Embed Size (px)
Citation preview

SURFACE AND INTERFACE ANALYSIS, VOL. 24, 51-58 (1996)
Surface Modification of Electroactive Polymer Films by Ozone Treatment
E. T. Kang,* K. G. Neob and X. Zbang Department of Chemical Engineering, National University of Singapore, Kent Ridge, Singapore 051 1
K. L. Tan Department of Physics, National University of Singapore, Kent Ridge, Singapore 051 1
D. J. Liaw Department of Chemical Engineering, National Taiwan Institute of Technology, Taipei, Taiwan 106, Republic of China
Surface modification of polypyrrole (PPY), polyaniline (PAN) and poly(3-alkylthiophene) films by ozone was studied by angle-resolved x-ray photoelectron spectroscopy (XPS). In the case of the N-containing polymers, oxida- tion occurs readily and mainly at the carbon atoms, resulting in the formation of C-0, C=O, COOH and even 0-COOH (especially for PAN) species. The carbon atoms of doped PPY and PAN films are significantly more resistant to ozone oxidation, but the samples suffer some loss of the dopant in the surface region. Ozone treatment does not involve the direct oxidation of the nitrogen heteroatoms to form the -NO, species, even at high extent of carbon oxidation. In addition, a substantial decrease in the intrinsic oxidation state ([=N- ]/[-NH- ] ratio) was observed in the 25% deprotonated PPY (DP-PPY) base, the 50% intrinsically oxidized emeraldine (EM) base and the 75% intrinsically oxidized nigraniliae (NA) base after ozone treatment and subsequent atmospheric expo- sure. In the case of the S-containing polymers, ozone treatment results predominantly in the oxidation of sulphur heteroatoms to give rise to the sulphone and peroxide species. The oxidized sulphur species, however, are readily reduced or consumed in a photochemical reaction.
~~
INTRODUCTION
During the past two decades, the synthesis and charac- terization of electroactive polymers have quickly become one of the most important research areas in polymer science.' Among the electroactive polymers, the N-containing polymers, such as polyaniline (PAN) and polypyrrole (PPY), and the S-containing polymers, such as polythiophene (PTH), have been of particular interest. They have controllable electrical conductivi- t ~ , ' - ~ environmental and interesting redox properties associated with chain heteroatoms.'-'' Some of these polymers also exhibits solution- and counterion-induced processability."-'6 Earlier surface studies on the polyacetylene films" have revealed much valuable information about the structural modifications of the film surfaces resulting from interactions with the environment. Owing to the reactive nature of the conju- gated polymer surface, most of the polyacetylene film surfaces are oxidized to some The forma- tion of covalent bonds between carbon and oxygen reduces the effective conjugation length of the polymer and renders it less conductive. The N-containing and S-containing electroactive polymers, on the other hand, do not interact strongly with oxygen in the atmo-
Th us to elucidate more precisely the surface oxidation processes of these polymers, a more drastic means of surface oxidation is needed. In this work, the surfaces of polyaniline, polypyrrole and poly(3-alkylthiophene) films are subjected to various
* Author to whom correspondence should be addressed.
CCC 0142-2421/96/0lOoSl-13 0 1996 by John Wiley & Sons, Ltd.
extents of oxidation by ozone. The ozone-treated sur- faces are subsequently analysed using XPS as a primary tool. Particular attention is given to the changes in the intrinsic redox states ([=N-I/[-NH-] ratios) of PAN and PPY as a result of ozone exposure.
EXPERIMENTAL
Polymer synthesis
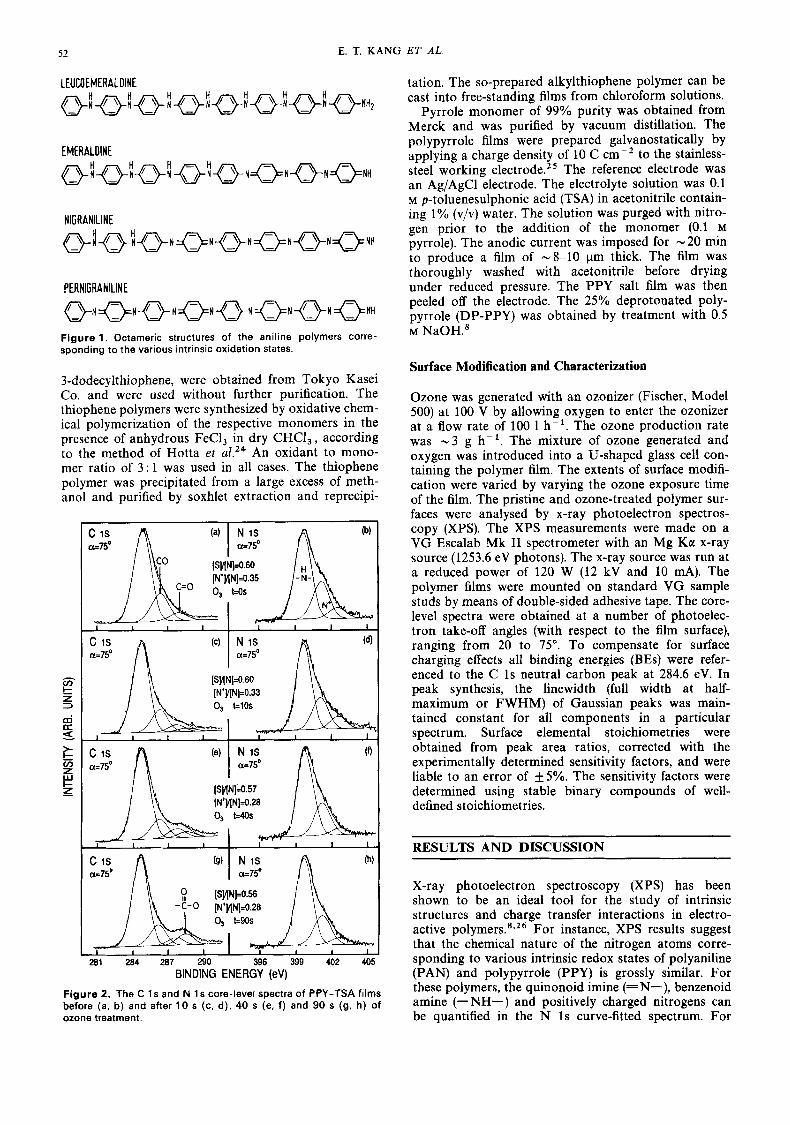
The chemical polymerization of aniline was carried out at - 5 "C using (NH4),S20, as the oxidant, similar to the method described in the literature,' except that 1 M HzS04 was used instead of 1 M HCl. The 50% intrinsi- cally oxidized emeraldine (EM) base was obtained by treating the polyaniline salt with excess 0.5 M NaOH. The base polymer was then cast into thin films of - 8-10 pm thick from N-methylpyrrolidinone (NMP) solution. The residual solvent trapped in the polymer films was removed by exhaustive pumping. The 75% intrinsically oxidized nigraniline (NA) film was obtained by subjecting the EM film to one cycle of acid-base treatment.22 The fully reduced leucoemeraldine (LM) base was obtained via the reduction of emeraldine films with phenylhydrazine, according to the method of Green and W ~ o d h e a d . ~ ~ The chemical structures, depicted in their respective octameric forms, for the various intrinsic oxidation states of PAN are shown in Fig. 1.
The 3-alkylthiophene monomers used in the present study, namely 3-hexylthiophene, 3-octylthiophene and
Receiued 5 M a y I995 Accepted 22 September 1995
E. T. KANG ET A L 52
LEUCOEMERALDINE
EMERALOINE
NlGRANlLlNE
PERNlGRANlLlNE
Figure 1. Octameric structures of the aniline polymers corre- sponding to the various intrinsic oxidation states.
3-dodecylthiophene, were obtained from Tokyo Kasei Co. and were used without further purification. The thiophene polymers were synthesized by oxidative chem- ical polymerization of the respective monomers in the presence of anhydrous FeCl, in dry CHCl,, according to the method of Hotta et uLz4 An oxidant to mono- mer ratio of 3 : 1 was used in all cases. The thiophene polymer was precipitated from a large excess of meth- anol and purified by soxhlet extraction and reprecipi-
281 284 287 290 396 399 402 405 BINDING ENERGY (eV)
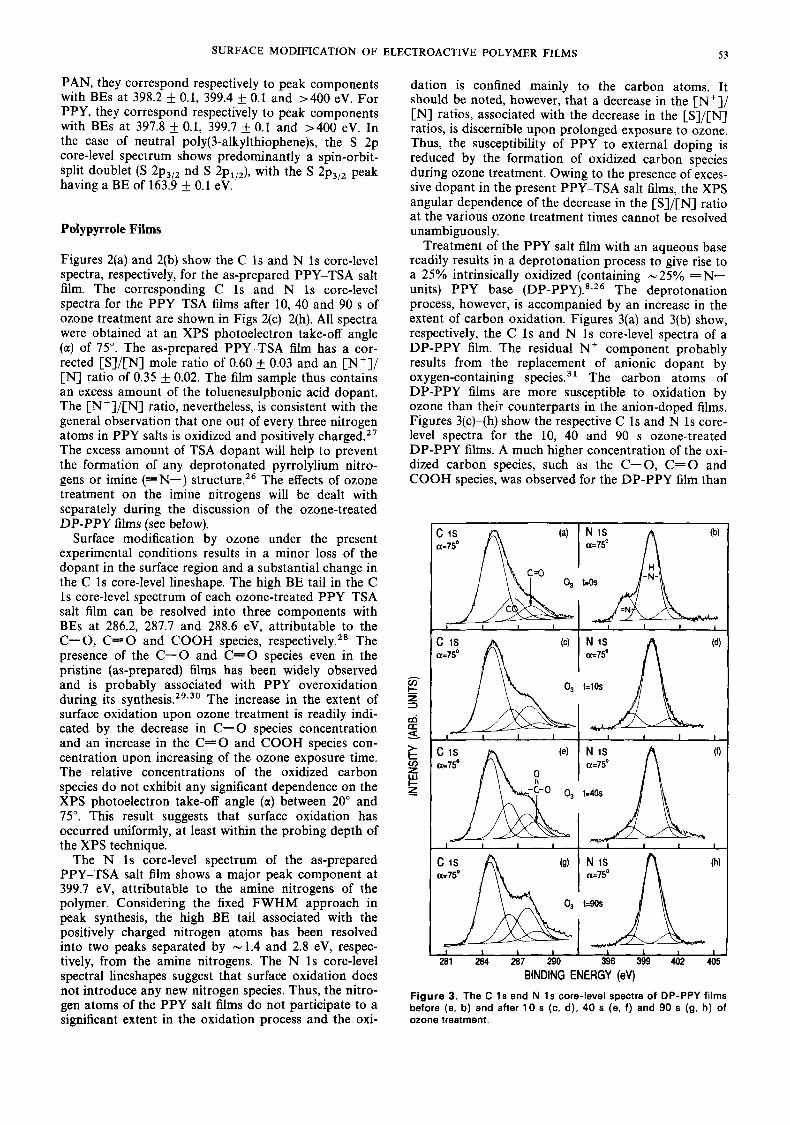
Figure 2. The C 1s and N 1s core-level spectra of PPY-TSA films before (a, b) and after 10 s (c, d), 40 s (e, f) and 90 s (9, h) of ozone treatment.
tation. The so-prepared alkylthiophene polymer can be cast into free-standing films from chloroform solutions.
Pyrrole monomer of 99% purity was obtained from Merck and was purified by vacuum distillation. The polypyrrole films were prepared galvanostatically by applying a charge density of 10 C cm ~ to the stainless- steel working ele~trode. '~ The reference electrode was an Ag/AgCl electrode. The electrolyte solution was 0.1 M p-toluenesulphonic acid (TSA) in acetonitrile contain- ing 1% (v/v) water. The solution was purged with nitro- gen prior to the addition of the monomer (0.1 M pyrrole). The anodic current was imposed for -20 min to produce a film of -8-10 pm thick. The film was thoroughly washed with acetonitrile before drying under reduced pressure. The PPY salt film was then peeled off the electrode. The 25% deprotonated poly- pyrrole (DP-PPY) was obtained by treatment with 0.5 M NaOH.'
Surface Modification and Characterization
Ozone was generated with an ozonizer (Fischer, Model 500) at 100 V by allowing oxygen to enter the ozonizer at a flow rate of 100 1 h-'. The ozone production rate was - 3 g hK1. The mixture of ozone generated and oxygen was introduced into a U-shaped glass cell con- taining the polymer film. The extents of surface modifi- cation were varied by varying the ozone exposure time of the film. The pristine and ozone-treated polymer sur- faces were analysed by x-ray photoelectron spectros- copy (XPS). The XPS measurements were made on a VG Escalab Mk I1 spectrometer with an Mg Ka x-ray source (1253.6 eV photons). The x-ray source was run at a reduced power of 120 W (12 kV and 10 mA). The polymer films were mounted on standard VG sample studs by means of double-sided adhesive tape. The core- level spectra were obtained at a number of photoelec- tron take-off angles (with respect to the film surface), ranging from 20 to 75". To compensate for surface charging effects all binding energies (BEs) were refer- enced to the C 1s neutral carbon peak at 284.6 eV. In peak synthesis, the linewidth (full width at half- maximum or FWHM) of Gaussian peaks was main- tained constant for all components in a particular spectrum. Surface elemental stoichiometries were obtained from peak area ratios, corrected with the experimentally determined sensitivity factors, and were liable to an error of f5%. The sensitivity factors were determined using stable binary compounds of well- defined stoichiometries.
RESULTS AND DISCUSSION
X-ray photoelectron spectroscopy (XPS) has been shown to be an ideal tool for the study of intrinsic structures and charge transfer interactions in electro- active polymers.8~26 For instance, XPS results suggest that the chemical nature of the nitrogen atoms corre- sponding to various intrinsic redox states of polyaniline (PAN) and polypyrrole (PPY) is grossly similar. For these polymers, the quinonoid imine (= N-), benzenoid amine (-NH-) and positively charged nitrogens can be quantified in the N 1s curve-fitted spectrum. For
53 SURFACE MODIFICATION OF ELECTROACTIVE POLYMER FILMS
PAN, they correspond respectively to peak components with BEs at 398.2 f 0.1, 399.4 & 0.1 and >400 eV. For PPY, they correspond respectively to peak components with BEs at 397.8 f 0.1, 399.7 f 0.1 and >400 eV. In the case of neutral poly(3-alkylthiophene)s, the S 2p core-level spectrum shows predominantly a spin-orbit- split doublet (S 2p,,, nd S 2p,/,), with the S 2p,,, peak having a BE of 163.9 f 0.1 eV.
Polypyrrole Films
Figures 2(a) and 2(b) show the C 1s and N 1s core-level spectra, respectively, for the as-prepared PPY-TSA salt film. The corresponding C 1s and N 1s core-level spectra for the PPY-TSA films after 10, 40 and 90 s of ozone treatment are shown in Figs 2(c)-2(h). All spectra were obtained at an XPS photoelectron take-off angle (a) of 75". The as-prepared PPY-TSA film has a cor- rected [S]/[N] mole ratio of 0.60 L 0.03 and an [N']/ [N] ratio of 0.35 f 0.02. The film sample thus contains an excess amount of the toluenesulphonic acid dopant. The [N']/[N] ratio, nevertheless, is consistent with the general observation that one out of every three nitrogen atoms in PPY salts is oxidized and positively ~harged . '~ The excess amount of TSA dopant will help to prevent the formation of any deprotonated pyrrolylium nitro- gens or imine (=N-) structure.26 The effects of ozone treatment on the imine nitrogens will be dealt with separately during the discussion of the ozone-treated DP-PPY films (see below).
Surface modification by ozone under the present experimental conditions results in a minor loss of the dopant in the surface region and a substantial change in the C 1s core-level lineshape. The high BE tail in the C 1s core-level spectrum of each ozone-treated PPY-TSA salt film can be resolved into three components with BEs at 286.2, 287.7 and 288.6 eV, attributable to the C-0, C - 0 and COOH species, respectively.28 The presence of the C - 0 and C=O species even in the pristine (as-prepared) films has been widely observed and is probably associated with PPY overoxidation during its s y n t h e s i ~ . ' ~ * ~ ~ The increase in the extent of surface oxidation upon ozone treatment is readily indi- cated by the decrease in C-0 species concentration and an increase in the C=O and COOH species con- centration upon increasing of the ozone exposure time. The relative concentrations of the oxidized carbon species do not exhibit any significant dependence on the XPS photoelectron take-off angle (a) between 20" and 75". This result suggests that surface oxidation has occurred uniformly, at least within the probing depth of the XPS technique.
The N 1s core-level spectrum of the as-prepared PPY-TSA salt film shows a major peak component at 399.7 eV, attributable to the amine nitrogens of the polymer. Considering the fixed FWHM approach in peak synthesis, the high BE tail associated with the positively charged nitrogen atoms has been resolved into two peaks separated by -1.4 and 2.8 eV, respec- tively, from the amine nitrogens. The N 1s core-level spectral lineshapes suggest that surface oxidation does not introduce any new nitrogen species. Thus, the nitro- gen atoms of the PPY salt films do not participate to a significant extent in the oxidation process and the oxi-
dation is confined mainly to the carbon atoms. It should be noted, however, that a decrease in the [Nil/ [N] ratios, associated with the decrease in the [S]/[N] ratios, is discernible upon prolonged exposure to ozone. Thus, the susceptibility of PPY to external doping is reduced by the formation of oxidized carbon species during ozone treatment. Owing to the presence of exces- sive dopant in the present PPY-TSA salt films, the XPS angular dependence of the decrease in the [S]/[N] ratio at the various ozone treatment times cannot be resolved unambiguously.
Treatment of the PPY salt film with an aqueous base readily results in a deprotonation process to give rise to a 25% intrinsically oxidized (containing - 25% = N- units) PPY base (DP-PPY).8*26 The deprotonation process, however, is accompanied by an increase in the extent of carbon oxidation. Figures 3(a) and 3(b) show, respectively, the C 1s and N 1s core-level spectra of a DP-PPY film. The residual N+ component probably results from the replacement of anionic dopant by oxygen-containing species.31 The carbon atoms of DP-PPY films are more susceptible to oxidation by ozone than their counterparts in the anion-doped films. Figures 3(c)-(h) show the respective C 1s and N 1s core- level spectra for the 10, 40 and 90 s ozone-treated DP-PPY films. A much higher concentration of the oxi- dized carbon species, such as the C-0, C=O and COOH species, was observed for the DP-PPY film than
c
I I I I I I I 1
281 284 287 290 396 399 402 405 BINDING ENERGY (eV)
Figure 3. The C 1s and N 1s core-level spectra of DP-PPY films before (a. b) and after 10 s (c, d), 40 s (a, f ) and 90 s (g, h) of ozone treatment.

E. T. KANG ET A t .
I I 1 I I 1 1
281 284 287 290 396 399 402 405 BINDING ENERGY (eV)
Figure 4. The C 1 s and N 1 s core-level spectra of NA films before (a, b) and after 10 s (c, d). 40 s (e, f) and 90 s (g, h) of ozone treatment.
for the corresponding PPY-TSA film at the same extent of ozone treatment.
Ozone treatment also has a more obvious effect on the nitrogen atoms of the DP-PPY film. The proportion of the imine nitrogens has been substantially reduced after the ozone treatment. Thus, the oxidation of carbon atoms in DP-PPY is accompanied by a decrease in the imine/amine nitrogen ratio of the polymer, with the most drastic decrease occurring during the initial 10 s of the ozone treatment. Similar behaviour has also been observed during the ozone treatment of polyaniline base (see Figs 4 and 5 below). The nitrogen atoms of DP-PPY films, however, do not interact directly with ozone to a significant extent. This conclusion is based on the fact that no -NO, species or the enhancement of the residual N + component was discernible in the N 1s core-level spectra, at least for the ozone treatment times used. Prolonged ozone treatment ( > 3 min) can result in the appearance of the NO, species with an N 1s core-level peak BE at -406.3 eV. However, a sub- stantial degree of oxidative degradation of the polymer has also occurred at this high level of ozone exposure.
Polyaniline
The effect of ozone treatment on the intrinsic redox states of electroactive polymers is best demonstrated in polyaniline (PAN). The aniline polymers have
I I I I 1 1 1 I I
281 284 287 290 396 399 402 405 BINDING ENERGY (eV)
Figure 5. C 1s and N 1 s core-level spectra of EM films before (a, b). and after 10 s (c, d). 40 s (e, f ) and 90 s (9, h) of ozone treatment.
the general formula [(- B- NH- B- NH-)), (-B-N=Q=N-),-,],, in which B and Q denote the C,H, rings in the benzenoid and quinonoid form, respectively. Thus, the intrinsic oxidation states of the polymers can range from that of the fully oxidized per- nigraniline (PNA, y = 0), through that of the 50% oxi- dized emeraldine (EM, y = 0.5) to that of the fully reduced leucoemeraldine (LM, y = 1). Figures 4(a) and 4(b) show, respectively, the C 1s and N 1s core-level spectra of the 75% intrinsically oxidized nigraniline (NA, y = 0.75 and containing 75% =N- units). C 1s core-level component at 286.1 eV is again attributable to the C-0 species and is consistent with the reactive nature of the conjugated polymer surface. A x-x* shake- up satellite structure at -290.6 eV is also discernible. The N 1s core-level lineshape is consistent with a 75% intrinsically oxidized polymer.
Figures 4(c)-(h) show the corresponding C 1s and N 1s core-level spectra of the NA films after being sub- jected to 10, 40 and 90 s of ozone treatment. As in the case of the PPY films, ozone treatment readily results in the extensive formation of oxidized carbon species in the NA polymer. The C 1s core-level spectra reveal that in addition to the formation of the C-0 and COOH species, a new, highly oxidized carbon species of the form - 0- COOH with a BE at 290.5 eVZ8 is also dis- cernible at high levels of ozone exposure. Thus, the carbon atoms of PAN are oxidized to an even higher state than those of PPY at the comparable level of

55 SURFACE MODIFICATION OF ELECTROACTIVE POLYMER FILMS
a=75O " I
a=79 I \
281 284 287 290 396 399 402 405 BINDING ENERGY (eV)
Figure 6. The C 1 s and N 1 s core-level spectra of LM films before (a, b) and after 10 s fc. d), 40 s (e, f) and 90 s (9, h) of ozone treatment.
ozone treatment. Again, the relative concentrations of the oxidized carbon species in the NA films do not exhibit any significant XPS angular dependence of cc values between 20" and 15".
Under the present experimental condition, no signifi- cant formation of -NO, species (N 1s BE > 403 eV)
was observed, suggesting that the nitrogen atoms of NA are not oxidized directly by ozone. On the other hand, however, the N 1s core-level spectra indicate a substan- tial reduction in the intrinsic oxidation state, or the [=N-]/[-NH-] ratio, of the polymer after even a low level of ozone exposure. The fact that the decrease in the proportion of imine nitrogens is associated pre- dominantly with an increase in the proportion of amine nitrogens, and to a lesser extent the proportion of posi- tively charged nitrogens, suggests that ozone treatment followed by atmospheric exposure must have resulted in the hydrolysis reaction. In PAN, the hydrolysis reaction has been known to result in the conversion of the quinonoid imine units to the benzenoid amine
Ozone treatment, followed by atmospheric exposure, readily gives rise to a large number of carbon- yl, hydroxyl and hydroxyl peroxide species which must be responsible for the simultaneous hydrolysis of the polymer surface. Alternatively, the presence of oxidized carbon species must have prevented the polymer surface from assuming a high intrinsic oxidation state, as a result of decrease in effective conjugation. The relatively small positive BE shift for the positively charged nitro- gen suggests that they must have resulted from charge transfer interactions with the oxidized carbon species, and not from the direct oxidation by ozone.
Similar oxidation behaviour involving carbon atoms and hydrolysis reaction involving nitrogen atoms upon ozone treatment were observed for the 50% intrinsically oxidized EM base films. Figures 5(a)-(h) show the respective C 1s and N 1s core-level spectra for the pris- tine and the 10, 40 and 90 s ozone-treated EM base films.
In the case of the fully reduced LM, the N Is, core- level lineshape suggests the presence of predominantly a single nitrogen species. In addition to the various oxi- dized carbon species, ozone treatment also results in the formation of some positively charged nitrogens, as indi- cated by the appearance of the high BE tail in the N Is core-level spectra for the 10, 40 and 90 s ozone-treated LM base films (Fig. 6). The relatively small positive BE shift and the lack of a direct dependence of their con- centrations on the ozone treatment times suggest that
281 284 287 290 396 399 402 405 BINDING ENERGY (eV)
Figure 7. The C 1 s and core-level spectra of EM-H,SO, films before (a, b) and after 40 s (c, d) of ozone treatment.

E. T. KANG ET AL.
$1 284 207 290 163 166 169 172 BINDING ENERGY (eV)
Figure 8. The C 1 s and S 2p core-level spectra of a pristine film (a, b), a 10 s ozone-treated film (c, d) and a 90 s ozone-treated poly(3-dodecylthiophene) film (e, f).
the positively charged nitrogens again do not result from direct oxidation of the nitrogen atoms by ozone. They are more appropriately associated with the weak charge transfer interactions between the amine nitro- gens of LM and the oxidized carbon species. Charge transfer interactions of the amine nitrogens of LM with protonic acids and electron acceptors have been widely reported.26
The reaction behaviour, in particular the lack of an appreciable increase in the intrinsic redox state, of LM in ozone differs from that observed when LM is exposed to a mild oxidizing agent. When LM is exposed to air for a long period of time (months), a small proportion
of the imine nitrogens becomes discernible in the N IS core-level spectrum. For LM dissolved in 0,-containing N-methylpyrrolidinone solution, small portions of the amine units are oxidized to the imine units. The oxida- tion process is accelerated in the presence of UV-visible light i l l ~ m i n a t i o n . ~ ~ However, the most drastic increase in the intrinsic oxidation state of LM was observed when LM was heated at 270°C in a 50% 0,-50% N, mixture, although the process also resulted in the for- mation of some positively charged nitrogen^.^^ Thus, in the presence of a strong oxidizing agent, such as ozone, excessive formation of the highly oxidized carbon species and the accompanied hydrolysis reaction must have curtailed the formation of the imine species. This conclusion is consistent with the substantial decrease in the [imine]/[amine] ratios of NA and EM base films after ozone treatment.
As in the case of PPY salt films, the EM-H,SO, salt films are substantially more resistant to surface oxida- tion by ozone than their corresponding base films. Figures 7(a)-(d) show the respective C Is and N 1s core- level spectra for a pristine EM-H,SO, salt film ([S]/ [N] mole ratio -0.38 for both a = 75" and CL = 20") and a 40 s ozone-treated EM-H,SO, salt film. After ozone treatment, the [S]/[N] mole ratio of the complex decreased to only -0.28 at a = 75" and 0.20 at a = 20". Thus, the formation of oxidized carbon species is accompanied by a decrease in the susceptibility of the polymer to protonation, with the outermost surface being most severely affected. This decrease in proto- nation level is again consistent with the observation that ozone treatment of NA and EM films is accompa- nied by a decrease in the [imine]/[amine] ratio (Figs 4 and 5 ) and the fact that protonation occurs prefer- entially at the imine nitrogens in the aniline polymers.
Poly(3-alkylthiophene) Films
In contrast to the oxidation phenomena observed in the N-containing polymers, exposure of the alkyl-
281 284 287 290 160 163 166 169 172 BINDING ENERGY (eV)
Figure 9. The C 1s and S 2p core-level spectra of a 40 s ozone-treated poly(3-hexylthiophene) film before (a, b) and after near-UV light illumination in water for -30 min (c, d).

51 SURFACE MODIFICATION OF ELECTROACTIVE POLYMER FILMS
substituted polythiophene films to ozone gives rise to only minor changes in the C 1s core-level lineshape of the polymer. However, a prominent new S 2p com- ponent at 168.5 eV, which is chemically shifted by +4.6 eV from that of the neutral thiophene peak, has appeared as a result of the surface oxidation. This high-BE component is associated with the formation of oxidized sulphur species, such as the sulphone or the peroxide species.36 It is enhanced upon increasing the ozone exposure time. Figures 8(a)-(f) show the respec- tive C 1s and S 2p core-level spectra for a pristine, a 10 s ozone treated and a 90 s ozone-treated poly(3- dodecylthiophene) film. Similar changes in the C Is and S 2p core-level spectra were observed for the poly(3- hexylthiophene) and poly(3-octylthiophene) films. In all cases, the C 1s and S 2p core-level lineshapes do not exhibit any significant XPS angular dependence for a values between 20" and 75". This result suggests that ozone has penetrated substantially and uniformly into the polymer films.
The oxidized sulphur species in each case can be readily reduced or consumed, for example, in a photo- chemical reaction. For the present ozone-treated poly(3- alkylthiophene) films, illumination of the films in an aqueous medium with near-UV light readily results in the loss of the oxidized sulphur species. The ease in the reduction of sulphone and sulphoxide species in the thiophene polymers has also been rep~r ted .~ ' Figures 9(a)-(d) show the respective C 1s and S 2p core-level spectra for a 40 s ozone-treated poly(3-hexylthiophene) film before and after near-UV light (wavelengths >290 nm) illumination in an aqueous medium for -30 min. Near-UV light illumination in an aqueous medium,
however, can also result in an increase in the amount of surface-oxidized carbon species [Fig. 9(c)]. The photo- chemical reaction of the oxidized thiophene units in an aqueous medium appears to differ from the photooxida- tion of alkanethiols and thiophenols in air.38 For the self-assembled monolayer of the thiols on gold, near-UV light illumination in air readily results in pho- tooxidation to form the sulphonate species.
~ ~~
CONCLUSION ~ ~~
Surface modification of N-containing and S-containing electroactive polymer film by ozone has been studied by XPS. Ozone treatment of doped and undoped PPY and PAN films readily results in the oxidation of the chain carbon atoms. The carbon atoms of the doped poly- mers, however, are substantially more resistant to oxi- dation by ozone, even though the dopant anions are associated mainly with the nitrogen atoms. No direct oxidation of the nitrogen atoms to -NO, species was observed at the extents of ozone treatment employed in the present work. However, ozone exposure and the accompanied carbon oxidation cause a substantial decrease in the intrinsic oxidation states (imine/amine ratios) of the base polymers, such as the 25% deproton- ated DP-PPY, the 50% intrinsically oxidized EM and the 75% intrinsically oxidized NA. The oxidation behaviour of poly(3-alkylthiophene) films under similar conditions involves predominantly the oxidation of the sulphur but not the carbon atoms and thus differs from that of the N-containing polymers.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
8.
9.
10. 11.
12 13
14
N. C. Billingham and P. D. Calvert, Adv. Polym. Sci. 90, 2 (1989). A. Ray, G. E. Asturias. D. L. Kershner, A. F. Richter, A. G. MacDiarmid and A. J. Epstein, Synth. Met. 29, El41 (1 989). A. F. Diaz and J. Bargon, in Handbook of Conducting Poly- mers, ed. by T. A. Skotheim, Vol. I, Chapt. 3. Marcel Dekker, New York (1986). G. Tourillon, in Handbook of Conducting Potymers, ed. by T. A. Skotheim, Vol. I, Chapt. 9. Marcel Dekker, New York (1 986). K. G. Neoh, E. T. Kang, S. H. Khor and K. L. Tan, Polym. Degrad. Stab. 27, 107 (1 990). J. C. Thieblemont, M. F. Planche, C. Petrescu, J. M. Bouvier and G. Bidan, Synth. Met. 59, 81 (1993). F. Mohammad, P. D. Calvert and N. C. Billingham, Synth. Met., 66, 33 (1994). K. L. Tan, B. T. G. Tan, E. T. Kang and K. G. Neoh, J. Chem. Phys. 94, 5382 (1991). A. Hugot-Le Goff and M. C. Bernad, Synth. Met. 60, 115 (1 993). M. S. A. Abdou and S. Holdcroft, Synth. Met. 60, 93 (1 993). S. Hotta, S. D. V. Rughooputh and A. J. Heeger, Synth. Met. 22,79 (1 987). Y . Cao, P. Smith and A. J. Heeger, Synth. Met. 48,91 (1 992). J-E. Osterholm, Y. Cao, F. Klaveffer and P. Smith, Synth. Met. 35,2901 (1 994). S. A. Chen and G. W. Hwang, J. Am. Chem. Soc. 116, 7939 I1 994).
15. A. G. MacDiarmid and A. J . Epstein, Synth. Met. 65, 3193
16. E. T. Kang, K. G. Neoh and K. L. Tan, Polymer 35, 3193 ( 1 994).
(1 994).
17. 18.
19.
20.
21.
22.
23.
24.
25. 26.
27.
28.
29.
30.
31.
32.
H. Munstedt, Polymer 29, 296 (1 988). W. Deits, P. Cukor, M. Rubner and H. Jopson, Synth. Met. 4, 199 (1 982). J. M. Pochan, H. W. Gibson, F. C. Bailey and D. F. Pochan, Polymer 21, 250 (1 980). P. Pfluger, M. Krounbi, G. B. Street and G. Weiser, J. Chem. Phys. 78, 321 2 (1 983). E. T. Kang, K. G. Neoh and K. L. Tan, Micromolecules 25, 6842 (1 992). E. T. Kang, K. G. Neoh and K. L. Tan, Surf. Interface Anal. 20, 833 (1 993). A. G. Green and A. E. Woodhead, J . Chem. SOC.. Chem. Commun. 2388 (1 91 0). S. Hotta, M . Soga and N. Sonoda, Synth. Met. 26, 267 (1 988). J. H. Lee and I. J. Chung, Synth. Met. 53, 245 (1 993). E. T. Kang, K. G. Neoh and K. L. Tan, Adv. Polym. Sci. 106, 135 (1993). A. Pion, 2. Kucharski, C. Budrowski, M. Zagorska, S. Krichene, J. Suwalski, G. Dehe and S. Lefrant, J. Chem. Phys. 83, 5923 (1985). G. E. Muilenberg (ed.), Handbook of X-ray Photoelectron Spectrscopy, p. 38. Perkin-Elmer, Eden Prairie, MN (1978). D. S. Park, Y. B. Shim and S. M. Park, J. Electrochem. Soc. 140,2749 (1993). J. C. Thieblemount, J. L. Gabelle and M. F. Planche, Synth. Met. 66, 243 (1 994). J. M. Ribo, A. Dicko, J. M. Tura and D. Bloor, Polymer 32, 728 (1991). M. Angelopoulous, G. E. Asturias, S. P. Ermer, A. Ray, E. M., Scherr and A. G. MacDiarmid, M o l . Cryst. tiq. Cryst. 160,151 (1 988).

58 E. T. KANG ET AL.
33. E. T. Kang, K. G. Neoh and K. L. Tan, Synfh. Met 68, 141
34. K. G. Neoh, E. T. Kang and K. L. Tan, Polymer 33, 2292
35. K. G. Neoh. E. T. Kang and K. L. Tan, Thermochim. Acfa 171,
36. A. Kaul and K. Udipi, Macromolecules 22, 1201 (1 989). 37. M. Hanack, G. Hieber, G. Dewald and H. Ritter, Synth. Met.
38. J. Huang and J. C. Hemminger, J. Am. Chem. SOC. 115, 3342
(1995).
(1992).
279 (1 990).
41-43, 507 (1 991 ).
(1993).