Embed Size (px)
Citation preview

S1
High Performance Solution-processed Solar Cells and Ambipolar behavior
in OFETs with Thienyl-BODIPY Scaffoldings
Thomas Bura,1 Nicolas Leclerc,2 Sadiara Fall3 Patrick Lévêque,3 Thomas Heiser,3 Pascal Retailleau,4 Sandra Rihn,1 Antoine Mirloup,1 and Raymond
Ziessel1
Supporting Information (53 pages)
Table of Contents Pages
1) General Methods S1
2) Synthetic Experimental Part S2
3) NMR Spectra traces S10
4) DSC traces for TB1-4 S39
5) X-ray diffraction data for TB2 S41
6) Atomic force microscopy investigations S41
7) Absorption and Fluorescence Spectra and Data (Table S1) S44
8) Cyclic voltammetry traces and Data (Table S2) S48
9) Devices preparation and charge mobilities (Table S3 + S4) S49
10) Additional References S53
1) General Methods All reactions were performed under an atmosphere of dried argon using standard Schlenk tube
techniques. All chemicals were used as received from commercial sources unless stated
otherwise. CH2Cl2 was distilled from P2O5 under an argon atmosphere. THF was distilled
from sodium and benzophenone under an Ar atmosphere. DMF was distilled from KOH
under an argon atmosphere. 1H NMR (300.1 MHz) and 13C NMR (75.5 MHz) spectra were
recorded at room temperature (rt) on a Bruker Advance 300 MHz spectrometer, 1H NMR
(200.1 MHz) and 13C NMR (50.5 MHz) spectra were recorded at rt on a Bruker Advance 200
MHz spectrometer using perdeuteriated solvents as internal standards. Chromatographic
purifications were performed using silica gel (40-63 µm). TLC was performed on silica gel
plates coated with fluorescent indicator.

S2
Absorption spectra were recorded on a Schimadzu UV-3000 absorption spectrometer. The
steady-state fluorescence emission and excitation spectra were obtained by using a HORIBA
JOBIN YVON FLUOROMAX 4. All fluorescence spectra were corrected. The fluorescence
quantum yield (φexp) was calculated from eq 1.1
(1)
Here, I denotes the integral of the corrected emission spectrum, OD is the optical density at
the excitation wavelength and η is the refractive index of the medium. The reference systems
used were rhodamine 6G (φref = 0.78) in air equilibrated water and
Tetramethoxydiisoindomethene-difluoroborate (φref = 0.51). 2 Luminescence lifetimes were
measured on an Edimburgh Instruments spectrofluorimeter equipped with a R928
photomultiplier and a PicoQuant PDL 800-D pulsed diode connected to a GwInstect GFG-
8015G delay generator. No filter was used for the excitation. Emission wavelengths were
selected by a monochromator. Lifetimes were deconvoluted with FS-900 software using a
light-scattering solution (LUDOX) for instrument response.
2) Synthetic Experimental Part All reagents thiophene, 2-bromothiophene, 2-(5-hexylthiophen-2-yl)thiophene, -(2-
methoxyethoxy)ethanol, 2-thienylethanol, were used directly as obtained commercially unless
otherwise noted. Compound 1,3 2,4 3,5 4,6 9,7 128 were prepared according to the respective
references.
General procedure A for the Vilsmeier reaction: In a Schlenk tube containing anhydrous
DMF at 0° was added 1.2 equivalent of POCl3. The mixture was stirred and then gradually
warmed at room temperature over a period of 1h. The thiophene derivative was added in the
mixture and then heated at 60°C during 12h. The solution was poured into ice-water. The pH
was adjusted to 7 with sodium hydroxide solution. The reaction mixture was extracted with
ether. The organic layer was washed three times with water and dried over MgSO4 or
absorbent cotton and the solvent was evaporated under reduced pressure and purified by
silica-gel column chromatography to afford the desired compound.

S3
General procedure B for the Knoevenagel condensation: In a round bottom flask equipped
with a Dean stark apparatus, thienyl-aldehyde 2.2 eq , piperidine (2 mL) and a crystal of p-
TsOH were added to a stirred solution of BODIPY (1eq) in toluene (20mL). The solution
was heated at reflux during 12 hours. After cooling to room temperature, the mixture was
washed three times with water. The organic phase was dried over MgSO4 or absorbent cotton
and the solvent was evaporated under reduced pressure. The resulting crude residue was
purified by silica-gel column chromatography to afford the desired compound.
Scheme S1: Synthesis of compound TB1.
NB
NFF
S S
I
NB
NFF
I
O
SToluene, piperidine
pTsOH
POCl3
DMF
S+
3
S
H
BuLi, THF
Bromohexyl
1 2 TB1
2-Hexylthienyl (1). The purification was carried out by chromatography on flash silica as
static phase with a mixture of petroleum ether as mobile phase and afforded 1 (92%). 1H
NMR (CDCl3, 300 MHz): 0.88-0.93 (m, 3H), 1.29-1.40 (m, 6H), 1.64-1.74 (m, 2H), 2.83 (t,
2H, 3J = 7.7 Hz), 6.78-6.79 (m, 1H), 6.91-6.94 (m, 1H), 7.11-7.12 (m, 1H).
5-Hexylthienyl-2-carbaldehyde (2). The purification was carried out by chromatography on
flash silica as static phase with a mixture of dichloromethane/petroleum ether 30/70 as mobile
phase and afforded 2 (61%). 1H NMR (CDCl3, 300 MHz): 0.85-0.90 (m, 3H), 1.24-1.41 (m,
6H), 1.64-1.74 (m, 2H), 2.86 (t, 2H, 3J = 7.6 Hz), 6.88-6.90 (m, 1H), 7.59-7.60 (m, 1H), 9.80
(s, 1H).
TB1 was prepared according to the general procedure B from 434.3 mg (0.965 mmol) of
compound 3 and 282.5 mg (1.439 mmol) of 5-hexylthienyl-2-carbaldehyde (2) dissolved in
toluene (20 mL). The purification was carried out by chromatography on flash silica as static
phase with a mixture of toluene/dichloromethane/petroleum ether (35/5/60) as mobile phase
and afforded TB1 as a dark purple powder (196.1 mg 25%). 1H NMR (CDCl3, 300 MHz):
0.88-0.93 (m, 6H), 1.33-1.42 (m, 12H), 1.45 (s, 6H), 1.67-1.74 (m, 4H), 2.83 (t, 4H, 3J = 7.6
Hz), 6.56 (s, 2H), 6.72 (s, 1H), 6.73 (s, 1H), 7.03 (s, 1H), 7.05 (s, 1H), 7.08 (d, 2H, 3J = 8.1
Hz), 7.27-7.42 (m, 4H), 7.84 (d, 2H, 3J = 8.1 Hz). 13C NMR (CDCl3, 75 MHz): 14.2, 14.8,

S4
15.0, 22.7, 28.9, 29.8, 30.8, 31.6, 31.7, 95.0, 117.2, 117.9, 121.4, 125.4, 129.0, 130.0, 130.4,
131.5, 132.8, 134.9, 138.1, 138.4, 140.1, 142.0, 142.4, 149.3, 153.3, 155.2. EI-MS, m/z (%):
806.1 ([M], 100). Anal. Calcd for C41H46BF2IN2S2 (Mr = 806.66): C, 61.05; H, 5.75; N, 3.47
Found: C, 60.81; H, 5.44; N, 3.21.
Scheme S2: Synthesis of compound TB2.
NB
NFF
S S
S S
I
NB
NFF
I
O
S
S
H
Toluene, piperidine
pTsOH
POCl3
DMF
S
S+
3 TB24
5-(5-Hexylthienyl-2-yl)thiophene-2-carbaldehyde (4). The purification was carried out by
chromatography on flash silica as static phase with a mixture of CH2Cl2/petroleum ether
(50/50) as mobile phase and afforded 4 as an yellow oil (1.1 g, 98 %).1H NMR (CDCl3, 200
MHz): 0.9 (t, 3H, 3J = 6.7Hz), 1.27-1.42 (m, 6H), 1.62-1.73 (m, 2H), 2.82 (t, 2H, 3J = 7.6Hz),
6.75 (d, 1H, 3J = 3.4 Hz), 7.16-7.20 (m, 2H), 7.65 (d, 1H, 3J = 3.9 Hz), 9.84 (s, 1H).
TB2 was prepared according to the general procedure B from 310 mg (0.68 mmol) of
compound 3 and 421 mg (1.51 mmol) of 5-(5-hexylthienyl-2-yl)thiophene-2-carbaldehyde
(4). The purification was carried out by chromatography on flash silica as static phase with a
mixture of dichloromethane/toluene/petroleum ether (20/30/50) as mobile phase and afforded
TB2 as a dark purple powder (460 mg, 70%). 1H NMR (CDCl3, 300 MHz): 0.91 (t, 6H, 3J =
6.6Hz), 1.26-1.43 (m, 12H), 1.47 (s, 6H), 1.69 (m, 4H), 2.82 (t, 4H, 3J = 7.4 Hz), 6.60 (s, 2H),
6.73 (d, 2H, 3J = 3,3 Hz), 7.04-7.12 (m, 8H), 7.30 (overlapping with solvent d, 2H, 3J = 16.0
Hz), 7.45 (d, 2H, 3J = 16.0 Hz), 7.85 (d, 2H, 3J = 8.1 Hz). 13C NMR (CDCl3, 50 MHz): 14.2,
15.1, 22.7, 28.9, 30.4, 31.7, 94.5, 118.0, 118.3, 123.9, 124.4, 125.2, 129.1, 129.8, 130.8,
133.6, 134.8, 135.0, 135.8, 138.4, 140.1, 140.7, 141.5, 146.6, 152.3. EI-MS, m/z (%): 970.1
([M], 100). Anal. Calcd for C49H50BF2IN2S4 (Mr = 970.91): C, 60.62; H, 5.19; N, 2.89;
Found: C, 60.52 ; H, 4.97 ; N, 2.70.

S5
Scheme S3: Synthesis of compound TB3.
HOO
O
NaH, CuI, Pyridine
S O O O S O O OSn
nBuLi, SnMe3Cl
S O O OS
S Br
Pd(PPh3)4, THF
5 6
S Br THF
POCl3
DMF
S O O OS
O
H NB
NFF
I
NB
NFF
S S
S S
O
O
O
O
I
OO
Toluene, piperidine
pTsOH
7
8 3 TB3
+
2-(2-(2-Methoxyethoxy)-ethoxy)thiophene (5):
Copper iodide (12.2 mmol) and sodium hydride (92 mmol) were added to a solution of 2-(2-
methoxyethoxy)ethanol (0.3 mol) in 20 mL of dry pyridine and allowed to stir 30 min at rt.
2-bromothiophene (61 mmol) was then added and the mixture was heated at 100°C for 7
days. The mixture was slowly cooled down to rt and then poured into a water-CH2Cl2
mixture. Then, the organic solution was extracted three times with HCl 10% to remove
pyridine and then water. The CH2Cl2 organic phase was dried over Na2SO4 and concentrated
under vacuum. The purification was carried out by chromatography on flash silica as static
phase with a mixture of cyclohexane / ethyl acetate (80/20) as mobile phase and afforded 5 as
an uncoloured oil (4.2 g, 34%). 1H NMR (CDCl3 300 MHz): 3.39 (s, 3H), 3.58 (dd ; 3J = 4.98 Hz, 3J = 6.39 Hz, 2H), 3.71
(dd ; 3J = 5.0 Hz, 3J = 6.36 Hz, 2H), 3.84 (t, 3J = 4.7 Hz, 2H), 4.2 (t, 3J = 4.7 Hz, 2H), 6.24
(dd, 3J = 5.82 Hz, 4J = 1.35 Hz, 1H), 6.55 (dd, 3J = 3.69 Hz, 4J = 1.29 Hz, 1H), 6.70 (q, 3J =
5.76 Hz, 3J = 3.75 Hz, 1H). 13C NMR (CDCl3, 50 MHz): 59.1, 69.4, 70.8, 71.9, 73.1, 105.2,
112.2, 124.6, 165.3. EI-MS, m/z (%): 202.1 ([M], 100). Anal. Calcd for C9H14O3S
(Mr = 202.27): C, 53.44; H, 6.98; Found C, 53.25; H, 6.84.
2-Trimethyltin-5-(2-(2-methoxyethoxy)-ethoxy)thiophene (6):
To a solution of compound 5 (13.8 mmol) in dry THF (40 mL) at -78°C, a solution of 2.5 M
of nBuLi in hexane (15.2 mmol) was added slowly. The resulting solution was then stirred at

S6
-78°C for 1h and then a 1.0 M solution of trimethyltin chloride in THF (18 mmol) was added.
The solution was warmed to rt and stirred overnight. Then the solution was poured into water
and extracted with ethylacetate. The organic layer was washed three times with water and
dried over Na2SO4. The organic layer was then concentrated under vacuum. The crude
product is used without further purification. 1H NMR (CDCl3, 300 MHz): 0.32 (s, 9H), 3.39 (s, 3H), 3.58 (dd, 3J = 4.89 Hz, 3J = 6.36 Hz,
2H), 3.71 (dd, 3J = 4.95 Hz, 3J = 6.45 Hz, 2H), 3.84 (t, 3J = 4.98 Hz, 2H), 4.21 (t, 3J = 4.65 Hz,
2H), 6.37 (d, 3J = 3.42 Hz, 1H), 6.79 (d, 3J = 3.51 Hz, 1H). 13C NMR (CDCl3, 50 MHz): -8.3,
59.1, 69.5, 70.8, 72.0, 73.2, 107.1, 123.0, 133.2, 170.7.
5-(2-(2-Methoxyethoxy)-ethoxy)-2,2’-bithiophene (7):
Compound 6 (6.7 mmol) and 2-bromothiophene (6.1 mmol) were dissolved in 25 mL of dry
THF and the solution was degassed with argon. Then, [Pd(PPh3)4] in catalytic amount was
added and the reaction mixture was stirred at 80°C for 48 hours under argon atmosphere.
Then, the solution was cooled to rt and filtered onto a celite pad. The organic layer was
washed three times with water and concentrated under reduced pressure. The purification was
carried out by chromatography on flash silica as static phase with a mixture of cyclohexane /
ethyl acetate (80/20) as mobile phase and afforded 7 as a pale yellow oil (1.23 g, 65%). 1H NMR (Acetone d6, 300 MHz): 3.31 (s, 3H), 3.52 (dd, 3J = 5.49 Hz, 3J = 6.66 Hz, 2H), 3.64
(dd, 3J = 5.04 Hz, 3J = 6.18 Hz, 2H), 3.81 (m, 2H), 4.24 (m, 2H), 6.28 (d, 3J = 3.90 Hz, 1H),
6.89 (d, 3J = 3.87 Hz, 1H), 7.03 (q, 3J = 5.10 Hz, 3J = 5.13 Hz, 1H), 7.09 (dd, 3J = 3.54 Hz, 4J=1.08 Hz, 1H), 7.31 (dd, 3J = 5.10 Hz, 4J = 1.17 Hz, 1H). 13C NMR (Acetone d6, 50 MHz):
58.8, 69.9, 71.2, 72.7, 74.1, 106.6, 122.4, 123.2, 124.4, 124.5, 128.7, 138.6, 165.3. EI-MS,
m/z (%): 284.1 ([M], 100). Anal. Calcd for C13H16O3S2 (Mr = 284.39): C, 54.90; H, 5.67;
Found: C, 54.68; H, 5.74.
5-(5-(2-(2-Methoxyethoxy)ethoxy)thiophen-2-yl)thienyl-2-carbaldehyde (8) was prepared
according to the general procedure A from 600 mg of 7 (2.11 mmol) and 0.3 mL of POCl3
(3.16 mmol). The purification was carried out by chromatography on flash silica as static
phase with a mixture of petroleum ether/ethyl acetate (50/50) as mobile phase to give an
yellow oil (480mg, 70%). 1H NMR (CDCl3, 200 MHz): 3.39 (s, 3H), 3.55-3.60 (m, 2H), 3.67-
3.72 (m, 2H), 3.83-3.87 (m, 2H), 4.22-4.26 (m, 2H), 6.21 (d, 1H, 3J = 4.1 Hz), 7.01-7.05 (m,
2H), 7.61 (d, 1H, 3J = 4.1 Hz), 9.80 (s, 1H). 13C NMR (CDCl3, 50 MHz): 59.2, 69.4, 71, 72.1,
73.3, 106.6, 122.6, 122.8, 124.6, 137.7, 140.5, 148.4, 166.9, 182.4. EI-MS, m/z (%): 312.1
([M], 100). Anal. Calcd for C14H16O4S2 (Mr = 312.4): C, 53.82; H, 5.16; Found: C, 53.69; H,
5.04.

S7
TB3 was prepared according to the general procedure B from 165 mg (0.36 mmol) of 3 and
250 mg (0.8 mmol) of 5-(5-(2-(2-methoxyethoxy)ethoxy)thiophen-2-yl)thiophene-2-
carbaldehyde. The purification was carried out by chromatography on flash silica as static
phase with a mixture of dichloromethane/ethyl acetate (80/20) as mobile phase to afford TB3
as a dark purple powder (510 mg, 62%). 1H NMR (CDCl3,300 MHz): 1.47 (s, 6H), 3.41 (s,
6H), 3.58-3.61 (m, 4H), 3.71-3.74 (m, 4H), 3.86-3.89 (m, 4H), 4.24-4.27 (m, 4H), 6.22 (d,
2H, 3J = 4 Hz), 6.59 s, 2H), 6.93-6.95 (m, 4H), 7.07-7.10 (m, 4H), 7.29 (overlapping with
solvent d, 2H, 3J = 16 Hz), 7.43 (d, 2H, 3J = 16 Hz), 7.85 (d, 2H, 3J = 8.2 Hz). EI-MS, m/z
(%): 1038.1 ([M], 100). Anal. Calcd for C47H46BF2IN2O6S4 (Mr = 1038.85): C, 54.34; H,
4.46 ; N, 2.70; Found: C, 54.17; H, 4.38; N, 2.52.
Scheme S4: Synthesis of compound TB4.
S Sn +S O O OI S O O O
S
Pd2dba3, P(o-tolyl)3 13
Toluene
SO
O
OO
OS
OH
NaH
THF
SO
O O+S
OO O
II2
HgO, benzene
OS
S OO
OH
POCl3
DMF
NB
NFF
I
Toluene, piperidine
pTsOH
O
S
S
O
O
O
H
+ NB
NFF
S S
S S
O
O
O
O
I
O O
TB4
3
9 10 11
12 14
Compound 9 : The purification was carried out by chromatography on flash silica as static
phase with a mixture of ethyl acetate/petroleum ether (50/50) as mobile phase and afforded 9
as an colorless oil. 1H NMR (CDCl3, 200 MHz): 2.45 (s, 3H), 3.35 s, 3H), 3.46-3.50 (m, 2H),
3.56-3.61 (m, 2H), 3.67-3.72 (m, 2H), 4.15-4.20 (m, 2H), 7.34 (d, 2H, 3J = 7.7 Hz), 7.80 (d,
2H, 3J = 8.2 Hz).
2-(2-(2-(2-Methoxyethoxy)ethoxy)ethyl)thiophene (10).
To a slurry of NaH (60 % in paraffine oil, 73 mg, 1.82 mmol) in anhydrous THF (5 mL), 2-
thienylethanol (260 µL, 1.56 mmol) was added under argon. The mixture was stirred at rt for

S8
45 min. Then the tosylate 9 (356 mg, 1.30 mmol) was dropwise added and the reaction
allowed to stir overnight. The reaction mixture at 0 °C was neutralized by a slow addition of 1
M solution of HCl, washed with H2O, extracted with ethyl acetate, dried over MgSO4 and
concentrated under reduced pressure. The purification was carried out by chromatography on
flash silica as static phase with a mixture of petroleum ether/ethyl acetate (8:2) as mobile
phase and afforded 10 (228.0 mg, 76%) as an yellow oil. 1H NMR (300 MHz, (CD3)2CO)
7.23 (dd, 1H, 3J = 5.1 Hz, 4J = 1.2 Hz), 6.93-6.90 (m, 2H), 3.67 (t, 2H, 3J = 6.6 Hz), 3.59-3.55
(m, 6H), 3.48-3.45 (m 2H), 3.28 (s, 3H), 3.06 (t, 2H, 3J = 6.6Hz). 13C NMR (75 MHz, CDCl3)
142.4, 127.4, 126.0, 125.9, 124.2, 72.7, 72.4, 71.2, 71.1, 71.0, 58.8, 31.0. EI-MS, m/z (%):
230.1 (100). Anal. Calcd for C11H18O3S (Mr = 230.32) C, 57.36; H, 7.88. Found: C, 57.01; H,
7.53.
1-(5-(2-(2-(2-Methoxyethoxy)ethoxy)ethyl)thiophen-2-yl)ethanone (11).
To a solution of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethyl)thienyl (2.47g, 10.73 mmol) in
benzene (110 mL) was added in small portions of mercuric oxide (2.44g, 11.27 mmol), and
iodine ( 2.86g, 11.27 mmol) at 0°C. The mixture was stirred for 4h at rt and then filtered
through a Celite pad. The filtrate was poured into water, and the aqueous layer was extracted
with ether. The organic extracts were washed with 5% aqueous Na2S2O3 and dried over
magnesium sulfate. The organic phase was concentrated under reduced pressure. The
purification was carried out by chromatography on flash silica as static phase with a mixture
of petroleum ether and ethyl acetate (6:4) as mobile phase to afford compound 11 (3.35 g,
87%) as a yellow oil. 1H NMR (200 MHz, CDCl3) 7.04 (d, 1H, 3J = 3.3 Hz), 6.54 (d, 1H, 3J =
3.3 Hz), 3.70-3.63 (m, 10H), 3.57-3.53 (m, 2H), 3.88 (s, 3H), 3.07 (t, 2H, 3J = 6.6 Hz). 13C
NMR (75.4 MHz, (CD3)2CO) 149.4, 137.5, 128.1, 126.0, 72.7, 71.9, 71.2, 71.1, 71.0, 58.9,
31.5. EI-MS, m/z (%): 356.0 (100). Anal. Calcd for C11H17IO3S (Mr = 356.22): C, 37.09; H,
4.81. Found: C, 36.90; H, 4.62.
Compound 12 : The crude product is used without further purification. 1H NMR (CDCl3, 300
MHz): 0.40 (s, 9H), 7.24-7.29 (overlapping with solvent m, 2H), 7.66-7.67 (m, 1H).
5-(2-(2-(2-Methoxyethoxy)ethoxy)ethyl)-2,2'-bithiophene (13):
2-trimethyltinthiophene (12) (1.68 mmol), 2-iodo-5-(2-(2-(2-methoxyethoxy)ethoxy)
ethyl)thiophene (0.9 mmol) and tri(o-tolyl)phosphine (67.5µmol) were dissolved in 5 mL of
dry toluene and the solution was degased with argon. Then, [Pd2dba3] (17.5 µmol) in catalytic
amount was added and the reaction mixture was stirred at 110°C for 24 hours under argon
atmosphere. Then, the solution was cooled to room temperature and filtered onto a celite pad.

S9
The organic layer was washed three times with water and concentrated under reduced
pressure. The purification was carried out by chromatography on flash silica as static phase
with a mixture of cyclohexane / ethyl acetate (80/20) as mobile phase to afford compound 11
as a pale yellow oil (177 mg, 63%). 1H NMR (CDCl3, 300 MHz): 3.07 (t, 2H, 3J = 6.8 Hz), 3.85 (s, 3H), 3.53-3.57 (m, 2H), 3.63-
3.76 (m, 8H), 6.74-6.76 (m, 1H), 6.97-7.01 (m, 2H), 7.09-7.11 (m, 1H), 7.16-7.19 (m, 1H).
13C NMR (CDCl3, 50 MHz): 30.7, 59.12, 70.5, 70.7, 71.8, 72.1, 123.2, 123.5, 123.9, 126,
127.7, 135.7, 137.9, 140.8. EI-MS, m/z (%): 312.1 ([M], 100). Anal. Calcd for C13H16O3S2
(Mr = 312.45): C, 57.66; H, 6.45; Found: C, 57.52; H, 6.51.
Compound 14 was prepared according to the general procedure A from 150 mg of 13 (0.476
mmol) and 66.6 µL of POCl3 (0.715 mmol). The purification was carried out by
chromatography on flash silica as static phase with a mixture of petroleum ether/ethyl acetate
(50/50) as mobile phase to give 14 as an yellow oil (91 mg, 56%). 1H NMR (200 MHz,
CDCl3) 9.80 (s, 1H), 7.61 (d, 1H, 3J = 4.0 Hz), 7.13-7.16 (m, 2H), 6.78 (d, 1H, 3J = 3.6 Hz),
3.73-3.60 (m, 8H), 3.54-3.50 (m, 2H), 3.35 (s, 3H), 3.06 (t, 2H, 3J = 6.4 Hz). 13C NMR (75
MHz, CDCl3) 182.5, 147.7, 144.2, 141.1, 137.5, 134.2, 126.6, 126.0, 123.6, 71.9, 71.3, 70.6,
70.4, 59.0, 30.8. EI-MS, m/z (%): 340.1 ([M], 100). Anal. Calcd for C16H20O4S2
(Mr = 340.46): C, 56.44; H, 5.92; Found: C, 56.21; H, 5.74.
TB4 was prepared according to the general procedure B from 55 mg (0.12 mmol) of 3 and 90
mg (0.27 mmol) of 5-(5-(2-(2-(2-methoxyethoxy)ethoxy)ethyl)thiophen-2-yl)thiophene-2-
carbaldehyde. The purification was carried out by chromatography on flash silica as static
phase with a mixture of dichloromethane/ethyl acetate (80/20) as mobile phase to afford TB4
as a dark purple powder. 1H NMR (CDCl3, 300 MHz): 1.47 (s, 6H), 3.10 (t, 4H, 3J = 6.7 Hz),
3.39 (s, 6H), 3.55-3.58 (m, 4H), 3.66-3.69 (m, 12H), 3.75 (t, 4H, 3J = 6.7 Hz), 6.60 (s, 2H),
6.80 (d, 2H, 3J = 3.6 Hz), 7.05-7.14 (m, 8H), 7.30 (overlapping with solvent d, 2H, 3J = 16,0
Hz), 7.45 (d, 2H, 3J = 16.0 Hz), 7.85 (d, 2H, 3J = 8.2 Hz). 13C NMR (CDCl3, 50 MHz): 15.1,
30.9, 59.2, 70.5, 70.7, 71.7, 72.1, 94.8, 118.0, 118.2, 124.1, 124.3, 126.4, 129.0, 129.7, 130.7,
133.6, 134.9, 135.6, 138.3, 139.8, 140.7, 141.5, 141.9, 152.2. EI-MS, m/z (%): 1094.1 ([M],
100). Anal. Calcd for C51H54BF2IN2O6S4 (Mr = 1094.96): C, 55.94; H, 4.97; N, 2.56; Found:
C, 55.78; H, 4.62; N, 2.40.
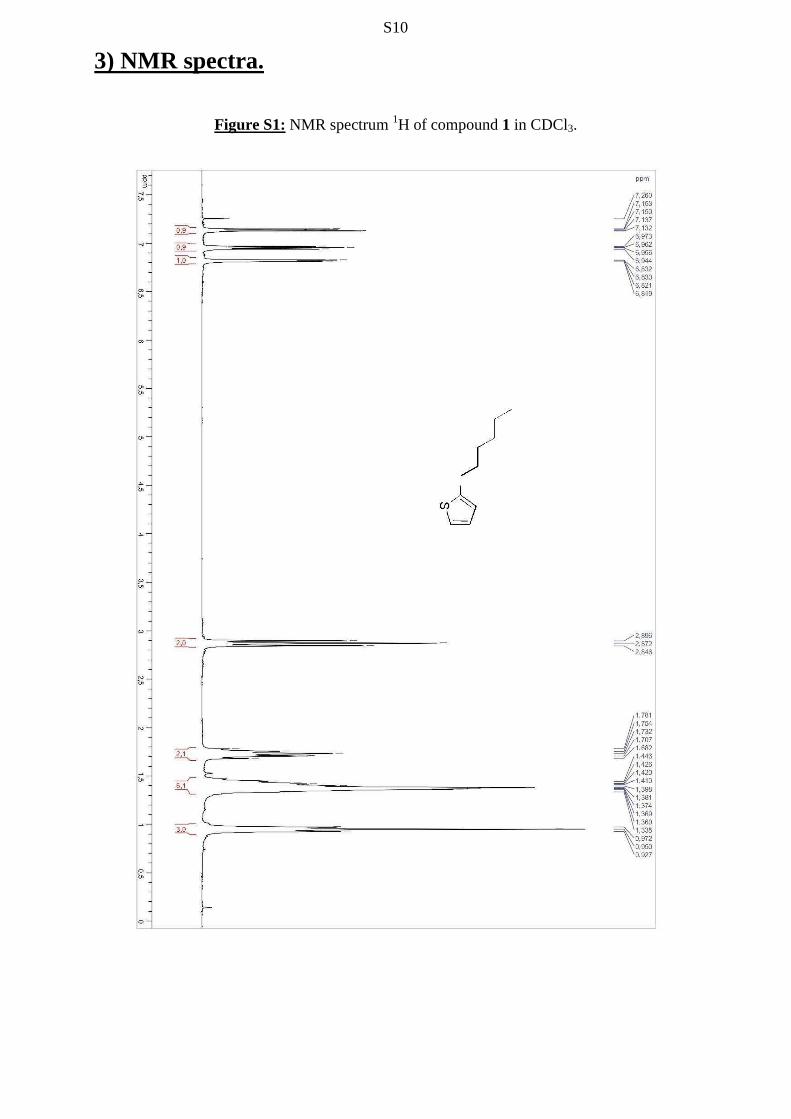
S10
3) NMR spectra.
Figure S1: NMR spectrum 1H of compound 1 in CDCl3.

S11
Figure S2: NMR spectrum 13C of compound 2 in CDCl3.

S12
Figure S3: NMR spectrum 1H of compound TB1 in CDCl3.

S13
Figure S4: NMR spectrum 13C of compound TB1 in CDCl3.

S14
Figure S5: NMR spectrum 1H of compound 4 in CDCl3.

S15
Figure S6: NMR spectrum 1H of compound TB2 in CDCl3.

S16
Figure S7: NMR spectrum 13C of compound TB2 in CDCl3.

S17
Figure S8: NMR spectrum 1H of compound 5 in CDCl3.
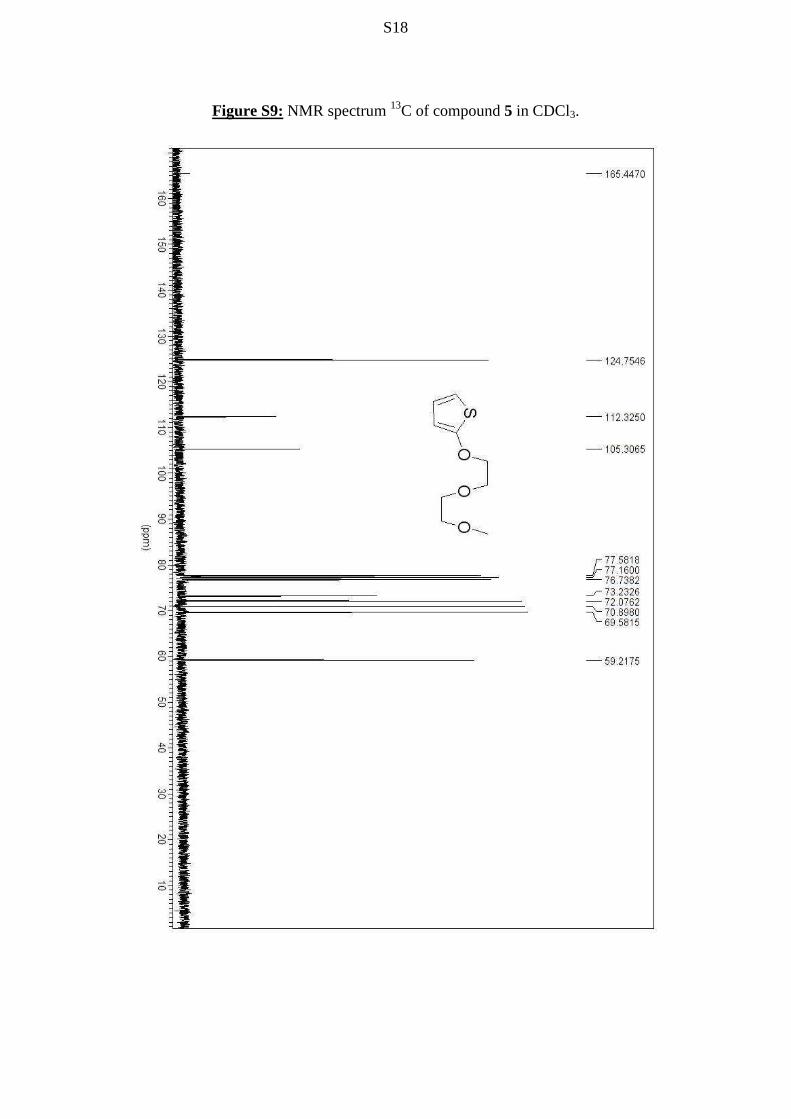
S18
Figure S9: NMR spectrum 13C of compound 5 in CDCl3.

S19
Figure S10: NMR spectrum 1H of compound 6 in CDCl3.

S20
Figure S11: NMR spectrum 13C of compound 6 in CDCl3.

S21
Figure S12: NMR spectrum 1H of compound 7 in (CD3)2CO.

S22
Figure S13: NMR spectrum 13C of compound 7 in (CD3)2CO.

S23
Figure S14: NMR spectrum 1H of compound 8 in CDCl3.

S24
Figure S15: NMR spectrum 13C of compound 8 in CDCl3.

S25
Figure S16: NMR spectrum 1H of compound TB3 in CDCl3.

S26
Figure S17: NMR spectrum 13C of compound TB3 in CDCl3.

S27
Figure S18: NMR spectrum 1H of compound 9 in CDCl3.

S28
Figure S19: NMR spectrum 1H of compound 10 in (CD3)2CO.

S29
Figure S20: NMR spectrum 13C of compound 10 in (CD3)2CO.

S30
Figure S21: NMR spectrum 1H of compound 11 in CDCl3.

S31
Figure S22: NMR spectrum 13C of compound 11 in (CD3)2CO.

S32
Figure S23: NMR spectrum 1H of compound 12 in CDCl3.

S33
Figure S24: NMR spectrum 1H of compound 13 in CDCl3.

S34
Figure S25: NMR spectrum 13C of compound 13 in CDCl3.

S35
Figure S26: NMR spectrum 1H of compound 14 in CDCl3.

S36
Figure S27: NMR spectrum 13C of compound 14 in CDCl3.

S37
Figure S28: NMR spectrum 1H of compound TB4 in CDCl3.

S38
Figure S29: NMR spectrum 13C of compound TB4 in CDCl3.

S39
4) DSC traces for TB n.
DSC. Differential scanning calorimetry analyses were performed on a DSC Q200 apparatus
from TA Instruments. The analyses were carried out under nitrogen at a heating rate of
10°C.min-1.
Figure S30: DSC thermogram for TB1 obtained at a rate of 10°C/min.
Figure S31: DSC thermogram for TB2 obtained at a rate of 10°C/min.

S40
Figure S32: DSC thermogram for TB3 obtained at a rate of 10°C/min.
Figure S33: DSC thermogram for TB4 obtained at a rate of 10°C/min.

S41
5) X-ray crystallographic data for TB 2.
Single crystal of TB2 was mounted in oil on a nylon loop and placed in the nitrogen cold
stream (193 K) of a Rigaku mm007 HF diffractometer equipped with a Rapid II curved
Image-Plate detector and a CuKα rotating anode source with Osmic confocal CMF optics. A
total of 159 images with 5° rotation per image and 60 second exposure per degree of
oscillation were measured according to a ω-scan profile data strategy derived by the
CrystalClear software package.9 Intensities were reduced and merged after empirical
absorption correction using Fs_process within CrystalClear. Resolution limit was set upon
I/σ(I) to 2θ = 108° (0.95 Å). The structure was solved by direct methods (SHELXS-97)10 and
refined on F2 by means of full-matrix least-squares methods (SHELXL-97)10. All non-
hydrogen atoms were refined anisotropically whereas hydrogen atoms were placed at the
calculated positions and refined using a riding model. ORTEP drawings were made using
ORTEP311 as implemented within PLATON12 and packing studies were carried out using
MERCURY.13 See supporting CIF file for further refinement details.
Results for compound TB2: C49 H50 B F2 I N2 S4, Mr= 970.86, dark squared thick plate, 0.26 x
0.25 x 0.07 mm, triclinic, space group P -1 (n° 2), a= 12.0762(8) Å, b= 13.2664(8) Å, c=
15.0653(11) Å, α= 80.999(6) °, β= 71.602(5) °, γ= 83.461(6) °, V= 2256.6(3) Å3, Z=2, ρcalcd=
1.429 g.cm-3, 2θmax = 107.98 °, 19611 measured reflections, 5424 independent, -12 ≤ h ≤ 12, -
12 ≤ k ≤ 13, -15 ≤ l ≤ 15, R(int) = 0.0768, µ= 7.658 mm-1, multi-scan absorption correction,
relative Tmin= 0.385 and Tmax= 1.000, 536 parameters were refined against all reflections, 6
anisotropic parameters restraints were used, R1= 0.094, wR2= 0.1658 (using all data) based
on observed F values, R1= 0.061, wR2= 0.1327 (3376 reflections with I>2σ(I)), ∆ρmin and
ρmax = -0.862 and 0.649 e.Å-3, GOF=1.115 based on F2.
6) Atomic Force Microscopy investigations.
The thin film morphology was investigated by tapping mode atomic force microscopy on the
Nanoscope IV system commercialized by Veeco®.
The investigated substrates, thin films composition and annealing conditions were identical to
those that lead to the optimized performances from solution chloroform, reported in table 1.
These are:
i) TB1:PCBM (1:2), 70°, 20 min, figure S34a
ii) TB2:PCBM (1:0.5), 120°, 20 min, figure S34b
iii) TB3:PCBM (1:2), without annealing, figure S34c
iv) TB4:PCBM (1:1), 80°C, 20 min, figure S34d

S42
Figure S34: Surface topography obtained by tapping mode AFM. (a) TB1:PCBM(1:2) after
70°C, 20 min; (b) TB2:PCBM(1:0.5) after 120°C 20 min; (c) TB3:PCBM(1:2); (d) TB4:PCBM
(1:1) after 80° 20min.
S34a
S34b

S43
S34c
S34d

S44
7) Absorption and Fluorescence Spectra
Table S1. Selected spectroscopic data for the novel dyes.
Dye λabs
(nm) ε (M-1.cm-1)
λem (nm)
Φf @ λexc
nma) τ
(ns)
kr b)
(107 s-1)
knrb)
(107 s-1)
solvent
TB1 667 146000 683 78%
@600nm 3.3 23.6 6.6 THF
TB2 714 104000 743 7.5%
@620nm 0.87 8.6 106 THF
TB3 724 97200 763 2.7%
@680nm 0.68 4 143 THF
TB4 713 98000 749 6.5%
@630nm 0.85 7.6 110 THF
a) Using Rhodamine 6G as reference, λex = 488 nm, Φ= 0.78 in water for TB1,14 and
tetramethoxydiisoindomethene-difluoroborate (φF = 0.51 in methanol) for the others.15
b) Calculated using the following equations : kr = ΦF /τF, knr = (1-ΦF)/ τF, assuming that the
emitting state is produced with unit quantum efficiency.

S45
Figure S35. Absorption (dark line), emission (λexc = 512 nm red line) and excitation (λemi =
680 nm green line) spectra of TB1 (c = 3 x 10-6 M in THF).
Figure S36. Absorption in thin film (red line) and solution in THF (blue line) of TB1.

S46
Figure S37. Absorption (dark line), emission (λexc = 680 nm red line) and excitation (λem =
800 nm green line) spectra of TB3 (c = 3 x 10-6 M in THF).
Figure S38. Absorption in thin film (red line) and solution in THF (blue line) of TB3.

S47
Figure S39. Absorption (blue line), emission (λexc = 630 nm red line) and excitation (λem =
780 nm purple line) spectra of TB4 (c = 3 x 10-6 M in THF).
Figure S40. Absorption in thin film (red line) and solution in THF (blue line) of TB4.

S48
8) Cyclic voltammetry traces and Data
Table S2. Selected electrochemical data for the novel dyes.
Compd ∆∆∆∆Eopt
eV
E1/2ox
V
E1/2red
V
ΕΕΕΕHOMO
eV
ΕΕΕΕLUMO
eV
∆∆∆∆E
eV
TB1 1.60 0.68 (62mV)
1.06 (80mV)
-0.97 (60mV)
-1.87 (irr) -5.46 -3.81 1.65
TB2 1.45
0.64 (64 mV)
0.82 (65mV)
1.47 (76mV)
-0.89 (61mV)
-1.62 (irr) -5.32 -3.86 1.46
TB3 1.42
0.57 (68mV)
0.65 (64mV)
1.2 (81mV)
-0.94 (69mV) -5.30 -3.85 1.45
TB4 1.48 0.63 (64mV)
0.82 (65mV)
-0.93 (64mV)
-5.34 -3.84 1.50
Figure S41. Cyclic voltamogramm of compound TB3 in dichloromethane at rt. 1.5 mM
substrate in 0.10 M Bu4NPF6.
Figure S42. Cyclic voltamogram of compound TB4 in dichloromethane at rt. 1.5 mM
substrate in 0.10 M Bu4NPF6.

S49
9) Device preparation and characterization
Field Effect Transistors elaboration.
Bottom contact field-effect transistors (FETs) were elaborated on commercially available
pre-patterned test structures whose source and drain contacts were composed of a 30 nm thick
gold layer on top of a 10 nm thick Indium Tin Oxide (ITO) layer. A 230 nm thick silicon
oxide was used as gate dielectric and n-doped (3x1017 /cm3) silicon crystal as gate electrode.
The channel length and channel width were 20 µm and 10 mm, respectively. The test
structures were cleaned in acetone and isopropyl alcohol and subsequently for 15 minutes in
an ultra-violet ozone system. Then, hexamethyldisilazane (HMDS) was spin coated (500 rpm
for 5 s and then 4000 rpm for 50 s) under nitrogen ambient and followed by an annealing step
at 130°C for 5 minutes. Finally, 4 mg/ml anhydrous chloroform TBn or TB2:PC61BM
solutions were spin coated (2200 rpm for 180s and 2500 rpm for 120s) to complete the FET
devices. The samples were then left overnight under vacuum (<10-6 mbar) to remove residual
solvent traces. Both, the FET elaboration and characterizations were performed in nitrogen
ambient. The transistor output and transfer characteristics were recorded using a Keithley
4200 semiconductor characterization system. The charge carrier mobility was extracted in the
saturation regime using the usual formalism.

S50
Table S3. Measured charge carrier mobility in the different dyes.
Dye µh (cm2/Vs) µe (cm2/Vs) TB1 1x10-4 / TB2 1x10-3 1x10-3 TB3 1x10-8 / TB4 6x10-7 /
Figure S43. Charge carrier mobilities calculated from FET’s transfer characteristics using
different TB2:PC61BM weight ratio.
1.E-07
1.E-06
1.E-05
1.E-04
1.E-03
1.E-02
1.E-01
0 1 2 3 4 5
Mo
bilit
y (c
m2 /
V.s)
TB:PC61BM wt. ratio
holes
electrons
1:0 1:0.8 1:4 0:1
10-1
10-2
10-3
10-4
10-5
10-6
10-7
Photovoltaic devices elaboration and characterization.
Bulk heterojunction devices were elaborated using the different synthesized molecules as
electron donor and PC61BM as electron acceptor. The standard device structure was the
following: ITO/PEDOT:PSS(~40 nm)/TBn:PC61BM/Al(~120 nm). Indium Tin Oxide coated
glass with a surface resistance lower than 20 Ω/sq was used as transparent substrate.
Substrates were cleaned sequentially by ultrasonic treatments in acetone, isopropyl alcohol,
and deionized water. After an additional cleaning for 30 minutes under ultra-violet generated
ozone, a highly conductive polyethylene dioxythiophene: polystyrene-sulphonate
PEDOT:PSS was spin coated (1500 rpm: 40 nm) from an aqueous solution and dried for 30
minutes at 120°C under vacuum before being transferred to the nitrogen filled glove box. The

S51
chloroform, chlorobenzene or chloroform/chlorobenzene molecule:PC61BM solutions were
stirred for at least 24 hours at 50°C before spin-coating. An extra stirring for 30 minutes at
100°C was added just before the active layer deposition. The molecule concentration of the
solution was varied from 5 mg/ml for chloroform solutions up to 40 mg/ml for chlorobenzene
solutions in order to obtain different active layer thicknesses and morphologies. The relative
TBn:PC61BM weight ratio was also varied from 1:0.2 to 1:3. The active layer spin coating
conditions were the same for all solvents: a first 180 seconds step (speed: 2200 rpm,
acceleration: 600 rpm/s) followed by a second 120 seconds step (speed: 2500 rpm,
acceleration: 600 rpm/s). Finally, a 120 nm thick aluminum layer was thermally evaporated
and used as cathode. The device active area was 9 mm2, while each sample included four
independent diodes. With the most promising molecule, an extra series of devices was
elaborated with and an additional Ca (20 nm) layer between the active layer and the
aluminum cathode. Current versus Voltage (J-V) characteristics were measured using a source
measurement unit Keithley 2400 under darkness and under AM1.5G (100 mW/cm2)
illumination. The standard illumination was provided by an Oriel 150 W (filtered Xe lamp)
solar simulator and the illumination power was calibrated using a THORLABS optical power
meter. External Quantum Efficiency (EQE) measurements were performed using a focused
light beam (on a standard silicon reference cell or on the solar cell under study) from a 250 W
halogen lamp passing through a filter wheel. The photovoltaic cells elaboration after substrate
preparation and the characterizations were performed in nitrogen ambient except the EQE
measurements.

S52
Table S4. Survey of the different experimental conditions tested for the photovoltaic devices
and the corresponding measurements (the results with a bold frame are included in the main
text). Dye TBn:PCBM
ratio Acceptor Voc (V) Jsc
(mA/cm2) FF (%)
PCE (%) Annealing Solvent Concentration
(Dye/ml)
d (nm)
TB1 1:0.8 PC61BM 0.73 3.6 27 0.7 70°C, 10m CHCl3 5mg/ml 110±10
TB1 1:1 PC61BM 0.68 3.4 28 0.6 70°C, 10m CHCl3 5mg/ml 110±10
TB1 1:2 PC61BM 0.76 5.8 31 1.4 70°C, 20m CHCl3 5mg/ml 110±10
TB1 1:3 PC61BM 0.74 4.9 32 1.2 70°C, 10m CHCl3/CB (1/1) 5mg/ml
TB1 1:2 PC61BM 0.74 4.5 31 1.0 70°C, 10m CB 10mg/ml
TB2 1:0.8 PC61BM 0.60 7.7 44 2.1 100°C, 20m CHCl3 5mg/ml 110±10
TB2 1:1.2 PC61BM 0.60 3.2 29 0.6 100°C, 20m CHCl3 5mg/ml 110±10
TB2 1:2 PC61BM 0.65 5.4 29 1.0 - CHCl3/CB (1/1) 8mg/ml
TB2 1:0.8 PC61BM 0.72 11.9 49 4.3 100°C, 10m CB 20mg/ml
TB2 1:0.8 PC61BM 0.74 14.3 42 4.5 100°C, 10m CB 40mg/ml 165±10
*TB 2 1:0.2 PC61BM 0.77 11.4 41 3.6 100°C, 10m CB 40mg/ml 165±10
*TB 2 1:0.5 PC61BM 0.70 14.3 47 4.7 100°C, 10m CB 40mg/ml 165±10
*TB 2 1:0.8 PC61BM 0.59 10.9 42 2.7 100°C, 10m CB 20mg/ml
*TB 2 1:0.2 PC71BM 0.73 9.3 39 2.7 100°C, 10m CB 40mg/ml 165±10
TB3 1:2 PC61BM 0.56 5.1 30 0.9 - CHCl3 5mg/ml 110±10
TB3 1:0.8 PC61BM 0.51 4.1 31 0.6 80°C, 10m CB 20mg/ml
TB3 1:1 PC61BM 0.50 4.7 32 0.7 80°C, 10m CB 20mg/ml
TB3 1:0.8 PC61BM 0.50 3.0 31 0.5 80°C, 10m CB 40mg/ml 165±10
TB3 1:1 PC61BM 0.51 3.3 33 0.6 80°C, 10m CB 40mg/ml 165±10
TB4 1:1 PC61BM 0.55 8.5 32 1.5 80°C, 20 m CHCl3 5mg/ml 110±10
TB4 1:1 PC61BM 0.48 6.1 30 0.9 80°C, 20 m CHCl3/CB (1/1) 5mg/ml
TB4 1:0.8 PC61BM 0.51 5.0 30 0.8 - CB 20mg/ml
TB4 1:0.8 PC61BM 0.57 2.3 30 0.4 - CB 40mg/ml 165±10
*Ca(20 nm)/Al(120 nm) cathode and annealing before cathode deposition

S53
10) Additional References
1 Lipset, F. R. Prog. Dielectr. 1967, 7, 217. 2 Olmsted, J. III, J. Phys. Chem. 1979, 83, 2581 3 S. Pu and al., J. Mol. Struc. 2009, 921, 89-100. 4 N. Leclerc and al. Chem. Mater., 2005, 17, 502-513. 5 T. Bura, R. Ziessel Org.Lett. 2011, 13, 3072-3075. 6 P. Frère and al. J. Org. Chem., 2003, 68, 7254-7265. 7 Rousseau, T.; Cravino, A.; Bura, T.; Ulrich, G.; Ziessel, R.; Roncali, J. Chem. Commun.
2009, 1673-1675. 8 Hou. Q. and al. J. Mater. Chem. 2002, 12, 2887-2892. 9 CrystalClear-SM Expert 2.0 r4 (Rigaku, 2009). 10 Sheldrick, G.M. (2008). Acta Cryst. A64, 112-122; Welter, R. Acta Cryst. 2006, A62,
s252.. 11 ) Burnett, Michael N. and Johnson, Carroll K.(1996). ORTEP-III: Oak Ridge Thermal
Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National
Laboratory Report ORNL-6895. 12 Spek, A. L. J. Appl. Cryst. 2003, 36, 7-13. 13 Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R.,
Towler, M. & van de Streek, J. J.Appl. Cryst. 2006, 39, 453-457. 14 Olmsted III, J. J. Phys. Chem. 1979, 83, 2581-2584. 15 Ulrich, G. ; Goeb, S. ; De Nicola, A. ; Retailleau, P. ; Ziessel R. Synlett. 2007, 1517-1520.

















![INSTITUTIONAL REPOSITORY SERVICES IN AZERBAIJAN… · Institutional Repositories ! ... Supporting education through learning materials ! ... Other Collections ! Personal Pages [57]](https://img.pdfslide.net/doc/110x75/5af73fb57f8b9a9e59907320/institutional-repository-services-in-azerbaijan-repositories-supporting.jpg)











