Embed Size (px)
Citation preview

ORIGINAL ARTICLESee Commentary on pages vii and xi
Suppression of the Immune Response Against ExogenousDesmoglein 3 in Desmoglein 3 Knockout Mice:An Implication for Gene Therapy
Manabu Ohyama,nzTakayuki Ota,n Miyo Aoki,n Kazuyuki Tsunoda,n Reiko Harada,z Shigeo Koyasu,wTakeji Nishikawa,n and Masayuki Amagain
Departments of nDermatology and wMicrobiology and Immunology, Keio University School of Medicine,Tokyo, Japan; zDivision of Dermatology,Tokyo Electric Power Company Hospital,Tokyo, Japan
Gene therapies for recessive genetic diseases mayprovoke unwanted immune responses against theintroduced gene product because patients, especiallythose with null mutation of a certain protein, have notolerance for the protein of interest.This study used des-moglein 3 knockout (Dsg3^/^) mice as a disease modelfor a genetic defect in DSG3, to investigate whethernonviral gene therapy induces an immune responseagainst Dsg3 and whether the reaction against Dsg3can be prevented. When mouse Dsg3 cDNA was in-jected in the skin of Dsg3^/^ mice, 50% of treatedDsg3^/^ mice developed anti-Dsg3 IgG, which can bindnative Dsg3 in vivo. To prevent this response, we usedan anti-CD40L monoclonal antibody, MR1, whichblocks the costimulatory interaction between CD40and CD40L. To evaluate the e¡ect of MR1, we graftedDsg3þ/þ skin on Dsg3^/^ mice, to mimic stable gene
transfer of Dsg3. After skin grafting, all the recipientDsg3^/^ mice were treated with either MR1 (n¼ 8) orcontrol hamster IgG (n¼ 8). All of the control IgG-trea-ted mice developed circulating anti-Dsg3 IgG about2 wk after grafting, and IgG deposition was observedon the surfaces of keratinocytes in the grafted Dsg3þ/þskin. Such anti-Dsg3 IgG production was signi¢cantlyprevented, however, when the recipient mice were trea-ted with MR1.These ¢ndings suggested that gene thera-pies for recessive diseases may provoke an immuneresponse against the transgene product, and that theCD40^CD40L interaction might be a reasonable targetfor e¡ective prevention of such undesirable immune re-sponses, leading, in turn, to a successful gene therapy.Key words: CD154/CD40 ligand/immunosuppression/recessivegenodermatosis/skin graft. J Invest Dermatol 120:610 ^615,2003
Skin is an attractive target for gene therapy, because it isreadily accessible for gene transfer, gene expression iseasily observed, and the skin can be removed when anunexpected side-e¡ect occurs (Vogel, 1993; Greenhalghet al, 1994; Hengge et al, 1995; 1996). As in other organs,
the genetic correction of recessive genodermatoses is of greatpotential value, as only palliative therapies are at present available(Khavari, 1998; 2000; Uitto and Pulkkinen, 2000). Therefore,attempts to develop gene therapies for forms of epidermolysisbullosa (Dellambra et al, 1998; Vailly et al, 1998; Seitz et al, 1999;Chen et al, 2000), X-linked ichthyosis (Jensen et al, 1993; Freiberget al, 1997), lamellar ichthyosis (Choate et al, 1996), and xerodermapigmentosum (Zeng et al, 1998) have been reported. In thesestudies, expression of the intended genes was successful and theimpaired protein functions were corrected. Several hurdlesremain to be cleared before practical gene therapy can be applied,however. One such problem is how to achieve long-term expres-sion of the introduced gene at therapeutic levels. This matter hasattracted great interest and has been investigated intensively. By
contrast, little attention has been paid to the immune reactionthat might be provoked against the introduced gene products.As skin is a primary immunologic barrier against foreign sub-
jects and contains many antigen-presenting cells (dendritic cells)(Tuting et al, 1998; Larregina and Falo, 2000), the immuneresponse against a transgene product, and the prevention of thisresponse, is a priority issue in gene therapy that is targeted onskin. This problem becomes particularly important when genetherapy is applied to correct a recessive genetic disease caused bynull mutation. In these patients, the immune system has neverencountered the missing protein. Accordingly, there is no toler-ance of what would be a novel protein in such patients. Conse-quently, any gene product introduced to correct a genetic defectmight be recognized as a foreign antigen, which would inevitablyresult in an unwanted immune response against the introducedgene product (Khavari, 1998; 2000). In contrast, patients withmissense mutations may still express endogenous gene productthat serves as self-protein to establish self-tolerance although itsfunction is altered. Despite the signi¢cance of this phenomenon,such immune responses in the skin, or in other organs, have notbeen satisfactorily studied. Several investigators have reported im-mune reactions against transgene products. As gene delivery wasperformed using viral vectors, however, it transpires that most ofthese documented reactions were against the transgene vehicles(Dai et al, 1995; Tripathy et al, 1996; Yang et al, 1996; Ilan et al,1997; Morral et al, 1997; Stein et al, 1999; 2000). This means that
Reprint requests to: Masayuki Amagai, M.D., Ph.D., Department ofDermatology, Keio University School of Medicine, 35 Shinanomachi,Shinjyuku-ku,Tokyo 160-8582, Japan; Email: [email protected]: Dsg, desmoglein.
Manuscript received July 17, 2002; revised October 13, 2002; accepted forpublication November 5, 2002
0022-202X/03/$15.00 � Copyright r 2003 by The Society for Investigative Dermatology, Inc.
610

the immune response elicited against a transgene product has notyet been evaluated. In addition, only a few studies have dealtwith the issue of immune provocation against a corrected self-antigen in a gene therapy model of recessive disease (Ilan et al,1997; Stein et al, 2000). Therefore, an adequate animal model andimmune assay are required in order to further evaluate and pre-vent such undesirable immune responses.Desmoglein 3 (Dsg3) is an epithelial type transmembrane des-
mosomal component that belongs to the cadherin gene super-family; it has been identi¢ed as the autoimmune target ofpemphigus vulgaris (Amagai et al, 1991; 1994). In 1997, the Dsg3knockout (Dsg3^/^) mouse was generated by genetic disruptionof DSG3 (Koch et al, 1997). The adult mouse develops crustederosions on traumatized skin, telogen-speci¢c hair loss, andweight loss due to decreased food intake as a result of painful ero-sions on the mucous membranes that are manifestations of Dsg3dysfunction. Recently, we intensively investigated the gross andhistopathologic phenotype of Dsg3^/^ mice (Ohyama et al,2002) in order to characterize their pathophysiology. In addition,we have developed an enzyme-linked immunosorbent assay(ELISA) using recombinant mouse Dsg3, which should prove tobe a valuable tool for detecting circulating anti-Dsg3 IgG in mice(Amagai et al, 2000).In this study, using the Dsg3^/^ mouse as a disease model for a
genetic defect inDSG3, we investigated whether an undesirable im-mune response against the transgene product of Dsg3 was actuallyprovoked after naked DNA injection of Dsg3 cDNA in Dsg3^/^mice. Then, we showed that blockading the CD40^CD40L inter-action prevented this undesirable immune response.
MATERIALS AND METHODS
Mice Dsg3^/^ mice were obtained by mating a male Dsg3^/^ mouseand female Dsg3þ/^ mice (Koch et al, 1997; Amagai et al, 2000). Thesemice have a mixed genetic background of 129/SV (H-2b) and C57BL/6 J(H-2b). The C57BL/6 J mice used as skin graft donors were purchasedfrom Japan Clea (Tokyo, Japan). All mice studies were approved by theanimal ethics review board of Keio University, Japan.
DNA constructs The plasmid pcDNA-mouse Dsg3 (mDsg3) wasconstructed by subcloning mDsg3 cDNA between the EcoR1 and Sph1sites of pcDNA1 containing the CMV promoter (Invitrogen, Carlsbad,CA). Dr. Jouni Uitto (Je¡erson Medical College, Philadelphia, PA)generously provided the cDNA.
Naked DNA injection Materials for naked DNA injection wereprepared and the injection was performed essentially as previouslydescribed (Hengge et al, 1995; 1996). The puri¢ed plasmid DNA wasdiluted to the indicated concentrations with phosphate-bu¡ered saline.Injections into the super¢cial dermis of the footpad or shaved trunk skinwere performed using syringes with ultra-¢ne 29G needles (U-100, JapanBecton Dickinson, Tokyo, Japan). The injected volume was 20^50 ml,depending on the site. Ten Dsg3^/^ mice were divided into four groupsto undergo gene therapy with pcDNA-mDsg3 naked DNA injection, atdoses of 50^200 mg per mouse per injection, and at injection frequenciesthat ranged from once every 2 wk to twice per week. The indicatedquantity of DNAwas dissolved in 100ml of phosphate-bu¡ered saline perinjection for each individual mouse. Two C57BL/6 J (Dsg3þ/þ ) micewere treated with 200 mg per mouse every 2 wk as controls. Serum wascollected at weekly intervals for further analysis. To determine whetheranti-Dsg3 IgG binds to the transgene product, pcDNA^mDsg3 (1 mg perml) was injected into the footpads of a Dsg3^/^ mouse that had producedanti-Dsg3 IgG after naked DNA injection of pcDNA^mDsg3. Biopsyspecimens from the footpads were taken 12 h after injection and frozensections were prepared. These were incubated with 100-fold diluted£uorescein isothiocyanate anti-mouse IgG antibody for 1 h, washed, andembedded for microscopic investigation.
Dsg3 expression assay To evaluate the potential of the constructedDNA to express Dsg3 in epidermis, plasmid solution of either pcDNA^mDsg3 or pcDNA1 (5 or 10 mg per ml for pcDNA^mDsg3, 10 mg per mlfor pcDNA1) was injected into one or two sites per footpad (six to eightsites per mouse) in Dsg3^/^ mice (n¼ 2 for each concentration of DNA).Skin biopsies were performed 18 h after injection. Samples were embedded
in OCT compound (Sakura Finetechnical, Tokyo, Japan) and cryostatsections of these samples were prepared for direct immuno£uorescencestudy. Sections were ¢rst incubated for 1 h with anti-mouse Dsg3monoclonal antibody, AK9 (Tsunoda et al, manuscript in preparation).Sections were then incubated for 1 h with 100-fold diluted £uoresceinisothiocyanate rabbit anti-mouse IgG antibody (Zymed Laboratories, SanFrancisco, CA). All these sections were examined using £uorescencemicroscopy (Eclipse E800, Nikon,Tokyo, Japan).
ELISA The production of anti-Dsg3 and anti-Dsg1 IgG was evaluated byELISA, using recombinant mouse Dsg3 and Dsg1, as previously reported(Ohyama et al, 2002). Brie£y, diluted sera (500-fold for sera of mice treatedwith naked DNA injection of Dgs3, 1000-fold for sera of Dsg3^/^ micewith Dsg3þ/þ skin graft) were incubated for 1 h on ELISA plates coatedwith 2.5 mg per ml mouse rDsg3, or rDsg1, expressing the entireextracellular domains using a baculovirus expression system (Amagai et al,2000). After washing, the plates were incubated with 5000-fold dilutedperoxidase-labeled goat anti-mouse IgG (Zymed Laboratories) for 1 h.After the second wash, color was developed using a solution containingequal amounts of tetramethylbenzidine and hydrogen peroxide (MedicalBiological Laboratories, Nagano, Japan) for 30 min, and stopped byadding 100 ml of 4 N H2SO4. The OD was measured at 450 nm using amicroplate reader (Bio-Rad Laboratories, Hercules, CA).We calculated themean and standard deviation for this ELISA using OD values of controlsera from 12 normal mice. Based on the meanþ 3SD, we set the cut-o¡values for de¢nite positive reactivity as 0.090 for Dsg3 IgG and 0.067 forDsg1 IgG. Sera were tested at least twice for Dsg3 ELISA and once forDsg1 ELISA.
Grafting Dsg3þ/þ skin onto Dsg3^/^ mice For the skin graftprocedure, 8� 8 mm pieces of dorsal skin were removed from sacri¢ced6- to 12-wk-old wild-type C57BL/6 J mice and placed in sterile saline.Seven 8- to 12-wk-old Dsg3^/^ mice, which have a mixed geneticbackground of 129/SV and C57BL/6 J, were carefully anesthetized withdimethyl ether and the dorsal skin was replaced with the skin graft. Thegraft was attached with nylon sutures. After topical application ofointment containing gentamicin sulfate (Schering-Plough, Osaka, Japan),the surgical site was dressed with gauze and an elastic bandage for 7^10 d.Typically, successful adaptation was obtained and grafts were maintained atthe surgical site for about 3 wk. Then, grafts began to necrotize and werespontaneously removed within 4 wk probably due to allo-reaction. Then asecond piece of Dsg3þ/þ skin was grafted onto the Dsg3^/^ mice toevaluate the in vivo reaction of anti-Dsg3 IgG. Serum samples from thesemice with grafts were collected once a week. Sera diluted 1 : 1000 wereanalyzed for anti-Dsg1 and anti-Dsg3 IgG with ELISA using recombinantmouse Dsg1 and Dsg3.
MR1/hamster IgG treatment Hamster anti-mouse CD40L mono-clonal antibody MR1was prepared according to the protocol described inthe original report (Noelle et al, 1992). Hybridomas producing MR1wereinjected intraperitoneally into hamsters and the collected ascites waspuri¢ed by ion-exchange high performance liquid chromatography toobtain MR1. The Dsg3^/^ mice with skin grafts were injectedintraperitoneally with either MR1 (n¼ 8) or control hamster IgG (n¼ 8)(Cappel Product, Aurora, OH).The dose of MR1was determined based onprevious reports (Durie et al, 1993;Yang et al, 1996; Stein et al, 1999; 2000) aswell as our pilot study. In our pilot study 125 mg to 1 mg per mouse weretested at various schedules, but 125^250 mg per mouse showed only partialsuppression.Therefore, expecting substantial suppression, we used 1mg permouse at day 0 and 0.5 mg per mouse at days 2, 4, 7, 14, 21, and 28. Serumsamples from these mice were collected once a week and analyzed withELISA using mouse rDsg3 and mouse rDsg1, as described above. Toevaluate whether intercellular deposition of IgG in the epidermis of theskin graft occurred, E2� 6 mm biopsy samples were taken from thesecond skin grafts, about 1 wk after grafting, and examined for IgGdeposition by direct immuno£uorescence. Seven grafts from eightMR1-treated mice and ¢ve grafts from eight control-IgG-treated micewere investigated in this study.
RESULTS
Development of anti-Dsg3 IgG in Dsg3^/^ mice after nakedDNA injection of Dsg3 cDNA We used Dsg3^/^ mice as amodel for recessive genodermatosis. To correct the defect inDsg3 expression, we introduced Dsg3 cDNA by naked DNAinjection. The proper expression of Dsg3 in the epidermis wascon¢rmed by staining skin sections from the injection site for
BLOCKING IMMUNE RESPONSE IN GENE THERAPY 611VOL. 120, NO. 4 APRIL 2003

Dsg3. Distinct positive staining for Dsg3 was observed on thesurfaces of keratinocytes at the sites injected with 5^10 mg per mlpcDNA�mDsg3 (Fig 1a), whereas no such cell surfacestaining for Dsg3 was observed at the sites injected withpcDNA1 (Fig 1b). We then determined whether Dsg3^/^mice develop anti-Dsg3 IgG after naked DNA injection ofpcDNA�mDsg3. Dsg3^/^ mice were injected with variousamounts of pcDNA�mDsg3 in various schedules (Table I), andserial serum samples were evaluated by mouse Dsg3 as well asmouse Dsg1 ELISA. Each mouse in the three groups of Dsg3^/^mice injected with 50 mg pcDNA�mDsg3 per mouse twice perweek (n¼ 2), or 100 mg per mouse once (n¼ 4) or twice (n¼ 2)developed anti-Dsg3 IgG at day 32. Although the anti-Dsg3 IgGlevel detected in these groups was relatively low, more distinctanti-Dsg3 IgG production was observed in two out of twoDsg3^/^ mice injected with 200 mg per mouse every 2 wk(Fig 2a). Therefore, anti-Dsg3 IgG production was observedaltogether in ¢ve of the 10 mice examined when 500 mg permouse or more total naked DNAwas injected. The sera of thesemice with anti-Dsg3 IgG showed no apparent reactivity againstmouse Dsg1 at a mean OD value of 0.04670.011 (n¼ 5). Incontrast to Dsg3^/^ mice, no anti-Dsg3 IgG production wasobserved in two wild-type mice (C57BL/6 J) injected with thesame amount of DNA (Fig 2a).Next, we determined whether the anti-Dsg3 IgG produced
after the naked DNA injection binds to the exogenous Dsg3expressed by naked DNA injection in Dsg3^/^ mice. Biopsieswere taken at the injection sites (footpad) of Dsg3^/^ miceproducing anti-Dsg3 IgG after naked DNA injection, and in vivoIgG deposition was examined by direct immuno£uorescence(Fig 2b). Obvious in vivo IgG deposition was noticed onkeratinocyte cell surfaces in six out of 36 biopsy sections. Thisindicated that the anti-Dsg3 IgG produced after the naked DNAinjection of Dsg3 cDNA could bind to gene therapy transgeneproducts in vivo.
Blockade of CD40L prevented anti-Dsg3 IgG production inDsg3^/^ mice with Dsg3þ/þ skin graft Achieving stablegene expression of the transgene is one of the ultimate goals ofgene therapy. To mimic the state of stable gene expression, wegrafted skin from wild-type Dsg3þ/þmice onto Dsg3^/^mice instead of injecting Dsg3 cDNA. In this model,production of anti-Dsg3 IgG was observed in all of the micetested 2 wk after skin grafting, and the titers increased thereafter(Fig 3, n¼ 7). The sera of these mice did not show any cross-
Figure1. Dsg3 expressed on keratinocyte cell surfaces followingnaked DNA injection. Positive staining for Dsg3 was observed on thesurfaces of keratinocytes at the site of pcDNA�mDsg3 injection into afootpad of a Dsg3^/^ mouse (a), but not at the site of control DNA injec-tion (b). Scale bar: 50 mm.
Table I. Production of anti-Dsg3 IgG in Dsg3^/^ mice after naked DNA injection of pcDNA�mDsg3
Dose per injection per mouse n Schedule Mice with anti-Dsg3 IgG Total DNA injecteda Time requiredb
50 mg 2 Twice per week 1 500 mg 32 d100 mg 4 Once per week 1 500 mg 32 d100 mg 2 Twice per week 1 1000 mg 32 d200 mg 2 Once every 2 wk 2 600/800 mg 35/56 daTotal DNA injected when anti-Dsg3 IgG production was observed.bTime required for development of anti-Dsg3 IgG.
Figure 2. Anti-Dsg3 IgG production in Dsg3^/^ mice after nakedDNA injection. (a) Anti-Dsg3 IgG production was measured by ELISAover time after naked DNA injection. Dsg3^/^ mice injected withpcDNA�mDsg3 (200 mg per mouse, every 2 wk) developed anti-Dsg3IgG. No such IgG elevation was observed in Dsg3þ/þ control mice, how-ever. (b) In vivo deposition of IgG in the epidermis was observed (arrow) atthe site of pcDNA�mDsg3 injection in a Dsg3^/^ mouse that was produ-cing anti-Dsg3 IgG. Scale bar: 50 mm.
612 OHYAMA ETAL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
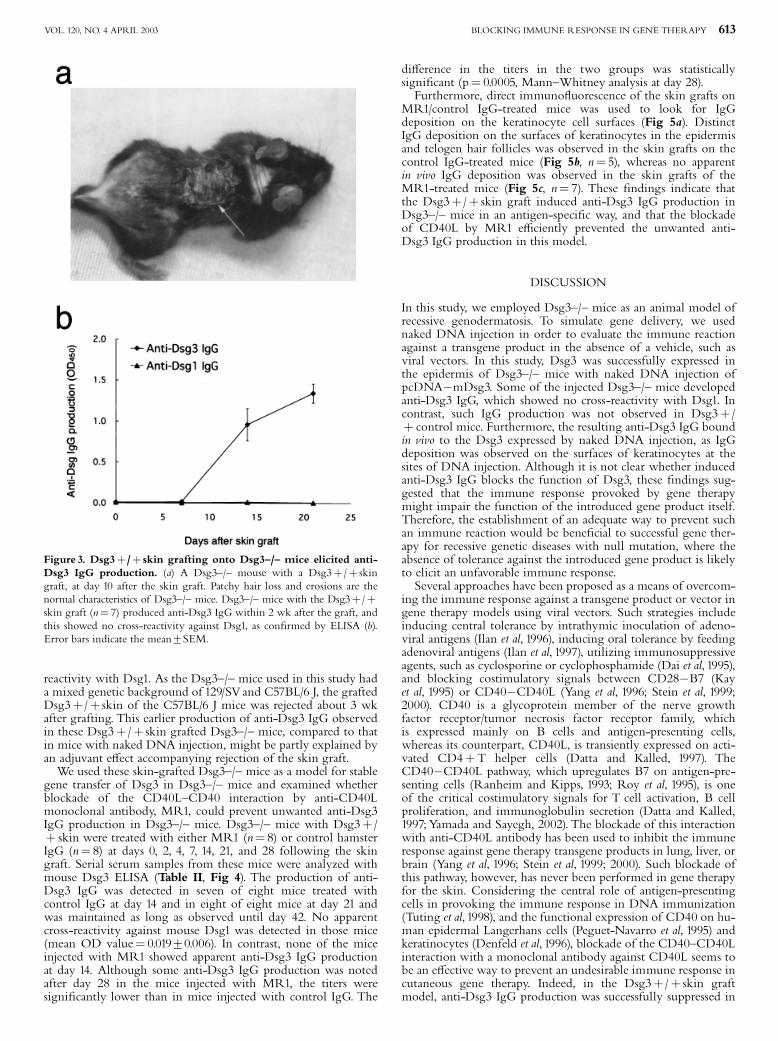
reactivity with Dsg1. As the Dsg3^/^ mice used in this study hada mixed genetic background of 129/SVand C57BL/6 J, the graftedDsg3þ/þ skin of the C57BL/6 J mice was rejected about 3 wkafter grafting. This earlier production of anti-Dsg3 IgG observedin these Dsg3þ/þ skin grafted Dsg3^/^ mice, compared to thatin mice with naked DNA injection, might be partly explained byan adjuvant e¡ect accompanying rejection of the skin graft.We used these skin-grafted Dsg3^/^ mice as a model for stable
gene transfer of Dsg3 in Dsg3^/^ mice and examined whetherblockade of the CD40L^CD40 interaction by anti-CD40Lmonoclonal antibody, MR1, could prevent unwanted anti-Dsg3IgG production in Dsg3^/^ mice. Dsg3^/^ mice with Dsg3þ/þ skin were treated with either MR1 (n¼ 8) or control hamsterIgG (n¼ 8) at days 0, 2, 4, 7, 14, 21, and 28 following the skingraft. Serial serum samples from these mice were analyzed withmouse Dsg3 ELISA (Table II, Fig 4). The production of anti-Dsg3 IgG was detected in seven of eight mice treated withcontrol IgG at day 14 and in eight of eight mice at day 21 andwas maintained as long as observed until day 42. No apparentcross-reactivity against mouse Dsg1 was detected in those mice(mean OD value¼ 0.01970.006). In contrast, none of the miceinjected with MR1 showed apparent anti-Dsg3 IgG productionat day 14. Although some anti-Dsg3 IgG production was notedafter day 28 in the mice injected with MR1, the titers weresigni¢cantly lower than in mice injected with control IgG. The
di¡erence in the titers in the two groups was statisticallysigni¢cant (p¼ 0.0005, Mann^Whitney analysis at day 28).Furthermore, direct immuno£uorescence of the skin grafts on
MR1/control IgG-treated mice was used to look for IgGdeposition on the keratinocyte cell surfaces (Fig 5a). DistinctIgG deposition on the surfaces of keratinocytes in the epidermisand telogen hair follicles was observed in the skin grafts on thecontrol IgG-treated mice (Fig 5b, n¼ 5), whereas no apparentin vivo IgG deposition was observed in the skin grafts of theMR1-treated mice (Fig 5c, n¼ 7). These ¢ndings indicate thatthe Dsg3þ/þ skin graft induced anti-Dsg3 IgG production inDsg3^/^ mice in an antigen-speci¢c way, and that the blockadeof CD40L by MR1 e⁄ciently prevented the unwanted anti-Dsg3 IgG production in this model.
DISCUSSION
In this study, we employed Dsg3^/^ mice as an animal model ofrecessive genodermatosis. To simulate gene delivery, we usednaked DNA injection in order to evaluate the immune reactionagainst a transgene product in the absence of a vehicle, such asviral vectors. In this study, Dsg3 was successfully expressed inthe epidermis of Dsg3^/^ mice with naked DNA injection ofpcDNA�mDsg3. Some of the injected Dsg3^/^ mice developedanti-Dsg3 IgG, which showed no cross-reactivity with Dsg1. Incontrast, such IgG production was not observed in Dsg3þ/þ control mice. Furthermore, the resulting anti-Dsg3 IgG boundin vivo to the Dsg3 expressed by naked DNA injection, as IgGdeposition was observed on the surfaces of keratinocytes at thesites of DNA injection. Although it is not clear whether inducedanti-Dsg3 IgG blocks the function of Dsg3, these ¢ndings sug-gested that the immune response provoked by gene therapymight impair the function of the introduced gene product itself.Therefore, the establishment of an adequate way to prevent suchan immune reaction would be bene¢cial to successful gene ther-apy for recessive genetic diseases with null mutation, where theabsence of tolerance against the introduced gene product is likelyto elicit an unfavorable immune response.Several approaches have been proposed as a means of overcom-
ing the immune response against a transgene product or vector ingene therapy models using viral vectors. Such strategies includeinducing central tolerance by intrathymic inoculation of adeno-viral antigens (Ilan et al, 1996), inducing oral tolerance by feedingadenoviral antigens (Ilan et al, 1997), utilizing immunosuppressiveagents, such as cyclosporine or cyclophosphamide (Dai et al, 1995),and blocking costimulatory signals between CD28�B7 (Kayet al, 1995) or CD40�CD40L (Yang et al, 1996; Stein et al, 1999;2000). CD40 is a glycoprotein member of the nerve growthfactor receptor/tumor necrosis factor receptor family, whichis expressed mainly on B cells and antigen-presenting cells,whereas its counterpart, CD40L, is transiently expressed on acti-vated CD4þT helper cells (Datta and Kalled, 1997). TheCD40�CD40L pathway, which upregulates B7 on antigen-pre-senting cells (Ranheim and Kipps, 1993; Roy et al, 1995), is oneof the critical costimulatory signals for T cell activation, B cellproliferation, and immunoglobulin secretion (Datta and Kalled,1997;Yamada and Sayegh, 2002). The blockade of this interactionwith anti-CD40L antibody has been used to inhibit the immuneresponse against gene therapy transgene products in lung, liver, orbrain (Yang et al, 1996; Stein et al, 1999; 2000). Such blockade ofthis pathway, however, has never been performed in gene therapyfor the skin. Considering the central role of antigen-presentingcells in provoking the immune response in DNA immunization(Tuting et al, 1998), and the functional expression of CD40 on hu-man epidermal Langerhans cells (Peguet-Navarro et al, 1995) andkeratinocytes (Denfeld et al, 1996), blockade of the CD40^CD40Linteraction with a monoclonal antibody against CD40L seems tobe an e¡ective way to prevent an undesirable immune response incutaneous gene therapy. Indeed, in the Dsg3þ/þ skin graftmodel, anti-Dsg3 IgG production was successfully suppressed in
Figure 3. Dsg3þ/þ skin grafting onto Dsg3^/^ mice elicited anti-Dsg3 IgG production. (a) A Dsg3^/^ mouse with a Dsg3þ/þ skingraft, at day 10 after the skin graft. Patchy hair loss and erosions are thenormal characteristics of Dsg3^/^ mice. Dsg3^/^ mice with the Dsg3þ/þskin graft (n¼ 7) produced anti-Dsg3 IgG within 2 wk after the graft, andthis showed no cross-reactivity against Dsg1, as con¢rmed by ELISA (b).Error bars indicate the mean7SEM.
BLOCKING IMMUNE RESPONSE IN GENE THERAPY 613VOL. 120, NO. 4 APRIL 2003

MR1-treated mice, whereas all of the control IgG-treated micedeveloped the undesired IgG within 2^3 wk, and the titer ofIgG was maintained at high levels for more than 6 wk. Further-more, no in vivo IgG deposition on keratinocyte surfaces wasobserved in the epidermis of skin grafts on the mice treated withMR1. The advantages of anti-CD40L therapy are that it does notrequire prior knowledge of the structure of the autoantigen andthat its e¡ect is relatively speci¢c because the CD40L molecule isexpressed mainly on activated T cells (Datta and Kalled, 1997).Unlike other immunosuppressive agents, such as corticosteroidsor cyclosporine, generalized immunosuppression was not observed
in previous murine studies of anti-CD40L antibody (Durie et al,1993; 1994; Mohan et al, 1995; Early et al, 1996; Gerriset et al, 1996).Although additional evidence is needed before it can be appliedin practice, the blockade of the CD40^CD40L interaction withmonoclonal antibody appears to be a valuable way to prevent anunwanted immune response against a transgene product, a re-sponse that may well be provoked in gene therapy.Taking all these ¢ndings into consideration, we conclude that
an unwanted immune response against a desired gene productmay be provoked in gene therapy for recessive disease, and thatadequate prevention of such an unfavorable immune reaction isrequired for the practical application of this promising therapy.
We would like to thank Dr. ShigeruTanaka for preparing the MR1 and Dr. JouniUitto for providing the mouse Dsg3 cDNA.We would also like to thank Mr.TakashiKimura and all of the technical assistants in the Research Laboratory at theTokyoElectric Power Company Hospital for their general research support, including animalcare, and Ms. Minae Suzuki for performing the immuno£uorescence sectioning.Thiswork was supported by Health Science Research Grants for Research on Speci¢cDiseases from the Ministry of Health, Labor, and Welfare, and by Grants-in-Aidof Scienti¢c Research from the Ministry of Education, Culture, Sports, Science, andTechnology of Japan.
Table II. Anti-Dsg3 IgG production (OD450) in MR1/control IgG treated Dsg3^/^ mice with Dsg3þ/þ skin graft
Time course (d)Mouse 0 7 14 21 28 35 42
MR1-treated mice# 1 0.014 0.011 0.009 0.002 0.017 0.003 ND# 2 0.013 0.016 0.025 0.014 0.042 0.047 0.620# 3 0.013 0.013 0.044 0.070 0.085 0.210 0.130# 4 0.013 0.007 0.049 0.043 0.600 0.780 0.110# 5 0.011 0.005 0.017 ND ND ND ND# 6 0.010 0.014 0.014 0.014 0.140 0.450 0.590# 7 0.010 0.005 0.002 0.027 0.017 0.049 ND# 8 0.005 0.003 0.009 0.098 0.600 0.600 0.007Meanþ SEM 0.011þ0.001 0.004þ 0.002 0.021þ0.006 0.038þ 0.012 0.210þ 0.094 0.310þ 0.11 0.290þ 0.130Control IgG-treated mice# 9 0.050 0.004 1.200 1.520 1.420 1.400 ND# 10 0.015 0.015 0.580 0.970 0.430 0.700 1.310# 11 0.014 0.012 0.530 1.060 1.300 1.300 1.300# 12 0.008 0.006 0.330 1.300 2.140 2.360 1.210# 13 0.006 0.014 1.160 0.790 0.570 0.600 1.860# 14 0.006 0.007 1.160 1.750 1.760 1.980 0.990# 15 0.008 0.005 0.920 1.300 1.380 1.610 ND# 16 0.004 0.011 0.084 1.520 1.170 1.110 NDMeanþ SEM 0.014þ 0.005 0.009þ 0.002 0.750þ 0.150 1.280þ 0.110 1.270þ 0.200 1.380þ 0.210 1.330þ 0.140
OD values above the cuto¡ values are in bold. ND indicates not determined.
Figure 4. MR1 e¡ectively suppressed the anti-Dsg3 IgG productionprovoked by grafting Dsg3þ/þ skin on Dsg3^/^ mice. All of thecontrol IgG-treated mice developed anti-Dsg3 IgG about 2 wk after graft-ing and this IgG production was maintained at high levels for more than 6wk. This immune reaction was e¡ectively suppressed when recipient micewere treated with MR1. Error bars indicate the mean7SEM. nMean wasobtained with ¢ve mice at day 42.
Figure 5. IgG deposition on the surfaces of keratinocytes in Dsg3þ/þgrafts on Dsg3^/^ mice and its e⁄cient prevention with MR1.Dsg3^/^ mice with Dsg3þ/þ skin grafts were treated with control ham-ster IgG (a, left) or MR1 (a, right). IgG deposition on keratinocyte cell sur-faces was detected in the grafts from control IgG-treated mice (b), whereasthis deposition was prevented in the grafts from MR1-treated mice (c).Scale bars: 1 cm for (a); 50 mm for (b), (c).
614 OHYAMA ETAL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY

REFERENCES
Amagai M, Klaus-Kovtun V, Stanley JR: Autoantibodies against a novel epithelialcadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 67:869^877,1991
Amagai M, HashimotoT, Shimizu N, NishikawaT: Absorption of pathogenic auto-antibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3)produced by baculovirus. J Clin Invest 94:59^67, 1994
Amagai M,Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, NishikawaT: Use of auto-antigen-knockout mice in developing an active autoimmune disease model forpemphigus. J Clin Invest 105:625^631, 2000
Chen M, O’Toole EA, Muellenho¡ M, Medina E, Kasahara N,Woodley DT: Devel-opment and characterization of a recombinant truncated type Vlll collagen‘minigene’ Implication for gene therapy of dystrophic epidermolysis bullosa.J Biol Chem 275:24429^24435, 2000
Choate KA, Medalie DA, Morgan JR, Khavari PA: Corrective gene transfer in thehuman skin disorder lamellar ichthyosis. Nat Med 2:1263^1267, 1996
Dai Y, Schwarz EM, Gu D, ZhangW-W, Sarvetnick N,Verma IM: Cellular and hu-moral immune responses to adenoviral vectors containing factor lX gene: tol-erization of factor lX and vector antigens allows for long-term expression. ProcNatl Acad Sci USA 92:1401^1405, 1995
Datta SK, Kalled SL: CD40^CD40 ligand interaction in autoimmune disease.Arthri-tis Rheum 40:1735^1745, 1997
Dellambra E,Vailly J, Pellegrini G, et al: Corrective transduction of human epidermalstem cells in laminin-5-dependent junctional epidermolysis bullosa. HumGeneTher 9:1359^1370, 1998
Denfeld RW, Hollenbaugh D, Fehrenbach A, et al: CD40 is functionally expressed inhuman keratinocytes. Eur J Immunol 26:2329^2334, 1996
Durie FH, Fava RA, FoyTM, Aru¡o A, Ledbetter JA, Noelle RJ: Prevention of col-lagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science261:1328^1330, 1993
Durie FH, Aru¡o A, Ledbetter J, Crassi KM, GreenWR, Fast LD, Noelle RJ: Anti-body to ligand of CD40, gp39, blocks the occurrence of the acute and chronicforms of graft-versus-host disease. J Clin Invest 94:1333^1338, 1994
Early GS, ZhaoW, Burns CM: Anti-CD40 ligand antibody treatment prevents thedevelopment of lupus-like nephlitis in a subset of New Zealand black� NewZealand white mice: response correlates with the absence of an anti-antibodyresponse. J Immunol 157:3159^3164, 1996
Freiberg RA, Choate KA, Deng H, Alperin ES, Shapiro LJ, Khavari PA: A modelof corrective gene transfer in X-linked ichthyosis. Hum Mol Genet 6:927^933,1997
Gerritse K, Laman JD, Noelle RJ, Aru¡o A, Ledbetter JA, BoersmaWJA, ClaassenE: CD40^CD40 ligand interactions in experimental allergic encephalomyelitisand multiple sclerosis. Proc Natl Acad Sci USA 93:2499^2504, 1996
Greenhalgh DA, Rothnagel JA, Roop DR: Epidermis: an attractive target tissue forgene therapy. J Invest Dermatol 103:63S^69S, 1994
Hengge UR, Chan EF, Foster RA,Walker PS, Vogel JC: Cytokine gene expressionin epidermis with biological e¡ects following injection of naked DNA. NatureGenet 10:161^166, 1995
Hengge UR,Walker PS, Vogel JC: Expression of naked DNA in human, pig, andmouse skin. J Clin Invest 97:2911^2916, 1996
Ilan Y, Attavar P, Takahashi M, et al: Induction of central tolerance by intrathymicinoculation of adenoviral antigens into the host thymus permits long-termgene therapy in gunn rats. J Clin Invest 98:2640^2647, 1996
IlanY, Prakash R, Davidson A, et al: Oral tolerization to adenoviral antigens permitslong-term gene expression using recombinant adenoviral vectors. J Clin Invest99:1098^1106, 1997
Jensen TG, Jensen UB, Jensen PK, Ibsen HH, Brandrup F, Ballabio A, Bolund L:Correction of steroid sulphatese de¢ciency by gene transfer into basal cells oftissue-cultured epidermis from patients with recessive X-linked ichthyosis. ExpCell Res 209:392^397, 1993
Kay MA, Holterman AX, Meuse L, Allen G, Ochs HD, Linsley PS, Wilson CB:Long-term hepatic adenovirus-mediated gene expression in mice followingCTLA4Ig administration. Nat Genet 11:191^197, 1995
Khavari PA: Gene therapy for genetic skin disease. J Invest Dermatol 110:462^467, 1998Khavari PA: Genetic correction of inherited epidermal disorders. Hum Gene Ther
11:2277^2282, 2000Koch PJ, Mahoney MG, Ishikawa H, et al: Targeted disruption of the pemphigus
vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocytecell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol137:1091^1102, 1997
Larregina AT, Falo LD: Generating and regulating immune responses through cuta-neous gene delivery. Hum GeneTher 11:2301^2305, 2000
Mohan C, Shi Y, Laman JD, Datta SK: Interaction between CD40 and its ligandgp39 in the development of murine lupus nephritis. J Immunol 154:1470^1480,1995
Morral N, O’nealW, Zhou H, Langston C, Beaudet A: Immune response to reporterproteins and high viral dose limit duration of expression with adenoviral vec-tors: comparison of E2a wild type and E2a deleted vectors. Hum GeneTher8:1275^1286, 1997
Noelle RJ, Meenakshi R, Shepherd DM, Stamenkovic I, Ledbetter JA, Aru¡o A: A39-kDa protein on activated helper T cells binds CD40 and transduces the sig-nal for cognate activation of B cells. Proc Natl Acad Sci USA 89:6550^6554, 1992
Ohyama M, Amagai M,Tsunoda K, et al: Immunologic and histopathologic charac-terization of active disease mouse model for pemphigus vulgaris. J Invest Der-matol 118:199^204, 2002
Peguet-Navarro J, Dalbiez-Gauthier C, Rattis FM, Van Kooten C, Banchereau J,Schmitt D: Functional expression of CD40 antigen on human epidermalLangerhans cells. J Immunol 155:4241^4247, 1995
Ranheim EA, Kipps TJ: Activated T-cells induce expression of B7/BB1 on normal orleukemic B cells through a CD40-dependent signal. J Exp Med 177:925^935,1993
Roy M, Aru¡o A, Ledbetter J, Linsley P, Kehry M, Noelle R: Studies on the inter-dependence of gp39 and B7 expression and function during antigen-speci¢cimmune responses. Eur J Immunol 25:596^603, 1995
Seitz CS, Giudice GI, Balding SD, Marinkovich MP, Khavari PA: BP180 gene deliv-ery in junctional epidermolysis bullosa. GeneTher 6:42^47, 1999
Stein CS, Ghodsi A, Derksen T, Davidson BL: Systemic and central nervous systemcorrection of lysosomal storage in mucopolysaccharidosis typeVII mice. J Virol73:3424^3429, 1999
Stein CS, Martins I, Davidson BL: Long-term reversal of hypercholesterolemia inlow density lipoprotein receptor (LDLR) de¢cient mice by adenovirus-mediated LDLR gene transfer combined with CD154 blockade. J Gene Med2:41^51, 2000
Tripathy SK, Black HB, Goldwasser E, Leiden JM: Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of repli-cation-defective adenovirus vectors. Nat Med 2:545^550, 1996
Tuting T, Storkus WJ, Falo LD: DNA immunization targeting the skin: molecularcontrol of adaptive immunity. J Invest Dermatol 111:183^188, 1998
Uitto J, Pulkkinen L: The genodermatoses: candidate disease for gene therapy. HumGeneTher 11:2267^2275, 2000
Vailly J, Gagnoux-Palacios L, Dell’Ambra E, et al: Corrective gene transfer of kerati-nocytes from patients with junctional epidermolysis bullosa restores assemblyof hemidesmosomes in reconstructed epithelia. GeneTher 5:1322^1322, 1998
Vogel JC: Keratinocyte gene therapy. Arch Dermatol 129:1478^1483, 1993Yamada A, Sayegh MH: The CD154�CD40 costimulatory pathway in transplanta-
tion.Transplantation 73:S36^S39, 2002YangY, Su Q, Grewal IS, Schilz R, Flavell RA,Wilson JM: Transient subversion of
CD40 ligand function diminishes immune response to adenovirus vectors inmouse liver and lung tissues. J Virol. 70:6370^6377, 1996
Zeng L, Sarasin A, Mezzina M: Retrovirus-mediated DNA repair gene transfer intoxeroderma pigmentosum cells. Cell BiolToxicol 14:105^110, 1998
BLOCKING IMMUNE RESPONSE IN GENE THERAPY 615VOL. 120, NO. 4 APRIL 2003





























