Embed Size (px)
Citation preview

1
Structure Comparison and Alignment
Modified from the slides by Ilya Shindyalov, SDSC/UCSD(http://www.sdsc.edu/pb/edu/pharm201/11/11.ppt)
and
Patrice Koehl, UCD(http://nook.cs.ucdavis.edu:8080/~koehl/Classes/CSB/CSB_Lecture6.ppt)

2
Agenda
• Concepts: – Similarity vs. Homology– Structure vs. Sequence Similarity– Alignment: Pairwise vs. Multiple– Complexity vs. Performance– Advantages vs. Bottlenecks
• Structural similarity measures• Structure Similarity: “How To” or “One of the problems
which would never be solved finally”?• In depth analysis of one algorithm (CE)• How new biological insights come from structure
similarity?

3
Similarity vs. Homology?
Similarity – statistically significant occurrence of identical or similarfeatures or a combination of them.
Concepts:
Homology – occurrence of identical or similar features as a result ofcommon ancestry.
Inferring Biological Meaning by Similarity assuming one of the following:
• Similarity implies homology. • Similarity can be used to transfer biological properties (e.g.
function) between molecules with characterized and uncharacterized biological properties, respectively.

4
1COL:A - COLICIN *A (C-TERMINAL DOMAIN) (PORE-FORMING DOMAIN)1CPC:L - C-PHYCOCYANIN
1COL:A 71 AMKINKADRDALVNAWKHVDAQDMANKLGNLSKAFK---------------VADVVMKVE 1CPC:L 29 ALVADGNKRMDVVNRITGNSS-TIVANAARSLFAEQPQLIAPGGNAYTSRRMAACLRDME
1COL:A 116 KVREKSIEGYETGNWGPLML-EVESWVLSG----IASSVALGIFSATLGAYALSLGV---1CPC:L 88 IILRYVTYAIFAGDASVLDDRCLNGLKETYLALGTPGSSVAVGVQKMKDAALAIAGDTNG
1COL:A 168 -PAIAVGIAGILLAAVVGAL 1CPC:L 148 ITRGDCASLMAEVASYFDKA
Similarity: Structure vs. Sequence?
Concepts:
Sequence Structure
Alignment – established equivalences between polypeptide residuesor the process of establishing such equivalences.
Representation of the alignment is generally irrelevant to the method of alignment or data used, while alignment itself is not. See below:
Alignment
Other properties/features

5
Similarity: Structure vs. Sequence?
Concepts: Structure superposition – placement of two or more 3D protein structures (as rigid bodies) in space, so that certain scoring function (RMSD, Intra-residue distance function) is optimized.
Often, only aligned residues are used in superposition.
State-of-the-art algorithm – (Kabsch, 1978).

6
Relationships between Protein Sequence and Structure
Similar Sequence Similar Structure
yes yes
no no
yes no
no yes
Comparison between two chains in PDB:

7The classic hssp curve from Sander and Schneider (1991) Proteins 9:56-68

8
Random 1000 structurally similar PDB polypeptide chains with z > 4.5 (CE)
Twilight Zone
Midnight Zone
Sequence vs Structure – 1000 Chains
alignment length
% s
eque
nce
iden
tity

9
1ABR:B - ABRIN-A1BAS:_ - BASIC FIBROBLAST GROWTH FACTOR (BFGF)
Seq. identity = 10% RMSD = 1.9Å

10
1NEU:_ - MYELIN P0 PROTEIN1DBK:L - FAB' FRAGMENT OF MONOCLONAL ANTIBODY DB3
Seq. identity = 14% RMSD = 1.9Å

11
Immunoglobulins 1MCO:L vs 3MCG:2 Seq. identity = 99%, RMSD = 5.5Å

12
Sequence versus Structure Alignment
• The protein sequence is a string of letters: there is an optimal solution (DP) to the problem of string matching, given a scoring scheme
• The protein structure is a 3D shape: the goal is to find algorithms similar to DP that finds the optimal match between two shapes.

13
Why structure comparison is important
• to automatically classify new protein structures• for functional assignment and hopefully new biology• for alignment of predicted structure against structural
templates• to establish improved sequence relationships not
possible from sequence alone• for protein engineering

14
Some Distinctions
• Pairwise alignments are different from multiple structure alignments
• Multiple structure alignment algorithms are rare and of questionable quality (see for example Nucleic Acids Research (2004), 32, W100-W103)
• Multiple structure alignments should not be confused with multiple pairwise alignments
• Here we focus on pairwise comparison and alignment

15
Protein Structure Comparison
• Global versus local alignment
• Measuring protein shape similarity
• Protein structure superposition
• Protein structure alignment

16
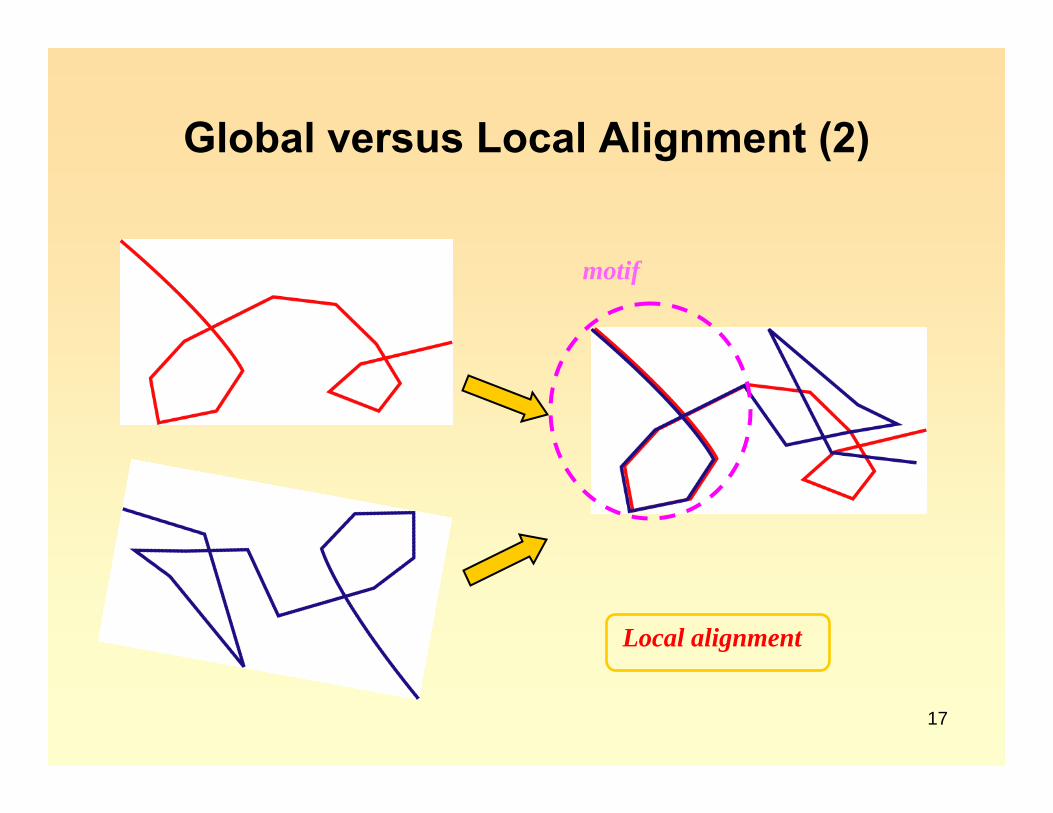
Global versus Local Alignment
Global alignment
17
Global versus Local Alignment (2)
Local alignment
motif

18
Measuring Protein Structure Similarity
• Visual comparison• Dihedral angle comparison• Distance matrix• RMSD (root mean square distance)
Given two “shapes” or structures A and B, we are interested in defining a distance, or similarity measure between A and B.
Is the resulting distance (similarity measure) D a metric?
D(A,B) ≤ D(A,C) + D(C,B)

19
Comparing Dihedral AnglesTorsion angles (φ,ψ ) are:
- local by nature- invariant upon rotation and translation of the molecule- compact (O(n) angles for a protein of n residues)
Add 1 degreeTo all φ, ψ
But…

20
Distance Matrix
1
2
3
4
6.0
8.1
5.9
03.85.98.14
3.803.86.03
5.93.803.82
8.16.03.801
4321

21
Distance Matrix (2)
• Advantages- invariant with respect to rotation and translation- can be used to compare proteins
• Disadvantages- the distance matrix is O(n2) for a protein with n
residues- comparing distance matrix is a hard problem- insensitive to chirality

22
Root Mean Square Distance (RMSD)
To compare two sets of points (atoms) A={a1, a2, …aN} and B={b1, b2, …,bN}:
-Define a 1-to-1 correspondence between A and B
for example, ai corresponds to bi, for all i in [1, N]
-Compute RMSD as:
∑=
=N
iii bad
NBARMSD
1),(1),(
d(ai, bi) is the Euclidian distance between ai and bi.

23
Protein Structure Superposition
• Simplified problem: we know the correspondence between set A and set B
• We wish to compute the rigid transformation Tthat best align a1 with b1, a2 with b2, …, aN with bN
• The error to minimize is defined as: ∑=
=
−N
iii
TbaT
1
2)(minε
Old problem, solved in Statistics,Robotics, Medical Image Analysis,…

24
Protein Structure Superposition (2)
• A rigid-body transformation T is a combination of a translation t and a rotation R: T(x) = Rx + t
• The quantity to be minimized is:
∑=
+−=N
iii
RttbRa
1
2
,minε

25
The Translation Part
( ) 021
=+−=∂∂ ∑
=
N
iii tbRa
tεε is minimum with respect
to t when:
Then: ∑∑==
+⎟⎠
⎞⎜⎝
⎛−=N
ii
N
ii baRt
11
If both data sets A and B have been centered on 0, then t = 0 !
Step 1: Translate point sets A and B such that their centroids coincide at the origin of the framework

26
The Rotation Part (1)
Let μA and μB be then barycenters of A and B, and “rename” A and Bto be centered on 0:
[ ][ ]BNBB
ANAA
N
iiB
N
iiA
bbbBaaaA
bN
aN
μμμμμμ
μ
μ
−−−=−−−=
=
=
∑
∑
=
=
......
1
1
21
21
1
1
Build covariance matrix: TABC =
x = 3x33xN
Nx3

27
The Rotation Part (2)
U and V are orthogonal matrices, and D is a diagonal matrix containing the singular values.U, V and D are 3x3 matrices
TUDVC =
Compute SVD (Singular Value Decomposition) of C:
Define S by:
⎩⎨⎧
−>
=otherwisediag
CifIS
}1,1,1{0)det(
ThenTUSVR =

28
2. Build covariance matrix:
The Algorithm
TABC =
TUDVC =
⎩⎨⎧
−>
=otherwisediag
CifIS
}1,1,1{0)det(
1. Center the two point sets A and B
3. Compute SVD (Singular ValueDecomposition) of C:
5. Compute rotation matrix
4. Define S:
TUSVR =
O(N) time!
6. Compute RMSD:
N
sdbaRMSD i
ii
N
ii
N
ii ∑∑∑
===
−+=
3
11
2
1
2 2''

29
Example 1: Align NMR Structures
Superposition of NMRModels
1AW6

30
Example 2: Calmodulin
Two forms ofcalcium-boundCalmodulin:
Ligand free
Complexedwithtrifluoperazine

31
Example 2: Calmodulin
Global alignment:RMSD =15 Å /143 residues
Local alignment:RMSD = 0.9 Å/ 62 residues

32
RMSD is not a Metric
cRMS = 2.8 ǺcRMS = 2.85 Ǻ

33
Structure Similarity: “How To” or “One of the Problems which would never be solved finally”?

34
How to build structural alignment statistics?
Parameters/factors to consider:
Distance: RMSD vs. intra-protein distances or contacts/interactions?
Gaps vs. unaccounted unaligned areas?
Similar sequence vs. similar structure?
Known functionally important residues vs. similarity in structure?
How to handle structural movements and flexible areas?
Baseline: what is random structure?

35
How to evaluate significance of the structural alignment?
For sequence alignment the baseline is random sequence. What is random structure?
Is this random?
Or this?

36
Approach of Levitt and Gerstein (1998)
1. Use a resource where structure similarity is defined – SCOP.
2. Find a scoring function which allows good separation of trueand false positives:
⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜
⎝
⎛
−
⎟⎟⎠
⎞⎜⎜⎝
⎛+
= ∑ 21
12
0
gap
ijstr
N
dd
MS
dij - distance between residues i and j in alignment;M = 25;d0 = 5 Å; Ngap – total number of gaps in local alignment (i.e. not counting terminal gaps);

37
Approach of Levitt and Gerstein (1998)

38
Maintain 9 of 10 interactionsRMSD 1.5 Å
Maintain 5 of 10 interactionsRMSD 0.5 Å
Biological vs Geometric Alignments Plastocyanin versus Azurin (from Godzik, 1996)

39
CDK2 in two functional states
while sequence is identical, there is a drastic movement of catalytic loop
structural alignment doesn’t align the catalytic loop

40
Current State of the Art
• There are many papers published on this, but relatively few have code to download or Web sites from which to perform comparisons
• All methods can identify obvious similarities between two structures
• Remote similarities are detected by a subset of methods – different remote similarities are recognized by different methods
• Good alignments are much harder to come by• Speed is a serious issue with some algorithms
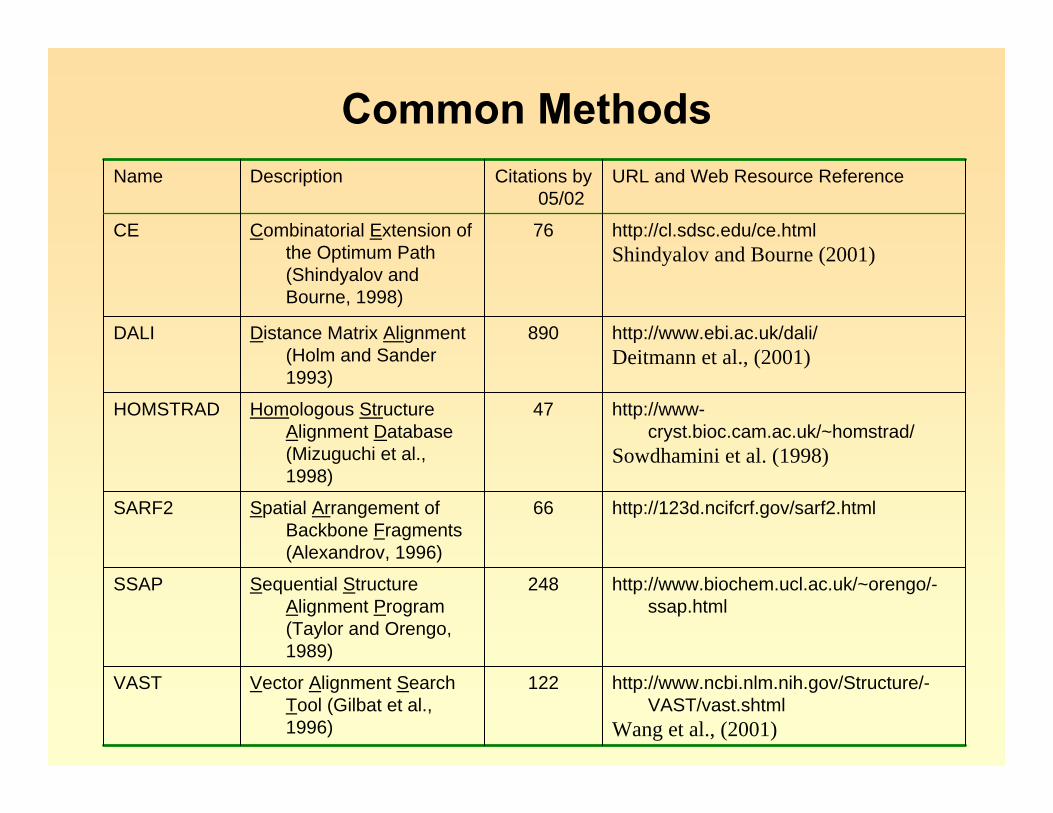
Common Methods
http://www.ncbi.nlm.nih.gov/Structure/-VAST/vast.shtml
Wang et al., (2001)
122Vector Alignment Search Tool (Gilbat et al., 1996)
VAST
http://www.biochem.ucl.ac.uk/~orengo/-ssap.html
248Sequential Structure Alignment Program (Taylor and Orengo, 1989)
SSAP
http://123d.ncifcrf.gov/sarf2.html66Spatial Arrangement of Backbone Fragments (Alexandrov, 1996)
SARF2
http://www-cryst.bioc.cam.ac.uk/~homstrad/
Sowdhamini et al. (1998)
47Homologous Structure Alignment Database (Mizuguchi et al., 1998)
HOMSTRAD
http://www.ebi.ac.uk/dali/Deitmann et al., (2001)
890Distance Matrix Alignment (Holm and Sander 1993)
DALI
http://cl.sdsc.edu/ce.htmlShindyalov and Bourne (2001)
76Combinatorial Extension of the Optimum Path (Shindyalov and Bourne, 1998)
CE
URL and Web Resource ReferenceCitations by 05/02
DescriptionName

42
Comparison of some methods (Novotny et al, 2004)















![Comparison of the Political Structure[1]](https://img.pdfslide.net/doc/110x75/577d2ae81a28ab4e1eaa68b5/comparison-of-the-political-structure1.jpg)