Embed Size (px)
Citation preview

Université de Lausanne - Faculté de biologie et de médecine
3e année
COURS INTEGRE D’HEMATOLOGIE
HEMOPATHIES MALIGNES (OMS 2001)
SYNDROMES
MYELODYSPLASIQUES
SYNDROMES MYELOPROLIFERATIFS
CHRONIQUES
Mars 2006 Dr Audrey Baur Chaubert, MER
Dr Pierre-Michel Schmidt, CC

1
CLASSIFICATION DES HEMOPATHIES MALIGNES
HEMOPATHIES MYELOIDES
SYNDROMES MYELOPROLIFERATIFS CHRONIQUES
SYNDROMES MYELODYSPLASIQUES
SYNDROMES MYELODYSPLASIQUES / MYELOPROLIFERATIFS
LEUCEMIES AIGUES MYELOIDES HEMOPATHIES LYMPHOIDES
LYMPHOMES MALINS, LEUCEMIES AIGUES LYMPHOBLASTIQUES
PROLIFERATION ET DIFFERENCIATION AU COURS DES
HEMOPATHIES MYELOIDES
CELLULE SOUCHE Mutation génétique Facteurs humoraux Interactions cellulaires Prolifération Différenciation
Syndromes myéloprolifératifs chroniques (SMP) + + Syndromes myélodysplasiques (SMD) SMD / SMP Leucémies aiguës myéloïdes + _ (LAM)

2
SYNDROMES MYELODYSPLASIQUES (SMD) Groupe hétérogène d'affections hématologiques, correspondant à une maladie clonale de la cellule souche hématopoïétique, caractérisée par une prolifération et une différenciation anormales d'une ou plusieurs lignées myéloïdes avec apoptose de nombreux précurseurs médullaires (hématopoïèse inefficace). Incidence 3/100.000 maladies des gens âgés : 20/100.000 chez les plus de 70 ans 10 - 20 % des patients seulement ont moins de 60 ans rares chez l'enfant.
Epidémiologie Le plus souvent inconnue. Instabilité génomique due à l'âge avancé ? Exposition au benzène Autres solvants ? Chimiothérapie (agents alkylants) Radiothérapie.
Contexte clinique Le plus souvent, maladies idiopathiques du patient âgé Syndromes myélodysplasiques primaires Après chimio- et / ou radiothérapie: SMD secondaires (“therapy-related”); groupe de
patients généralement plus jeunes. Manifestations habituelles Sang périphérique: cytopénie(s) intéressant une ou plusieurs lignées Contraste frappant avec une moelle hématopoïétique généralement hypercellulaire
("cytopénie(s) à moelle riche"). Plus rarement, la moelle est hypoplasique Evolution possible en leucémie aiguë (20 à 30 % de tous les patients) Décès survenant le plus souvent par infection ou hémorragie
(insuffisance médullaire) Mauvaises réponse et tolérance à la chimiothérapie.
Anomalies chromosomiques 50 à 90 % des cas. Plus fréquentes dans les SMD secondaires Souvent à proximité d’oncogènes ou de facteurs hématopoïétiques de croissance
Par exemple : 5q - et gènes pour GM-CSF (Granulocyte-Macrophage Colony-Stimulating
Factor) monosomie 7 et érythropoïétine
Les anomalies chromosomiques complexes sont de mauvais pronostic.

3
Rôle de l'apoptose extensive ou mort cellulaire prématurée intramédullaire et du contrôle moléculaire dans la pathogenèse des SMD Voir tableaux 1 et 2 Apoptose : Mort programmée ou suicide des cellules. Morphologiquement, condensation chromatinienne, désintégration nucléaire et
rétraction des cellules Les cellules apoptotiques n’induisent pas de réponse inflammatoire, mais sont la
cible de macrophages L'apoptose joue un rôle dans un grand nombre de processus physiologiques, tels
que développement embryonnaire, maintenance des tissus, sélection clonale dans le système immun
Différents gènes ou produits de gènes sont impliqués dans le contrôle ou la participation à l’apoptose :
p53: peut induire l’apoptose des cellules myéloïdes c-Myc: peut induire à la fois des signaux de prolifération et de mort
cellulaire (suivant la présence ou l'absence d'autres facteurs) Bcl-2 family: plusieurs gènes en relation avec la protéine Bcl-2 ont été
identifiés; certains agissent dans le sens d'une suppression de l'apoptose.
Selon des travaux récents sur les SMD, une cytopénie sanguine périphérique combinée à une moelle normo - ou hypercellulaire peut s’expliquer par une apoptose exagérée. Les études de cinétique cellulaire et d’apoptose dans les SMD ont montré un taux augmenté de prolifération coïncidant avec un taux très élevé d'apoptose. L'apoptose extensive annulerait le haut taux de croissance et de prolifération, entraînant une hématopoïèse inefficace. SMD et progression leucémique : La progression leucémique pourrait résulter de clones néoplasiques ayant échappé au contrôle apoptotique. Les mécanismes moléculaires permettant le déclenchement de l’apoptose exagérée ou la progression leucémique n'ont pas été élucidés. Classification des SMD : basée prioritairement sur la morphologie (anomalies de maturation = signes de myélodysplasie, pourcentage de blastes sanguins et médullaires, présence ou non de sidéroblastes en couronne), monocytose périphérique (< 1G/l). Voir tableau 3

4
Le diagnostic de SMD repose sur une combinaison de données : Status clinique Evaluation morphologique du frottis sanguin périphérique et de la ponction-biopsie
de moelle osseuse (cytologie et histologie médullaires) Analyse cytogénétique.
Le pronostic de SMD s'établit sur divers critères : Sévérité de la ou des cytopénie(s) Age Sous-groupes OMS Cytogénétique.
Voir tableau 4 Diagnostic différentiel des SMD Le plus souvent un diagnostic par exclusion, après avoir éliminé d’autres causes de cytopénie (s). A exclure : Désordres nutritionnels: carences (effets réversibles) :
Des modifications myélodysplasiques sévères non néoplasiques peuvent résulter d’un déficit en vitamine B12 ou en acide folique. Modifications dysplasiques particulièrement marquées de la lignée érythroïde. Examens de laboratoire ou essai thérapeutique de substitution nécessaires
Effets toxiques réversibles de médicaments (chimiothérapie), abus d’alcool Maladies infectieuses (HIV, TBC, Parvovirus).
Voir tableau 5 Un diagnostic de SMD doit être posé avec prudence: revue soignée de l’anamnèse, de la présentation clinique et des données hématologiques. Traitement symptomatique : transfusions de globules rouges / de plaquettes. Antibiotiques. Facteurs de croissance: Erythropoïétine, G-CSF (Granulocyte Colony- Stimulating Factor). (Poly-) chimiothérapie. Greffe de moelle allogénique (sujet jeune). Immunosuppression (SAL = Sérum Anti-Lymphocytaire) dans les SMD hypoplasiques.

5
SYNDROMES MYELOPROLIFERATIFS CHRONIQUES (SMP) Affections de l’adulte. Rares chez l’enfant Groupe de maladies hématologiques proches parentes, avec des caractéristiques
communes. Possibilité de chevauchement entre les entités: intérêt d’un terme général
Maladie clonale d’une cellule souche hématopoïétique, se traduisant par une prolifération exagérée, chronique et irréversible d’une ou plusieurs lignées myéloïde(s) (érythroïde, granulocytaire, mégacaryocytaire) avec différenciation (maturation)
Des anomalies fonctionnelles peuvent être observées (par ex. anomalies de la fonction plaquettaire)
En cours d’évolution, une transformation en leucémie aiguë est possible : prolifération de cellules progénitrices blastiques et arrêt de différenciation.
Les 5 types principaux de SMP sont :
La leucémie myéloïde chronique La myélofibrose chronique idiopathique (avec hématopoïèse extramédullaire) La polycythaemia vera (Maladie de Vaquez) La thrombocytémie essentielle Le syndrome hyperéosinophile ou leucémie chronique à éosinophiles.
Caractères communs des SMP
Hyperplasie myéloïde et fibrose de la moelle osseuse Reprise de l'hématopoïèse dans les os longs (par ex. fémur) Hématopoïèse extra-médullaire ou "métaplasie myéloïde" dans la rate, le foie et
parfois d'autres organes Hépatosplénomégalie Erythroblastomyélémie Intrication de phénomènes thrombotiques et hémorragiques Complications infectieuses Transformation en leucémie aiguë.
Cultures cellulaires: croissance spontanée de l'hématopoïèse, en l'absence de facteurs de croissance. Seule la leucémie myéloïde chronique est caractérisée par une anomalie chromosomique précise :Chromosome Philadelphie (Ph): t (9 ;22) (q34 ; q11). Cette translocation entraîne une fusion de séquences du gène BCR du chromosome 22 avec des régions du gène ABL du chromosome 9.

6
Myélofibrose chronique idiopathique (avec hématopoïèse extramédullaire)
Biologie
maladie clonale de la cellule souche réaction fibreuse liée à la libération de PDGF = Platelet-Derived Growth Factor
par les mégacaryocytes et plaquettes anormaux; multiplication des fibroblastes médullaires et augmentation de la synthèse du collagène.
Présentation clinique "De novo" ou peut compliquer un autre SMP (surtout la polycythaemia vera) sang périphérique suggestif (anisocytose, poïkilocytose, érythrocytes "en
larmes" ou dacryocytes, érythroblastes, myélémie, (érythroblastomyélémie), anisocytose plaquettaire.
Examen physique (selon diverses séries) : splénomégalie 90 -100 % hépatomégalie 50 - 90 % adénopathies 5 - 20 % douleurs osseuses 1 - 10 %
Survie très variable (6 mois à 9 ans, médiane 3-5 ans) selon une étude portant sur 51 patients
Causes de mort: infections, thromboses, hémorragies, insuffisance cardiaque, transformation en leucémie aiguë.
Histologie de la moelle osseuse (biopsie): 3 stades évolutifs :
moelle hyperplasique; quantité anormalement élevée de mégacaryocytes, souvent en amas; fibrose réticulinique
raréfaction de l'hématopoïèse; fibrose collagène atrophie de la moelle: subsistent quelques mégacaryocytes
dystrophiques. En plus de la fibrose, remaniement osseux sclérosant, mutilant.
Polycythaemia vera (Maladie de Vaquez)
Caractéristiques Syndrome myéloprolifératif chronique caractérisé par une augmentation de la
production érythrocytaire Production érythrocytaire indépendante des mécanismes qui règlent
habituellement l’érythropoïèse Croissance spontanée des cultures érythroïdes in vitro Augmentation de la masse des globules rouges (hématocrite) et de l’Hb Atteinte sang et moelle prédominante Atteinte du foie et de la rate (hématopoïèse extramédullaire) tardivement
A exclure: érythrocytose secondaire - hypoxie - sécrétion inappropriée d’érythropoïétine par certaines tumeurs (reins,
foie, cervelet)

7
Clinique Age médian 60 ans Symptômes liés à l ’augmentation de la masse sanguine (hypertension, thrombose,
maux de tête, troubles visuels…)
Deux phases dans l’évolution clinique : 1. Phase polycythémique, associée à l’augmentation de la masse des globules
rouges 2. Phase « post-polycythémie » avec cytopénie, hématopoïèse inefficace, fibrose de
la moelle et augmentation de la taille de la rate
Examen de la moelle osseuse 1. Phase polycythémique : moelle hypercellulaire, hyperplasie de la lignée
érythroïde et parfois des autres lignées (le plus souvent mégacaryocytaire) 2. Phase « post-polycythémie » : moelle fibreuse semblable à l’image observée
dans la myélofibrose chronique idiopathique
Pronostic Avec les traitements actuels, survie médiane de 10 ans. Décès généralement liés aux phénomènes thrombo-emboliques ou hémorragiques. 2 -3 % des patients développent un syndrome myélodysplasique ou une leucémie aiguë. En résumé, le diagnostic de SMP repose sur une combinaison de données : Status clinique Evaluation morphologique du frottis sanguin périphérique, de la ponction biopsie de
moelle osseuse (cytologie et histologie médullaires) Analyse cytogénétique (leucémie myéloïde chronique et chromosome Ph) Cultures érythroïdes spontanées en absence d'érythropoïétine (polycythaemia vera).
Diagnostic différentiel Hyperplasie médullaire réactionnelle sur pertes sanguines aiguës, destruction et / ou
séquestration périphérique (anémies hémolytiques, auto-immunité, hypersplénisme) Rupture de la "barrière médullo-sanguine" et érythroblastomyélémie lors d'infiltration
de la moelle osseuse par une tumeur maligne non hématologique (métastases). Un diagnostic de SMP doit être posé avec prudence: revue soignée de l’anamnèse, de la présentation clinique et des données hématologiques. Référence bibliographique: Jaffe E.S., Harris N.L., Stein H., Vardiman J.W.: Pathology & Genetics. Tumors of Haematopoietic and Lymphoid Tissues, IARCPress, Lyon, 2001.
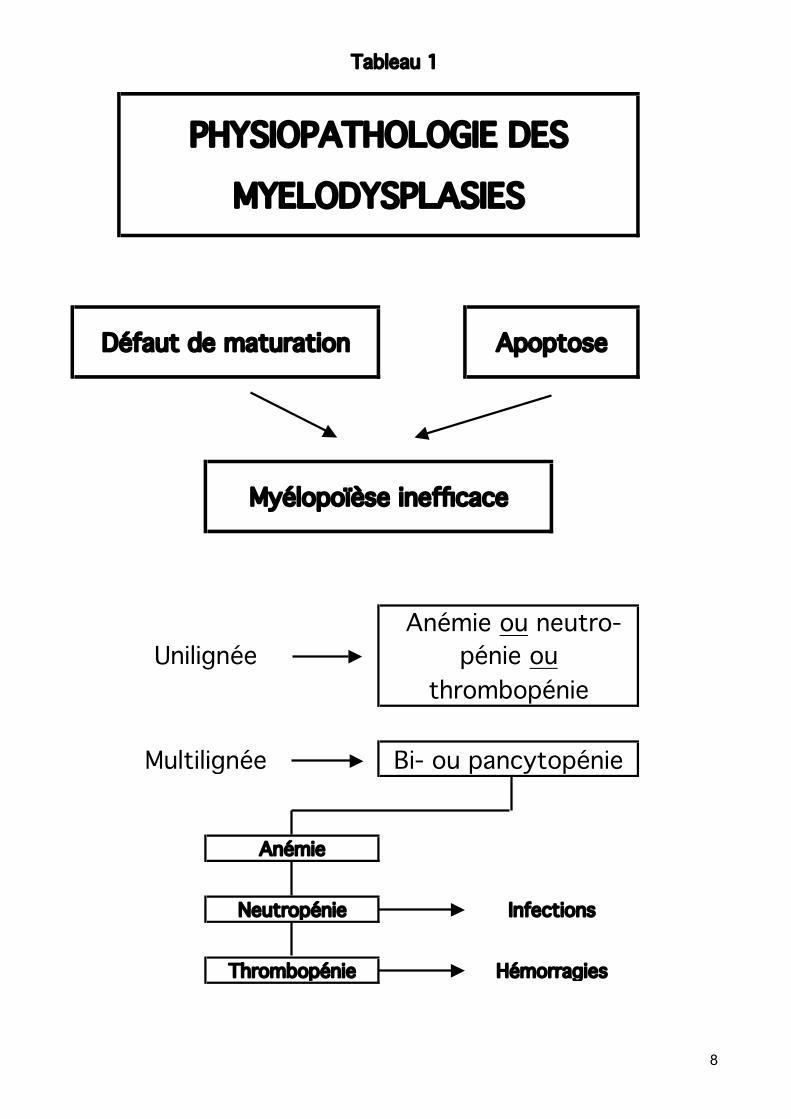
Tableau 1
Thrombopénie Hémorragies
Multilignée Bi- ou pancytopénie
Anémie
Neutropénie Infections
thrombopénie
PHYSIOPATHOLOGIE DES
MYELODYSPLASIES
Unilignée pénie ou
Défaut de maturation Apoptose
Myélopoïèse inefficace
Anémie ou neutro-
8
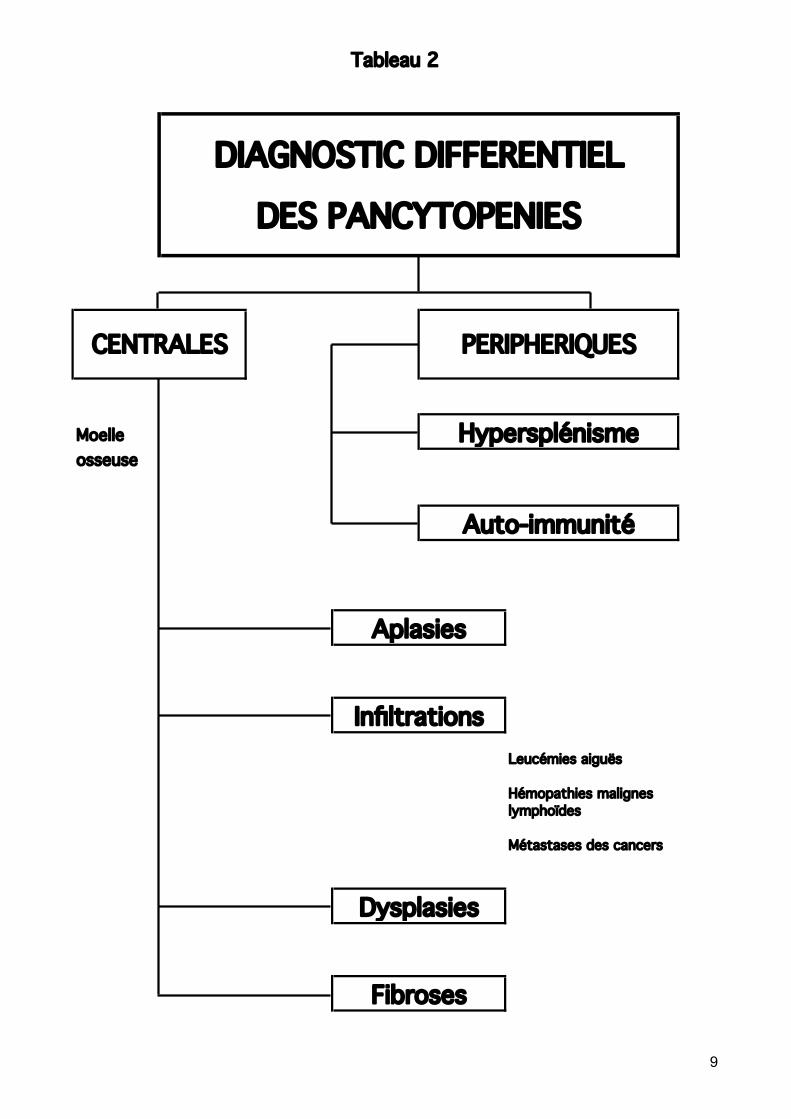
Tableau 2
Moelleosseuse
Leucémies aiguës
Hémopathies malignes lymphoïdes
Métastases des cancers
Aplasies
Fibroses
Dysplasies
Infiltrations
Hypersplénisme
Auto-immunité
DIAGNOSTIC DIFFERENTIEL
DES PANCYTOPENIES
CENTRALES PERIPHERIQUES
9

Tableau 3
10
CLASSIFICATION DES SYNDROMES MYELODYSPLASIQUES (OMS, 2001)1
Critères diagnostiques
Présence de signes de dysplasie unilignée multilignée
Pourcentage de blastes sanguins et médullaires
Présence ou non de sidéroblastes pathologiques.
Classification
Anémie réfractaire (AR)
Anémie réfractaire avec sidéroblastes en couronne (ARS)
Cytopénie réfractaire avec dysplasie multilignée (CRDM)
Cytopénie réfractaire avec dysplasie multilignée et sidéroblastes en couronne (CRDM-RS)
Anémie réfractaire avec excès de blastes (AREB)
AREB-1 (blastes médullaires: 5-9 %)
AREB-2 (blastes médullaires: 10-19 %)
Syndromes myélodysplasiques non classables
Syndrome myélodysplasique avec anomalie isolée 5q-(syndrome 5 q-).
1Jaffe E.S., Harris N.L., Stein H., Vardiman J.W.: Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues, IARCPress, Lyon, 2001.

Tableau 4
11
FACTEURS PRONOSTIQUES DES SYNDROMES
MYELODYSPLASIQUES
Age ( 60 ans) Scores pronostiques (cytopénies - % de blastes médullaires - cytogénétique) Pourcentage élevé de précurseurs médullaires exprimant CD34 en histologie.
COMPLICATIONS – EVOLUTION – SURVIE
Complications Infections récurrentes
Manifestations hémorragiques Troubles immunitaires.
Evolution - Survie
Leucémie aiguë Médiane de survie
AR 6 % 66 mois ARS 1-2 % 72 mois CRMD 11 % 33 mois AREB-1 25 % 18 mois AREB-2 33 % 10 mois.
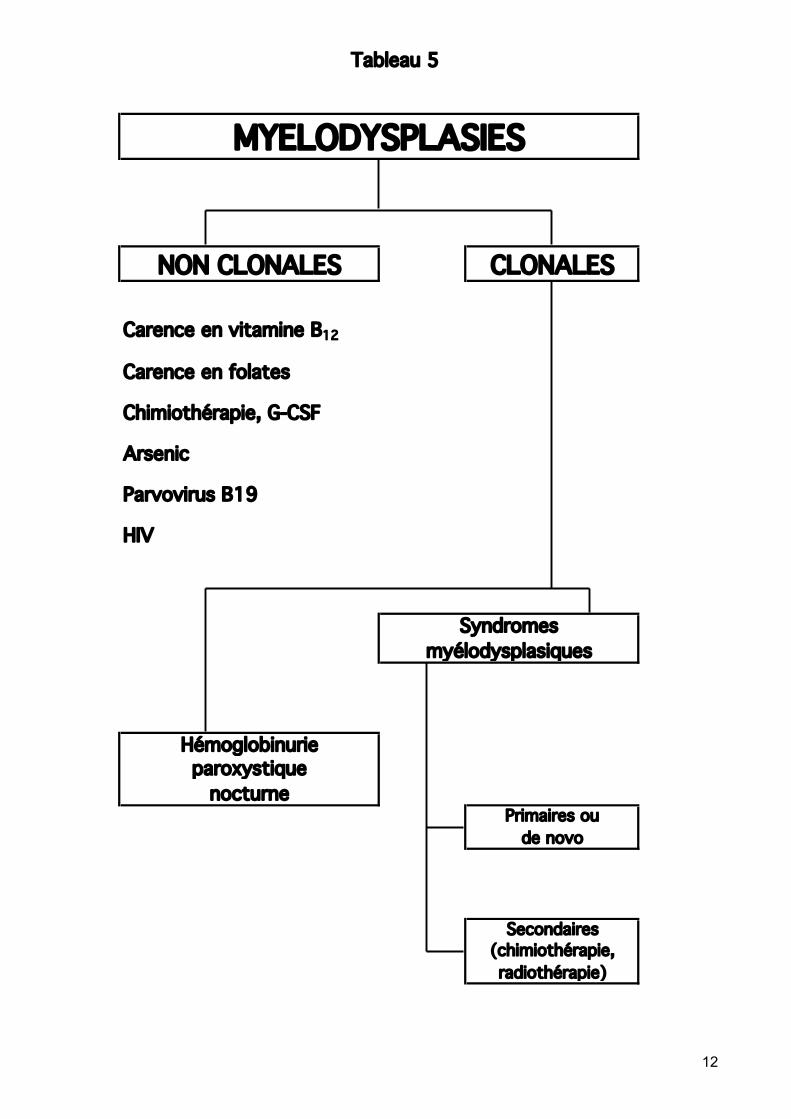
Tableau 5
Carence en vitamine B12
Carence en folates
Chimiothérapie, G-CSF
Arsenic
Parvovirus B19
HIV
MYELODYSPLASIES
Primaires ou
Syndromesmyélodysplasiques
NON CLONALES CLONALES
Hémoglobinurieparoxystique
nocturne
de novo
Secondaires(chimiothérapie,radiothérapie)
12
Tableau 6
Examen de la moelleosseuse
Précurseur immature deslignées granuleuses
Variété d'hémopathie
maligne lymphoïde
CST =cellule-souchetotipotente
ERYTHROCYTES PLAQUETTES NEUTROPHILES
EOSINOPHILES MONOCYTES
Hématopoïèse des lignées érythroïde, granuleuse,monocytaire et mégacaryocytaire
CSM = CS MYELOÏDE CSL = CS LYMPHOÏDE
MYELOPOÏESE
BASOPHILES
MYELOCYTE
MYELOME
MYELOÏDE
Attention !
MYELO -
MYELOGRAMME
PLASMOCYTAIRE
13

14
Tableau 7
INDICATIONS A L'EXAMEN COMBINE DE LA MOELLE OSSEUSE (aspiration et biopsie)
Diagnostic, bilan, suivi de :
Syndromes myéloprolifératifs, myélodysplasiques, myélodysplasiques / myéloprolifératifs
Leucémies aiguës
Hémopathies malignes lymphoïdes
Recherche de :
Processus infectieux (granulomes, micro-organismes)
Métastases ostéo-médullaires
Investigation de cytopénie(s)

15
Tableau 8
HEMOPATHIES MALIGNES – LES OUTILS DIAGNOSTIQUES
CLINIQUE
MORPHOLOGIE
CYTOLOGIE HISTOLOGIE CYTOCHIMIE
MARQUEURS IMMUNOLOGIQUES
GENETIQUE
CYTOGENETIQUE Ex.: LMC1 : t(9;22)
BIOLOGIE MOLECULAIRE Ex.: LMC1 : BCR /ABL
CULTURES CELLULAIRES.
1LMC = Leucémie myéloïde chronique.





























