Embed Size (px)
Citation preview

Supporting Information
© Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006

1
Supporting Information
Enantioselective Cyclopropanation with TADDOL-Derived Phosphate Ligands
Arnaud Voituriez and André B. Charette*
Département de Chimie, Université de Montréal, P.O. Box 6128, Station Downtown, Montréal, QC, H3C 3J7 (Canada)
Tel.: (+1) 514-343-2432
Fax: (+1) 514-343-5900
E-mail: [email protected]
General: All non-aqueous reactions were run under an inert atmosphere (argon) with rigid
exclusion of moisture from reagents and glassware by using standard techniques for manipulating
air-sensitives compounds. All glassware was stored in the oven and/or was flame-dried prior to use
under an inert atmosphere. Anhydrous solvents were obtained either by filtration through drying
columns (THF, Et2O, CH2Cl2 and toluene) on a GlassContour system (Irvine, CA) or by distillation
over CaH2 (DCE). Analytical thin-layer chromatography (TLC) was performed on precoated, glass-
backed silica gel (Merk 60 F254). Visualization of the developed chromatogram was followed by
UV absorbance, aqueous cerium molybdate or aqueous potassium permanganate. Flash column
chromatography was performed using 230-400 mesh silica (EM Science or Silicycle) of the
indicated solvent system according to standard techniques. Infrared spectra were taken on a Perkin
Elmer Spectrum One FTIR and are reported in reciprocal centimeters (cm-1). Nuclear magnetic
resonance spectra (1H, 13C, 31P) were recorded either on Bruker AV 300, AMX 300, AV 400 or
ARX 400 spectrometers. Chemical shifts for 1H NMR spectra are recorded in parts per million from
tetramethylsilane with the solvent resonance as the internal standard (chloroform, δ, 7.24 ppm).
Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q =
quartet, m = multiplet and br = broad), coupling constant in Hz and integration. Chemical shifts for 13C NMR spectra are recorded in parts per million from tetramethylsilane using the central peak of
CDCl3 (77.16 ppm) as the internal standard. Optical rotations were determined with a Perkin-Elmer
341 polarimeter at 589 nm. Data are reported as follows: [α]Dtemp, concentration (c in g/100 mL)
and solvent. High resolution mass spectra were performed on a LC-MSD-Tof instrument from
Agilent Technologies. Sodium adducts [M+Na]+ was used for empirical formula confirmation.

2
Analytical Supercritical Fluid Chromatography was performed on a Berger SFC Analytical
Instrument equipped with a diode array UV detector. Data are reported as follows: column type,
temperature, pressure, eluent, flow rate, retention time (tr).
Diethylzinc was purchased neat from Akzo Nobel and used without purification. All others starting
materials were purchased from Aldrich or Alfa Aesar. Phosphorus trichloride, triethylamine, DBU,
diiodomethane and 3-hydroxypropionitrile were distilled prior to use. Unless otherwise stated, the
other commercial reagents were used without purification. Racemic samples for SFC analysis were
prepared using achiral cyclopropanating reagents developed in our group.[5],[13a] The TADDOLs 1a-
e were synthesized according to the literature procedure.[9]
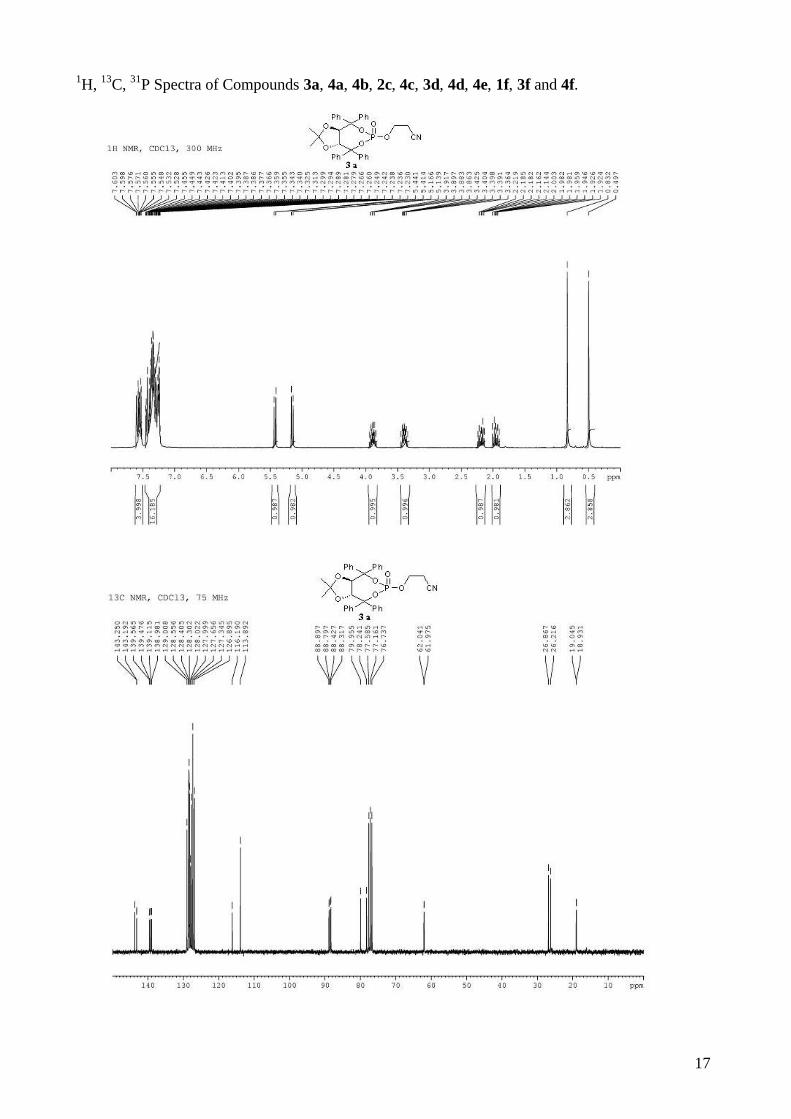
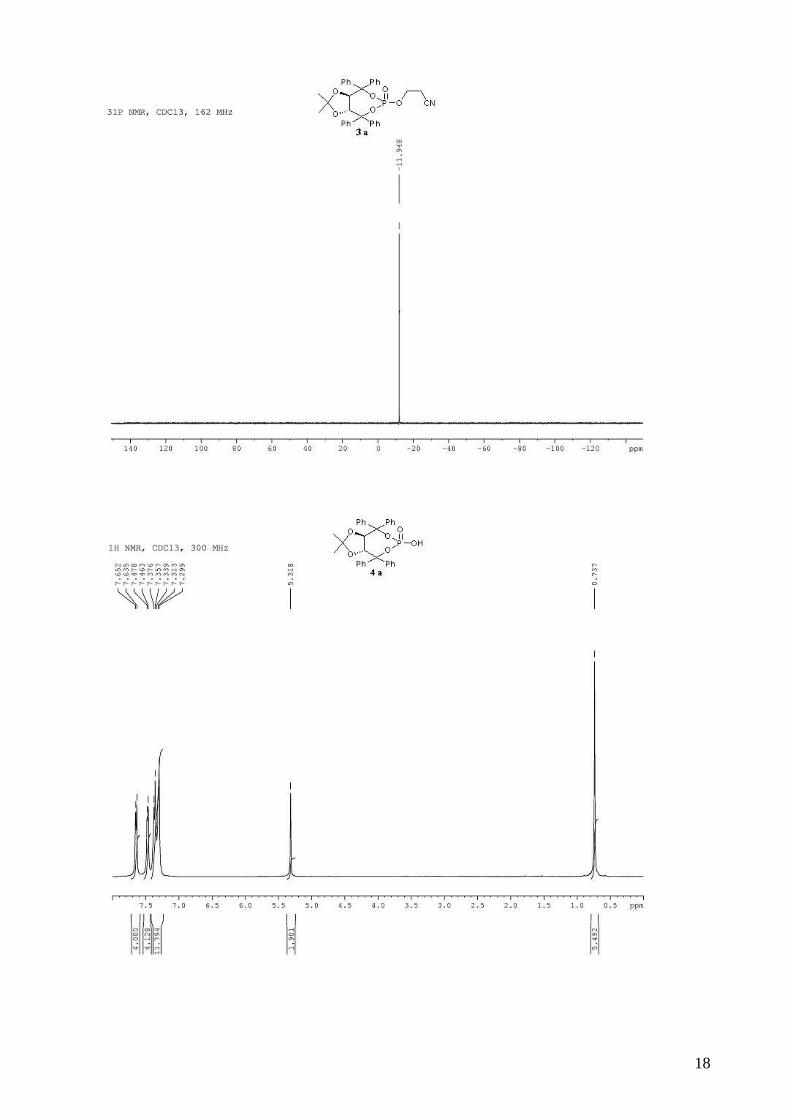
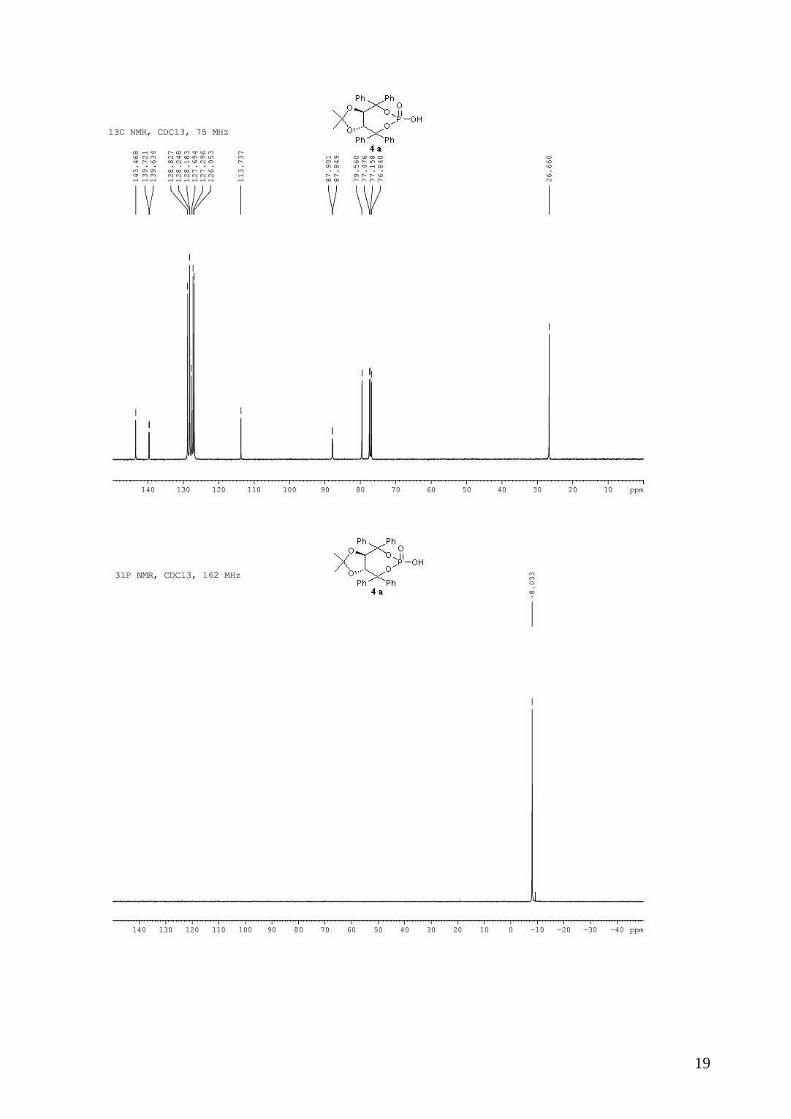
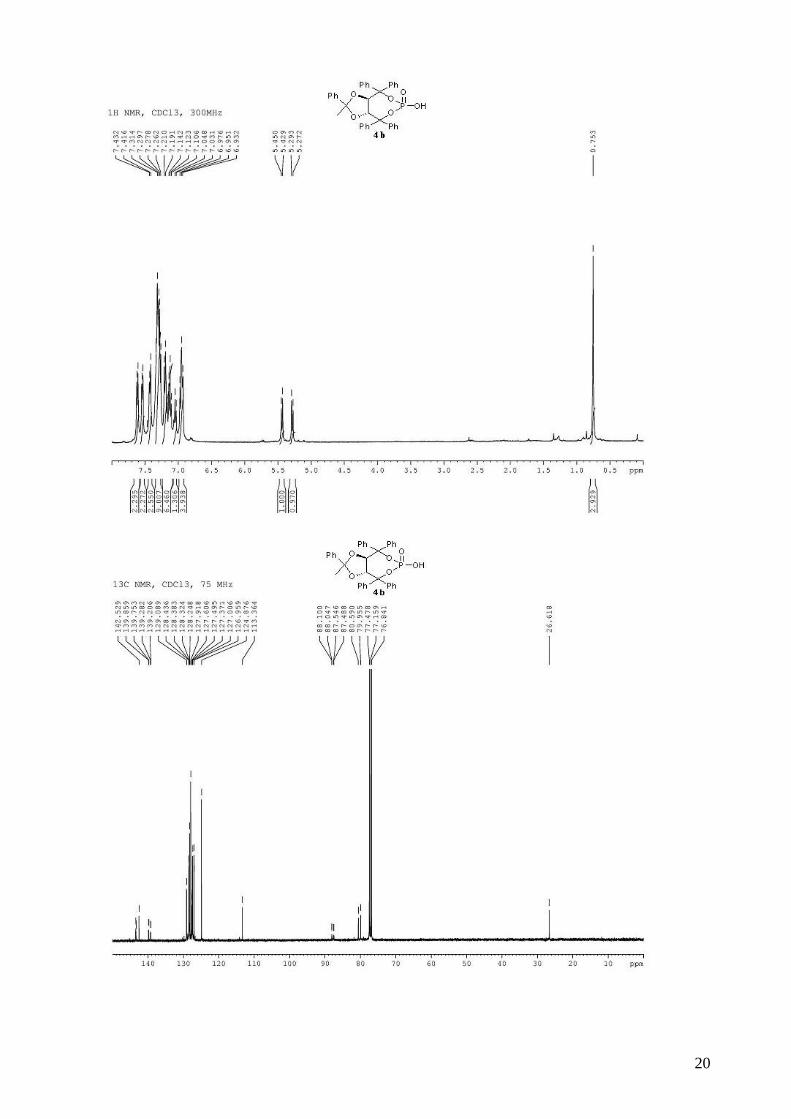
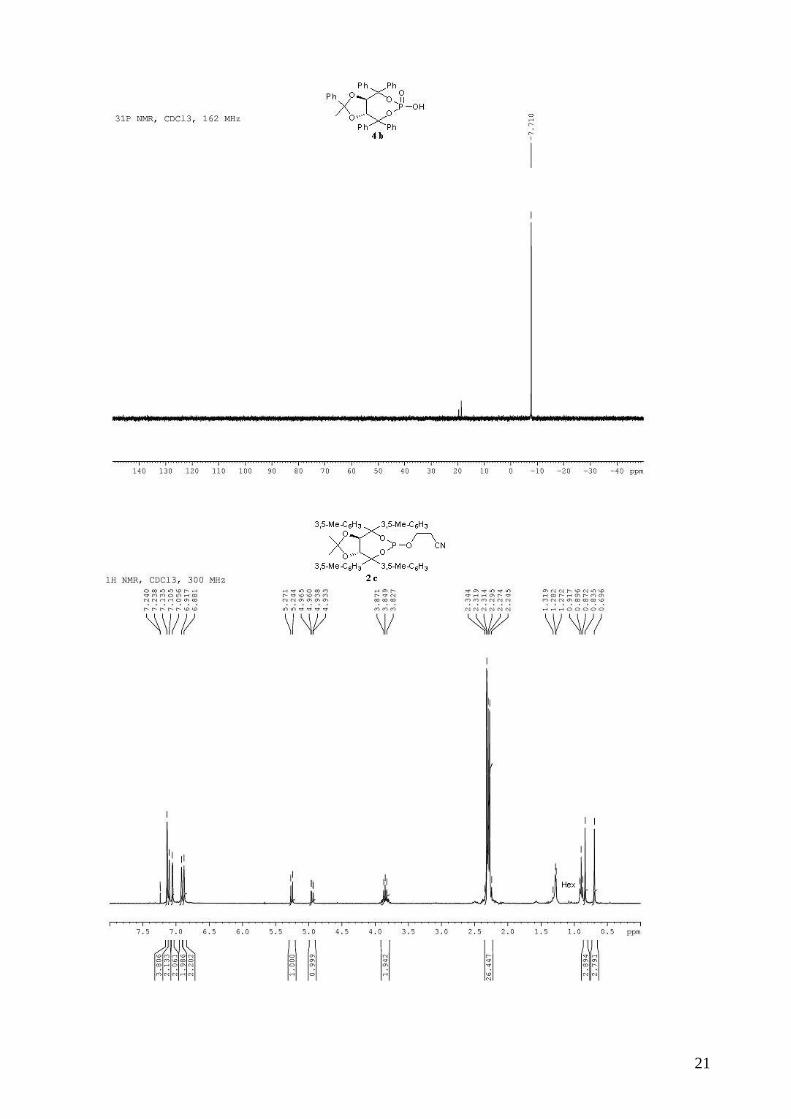
(3aR,8aR)-2-methyl-2,4,4,8,8-pentaphenyltetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-ol 6-oxide (4b). To a stirred solution of (R,R)-(Ph,Me)-TADDOL-
(Ph) 1b (529 mg, 1.00 mmol) and triethylamine (472 µL, 3.40 mmol, 3.4 equiv) in dry THF (5 mL)
at 0 °C was added dropwise PCl3 (92 µL, 1.05 mmol, 1.05 equiv). The resulting mixture was stired
at 0 °C for 30 min. The 3-hydroxypropionitrile (75 µL, 1.1 mmol, 1.1 equiv) in dry THF (5 mL)
was then added dropwide via cannula. The reaction mixture was allowed to warm to room
temperature and stirred for 2 h. The reaction mixture was diluted with Et2O and the
triethylammonium chloride salts were filtered through a celite pad. The solvent was removed in
vacuo. The obtained light yellow solid was directly used without purification in the oxidation step.
To the crude phosphite in CH2Cl2 (10 mL) was added 30% aqueous H2O2 (700 µL, 6.2 mmol, 6.2
equiv). The biphasic mixture was stirred vigorously for 30 min and then quenched by the addition
of 20 mL of saturated aqueous NaHCO3. The mixture was extracted with CH2Cl2 and the combined
organic extracts were washed with saturated aqueous NaCl, dried (MgSO4), filtered and
concentrated. The purification by flash chromatography failed. The crude product was used without
purification. To a stirred solution of the crude phosphate (1.00 mmol) in dry CH2Cl2 (10 mL) was
added, dropwise at room temperature, DBU (150 µL, 1.0 mmol, 1.0 equiv). The solution was stirred
5 min at room temperature and when the reaction was complete by TLC, AcOH (50 µL) was added,
followed by H2O (10 mL). The organic layer was then washed two times by a 0.3M HCl solution,
saturated aqueous NaCl, and dried (MgSO4), filtered and concentrated. The resulting light yellow
solid was recrystallized (CH2Cl2/ hexane) and dried several days on vacuum pump (442 mg, 75%
for three steps), and used directly in asymmetric cyclopropanation. mp = 130-132 °C; 1H NMR
(400 MHz, CDCl3) δ 7.66-7.58 (m, 2H, H-Ph), 7.56-7.52 (m, 2H, H-Ph), 7.46-7.40 (m, 2H, H-Ph),
7.38-7.24 (m, 8H, H-Ph), 7.23-7.10 (m, 6H, H-Ph), 7.08-7.02 (m, 1H, H-Ph), 7.00-6.92 (m, 4H, H-
Ph), 5.44 (d, J = 8.3 Hz, 1H, -CH-CPh2-), 5.28 (d, J = 8.3 Hz, 1H, -CH-CPh2-), 0.75 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 143.5, 143.4, 143.3, 142.5, 139.8 (d, JP-C = 10.6 Hz), 139.2 (d, JP-C

3
= 7.6 Hz), 129.1, 128.4, 128.4, 128.3, 128.2, 127.9, 127.9, 127.6, 127.5, 127.4, 127.0, 127.0, 124.9,
113.4, 88.1 (d, JP-C = 5.3 Hz), 87.5 (d, JP-C = 5.8 Hz), 80.6, 80.0, 26.6; 31P NMR (162 MHz,
CDCl3) δ -7.09; IR: νmax = 3060, 2987, 2927, 1600, 1494, 1264, 1155, 1009, 997, 944, 737, 694
cm-1; HRMS (ESI) Calcd. For C36H31O6NaP [M+Na]+: 613.1750. Found: 613.1747.
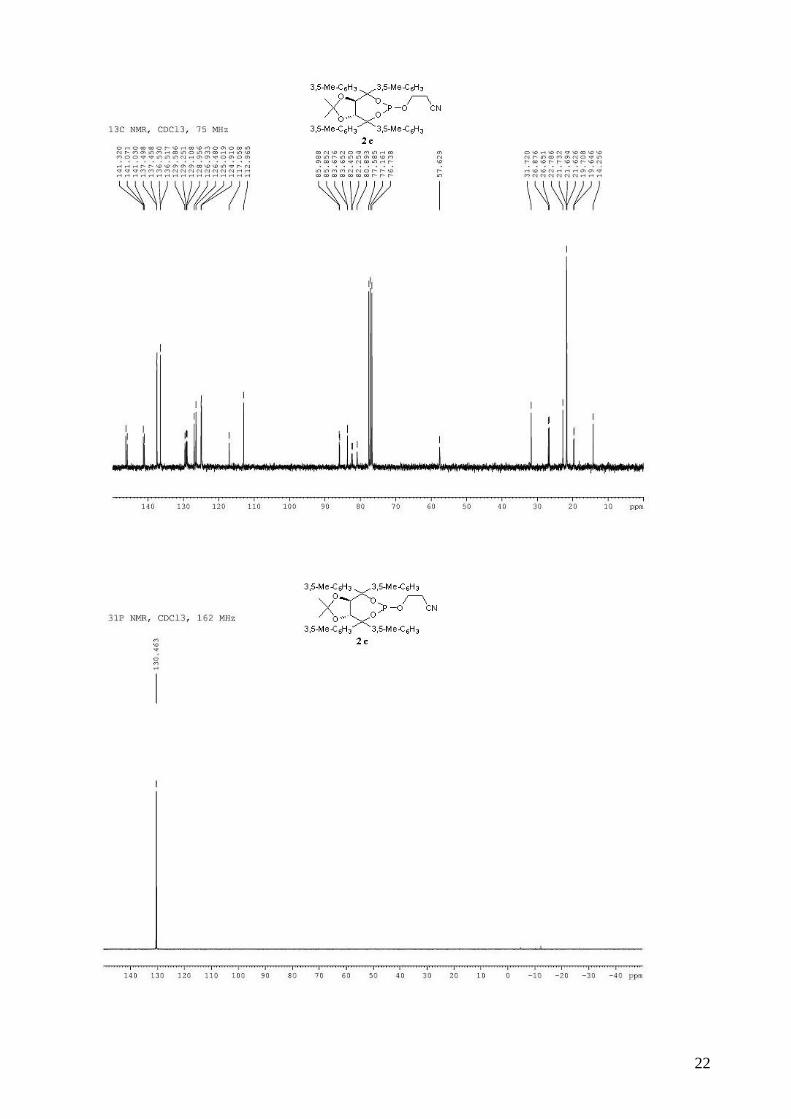
3-{[(3aR,8aR)-4,4,8,8-tetrakis(3,5-dimethylphenyl)-2,2-dimethyltetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-yl]oxy}propanenitrile (2c). To a stirred solution of (R,R)-(Me,Me)-
TADDOL-(3,5-Me-C6H3) 1c (1.29 g, 2.23 mmol) and triethylamine (1.05 mL, 7.58 mmol, 3.4
equiv) in dry THF (10 mL) at 0 °C was added dropwise PCl3 (204 µL, 2.34 mmol, 1.05 equiv). The
resulting mixture was stired at 0 °C for 1 h. The 3-hydroxypropionitrile (167 µL, 2.45 mmol, 1.1
equiv) in dry THF (10 mL) was then added dropwide via cannula. The reaction mixture was
allowed to warm to room temperature and stirred overnight. The reaction mixture was diluted with
Et2O and the triethylammonium chloride salts were filtered through a celite pad. The solvent was
removed in vacuo. The obtained light yellow solid was purified by flash chromatography (hexane /
Et2O / TEA: 60 / 40 / 1) afforded 1.04 g of 2c (69%) of a white solid. The purification at this step of
the synthesis of the ligand was possible, but the compounds were easier to handle and purify after
the oxidation step. Rf 0.70 (hexane / Et2O: 60 / 40); 1H NMR (300 MHz, CDCl3) δ 7.13 (s, 4H, H-
Ar), 7.10 (s, 2H, H-Ar), 7.05 (s, 2H, H-Ar), 6.91 (s, 2H, H-Ar), 6.88 (s, 2H, H-Ar), 5.25 (d, J = 8.1
Hz, 1H, -CH-CAr2-), 4.94 (d, J = 8.2 Hz, 1H, -CH-CAr2-), 3.90-3.80 (m, 2H, -OCH2CH2CN), 2.35-
2.20 (m, 26H, -OCH2CH2CN and 8 x CH3), 0.83 (s, 3H, CH3), 0.69 (s, 3H, CH3); 13C NMR (75
MHz, CDCl3) δ 146.3, 145.9 (d, JP-C = 1.9 Hz), 141.3, 141.0, 141.0, 137.5, 137.5, 136.5 (d, JP-C =
1.0 Hz), 129.6, 129.3, 129.1, 129.0, 126.9, 126.5, 125.0, 124.9, 117.1, 113.0, 85.9 (d, JP-C = 10.2
Hz), 83.6 (d, JP-C = 1.8 Hz), 82.3 (d, JP-C = 14.7 Hz), 80.9, 57.7, 26.7 (d, JP-C = 16.9 Hz), 21.7, 21.7,
21.6, 19.6 (d, JP-C = 4.7 Hz); 31P NMR (162 MHz, CDCl3) δ 130.46; IR: νmax = 2917, 2253 (CN),
1599, 1455, 1380, 1265, 1216, 1164, 1038, 981, 854, 741 cm-1.
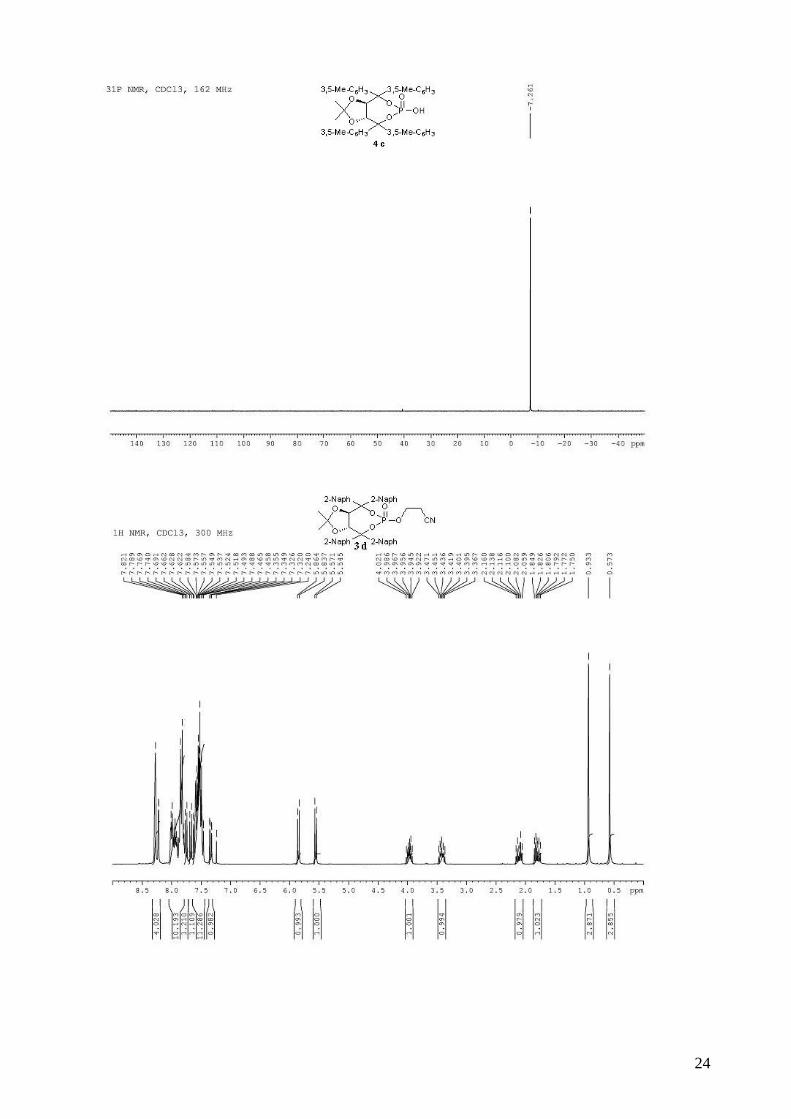
(3aR,8aR)-4,4,8,8-tetrakis(3,5-dimethylphenyl)-2,2-dimethyltetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-ol 6-oxide (4c).
To the phosphine 2c (995 mg, 1.47 mmol) in CH2Cl2 (14 mL) was added 30% aqueous H2O2 (750
µL, 6.62 mmol, 4.5 equiv). The biphasic mixture was stirred vigorously for 45 min and then
quenched by the addition of 100 mL of saturated aqueous NaHCO3 solution. The mixture was
extracted with CH2Cl2 and the combined organic extracts were washed with saturated aqueous
NaCl, dried (MgSO4), filtered and concentred. To a stirred solution of crude 3c (1.02 g, 1.47
mmol) in dry CH2Cl2 (18 mL) was added, dropwise at room temperature, DBU (231 µL, 1.54

4
mmol, 1.05 equiv). The solution was stirred 30 min at room temperature and when the reaction was
complete by TLC, AcOH (85 µL) was added, followed by H2O (20 mL). The organic layer was
then washed two times by a 0.3M solution of HCl, saturated aqueous NaCl, and dried (MgSO4),
filtered and concentrated. The resulting white solid was dried several days on vacuum pump (780
mg, 83% for two steps), and can be used directly in asymmetric tests. mp = 158-160 °C; 1H NMR
(400 MHz, CDCl3) δ 7.16 (s, 4H, H-Ar), 7.00 (s, 4H, H-Ar), 6.91 (s, 2H, H-Ar), 6.84 (s, 2H, H-Ar),
5.17 (s, 2H, -CH-CAr2-), 2.27 (s, 12H, 4 x CH3), 2.21 (s, 12H, 4 x CH3), 0.69 (s, 6H, 2 x CH3); 13C
NMR (100 MHz, CDCl3) δ 143.3, 139.7 (d, JP-C = 9.5 Hz), 137.3, 136.4, 129.9, 129.1, 126.4,
124.8, 113.8, 88.0 (d, JP-C = 6.4 Hz), 79.9, 26.8, 21.6, 21.5; 31P NMR (162 MHz, CDCl3) δ -7.26;
IR: νmax = 2988, 2918, 1603, 1456, 1371, 1261, 1235, 1163, 1013, 924, 854, 749 cm-1; HRMS
(ESI) Calcd. for C39H45O6NaP [M+Na]+: 663.2845. Found: 663.2854; [α]D20 = -199.7 (1.35,
CHCl3).
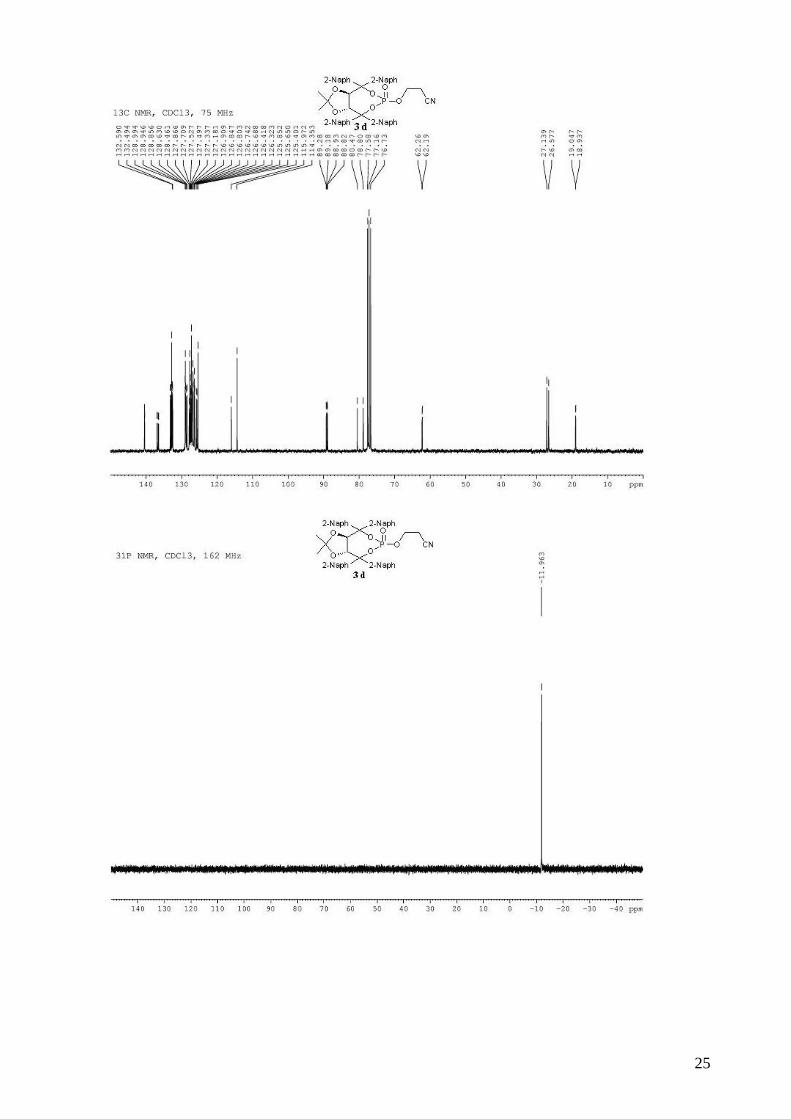
3-{[(3aR,8aR)-2,2-dimethyl-4,4,8,8-tetra-2-naphthyl-6-oxidotetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-yl]oxy}propanenitrile (3d). To a stirred solution of (R,R)-(Me,Me)-
TADDOL-(2-naphthyl) 1d (3.00 g, 4.50 mmol) and triethylamine (2.12 mL, 15.3 mmol, 3.4 equiv)
in dry THF (19 mL) at 0 °C was added dropwise PCl3 (412 µL, 4.72 mmol, 1.05 equiv). The
resulting mixture was stired at 0 °C for 1 h. The 3-hydroxypropionitrile (338 µL, 4.95 mmol, 1.1
equiv) in dry THF (19 mL) was then added dropwide via cannula. The reaction mixture was
allowed to warm to room temperature and stirred for 2 h. The reaction mixture was diluted with
Et2O and the triethylammonium chloride salts were filtered through a celite pad. The solvent was
removed in vacuo. The obtained light yellow solid 2d was directly used without purification in the
oxidation step. To the crude phosphine in CH2Cl2 (40 mL) was added 30% aqueous H2O2 (2.75 mL,
24.3 mmol, 5.4 equiv). The biphasic mixture was stirred vigorously for 20 min and then quenched
by the addition of 50 mL of saturated aqueous NaHCO3 solution. The mixture was extracted with
CH2Cl2 and the combined organic extracts were washed with saturated aqueous NaCl, dried
(MgSO4), filtered and concentrated. Purification by flash chromatography (hexane / Et2O: 80 / 20 to
pure Et2O) afforded 3.08 g (88% for two steps) of compound 3d as a white solid. Rf 0.50 (Et2O
pure); mp = 124-126 °C; 1H NMR (300 MHz, CDCl3) δ 8.30-8.20 (m, 4H, H-Ar), 8.04-7.78 (m,
10H, H-Ar), 7.75 (d, J = 8.8 Hz, 1H, H-Ar), 7.68 (d, J = 8.8 Hz, 1H, H-Ar), 7.65-7.44 (m, 11H, H-
Ar), 7.34 (dd, J = 8.8 Hz, 1.8 Hz, 1H, H-Ar), 5.85 (d, J = 8.0 Hz, 1H, -CH-CAr2-), 5.56 (d, J = 8.0
Hz, 1H, -CH-CAr2-), 4.00-3.92 (m, 1H, -OCHHCH2CN), 3.47-3.36 (m, 1H, -OCHHCH2CN), 2.15-
2.02 (m, 1H, -OCH2CHHCN), 1.86-1.75 (m, 1H, -OCH2CHHCN), 0.93 (s, 3H, CH3), 0.57 (s, 3H,
CH3); 13C NMR (75 MHz, CDCl3) δ 140.5, 140.4, 140.4, 140.3, 136.9 (d, JP-C = 7.0 Hz), 136.5 (d,

5
JP-C = 9.9 Hz), 133.2, 133.0, 132.8, 132.8, 132.6, 132.6, 132.5, 129.0, 128.9, 128.9, 128.6, 128.5,
127.9, 127.7, 127.5, 127.5, 127.3, 127.2, 126.9, 126.8, 126.8, 126.7, 126.7, 126.4, 126.3, 125.8,
125.6, 125.4, 116.0, 114.3, 89.2 (d, JP-C = 7.4 Hz), 88.9 (d, JP-C = 8.2 Hz), 80.5, 78.8, 62.2 (d, JP-C =
4.9 Hz), 27.1, 26.6, 19.0 (d, JP-C = 8.3 Hz); 31P NMR (162 MHz, CDCl3) δ -11.96; IR: νmax =
3057, 2990, 2935, 2255 (CN), 1599, 1505, 1383, 1355, 1287, 1163, 1103, 1000, 949, 733 cm-1;
HRMS (ESI) Calcd. For C50H40NO6NaP [M+Na]+: 804.2486. Found: 804.2489; [α]D20 = -258.9
(2.44, CHCl3).
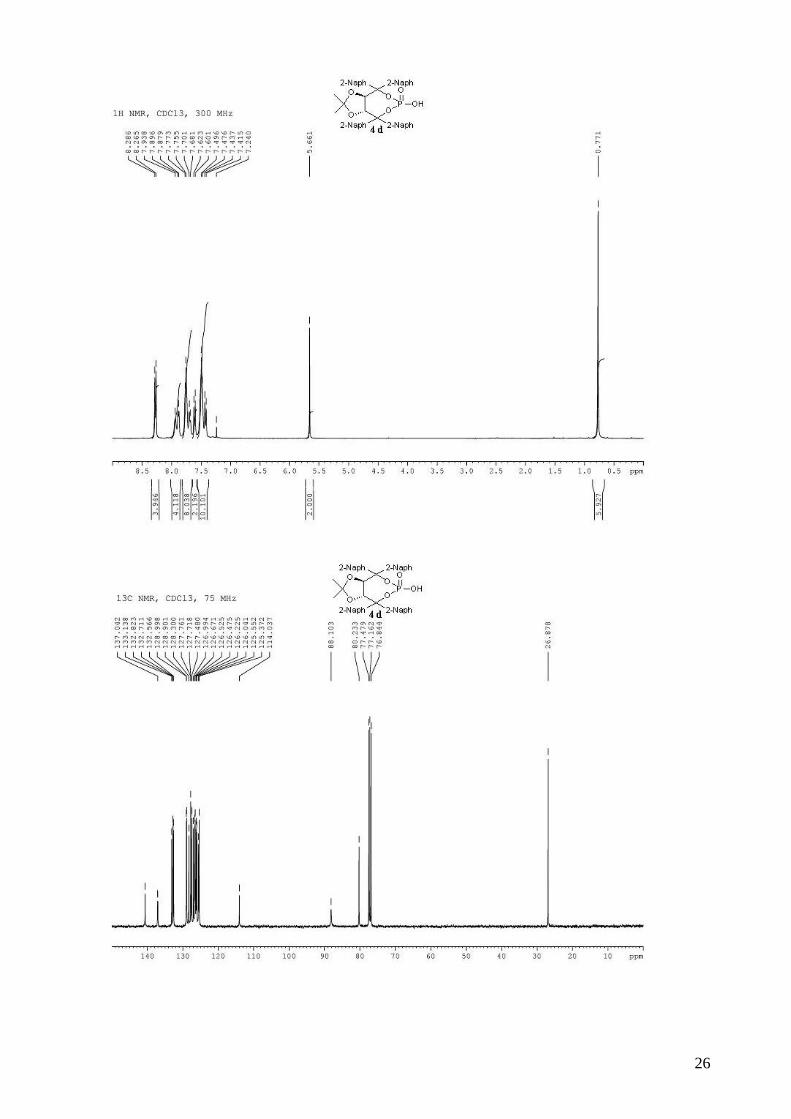
(3aR,8aR)-2,2-dimethyl-4,4,8,8-tetra-2-naphthyltetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-ol 6-oxide (4d). To a stirred solution of 3d (5.37 g, 6.87 mmol) in dry
CH2Cl2 (80 mL) was added, dropwise at room temperature, DBU (1.08 mL, 7.21 mmol, 1.05
equiv). The solution was stirred 30 min at room temperature and when the reaction was complete
by TLC, AcOH (0.4 mL) was added, followed by H2O (80 mL). The organic layer was then washed
two times by a 0.3M HCl solution, saturated aqueous NaCl, and dried (MgSO4), filtered and
concentrated. The resulting white solid was dried several days on vacuum pump (4.74g, 95%), and
can be used directly in asymmetric tests. mp = 186-188 °C; 1H NMR (400 MHz, CDCl3) δ 8.32-
8.23 (d, J = 8.3 Hz, 4H, H-Ar), 7.98-7.86 (m, 4H, H-Ar), 7.80-7.66 (m, 8H, H-Ar), 7.65-7.58 (m,
2H, H-Ar), 7.64-7.38 (m, 10H, H-Ar), 5.66 (s, 2H, -CH-CAr2-); 0.77 (s, 6H, 2 x CH3); 13C NMR
(100 MHz, CDCl3) δ 140.6, 137.1 (d, JP-C = 9.1 Hz), 133.1, 132.8, 132.7, 132.6, 129.0, 128.9,
128.3, 127.8, 127.7, 127.5, 127.0, 126.7, 126.5, 126.5, 126.2, 126.0, 125.5, 125.4, 114.0, 88.1, 80.2,
26.9; 31P NMR (162 MHz, CDCl3) δ -7.24; IR: νmax = 3057, 2989, 2934, 1598, 1506, 1372, 1236,
1163, 1007, 737 cm-1; HRMS (ESI) Calcd. for C47H37O6NaP [M+Na]+: 751.2219. Found:
751.2224; [α]D20 = -283.5 (2.32, CHCl3).
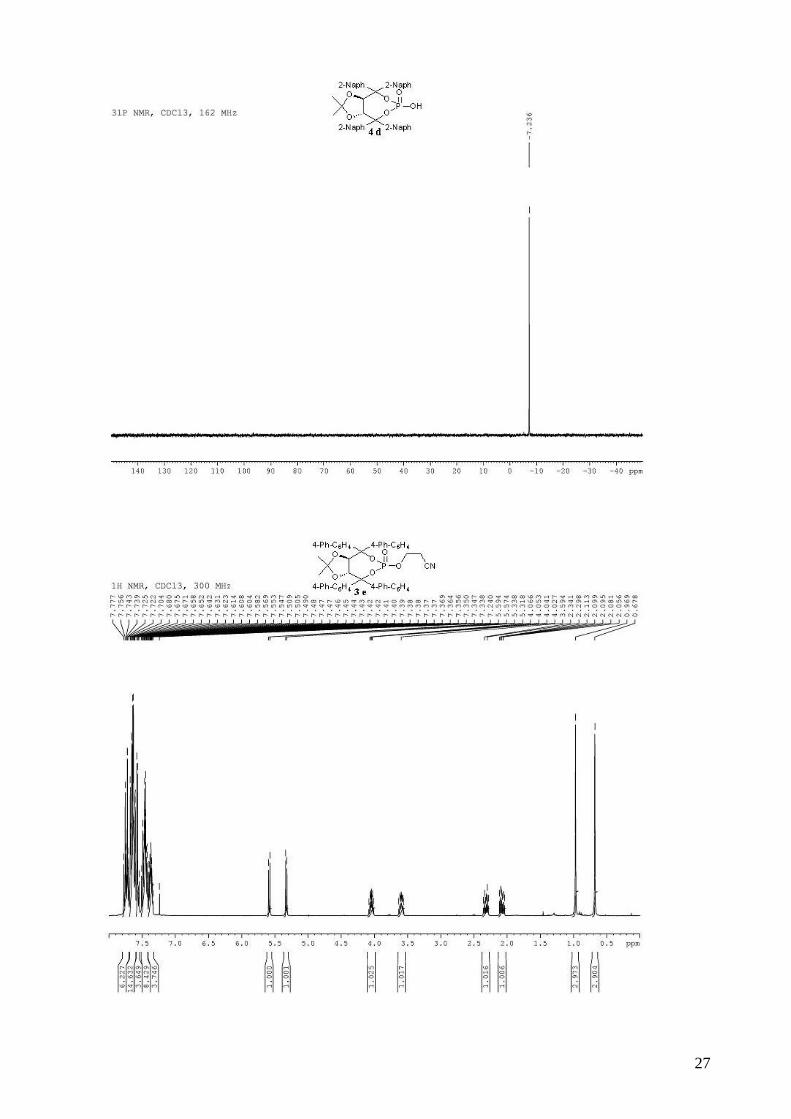
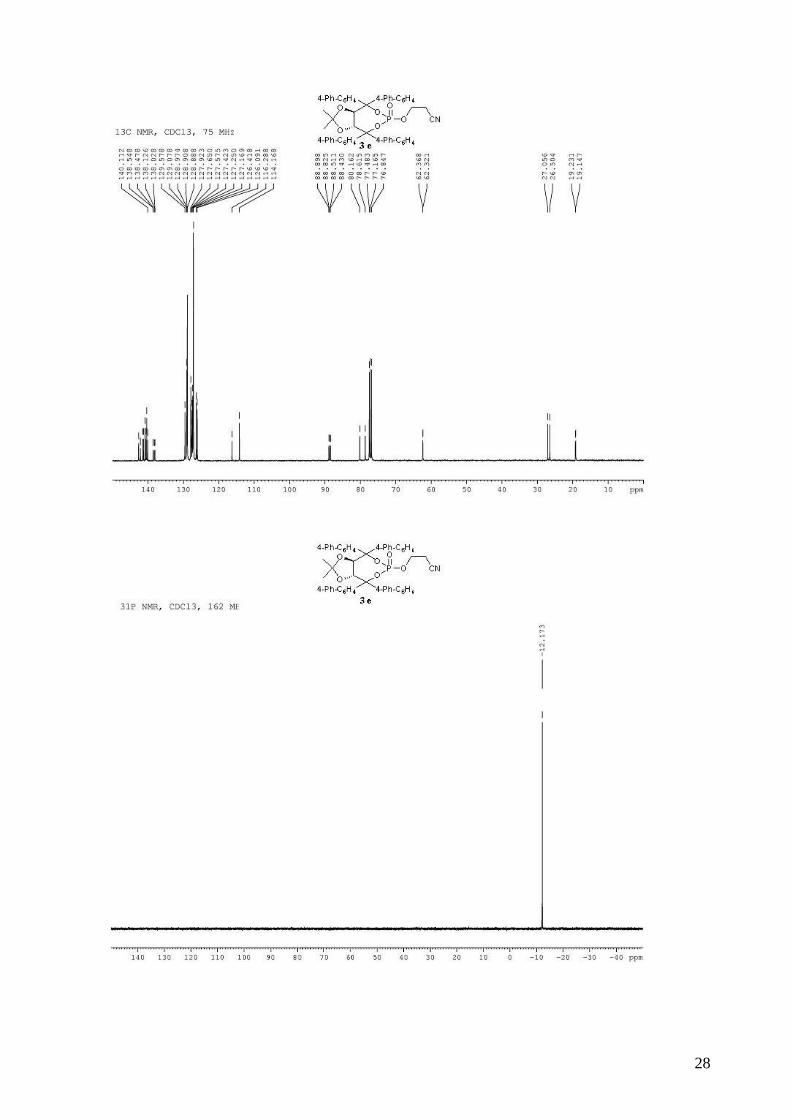
3-{[(3aR,8aR)-4,4,8,8-tetra-1,1’-biphenyl-4-yl-2,2-dimethyl-6-oxidotetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-yl]oxy}propanenitrile (3e). To a stirred solution of (R,R)-(Me,Me)-
TADDOL-(4-Ph-C6H4) 1e (1.67 g, 2.17 mmol) and triethylamine (1.02 mL, 7.36 mmol, 3.4 equiv)
in dry THF (10 mL) at 0 °C was added dropwise PCl3 (199 µL, 2.27 mmol, 1.05 equiv). The
resulting mixture was stired at 0 °C for 1 h. The 3-hydroxypropionitrile (178 µL, 2.60 mmol, 1.2
equiv) in dry THF (10 mL) was then added dropwide via cannula. The reaction mixture was
allowed to warm to room temperature and stirred for 2 h. The reaction mixture was diluted with
Et2O and the triethylammonium chloride salts were filtered through a celite pad. The solvent was
removed in vacuo. The obtained light yellow solid 2e was directly used without purification in the
oxidation step. To the crude phosphite in CH2Cl2 (15 mL) was added 30% aqueous H2O2 (1.13 mL,

6
9.96 mmol, 4.6 equiv). The biphasic mixture was stirred vigorously for 20 min and then quenched
by the addition of 20 mL of saturated aqueous NaHCO3. The mixture was extracted with CH2Cl2
and the combined organic extracts were washed with saturated aqueous NaCl, dried (MgSO4),
filtered and concentrated. Purification by flash chromatography (hexane / Et2O: 80 / 20 to pure
Et2O) afforded 940 mg (49% for two steps) of compound 3e as a white solid. Rf 0.64 (Et2O pure);
mp = 186-188 °C; 1H NMR (300 MHz, CDCl3) δ 7.78-7.70 (m, 6H, H-Ar), 7.70-7.54 (m, 18H, H-
Ar), 7.52-7.41 (m, 8H, H-Ar), 7.40-7.34 (m, 4H, H-Ar), 5.58 (d, J = 8.1 Hz, 1H, -CH-CAr2-), 5.32
(d, J = 8.1 Hz, 1H, -CH-CAr2-), 4.08-3.98 (m, 1H, -OCHHCH2CN), 3.62-3.58 (m, 1H, -
OCHHCH2CN), 2.35-2.28 (m, 1H, -OCH2CHHCN), 2.12-2.04 (m, 1H, -OCH2CHHCN), 0.97 (s,
3H, CH3), 0.68 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ 142.6, 142.2 (d, JP-C = 4.0 Hz), 141.5,
141.3, 140.9, 140.6, 140.3, 140.1, 138.5 (d, JP-C = 6.9 Hz), 138.1, (d, JP-C = 9.8 Hz), 129.6, 129.1,
129.0, 128.9, 127.9, 127.6, 127.6, 127.4, 127.2, 127.2, 126.4, 126.1, 116.3, 114.2, 88.9 (d, JP-C =
7.3 Hz), 88.5 (d, JP-C = 8.2 Hz), 80.2, 78.6, 62.3 (d, JP-C = 4.7 Hz), 27.1, 26.5, 19.2 (d, JP-C = 8.5
Hz); 31P NMR (162 MHz, CDCl3) δ -12.17; IR: νmax = 3031, 2989, 2247 (CN), 1599, 1486, 1383,
1264, 1028, 1014, 845, 734, 695 cm-1; HRMS (ESI) Calcd. For C58H48NO6NaP [M+Na]+:
908.3111. Found: 908.3110; [α]D20 = -177.9 (1.64, CHCl3).
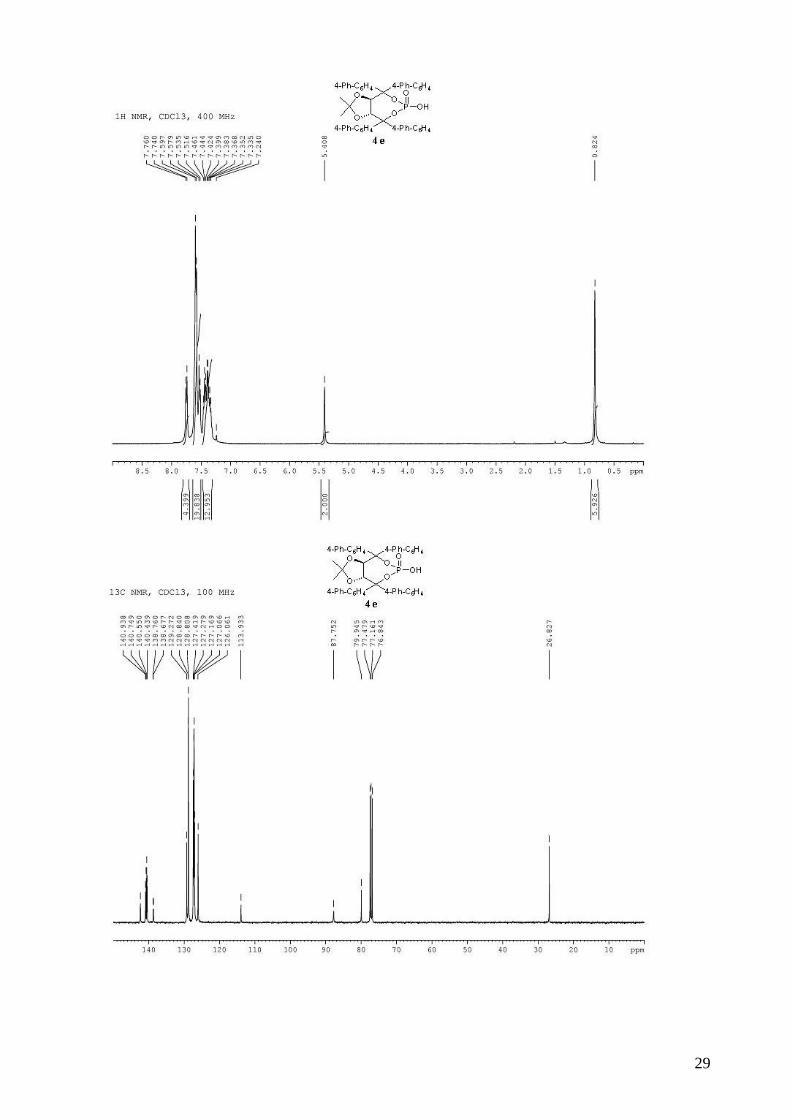
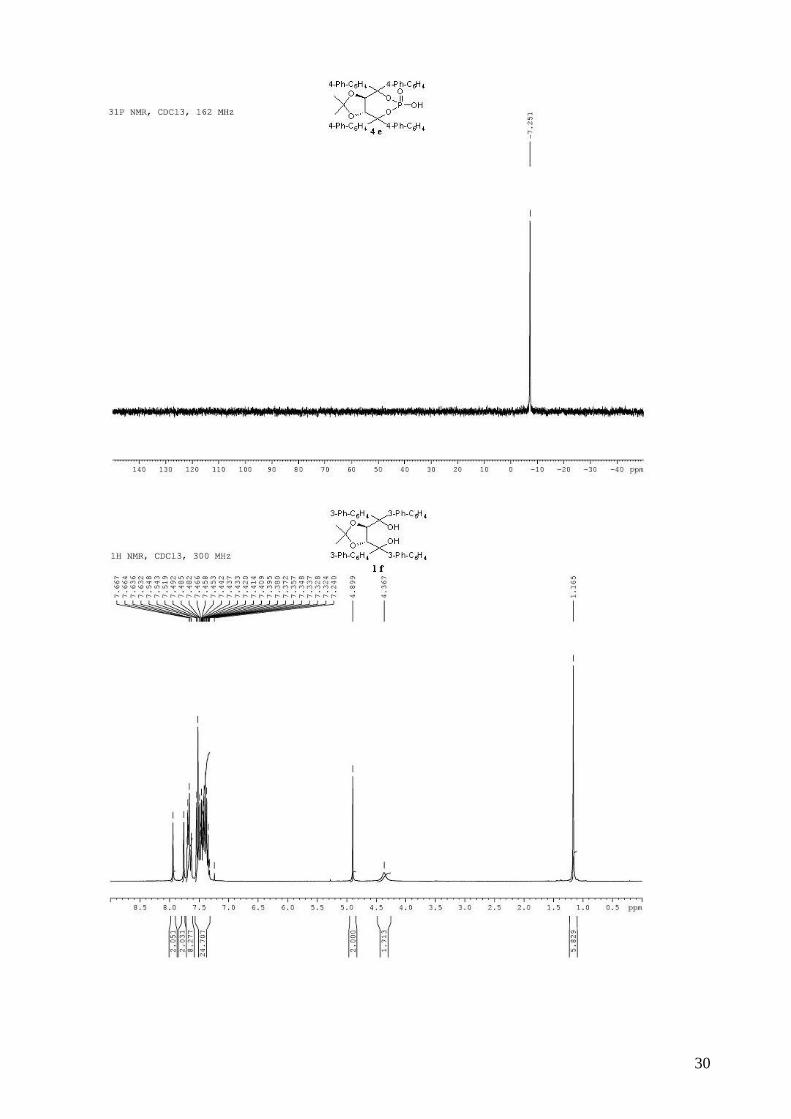
(3aR,8aR)-4,4,8,8-tetra-1,1’-biphenyl-4-yl-2,2-dimethyltetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-ol 6-oxide (4e). To a stirred solution of 3e (860 mg, 0.97 mmol) in
dry CH2Cl2 (12 mL) was added, dropwise at room temperature, DBU (153 µL, 1.02 mmol, 1.05
equiv). The solution was stirred 15 min at room temperature and when the reaction was complete
by TLC, AcOH (55 µL) was added, followed by H2O (10 mL). The organic layer was then washed
two times by a 0.3M HCl solution, saturated aqueous NaCl, and dried (MgSO4), filtered and
concentrated. The resulting white solid was dried several days on vacuum pump (805 mg, 99%),
and can be used directly in asymmetric tests. mp > 190 °C; 1H NMR (400 MHz, CDCl3) δ 7.80-
7.65 (m, 4H, H-Ar), 7.60-7.26 (m, 32H, H-Ar), 5.41 (s, 2H, -CH-CAr2-), 0.82 (s, 6H, 2 x CH3); 13C
NMR (100 MHz, CDCl3) δ 140.9, 140.7, 140.6, 140.4, 138.8, 138.7, 129.3, 128.8, 128.8, 127.4,
127.3, 127.2, 127.1, 126.0, 113.9, 87.8, 79.9, 26.8; 31P NMR (162 MHz, CDCl3) δ -7.25; IR: νmax =
3030, 2989, 2934, 1599, 1486, 1260, 1215, 1165, 1003, 744 cm-1; HRMS Calcd. for C55H45O6NaP
[M+Na]+: 855.2845. Found 855.2846; [α]D20 = -183.1 (1.03, CHCl3).
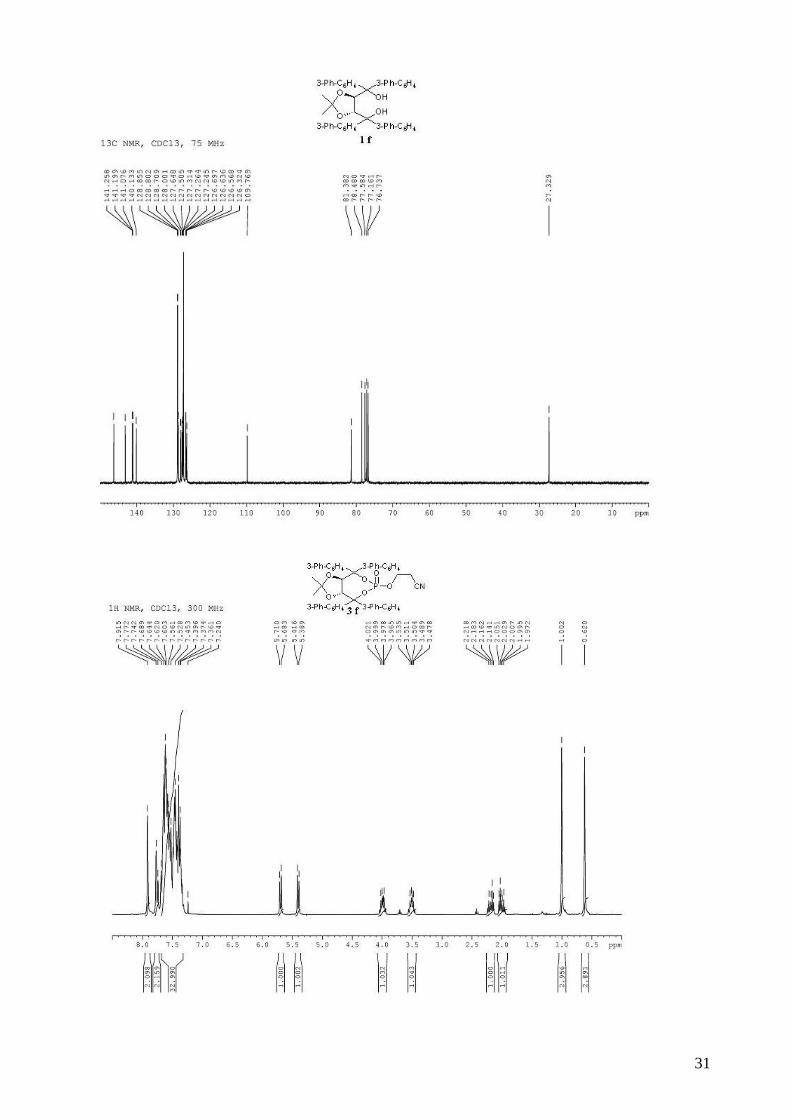
((4R,5R)-2,2-dimethyl-1,3-dioxolane-4,5-diyl)bis(dibiphenyl-3-ylmethanol) (1f). (R,R)-diethyl
O,O-isopropylidenetartrate (1.82 g, 7.39 mmol) in THF (15mL) was added dropwise to a solution
of (naphthalen-1-yl)magnesium bromide (32.5 mmol, prepared from 7.58 g of 2-bromonaphthalene

7
and 808 mg of Mg) in THF (15 mL). After workup, purification by flash chromatography (hexane /
Et2O: 80 / 20) afforded 3.91 g (69%) of compound 1f as a white solid. Rf 0.41 (Et2O / hexane: 70 /
30); mp = 108-110 °C; 1H NMR (300 MHz, CDCl3) δ 7.96-7.92 (m, 2H, H-Ar), 7.78-7.74 (m, 2H,
H-Ar), 7.72-7.62 (m, 8H, H-Ar), 7.56-7.30 (m, 24H, H-Ar), 4.90 (s, 2H, -CH-CAr2-), 4.36 (br s,
2H, -OH), 1.16 (s, 6H, -CH3); 13C NMR (100 MHz, CDCl3) δ 146.3, 143.2, 141.3, 141.2, 141.1,
140.1, 128.9, 128.8, 128.7, 128.0, 127.6, 127.5, 127.3, 127.3, 127.2, 126.7, 126.6, 126.6, 126.3,
109.8, 81.4, 78.5, 27.3; IR: νmax = 3305, 3030, 2986, 1597, 1478, 1413, 1264, 1165, 1044, 883, 734,
697 cm-1; HRMS Calcd. for C55H46O4Na [M+Na]+: 793.3288. Found 793.3288; [α]D20 = -31.6
(1.62, CHCl3).
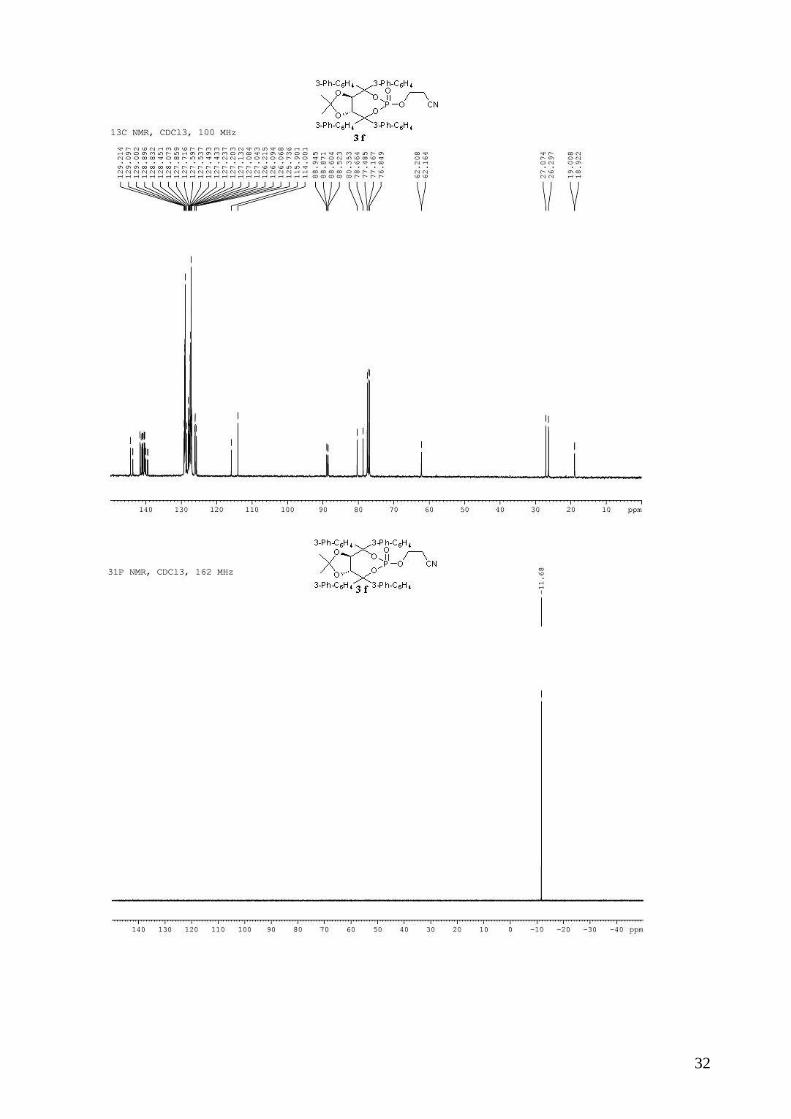
3-{[(3aR,8aR)-4,4,8,8-tetra-1,1’-biphenyl-3-yl-2,2-dimethyl-6-oxidotetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-yl]oxy}propanenitrile (3f). To a stirred solution of (R,R)-(Me,Me)-
TADDOL-(3-Ph-C6H4) 1f (3.47 g, 4.50 mmol) and triethylamine (2.12 mL, 15.3 mmol, 3.4 equiv)
in dry THF (14 mL) at 0 °C was added dropwise PCl3 (412 µL, 4.72 mmol, 1.05 equiv). The
resulting mixture was stired at 0 °C for 1 h. The 3-hydroxypropionitrile (400 µL, 5.85 mmol, 1.3
equiv) in dry THF (14 mL) was then added dropwide via cannula. The reaction mixture was
allowed to warm to room temperature and stirred for 2 h. The reaction mixture was diluted with
Et2O and the triethylammonium chloride salts were filtered through a celite pad. The solvent was
removed in vacuo. The obtained light yellow solid was directly used without purification in the
oxidation step. To the crude phosphite in CH2Cl2 (30 mL) was added 30% aqueous H2O2 (2.85 mL,
25.2 mmol, 5.6 equiv). The biphasic mixture was stirred vigorously for 20 min and then quenched
by the addition of 40 mL of saturated aqueous NaHCO3. The mixture was extracted with CH2Cl2
and the combined organic extracts were washed with saturated aqueous NaCl, dried (MgSO4),
filtered and concentrated. Purification by flash chromatography (hexane / Et2O: 40 / 60 to pure
Et2O) afforded 2.36 g (59% for two steps) of compound 3f as a white solid. Rf 0.67 (Et2O pure);
mp = 112-114 °C; 1H NMR (300 MHz, CDCl3) δ 7.91 (s, 2H, H-Ar), 7.65-7.20 (m, 34H, H-Ar),
5.69 (d, J = 8.0 Hz, 1H, -CH-CAr2-), 5.40 (d, J = 8.0 Hz, 1H, -CH-CAr2-), 4.02-3.95 (m, 1H, -
OCHHCH2CN), 3.53-3.46 (m, 1H, -OCHHCH2CN), 2.22-2.12 (m, 1H, -OCH2CHHCN), 2.05-1.95
(m, 1H, -OCH2CHHCN), 1.00 (s, 3H, CH3), 0.62 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ
144.4, 143.7 (d, JP-C = 4.6 Hz), 141.6, 141.4, 141.1, 140.9, 140.8, 140.5, 140.4, 140.3, 140.1 (d, JP-C
= 6.3 Hz), 139.5 (d, JP-C = 10.0 Hz), 129.2, 129.1, 129.0, 129.0, 128.9, 128.9, 128.5, 128.1, 127.9,
127.7, 127.6, 127.6, 127.5, 127.4, 127.2, 127.2, 127.1, 127.1, 127.0, 126.2, 126.1, 126.1, 125.7,
115.9, 114.0, 88.9 (d, JP-C = 7.4 Hz), 88.5 (d, JP-C = 8.1 Hz), 80.4, 78.7, 62.2 (d, JP-C = 4.4 Hz),
27.1, 26.3, 19.0 (d, JP-C = 8.6 Hz); 31P NMR (162 MHz, CDCl3) δ -11.68; IR: νmax = 3056, 3031,

8
2989, 2255 (CN), 1598, 1478, 1452, 1414, 1276, 1264, 1007, 939, 763, 698 cm-1; HRMS (ESI)
Calcd. for C58H48NO6NaP [M+Na]+: 908.3111. Found: 908.3128; [α]D20 = -196.5 (1.79, CHCl3).
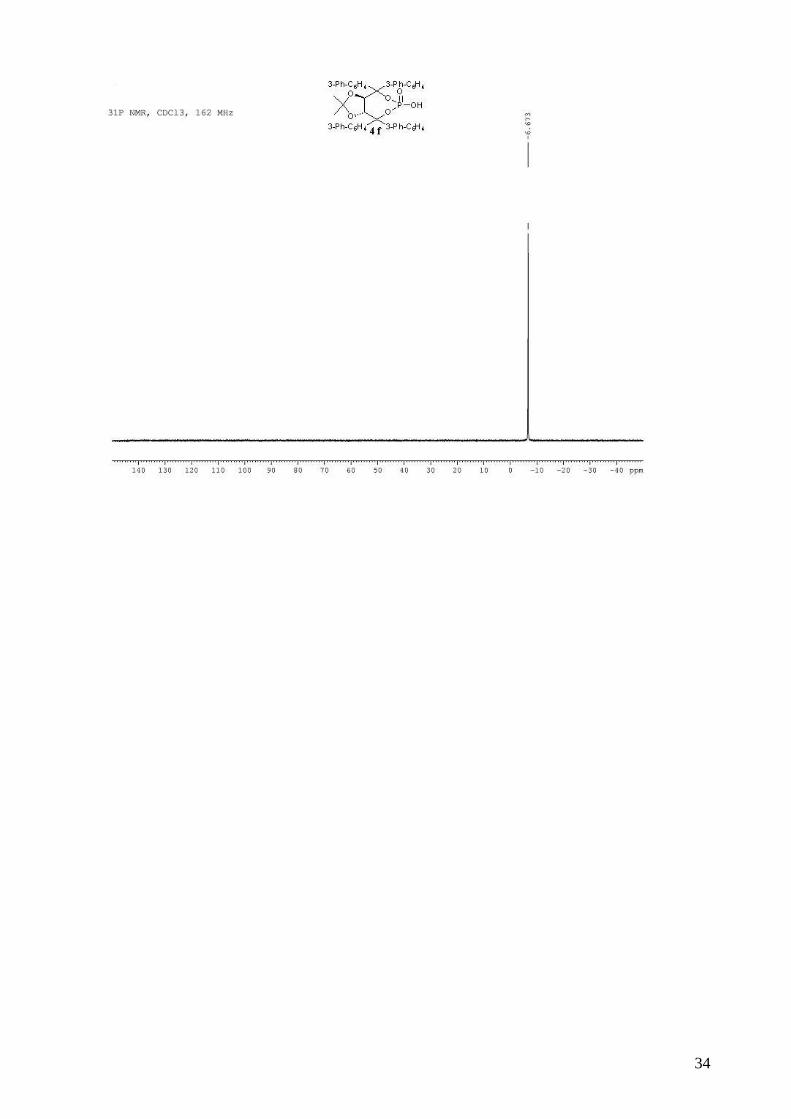
(3aR,8aR)-4,4,8,8-tetra-1,1’-biphenyl-3-yl-2,2-dimethyltetrahydro[1,3]dioxolo[4,5-
e][1,3,2]dioxaphosphepin-6-ol 6-oxide (4f).
To a stirred solution of 3f (2.20 g, 2.48 mmol) in dry CH2Cl2 (30 mL) was added, dropwise at room
temperature, DBU (391 µL, 2.61 mmol, 1.05 equiv). The solution was stirred 30 min at room
temperature and when the reaction was complete by TLC, AcOH (0.14 mL) was added, followed
by H2O (50 mL). The organic layer was then washed two times by a 0.3M solution of HCl,
saturated aqueous NaCl, and dried (MgSO4), filtered and concentrated. The resulting white solid
(1.88 g, 91%) was dried several days on vacuum pump, and can be used directly in asymmetric
tests. This compound was kept several months at -20 °C without degradations. mp = 160-162 °C; 1H NMR (400 MHz, CDCl3) δ 8.00-7.94 (m, 2H, H-Ar); 7.80-7.74 (m, 2H, H-Ar); 7.70-7.50 (m,
16H, H-Ar), 7.48-7.27 (m, 16H, H-Ar), 5.56 (s, 2H, -CH-CAr2-), 0.82 (s, 6H, 2 x CH3); 13C NMR
(100 MHz, CDCl3) δ 143.8, 141.2, 141.1, 140.1, 140.1, 128.9, 128.8, 128.1, 127.7, 127.4, 127.3,
127.3, 127.2, 126.7, 126.1, 125.7, 113.9, 88.0, 79.9, 26.7; 31P NMR (162 MHz, CDCl3) δ -6.67;
IR: νmax = 3060, 2989, 1598, 1478, 1416, 1264, 1015, 736, 702 cm-1; HRMS (ESI) Calcd for
C55H45O6NaP [M+Na]+: 855.2845. Found: 855.2878; [α]D20 = -153.3 (2.76, CHCl3).
General procedure for the cyclopropanation using chiral phosphates ligands derived from
TADDOL 4a-f:
To a solution of the ligand (0.31 mmol, 1.25 equiv) in DCE (1.5 mL) at -15 °C was added ZnEt2
(32 µL, 0.31 mmol, 1.25 equiv) dropwise. This solution was stirred for 15 min after which CH2I2
(25 µL, 0.31 mmol, 1.25 equiv) was added dropwise. This solution was stirred for an additional 20
min A solution of substrate (0.25 mmol, 1.0 equiv) in DCE (1.0 mL) was added and the resulting
solution was stirred for 48 h. The reaction was quenched with saturated NH4Cl solution, washed
with saturated aqueous NaCl, dried (MgSO4), filtered and concentrated. The crude product was
purified by flash chromatography to afford the pure desired cyclopropane derivative. All the
cyclopropanes synthesized have spectral and physical data identical to the data reported in the
literature. The enantiomeric excess was determined by chiral SFC analysis for the corresponding
cyclopropane of substrate A, C, D and E, using the corresponding racemic mixture as standard. The
data are reported as follows: column type, temperature, pressure, eluent, flow rate, retention time
(tr). The enantiomeric excess was determined by chiral HPLC analysis for the corresponding
cyclopropane of substrate B. The data are reported as follows: column type, eluent, flow rate,

9
retention time (tr). The enantiomeric excess was determined by chiral GC analysis for the
corresponding cyclopropane of substrate F. The data are reported as follows: column type,
temperature, pressure, retention time (tr).
10
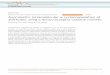
Substrate A: [(2-(benzyloxy)methyl)cyclopropyl]benzene. SFC (Chiralcel OJ, 40 °C, 180 bars,
3% MeOH, 1.3 mL/min), tr (minor) 23.8 min, tr (major) 26.2 min.
28272625242322
550
500
450
400
350
300
250
200
150
100
50
0
Start [Min] Time [Min] End [Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
22.49 23.24 25.07 50.05 561.7 611.9 50.051 25.07 25.86 28.69 49.95 448.4 610.6 49.949
100 1010.1 1222.5 100
29282726252423
360
340
320
300
280
260
240
220
200
180
160
140
120
100
80
60
40
20
0
Start [Min] Time [Min] End [Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
22.86 23.84 25.24 15.18 78 79.6 15.177 25.24 26.23 29 84.82 343.6 444.7 84.823
100 421.5 524.3 100
11
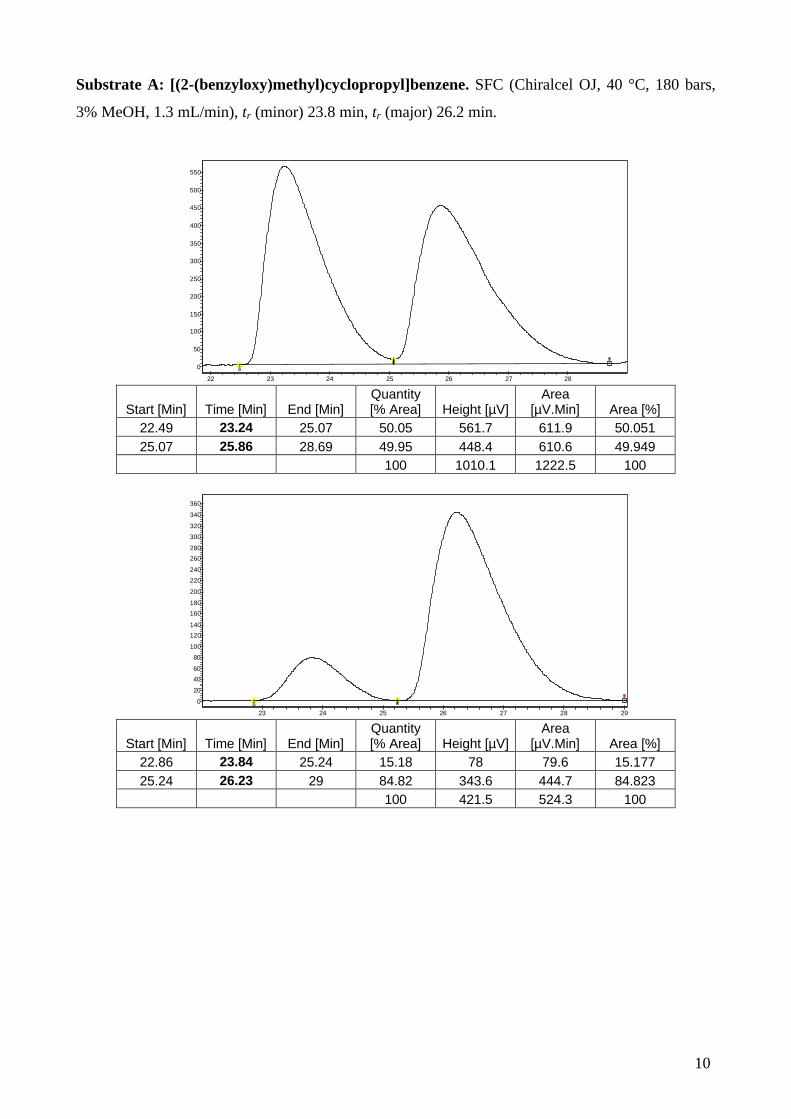
Substrate B: [(2-methoxymethyl)cyclopropyl]benzene: HPLC (Chiralcel OJ, hexane / i-PrOH :
98 / 2, 1.0 mL/min), tr (major) 7.5 min, tr (minor) 9.1 min.
Time [Min] Width [min] Area Area [%] 7.298 0.372 40808.957 49.831
10.314 0.498 41084.996 50.169 100
Time [Min] Width [min] Area Area [%] 7.476 0.4350 6387.853 86.321 9.110 0.4105 1012.265 13.679
100
12
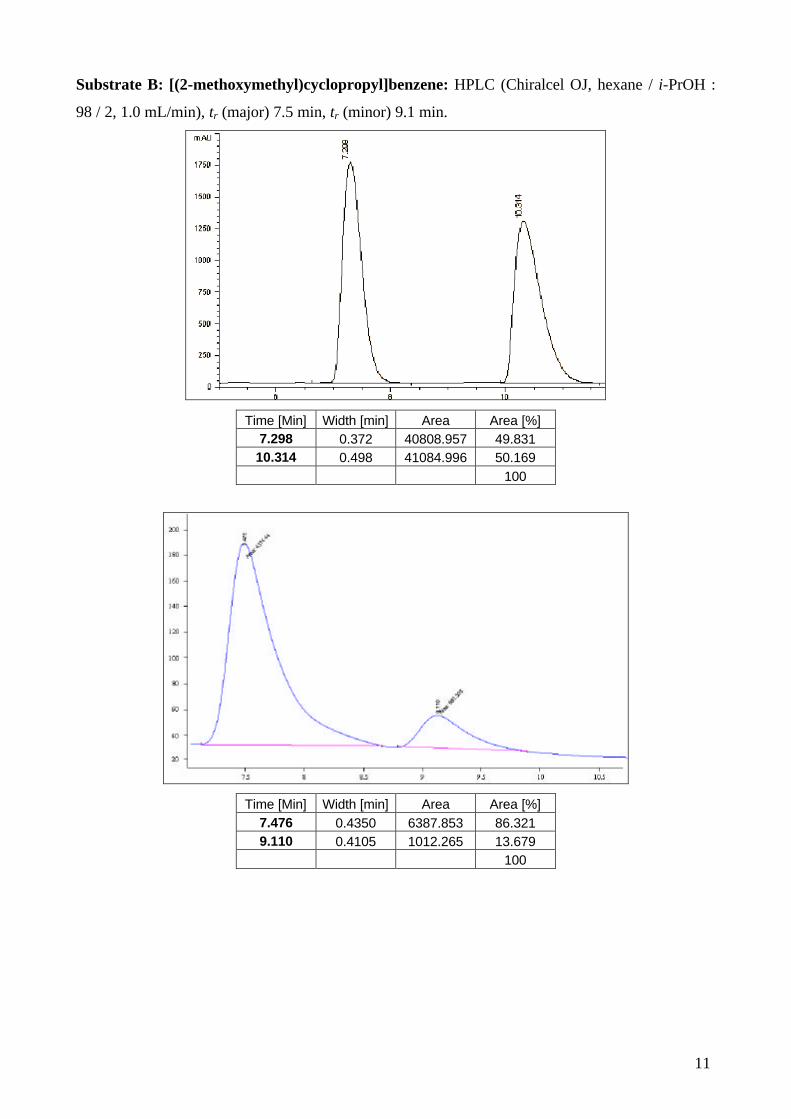
Substrate C: [(2-(benzyloxy)methyl)cyclopropyl]-4-methoxybenzene. SFC (Chiralcel OJ, 40 °C,
180 bars, 5% MeOH, 2.0 mL/min), tr (minor) 14.4 min, tr (major) 18.0 min.
21201918171615141312
210
200
190
180
170
160
150
140
130
120
110
100
90
80
70
60
50
40
30
20
Start [Min] Time [Min] End [Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
13.65 14.37 15.87 50.66 185.5 134.5 50.658 17.35 18.3 20.2 49.34 135.6 131 49.342
100 321 265.5 100
21201918171615141312
300
280
260
240
220
200
180
160
140
120
100
80
60
40
20
0
Start [Min] Time [Min] End [Min] RT Offset
[Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
13.78 14.41 15.36 0 16.21 81.5 56.5 16.208 17.28 18.01 19.94 0 83.79 304.4 292.3 83.792
100 385.9 348.8 100
13
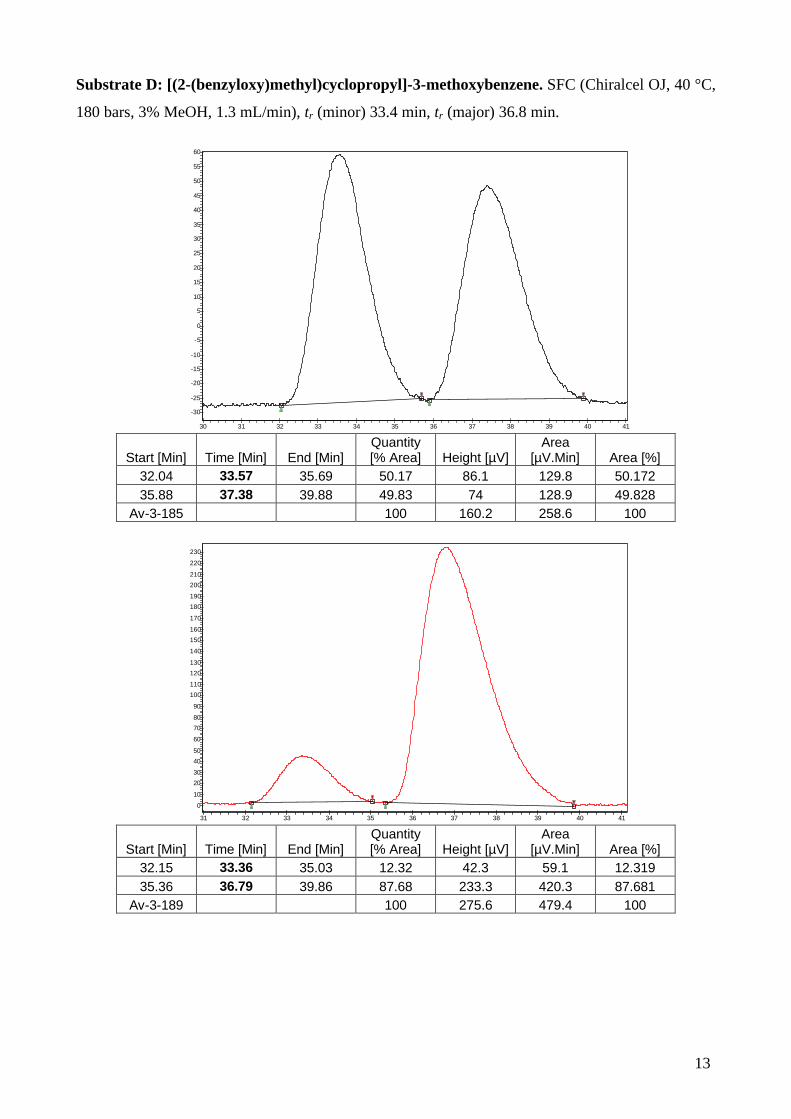
Substrate D: [(2-(benzyloxy)methyl)cyclopropyl]-3-methoxybenzene. SFC (Chiralcel OJ, 40 °C,
180 bars, 3% MeOH, 1.3 mL/min), tr (minor) 33.4 min, tr (major) 36.8 min.
414039383736353433323130
60
55
50
45
40
35
30
25
20
15
10
5
0
-5
-10
-15
-20
-25
-30
Start [Min] Time [Min] End [Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
32.04 33.57 35.69 50.17 86.1 129.8 50.172 35.88 37.38 39.88 49.83 74 128.9 49.828
Av-3-185 100 160.2 258.6 100
4140393837363534333231
230
220
210
200
190
180
170
160
150
140
130
120
110
100
90
80
70
60
50
40
30
20
10
0
Start [Min] Time [Min] End [Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
32.15 33.36 35.03 12.32 42.3 59.1 12.319 35.36 36.79 39.86 87.68 233.3 420.3 87.681
Av-3-189 100 275.6 479.4 100
14
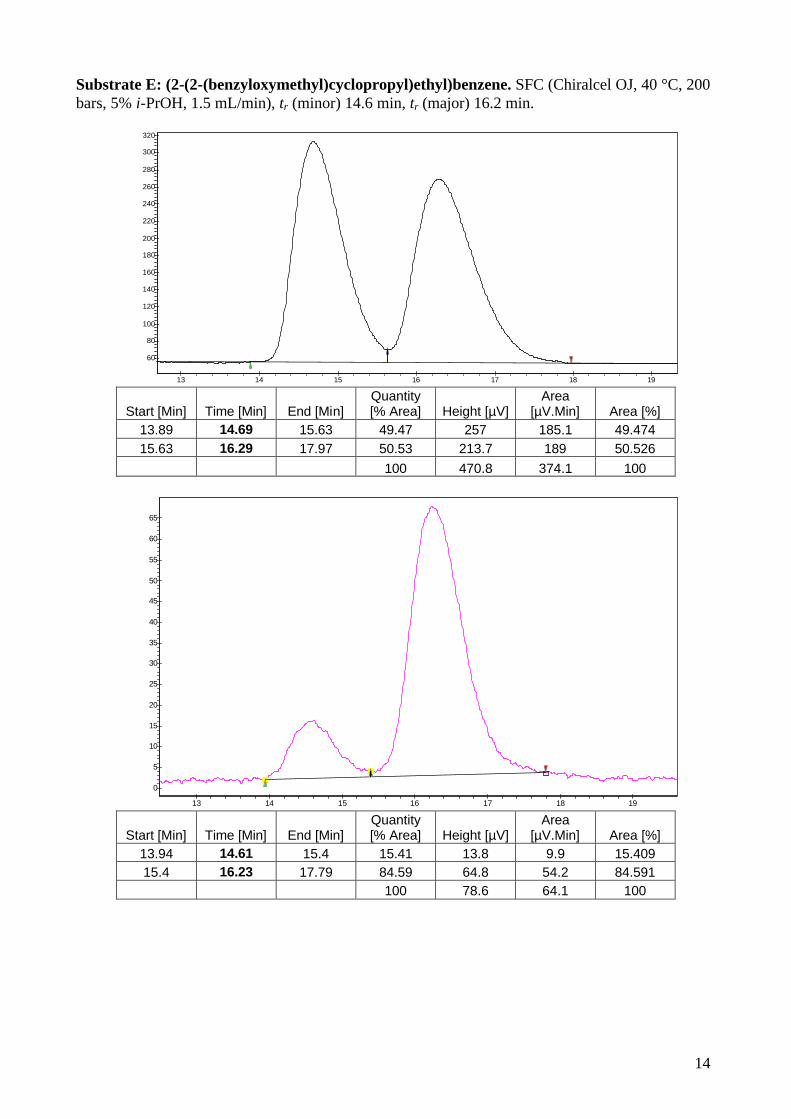
Substrate E: (2-(2-(benzyloxymethyl)cyclopropyl)ethyl)benzene. SFC (Chiralcel OJ, 40 °C, 200 bars, 5% i-PrOH, 1.5 mL/min), tr (minor) 14.6 min, tr (major) 16.2 min.
19181716151413
320
300
280
260
240
220
200
180
160
140
120
100
80
60
Start [Min] Time [Min] End [Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
13.89 14.69 15.63 49.47 257 185.1 49.474 15.63 16.29 17.97 50.53 213.7 189 50.526
100 470.8 374.1 100
19181716151413
65
60
55
50
45
40
35
30
25
20
15
10
5
0
Start [Min] Time [Min] End [Min] Quantity [% Area] Height [µV]
Area [µV.Min] Area [%]
13.94 14.61 15.4 15.41 13.8 9.9 15.409 15.4 16.23 17.79 84.59 64.8 54.2 84.591
100 78.6 64.1 100
15
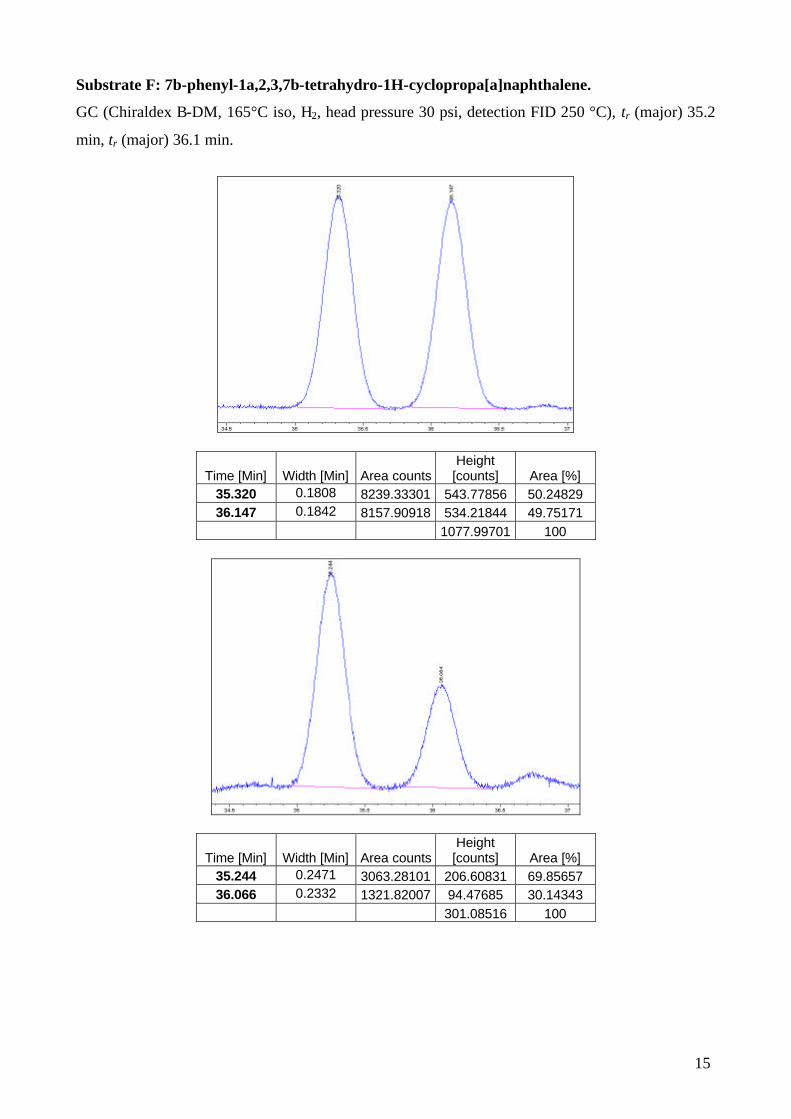
Substrate F: 7b-phenyl-1a,2,3,7b-tetrahydro-1H-cyclopropa[a]naphthalene.
GC (Chiraldex B-DM, 165°C iso, H2, head pressure 30 psi, detection FID 250 °C), tr (major) 35.2
min, tr (major) 36.1 min.
Time [Min]
Width [Min] Area counts Height
[counts] Area [%] 35.320 0.1808 8239.33301 543.77856 50.24829 36.147 0.1842 8157.90918 534.21844 49.75171
1077.99701 100
Time [Min]
Width [Min] Area counts Height
[counts] Area [%] 35.244 0.2471 3063.28101 206.60831 69.85657 36.066 0.2332 1321.82007 94.47685 30.14343
301.08516 100
16
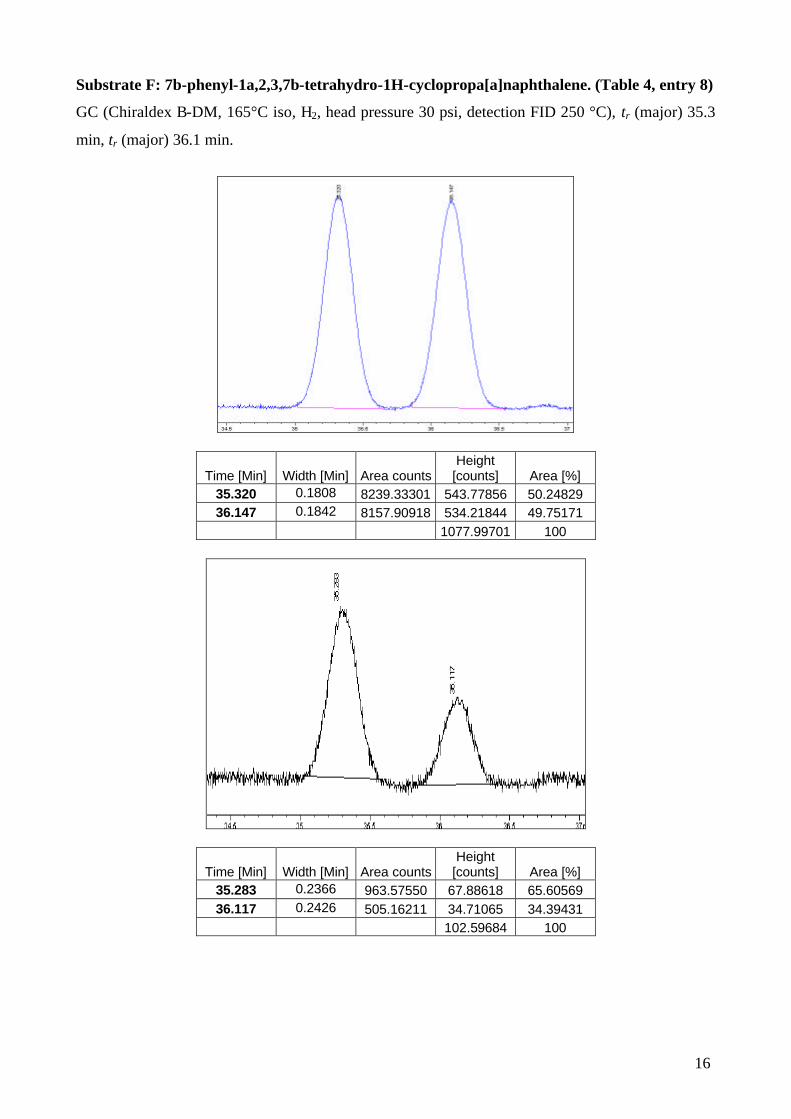
Substrate F: 7b-phenyl-1a,2,3,7b-tetrahydro-1H-cyclopropa[a]naphthalene. (Table 4, entry 8)
GC (Chiraldex B-DM, 165°C iso, H2, head pressure 30 psi, detection FID 250 °C), tr (major) 35.3
min, tr (major) 36.1 min.
Time [Min]
Width [Min] Area counts Height
[counts] Area [%] 35.320 0.1808 8239.33301 543.77856 50.24829 36.147 0.1842 8157.90918 534.21844 49.75171
1077.99701 100
Time [Min]
Width [Min] Area counts Height
[counts] Area [%] 35.283 0.2366 963.57550 67.88618 65.60569 36.117 0.2426 505.16211 34.71065 34.39431
102.59684 100
17
1H, 13C, 31P Spectra of Compounds 3a, 4a, 4b, 2c, 4c, 3d, 4d, 4e, 1f, 3f and 4f.
18
19
20
21
22

23
24
25
26
27
28
29
30
31
32

33
34