Embed Size (px)
DESCRIPTION
THIS STUDY ABOUT QUERCETINS EFFECTS ON THE HUMAN CELL LINES
Citation preview

WILLIAM BOYD MAYER, JR. The Effects of Iron and Quercetin on Apoptosis in a Human Hepatoma Cell Line (Under the Direction of JOAN G. FISCHER)
Quercetin, a flavonoid, is cytotoxic to human tumor cells at high concentrations.
The objective of the present study was to determine if cellular iron status influences
quercetin-stimulated cytotoxicity and apoptosis in a human hepatoblastoma cell line. To
test the hypothesis, cell viability, lipid peroxidation, and apoptosis were measured in
HepG2 cells following treatment with various levels of iron and quercetin. The data
suggest that Fe concentration may influence quercetin’s effect on cell viability and
apoptosis in HepG2 cells.
INDEX WORDS: Quercetin, Iron, Apoptosis, Liver cells, Oxidative stress,
Reactive oxygen species

THE EFFECTS OF IRON AND QUERCETIN ON APOPTOSIS IN A HUMAN
HEPATOMA CELL LINE
by
WILLIAM BOYD MAYER, JR.
B.S., Clemson University, 1995
A Thesis Submitted to the Graduate Faculty of The University of Georgia in Partial
Fulfillment of the Requirements for the Degree
MASTER OF SCIENCE
ATHENS, GEORGIA
2001

© 2001
William Boyd Mayer, Jr.
All Rights Reserved

THE EFFECTS OF IRON AND QUERCETIN ON APOPTOSIS IN A HUMAN
HEPATOMA CELL LINE
by
WILLIAM BOYD MAYER, JR.
Approved:
Major Professor: Joan G. Fischer Committee: Mary Ann Johnson Arthur Grider Electronic Version Approved:
Gordhan L.Patel Dean of the Graduate School The University of Georgia December 2001

ACKNOWLEDGEMENTS
I am truly grateful to the many individuals who contributed their time and efforts
to my development as a graduate student. First, I would like to express my appreciation
to Dr. Joan Fischer. As a mentor, she taught me with patience, always providing support
and direction. She was a perfect role model, both as a professional and as a person.
I would also like to thank my committee members, Drs. Johnson and Grider. For
my entire graduate career, they were instrumental in helping to improve my research and
analytical skills. As well, they opened their labs to me for use of much-needed
equipment.
In our lab, Joyce Power was my supervisor, life management consultant, and
mother-figure. Her friendship and kindness will always be a special part of my
experience at the University of Georgia.
Dr. Michael Mouat was an invaluable resource to my project. I am thankful not
only for his expert advice, but also for his hospitality and gracious attitude since the
beginning of my career.
I would also like to thank Dr. Barbara Grossman and Mrs. Linda Azain. Both
provided incredible examples as instructors, and gave me the opportunity to develop my
creativity and public speaking skills.
Thanks to Dr. Rick Lewis, Chris Modlesky, and Jean Edmonds. They provided
their time and lab equipment to help with my research.
iv

v
I would like to thank my entire family in both Mt. Pleasant and Augusta. They
provided me with the unconditional support and constant encouragement I needed to
achieve my goals.
Most of all, I am grateful to my wife, Mary Dickey. She has given so much to
support me in school and in my life.

TABLE OF CONTENTS
Page
ACKNOWLEDGEMENTS............................................................................................... iv
LIST OF FIGURES ......................................................................................................... viii
LIST OF TABLES............................................................................................................. ix
CHAPTER
I INTRODUCTION...................................................................................................1
II LITERATURE REVIEW........................................................................................4
Cancer ..........................................................................................................4
Cell Proliferation/Division...........................................................................6
Apoptosis .....................................................................................................7
Measures of Apoptosis...............................................................................11
Reactive Oxygen Species...........................................................................15
Iron and Oxidative Stress...........................................................................19
Fruits and Vegetables and Cancer Prevention ...........................................21
Polyphenols................................................................................................23
Quercetin....................................................................................................30
III IRON STATUS OF THE CELL MAY ALTER QUERCETIN-INDUCED
APOPTOSIS OF HUMAN HEPATOMA CELLS ...............................................38
Abstract ......................................................................................................38
Introduction................................................................................................39
vi

vii
Methods .....................................................................................................41
Results........................................................................................................50
Discussion..................................................................................................52
Figures .......................................................................................................60
Tables.........................................................................................................64
IV SUMMARY
Major Findings...........................................................................................67
Implications ...............................................................................................68
Study Limitations.......................................................................................68
Further Research ........................................................................................69
REFERENCES ......................................................................................................71

LIST OF FIGURES
CHAPTER III
Figure 1: Cellular Iron Concentration Following 24 Hour Iron + 24 Hour Quercetin Treatments.........................................................60
Figure 2: Lipid Peroxidation......................................................................61
Figure 3: Cell Viability Measured by Trypan Blue Exclusion ..................62
Figure 4: Mean Percent Increase in Quercetin-Induced Apoptotic DNA Fragmentation Above Control Within Each Iron Treatment ....................................................................................63
viii

LIST OF TABLES
CHAPTER II
Table 1: Previous Studies Demonstrating the Effect of Flavonoids Alone or in Combination with Redox Metals on Cell Lines or Specific Cell Components ........................................................37
CHAPTER III
Table 2: Study Design - Assays and Treatment Groups............................47
Table 3: General Timeline for Assays .......................................................48
Table 4: DNA Fragmentation: Chi Square Analysis .................................64
Table 5: Percent Cells with Caspase Activity............................................65
Table 6: Percent of Cells with Apoptotic Morphology (Chromatin Condensation)............................................................66
ix

CHAPTER I
INTRODUCTION Iron is a redox active metal, meaning that it may readily alternate between ferrous
and ferric states, accepting or donating an electron to a wide range of biological
substances. Through this mechanism, iron catalyzes a variety of damaging reactions
within the cell (McCord 1998). Iron can catalyze the formation of the hydroxyl radical
from H2O2 as well as the decomposition of lipid hydroperoxides to alkoxyl, peroxyl, and
other free radicals (Ibrahim 1997). Therefore, iron is a key factor in the establishment of
a prooxidant status in the cell (Meneghini 1997). Previous studies in our laboratory and
others have suggested that iron status of a cell may alter apoptosis (Stimson 1998; Wang
et al. 1999; Whittaker et al. 1996). Further, studies by Cermak et al. (1993), proposed
that acute exposure of intestinal tumor cells to iron may increase their susceptibility to
oxidant-mediated lysis.
Apoptosis is described as a sort of cellular suicide (Samali 1996) in which a cell
takes an active role in its own demise. It consists of a preprogrammed cascade of
metabolic events that occurs in response to various environmental inducers or a
combination of intrinsic factors, ultimately leading to the disintegration of the cell
(Darzynkiewiez 1995). Studies have shown that cell death caused by apoptosis may be
the mechanism by which some chemotherapeutic agents function.
It has also been proposed that oxidative stress is a mediator of apoptosis in eukaryotic
cells via several different mechanisms (Buttke and Sandstrom 1994). Oxidative stress
1

2
may induce apoptosis by damaging DNA. This may occur via metal-catalyzed oxidation
with subsequent generation of reactive intermediates such as hydrogen peroxide and the
hydroxyl radical (Duthie et al. 1997). In addition, oxidative stress may lead to the
formation of oxidized lipids in cell membranes, which may induce apoptosis (Buttke and
Sandstrom, 1994).
Recent studies (Kuo 1996) have shown that apoptosis was the mechanism of cell
death when flavonoids were added to neoplastic cell lines. Some phenolic antioxidants,
such as flavonoids, can reduce ferric iron to ferrous iron, thereby facilitating free radical
damage to carbohydrates, lipids, proteins, and DNA (Smith et al. 1992). Free iron may
also be released from ferritin by an appropriate reducing agent such as quercetin (Cermak
1993). Sahu and Washington (1992) found that increasing quercetin levels in the
presence of iron significantly increased lipid peroxidation and nuclear DNA damage in
rat liver nuclei. Therefore, tumor cells exposed to high iron levels in the presence of a
reducing agent, such as quercetin, could theoretically undergo apoptosis.
The purpose of this study was to determine whether a flavonoid common to many
foods (quercetin) could induce apoptosis in the HepG2 cell line, a liver hepatoma cell
line, and whether cellular iron status alters apoptosis.
It was hypothesized that 1) cellular iron status would increase with higher media
iron concentrations, and that addition of quercetin to iron-loaded cells would have no
effect on cellular iron, 2) addition of quercetin to iron-loaded cells would increase lipid
peroxidation, 3) cellular iron concentration would influence the prooxidative ability of
quercetin to decrease cell viability, 4) cellular iron concentration would influence the pro-
oxidative ability of quercetin to induce specific apoptotic changes in the cell measured as

3
200-bp DNA nucleosome fragmentation (ladder effect), caspase activation, and
chromatin condensation.
Major findings were 1) cellular iron status increased with iron loading over a 24
hour incubation period, 2) addition of quercetin to iron-loaded cells had no effect on
cellular iron, 3) iron had a positive effect on lipid peroxidation, while quercetin did not,
4) 80 µM concentrations of quercetin tended to decrease cell viability in iron-loaded
cells, 5) an 80 µM concentration of quercetin was consistently associated with an
increase in apoptosis-induced DNA fragmentation, and increases were greater in iron-
loaded cells, but the iron effect was not significant, 6) chromatin condensation was
affected by quercetin, but not iron, and 7) caspase activity was significantly increased in
cells supplemented with 80 µM quercetin, with the greatest increases seen in iron-
loaded cells.

CHAPTER II
LITERATURE REVIEW
CANCER
More than 1 million new cases of cancer and over 500,000 deaths occur each
year in the U.S. due to cancer (Breslow 1997). Moreover, the number of Americans
ever diagnosed with any invasive (metastatic) cancer could be over 11 million by
2020. Finally, people diagnosed with certain cancers are at increased risk of
additional cancers (Polednak 1997). Overall, rates of cancer are quite varied among
U.S. racial and ethnic populations. Considering all American men, cancer rates are
highest among African Americans (560 cases per 100,000 people) and whites (469
cases per 100,000 people). Incidence was lower among women in every racial and
ethnic group than among the men (Breslow 1997). Cancer is a group of diseases
characterized by uncontrolled growth and proliferation of abnormal cells. It is the
result of both genetic and environmental factors, which may act together or in
sequence to initiate carcinogenesis (American Cancer Society 1998). In addition to
elucidating some of the genetic and environmental causes of cancer, current research
has also gone further to uncover the mechanisms by which carcinogenesis occurs.
Cancer is caused by aberrant DNA. First, genetic mutations in critical genes can lead
to tumors. In fact, in approximately one-half of human tumors, mutations exist in the
tumor-suppressor gene, p53. This is important because inactivation of the p53 gene
allows for uncontrolled cell division. Another mechanism involved in carcinogenesis
4

5
is DNA lesions. This refers to damaged bases or chromosome breaks that have a
certain probability of producing a mutation upon cell division. The “effectiveness” of
a particular lesion depends on the rate of excision by DNA repair enzymes, and on the
probability that a mutation will occur when the cell divides. Cellular apoptosis or
“programmed cell death” provides the body with a mechanism for removal of cells
with damaged DNA. The effect that a particular insult has on the cell is dependent on
the level of each of these defenses, which in turn are actually dependent on the past
history of exposure. These defenses can also be disabled by lack of micronutrients
such as antioxidants in the diet.
On a molecular level, antioxidant deficiency is considered to be a risk factor for
cancer, as it can lead to “unrepaired or misrepaired endogenous oxidative DNA
damage” (Sahu and Washington 1992) and membrane lipid peroxidation. Free
radicals may mediate many events in carcinogenesis by producing damage to DNA,
lipids, proteins, and carbohydrates. External sources of free radical damage include
polycyclic aromatic hydrocarbons in food, polluted air, cigarette smoke, background
radiation, and internal sources include oxidative transformations in prostaglandin
synthesis, redox cycling of quinones, and oxidative phosphorylation (Block 1992).
Still another mechanism that may underlie excess free radical production is
chronic infection and inflammation. Leukocytes and other phagocytic cells combat
bacteria, parasites, and virus-infected cells by destroying them with nitrogen oxide and
superoxide. Both of these compounds can lead to formation of peroxynitrite,
hypochlorite, and hydrogen peroxide, all of which are mutagenic oxidizing agents.
So, while these oxidants protect the body from immediate harm, they can cause

6
oxidative damage to DNA, DNA mutation, and chronic cell death with compensatory
cell division. All of these processes then contribute to cancer development.
Cancer, one of the three major diseases in the U.S, is becoming more common
in the Western world and also in many populations of developing countries. With
modern medicine, some treatments have met with success, but they have been very
expensive. It is becoming evident that the most cost-effective control of these diseases
is through prevention. This is mainly achieved through the diet and other lifestyle
changes (Argiles 1998). In addition, cancer chemoprevention is a new field that has
great potential to affect cancer incidence rates.
CELL PROLIFERATION / DIVISION
A major factor in mutagenesis, and hence cancer development, is cell division.
When the cell divides, a DNA lesion can produce a point mutation, a single base
substitution, a deletion, or a translocation. Therefore, a critical factor in the mutagenic
effect of an agent is the increment of division that it produces in addition to cell
division in those cells that are important - the stem cells. Stem cells are not discarded,
as daughter cells are. An increase in the rate of cell division of stem cells increases
the mutation, leading to cancer. Increased cell division can be caused by a wide array
of agents, including increased levels of a particular hormone, excess calories, chronic
inflammation, or chemical exposure (Ames 1995). It is easy to see that without
continuous aid from antioxidants and radical scavengers, and endogenous defense
mechanisms, survival would cease (Block 1992). Another important protective
mechanism is cell cycle checkpoints. These checkpoints prevent cells with too many
DNA lesions from dividing (Darzynkiewicz 1995).

7
Most recently, the view of cancer as a disease of cell proliferation has been
challenged. Cell death studies have now shown how cell populations are maintained,
and how defects in cell regulation may contribute to the development of malignancy.
Therefore, focus has shifted to include cell death inducers and repressors. Defects in
either may produce deleterious results, especially considering the fact that increasing
the life span of a cell also increases the probability of acquiring mutations. When cell
proliferation exceeds cell death, researchers believe that this is 1) a result of
overexpression of a cell death repressor protein, or 2) resistance to conditions that
would normally kill the cell. Reduced cell death may also be caused by loss-of -
function mutations in cell death inducer proteins (Martin 1997).
APOPTOSIS
Apoptosis, or “programmed cell death,” is described as a sort of cellular suicide.
The term “apoptosis” comes from a Greek word that describes the process of leaves
falling from a tree, or petals from a flower (Samali 1996). It is well established that
during this process, a cell takes an active role in its own demise. A preprogrammed
cascade of metabolic events occurs in response to various environmental inducers or
by a combination of intrinsic factors, ultimately leading to the disintegration of the cell
(Darzynkiewicz 1995). As a mechanism, apoptosis is believed to be evolutionarily
conserved due to the fact that it occurs in both nematodes and vertebrates (Clutton
1997).
Apoptosis is also important in many physiologic processes, especially during
embryonic and fetal development (Kuo 1996). Some examples of this process relative
to development are regression of the Mullerian duct in male embryos, the removal of

8
interdigital webs, and amphibian tail regression during metamorphosis (Columbano
1995). Other processes that require apoptosis are: establishment of immune self-
tolerance, killing of cells by cytotoxic immune cells (King and Cidlowski 1995),
hormone-dependent atrophy, and even tumor growth. Apoptosis is the mechanism in
each of these processes that regulates cell number and destroys damaged cells (Kuo
1996).
A variety of stimuli can induce apoptosis, such as DNA damage, growth factor
withdrawal, calcium ion influx, and viral infection (King and Cidlowski 1995). In
vivo, apoptosis is detected primarily in proliferating tissues (Meikrantz and Schlegel
1995). Apoptotic bodies are accumulations of separate, intact pieces of the cell body
that result from the effect of specific destructive molecules which dismantle the cell’s
interior (Martin 1997). These compact bodies can be found anywhere in the body
where cells are dividing. Examples include intestinal crypts, the epithelium of the
adrenal cortex, in differentiating spermatogonium, and in the germinal centers of
lymph nodes (Meikrantz and Schlegel 1995). Apoptosis is also evident after periods
of rapid proliferation, such as in mammary tissue following weaning, in the
endometrium at estrus, and during ovarian follicular atresia. It is therefore expected
that apoptosis is a hallmark of malignancies, as it occurs at increased frequency in
preneoplastic tissues. In this case, apoptosis is thought to serve as a balance for an
increase in cell number due to proliferation with cell loss caused by death (Meikrantz
and Schlegel 1995).
Cells undergoing apoptosis share a few morphological features that are typically
characteristic of mitosis. Both apoptotic and mitotic cells are characterized by lack of

9
adhesion, rounded morphology, and reduced cell volume. Also, both mitotic and
apoptotic cells condense their chromatin in disassembling the nuclear lamina (King
and Cidlowski 1995). In spite of these similarities, several differences do exist. First,
apoptotic cells do not assemble a mitotic spindle. Also, apoptotic cells exhibit rapid
cytoplasmic blebbing – zeisosis, or “bubbling” of the cell membrane - while mitotic
cells do not. Finally, in most cases, apoptotic cells cleave their own DNA into
approximately 200 bp fragments (King and Cidlowski 1995). Overall, apoptosis plays
an opposing role to mitosis in the maintenance of cell populations. Therefore, cell
deletion occurs at an increased rate in rapidly dividing tissues (Columbano 1995).
It has been proposed that there are two phases in the process of apoptosis: an
activation phase and an execution phase. The activation phase is composed of the
intracellular pathway that activates apoptosis. The execution phase is composed of the
molecular mechanisms of apoptosis, which may involve proteases and nucleases.
Reactive oxygen species are most likely to induce the activation phase by acting as
cytoplasmic messengers in cell signal transduction (Rollet-Labelle 1998). The
specific mechanisms of apoptosis involves several key events, which include rapid cell
dehydration, increase in free calcium concentration, activation of an endonuclease
which has affinity to internucleosomal DNA sections, and activation of
transglutaminase activity. The biochemical events of apoptosis coincide with changes
in cell morphology. The loss of intracellular water is reflected by cell shrinkage and
condensation of the cytoplasm. Condensation of chromatin occurs, starting from the
periphery of the nucleus, followed by nuclear fragmentation. Next, apoptotic bodies,
or regions of cytoplasm that harbor intact and still functionally active organelles,

10
together with fragmentation of nuclei (wrapped in fragments of the plasma membrane)
detach from the cell. Finally, apoptotic bodies are incorporated in the tissue by
neighboring phagocytic cells. Again, there is no leakage of the cytoplasmic contents
into the intercellular space, so tissue inflammation or scarring does not occur
(Darzynkiewicz 1995). Overall, there is a marked increase in transglutaminase
mRNA, protein and enzyme activity, and protein-bound lysine. The transglutaminase
activity may stabilize the apoptotic cells and form a shell around the cell, thereby
inhibiting membrane leakage (Samali 1996).
As apoptosis is a programmed cell death, it is the result of activation of specific
mechanisms that systematically destroy integral cellular structures (Jacob 1997).
However, another form of cell death exists in response to an acute departure from
normal physiologic conditions (Martin 1997). This sudden cell death is called
necrosis, a result of any environmental stress that directly overwhelms cellular defense
mechanisms (Jacob 1997). Ultimately, it is the intensity of the insult that determines
whether apoptosis or necrosis occurs (Stoian 1996).
The main differences between apoptosis and necrosis are evident in the events
that occur in each respective process. In necrosis, the cell membrane is damaged; the
cell swells (due to an influx of water) and lyses (Rollet-Labelle 1998). Lysis exposes
intracellular contents to the extracellular environment, many of which are toxic when
not contained within the cell. As neighboring cells are damaged by these exposed
toxins, a chain reaction of death radiates from the original necrotic cell. To further
exacerbate this chain reaction, neutrophils and inflammatory macrophages are
attracted to the dying cells, and release even more damaging toxic enzymes. In

11
apoptosis, cell shrinkage, chromatin condensation, and chromatin DNA fragmentation
occur (Jiang 1996). The changes in the plasma membrane of the cell attract
phagocytes (Martin 1997), and the cell is subsequently engulfed by these phagocytes
(Rollet-Labelle 1998). Therefore, as opposed to necrotic cells, apoptotic cells do not
release their contents into the intracellular matrix, so the inflammation response that
accompanies necrosis does not occur. This is an important factor that distinguishes
apoptosis as an “altruistic” form of cell death. In other words, damaged or injured cells
commit suicide to allow adjacent cells to proliferate without being affected by the
otherwise necrotic death of neighboring cells (Samali 1996).
MEASURES OF APOPTOSIS
Nuclear Morphology
“The main component of apoptotic cells is the appearance of highly condensed
chromatin, segregated into sharply defined bodies within an intact nuclear envelope”
(Meikrantz and Schlegel 1995). Key structural proteins that maintain the integrity of
the nuclear matrix are a group of cellular substrates affected by caspases during
apoptosis. Selective proteolysis of these proteins results in the disintegration of the
nuclear envelope (Lockshin 1998). As the nuclear envelope breaks down, chromatin
strands become detached and condense into “crescentic caps or toroids at the
periphery of the nucleus” (Martin 1997). These variations in cellular morphology can
be seen with Hoechst 33342, a cell permeable bisbenzimide adenine-thymine specific
fluorescent dye that binds to the minor groove of DNA (Zhang 1999). It stains
chromatin, a chain of nucleosomes joined by linker DNA (naked DNA; Voet and Voet

12
1990) and allows for the quantification of cells with the abnormal nuclear morphology
(chromatin condensation) characteristic of apoptosis.
DNA fragmentation
During apoptosis, endonucleases digest DNA. Therefore, detection of DNA
fragments is a commonly used assay for apoptosis (Meikrantz and Schlegel 1995).
The characteristic ladder pattern of DNA produced during the late stages of apoptosis
is caused by cleavage of internucleosomal linker DNA (Samali 1996). In contrast,
necrosis is identified by a diffuse smear on the gel, indicating random DNA
breakdown (Columbano 1995). Specifically, three patterns of the DNA degradation
are indicated in apoptosis: single strand nicks, larger DNA fragmentation of 50-200
kbp, and nucleosome size fragments of 180-200 bp in size (Samali 1996). Standard
gel electrophoresis is one method of identifying DNA ladders. In situ, a terminal
deoxynucleotidyl transferase mediated dUTP nick end-labeling (TUNEL) assay can be
used to stain apoptotic cells. This technique labels 3’-OH ends of DNA that have been
exposed during apoptotic DNA cleavage (Allen 1997). ELISA, or Enzyme-Linked
ImmunoSorbent Assay also can be used to quantify the hallmark 180-200 bp
nucleosomes produced by the cleavage of internucleosomal linker DNA (DNA not
associated with histones; Voet and Voet, 1990). This quantification is achieved with a
variety of techniques, an example being the Direct Sandwich ELISA. This particular
ELISA involves the passive adsorption of a monoclonal anti-histone antibody to a
solid phase, usually a plastic microtiter plate module, followed by addition of an
antigen (the histone-linked nucleosomes). The bound antigen is then “sandwiched”
between the solid phase antibody and an enzyme-labeled antibody conjugate. The

13
amount of conjugate retained within the immunocomplex after a washing procedure is
determined photometrically after binding with an ABTS (2,2’-azino-di-[3-
ethylbenzthiazoline sulfonate]) substrate. A spectrophotometer passes a specific
wavelength of light through the retained immunocomplexes, measuring the amount of
absorption of light (Crowther 1995).
Caspase Detection
Caspases are a family of proteases that have recently received much attention,
due to their suspected roles in programmed cell death. Caspases are present in healthy
cells as zymogens, or inactive configurations (Martin 1997), and are activated through
limited proteolysis by the removal of a pro-peptide (Slee 1999). Caspase activity is
triggered by specific stimuli. These stimuli can be organized into three broad
categories including 1) cellular death receptors, 2) the cytotoxic contents of certain
lymphocyte cell granules, and 3) stimuli that provoke generalized cell damage. The
third category of caspase stimuli includes cytotoxic drugs, radiation, heat shock,
survival factor deprivation, and other cellular stresses (Slee 1999).
Once activated by a stimulus, caspases cleave other caspases in a cascade of
activation. Most notably, many substrate proteins within the cell are also cleaved
during the cascade, leaving the hallmark structural changes of apoptosis (Martin
1997). During apoptotic cell death, caspases cleave approximately 200 polypeptides
of the cellular proteome. Approximately 70 of these caspase targets have been
identified. Therefore, genomic disassembly and subsequent cleavage into
oligonucleosomal fragments are accepted evidence of apoptosis (Nicholson 1999).

14
To ensure their efficacy, caspases are also responsible for halting normal DNA
repair processes by inactivating two key proteins involved in the homeostatic
maintenance of the genomic integrity – poly(ADP-ribose) polymerase (PARP) and
DNA-PKcs (catalytic subunit of DNA-dependent protein kinase). Obviously, if these
proteins remain activated, they would counteract any proteolysis events in the genome.
Simultaneously, caspases mediate the disablement of an inhibitor of an apoptosis-
dedicated endonuclease. In summary, results of the cleavage events are to 1) disable
DNA maintenance and repair mechanisms, 2) stop cell cycle progression, 3) render
caspase inhibitors inactive, 4) identify the dying cell as apoptotic, with the
consequence of phagocytosis. Finally, through proteolysis, caspases are also involved
in the disassembly of the cytoskeleton and nuclear scaffold (Nicholson 1999).
The tripartite structure of caspases consists of a prodomain, which varies among
the 11 known human caspases. Additionally, one large and one small subunit
comprise the active form of the enzyme. These subunits are released from the
proenzyme by cleavage at Asp (P1)-X(P1) bonds. This cleavage site enables the
caspases to auto-activate or be activated by other caspases. The proteolytic
components of the caspases include active site Cys and His residues. The active site
of caspases recognizes a very specific tetrapeptide sequence within a target substrate
polypeptide, which facilitates inhibitor and synthetic substrate design. Irreversible
caspase inhibitors, such as fluouromethylketones, may be labeled with a fluorogenic
agent, such as carboxyfluorescein. This cell permeable inhibitor binds covalently to a
wide range of active caspases to allow detection of caspase activity by flow cytometry,
fluorescence microscopy, or fluorescence spectroscopy (Nicholson 1999).

15
REACTIVE OXYGEN SPECIES
“Thirst” for electrons by molecules causes formation of a variety of reactive
oxygen intermediates. Reactive oxygen intermediates are oxygen species that possess
unpaired electrons or the ability to abstract electrons from other molecules. Reactive
oxygen species easily react with cellular macromolecules. This reaction can cause
immediate damage, or it may initiate a chain reaction where the free radical is passed
from one macromolecule to another, damaging cellular structures (Buttke and
Sandstrom 1994).
Under normal physiological conditions, reactive oxygen species can be formed.
For example, superoxide and hydrogen peroxide are produced as by-products of the
catalytic action of oxidases. These may also be formed by the leakage of electrons
from cellular electron-transferring chains (de Groot and Rauen 1998). More
physiological situations that produce free radicals include ubiquinone oxidation in
mitochondria, cytochrome p450 oxidation in microsomal membranes, reaction of
superoxide with hydrogen peroxide in the presence of free iron, prostaglandin
synthesis, respiratory bursts of phagocytic cells, xanthine oxidase activity, and
arginine oxidation by nitric oxide synthesis (Stoian 1996). Clearly, reactive oxygen
species are present in abundance in the normal human body. They can also be created
by external factors such as ionizing radiation (de Groot and Rauen 1998).
Of course, cells provide their own protection against reactive oxygen species.
Organisms have developed a variety of defenses to protect themselves against reactive
oxygen species. Superoxide dismutase, catalase, and glutathione (GSH) peroxidase
are some enzymes that perform this function. Also, some non-enzymatic compounds

16
protect against reactive oxygen species. Examples are glutathione, ascorbic acid, lipid
soluble alpha tocopherol, and some phytochemicals (de Groot and Rauen 1998).
Common examples of reactive oxygen species include hydrogen peroxide, the
superoxide and hydroxyl radicals, nitric oxide, singlet oxygen, and hypochlorous acid.
Although these reactive molecules are capable of attacking all biological molecules,
they tend to differ in their reactivities with regards to cellular molecules. The
hydroxyl radical attacks almost all biological molecules, while others like hydrogen
peroxide, singlet oxygen, and nitric oxide are less reactive. However, it should be
noted that the “low” reactivity species can be converted to highly reactive species.
Particularly, reaction of hydrogen peroxide with the lower valence states of iron and
copper can lead to the formation of the hydroxyl radical, or species of similar
reactivity, such as Fe2+O (ferryl ion) or Cu(OH)2+ (de Groot and Rauen 1998). Of the
above mentioned species, the hydroxyl radical is definitely thought to be the most
potent. This radical can attack every class of biological macromolecule. It can
depolymerize polysaccharides, cause DNA strand breaks, inactivate enzymes, and
initiate lipid peroxidation. Most of the hydroxyl radicals generated in vivo come from
metal ion-catalyzed breakdown of hydrogen peroxide. The extremely reactive
hydroxyl radical, once formed, can then initiate the process of lipid peroxidation
through abstracting a hydrogen atom from an unsaturated aliphatic lipid side chain.
This gives rise, by an autocatalytic chain reaction, to a lipid hydroperoxide (Haslam
1996). In vivo, it is estimated that generation of a single hydroxyl radical creates a
chain of 10 to 15 lipid peroxidation cycles until the chain is terminated, usually by
vitamin E. In addition, the general model of DNA modifications indicates the

17
hydroxyl radical is a common attacking species (Meneghini 1997). Therefore, it is the
action of the hydroxyl radical that may produce the greatest physiological damage to
human body components (McCord 1998).
Another radical worthy of mention is the superoxide radical. This free radical is
derived from molecular oxygen by the addition of a single electron. A wide variety of
pathological situations can produce the superoxide radical, including all infectious
diseases, and all diseases involving ischemia and reperfusion. Many researchers
believe that the most deleterious effect produced by the superoxide radical is the fact
that it causes reductive release of iron from ferritin. It is hypothesized that the
superoxide radical enters the ferritin core through hydrophilic channels. This is
followed by reduction of Fe3+ to Fe2+. This, in turn, allows release of iron from the
ferritin core. The combined presence of the superoxide radical and redox-active iron
can prove devastating to the cell in terms of maintaining membrane structure,
function, and viability (McCord 1998).
It is widely theorized that these reactive species may contribute to an array of
diseases, including cancer, atheroslerosis, Parkinson’s disease, Alzheimer’s dementia,
and reperfusion injury (de Groot and Rauen 1998). Reactive oxygen species generally
cause damage by reacting with essential cell and tissue molecules such as proteins,
lipids, and DNA. Furthermore, a concurrent decrease in cellular antioxidant capacity
might allow even more damage (de Groot and Rauen 1998). With regards to DNA,
reactive oxygen species can initiate many types of modifications. The most common
of these are approximately 20 possible base modifications. Also, strand cleavage is
another critical DNA lesion produced by reactive oxygen species (Meneghini 1997).

18
It has been shown that reactive oxygen species play a role as mediation molecules.
For example, nitric oxide acts as a neurotransmitter and has been shown to be an
endothelium-derived relaxing factor responsible for vasodilation. The generation of
these compounds is also important in signal transduction and microbicidal pathways
(de Groot and Rauen 1998).
Recently, investigators have suggested that reactive oxygen species play a
critical role in regulating apoptosis. Researchers have demonstrated that well-known
pathways of apoptosis are associated with moderate accumulation of reactive oxygen
species (McConkey 1998). Four observations that support this relationship are: 1)
antioxidants can inhibit apoptosis - which is not beneficial in destruction of tumor
cells, 2) generation of hydrogen peroxide has been detected in cells undergoing
apoptosis, 3) extracellular addition of reactive oxygen intermediates can stimulate
apoptosis, and 4) downregulation of intracellular antioxidant levels can induce
apoptosis. In addition, it is also thought that higher concentrations of reactive oxygen
species can induce necrosis, while lower concentrations stimulate apoptosis (Higuchi
1998).
There are several proposed mechanisms by which reactive oxygen species might
facilitate activation-induced cell death. First, reactive oxygen intermediate-mediated
damage has been shown to activate poly-ADP ribose transferase, which is associated
with apoptosis. Apparently, the polymerization of ADP-ribose to proteins results in a
rapid decline in cellular NAD/NADH stores, and a decrease in ATP. Both of these
conditions can induce apoptosis. Another mechanism states that oxidative stress may
cause formation of oxidized lipids in cell membranes. Reactive oxygen intermediates

19
react with polyunsaturated fatty acids and cholesterol in cell membranes, thereby
inducing apoptosis. Finally, it may also be possible that intracellular reactive oxygen
intermediates may activate apoptosis genes, most likely via an oxidative stress-
responsive nuclear transcription factor (Buttke and Sandstrom 1994).
IRON AND OXIDATIVE STRESS
Physiologically, iron is essential, but it also can be dangerous if present in a free
state. Human beings have evolved mechanisms for absorbing iron with 10%
efficiency. In humans, iron homeostasis is controlled by the rate of erythropoiesis and
the size of the iron stores (Khumalo 1998). The average adult male contains 4.5 grams
of iron, absorbs approximately 1 mg per day from the diet, and excretes about 1 mg
per day when in iron balance. Plasma iron turnover equals about 35 mg per day, with
slight disturbances of iron metabolism leading to iron overload or deficiency (Haslam
1996). There is no homeostatic mechanism to eliminate excess iron. Iron is rarely
present as a free ion, but instead, is bound to an array of active proteins such as
hemoglobin, myoglobin, ferritin, and transferrin (Jacob 1997). Cells store any excess
iron in the proteins ferritin and hemosiderin, predominantly in the liver. Therefore, as
long as the body is healthy, stored iron poses no great threat. However, under disease
conditions, it produces multiple possibilities for harmful superoxide production. Once
iron is freed in the presence of superoxide and its dismutation product, hydrogen
peroxide, the hydroxyl radical may be formed by the Haber-Weiss reaction (McCord
1998).
Although the transport protein, transferrin, has an exceptionally strong affinity
for ferric iron, iron concentration can also be locally and transiently increased through

20
tissue and blood degradation when injury occurs (Jacob 1997). At this point, iron can
promote free radical production, lipid peroxidation, and can initiate inflammatory
processes (Kato 1996) in conditions such as organ transplantation, carcinogenesis,
and aging (Kawabata 1997).
Many studies have indicated that transition metals are a major factor in the
initiation and propagation of peroxidative damage induced by free radicals (Ibrahim
1997). Iron is a redox active metal, meaning that it may readily alternate between
ferrous and ferric states, accepting or donating an electron to a wide range of
biological substances. Through this mechanism, iron catalyzes a variety of damaging
reactions within the cell (McCord 1998). In fact, sampling of chronic wounds (venous
stasis ulcers) shows a high local concentration of free iron ions. This lends credibility
to the idea that blood cells from a hemorrhage decompose and liberate their iron.
Then, the local increase in iron concentrations increases cellular iron concentration,
leading to excess production of reactive oxygen species (Jacob 1997).
The reactions by which some reactive species are formed are quite well known,
and include the Haber-Weiss and the Fenton reactions:
O2.- + Fe3+ O2 + Fe2+
O2.- + 1 e- + 2H+ H2O2
H2O2 + Fe2+ OH. + OH- + Fe3+
Net: O2.- + H2O2 O2 + OH. + OH. (Haber-Weiss reaction)
(Halliwell and Gutteridge 1999). On a molecular level, iron can catalyze the
decomposition of lipid hydroperoxides to generate alkoxyl, peroxyl, and other free
radicals (Ibrahim 1997). Therefore, iron may be a key factor in the establishment of a

21
prooxidant status in the cell (Meneghini 1997). The transition metals (vanadium, iron,
manganese, copper, and cobalt) are distinguished from other metals found in living
systems in that they can act as pi electron donors, and are redox active (Haslam 1996).
FRUITS AND VEGETABLES AND CANCER PREVENTION
An increasing amount of evidence indicates that nutrition is one of the most
important factors affecting health. Five of the ten leading causes of death for
Americans are associated with diet: heart disease, some cancers, stroke, diabetes, and
atherosclerosis (Glanz 1997). It has been estimated that about 30-35% of all cancers
can be attributed to diet. Some of the major hypotheses in this area are: high fat
consumption is a risk factor for prostate and colorectal cancers; high alcohol intake is
linked to oral, breast, or liver cancers; and low fiber intake is correlated with elevated
colon cancer risk. Conversely, another well-supported hypothesis is that high
consumption of fruits and vegetables is protective against many cancers (Steinmetz
and Potter 1996).
Inadequate consumption of fruits and vegetables is cited in almost 200 studies in
the epidemiological literature as correlating with increased cancer incidence (Block
1992). Cohort studies, case control studies, and animal studies have all shown an
inverse relationship between fruit and vegetable intake and risk of cancer (Steinmetz
and Potter 1996). Adequate intakes of fruits and vegetables and other fiber-rich foods
impact the incidence of cancers of the breast, colon, rectum, lung, and prostate (Glanz
1997). This suggests that the amount of cancer that could be prevented by increasing
fruit and vegetable consumption may be quite large, due to the fact that low intake is
so widespread. Furthermore, the quarter of the U.S. population with the lowest fruit

22
and vegetable intake has almost two times the rate of cancer when compared to the
quarter of the population with high fruit and vegetable consumption (Ames 1995).
Knowledge of how compounds in fruits and vegetables may lower cancer risk,
or be chemotherapeutic, is critical to our understanding of the effect of nutrition in
cancer prevention and treatment. It is believed that many of the compounds found in
fruits and vegetables protect against cancer via an antioxidant mechanism.
Historically, eating certain fruits and vegetables was thought to prevent or cure illness
ranging from headaches to heart disease. Early medicine was centered in large part
around prescription of specific food concoctions (particularly plant foods) for certain
illnesses. In recent years, researchers have evaluated which specific fruits and
vegetables have most often been found to be inversely associated with risk of any
cancer. Eighty-five percent of studies have shown that raw vegetables play a very
important role in preventive nutrition. Seventy percent of studies have also shown a
beneficial effect for allium vegetables (onions and leeks), carrots, green vegetables,
cruciferous vegetables, and tomatoes. An incredible number of substances in fruits
and vegetables have been shown or postulated to have anticarcinogenic properties
(Steinmetz and Potter 1996). If scientists are able to identify specific protective
compounds in foods, they may vary with the stage of the cancer process, but they
should also differ by sex, age, and across differing genetic metabolic profiles. The
effects of these foods may vary by organ site (Potter 1996).
Block (1992) cites approximately 120 of 130 studies conducted that show a
statistically significant reduction of risk of cancers of the lung, larynx, esophagus, oral
cavity, pancreas, stomach, rectum, cervix, colon, ovary, endometrium, breast, and

23
bladder with increased antioxidant intake. As a result of data such as these, it has been
hypothesized that the food supply may provide sufficient antioxidants to prevent some
of the events of cancer initiation and propagation. To do this, antioxidants must be
present at levels in proportion to the level of the agent that is causing oxidative
damage. However, individuals, or even populations, vary in the level of such
stressors. Therefore, no individual level will be sufficient at all times and for persons.
Furthermore, an effective level may differ depending on the cancer site.
POLYPHENOLS
Many natural substances in fruits and vegetables have been identified which
affect the process of carcinogenesis (Ren and Lien 1997). In the search for
chemopreventive agents, hundreds of compounds have been isolated from plant foods.
Some of the better studied compounds are carotenoids, vitamins C and E, selenium,
phytoestrogens, saponins, organic sulfur compounds, indoles, unsaturated fatty acids,
and polyphenols (Ren and Lien 1997; Steinmetz and Potter 1996).
Scientists have been interested in plant polyphenols for several decades. Their
role in plant physiology is essential, as they are involved in growth, reproduction, and
resistance to pathogens and predators. Polyphenols are widespread in plant foods and
beverages. The levels of each compound may vary greatly between varieties.
Polyphenols contained in plant foods depend upon genetic factors and environmental
conditions. Germination, degree of ripeness, variety, processing, and storage are also
factors that can influence the levels of these compounds in plants. Polyphenols are
found in virtually all plant organs. Therefore, they are a significant part of the human
diet. In the past, much of the nutritional focus was on the ability of some polyphenols

24
to bind and precipitate macromolecules (dietary protein, carbohydrates, and digestive
enzymes), altering the digestibility of food (Bravo 1998). Recently, the protective
effects of polyphenols are believed to be closely related to their status as antioxidants
(Yang 1997; Haslam 1996). Epidemiological evidence shows that consumption of
polyphenol-rich items in the diet is associated with an increase in plasma antioxidant
potential, thereby increasing resistance to oxidative stress (Haslam 1996). In fact, a
substantial body of evidence now exists that suggests that plant foods are a rich source
of molecules that quench the damaging effects of reactive oxygen species (Fenech
1997).
Antioxidants can be classified according to their actions: free radical
terminators, chelators of metal ions capable of catalyzing lipid oxidation, or as oxygen
scavengers that react with oxygen in closed systems. Phenolic antioxidants generally
fall in the category of free radical terminators (Shahidi 1992). The phenoxy radical
that occurs during the free radical termination process involving polyphenols cannot
initiate a new free radical or be subject to rapid oxidation by a chain reaction.
Therefore, phenolic antioxidants are very good hydrogen and electron donors. Also,
their radical intermediates are relatively stable due to resonance delocalization and
lack of suitable sites for attack by molecular oxygen (Shahidi 1992).
In general, “phenolic compounds” describes a wide range of plant substances
that have one or more aromatic rings with one or more hydroxyl substituents.
Phenolic compounds are often found as glycosides (with a sugar molecule attached),
and therefore tend to be water-soluble (Croft 1998 ). They have a molecular weight
range of 500-4000, possess 12-16 different phenolic groups and 5-7 different aromatic

25
rings per 1000 relative molecular mass; the ability to precipitate some alkaloids,
gelatin, and other proteins from solution; and they are categorized under 2 main
structural themes: galloyl and hexahydroxydiphenoyl esters and their derivatives, and
condensed proanthocyanidins (Haslam 1996).
Some phenolic flavonoids are more protective in vitro than ascorbate or vitamin
E (Fenech 1997). Non-flavonoid phenolic acids may also be suitable as antioxidants.
In particular, those possessing a catechol-type structure, such as caffeic acid, are
implicated (Croft 1998). Additionally, recent studies have shown that simple cell-
derived phenolic acids, such as 3-hydroxyxanthranilic acid may act as co-antioxidants
for alpha-tocopherol, causing inhibition of lipoprotein and plasma lipid peroxidation
(Croft 1998). Most plant polyphenols are known to inhibit lipid peroxidation and
lipoxygenases in vitro, and they have shown an ability to scavenge hydroxyl,
superoxide, and peroxyl radicals, all of which are important factors in cellular
prooxidant states (Haslam 1996).
Some studies have suggested a protective role for polyphenols, but some
polyphenols are better studied than others. Specifically, tea has been shown to have
an inhibitory action against experimental carcinogenesis in many animal cancer
models such as the lung, skin, liver, esophagus, and stomach (Yang 1997). Green tea
polyphenols have been shown to prevent metabolic activation of carcinogens, inhibit
tumor promotion and cell proliferation, and act as agents that induce antioxidant or
phase II enzymes (Leanderson 1997). Green tea contains certain polyphenolic
compounds, commonly known as catechins, and theaflavins (Yang 1998). The
catechins have been shown to inhibit lung, esophageal, gastric, and skin tumorigenesis

26
in animals. However, the relationship between green tea polyphenols to the same
cancers in humans remains to be elucidated (Yang 1997).
Some studies have indicated other specific polyphenols as having a beneficial
effect. In a study by Yang (1998) epigallocatechin-3-gallate is a polyphenol that may
be an important antioxidant, and possibly a cancer preventive compound. This
conclusion was reached by the fact that the polyphenol under study possessed growth
inhibitory activities against human lung tumor cell lines. This same study also
indicated that theaflavins caused growth inhibition in lung cancer cells (Yang 1998).
Another study, by Kaul and Khanduja (1998), utilized topical application of
polyphenolic compounds to inhibit skin tumor promotion. The researchers
hypothesized that plant polyphenols may reduce tumorigenesis by halting arachidonic
acid metabolism, as well as superoxide radical generation by known tumor promoters.
Red wine phenolic compounds have also been implicated in preventing lipid
oxidation (Fenech 1997), and caffeic acid has also been shown to act in a prooxidative
manner with copper- induced oxidation of LDL in the propagation phase (Croft 1998).
Overall, epidemiological evidence points to a reduced risk of certain degenerative
diseases by the consumption of beverages containing polyphenols. Specifically, green
tea and red wine are rich sources of these compounds. However, more research is
needed to elucidate the absorption, penetration, and metabolism of plant polyphenols
in the human body. Until this occurs, the evidence for their beneficial effects remains
based upon epidemiological correlation rather than strict scientific observation
(Haslam 1996).

27
Flavonoids
Among polyphenols, flavonoids are some of the most prevalent and important
low molecular weight phenolics (Bravo 1998). They are abundant in our food supply,
being found in fruits, leafy vegetables, roots, tubers, bulbs, herbs, spices, legumes, tea,
coffee, red wine, and beer (Sahu and Washington 1992). In addition to
phenylpropanoids or hydroxycinnamic acid derivatives, flavonols are found in almost
every plant. The concentration of flavonoids in these plant products is much greater in
the leaves and peels as compared to the deeper tissues (Stavric and Matula 1992). In
plants, the purpose of flavonoids is to protect the plant against fungal parasites,
herbivores, pathogens, and oxidative cell injury. Flavonoids also produce the stimuli
to assist in pollination and guide insects to their food source (Cook and Samman
1996).
Flavonoids also contribute to the flavor and color of many fruits, vegetables, and
other products, such as wine and tea (Croft 1998). More than 4000 varieties of
flavonoid compounds have been identified in vascular plants. These vary in quality
and type according to variations in plant growth, conditions, and maturity. Only a
small number of plant species have been systematically examined for their flavonoid
content. So, as yet, the indentification and quantification of every flavonoid is still
incomplete (Cook and Samman 1996). Major classes of flavonoids include flavanols,
flavones, flavanones, catechins (or flavanols), anthocyanidins, isoflavones,
dihydroflavonols, and chalcones (Cook and Samman 1996). These compounds are all
structurally related to the parent compound, 2-phenyl-benzopyrene (Yang 1997). The
most commonly occurring flavones and flavonols are those with dihydroxylation in

28
the 3’ and 4’ positions of the B ring, as well as those with a single B ring hydroxyl
group in the 4’ position (Rice-Evans 1996). The flavanols, especially catechin and the
catechin gallate ester family, and the flavonols quercetin, kaempferol, and their
glycosides, are all found in green and black tea and red wine. Quercetin is also a
major component of onions and apples, while myricetin and quercetin are both found
in berries. The flavanones are mostly found in citrus fruits (Rice-Evans 1996). The
role of many polyphenolic compounds, including flavonoids, has yet to be elucidated
(Bravo 1998). It is vital to attempt to understand the bioavailability of polyphenols,
their mechanisms of action, and possible relationships with other components of the
diet.
Originally, studies suggested that Americans consume approximately 1 gram of
polyphenols in their diets on a daily basis. Of this 1 gram, approximately 170 mg are
4-oxo-flavonoids (i.e. flavones, flavanone, flavonols, and chalcones). In addition,
most of the 4-oxo-flavonoids are probably composed of flavones (apigenin glycoside
and luteolin glycosides); and flavonols (kaempferol glycosides and quercetin
glycosides) (Brown 1980). However, more recent evidence has suggested that this
estimate is probably too high. Hertog (1993) documented that Dutch intake of
flavonols and flavones was 23 mg/day (as aglycones). Also, estimates of 115 mg/day
as aglycones are given for the United States (Hollman and Katan 1997). Chronic
pharmacologic doses of flavonoids have been tested, with deleterious effects including
acute renal failure, hemolytic anemia, thrombocytopenia, hepatitis, fever, and skin
reactions (Cook and Samman 1996). It is important to note, though, that flavonoids

29
are unlikely to be consumed in toxic amounts as part of a balanced diet (Cook and
Samman 1996).
Flavonoids possess a variety of biological effects, including antibacterial,
antiviral, anti-inflammatory, antiallergenic, and vasodilatory actions. They are also
shown to inhibit platelet aggregation, and decrease capillary permeability and fragility
(Cook and Samman 1996). Concentrated forms of flavonoids have been used for
centuries to treat various human ailments including inflammation, allergy, headache,
cancer, viral infection, colds, bee stings, and gastric and duodenal ulcers (Cook and
Samman 1996). In conventional medicine, flavonoids have been used for over 40
years to treat peripheral circulation disorders. Also, more than 100 different
preparations such as cianidol, diosmetin, hesperidin, leucocianidin, rutin, and
troxerutin are produced and sold in France in Switzerland. However, it should be
noted that many of these preparations have yet to be tested in clinical investigations
(Cook and Samman 1996). Flavonoids and other plant phenolics have also been
shown to be inhibitors of certain enzyme systems under some conditions. Among
these are phospholipase A2, cyclooxygenase, lipoxygenase, glutathione reductase, and
xanthine oxidase (Rice-Evans 1996).
Flavonoids are low molecular weight polyphenolic substances based on the
flavan nucleus. The common structure of flavonoids is C6-C3-C6 (diphenylpropane):
two aromatic rings linked through three carbons to form an oxygenated heterocycle
(Bravo 1998). The basic three phenolic rings are referred to as A, B, and C (pyran)
rings (Cook and Samman 1996). The structure of flavonoids varies widely within
major classifications. Hydrogenation, hydroxylation, methylation, malonylation,

30
sulphation, and glycosation are all substitutions that can occur (Cook and Samman
1996). Flavonoids are reputed to be efficient radical scavengers. Researchers have
developed three criteria for effective radical scavenging among the flavonoid group.
These are 1) the o-dihydroxy structure in the B ring. This gives higher stability to the
radical form and participates in electron delocalization. 2) The 2,3 double bond in
conjugation with a 4-oxo function in the C ring is responsible for electron
delocalization from the B ring. Electron delocalization of the aromatic nucleus
confers antioxidative potency. 3) The 3- and 5- OH groups with 4-oxo function in the
A and C rings are required for maximum radical scavenging potential (Rice-Evans
1996). Glycosylation in flavonoids generally occurs in the 3 position and also in the 7
position. Glucose is usually the most common sugar residue, but others include
galactose, rhamnose, and xylose (Rice-Evans 1996). Within the flavonoid group,
flavones, flavanols, and their glycosides are the most common. Quercetin - one of the
most potent and widely examined flavonoids - is part of the flavonol group (Bravo
1998).
QUERCETIN
Quercetin absorption
Studies of the absorption of flavonoids in humans are not conclusive at this time.
There is evidence that flavonoids are absorbed in significant quantities and that they
can be absorbed as both free aglycone and glycoside forms. Both forms have been
found in blood and urine. Some research indicates that peak absorption may be 2-3
hours after ingestion (Manach 1998).

31
Naturally occurring flavonoids are usually glycosylated. This is important in the
absorptive process. Unabsorbed flavonoids from the small intestine, as well as
absorbed, conjugated flavonoids secreted from the gallbladder reach the colon. Here,
both groups of flavonoids undergo microbial degradation, stripping them of their sugar
moieties, glucuronic acids and sulfates. Their absorption (flavonoids) is determined
by the hydroxylation pattern. 5, 7, and 3’, 4’ hydroxylated compounds are susceptible
to hydrolysis and heterocyclic ring cleavage by microbiological degradation in the
colon (Croft 1998). Absorption is then possible due to the fact that the resulting
aglycones are less polar. As a result of this structure-dependent hydrolysis, biological
effects of flavonoids predicted from in vitro studies may be different in vivo,
depending on the structure of the parent compounds (Hollman and Katan 1997).
Manach et al. showed that quercetin and rutin are both absorbed by rats in a 1995
study. Rats were fed diets containing various levels of quercetin and rutin, the main
glycoside form of quercetin. Measurement of cecal contents indicated similar
quantities of quercetin and rutin in rats fed the same molar amounts of each flavonoid.
Further, quercetin and rutin diets led to similar levels of plasma metabolites. It was
therefore concluded that the small intestine was not an effective site for absorption
(Manach 1995). Quercetin showed a high affinity for albumin (Manach 1995). They
concluded that the strong binding of quercetin metabolites to albumin may affect
quercetin’s actions within a physiological context, and that any effects in vivo are due
to the quercetin-albumin complex, rather than unbound quercetin (Manach 1995).
In a subsequent rodent study, Manach et al. (1997) found that quercetin was
indeed absorbed in the small intestine, while rutin was not. They also found that

32
absorption of quercetin was less efficient in rats previously maintained on a quercetin-
containing diet. Therefore, a steady intake of quercetin could lower the rate of
digestive absorption, as is the case with other micronutrients (Manach 1997).
Moving to the human model, Manach et al. (1998), studied plasma
concentrations of healthy volunteers following a meal composed mainly of plant
products high in quercetin. They observed a significant increase in conjugated
quercetin derivatives in the blood at 3 and 7 hour intervals following the meal, which
returned to basal levels or below after about 20 hours of no further quercetin
consumption. The relatively rapid increase in absorption, evidenced by the plasma
concentrations, suggests that absorption probably occurred in the proximal intestine.
Within the same study, the conjugated derivatives found in plasma were measured for
antioxidant capacity. Although their activity was half that of the aglycone form, Cu2+-
induced oxidation of human LDL was still significantly delayed. Overall, the
investigators concluded that beneficial effects of quercetin and other flavonoids would
depend on regular consumption, and that any physiological effects in vivo may be due
to a synergism with other compounds in fruits and vegetables (Manach 1998).
In another human study, Hollman (1995) studied the absorption of flavonoids
from foods (mostly flavonoid glycosides) in 30 ileostomy patients. Logically, without
colonic microbial degradation, the flavonoids could reach the end of the small
intestine as glycosides. Results showed that quercetin glycosides from onions were
more readily absorbed (52% of ingested amounts) than the aglycone forms (24% of
ingested amount). Based on these results, Hollman concluded that glycosides may be
absorbed in humans without prior hydrolization by colonic microbes (Hollman 1995).

33
Manach et al. (1998) agreed with Hollman that the glycosylated forms may have been
more easily absorbed. Recent studies have confirmed that quercetin glycosides are
hydrolyzed and absorbed in the small intestine (Erland et al. 2000; Wallen et al. 2000).
Flavonoids are metabolized in two main body compartments: the liver and the colon.
The liver, where absorbed flavonoids and their absorbed colonic metabolites are
received, is considered to be the main site of quercetin metabolism (Hollman and
Katan 1997). Biotransformation enzymes produce methoxy-, gluco-, and sulfo-
conjugations of flavonoids. Glucuronides and sulfates of quercetin and quercetin
aglycone are found in plasma of human subjects fed either quercetin or quercetin
glycosides (Manach 1997). Following transformation, a large amount of flavonoid
metabolites are excreted in the urine (Hollman and Katan 1997).
Quercetin - an antioxidant and a prooxidant
Oxidative stress occurs when antioxidant defenses are unable to overcome the
production of free radicals and may contribute to many different conditions including
atherosclerosis, cancer, and chronic inflammation. Therefore, the antioxidant capacity
of flavonoids is of great interest (Croft 1998). Flavonoids act as antioxidants by
breaking the free radical chain reaction. An antioxidant, by definition, is a substance,
when present at low concentrations relative to an oxidizable substrate, can
significantly delay or prevent oxidation of the substrate. In addition, the resulting
radical must be stable enough to prevent it from acting as a chain-propagating radical
(Croft 1998). Flavonoids quench the hydroxyl and superoxide ions, which are highly
reactive species involved in lipid peroxidation (Morel 1993). It is also proposed that
flavonoids terminate the propagation phase of lipid peroxidation by donating hydrogen

34
atoms to the peroxy radical, thereby forming a flavonoid radical. The flavonoid
radical then reacts with free radicals and terminates the propagation chain (Cook and
Samman 1996).
Not only are flavonoids soluble, chain-breaking inhibitors of the peroxidation
process, they are also thought to be potent metal chelators (Morel 1993). Through this
action, they can inhibit the Fenton reaction, which is driven by the superoxide anion
(Cook and Samman 1996). In fact, Morel et al. (1993) demonstrated iron removal
from iron-loaded hepatocyte cultures by catechin, quercetin, and diosmetin.
Under certain conditions – such as high phenolic concentrations, high pH, or
presence of metal ions – phenolic antioxidants can autooxidize and behave as
prooxidants (Bravo 1998). Flavonoids have been shown to produce reactive oxygen
species in vitro in the presence of reduced metals such as iron and copper (Sahu 1994).
Some flavonoids are able to mobilize iron from ferritin, and are capable of reducing
ferric iron to the more reactive ferrous iron (Morel 1993). A recent study by Cao et al.
(1997) used three different oxidation systems. In these, flavonoids had strong
antioxidant activity against peroxyl radicals generated from AAPH and also against
hydroxyl radicals. However, they were pro-oxidative with respect to copper. It was
concluded that the flavonoids must be able to reduce Cu2+ to Cu+, thereby allowing the
formation of initiating radicals (Croft 1998). This prooxidative effect has been shown
in numerous studies with one flavonoid in particular. Quercetin has been found to be
mutagenic in almost every in vitro mutagenicity test (Canada 1990). Because
quercetin is mutagenic only under aerobic conditions, active oxygen species are
strongly implicated (Sahu and Washington 1992). In a study by Miura et al. (1998),

35
quercetin was the most potent in generating hydrogen peroxide at pH 7.4.
Polyphenols that are similar to quercetin have been shown to generate active oxygen
species by autooxidation. Examples are pyrogallol, which generates the superoxide
dical by its autooxidation. Hydroquinone and catechol – other examples of
polyphenols – produce hydrogen peroxide and the superoxide radical at physiological
pH. In the presence of iron, these two reactive oxygen species generate the hydroxyl
radical (Sahu and Washington 1991).
Many flavonoids may cause lipid peroxidation or may accelerate oxidative
damage to non-lipids, such as carbohydrates and DNA. The products of this oxidative
damage can be cytotoxic (Smith 1992). For example, quercetin has also been found to
induce single strand DNA breaks in isolated rat liver nuclei, in calf thymus DNA, and
in open plasmid DNA (Duthie 1997). In a lipid system, quercetin exhibited
prooxidative effects at higher doses (Stadler 1995). Finally, from the evidence shown
in Table 1, it is obvious that flavonoids cannot be simply classified as antioxidants.
They must be examined more thoroughly for their pro-oxidant properties (Laughton
1989). The prooxidative properties of flavonoids may be beneficial. Free radical
production may stimulate apoptosis of tumor cell lines under certain circumstances. It
is now well accepted that the majority of chemo- and radiotherapeutic agents destroy
malignant cells by initiating apoptosis (Martin 1997). Evidence is also accumulating
that the efficiency of anti-tumor agents is related to the ability of the target tumor cells
to respond by apoptosis. Recent studies by Kuo (1996) have shown that apoptosis was
the mechanism of cell death when flavonoids were added to the neoplastic cell lines

36
CaCo2 and HT29. Kuo also found that curcumin - a popular spice - exhibits anti-
tumor activity by initiating apoptosis in human tumor cells (Kuo 1996).
Cermak et al. (1993) suggests that acute exposure of intestinal tumor cells to
iron may increase their susceptibility to oxidant-mediated lysis. This study postulates
that free iron may be released from ferritin by an appropriate reducing agent (e.g.,
quercetin) (Cermak 1993). It is therefore reasonable to think that if tumor cells were
exposed to high iron levels and an appropriate reducing agent, such as quercetin, that
oxidative stress might induce apoptosis in these cells.
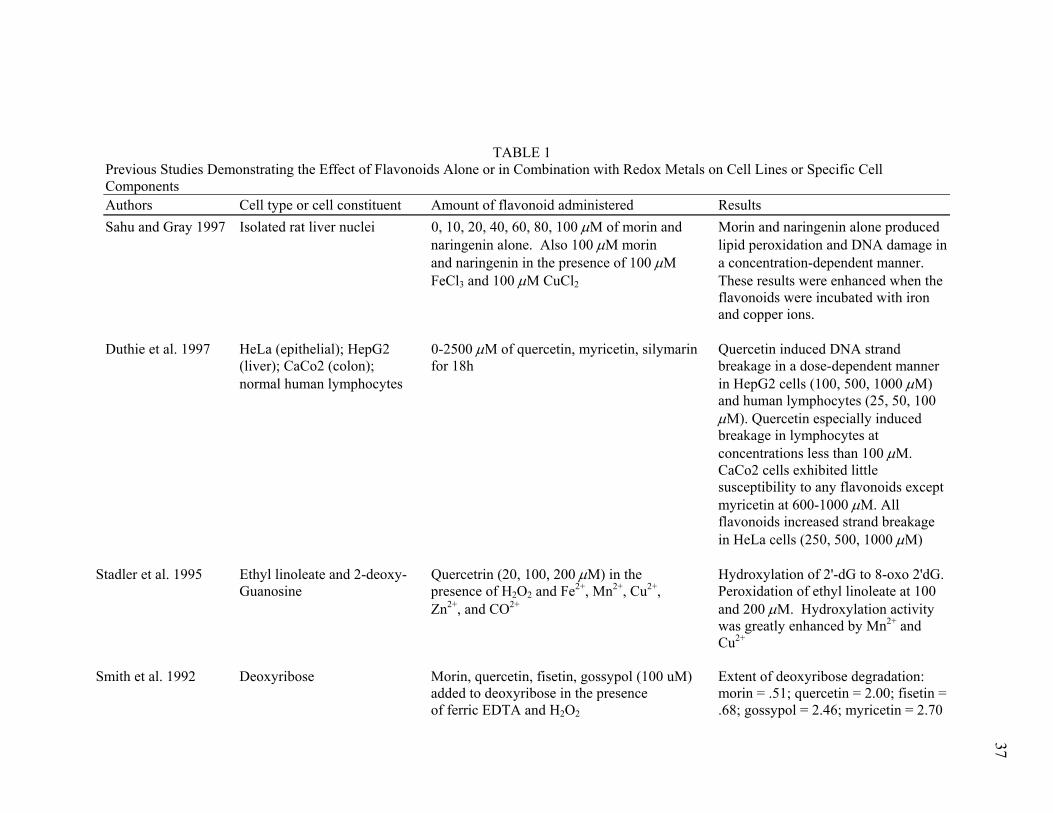
TABLE 1 Previous Studies Demonstrating the Effect of Flavonoids Alone or in Combination with Redox Metals on Cell Lines or Specific Cell Components Authors Cell type or cell constituent Amount of flavonoid administered Results Sahu and Gray 1997 Isolated rat liver nuclei 0, 10, 20, 40, 60, 80, 100 µM of morin and Morin and naringenin alone produced naringenin alone. Also 100 µM morin lipid peroxidation and DNA damage in and naringenin in the presence of 100 µM a concentration-dependent manner. FeCl3 and 100 µM CuCl2 These results were enhanced when the flavonoids were incubated with iron and copper ions. Duthie et al. 1997 HeLa (epithelial); HepG2 0-2500 µM of quercetin, myricetin, silymarin Quercetin induced DNA strand (liver); CaCo2 (colon); for 18h breakage in a dose-dependent manner normal human lymphocytes in HepG2 cells (100, 500, 1000 µM)
and human lymphocytes (25, 50, 100 µM). Quercetin especially induced breakage in lymphocytes at concentrations less than 100 µM. CaCo2 cells exhibited little susceptibility to any flavonoids except myricetin at 600-1000 µM. All flavonoids increased strand breakage in HeLa cells (250, 500, 1000 µM) Stadler et al. 1995 Ethyl linoleate and 2-deoxy- Quercetrin (20, 100, 200 µM) in the Hydroxylation of 2'-dG to 8-oxo 2'dG. Guanosine presence of H2O2 and Fe2+, Mn2+, Cu2+, Peroxidation of ethyl linoleate at 100 Zn2+, and CO2+ and 200 µM. Hydroxylation activity was greatly enhanced by Mn2+ and Cu2+ Smith et al. 1992 Deoxyribose Morin, quercetin, fisetin, gossypol (100 uM) Extent of deoxyribose degradation: added to deoxyribose in the presence morin = .51; quercetin = 2.00; fisetin = of ferric EDTA and H2O2 .68; gossypol = 2.46; myricetin = 2.70
37

CHAPTER III
IRON STATUS OF THE CELL MAY ALTER QUERCETIN-INDUCED APOPTOSIS
OF HUMAN HEPATOMA CELLS
ABSTRACT
Quercetin, a flavonoid, is cytotoxic to human tumor cells at high concentrations.
The objective of the present study was to determine if cellular iron status influences
quercetin-stimulated cytotoxicity and apoptosis in a human hepatoblastoma cell line.
HepG2 cells were incubated in cell culture media supplemented with 0, 10 or 25 µM Fe
(added as ferric citrate) for 24 hours to achieve varying Fe concentrations. Fe-
supplemented media was then removed and cells were washed. Cells were subsequently
incubated with media containing 0, 40 or 80 µM quercetin in 0.5% DMSO for an
additional 24 hours. Following incubation with quercetin, lipid peroxidation was
measured with thiobarbituric reactive substances assay, cell viability was assessed with
the Trypan Blue exclusion assay, and apoptosis was assessed by enzyme immunoassay,
chromatin condensation, and caspase activity. At least three replicates were obtained for
each assay. Supplementation with 10 and 25 µM Fe resulted in cellular Fe levels that
were 3- and 6- times those of control, as determined with atomic absorption
spectrophotometry (AAS). Subsequent incubation with quercetin did not alter the Fe
content of cells, as found with AAS. Lipid peroxidation was increased with increasing
media iron concentrations, with quercetin showing no contribution to this effect. Cell
38

39
viability was significantly reduced only after incubation with 25 µM Fe followed by 80
µM quercetin (69% vs 78%, respectively, for treated vs control). Cellular apoptosis was
not consistently enhanced by incubation with 40 µM quercetin. Increases in cellular
apoptosis after incubation with 80 µM quercetin were greatest in iron loaded cells (10
µM and 25 µM Fe levels). Apoptotic morphology, assessed as chromatin condensation,
was only affected by quercetin. No synergism between iron and quercetin was suggested.
Caspase activity was decreased by quercetin at control iron levels, but increased by
quercetin in cells treated with the highest levels of iron. Overall, the data suggest that Fe
concentration may influence quercetin's effect on cell viability and apoptosis in HepG2
cells.
INTRODUCTION
Iron is a redox active metal, meaning that it may readily alternate between ferrous
and ferric states, accepting or donating an electron to a wide range of biological
substances. Through this mechanism, iron catalyzes a variety of damaging reactions
within the cell (McCord 1998). Iron can catalyze the formation of the hydroxyl radical
from H2O2 as well as the decomposition of lipid hydroperoxides to alkoxyl, peroxyl, and
other free radicals (Ibrahim 1997). Therefore, iron is a key factor in the establishment of
a prooxidant status in the cell (Meneghini 1997). Previous studies in our laboratory and
others have suggested that iron status of a cell may alter apoptosis (Stimson 1998; Wang
et al. 1999; Whittaker et al. 1996). Further, studies by Cermak et al. (1993), proposed
that acute exposure of some tumor cells to iron may increase their susceptibility to
oxidant-mediated lysis.

40
Apoptosis is described as a sort of cellular suicide (Samali 1996) in which a cell
takes an active role in its own demise. It consists of a preprogrammed cascade of
metabolic events that occurs in response to various environmental inducers or a
combination of intrinsic factors, ultimately leading to the disintegration of the cell
(Darzynkiewiez 1995). Studies have shown that cell death caused by apoptosis may be
the mechanism by which some chemotherapeutic agents function.
It has also been proposed that oxidative stress is a mediator of apoptosis in
eukaryotic cells via several different mechanisms (Buttke and Sandstrom 1994).
Oxidative stress may induce apoptosis by damaging DNA. This may occur via metal-
catalyzed oxidation with subsequent generation of reactive intermediates such as
hydrogen peroxide and the hydroxyl radical (Duthie et al. 1997). In addition, oxidative
stress may lead to the formation of oxidized lipids in cell membranes, which may induce
apoptosis (Buttke and Sandstrom, 1994).
Recent studies (Kuo 1996) have shown that apoptosis was the mechanism of cell
death when flavonoids were added to neoplastic cell lines. It has been proposed that
some phenolic antioxidants, such as flavonoids, can reduce ferric iron to ferrous iron,
thereby facilitating free radical damage to carbohydrates, lipids, proteins, and DNA
(Smith 1992). Free iron may also be released from ferritin by an appropriate reducing
agent such as quercetin (Cermak 1993). Sahu and Washington (1992) found that
increasing quercetin levels in the presence of iron significantly increased lipid
peroxidation and nuclear DNA damage in rat liver nuclei. Therefore, tumor cells
exposed to high iron levels in the presence of a reducing agent, such as quercetin, could
theoretically undergo apoptosis.

41
The purpose of this study was to determine whether a flavonoid common to many
foods (quercetin) could induce apoptosis in the HepG2 cell line, a liver hepatoma cell
line, and whether cellular iron status alters apoptosis.
It was hypothesized that 1) cellular iron status would increase with higher media
iron concentrations, and that addition of quercetin to iron-loaded cells would have no
effect on cellular iron, 2) addition of quercetin to iron-loaded cells would increase lipid
peroxidation, 3) cellular iron concentration would influence the prooxidative ability of
quercetin to decrease cell viability, and 4) cellular iron concentration would influence the
pro-oxidative ability of quercetin to induce specific apoptotic changes in the cell
measured as: 200-bp DNA nucleosome fragmentation (ladder effect), caspase activation,
and chromatin condensation.
METHODS
Cell culture, media, and experimental design
Cells used in this study were human hepatoma cells (HepG2; ATCC, Rockville,
MD). Cells were grown in filtered Eagle’s minimum essential medium (MEM) with
2mM L-glutamine and Earle’s balanced saline solution (BSS; Sigma, St. Louis, MO)
with 1.5g/L sodium bicarbonate (Sigma, St. Louis, MO), 1.0 mM/L sodium pyruvate
(Sigma, St. Louis, MO), 0.1 mM/L nonessential amino acids (Sigma, St. Louis, MO), 10
mg streptomycin + 10,000 units penicillin/ml (Sigma, St. Louis, MO), with 10% fetal
bovine serum (FBS; Equitech-Bio, Inc., Kerrville, TX). All assays were completed using
FBS from the same lot. HepG2 cells were maintained in 75 cm2 canted-neck flasks in 15
ml of media with 1:4 passage occurring once per week. Cells were incubated at 37oC,

42
5% CO2 (Harris Model# HWO701T-ABA, Norwalk, CT). Studies were conducted on
passages 78-163.
When cells were approximately 100% confluent and four days after passage, cells
were incubated in 0, 10, 25, and 50 µM /L iron-loaded media. Following a 24-hour
incubation, cells were washed with Hanks BSS (.03% EDTA) to remove iron-loaded
media. Media with 0, 40, 80 µM quercetin in .2% DMSO was added to cells. Cells were
incubated for another 24 hours, and harvested for assays for cell viability and apoptosis.
Each batch of iron and quercetin loaded media was tested for iron content using a
Perkin Elmer Model# 5000 atomic absorption spectrophotometer (Perkin Elmer Corp,
Shelton, CT; percent recovery = 90%; n=16). Measured Fe content of media was 3.5,
5.6, 10.4 µM for 0, 10, 25 µM /L media, respectively; n = 6 replicates. Iron content of
media was consistent across media batches. It was determined that the 50 µM /L iron-
loaded media did not significantly increase cellular iron levels over the levels achieved
with 25 µM /L iron-loaded media. Therefore, the 25 µM /L iron-loaded media was
chosen as the highest level for experiments.
Cellular iron concentration
Harvested cells in solution were centrifuged (Beckman J2HS, JS-7.5 swinging
bucket rotor, Palo Alto, CA) at 750 X G for 5 min. The supernatant was discarded, and
the pellet was resuspended in 3 ml PBS. Centrifugation/resuspension were repeated
twice, with resuspension in 1 ml PBS after the final centrifugation. The 1 ml suspension
was centrifuged once more to pellet the cells. The supernatant was discarded. HCl (.17
ml) was added to each pellet, and vortexed. 1.5 ml dH2O was added to each tube. Iron

43
concentration was determined with an atomic absorption spectrophotometer and
expressed as umol per million cells; n = 3 replicates.
Cell viability
The Trypan Blue assay was used to measure cell viability. Five ml cell
suspensions of each treatment group were pipetted from each flask into a test tube. All
tubes were centrifuged at 750 X G for 5 min. The supernatant was discarded. Next, 3
mL Hanks BSS was added to each cell pellet. In a separate tube, .5 ml of .4% Trypan
Blue solution (Sigma, St. Louis, MO) and .3 mL of Hanks BSS were combined.
Immediately before counting, cell suspensions were aspirated to obtain an even
distribution of cells. .2 ml of the cell suspension was then added to the Trypan
Blue/Hanks BSS solution and was incubated for 5 minutes before counting to avoid
overexposure of the stain. Each tube was mixed thoroughly by gentle thumping until
most clumps disappeared. A 10 µL aliquot of the Trypan Blue/cell suspension was added
to a hemocytometer, and cells were counted under an Olympus IMT-2 Inverted Research
Microscope (Olympus America Inc., Melville, NY). Cell viability was calculated as total
viable cells (unstained)/total cells (stained and unstained) x 100; n = 4 replicates.
Lipid peroxidation
The thiobarbituric acid reactive substances (TBARS) assay was used to assess
lipid peroxidation (n = 3 replicates). TBARS was measured as described by Buege and
Aust (1978). Briefly, washed cell suspensions were treated with TBA-TCA-HCl
(thiobarbituric acid-trichloracetic acid-hydrochloride acid), placed in boiling water for 15
minutes and then centrifuged at for 15 minutes (4000 x g, Beckman J2HS, JS-7.5
swinging bucket rotor, Palo Alto, CA). The supernatant was collected and absorbance at

44
535 nm was read using a Beckman DU-650 spectrophotometer (Beckman Instruments,
Fullerton, CA). Protein concentration was determined as described by Lowry et al.
(1951).
DNA fragmentation
An Enzyme-Linked Immunosorbent Assay (ELISA) was used to assess apoptosis
(Cell Death Detection ELISA, Boehringer Mannheim, GmbH, Germany) using a
microplate reader (Dynex Technologies, Chantilly, VA). Cell suspensions from 3 parent
flasks (0,10, 25 µM Fe levels) were counted immediately following passage procedure
each week prior to the assay. Resulting cell numbers were used to determine dilution
factor for the subsequent ELISA. Cell suspensions containing 105 cells/ml were used for
the assay.
Cell suspensions were centrifuged, and the cell pellet was resuspended in 1 ml
culture medium. After a second centrifugation, each cell pellet was resuspended with
500 µL incubation buffer containing Tween 20 or 80 and incubated approximately 30
minutes at 4oC to induce lysis. The lysate was centrifuged at 15,000 rpm (20,000 x G)
for 10 min. 50 µL of the supernatant (cytoplasmic fraction) was carefully removed and
transferred to duplicate eppendorf tubes. The supernatant was stored at - 20oC until the
following day.
100 µL coating anti-histone (monoclonal mouse antibody) solution was pipetted
into each well of a microtiter plate (MTP). MTP were covered tightly with an adhesive
cover foil and incubated overnight at 4oC. Maximum incubation time was 18 hours.
The coating solution was then removed, and 200 µL incubation buffer containing
Tween 20 or 80 was then pipetted into each well of the MTP. The MTP were covered

45
tightly with the adhesive cover foil and incubated for 30 min at room temperature. The
coating solution was then removed, and the MTP was washed three times.
Samples were diluted to obtain 1 x 104 cell equivalents/ml. Each sample was
vortexed after dilution. 100 µL of each sample solution was pipetted into the MTP. For
determination of the background of the immunosassay, 100 µL of incubation buffer were
pipetted into 2 additional wells. MTP were covered tightly with adhesive foil and
incubated for 90 minutes at room temperature. The washing procedure was repeated for
all samples and blanks.
The conjugate solution - anti-DNA peroxidase (monoclonal mouse antibody)
(10:1 dilution) was added to each well of the MTP, except the blank positions. The MTP
were covered tightly with adhesive foil and incubated for 90 minutes at room
temperature. The washing procedure was repeated. A substrate solution containing 2,2’-
azino-di[3-ethylbenzthiazoline sulfonate] was pipetted into each well of the MTP and
incubated on a plate shaker at 250 rpm for 15 minutes until the color development was
sufficient for a photometric analysis on a microplate reader at 405 nm against the
substrate reaction solution as the blank; n = 4 replicates.
Nuclear morphology
Apoptosis assessed with Hoechst staining (Intergen Company, Purchase, NY)
was used to examine nuclear morphology and chromatin condensation in the 0 and 80
µM /L quercetin groups with 0 and 25 µM /L added iron. 100 µL cell suspensions were
diluted with 200 µL media for treatment with 1.5 µL Hoechst 33342 stain. Hoechst-
stained cells were counted using a 480 nm UV filter. Apoptotic morphology was
assessed using 100x objective. This allowed more detailed view of chromatin assembly,

46
while allowing a minimal path of light to hit cells, thereby avoiding fading before
morphology could be determined. Cells showing apoptotic morphology were expressed
as a percentage of total cells; n = 3 replicates.
Caspase activity
An Intergen Caspatag Fluorescein Caspase Activity Kit (Intergen Company,
Purchase, NY) kit was used to determine caspase activity in the 0 and 80 µM/L quercetin
groups with 0 and 25 µM/L added iron. 300 µL cell suspensions were incubated with a
carboxyfluourescein (FAM) derivative of benzyloxycarbonyl valylalanyl aspartic acid
flouromethyl ketone (zVAD-FMK) (FAM-VAD-FMK fluorescein labeled inhibitor).
Caspase activity was determined with fluorescence microscopy using a Nikon Contrast-2
(Nikon Instruments Inc., Melville, NY) with a 520 nm filter. Caspase-positive cells were
counted, followed by dead (propidium iodide-labeled) cells in a hemocytometer. A ratio
of caspase positive cells to total cells is presented; n = 5 replicates.
Statistical analysis
Treatment means, standard error of the mean, analysis of variance (ANOVA),
least significant difference tests, tests for normality, and non parametric analysis were
determined using the statistical package SAS (SAS version 6.10, SAS Institute, Cary,
NC). The overall effects and interactions of iron, and quercetin on cellular iron
concentration, lipid peroxidation, and quercetin-induced DNA fragmentation were
determined with ANOVA. Fisher’s Least Significance Difference test was used to assess
the difference between means for treatments. Differences among treatment groups were
considered significant if p < 0.05. The apoptosis data did not appear to be normally

47
distributed, and were analyzed using non-parametric methods. However, ANOVA and
Kruskall-Wallace tests produced similar results.
TABLE 2
Study Design - Assays and Treatment Groups Iron incubation: 24 hr (followed by wash) Quercetin incubation: 24 hr Assays: Cellular Iron Status (n=3 replicates) Cell Viability - Trypan Blue Exclusion (n=3-4 replicates) Lipid Peroxidation - TBARS (n=3 replicates) Apoptosis:
- DNA Fragmentation ELISA (n=4 replicates) - Caspase Activity (n=5 replicates) - Nuclear Morphology (Hoechst 33342) (n=3 replicates)
0 µM Fe
0µM 40 µM 80 µM
Quercetin Quercetin Quercetin
10 µM
Fe
0 µM 40 µM 80 µM Quercetin Quercetin Quercetin
25 µM Fe
0 µM 40 µM 80 µM Quercetin Quercetin Quercetin
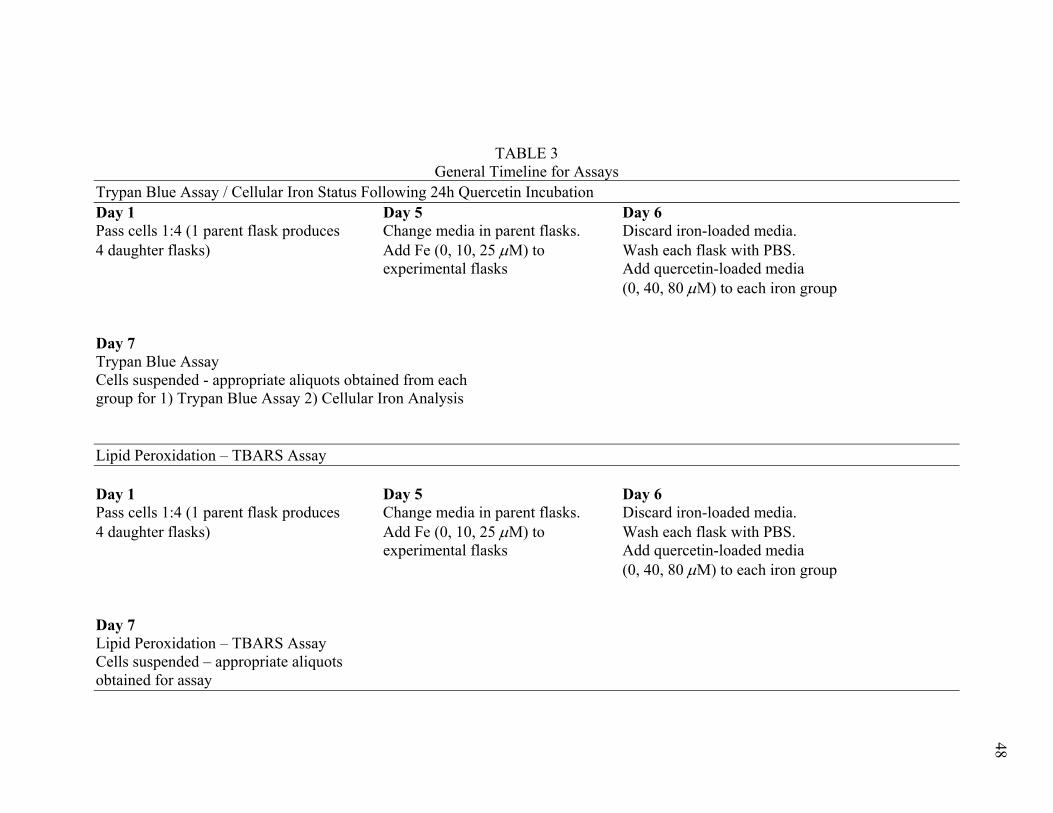
TABLE 3 General Timeline for Assays
Trypan Blue Assay / Cellular Iron Status Following 24h Quercetin Incubation Day 1 Day 5 Day 6 Pass cells 1:4 (1 parent flask produces Change media in parent flasks. Discard iron-loaded media. 4 daughter flasks) Add Fe (0, 10, 25 µM) to Wash each flask with PBS. experimental flasks Add quercetin-loaded media (0, 40, 80 µM) to each iron group Day 7 Trypan Blue Assay Cells suspended - appropriate aliquots obtained from each group for 1) Trypan Blue Assay 2) Cellular Iron Analysis Lipid Peroxidation – TBARS Assay Day 1 Day 5 Day 6 Pass cells 1:4 (1 parent flask produces Change media in parent flasks. Discard iron-loaded media. 4 daughter flasks) Add Fe (0, 10, 25 µM) to Wash each flask with PBS. experimental flasks Add quercetin-loaded media (0, 40, 80 µM) to each iron group Day 7 Lipid Peroxidation – TBARS Assay Cells suspended – appropriate aliquots obtained for assay
48

DNA Fragmentation ELISA Day 1 Day 5 Day 6 Pass cells 1:4 (1 parent flask produces Change media in parent flasks. Discard iron-loaded media. 4 daughter flasks). Obtain cell count Add Fe (0, 10, 25 µM) to Wash each flask with PBS. from parent flasks to calculate dilutions experimental flasks Add quercetin-loaded media for assay (0, 40, 80 µM) to each iron group Day 7 Day 1 DNA Fragmentation Working Procedure of ELISA. Samples (180-200 bp nucleosomes) Pass cells 1:4. Obtain cell count obtained and stored at –20oC for from parent flasks to calculate working procedure of assay (12-18h incubation) dilutions for next week’s ELISA Caspase Activity / Chromatin Condensation Assays Day 1 Day 5 Day 6 Pass cells 1:4 (1 parent flask produces Change media in parent flasks. Discard iron-loaded media. 4 daughter flasks) Add Fe (0, 10, 25 µM) to Wash each flask with PBS. experimental flasks Add quercetin-loaded media (0, 40, 80 µM) to each iron group Day 7 Caspase activity/Hoechst stain Appropriate aliquots obtained from each group. For caspase activity, aliquots were not diluted. For Hoechst stain, separate aliquots were obtained from same experimental flasks and diluted.
49

50
RESULTS Cellular iron concentration
Cellular iron status increased as the media iron concentration increased from 0 to
25 µM (P = 0.0001). Iron was increased 3-fold between the 0 and 10 µM group, and 6-
fold between the 0 and 25 µM group (for the 0, 10, and 25 µM groups, mean cellular Fe
post-treatment was .0047 µM/106 cells, .0129 µM/106 cells, and .028 µM/106 cells,
respectively). Quercetin treatment had no effect on cellular iron (Fig. 1).
Lipid peroxidation (TBARS)
Iron concentration had an overall positive effect on lipid peroxidation (P<0.03).
Among the treatment groups, as iron increased, lipid peroxidation - indicated by the
formation of thiobarbituric acid reactive substances – increased. However, quercetin had
no significant effect on lipid peroxidation, either alone, or in combination with iron (Fig.
2). When no iron was added to the media, TBARS concentration was 0.1012 nmol
MDA/mg protein. Adding 10 µM and 25 µM iron increased TBARS 34% and 96%,
respectively. Of note, the 25 µM Fe / 40 µM quercetin group resulted in the highest
TBARS concentration.
Cell viability
As determined with the Trypan Blue assay, quercetin and iron alone had no
significant effect on cell viability. However, there was a weak trend toward a quercetin
effect (P=.0984) and a strong trend towards an iron x quercetin interaction (P=.0563)
(Fig. 3). Within the 0 µM Fe treatment group, cell viability was slightly increased with
each increase in media quercetin concentration. In contrast, within the 10 and 25 µM Fe
treatment groups, mean cell viability decreased with increasing media quercetin

51
concentrations. The cells treated with 25 µM Fe and 80 µM quercetin was the only group
with a significantly lower mean cell viability than controls.
DNA fragmentation
The average percent increase in DNA fragmentation that occurred with the
addition of 40 and 80 µM quercetin is shown in Figure 4, and the median changes in
DNA fragmentation are shown in Table 4. Iron concentration did not have a significant
effect on quercetin-induced DNA fragmentation. Quercetin level had a significant effect
on apoptotic DNA fragmentation (p<0.02). However, it was apparent that the addition of
40 µM quercetin did not consistently increase DNA fragmentation above control levels,
regardless of iron concentration. Also, apoptotic DNA fragmentation in cells treated with
80 µM quercetin was consistently increased only in iron-loaded cells.
The DNA fragmentation assay suggested that 80 µM quercetin was required to
have a consistent effect on apoptosis. Therefore, only 80 µM quercetin was used in the
final two apoptotic assays. Also, only two iron concentrations (0 and 25 µM were chosen
to determine if there was an iron effect.
Chromatin condensation
Apoptosis was also assessed with light microscopy examination of chromatin
condensation (Table 6). Both ANOVA and Chi-Square analyses showed a weak trend
toward an effect of quercetin on apoptosis. No synergism between iron and quercetin
was indicated to increase apoptosis, which does not support the apoptotic pattern as
measured with DNA fragmentation.

52
Caspase activity
Caspase activity was only measured with two concentrations of iron (0 and 25
µM) and quercetin (0 and 80 µM). Data are presented in Table 5. Results in the 0 µM
quercetin treatment groups showed no significant difference in percent of cells with
caspase activity between 0 µM Fe addition and 25 µM Fe addition. The data suggests a
weak interaction between iron and quercetin. 80 µM quercetin decreased caspase activity
in cells with normal iron concentrations, but increased caspase activity in cells with high
iron concentrations (ANOVA: Fe x Q: P=.0362).
DISCUSSION
The present study was conducted to determine if cellular iron status influences
quercetin-stimulated cytotoxicity and apoptosis in a human hepatoblastoma cell line. The
main findings were that 1) high cellular iron elevated lipid peroxidation, but quercetin
had no effect on peroxidation alone or in combination with iron, 2) cell viability was
decreased in groups treated with both high levels of iron and high levels of quercetin, and
3) high levels of iron and quercetin together triggered apoptosis, evidenced by increased
apoptotic DNA fragmentation and caspase activity.
Atomic absorption spectrophotometry revealed that increasing media iron content
increased cellular iron in a dose dependent manner. Further, cellular iron status was not
effected by addition of quercetin (Fig. 1). Cellular iron status in cells treated with 10 and
25 µM iron was three and six times higher, respectively, than the controls. This is very
similar to the increases in liver iron achieved by increasing dietary iron to 650 and 1500
mg Fe/kg diet in previous rat studies in our laboratory (Yin 1999). In acute iron
overload, free iron may be more prevalent than in chronic iron overload because ferritin

53
and transferrin synthesis may not be sufficient to accommodate the rapid excess of iron
(Cermak 1993). However, free iron was not assessed in this study, so it was unclear if
iron was free or bound.
The effect of quercetin on cellular iron concentrations was also examined. It is
well established that polyphenols as a group, and hence flavonoids, exert antioxidant
activity, in part, through their ability to chelate metal ions known to induce Fenton-type
reactions, such as iron and copper (Bravo 1998; Croft 1998). Obviously, this property
could prevent some free quercetin from being absorbed by the HepG2 cells if iron-loaded
media were used concurrently with quercetin loading. Therefore, before addition of
quercetin-treated media to cells, iron-loaded media was removed from the culture flasks,
and cells were washed. Some research has suggested that extracellular quercetin may
have the ability to lower cellular iron concentrations (Morel 1993). In our laboratory,
Stimson (1998) found that diets with 1% quercetin supplementation significantly
decreased liver iron concentrations in rats fed medium (424 mg Fe/kg diet) and high iron
(664 mg/kg diet) diets. However, it was not clear if this action took place in the lumen of
the intestine at the cellular level. While cellular iron was not altered by quercetin in the
present study, it is possible that a 24 hour incubation with quercetin was not quite
sufficient to liberate enough iron from the cell. Perhaps a longer incubation period with
the quercetin would be necessary to induce a change in cellular iron status.
Many researchers (Cermak 1993; Morel 1993; Jacob 1997; McCord 1998;
Halliwell and Gutteridge 1999), as well as previous studies (Stimson 1998) in our
laboratory have indicated that iron status of a cell may contribute to oxidative stress.
Similarly, in the present study, increased cellular iron significantly increased lipid

54
peroxidation, seen as higher TBARS concentrations. This is in accordance with animal
studies in our lab, where increased dietary iron increased lipid peroxidation in rat liver
(Yin 1999). The results of the present study also coincide with the observations of
Halliwell and Gutteridge’s review (1999), where they discuss that free iron or low
molecular weight iron chelates have the ability to form oxygen radicals that can initiate
and propagate lipid peroxidation. Two possible modes of action theorized are 1)
formation of hydroxyl radicals through a Fenton reaction that stimulates lipid
peroxidation, and 2) ability to decompose lipid peroxides to form alkoxyl and peroxyl
radicals - propagating further lipid peroxidation.
I further hypothesized that addition of high concentrations of quercetin to iron-
loaded cells would increase oxidative stress, thereby increasing lipid peroxidation, which
is one trigger of apoptosis. This result was not observed in my study, as quercetin
increased apoptosis, but had no significant effect on lipid peroxidation. However, it is
interesting to note that the highest lipid peroxidation values - although non-significant -
were found to be associated with quercetin. In a related study, Laughton et al. (1989)
found that addition of the flavonoids quercetin and myricetin to Fe3+ (100 µM /L as ferric
chloride) or Fe3+ EDTA (100 µM) consistently accelerated bleomycin-dependent DNA
degradation at concentrations from 0-75 µM, but had no effect on microsomal lipid
peroxidation at concentrations of 0-50 µM. This may suggest that the prooxidative
actions of flavonoids in conjunction with iron may be site and model specific.
The Trypan Blue assay is a simple assay of cell viability. It does not discriminate
between cell death caused by apoptosis or necrosis. It does, however, provide an initial
insight into the effects of quercetin and iron on cell viability. It has been proposed that

55
flavonoids may be protective by increasing apoptosis in damaged cells (Kuo 1996). In
support of this, Kuo (1996) found that quercetin and other flavonoids decreased cell
viability through apoptosis in Caco-2, HT-29, and IEC-6 cells. Regarding iron’s effect
on cell viability, Cermak et al. (1993) suggested that acute exposure of intestinal tumor
cells to iron might increase their susceptibility to oxidant-mediated lysis. Taken together,
these experiments point to a possibility of both quercetin and iron reducing cell viability
in tumor cells. While our results were not statistically significant, we did notice a strong
trend (P=.0563) towards an iron and quercetin interaction in decreasing cell viability.
This did suggest that iron and quercetin may interact to damage cell components, but this
assay could not distinguish between apoptosis and necrosis.
I hypothesized that at high concentrations, quercetin could act as a prooxidant,
particularly in the presence of added iron, thus enhancing apoptosis of HepG2 cells. It is
known that reactive oxygen species can trigger measurable apoptotic changes within
cells. Indeed, a possible trigger of the observed apoptotic DNA fragmentation may
specifically be oxidant-induced DNA damage. DNA appears to be extremely susceptible
to attack by certain radical species. For example, due to its high reactivity, the hydroxyl
radical can attack sugars, purines, and pyrimidines of DNA (Halliwell and Gutteridge
1999). Studies by Sahu and Washington (1991) and Sahu and Gray (1997) suggest that
the autooxidation of flavonoids catalyzed by iron and copper ions can result in reactive
oxygen species formation, causing DNA damage. These researchers proposed that lipid
peroxidation is the first consequence of formation of the reactive oxygen species. DNA
is located in close proximity to the nuclear membrane - hence lipid peroxidation could
also effect the DNA inside the nuclear membrane, as the levels of lipid peroxides and

56
intermediate radicals are then amplified by a chain reaction (Sahu and Gray 1997). The
same researchers also proposed that reactive oxygen species may be produced in close
proximity to DNA-bound metal ions...“as iron is an essential trace element in cells and is
endogenously associated with DNA, agents capable of producing H2O2 inside the cell can
produce metal-catalyzed, site-specific oxidative DNA damage” (Sahu and Gray 1997).
Overall, their studies demonstrated that both free radicals and lipid peroxides can damage
DNA, regardless of the sequence of production.
Finally, to again reinforce the possibility of DNA damage as an apoptotic trigger,
the same study by Sahu and Washington (1991), indicated that quercetin at 0-100 µM
decreased double stranded DNA content in isolated rat liver nuclei in a concentration-
dependent manner after a 30-minute incubation. When iron and copper ions were added
at equal molar ratios with the same 30-minute incubation period, the loss of double
stranded DNA was even greater.
Regarding mechanism, flavonoids may increase free radical damage to DNA
through a reduction of Fe3+ to Fe2+ (Smith 1992). In particular, when flavonoids
gossypol, quercetin, or myricetin were added to reaction mixtures containing Fe3+-EDTA
and H2O2 at pH 7, they produced an increase in deoxyribose degradation. DNA
degradation was almost completely halted by catalase, and other hydroxyl radical
scavengers. Therefore, the deoxyribose degradation was attributed to hydroxyl radical
formation (Laughton 1989). However, it should be noted that inclusion of the same
flavonoids in reaction mixtures containing H2O2 and Fe3+-citrate had no effect on
increasing hydroxyl radical generation. It is possible that the form of iron or iron
complex may have an effect on its interaction with the flavonoids, and may explain

57
differences between our results and the results of others. Comparing these studies to ours
with DNA damage as the main possible trigger is speculative, as we did not include
assays that measured DNA damage as an endpoint. In the current study, while an iron
and quercetin interaction was not found to be significant, the greatest quercetin-induced
increases in DNA fragmentation were seen only in the iron-loaded cells. Clearly, our
study evaluated a different endpoint. However, as DNA damage is a known initiator of
apoptosis (King and Cidlowski 1995), and it could be the mechanism for quercetin-
induced apoptotic DNA fragmentation we found in iron loaded cells.
Caspase activity is an assay of early apoptotic changes within the cell. We
proposed that an iron and quercetin interaction would synergistically increase caspase
activity. Our results indicated that iron alone had no effect on caspase activity.
However, the effect of quercetin depended on cellular iron status. First, in cells that were
not iron-loaded, quercetin treatment had a protective effect, lowering caspase activity. In
the cells treated with both high levels of iron and quercetin, caspase activity was
increased. The redox status of the cell has a complex effect on apoptosis. Hampton
(1998) reviewed studies showing that the addition of exogenous oxidants induces
apoptotic caspase activity. It is possible that the levels of quercetin and iron used in the
current study functioned within this “apoptotic window” to induce a concentration of
reactive oxygen species that effectively increased caspase activity. The redox
environment of the cell may also explain why a protective effect was seen in the control
cells with quercetin loading. At a quercetin concentration of 80µM, without cellular iron
overload, quercetin may have acted as an antioxidant, scavenging radical species, thereby

58
preventing the reducing environment necessary to enhance reactive oxygen species
production.
Chromatin condensation, a result of the proteolytic action of caspases, is a late
stage indicator of apoptosis. It can be used to reinforce the evidence of caspase activity,
as theoretically, levels of chromatin condensation should be somewhat proportional to
caspase activity and show trends similar to those shown with the DNA fragmentation
assay. Our results from examining chromatin condensation were similar to the results
obtained in the DNA fragmentation assay in that only the cells loaded with quercetin
showed a trend towards increasing apoptosis. However, chromatin condensation did not
necessarily coincide with the results of our caspase activity assay and did not confirm an
iron and quercetin interaction toward increasing numbers of cells with apoptotic
morphology. Only a weak trend was noticed towards a positive quercetin effect
(P=.0773) on increasing numbers of cells with apoptotic morphology. One possible
explanation for a positive iron and quercetin interaction on caspase activity not being
reinforced by a proportional effect on apoptotic morphology comes from the fact that the
cells examined for chromatin condensation were coincubated with propidium iodide (PI),
an indicator of dead cells. While staining with propidium iodide may have allowed
detection of dead cells, it did not enable inference into the mode of cell death. It is
possible that a number of the PI-labeled cells that were already dead by the time of the
assay may have died by apoptosis. A possible way of discerning dead necrotic cells from
dead apoptotic cells would be to discriminate apoptotic cells by the cellular markers that
attract phagocytic macrophages. Overall, difference in timing between caspase activation
and physical manifestation of that activity (chromatin condensation) may be the reason

59
for the difference between results for the caspase activity assay and the Hoechst
procedure.
In conclusion, this study indicates that the level of quercetin needed to increase
apoptosis is well above levels that may be achieved by diet alone. Additionally, iron may
enhance the susceptibility of cells to quercetin-induced apoptosis.
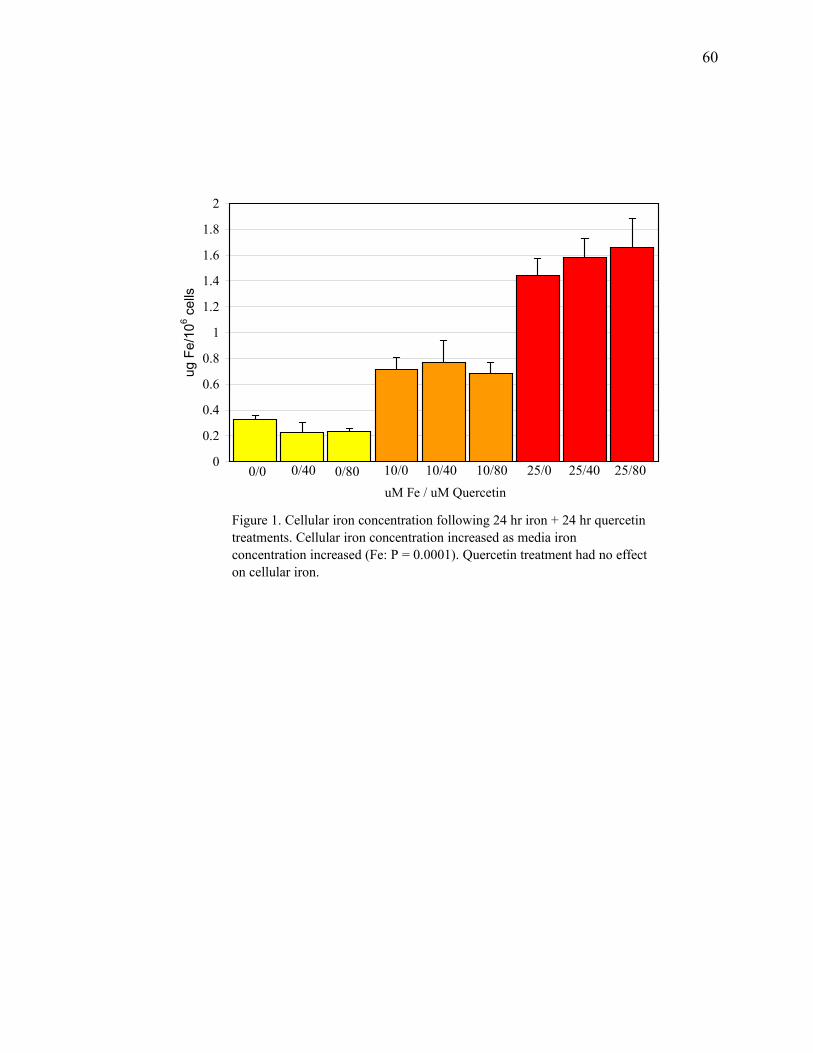
60
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
uM Fe / uM Quercetin
ug F
e/10
6 cel
ls
Figure 1. Cellular iron concentration following 24 hr iron + 24 hr quercetin treatments. Cellular iron concentration increased as media iron concentration increased (Fe: P = 0.0001). Quercetin treatment had no effect on cellular iron.
0/0 0/80 10/0 10/40 10/80 25/0 25/40 25/800/40
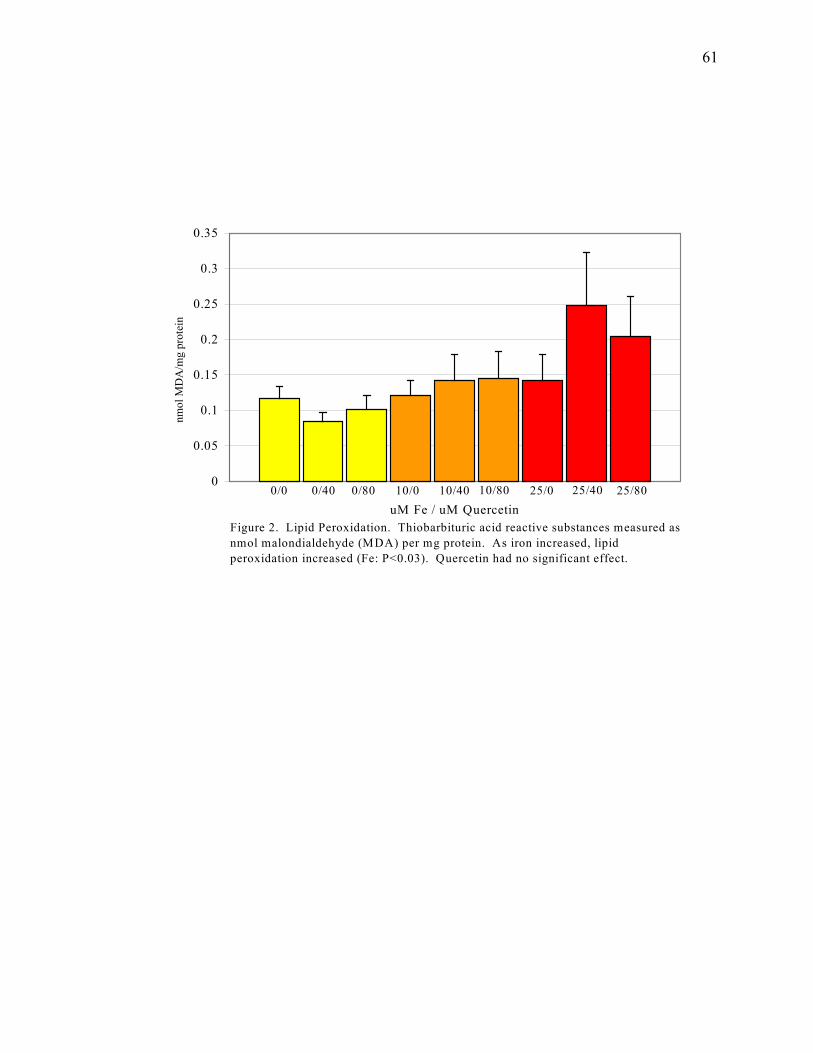
61
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
uM Fe / uM Quercetin
nmol
MD
A/m
g pr
otei
n
Figure 2. Lipid Peroxidation. Thiobarbituric acid reactive substances measured as nmol malondialdehyde (MDA) per mg protein. As iron increased, lipid peroxidation increased (Fe: P<0.03). Quercetin had no significant effect.
0/0 0/40 0/80 10/0 10/40 10/80 25/0 25/40 25/80
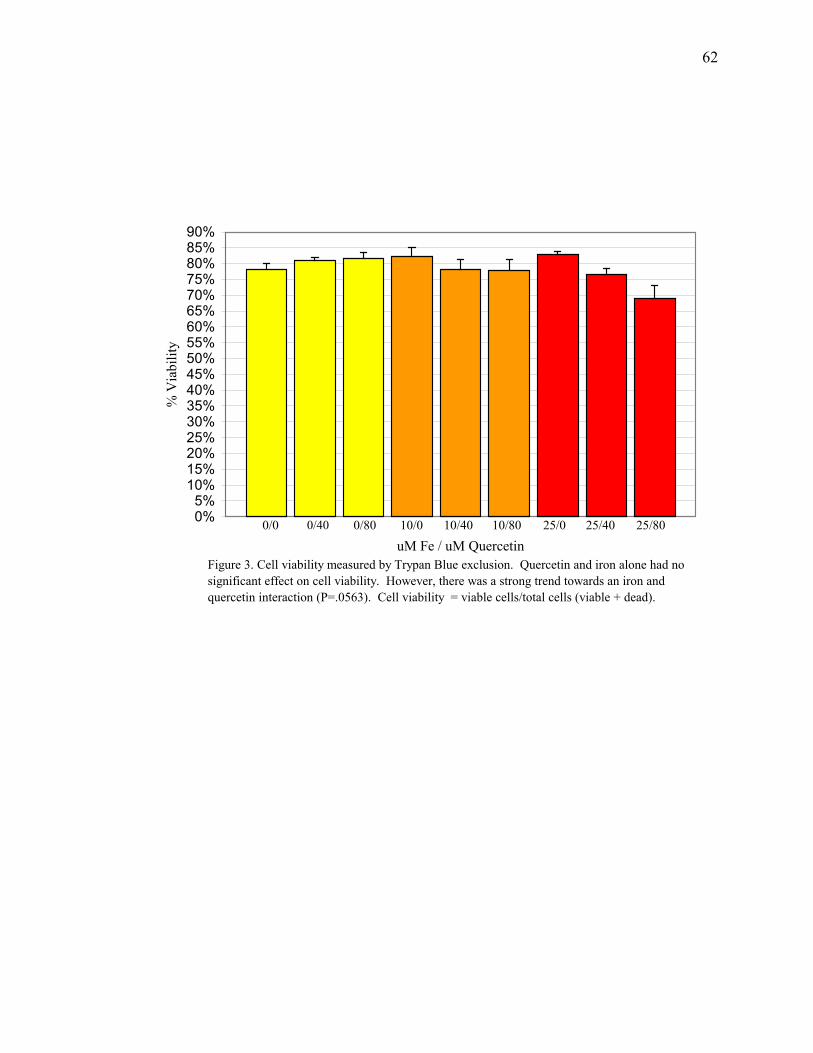
62
0%5%
10%15%20%25%30%35%40%45%50%55%60%65%70%75%80%85%90%
uM Fe / uM Quercetin
% V
iabi
lity
Figure 3. Cell viability measured by Trypan Blue exclusion. Quercetin and iron alone had no significant effect on cell viability. However, there was a strong trend towards an iron and quercetin interaction (P=.0563). Cell viability = viable cells/total cells (viable + dead).
0/0 0/80 10/0 10/40 10/80 25/0 25/40 25/800/40

63
0
20
40
60
80
100
120
140
160
um Fe / uM Quercetin
% in
crea
se in
180
-200
bp
frag
men
tatio
n ab
ove
cont
rol
0/40 0/80 10/40 10/80 25/40 25/80
Figure 4. Apoptotic DNA fragmentation=180-200 bp nucleosomes above control. Iron did not have a significant effect on quercetin-induced DNA fragmentation. However, DNA fragmentation was consistently increased only by 80 uM quercetin (p<0.02).
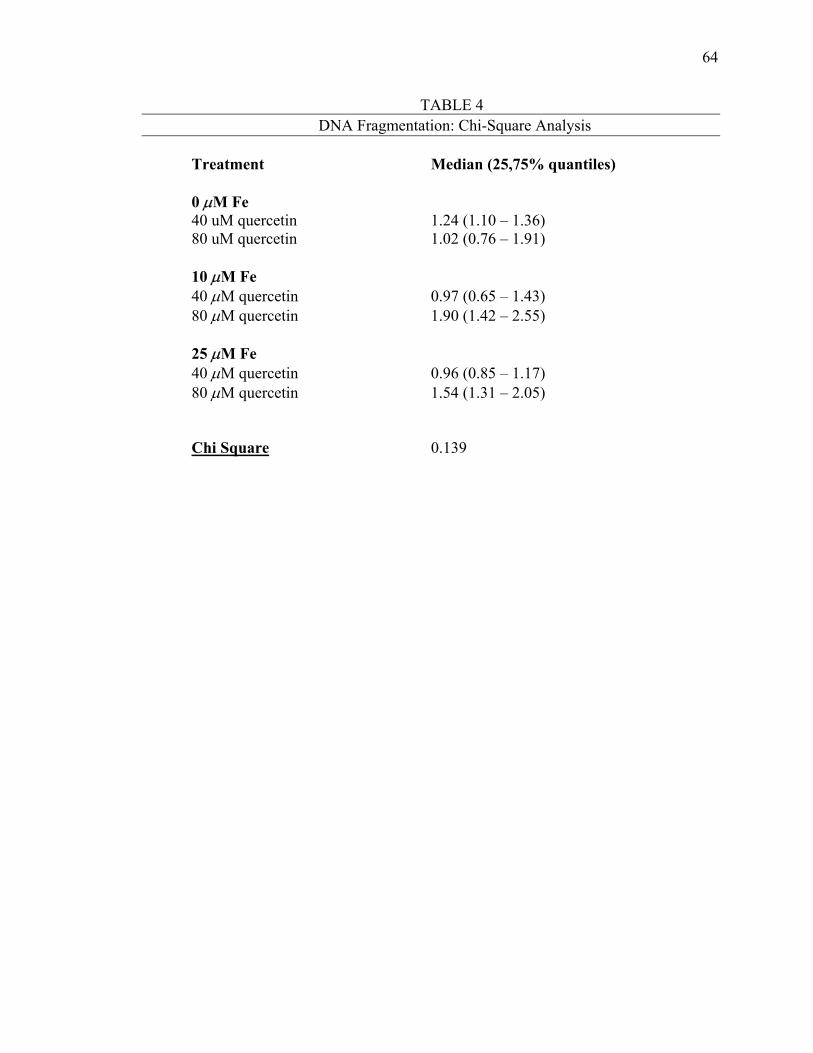
64
TABLE 4 DNA Fragmentation: Chi-Square Analysis
Treatment Median (25,75% quantiles) 0 µM Fe 40 uM quercetin 1.24 (1.10 – 1.36) 80 uM quercetin 1.02 (0.76 – 1.91) 10 µM Fe 40 µM quercetin 0.97 (0.65 – 1.43) 80 µM quercetin 1.90 (1.42 – 2.55) 25 µM Fe 40 µM quercetin 0.96 (0.85 – 1.17) 80 µM quercetin 1.54 (1.31 – 2.05) Chi Square 0.139

65
TABLE 5 Percent Cells with Caspase Activity
Treatment Mean1 Median2 Range 0 µM Fe 0 µM quercetin 0.2662 + 0.0868a,b 0.3407 (0.1271-0.4178) (0-0.4454) 80 µM quercetin 0.0561 + 0.0345b 0 (0-0.1323) (0-0.1484) 25 µM Fe 0 µM quercetin 0.2350 + 0.0892a,b 0.1832 (0.1087-0.4367) (0-0.4464) 80 µM quercetin 0.5083 + 0.1673a 0.5175 (0.3125-0.7519) (0-0.9597) P-value Fe 0.0638 Quercetin 0.7686 Fe/quercetin 0.0362 Chi Square 0.1398
1Mean + SEM (n=5 trials) 2Median (25,75 quantiles)
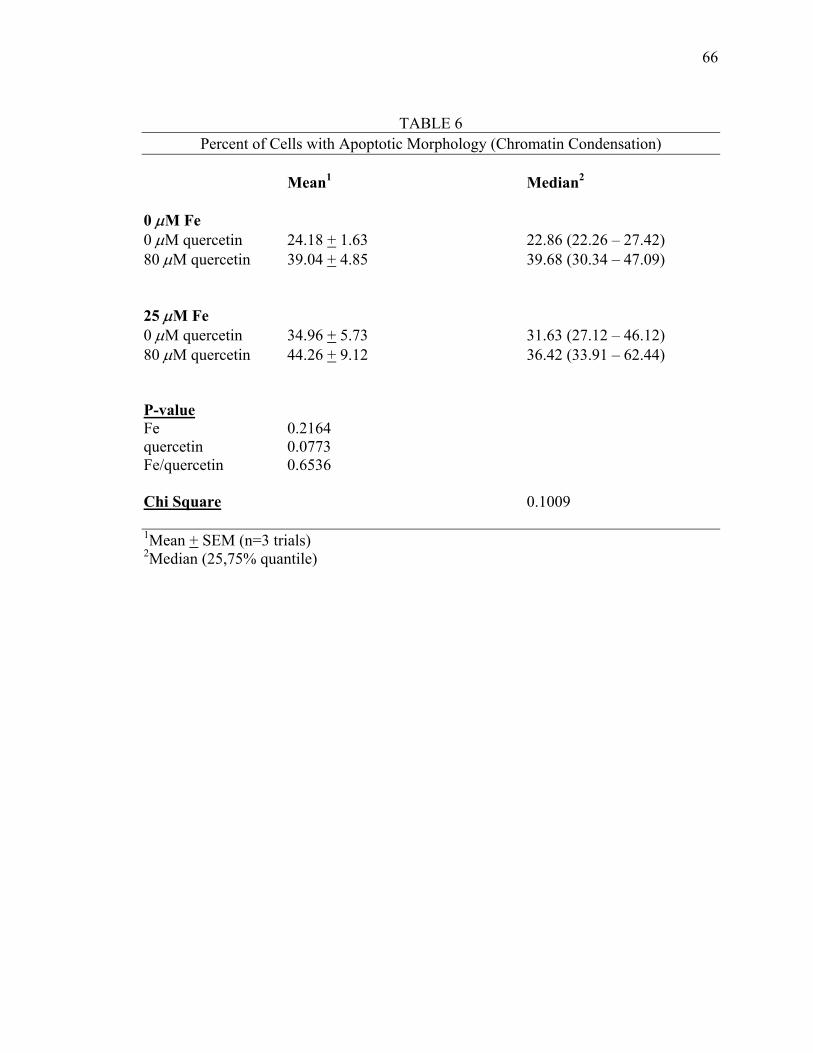
66
TABLE 6
Percent of Cells with Apoptotic Morphology (Chromatin Condensation) Mean1 Median2 0 µM Fe 0 µM quercetin 24.18 + 1.63 22.86 (22.26 – 27.42) 80 µM quercetin 39.04 + 4.85 39.68 (30.34 – 47.09) 25 µM Fe 0 µM quercetin 34.96 + 5.73 31.63 (27.12 – 46.12) 80 µM quercetin 44.26 + 9.12 36.42 (33.91 – 62.44) P-value Fe 0.2164 quercetin 0.0773 Fe/quercetin 0.6536 Chi Square 0.1009 1Mean + SEM (n=3 trials) 2Median (25,75% quantile)

CHAPTER IV
SUMMARY
MAJOR FINDINGS
The purpose of this study was to determine whether a flavonoid common to many
foods (quercetin) could induce apoptosis in the HepG2 cell line, a liver hepatoma cell
line, and whether cellular iron status alters apoptosis. It was hypothesized that 1) cellular
iron status would increase with higher media iron concentrations, and that addition of
quercetin to iron-loaded cells would have no effect on cellular iron, 2) addition of
quercetin to iron-loaded cells would increase lipid peroxidation, 3) cellular iron
concentration would influence the prooxidative ability of quercetin to decrease cell
viability, 4) cellular iron concentration would influence the prooxidative ability of
quercetin to induce specific apoptotic changes in the cell measured as: 200-bp DNA
nucleosome fragmentation (ladder effect), caspase activation, and chromatin
condensation.
Major findings were 1) cellular iron status increased with iron loading over a 24
hour incubation period, 2) addition of quercetin to iron-loaded cells had no effect on
cellular iron, 3) iron had a positive effect on lipid peroxidation, while quercetin did not,
4) 80 µM concentrations of quercetin tended to decrease cell viability in iron-loaded
cells, 5) 80 µM concentration of quercetin was consistently associated with an increase in
apoptosis-induced DNA fragmentation, and increases were greater in iron-loaded cells,
67

68
but the iron effect was not significant, 6) chromatin condensation was affected by
quercetin, but not iron, and 7) finally, caspase activity was significantly increased in iron-
loaded cells supplemented with 80 µM quercetin.
IMPLICATIONS
This study suggested that the level of quercetin needed to increase apoptosis is
well above levels that may be achieved by diet alone. It also indicates that iron may
enhance the susceptibility of cells to quercetin-induced apoptosis. The majority of
chemo- and radiotherapeutic agents destroy malignant cells by initiating apoptosis
(Martin 1997). Apoptosis is an ideal way to reduce tumor cell growth, because it induces
damaged or injured cells to commit suicide, allowing adjacent cells to proliferate without
being affected by the otherwise necrotic death of neighboring cells (Samali 1996).
Some predictions indicate that the prevalence of Americans ever diagnosed with
invasive cancer could be over 11 million by 2020 (Polednak 1997). Regarding modern
medicine, cancer treatments have met with success, but it has been very expensive
(Argiles 1998). Therefore, more cost-effective therapies need to be researched and
implemented. This study used an inexpensive and readily available combination of
components to produce apoptosis - an effective and established method of chemotherapy.
STUDY LIMITATIONS
Cellular iron levels were measured to ensure consistent increases in cellular iron
status with increased iron media levels. As well, media iron concentrations were
determined to ensure consistency among media batches. Due to the scope of this study,
though, quercetin levels in the media and the cells - following supplementation - could
not be measured. Obviously, knowledge of cellular quercetin status post-

69
supplementation, as well as the prominent areas of quercetin accumulation within the
cells, would provide further insight into specific cellular components that may be affected
by this compound. Further, the aglycone form of quercetin was used in this study, while
quercetin metabolites are most prevalent in human tissues. Also, only one incubation
time was tested for both iron and quercetin. Longer incubation time with either iron or
quercetin, or both, could have produced different results. Another important limitation
again occurs due to the scope of this study. More replicates of each assay would have
provided a stronger basis for discussion of assay results. Finally, although our model was
a human cancer cell line, we could not duplicate the complex environment that occurs in
vivo. Also, the levels of quercetin used were pharmacologic - they are higher than any
level that could be achieved by diet alone. However, as a neoplastic human cell line, our
model provides a foundation for additional chemotherapy research in human studies.
FURTHER RESEARCH
This study builds on a growing area of research into the dual roles of flavonoids.
Where much prior research has focused on the antioxidant capabilities of flavonoids like
quercetin, in conjunction with redox metal-catalyzed oxidative stress, this study further
examined the prooxidative effects produced by combining specific amounts of flavonoids
and redox metals. First, longer incubation times should be tested. Before assaying for
apoptosis, it would be helpful to have additional measures of oxidative stress produced by
each respective treatment. Further, identification of the specific reactive oxygen species
produced by iron and quercetin interactions would provide more insight into the
mechanism behind apoptosis in this system.

70
This study used the endpoints of 180-200 bp DNA fragmentation, chromatin
condensation, and general caspase activity to specifically determine apoptosis. Further
investigation into the intervals at which these endpoints are produced might indicate a
better timepoint by which to conduct the respective assays. As well, other assays such as
the TUNEL and electrophoretic assays, could be utilized to further reinforce the presence
of cellular apoptosis.
This study also indicated that a high level of quercetin is necessary to induce
apoptosis - more than diet can provide. Building from the maximum dose used - 80 µM -
future investigation of lower dose levels could determine the safest, most effective
pharmacologic range of quercetin concentrations that could induce apoptosis in tumor
cells. Finally, as iron status of the cell may influence the efficacy of quercetin
supplementation, iron loading must be refined to specifically target tumor cells to avoid
affecting healthy cells.

REFERENCES
American Cancer Society. (1998) Cancer Facts and Figures - 1998. American Cancer Society. (2000) Nutrition During and After Cancer Treatment: A guide for informed choices by cancer survivors. Draft. Ames, B.N., Gold, L.S., and Willett, W.C. (1995) The causes and prevention of cancer. Proc Natl Acad Sci USA 92: 5258-5265. Argiles, J.M., Carbo, N., Costelli, P., Lopez-Soriano, F.J. (1998) Prevention of cancer and cardiovascular diseases: a common strategy? Med Res Rev 18: 139-148. Barisani, D., Cairo, G., Ginelli, E. Marozzi, A., and Conte, D. (1999) Nitric oxide reduces nontransferrin-bound ion transport in HepG2 cells. Hepatology 29: 464-470. Block, G. (1992) The data support a role for antioxidants in reducing cancer risk. Nutr Rev 50: 207-213. Bravo, L. (1998) Polyphenols: chemisty, dietary sources, metabolism, and nutritional significance. Nutr Rev 56: 317-333. Breslow, R.A., Sorkin, J.D., Frey, C.M., and Kessler, L.G. (1997) American’s knowledge of cancer risk and survival. Prev Med 26: 170-177. Brown, J.P. (1980) A review of the genetic effects of naturally occurring flavonoids, anthroquinones, and related compounds. Mut Res 75: 243-277. Buege, J.A., and Aust, S.D. (1978) Microsomal lipid peroxidation. Meth Enzymol 52: 302-319. Buttke, T.M., and Sandstrom, P.A. (1994) Oxidative stress as a mediator of apoptosis. Immun Today 15: 7-10. Canada, A.T., Giannella, E., Nguyen, T.D., and Mason, R.P. (1990) The production of reactive oxygen species by dietary flavonols. Free Rad Biol Med 9: 441-449. Cao, G., Sofic, E., Prior, R.L. (1997) Antioxidant and pro-oxidant behaviour of flavonoids: structure-activity relationships. Free Rad Biol Med 22: 749-760. Cermak, J., Balla, J., Jacob, H.S., Balla, G., Enright, H., Nath, K., and Vercellotti, G.M. (1993) Tumor cell heme uptake induces ferritin synthesis resulting in altered oxidant sensitivity: possible role in chemotherapy efficacy. Cancer Res 53: 5308-5313.
71

72
Clutton, S. (1997) The importance of oxidative stress in apoptosis. Br Med Bull 53: 662-668 Columbano, A. (1995) Cell death: current difficulties in discriminating apoptosis from necrosis in the context of pathological processes in vivo. J Cell Biochem 58: 181-190. Cook, N.C. and Samman, S. (1996) Flavonoids – chemistry, metabolism, cardioprotective effects, and dietary sources. J Nutr Biochem 7: 66-76. Croft, K.D. (1998) The chemistry and biological effects of flavonoids and phenolic acids. Ann NY Acad Sci 854: 435-442. Cragg, L., Hebbel, R.P., Miller, W., Solovey, A., and Enright, H. (1998) The iron chelator L1 potentiates oxidative DNA damage in iron-loaded liver cells. Blood 92: 632-638. Crowther, J.R. (1995) ELISA: Theory and Practice. New Jersey. Humana Press Inc. Darzynkiewicz, Z. (1995) Apoptosis in antitumor strategies: modulation of cell cycle or differentiation. J Cell Biochem 58: 151-159. de Groot, H. and Rauen, U. (1998) Tissue injury by reactive oxygen species and the protective effects of flavonoids. Fundam Clin Pharmacol 12: 249-255. Duthie, S.J., Johnson, W., and Dobson, V.L. (1997) The effect of dietary flavonoids on DNA damage (strand breaks and oxidized pyrimidines) and growth in human cells. Mut Res 390: 141-151. Fenech, M., Stockley, C., Aitken, C. (1997) Moderate wine consumption protects against hydrogen peroxide-induced DNA damage. Mutagenesis 12: 289-296. Fotsis, T., Pepper, M.S., Aktas, E., Breit, S., Raska, S., Adlercreutz, H., Wahala, K., Montesano, R., and Schweigerer, L. (1997) Flavonoids, dietary-derived inhibitors of cell proliferation and in vitro angiogenesis. Cancer Res 57: 2916-2921. Glanz, K. (1997) Behavioral research contributions and needs in cancer prevention and control: dietary change. Prev Med 26: S43-S55. Glanz, K., Basil, M., Maibach, E., Goldberg, J., andSnyder, D. (1998) Why Americans eat what they do: taste nutrition, cost, convenience, and weight control concerns as influences on food consumption. J Am Diet Assoc 98: 1118-1126. Halliwell, B. and Gutteridge, J.M.C. (1999) Free radicals in biology and medicine. New York. Oxford University Press. Hampton, M.B., Fadeel, B., and Orrenius, S. (1998) Redox regulation of the caspases during apoptosis. Ann NY Acad Sci 854: 328-335.

73
Harnack, L., Block, G., Subar, A., Lane, S., and Brand, R. (1997) Association of cancer prevention-related nutrition knowledge, beliefs, and attitudes to cancer prevention dietary behavior. J Am Diet Assoc 97: 957-965. Haslam, E. (1996) Natural polyphenols (vegetable tannins) as drugs: possible modes of action. J Nat Prod 59: 205-215. Hertog, M.G.L, Hollman, P.C.H., and Katan, M.B. (1993) Intake of potentially anticarcinogenic flavonoids and their determinants in adults in The Netherlands. Nutr Cancer 20: 21- 29. Higuchi, M., Toshie, H., Proske, R.J., and Yeh, E.T.H. (1998) Regulation of reactive oxygen species-induced apoptosis and necrosis by caspase 3-like proteases. Oncogene 17: 2753-2760. Hiroaka, W., Vazquez, N., Nieves-Neira, W., Chanock, S.J., and Pommier, Y. (1998) Role of oxygen radicals generated by NADPH oxidase in apoptosis induced in human leukemia cells. J Clin Inves 102: 1961-1968. Hollman, P.C.H., de Vries, J.H.M., van Leeuwen, S.D., Mengelers, M.J.B., and Katan, M.B. (1995) Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers. Am J Clin Nutr 62: 1276-1282. Hollman, P.C.H., and Katan, M.B. (1998) Absorption, metabolism, and bioavailability of flavonoids. In: Flavonoids in Health and Disease. (Rice-Evans, C.A., and Packer, L., eds.) pp. 483-523. Marcel Dekker. New York. Ibrahim, W., Lee, U., Yeh, C., Szabo, J., Bruckner, G., and Chow, C.K. (1997) Oxidative stress and antioxidant status in mouse liver: effects of dietary lipid, vitamin E, and iron. J Nutr 127: 1401-1406.
Jacob, A.K., Hotchkiss, R.S., DeMeester, S.L., Hiramatsu, M., Karl, I.E., Swanson, P.E., Cobb, J.P., and Buchman, T.G. (1997) Endothelial cell apoptosis is accelerated by inorganic iron and heat via and oxygen radical dependent mechanism. Surgery 122: 243-254. Jiang, M.C., Yang-Yen, H.F., Yen, J.J.Y., and Lin, J.K. (1996) Curcumin induces apoptosis in immortalized NIH 3T3 and malignant cancer cell lines. Nutr Cancer 26: 111-120. Kato, J., Kobune, M., Kohgo, Y., Sugawara, N., Hisai, H., Nakamura, T., Sakamaki, S., Sawada, N., and Nitsu, Y. (1996) Hepatic iron deprivation prevents spontaneous development of fulminant hepatitis and liver cancer in Long- Evans Cinnamon rats. J Clin Invest 98: 923-929.

74
Kaul, A. and Khanduja, K.L. (1998) Polyphenols inhibit promotional phase of tumorigenesis: relevance of superoxide radicals. Nutr Cancer 32: 81-85. Kawabata, T., Ma, Y., Yamador, I., and Okada, S. (1997) Iron-induced apoptosis in mouse renal proximal tubules after an injection of a renal carcinogen, iron-nitriloacetate. Carcinogenesis 18: 1389-1394. Khumalo, H., Gomo, A.R., Moyo, V.M., Gordeuk, V.R., Saungweme, T., Rouault, T.A., and Gangaidzo, I.T. (1998) Serum transferrin receptors are decreased in the presence of iron overload. Clin Chem 44: 40-44. King, K.L., and Cidlowski, J.A. (1995) Cell cycle and apoptosis: common pathways to life and death. J Cell Biochem 58: 175-180. Krebs-Smith, S. (1998) Progress in improving diet to reduce cancer risk. Cancer 83: 1425-1432. Kuo, M.L., Huang, T.S., and Lin, J.K. (1996) Curcumin, an antioxidant and anti-tumor promoter, induces apoptosis in human leukemia cells. Biochem et Biophys Acta 1317: 95-100. Landis, S.H., Murray, T., Bolden, S., and Wingo, P.A. (1998) Cancer Statistics, 1998. CA Cancer J Clin 48: 6-29. Laughton, M.J., Halliwell, B., Evans, P.J., and Hoult, J.R.S. (1989) Antioxidant and pro-oxidant actions of the plant phenolics quercetin, gossypol, and myricetin: effects on lipid peroxidation, hydroxyl radical generation and bleomycin-dependent damage to DNA. Biochem Pharm 38: 2859-2865. Lichtenstein, A.H., Kennedy, E., Barrier, P., Danford, D., Ernst, N.D., Grundy, S.M., Leveille, G.A., Van Horn, L., Williams, C.L., and Booth, S.L. (1998) Dietary fat consumption and health. Nutr Rev 56: S3-S28. Leanderson, P., Faresjo, A.O., and Tagesson, C. (1997) Green tea polyphenols inhibit oxidant-induced DNA strand breakage in cultured lung cells. Free Rad Biol Med 23: 235-242. Lockshin, R.A., Zakeri, Z., Tilly, J.L. (1998) When Cells Die: A comprehensive evaluation of apoptosis and programmed cell death. New York. Wiley-Liss, Inc. Lowry, O.H., Rosenbrough, N.J., Farr, A.L., and Randall, R.J. (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275 Manach, C., Morand, C., Crespy, V., Demigne, C., Texier, O., Regerat, F., and Remesy, C. (1998) Quercetin is recovered in human plasma as conjugated derivatives which retain antioxidant properties. FEBS Lett 426: 331-336.

75
Manach, C., Morand, C., Demigne, C., Texier, O., Regerat, F., and Remesy, C. (1997) Bioavailability of rutin and quercetin in rats. FEBS Lett 409: 12-16. Manach, C., Morand, C., Texier, O., Favier, M.L., Agullo, G., Demigne, C., Regerat, F., and Remesy, C. (1995) Quercetin metabolites in plasma of rats fed diets containing rutin or quercetin. J Nutr 125: 1911-1922. Martin, S.J. (1997) Apoptosis and Cancer. New York. Karger Landes Systems. McClelland, J.W., Denmark-Wahnefried, W., Mustian, R.D., Cowan, A.T., and Campbell, M.K. (1998) fruit and vegetable consumption of rural african americans: baseline survey results of the black churches united for better health 5 A Day project. Nutr Cancer 30: 148-157. McConkey, D.J. (1998) Biochemical determinants of apoptosis and necrosis. Tox Lett 99: 157-168. McCord, J.M. (1998) Iron, free radicals, and oxidative injury. Sem Hematol 35: 5-12. Meikrantz, W., and Schlegel, R. (1995) Apoptosis and the cell cycle. J Cell Biochem 58:160-174. Meneghini, R. (1997) Iron homeostasis, oxidative stress, and DNA damage. Free Rad Biol Med 23: 783-792. Miura, Y.H., Tomita, I., Watanabe, T., Hirayama, T., and Fukui, S. (1998) Active oxygens generation by flavonoids. Biol Pharm Bull 21: 93-96. Morel, I., Lescoat, G., Cogrel, P., Sergent, O., Pasdeloup, N., Brissot, P., Cillard, P., and Cillard, J. (1993) Antioxidant and iron-chelating activities of the flavonoids catechin, quercetin, and diosmetin on iron-loaded rat hepatocyte cultures. Biochem Pharmacol 45: 13-19. Musonda, C.A., and Chipman, J.K. (1998) Quercetin inhibits hydrogen peroxide (H2 O2)- induced NF-KB DNA binding activity and DNA damage in HepG2 cells. Carcinogenesis 19: 1583-1589. Nicholson, D.W. (1999) Caspase structure, proteolytic substrates, and function during cell death. Cell Death and Diff 6: 1028-1042. Parker, S.L., Davis, K. J., Wingo, P., Ries, L.A.G., and Heath, C.W. (1998) Cancer statistics by race and ethnicity. Cancer J Clin 48: 31-48. Perieira, M.A., Grubbs, C.J., Barnes, L.H., Hong, L., Olson, G.R., Eto, Isao, Juliana, M., Whitaker, L.M., Kelloff, G.J., Steele, V.E., and Lubet, R.A. (1996) Effects of the phytochemicals, curcumin and quercetin, upon azoxymethane-induced colon cancer

76
and 7,12-dimethylbenz[a]anthracene-induced mammary cancer in rats. Carcinogenesis 17: 1305-1311. Polednak, A.P. (1997) Estimating the prevalence of cancer in the United States. Cancer 80: 136-141. Potter, J.D. (1996) Food and phytochemicals, magic bullets and measurement error: a commentary. Am J Epidem 144: 1026-1027. Ren, S.R. and Lien, E.J. (1997) Natural products and their derivatives as cancer chemopreventive agents. Progress Drug Res 48: 147-171. Rice-Evans, C.A., Miller, N.J., Paganga, G. (1996) Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Rad Biol Med 20: 933-956. Rollet-Labelle, E., Grange, M., Elbim, C., Marquetty, C., Gougerot-Pocidalo, M., Pasquier, C. (1998) Hydroxyl radical as a potential intracellular mediator of polymorphonuclear neutrophil apoptosis. Free Rad Biol Med 24: 563-572. Sahu, S.C. (1994) Dual role of flavonoids in mutagenesis and carcinogenesis. Environ Carcino and Ecotox Revs 12: 1-21. Sahu, S.C., and Gray, G.C. (1997) Lipid peroxidation and DNA damage induced by morin and naringenin in isolated rat liver nuclei. Food and Chem Tox 35: 443-447. Sahu , S.C., and Washington, M.C. (1992) Effect of ascorbic acid and curcumin on quercetin-induced nuclear DNA damage, lipid peroxidation and protein degradation. Cancer Lett 63: 237-241. Sahu, S.C., and Washington, M.C. (1991) Quercetin-induced lipid peroxidation and DNA damage in isolated rat liver nuclei. Cancer Lett 58: 75-79. Samali, A., Gorman, A.M., and Cotter, T.G. (1996) Apoptosis – the story so far. Experientia 52: 933-941. Shahidi, F., Janitha, P.K., and Wanasundra, P.D. (1992) Phenolic antioxidants. Crit Rev Food Sci Nutr 32: 67-103. Simandan, T, Sun, J., and Dix, T.A. (1998) Oxidation of DNA bases, deoxyribonucleosides and homopolymers by peroxyl radicals. Biochem J 335: 233-240. Slee, E.A., Adrain, C., and Martin, S.J. (1999) Serial killers: ordering caspase activation events in apoptosis. Cell Death and Diff 6: 1067-1074.

77
Smith, C. Halliwell, B., and Aruoma, O.I. (1992) Protection by albumin against the pro-oxidant actions of phenolic dietary components. Food Chem Toxic 30: 483-489. Stadler, R.H., Markovic, J., and Turesky, R.J. (1995) In vitro anti- and pro-oxidative effects of natural polyphenols. Biol Trace Elem Res 47: 299-305. Stavric, B., and Matula, T.I. (1992) Flavonoids in foods : their significance for nutrition and health. In: Lipid Soluble Antioxidants: Biochemistry and Clinical Applications (Ong, A.S.H., and Packer, L. eds.) pp. 274-293. Birkhauser Verlag. Basel, Switzerland. Steinmetz, K.A., and Potter, J.D. (1996) Vegetables, fruit, and cancer prevention: a review. J Am Diet Assoc 96: 1027-1039. Stimson, J.L.P. (1998) The effects of iron and quercetin on lipid peroxidation and apoptosis in colon. M.S. Thesis, University of Georgia. Stoian, I., Oros, A., and Moldoveanu, E. (1996) Apoptosis and free radicals. Biochem Mol Med 59: 93-97. Tsuda, H., Uehara, N., Iwahori, Y., Asamoto, M., Iigo, M., Nagao, M., Matsumoto, K., Ito, M., and Hirono, I. (1994) Chemopreventive effects of B-carotene, a- tocopherol and five naturally occurring antioxidants on initiation of hepatocarcinogenesis by 2-amino-3-methylimidazo[4,5f]quinoline in the rat. Jpn J Cancer Res 85: 1214-1219. Verma, A.K. (1992) Modulation of mouse skin carcinogenesis and epidermal phospholipid biosynthesis by the flavonoid quercetin. In: Phenolic Compounds in Food and Their Effects on Health II. (Huang, M-T., Ho, C-T. and Lee, C.Y. eds.) pp. 251-264. American Chemical Society. Washington, D.C. Voet, D., and Voet, J.G. (1990) Biochemistry. New York. John Wiley and Sons. Wang, G.S., Eriksson, L.C., Xia, L., Olsson, J.M., and Stal, P. (1999a) Dietary iron overload inhibits carbon tetrachloride-induced promotion in chemical hepatocarcinogenesis: effects on cell proliferation, apoptosis, and antioxidation. J Hepatol 30: 689-698. Whittaker, P., Hines, F.A., Robl, M.G., Dunkel, V.C. (1996) Histopathological evaluation of liver, pancreas, spleen, and heart from iron-overloaded Sprague-Dawley rats. Toxicol Path 24: 558-563. Yang, C.S., Lee, M., Chen, L., and Yang, G. (1997) Polyphenols as inhibitors of carcinogenesis. Environ Health Perspect 105: 971-976

78
Yang, C.S., Yang, G., Landau, J.M., and Liao, J. (1998) Tea and tea polyphenols inhibit cell hyperproliferation, lung tumorigenesis, and tumor progression. Exp Lung Res 24: 629-639. Yin, T. (1999) The effects of dietary iron intake and WY-14,643, a peroxisome proliferator, on liver oxidative status and protein kinase C activation. M.S. Thesis, University of Georgia. Zhang, X., Chen, J., Davis, B., and Kiechle, F. (1999) Hoechst 33342 induces apoptosis in HL-60 cells and inhibits topoisomerase I in vivo. Arch Path Lab Med 123: 921-927.





























