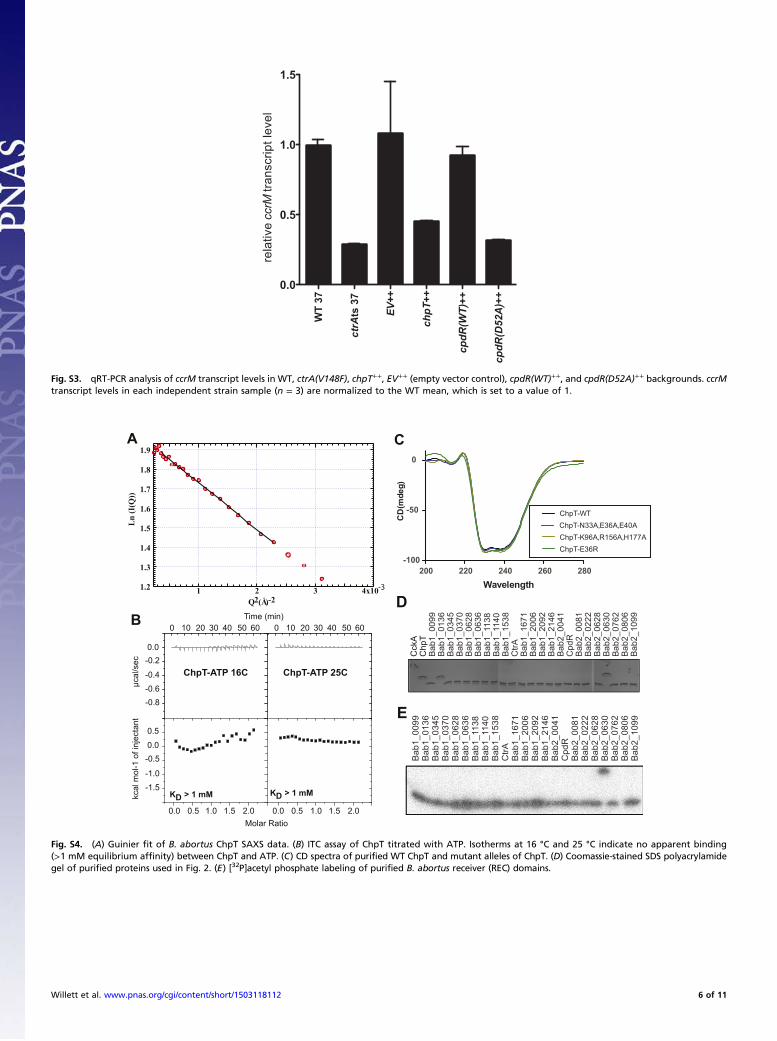
WT
37
ctrA
ts 3
7
EV+ +
chpT
++
cpdR(WT)
++
cpdR(D52A)+
+
0.0
0.5
1.0
1.5
rela
tive
ccrM
tran
scrip
t lev
el
Fig. S3. qRT-PCR analysis of ccrM transcript levels in WT, ctrA(V148F), chpT++, EV++ (empty vector control), cpdR(WT)++, and cpdR(D52A)++ backgrounds. ccrMtranscript levels in each independent strain sample (n = 3) are normalized to the WT mean, which is set to a value of 1.
A
KD > 1 mM
ChpT-ATP 25CChpT-ATP 16C
Time (min)
Molar Ratio
kcal
mol
-1 o
f inj
ecta
ntμc
al/s
ec
0 10 20 30 40 50 60 0 10 20 30 40 50 60
0.0 0.5 1.0 1.5 2.0 0.0 0.5 1.0 1.5 2.0
0.5
0.0-0.5
-1.0
-1.5
0.0-0.2
-0.4
-0.6-0.8
KD > 1 mM
B
C1.9
1.8
1.7
1.6
1.5
1.4
1.3
1.2
Ln (I
(Q))
4x10321 -3
Q2 (Å-2)
200 220 240 260 280-100
-50
0
ChpT-WTChpT-N33A,E36A,E40AChpT-K96A,R156A,H177AChpT-E36R
Wavelength
CD
(mde
g)
D
E
Cck
AC
hpT
Bab
1_00
99B
ab1_
0136
Bab
1_03
45B
ab1_
0370
Bab
1_06
28B
ab1_
0636
Bab
1_11
38B
ab1_
1140
Bab
1_15
38C
trAB
ab1_
1671
Bab
1_20
06B
ab1_
2092
Bab
1_21
46B
ab2_
0041
Bab
2_06
30
Bab
2_08
06B
ab2_
0762
Bab
2_06
28B
ab2_
0222
Bab
2_00
81C
pdR
Bab
2_10
99
Bab
1_00
99B
ab1_
0136
Bab
1_03
45B
ab1_
0370
Bab
1_06
28B
ab1_
0636
Bab
1_11
38B
ab1_
1140
Bab
1_15
38C
trAB
ab1_
1671
Bab
1_20
06B
ab1_
2092
Bab
1_21
46B
ab2_
0041
Bab
2_06
30
Bab
2_08
06B
ab2_
0762
Bab
2_06
28B
ab2_
0222
Bab
2_00
81C
pdR
Bab
2_10
99
Fig. S4. (A) Guinier fit of B. abortus ChpT SAXS data. (B) ITC assay of ChpT titrated with ATP. Isotherms at 16 °C and 25 °C indicate no apparent binding(>1 mM equilibrium affinity) between ChpT and ATP. (C) CD spectra of purified WT ChpT and mutant alleles of ChpT. (D) Coomassie-stained SDS polyacrylamidegel of purified proteins used in Fig. 2. (E) [32P]acetyl phosphate labeling of purified B. abortus receiver (REC) domains.
Willett et al. www.pnas.org/cgi/content/short/1503118112 6 of 11

A
DC
GOL
B
Hydrogen BondSalt Bridges
S26
N33
E36
E40
K96
R156
H177
N33
E36
E40
K96
R129
R153
H177
D8
D9
S15
D31
N53
P55
F103
H104
K105
D9
E34
D56
F103
H104
K105
ChpTA
ChpTB
CtrAC
CtrAD
S26
N33
E36
E40
K96
R156
H177
N33
E36
E40
K96
R129
R153
H177
D8 D9
S15
D31
N53
P55
F103
H104
K105
D9E34
D56
F103
H104
K105
ChpT (A)
CtrA (C)
CtrA (D)
ChpT (B)
Fig. S5. (A) Simulated annealing composite omit map (contoured at ±2σ) of a region surrounding a glycerol molecule (GOL; colored green) in the bindingpocket of the ChpT CA domain. (B) Interaction map of polar contacts between ChpT monomers (A and B; blue) and CtrAREC domains (C and D; red), hydrogenbonds (black lines), and salt bridges (dotted red lines). Magnified view of ChpT–CtrA complex structure showing (C) ChpT(A)–CtrA(C) and (D) ChpT(B)–CtrA(D)interacting residues.
Table S1. Deletion statistics for the essential B. abortus chpTand cckA genes
Strain Results of chpT deletion
WT sacB inactivation ΔchpT2308 81 17 02308 PchpT-chpT 57 6 8
Results of cckA deletionWT sacB inactivation ΔcckA
2308 58 42 02308 PcckA-cckA 41 37 21
Gene deletion by double-crossover recombination, using sacB counter se-lection on sucrose, was attempted in WT B. abortus strain 2308 in the absence(n = 98 total colonies screened) and presence (n = 71 total colonies screened)of a complementing copy of chpT on a replicating plasmid (PchpT-chpT).Deletion of cckA was attempted on WT B. abortus strain 2308 in the absence(n = 100 total colonies screened) and presence (n = 99 total colonies screened)of a complementing copy of cckA on a replicating plasmid (PcckA-cckA). Thetable reports the number of colonies identified as WT revertants after sucrosecounterselection (WT), the number of colonies with random sacB inactiva-tion, and the number of colonies harboring an in-frame deletion of chpT(ΔchpT) or cckA (ΔcckA). We only identified colonies harboring an in-framedeletion when the complementing copy of chpT or cckA was present.
Willett et al. www.pnas.org/cgi/content/short/1503118112 7 of 11

Table S2. Crystallographic data and refinement statistics
Statistics ChpT ChpT–CtrA
Data collection statisticsResolution range, Å 35.42–1.66 (1.719–1.66) 17.88–2.742 (2.839–2.742)Unique reflections 50,679 (5,049) 32,556 (3,179)Rmerge 0.043 0.086I/σI 27.66 (2.68) 23.30 (2.52)Redundancy 7.5 11.8Completeness, % 99.96 (100.00) 99.80 (98.03)
Refinement statisticsSpace group P 43 P 3221Unit cell 70.84 70.84 87.01 (90 90 90) 124.951 124.951 136.332 (90 90 120)Rfree reflections 3,925 2,007Rwork 0.1624 (0.2455) 0.1753 (0.2337)Rfree 0.1906 (0.2620) 0.2216 (0.2875)Average B-factor 27.3 65.4RMS (bonds) 0.007 0.009RMS (angles) 1.11 1.22
Ramachandran analysisFavored, % 98.3 98.0Allowed, % 1.7 1.8Outliers, % 0.0 0.2
Willett et al. www.pnas.org/cgi/content/short/1503118112 8 of 11

Table S3. Strains used in this study
FC no. E. coli strain Notes
2271 Rosetta / pET28a-RR0099 Expresses soluble REC domain - Bab1_00992272 Rosetta / pET28a-RR0136 Expresses soluble REC domain - Bab1_01362273 Rosetta/pET28a-RR0345 Expresses soluble REC domain - Bab1_03452274 Rosetta/pET28a-RR0370 Expresses soluble REC domain - Bab1_03702275 Rosetta/pET28a-RR0628 Expresses soluble REC domain - Bab1_06282276 Rosetta/pET28a-RR0636 Expresses soluble REC domain - Bab1_06362277 Rosetta/pET28a-RR1138 Expresses soluble REC domain - Bab1_11382278 Rosetta/pET28a-RR1140 Expresses soluble REC domain - Bab1_11402279 Rosetta/pET28a-RR1538 Expresses soluble REC domain - Bab1_15382280 Rosetta/pET28a-RR1614 Expresses soluble REC domain - ctra - Bab1_16142281 Rosetta/pET28a-RR1671 Expresses soluble REC domain - Bab1_16712282 Rosetta/pET28a-RR2006 Expresses soluble REC domain - Bab1_20062283 Rosetta/pET28a-RR2092 Expresses soluble REC domain - Bab1_20922284 Rosetta/pET28a-RR2146 Expresses soluble REC domain - Bab1_21462285 Rosetta/pET28a-RR0041 Expresses soluble REC domain - cpdr - Bab2_00412286 Rosetta/pET28a-RR0042 Expresses soluble REC domain - Bab2_00422287 Rosetta/pET28a-RR0081 Expresses soluble REC domain - Bab2_00812288 Rosetta/pET28a-RR0222 Expresses soluble REC domain - Bab2_02222289 Rosetta/pET28a-RR0628 Expresses soluble REC domain - Bab2_06282290 Rosetta/pET28a-RR0630 Expresses soluble REC domain - Bab2_06302291 Rosetta/pET28a-RR0762 Expresses soluble REC domain - Bab2_07622292 Rosetta/pET28a-RR0806 Expresses soluble REC domain - Bab2_08062293 Rosetta/pET28a-RR1099 Expresses soluble REC domain - Bab2_10992294 Rosetta/pET28a-chpT Expresses full-length chpt - Bab1_16132295 Rosetta/pET28a-chpT(N33A,E36A,E40A) Site-directed mutant of FC22942296 Rosetta/pET28a-chpT(K96A,R156A,H177A) Site-directed mutant of FC22942297 TOP10/pNPTS138::ctrA(V148F) For making V148F mutant in Brucela2298 Rosetta/pET28a-chpT(N33R) Site-directed mutant of FC22942299 Rosetta/pET28a-chpT(E36R) Site-directed mutant of FC22942300 Rosetta/pET28a-chpT(E40R) Site-directed mutant of FC22942301 Rosetta/pET28a-ctrA(S15R) Site-directed mutant of FC22802302 Rosetta/pET28a-cpdR(F16R) Site-directed mutant of FC22852303 Rosetta/pET28a-cckAsoluble Expresses soluble fragment of cckaCBA no. Brucella strain21 B. abortus 2308 WT Brucella strain25 2308 pSRK- WT Brucella with empty pSRK plasmid113 2308 pSRK-chpT WT Brucella expressing chpT-WT from pSRK114 2308 pSRK-chpT(E36R) WT Brucella expressing chpT(E36R) from pSRK115 2308 ctrA(V148F) Temperature-sensitive allele of ctrA116 2308 pSRK-cpdR(WT) WT Brucella expressing cpdR-WT117 2308 pSRK-cpdR(D52A) WT Brucella expressing cpdR-D52A
“FC” and “CBA” refer to S.C. laboratory strain prefixes.
Willett et al. www.pnas.org/cgi/content/short/1503118112 9 of 11

Table S4. Primers used in this study
Name Sequence 5′→3′ Notes
RR0099-BamHI F AAGGATCCTTGATGAACGATCTCGAACATAGG BAB1_0099; for cloning BamHI/HindIIIfragment into pET28a foroverexpression; FL protein
RR0099-HindIII R AAAGCTTTCAGGCGACGCTGGATGA
RR0136 - BamHI - F GGATCCATGGAAGACTTGAAGCA BAB1_0136 - for cloning BamHI/HindIIIfragment into pET28afor overexpression
RR0136 - HindIII - R AAGCTTTCAGCGCGGCGCGCGCT
RR0345 - BamHI- F GGATCCATGCATTTCATTATCGC BAB1_0345 - for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–121
RR0345 - HindIII - R AAGCTTtcaGCCGGCAAGCACCGCACCCA
RR0370 - BamHI - F GGATCCATGACGAATGAATGCAA BAB1_0370; for cloning BamHI/HindIIIfragment into pET28a for overexpression;
amino acids 1–126RR0370 - HindIII - R AAGCTTtcaGCCGGAGGCGACGGCAT
RR0628 - BamHI - F GGATCCTTGCGTATCCTGATTGTTGA BAB1_0628; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–120
RR0628 - HindIII - R AAGCTTtcaGGCAGCACGGCGGATCA
RR0636 - BamHI - F GGATCCATGAAAATTCTCGTTATC BAB1_0636; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–118
RR0636 - HindIII - R AAGCTTtcaGCGGCGCTGCAAGACCTCGACAC
RR1138 - BamHI - F GGATCCATGGCGGCCGATATTCT BAB1_1138; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–120
RR1138 - HindIII - R AAGCTTtcaGGTTTCCAGCGCCCGTT
RR1140 - BamHI - F GGATCCATGATTGCAGGGCGTCCGA BAB1_1140; for cloning BamHI/HindIIIfragment into pET28a for overexpression:amino acids 1–120
RR1140 - HindIII - R AAGCTTtcaTTCCGAGAGCGCGCGCC
RR1538 - BamHI - F GGATCCATGGGATCGAAAGACGC BAB1_1538; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–137
RR1538 - HindIII - R AAGCTTtcaACGGCGCAGGATATTGT
RR1614 - BamHI -F GGATCCATGCGCGTCCTTTTGAT BAB1_1614; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–118
RR1614 - XhoI - R CTCGAGtcaGCGGCGGACGATCGCATGGATACGG
RR1671 - BamHI - F GGATCCCGTCTGATGATCATTGAGGATG BAB1_1671; for cloning BamHI/XhoIfragment into pET28a for overexpression;amino acids 141–264
RR1671 - XhoI - R CTCGAGTCAGGCAGCAACTTGCGA
RR2006 - BamHI - F GGATCCATGACAGGCCGTACTAT BAB1_2006; for cloning BamHI/XhoIfragment into pET28a for overexpression;amino acids 1–122
RR2006 - XhoI - R CTCGAGtcaCTGGCGCAACTGCGCGC
RR2092 - BamHI - F GGATCCATGAAGGAAGCTTCGGC BAB1_2092; for cloning BamHI/XhoIfragment into pET28a for overexpression;amino acids 1–128
RR2092 - XhoI - R CTCGAGtcaGCGCGCCGCCACGCGGC
RR2146 - BamHI - F GGATCCATGTCCCAAGTACCCCGA BAB1_2146; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–124
RR2146 - HindIII - R AAGCTTtcaCGCCTTGACGCGGGCAA
RR0041 - BamHI - F GGATCCATGAACTGCGCATATGA BAB2_0041; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–126
RR0041- HindIII - R AAGCTTtcaTGTCCGGATCGCCTGTT
RR0042- BamHI - F GGATCCATGAAGAGAATCCTTCTAGCTGA BAB2_0042; for cloning BamHI/HindIIIfragment into pET28a foroverexpression; FL protein
RR0042 - HindIII - R AAGCTTTCAGGCGGCGATCAGCA
RR0081 - BamHI -F GGATCCATGGCAACGCGCATTCT BAB2_0081; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–121
RR0081 - HindIII - R AAGCTTtcaGAGTTTCAACACATTTGC
RR0222 - BamHI - F GGATCCATGAGAATTATCCTCAT BAB2_0222; for cloning BamHI/HindIIIfragment into pET28afor overexpression
RR0222- HindIII - R AAGCTTtcaGCGCCGCGCTACCGCGT
RR0628 - BamHI -F GGATCCATGACGAAAAGCGTAAT BAB2_0628; for cloning BamHI/HindIIIfragment into pET28a for overexpression;FL protein
RR0628 - HindIII - R AAGCTTCTAGGCATCGCCCAGAT
RR0630 - BamHI - F GGATCCATGACAGCAAGGATTCTCGTCG BAB2_0630: for cloning BamHI/XhoIfragment into pET28a foroverexpression: For amplificationof both REC domains together
RR0630 - XhoI - R CTCGAGtcaGCGCTTGATCTGTGTGC
Willett et al. www.pnas.org/cgi/content/short/1503118112 10 of 11

Table S4. Cont.
Name Sequence 5′→3′ Notes
RR0762 - BamHI - F GGATCCTTGAAAGAAGACGCCCACATTC BAB2_0762; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–121
RR0762 - HindIII - R AAGCTTtcaACGGCGAAGCACGGCGC
RR0806 - BamHI - F GGATCCATGAATATTTCTCCACA BAB2_0806; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–157
RR0806 - HindIII - R AAGCTTtcaGCCAGCCATGGCAAGCCGA
RR1099 - BamHI - F GGATCCATGATTGTTGTCGTTGACGACA BAB2_1099; for cloning BamHI/HindIIIfragment into pET28a for overexpression;amino acids 1–106
RR1099 - HindIII - R AAGCTTtcaGCGGACATGAACGGGCTT
Hpt1613 - BamHI - F GGATCCATGTCCTTGCCCGTTAC Bab1_1613, ChpT; for cloning BamHI/HindIIIfragment into pET28a for overexpression;expresses entire protein
Hpt1613 - Hindiii - R AAGCTTTCATTCCGCGGAAAAGACGATAT
dHpt1613-Up-BamHI-F GGATCCAGCGATGAAGGAGACAAACAGA For deletion of B. abortus Hpt 1613dHpt1613-Up-R ccgcggaCAAGGACATGGCGCTTT
dHpt1613-Dn-F ATGTCCTTGTCCGCGGAATGAATTGA
dHpt1613-Dn-SalI-R AAGTCGACACGATTATATGACCAAG
CckA-kinase only-BamHI-F ATATGGATCCGCGCTGGAAAACCAGAT For cloning BamHI/HindIII fragmentinto pET28a for overexpression;for soluble form of CckA
CckA-FL-HindIII-R ATATAAGCTTTCAATCCTGTTTCTCCAG
ChpT-F-NdeI-Brucella-o/E TATACATATGTCCTTGCCCGTTAC Site-directed mutagenesis primersChpT-R-KpnI-Brucella-o/E ATATGGTACCTCATTCCGCGGAAAAGACGATAT
ChpT-E40A-R catcggcgccacctgcttccagcaattcc
ChpT-E40A-F ggaattgctggaagcaggtggcgccgatg
ChpT-N33A,E36A-R cttcttccagcaatgccagaccagcattgatcgcaccgaccggt
ChpT-N33A,E36A-F accggtcggtgcgatcaatgctggtctggcattgctggaagaag
ChpT-K96A-R aggtgaattccggcgcttcgttcctgaaatattcagtggcga
ChpT-K96A-F tcgccactgaatatttcaggaacgaagcgccggaattcacct
ChpT-R156A-R cggcggcacggctagcatccggcctttgac
ChpT-R156A-F gtcaaaggccggatgctagccgtgccgccg
ChpT-H177A-R gggctgtacggaagcggcgtcgatgggc
ChpT-H177A-F gcccatcgacgccgcttccgtacagcc
chpT-R-HA+XbaI tctagaTCAAGCGTAATCTGGAACATCGTATGGGTATTCCGCG-
GAAAAGACGATAT
CtrA-V148F-R ccggtcagatgaaagcgctggcctgcg
CtrA-V148F-F cgcaggccagcgctttcatctgaccgg
ChpT-N33R-R gcaattccagacccctattgatcgcaccgaccggtgag
ChpT-N33R-F ctcaccggtcggtgcgatcaataggggtctggaattgc
ChpT-E36R-R gccaccttcttccagcaatctcagaccattattgatcgca
ChpT-E36R-F tgcgatcaataatggtctgagattgctggaagaaggtggc
ChpT-E40R-R atcggcgccacctctttccagcaattccagaccattattg
ChpT-E40R-F caataatggtctggaattgctggaaagaggtggcgccgat
ctrA-s15R-R ttgagcatcaactcaatcctctgtgcgatagcact
ctrA-s15R-F agtgctatcgcacagaggattgagttgatgctcaa
cpdR-F16R-R ccagcgcttttacgaggcgacggcgcatatcgttatcg
cpdR-F16R-F cgataacgatatgcgccgtcgcctcgtaaaagcgctgg
rplK_qPCR_F CCGGTGACCTACTTCCTCAA Housekeeping control for qPCRrplK_qPCR_R ATCGAACGGGCAGAACCT
ccrM_qPCR_F GGCTCGACTCCATCATCAAA For qPCR to analyze B. abortusccrM (bab1_0516). Yields109-bp PCR product
ccrM_qPCR_R CCGCCAAGCTGGAGATTATAG
Willett et al. www.pnas.org/cgi/content/short/1503118112 11 of 11





























