Embed Size (px)
Citation preview

RESEARCH PAPER
Synthesis and characterization of novel mesocompositesCo3O4 and CuO@OMS (ordered mesoporous silica)as active catalysts for hydrocarbon oxidation
Cezar Comanescu
Received: 31 October 2013 / Accepted: 5 February 2014
� Springer Science+Business Media Dordrecht 2014
Abstract Novel metal nanoporous transition metal
oxides MxOy (Co3O4, CuO) have been synthesized by
thermal decomposition of inorganic salts precursors
(acetates, nitrates) impregnated into hexagonal mes-
oporous silica (OMS, ordered mesoporous silica) of
SBA-15 type (prepared in-house) at different precur-
sor loadings, the mesocomposites thus obtained being
monitored after each impregnation–calcination step
by small and wide angle powder XRD. The pore size
for the ordered silica host range from 5.08 to 7.06 nm.
Retention of the hexagonal silica framework has been
observed in spite of the temperatures up to 500 �C.
Mesoporous Co3O4 has been obtained by leaching the
silica through overnight HF dissolution, which par-
tially preserved the small-range ordering found in the
parent Co3O4@OMS composite prior to leaching.
Both Co3O4 (meso) and Co3O4@SBA-15 have been
tested in methane oxidation and were found to be
superior to the bulk Co3O4 performance, with
mesoporous Co3O4 being able to fully oxidize meth-
ane to CO2 and H2O at 350 �C, while Co3O4@OMS
exhibits a lower activity with 20 % conversion at
350 �C. CuO@OMS shows the lowest activity, with
only *13 % conversion at 500 �C.
Keywords Nanoporous � Mesocomposite �Nanocasting � Catalytic activity � SBA-15 �XRD
Abbreviations
OMS Ordered mesoporous silica
TM Transition metal
XRD X-ray diffraction
SAXS Small angle XRD
WAXRD Wide angle XRD
SEM Scanning electron microscopy
TEM Transmission electron microscopy
SAED Selected area electron diffraction
ICDD International center for diffraction data
Introduction
Nanostructured metal oxides have attracted consider-
able attraction during the last decade due to useful
properties which made them excellent candidates for
catalysis, applicative electrochemistry (Maruyama and
Arai 1996) and gas sensor devices (Li et al. 2005).
Nanosized Co3O4 particles have been synthesized
recently through several synthesis routes, like sol–gel
method, template-assisted growth, hydrothermal,
Electronic supplementary material The online version ofthis article (doi:10.1007/s11051-014-2323-4) contains supple-mentary material, which is available to authorized users.
C. Comanescu
Inorganic Chemistry Department, University Politehnica
of Bucharest, Polizu St., No. 1–7, 011061 Bucharest,
Romania
C. Comanescu (&)
National Institute of Materials Physics, Atomistilor St.,
No. 105 bis, 077125 Magurele, Romania
e-mail: [email protected]
123
J Nanopart Res (2014) 16:2323
DOI 10.1007/s11051-014-2323-4

precipitation, and spray pyrolysis (Kandalkar et al.
2008; Shinde et al. 2006; Armelao et al. 2001; Wang
et al. 2009). Various morphologies can be attained
ranging from particles to rods and wires, depending on
the synthesis procedure (Shu et al. 2009). Thermal and
chemical stability as well as surface area and particle
size distribution greatly affect the performance of
Co3O4 in various catalytic applications while modify-
ing the magnetic properties compared to those of the
bulk cobalt oxide (Benitez et al. 2011).
The synthesis route can modify substantially the
properties of the metal oxide material (size, shape,
stability) and greatly alter its reactivity. Among the
synthesis routes afore-mentioned, nanocasting is con-
ceptually and practically the method that can afford
high-quality metal oxides because the sacrificial hard-
template is a highly ordered material to begin with,
and the metal oxide precursor can fill the pores and
decompose in an ordered framework, while simple
decomposition of the metal salt precursor would
typically lead to bulk, amorphous metal oxide phase.
Cobalt oxide Co3O4 has been explored in the past
(reports go back to 1968) as a catalyst for methane
combustion with moderate results, due to the high
temperature (and implicitly high energy) required to
achieve complete conversion (Levy 1968). Researches
focused on Pd/Al2O3 catalysts that start converting
methane at 300 �C and reach 100 % conversion at
480 �C (Baldwin and Burch 1990). It was found that
CH4 reduces PdO formed upon reaction of Pd with O2,
and that the H and CHx (x = 1–3) fragments formed
by CH4 dissociative adsorption need small quantities
of Pd metal for enhanced combustion. However,
metallic Pd was found inactive, and the presence of
crystalline PdO was thought to be beneficial to easier
reduce than the amorphous variant. A cooperative Pd–
PdO activity was employed by the authors, with Pd
active in dissociative adsorption of methane, and
PdO—formed fast and covering the metal surface in
various thicknesses—enhances the low temperature
methane oxidation due to its easier reducibility. O2 is
adsorbed stronger than CH4 onto alumina support,
which may inhibit reaction under oxidizing conditions
(Carstens et al. 1998; Hicks et al. 1990).
The size of the Pd catalyst was also found to greatly
influence the turnover frequency, from 0.02 (small
particles) to 1.3 s-1 (large Pd particles), which
suggests that in methane oxidation the catalyst struc-
ture is important, due to different reactivity of the
adsorbed oxygen species (more reactive on smaller
particles with a larger number of active sites and
defects) (Hicks et al. 1990; Cullis and Willatt 1983).
However, even if Pd has long been employed for
successful methane combustion, the precise nature of
the catalyst site is still under debate, and it was
accepted that higher crystallinity of PdO leads to
higher activity when compared to dispersed PdO
(Hicks et al. 1990). The differences observed by
various groups reveal a complicated behavior for Pd/
Al2O3, dependent on preparative routes and reaction
conditions.
Water as well as higher concentrations of CO2 have
been reported to inhibit the oxidation, with the former
being responsible of Pd(OH)2 formation at the surface,
thus rendering the active sites inactive (catalyst poison-
ing). It seems important that water should not be present
in the catalyst at the beginning of the reaction, since the
poisoning effect should be alleviated at higher
([300 �C) temperatures (Ribeiro et al. 1994).
Other oxides have been employed for methane
oxidation, like titania or tin oxide, albeit with lower
activity than Pd/Al2O3 (Cullis and Willatt 1983). Early
reports of Co3O4 used in conjunction with Pd go back to
1999 (Li and Hoflund 1999), when Li et al. showed Pd/
Co3O4 to exhibit a higher activity in methane oxidation
than the standard Pd/Al2O3. Noble metals (Au) copre-
cipitated on transition metal oxide supports also exhib-
ited unexpectedly high activity (starting above 250 �C),
although the studies were complicated by the fact that
even the support (Co3O4, NiO, MnOx, etc.) acted as
active sites at higher temperatures. Higher oxidation
state of Au was suggested to be responsible for
increasing oxidation activity (Waters et al. 1995).
One of the main drawbacks of these oxides is their
low specific surface area which hinders their use as
catalysts (Pena and Fierro 2001). Other metal oxides
(CeO2) have been investigated due to their capacity to
store oxygen but also as combination with other oxides
as binary oxides (Pengpanich et al. 2002; Zamar et al.
1995). The role played by metal oxides could be due to
generation and participation of surface oxygen species
and vacancies (Zhu et al. 2005). The main advantages
of using metal oxides instead of noble metals are the
lower cost of raw materials used, higher thermal
stability and reducing the NOx emissions coming from
the N2 bounded to the fuel. However, among the
disadvantages one may mention the lower selectivity
and thus the higher ignition temperature.
2323 Page 2 of 21 J Nanopart Res (2014) 16:2323
123

In the present study, we present the results obtained
by employing neat Co3O4 nanoparticles as well as
Co3O4(nano)@OMS, and we compare the perfor-
mance of these catalysts in methane combustion. We
also prepared a mesoporous CuO@OMS to check the
relative activity that is achieved by another transition
metal oxide embedded in mesoporous silica at similar
concentration, reaction, and condition routes. Excel-
lent selectivity was achieved by both Co-based
catalysts, the final reactivity order being Co3O4(na-
no) [ Co3O4@OMS [ CuO@OMS. This result falls
in line with the findings of McCarthy et al., who
studied and established the following single metal-
oxide catalyst performances in CH4 combustion
to decrease in the order Co3O4 [ CuO [ NiO [Mn2O3 [ Cr2O3 (McCarthy et al. 1997). One major
drawback of using neat metal oxides is that high
temperatures required to oxidize methane lead to
sintering and finally to catalyst deactivation (Zavyal-
ova et al. 2007). A mean to bypass this shortcoming is
to use a support for stabilizing and dispersing the metal
catalyst (Milt et al. 2002).
The catalytic hydrocarbon combustion is a
‘‘greener’’ alternative to classical flame-combustion
(Saracco et al. 1996). The first representative of the
alkane homologes—CH4 (methane)—has an ignition
point (consumption of 10 % of the fuel itself) of about
400 �C, which led scientists to envision new catalysts
featuring: high activity, long-term stability, low igni-
tion temperature and cost (Yisup et al. 2005).
While dispersed noble metals (Pd, Pt) on support
materials have been investigated in the past (Burch
and Loader 1994; Yang et al. 2000; Sekizawa et al.
1996), the cost, extreme functioning regime, and
sintering susceptibility plagued their use as hydrocar-
bon oxidation catalysts. A cheaper route was recently
introduced, which employs the use of perovskite-type
mixed oxides (Co, Cr), but the low specific surface
area was a major drawback, as this translated in lower
catalytic activity (Pena and Fierro 2001).
Metal oxides are a promising alternative to noble
metal catalysts used in the past due to some specific
features: they can store oxygen, can participate in
generating surface oxygen species and vacancies and
exhibit high thermal stability, albeit at the cost of
lower selectivity and rather high ignition temperature
(Zhu et al. 2005).
The large pore diameter of mesoporous materials
(Tanev and Pinnavaia 1996; Zhang et al. 1997)
enables the catalysis of large molecules or incorpora-
tion of large active species; among these materials
SBA-15 attracts more attention owing to its excellent
hydrothermal stability, less condensed structure, and
unique micro-mesoporosity, which makes it a para-
mount candidate for supports or catalyst. However,
siliceous SBA-15 lacks active sites due to its chemical
composition; hence it is necessary to introduce other
active species in the mesopores. Obtaining mesopor-
ous materials at conditions near ambient opens new
perspectives toward an easier synthetic procedure.
Moreover, lack of thermal treatment (replaced with
solvent extraction in H2O–EtOH solution) and regen-
eration of the surfactant (from the ethanol solution)
would contribute to a new cost-effective route to
mesoporous siloxanes.
Driven by the interesting properties of mesoporous
materials as supports for active catalysts, we have
synthesized Co3O4 nanoparticles by varying the type
and quantity of the co-precursor (Co(NO3)2 or
Co(CH3COO)2) and the infiltration route of the
precursor into the mesoporous OMS support (plain
mixing vs. incipient wetness method). The new
materials have been characterized by powder XRD
(low and wide angle), N2-sorption isotherms, scanning
electron microscopy (SEM) and transmission electron
microscopy (TEM), while the actual catalyst (Co3O4
or CuO) surface loading was assessed by energy-
dispersive X-ray spectroscopy (EDAX).
While a recent report shows that nanostructured
Co3O4 can be obtained by direct soft-templating
method using P123 as anionic surfactant and precise
reaction conditions (Dahal et al. 2012), the hard-
template method might still be more attractive because
it offers the possibility to reproducibly synthesize the
OMS first and thus gives a better control over the final
structural features of the mesoporous composite
Co3O4@OMS and implicitly that of mesoporous
Co3O4 obtained after HF or KOH silica leaching.
Materials and methods
Chemicals and reagents
All reagents used: TEOS (tetraetoxysilane, 98 %
purity, Sigma-Aldrich), Pluronic P123 (Sigma-
Aldrich), CH3CH2OH (94–96 %, Alfa Aesar), NaCl
(ACS reagent,[99 %, Sigma-Aldrich), and HCl conc.
J Nanopart Res (2014) 16:2323 Page 3 of 21 2323
123

(36–38 wt%, Fluka) were analytical grade and used as
received without further purification.
Nanomaterials studied
The SBA-15 type mesoporous silica has been prepared
following an optimized route which includes NaCl
addition for improved mesophase formation (Comane-
scu and Guran 2011). In short, NaCl addition and pH
adjustments were employed for an assisted mesophase
formation at temperatures lower than those typically
required for standard SBA-15 synthesis (100–145 �C)
(Zhao et al. 2000). Over a vigorously stirring solution
of Pluronic P123 [PEO20PPO70PEO20]—nonionic
block copolymer, poly(ethylene glycol)–poly(propyl-
ene glycol)–poly(ethylene glycol), small amounts of
salt (NaCl) and concentrated (37 wt%) HCl, tetrae-
thosysilane TEOS (Si(OCH2CH3)4) was added drop-
wise. The reaction mixture containing partially
hydrolyzed siloxanic precursor was sealed in a Teflon
bomb and heated in an oven at temperatures up to
90 �C. This hydrothermal treatment was carried out
for 24 h, time after which the mixture was filtered and
washed with deionized water and ethanol (for removal
of any traces of salt). The composite containing the
P123 was dried overnight at 60 �C and calcination at
550 �C (1�/min during heating) for 6 h (complete
surfactant removal) followed by natural cooling to
room temperature. This led to the final OMS samples
used herein.
A reference sample (following literature procedure,
sample S10) was prepared by stirring the reaction
mixture at 35 �C for 20 h (TEOS prehydrolysis)
followed by hydrothermal treatment at 90 �C for
24 h. While the phase separation can be detected
macroscopically in about 1 h, using a ratio ri = 0.08
NaCl (sample S12), the precipitation occurs much
faster, after 12–15 min. The initial synthesis condi-
tions are 1TEOS:0.016P123:5.9HCl:riNaCl:194H2O.
The salting effect was employed in this synthesis, as
the TEOS condensation rate will increase with NaCl
addition, still following a similar route to nonpromot-
ed condensation. S31 was prepared using a TEOS:-
NaCl ratio 1:0.16, while for S34 a 1:0.24 ratio was
utilized (Table 1).
A number of three composites have been obtained
using inorganic salts impregnated into mesoporous
silica (SBA-15 type) and subsequent thermal treat-
ment up to 550 �C. As inorganic sources, we used
Co(NO3)2, Co(CH3COO)2, and Cu(CH3COO)2. A
comparison was made between the decomposition
products of Co(NO3)2 and Co(CH3COO)2 into the
channels of mesoporous silica based on powder XRD
pattern data. We have also compared the ‘‘just
mixing’’ impregnation procedure and the ‘‘incipient
wetness’’ method (see Supplementary Fig. S2 for
impregnation procedure details).
The solubility of Co(CH3COO)2 in EtOH was
assessed in-house and found to be 28.8 g Co(CH3-
COO)2 in 1 L EtOH (0.16 M) at ambient temperature
and pressure. 8 mL of a saturated solution containing
the Co2? metal precursor was used for two steps
(2 9 4 mL) incipient wetness impregnation in 0.5 g
silica S31.
Experiments have shown, however, that using a
concentrated (e.g., saturated) Co2? precursor solution
leads to considerable amount of amorphous cobalt
oxide forming outside of the siloxanic mesopores.
Using lower concentration of Co(NO3)2 or Co(OAc)2
in ethanol solutions can help circumvent the crystal-
lization of cobalt oxide outside the siloxanic materials
pores and thus yielding higher quality materials, with
Table 1 Highlights for synthesis of mesoporous OMS samples S10, S12, S31, S34
Sample ri ([NaCl]) Temperature of TEOS
prehydrolysis (�C)
Observations
S10 (reference
sample)
– 35 �C, 20 h, then 90 �C
for 24 h
No reaction appears after TEOS
addition
into acidic NaCl solution
Macroscopically separation of
phases occurs within 1 h
S12 0.08 35 �C, 20 h then 90 �C,
24 h
Reaction proceeds faster,
commencing with phase separation
A white precipitate formes in
less than 150
S31 0.16 35 �C, 24 h then 90 �C,
24 h
Faster phase separation White precipitate formes after
100
S34 0.24 35 �C, 20 h then 90 �C
for 24 h
Fastest visible macroscopical
separation
While precipitate in less than 100
2323 Page 4 of 21 J Nanopart Res (2014) 16:2323
123

better dispersion of the metal oxide inside the silica
framework and higher stability. Perhaps an aspect
easily overlooked is that the efficiency of metal salt
precursor infiltrating the mesopores is enhanced due to
interaction of the silanol groups (–Si(OH)) present in
considerable amounts in the pore walls, and the Co2?
cation. This coordination mode of cobalt (II) helps
accommodating even more metal salt precursor solu-
tion (Fig. 1).
Materials characterization
Characterization of the obtained composites has been
carried out using: N2-sorption measurements, low
angle SAXS and wide-angle WAXRD powder dif-
fraction, TEM, SEM, and energy-dispersive X-ray
spectroscopy (EDXS).
Porosity assessment (BET Brunauer–Emmett–
Teller N2 isotherms) was done at 77 K over the relative
pressure range 0.01–0.995 using a Quantachrome
NovaWin 1200e analyzer. Samples were degassed
prior to loading into the sample holder at 80 �C
overnight under vacuum (10-4 bar). Pore size distri-
bution (PSD) was computed based on DFT or BJH
model, pore volume was computed at the maximum
achieved relative pressure (typically P/P0 = 0.99),
and the surface area was computed in the relative
pressure range 0.05–0.30 following the recommenda-
tion for mesoporous materials. P/P0 range is 0.05–0.35
but, however, the liner region of the 1/[W(P0/P) - 1]
versus P/P0 tends to shift to lower relative pressure for
mesoporous materials and even more so, for micropo-
rous materials (Brunauer et al. 1938).
Small angle and wide angle X-ray powder diffrac-
tion (SAXS and WAXRD) patterns were collected on a
Bruker D8 Advance and Philips PW 1820/00 diffrac-
tometers (CuKa radiation, 40 mA, 40 kV); SAXS
measurements were collected using 2h angles between
0.7� and 3� with a scanspeed of 1 and a 0.005 increment.
Transmission electron microscopy (TEM) TEM
images were collected on a Tecnai 12 (FEI), a new
generation of transmission electron microscopes oper-
ating at an accelerating voltage of 40 kV (possible
values range 20–120 kV), with a maximum possible
magnification 1209 to 300 0009. The micrographs
cover the 5–100 nm range to show the mesocomposite
structuring. Samples were ultrasonicated in C2H5OH,
and a drop of this solution was dried on a carbon
coated microgrid prior to the measurements.
Scanning electron microscopy (SEM) The images
were recorded on a scanning electron microscope
EVO 40 Carl Zeiss, equipped with a Pentafet Link
EDXS microanalysis system. All samples were cov-
ered with a thin gold layer prior to imaging.
Energy-dispersive X-ray spectroscopy (EDXS) was
used to assess the surface elemental composition of the
as-obtained mesocomposites.
Catalytic activity. Catalytic performance was
assessed in direct CH4 oxidation on a ‘‘U’’-shaped
tubular reactor made of quartz, operating at atmospheric
pressure, loaded typically with 0.1 g of Co3O4@OMS
or Co3O4-meso, deposited as a powder on the quartz
wool, in the temperature range 250–550 �C. Before the
catalytic process, the catalyst was flushed with air
(100 mL/min) for 1 h at 550 �C to remove adsorbed
species and then was cooled to 250 �C. Evaluation of
catalytic activity was done by feeding the reactor a
mixture of 10 % CH4 in N2 and air flowing at 100 mL/
min (CH4/O2:1/5); the temperature was raised from 250
to 550 �C in 50� steps. CO2 and hydrocarbons were
checked using a Porapaq QS 80/100 and Molesieve 5A
80/100 column, mounted on a DANI GC 1000
chromatograph equipped with a TCD detector.
Results and discussion
Structural characterization of mesoporous support
OMS. Three representative mesoporous silica
Fig. 1 Co2? in Td coordination environment to four silanol
groups
J Nanopart Res (2014) 16:2323 Page 5 of 21 2323
123
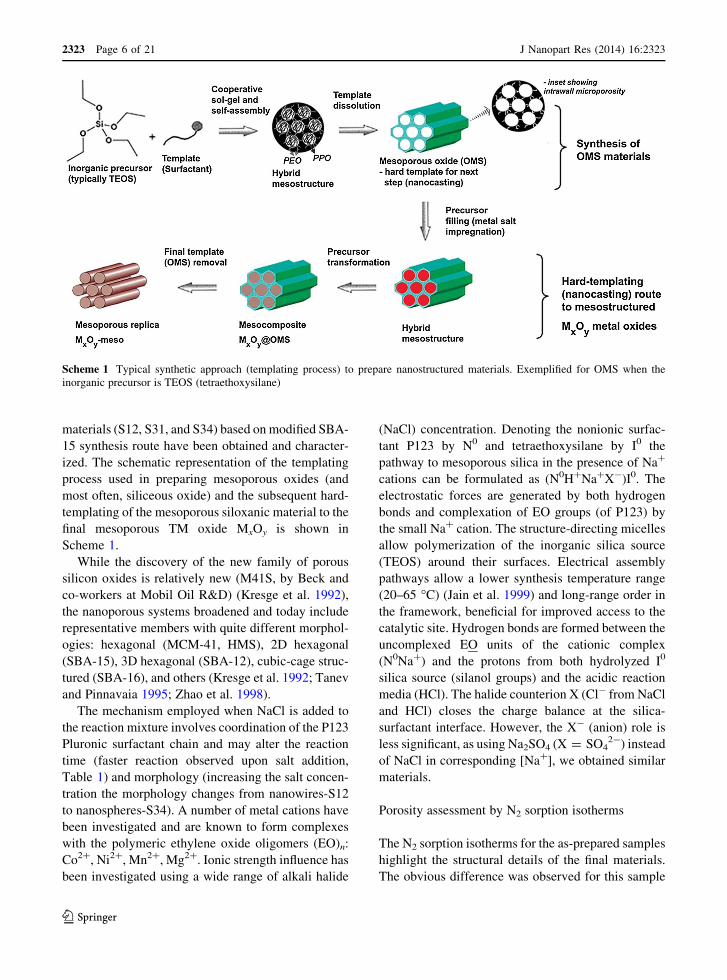
materials (S12, S31, and S34) based on modified SBA-
15 synthesis route have been obtained and character-
ized. The schematic representation of the templating
process used in preparing mesoporous oxides (and
most often, siliceous oxide) and the subsequent hard-
templating of the mesoporous siloxanic material to the
final mesoporous TM oxide MxOy is shown in
Scheme 1.
While the discovery of the new family of porous
silicon oxides is relatively new (M41S, by Beck and
co-workers at Mobil Oil R&D) (Kresge et al. 1992),
the nanoporous systems broadened and today include
representative members with quite different morphol-
ogies: hexagonal (MCM-41, HMS), 2D hexagonal
(SBA-15), 3D hexagonal (SBA-12), cubic-cage struc-
tured (SBA-16), and others (Kresge et al. 1992; Tanev
and Pinnavaia 1995; Zhao et al. 1998).
The mechanism employed when NaCl is added to
the reaction mixture involves coordination of the P123
Pluronic surfactant chain and may alter the reaction
time (faster reaction observed upon salt addition,
Table 1) and morphology (increasing the salt concen-
tration the morphology changes from nanowires-S12
to nanospheres-S34). A number of metal cations have
been investigated and are known to form complexes
with the polymeric ethylene oxide oligomers (EO)n:
Co2?, Ni2?, Mn2?, Mg2?. Ionic strength influence has
been investigated using a wide range of alkali halide
(NaCl) concentration. Denoting the nonionic surfac-
tant P123 by N0 and tetraethoxysilane by I0 the
pathway to mesoporous silica in the presence of Na?
cations can be formulated as (N0H?Na?X-)I0. The
electrostatic forces are generated by both hydrogen
bonds and complexation of EO groups (of P123) by
the small Na? cation. The structure-directing micelles
allow polymerization of the inorganic silica source
(TEOS) around their surfaces. Electrical assembly
pathways allow a lower synthesis temperature range
(20–65 �C) (Jain et al. 1999) and long-range order in
the framework, beneficial for improved access to the
catalytic site. Hydrogen bonds are formed between the
uncomplexed EO units of the cationic complex
(N0Na?) and the protons from both hydrolyzed I0
silica source (silanol groups) and the acidic reaction
media (HCl). The halide counterion X (Cl- from NaCl
and HCl) closes the charge balance at the silica-
surfactant interface. However, the X- (anion) role is
less significant, as using Na2SO4 (X = SO42-) instead
of NaCl in corresponding [Na?], we obtained similar
materials.
Porosity assessment by N2 sorption isotherms
The N2 sorption isotherms for the as-prepared samples
highlight the structural details of the final materials.
The obvious difference was observed for this sample
Scheme 1 Typical synthetic approach (templating process) to prepare nanostructured materials. Exemplified for OMS when the
inorganic precursor is TEOS (tetraethoxysilane)
2323 Page 6 of 21 J Nanopart Res (2014) 16:2323
123
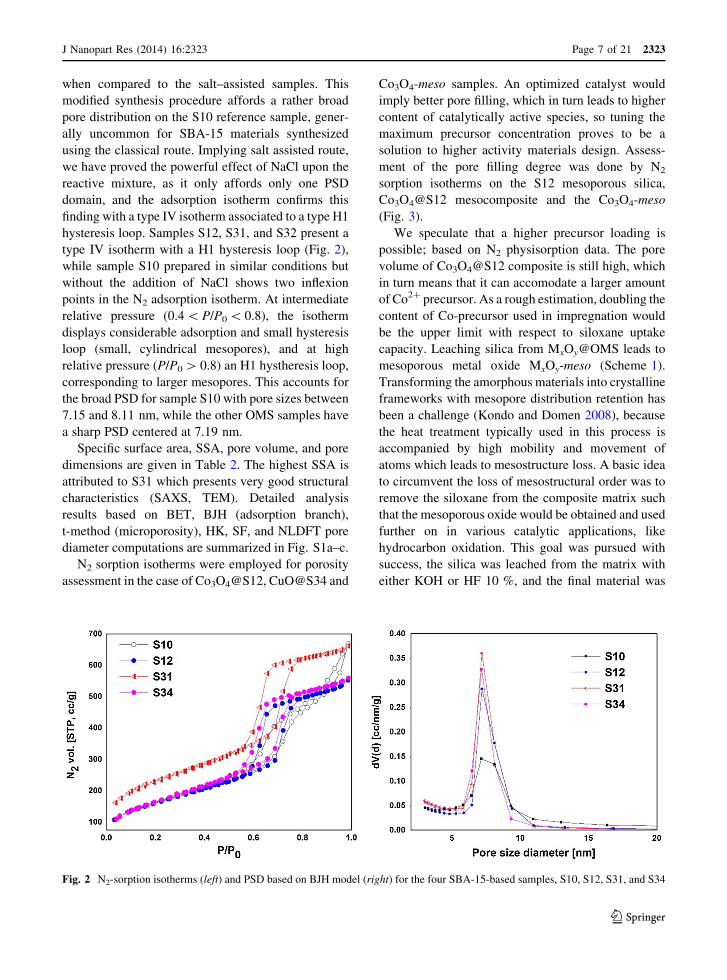
when compared to the salt–assisted samples. This
modified synthesis procedure affords a rather broad
pore distribution on the S10 reference sample, gener-
ally uncommon for SBA-15 materials synthesized
using the classical route. Implying salt assisted route,
we have proved the powerful effect of NaCl upon the
reactive mixture, as it only affords only one PSD
domain, and the adsorption isotherm confirms this
finding with a type IV isotherm associated to a type H1
hysteresis loop. Samples S12, S31, and S32 present a
type IV isotherm with a H1 hysteresis loop (Fig. 2),
while sample S10 prepared in similar conditions but
without the addition of NaCl shows two inflexion
points in the N2 adsorption isotherm. At intermediate
relative pressure (0.4 \ P/P0 \ 0.8), the isotherm
displays considerable adsorption and small hysteresis
loop (small, cylindrical mesopores), and at high
relative pressure (P/P0 [ 0.8) an H1 hystheresis loop,
corresponding to larger mesopores. This accounts for
the broad PSD for sample S10 with pore sizes between
7.15 and 8.11 nm, while the other OMS samples have
a sharp PSD centered at 7.19 nm.
Specific surface area, SSA, pore volume, and pore
dimensions are given in Table 2. The highest SSA is
attributed to S31 which presents very good structural
characteristics (SAXS, TEM). Detailed analysis
results based on BET, BJH (adsorption branch),
t-method (microporosity), HK, SF, and NLDFT pore
diameter computations are summarized in Fig. S1a–c.
N2 sorption isotherms were employed for porosity
assessment in the case of Co3O4@S12, CuO@S34 and
Co3O4-meso samples. An optimized catalyst would
imply better pore filling, which in turn leads to higher
content of catalytically active species, so tuning the
maximum precursor concentration proves to be a
solution to higher activity materials design. Assess-
ment of the pore filling degree was done by N2
sorption isotherms on the S12 mesoporous silica,
Co3O4@S12 mesocomposite and the Co3O4-meso
(Fig. 3).
We speculate that a higher precursor loading is
possible; based on N2 physisorption data. The pore
volume of Co3O4@S12 composite is still high, which
in turn means that it can accomodate a larger amount
of Co2? precursor. As a rough estimation, doubling the
content of Co-precursor used in impregnation would
be the upper limit with respect to siloxane uptake
capacity. Leaching silica from MxOy@OMS leads to
mesoporous metal oxide MxOy-meso (Scheme 1).
Transforming the amorphous materials into crystalline
frameworks with mesopore distribution retention has
been a challenge (Kondo and Domen 2008), because
the heat treatment typically used in this process is
accompanied by high mobility and movement of
atoms which leads to mesostructure loss. A basic idea
to circumvent the loss of mesostructural order was to
remove the siloxane from the composite matrix such
that the mesoporous oxide would be obtained and used
further on in various catalytic applications, like
hydrocarbon oxidation. This goal was pursued with
success, the silica was leached from the matrix with
either KOH or HF 10 %, and the final material was
Fig. 2 N2-sorption isotherms (left) and PSD based on BJH model (right) for the four SBA-15-based samples, S10, S12, S31, and S34
J Nanopart Res (2014) 16:2323 Page 7 of 21 2323
123
analyzed by X-ray diffraction to confirm the Co3O4
phase in Co3O4@OMS. The final SBET of mesoporous
Co3O4 was determined to be 105 m2/g and
Vt = 0.24 cm3/g, with d0 = 3.4 nm, and were
deduced using the BJH method applied on the
absorption branch of the isotherm.
The pore characteristics for CuO–S34 composite
were deduced from the N2 sorption isotherms (Fig. 3).
The N2 sorption isotherm presents a broader
hysteresis for CuO–S34 composite consistent with a
broader PSD (Fig. 4). We observe *50 % pore filling
upon the incipient wetness method of S34 silica with
Cu2? precursor and subsequent thermal treatment,
pore volume decreases accordingly from 1.16 to
0.51 cc/g due to partial filling of silica pores with
CuO (Table 3).
The available mesoporosity could be used to further
tune the highest CuO content for a given mesoporous
sieve as support. The PSD confirms that there is not
much contraction occurring within the mesopores
upon CuO formation inside the silica host (Fig. 4,
right) The mean pore size, however, decreases to
2.9 nm, which means that smaller mesopores account
for the decrease in the pore size (CuO formed within
silica pores). DFT confirms the presence of smaller
mesopores by the broad shoulder at *1.8 nm,
consistent with formation of CuO within the mesop-
ores (Fig. 5).
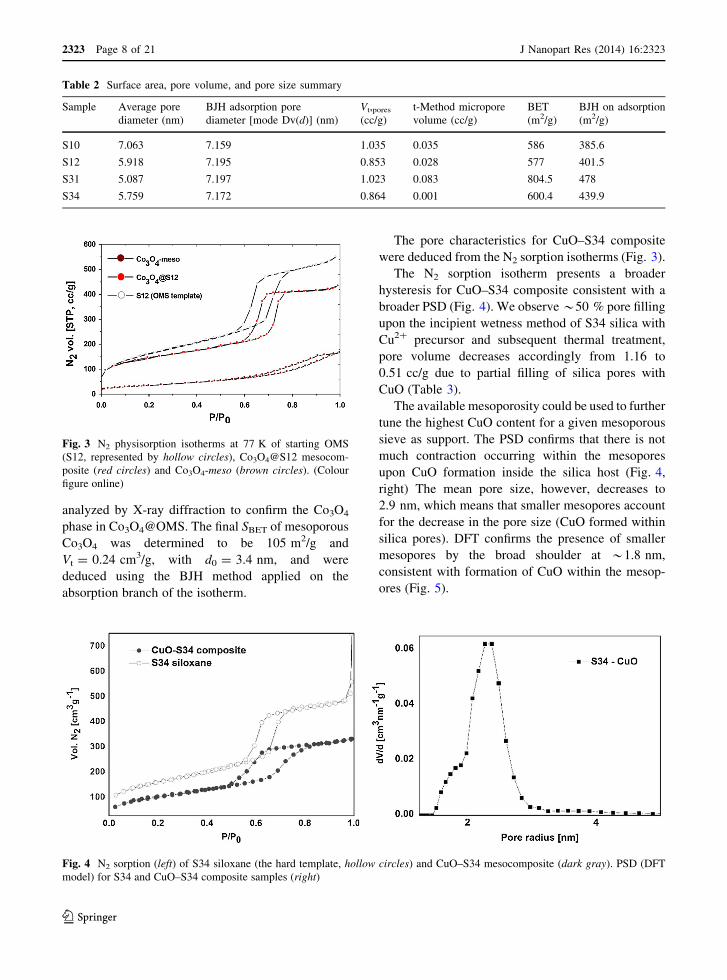
Table 2 Surface area, pore volume, and pore size summary
Sample Average pore
diameter (nm)
BJH adsorption pore
diameter [mode Dv(d)] (nm)
Vt,pores
(cc/g)
t-Method micropore
volume (cc/g)
BET
(m2/g)
BJH on adsorption
(m2/g)
S10 7.063 7.159 1.035 0.035 586 385.6
S12 5.918 7.195 0.853 0.028 577 401.5
S31 5.087 7.197 1.023 0.083 804.5 478
S34 5.759 7.172 0.864 0.001 600.4 439.9
Fig. 3 N2 physisorption isotherms at 77 K of starting OMS
(S12, represented by hollow circles), Co3O4@S12 mesocom-
posite (red circles) and Co3O4-meso (brown circles). (Colour
figure online)
Fig. 4 N2 sorption (left) of S34 siloxane (the hard template, hollow circles) and CuO–S34 mesocomposite (dark gray). PSD (DFT
model) for S34 and CuO–S34 composite samples (right)
2323 Page 8 of 21 J Nanopart Res (2014) 16:2323
123

SAXS and WAXRD
XRD patterns are indexed to reflexions on the strong
(100) and weaker (110) and (200) planes which
suggest a P6mm group, specific to SBA-15 materials.
This corresponds to high ordering siloxanic materials
(Fig 2). While d(100) gives ‘‘a’’ (lattice parameter),
analysis of N2 adsorption isotherms determines mean
pore diameter (Table 4). These results are correlated
by the siloxanic wall thickness, which is the difference
between cell parameter ‘‘a’’ and mean pore diameter
‘‘wd.’’
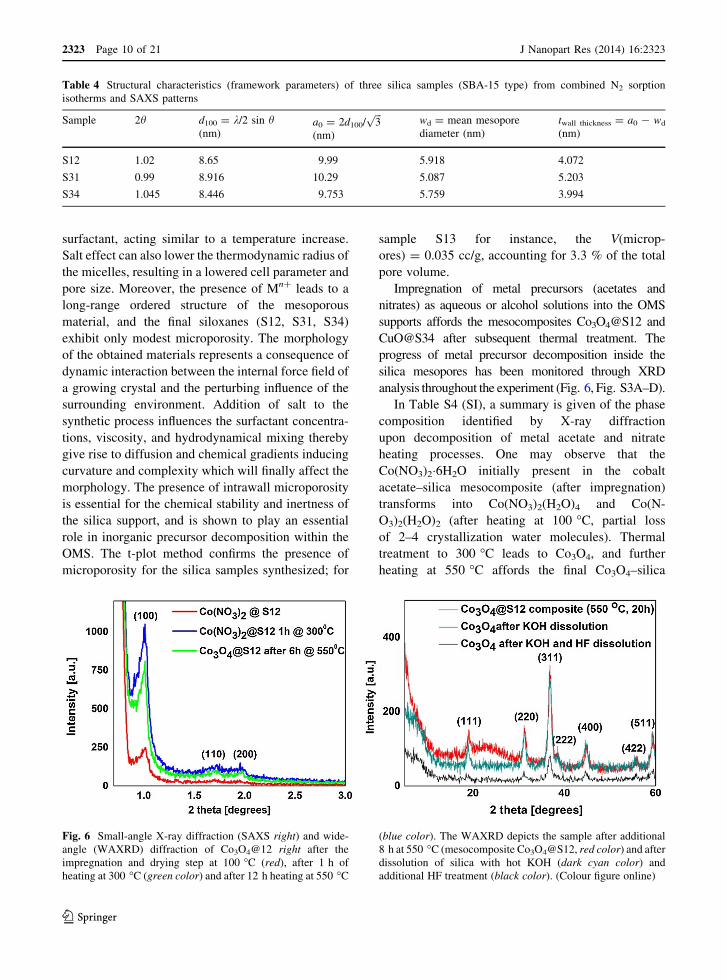
The material exhibiting the thickest walls is S31,
and this implies that such material has a higher thermal
and hydrolytic stability than the other two samples
presented (S12 and S34). However, the results present
all obtained OMS materials as having very good
physical properties.
Studies have shown that SBA-15 materials contain
intrawall micropores that can interconnect the ordered
primary mesopores, and originates from the hydro-
philic part of the triblock copolymer (EO) trapped
within the silica walls. Galarneau and Fajula showed
that the size of these intrawall pores in the SBA-15
materials is a function of the hydrothermal treatment
temperature (Galarneau et al. 2001). Large intercon-
necting mesopores were observed when SBA-15
materials were aged at 140 �C. On the other hand,
the framework pores of materials aged at 35 �C were
mainly constituted by small intrawall mesopores
(2 \ Dp \ 3 nm) and micropores (Dp \ 2 nm). The
PSDs of the framework pores also showed that a
significant decrease of the intrawall pore volume
occurred upon calcination between 250 and 550 �C.
Furthermore, as for the primary mesopores, the
decrease of this volume associated with intrawall
pores during calcination was more significant for
SBA-15 aged at lower temperatures. For materials
aged at 35 �C, the specific surface area associated with
the intrawall micropores corresponds to about 40 % of
the entire surface area, whereas the specific surface
area of the material aged at 140 �C mostly corre-
sponded to pores wider than 6 nm. Substantial lattice
shrinkage between 250 and 550 �C, may usually result
in reduced specific surface areas, while smaller lattice
contraction occurs for materials aged at higher tem-
peratures. Tuning the NaCl to TEOS initial ratio ri, one
can reduce the critical micelle concentration of the
Table 3 Textural characteristics of S34 and CuO–S34
deduced from N2 adsorption isotherms (DFT model)
Sample SBET
(m2/g)
C constant Pore volume
(cm3/g)
Mean pore
width (nm)
S34 550.6 290 1.16 4.2
CuO–S34 352.1 120 0.51 2.9
Fig. 5 SAXS (left) and WAXRD (right) data for OMS samples
showing three resolved peaks indexed to (100), (110), and (200)
reflexion planes. The wide angle X-ray diffraction (WAXRD)
confirms the amorphous siloxanic framework at larger angles
(typically around 2h = 23�)
J Nanopart Res (2014) 16:2323 Page 9 of 21 2323
123
surfactant, acting similar to a temperature increase.
Salt effect can also lower the thermodynamic radius of
the micelles, resulting in a lowered cell parameter and
pore size. Moreover, the presence of Mn? leads to a
long-range ordered structure of the mesoporous
material, and the final siloxanes (S12, S31, S34)
exhibit only modest microporosity. The morphology
of the obtained materials represents a consequence of
dynamic interaction between the internal force field of
a growing crystal and the perturbing influence of the
surrounding environment. Addition of salt to the
synthetic process influences the surfactant concentra-
tions, viscosity, and hydrodynamical mixing thereby
give rise to diffusion and chemical gradients inducing
curvature and complexity which will finally affect the
morphology. The presence of intrawall microporosity
is essential for the chemical stability and inertness of
the silica support, and is shown to play an essential
role in inorganic precursor decomposition within the
OMS. The t-plot method confirms the presence of
microporosity for the silica samples synthesized; for
sample S13 for instance, the V(microp-
ores) = 0.035 cc/g, accounting for 3.3 % of the total
pore volume.
Impregnation of metal precursors (acetates and
nitrates) as aqueous or alcohol solutions into the OMS
supports affords the mesocomposites Co3O4@S12 and
CuO@S34 after subsequent thermal treatment. The
progress of metal precursor decomposition inside the
silica mesopores has been monitored through XRD
analysis throughout the experiment (Fig. 6, Fig. S3A–D).
In Table S4 (SI), a summary is given of the phase
composition identified by X-ray diffraction
upon decomposition of metal acetate and nitrate
heating processes. One may observe that the
Co(NO3)2�6H2O initially present in the cobalt
acetate–silica mesocomposite (after impregnation)
transforms into Co(NO3)2(H2O)4 and Co(N-
O3)2(H2O)2 (after heating at 100 �C, partial loss
of 2–4 crystallization water molecules). Thermal
treatment to 300 �C leads to Co3O4, and further
heating at 550 �C affords the final Co3O4–silica
Table 4 Structural characteristics (framework parameters) of three silica samples (SBA-15 type) from combined N2 sorption
isotherms and SAXS patterns
Sample 2h d100 = k/2 sin h(nm)
a0 = 2d100/ffiffiffi
3p
(nm)
wd = mean mesopore
diameter (nm)
twall thickness = a0 - wd
(nm)
S12 1.02 8.65 9.99 5.918 4.072
S31 0.99 8.916 10.29 5.087 5.203
S34 1.045 8.446 9.753 5.759 3.994
Fig. 6 Small-angle X-ray diffraction (SAXS right) and wide-
angle (WAXRD) diffraction of Co3O4@12 right after the
impregnation and drying step at 100 �C (red), after 1 h of
heating at 300 �C (green color) and after 12 h heating at 550 �C
(blue color). The WAXRD depicts the sample after additional
8 h at 550 �C (mesocomposite Co3O4@S12, red color) and after
dissolution of silica with hot KOH (dark cyan color) and
additional HF treatment (black color). (Colour figure online)
2323 Page 10 of 21 J Nanopart Res (2014) 16:2323
123
composite Co3O4@S13 (‘‘plain mixing’’). The
decomposition pathway is similar when Co(N-
O3)2�6H2O is impregnated over six steps into silica
host (Co3O4@S12, incipient wetness impregnation
method), with the main advantage that decomposi-
tion of metal nitrate outside of the silica pores is
circumvented due to progressive filling-decomposi-
tion cycles. Incipient wetness method was chosen
as the method of choice for inorganic precursor
incorporation into silica host. Decomposition of the
Co(CH3COO)2 impregnated into S31 mesoporous
silica shows after the first heating step at 100 �C
the presence of cobalt acetate hydroxide, while
heating to 300 �C leads to Co3O4 (and Co2O3-
traces), and the heating step at 550 �C produces the
final composite Co3O4@S31, where the Co3O4 is
the main phase. However, decomposition of the
cobalt (II) acetate leads to a number of impurities
identified by powder XRD (Co(OH)2, CoO(OH)
and Co2SiO4), and so the Co(NO3)2 remains the
Co(II) precursor of choice. Decomposition of the
Cu(CH3COO)2 into S34 silica leads to CuO after
the thermal treatment at 300 �C (along with traces
of cuprite Cu2O), CuO (tenorite) being the final
phase present in the CuO@S34 mesocomposite.
SAXS measurements confirm that the mesopores
are not affected by the thermal treatment at 550 �C, as
evidenced in the case of the CuO@S34 composite
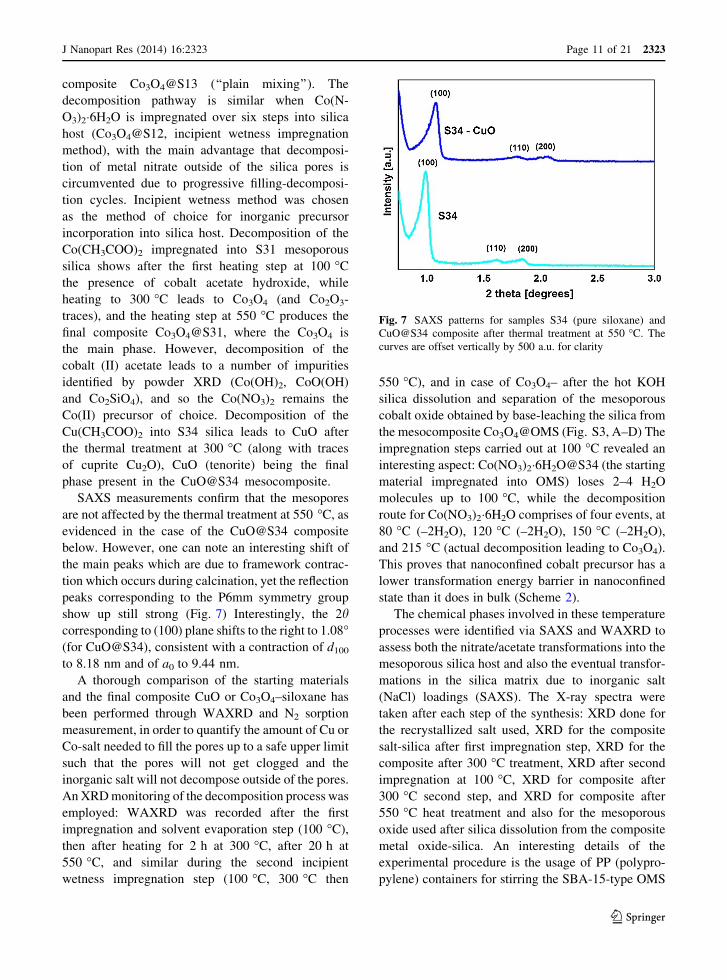
below. However, one can note an interesting shift of
the main peaks which are due to framework contrac-
tion which occurs during calcination, yet the reflection
peaks corresponding to the P6mm symmetry group
show up still strong (Fig. 7) Interestingly, the 2hcorresponding to (100) plane shifts to the right to 1.08�(for CuO@S34), consistent with a contraction of d100
to 8.18 nm and of a0 to 9.44 nm.
A thorough comparison of the starting materials
and the final composite CuO or Co3O4–siloxane has
been performed through WAXRD and N2 sorption
measurement, in order to quantify the amount of Cu or
Co-salt needed to fill the pores up to a safe upper limit
such that the pores will not get clogged and the
inorganic salt will not decompose outside of the pores.
An XRD monitoring of the decomposition process was
employed: WAXRD was recorded after the first
impregnation and solvent evaporation step (100 �C),
then after heating for 2 h at 300 �C, after 20 h at
550 �C, and similar during the second incipient
wetness impregnation step (100 �C, 300 �C then
550 �C), and in case of Co3O4– after the hot KOH
silica dissolution and separation of the mesoporous
cobalt oxide obtained by base-leaching the silica from
the mesocomposite Co3O4@OMS (Fig. S3, A–D) The
impregnation steps carried out at 100 �C revealed an
interesting aspect: Co(NO3)2�6H2O@S34 (the starting
material impregnated into OMS) loses 2–4 H2O
molecules up to 100 �C, while the decomposition
route for Co(NO3)2�6H2O comprises of four events, at
80 �C (–2H2O), 120 �C (–2H2O), 150 �C (–2H2O),
and 215 �C (actual decomposition leading to Co3O4).
This proves that nanoconfined cobalt precursor has a
lower transformation energy barrier in nanoconfined
state than it does in bulk (Scheme 2).
The chemical phases involved in these temperature
processes were identified via SAXS and WAXRD to
assess both the nitrate/acetate transformations into the
mesoporous silica host and also the eventual transfor-
mations in the silica matrix due to inorganic salt
(NaCl) loadings (SAXS). The X-ray spectra were
taken after each step of the synthesis: XRD done for
the recrystallized salt used, XRD for the composite
salt-silica after first impregnation step, XRD for the
composite after 300 �C treatment, XRD after second
impregnation at 100 �C, XRD for composite after
300 �C second step, and XRD for composite after
550 �C heat treatment and also for the mesoporous
oxide used after silica dissolution from the composite
metal oxide-silica. An interesting details of the
experimental procedure is the usage of PP (polypro-
pylene) containers for stirring the SBA-15-type OMS
Fig. 7 SAXS patterns for samples S34 (pure siloxane) and
CuO@S34 composite after thermal treatment at 550 �C. The
curves are offset vertically by 500 a.u. for clarity
J Nanopart Res (2014) 16:2323 Page 11 of 21 2323
123

at room temperature after having added dropwise the
Co(NO3)2 ethanol precursor solution (incipient wet-
ness method) for 5 min. The hydrophobic surface (as
opposed to that of glass) of polypropylene afforded a
reasonable loading of 15.5 % as confirmed by EDAX
measurements for the final mesocomposite.
Based on the thermal decomposition of the metal salt
precursor, one can conclude that simple mixing of the
Co(NO3)2 or Co(OAc)2 solution (in water or ethanol) in
one step followed by drying at 100 �C, heating at
300 �C for 2 h and additional 20 h at 550 �C does lead
to lower quality nanomaterials. The reason is that a fair
amount of the metal oxide would form outside of the
silica pores, and as such, will behave differently than
the oxide confined in the SBA-15 mesopores, where it
originally formed by thermal decomposition of either
the cobalt nitrate or acetate. Increasing the number of
incipient wetness steps greatly enhances the quality of
the final mesocomposites (a table summarizing the
transformations identified during metal salt decompo-
sition under the experimental conditions employed here
is given in SI, Table S4). It is noteworthy to observe that
even after calcination step at 300 �C, the metal oxide
clearly shows up in the XRD patterns, consistent with
Co3O4 and CuO XRD patterns, respectively. The
framework strengthening step at 550 �C affords the
final mesocomposites.
EDAX
The EDAX measurements (Fig. 8) for the samples
Co3O4@S12 and CuO@S34 conducted in a joint
SEM-EDAX experiment confirm the presence of the
metal oxide at the surface of the composite. This
allows us to compute the percent of metal oxide
present in the composite (Table 5).
EDAX results show a percent composition based on
the elements identified. For instance, the mesocom-
posite CuO@OMS has the composition xCuO�ySiO2.
The EDAX gives the content in Cu (%) and Si (%),
which are based on the composite formulation
(MCuO = 79.54 g mol-1, MSiO2 = 60.08 g mol-1;
ACu = 63.54 g mol-1; ASi = 28.09 g mol-1):
% Cu ¼ 63:54� x
79:54� xþ 60:08� y� 100 and
% Si ¼ 28:09� y
79:54� xþ 60:08� y� 100:
ð1Þ
The proprietary software operating the joint SEM–
EDS apparatus outputs the following data upon
acquisition (Table 5).
For CuO@OMS sample, the ratio is effectively given
as 90.78 % SiO2, 9.22 % CuO. In the case of
Co3O4@OMS, we started from the mol%: 85.54 %
SiO2, 14.46 % CoO (the real form of cobalt is, however,
Co3O4 and not CoO as assumed by software), this means
that the Co3O4 content in 100 g composite
Co3O4@OMS will be: (1/3)*(14.46/74.93)*240.79 =
15.49 g, hence 15.49 wt% (MCoO = 74.96 g mol-1,
MCo3O4 = 240.79 g mol-1). The final composition
reflecting the wt% in SiO2 and metal oxide, respectively,
is given in Table 6.
More than 15 wt% Co3O4 has been incorporated
into the silica host in case of Co3O4@S12, while a
more conservative, 9.2 % CuO was found in
CuO@S34. The metal oxide loading will show a
direct correlation with the catalytic performance of the
as-synthesized samples.
Fig. 8 EDAX measurements for samples Co3O4–S12 (left) and CuO–S34 (right). The values on the abscissa are given in keV for both
samples
Co(NO3)2.6H2O Co(NO3)2.4H2O Co(NO3)2.2H2O Co(NO3)2 1/3 Co3O4 + 1/3 N2O4 +2/3 N2O5- 2 H2O - 2 H2O - 2 H2O
80 oC 120 oC 150 oC 215 oC
confined in OMS, 100oC
- (2-4) H2O
Scheme 2 Decomposition pathway for Co(NO3)2�6H2O in bulk and confined state
2323 Page 12 of 21 J Nanopart Res (2014) 16:2323
123

Morphology
Transmission electron microscopy (TEM)
The TEM images for sample S31 are shown in Fig. 9.
Looking at the siloxanic SBA-15 through a direction
perpendicular to the pore axis a parallel pattern is
observed (Fig. 9a, c, d).
At 10 nm scale, the channels appear parallel (view
perpendicular to the electron beam), and the pore
diameter (*7.2 nm, Fig. 9c) confirms the PSD deduced
from N2 sorption isotherms. TEM micrographs prove a
hexagonal ordering of the silica framework, catalyzed by
the NaCl addition; during the end of the mesophase
formation, an aliquot was taken and checked under the
transmission electron microscope, confirming the hex-
agonal ordering of the framework (black arrows point to
the channels in Fig. 9f). The honeycomb structure
characteristic to the SBA-15 type of materials can be
seen clearly in Fig. 9b, e.
Examination by TEM imaging (Figs. 10, 11) of the
metal oxide-silica mesocomposites reveals that the
hexagonal arrangement of the silica is preserved
during and after pores filling with inorganic precursor,
a result confirmed by SAXS diffraction data at low
angle. The differences in electron density make the
silica support show lighter contrast, and the metal
oxide (Co3O4, CuO) appear as areas of darker contrast.
Highly dispersed cobalt oxide nanoparticles can be
observed throughout the Co3O4@OMS as patches of
different size and shape. Interestingly, the diameter of
some patches exceeds that of the OMS silica host. This
is due to the micropores in the SBA-15 type of silica
(S12) that interconnect the mesopores, and allow the
extended growth of the crystalline cobalt oxide. TEM
micrographs show both empty mesopores and cobalt
oxide patches which grew in multiple neighboring
mesopores due to their inherited microporosity. When
the channels are perpendicular to the electron beam,
they appear as parallel, highly ordered channels
(Fig. 10c). The cobalt oxide loading is visible as
darker regions apparently filling up the mesopores
(Fig. 10c). Larger patches occur within the composite
and present different morphologies, depending on the
extent of cobalt oxide crystallite growth due to cobalt
nitrate infiltration in the silica channels (meso- and
micropores) and subsequent decomposition to yield
Co3O4. These patches of cobalt oxide are well
Table 5 Computation details for CuO and Co3O4 contents in the corresponding composites
EDAX ZAF quantification (standardless)
Oxides
SEC table: default
Co3O4@OMS
Elem wt% mol% K-ratio Z A F
SiO2 85.54 88.06 0.2306 0.9930 0.5807 1.0000
CoO 14.46 11.94 0.1001 0.8737 1.0070 1.0000
Total 100.00 100.00
kV 29.00, tilt 0.00, take-off, 32.09, Tc 50.0, det
type UTW, sapphire res 134.40, Lsec 105
CuO@OMS
Elem wt% mol% K-ratio Z A F
SiO2 90.78 92.88 0.2514 0.9888 0.5990 1.0000
CuO 9.22 7.12 0.0642 0.8602 1.0139 1.0000
Total 100.00 100.00
kV 29.00, tilt 0.00, take-off, 32.09, Tc 50.0,
det type UTW, sapphire res 134.40, Lsec 79
Table 6 EDAX analysis results on mesocomposites
Co3O4@S12 and CuO@S34
Sample SiO2 wt% MxOy wt%
Co3O4–S12 84.54 15.49 wt% Co3O4
CuO–S34 90.78 9.22 wt%–CuO
J Nanopart Res (2014) 16:2323 Page 13 of 21 2323
123
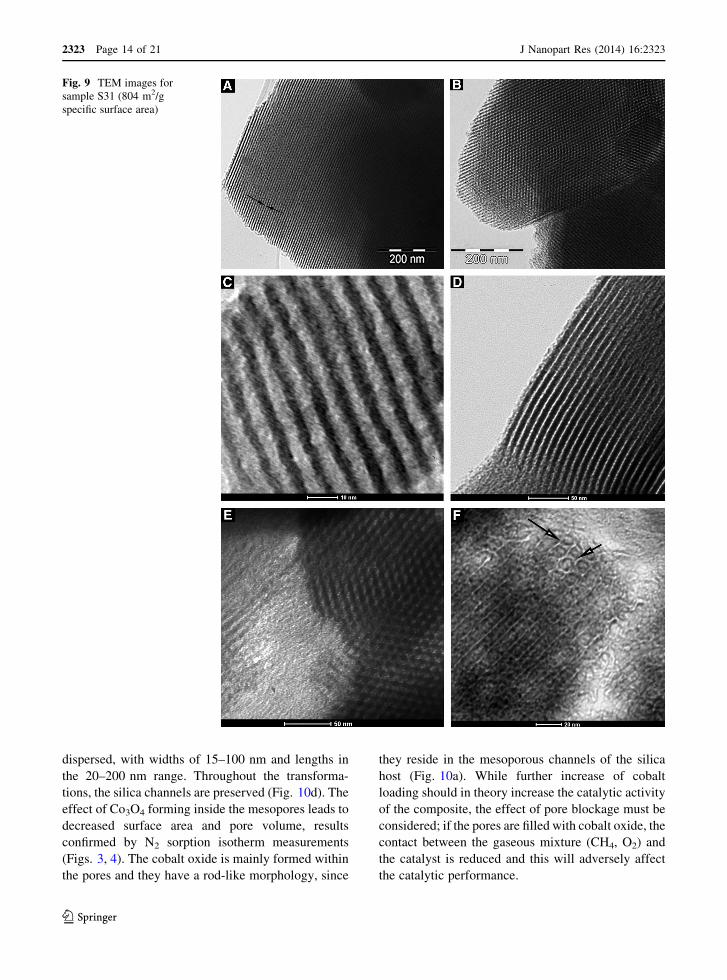
dispersed, with widths of 15–100 nm and lengths in
the 20–200 nm range. Throughout the transforma-
tions, the silica channels are preserved (Fig. 10d). The
effect of Co3O4 forming inside the mesopores leads to
decreased surface area and pore volume, results
confirmed by N2 sorption isotherm measurements
(Figs. 3, 4). The cobalt oxide is mainly formed within
the pores and they have a rod-like morphology, since
they reside in the mesoporous channels of the silica
host (Fig. 10a). While further increase of cobalt
loading should in theory increase the catalytic activity
of the composite, the effect of pore blockage must be
considered; if the pores are filled with cobalt oxide, the
contact between the gaseous mixture (CH4, O2) and
the catalyst is reduced and this will adversely affect
the catalytic performance.
Fig. 9 TEM images for
sample S31 (804 m2/g
specific surface area)
2323 Page 14 of 21 J Nanopart Res (2014) 16:2323
123

The presence of CuO within the OMS host is
apparent in Fig. 11a, where extended regions of high
contrast are present, and can be attributed to the metal
oxide showing darker contrast compared to silica due
to electron density difference. The OMS mesopores
are unaffected by the CuO loading and thermal
treatment, and appear as parallel channels (Fig. 11b,
c, d) when the electron beam is perpendicular to the
pores. Moreover, there is little framework contraction
during the thermal treatment. There are, however,
some patches of CuO formed on the external area
(Fig. 11c). When the channels run parallel to the
electron beam, the honeycomb structure is visible, and
cobalt oxide seems to close the channels (black
arrows, Fig. 11b).
Scanning electron microscopy (SEM)
The obtained materials feature interesting structural
characteristics, as evidenced by the SEM images. The
mesotructures reported herein have either hexagonal
nanowire array (Co3O4–S12) or nanosphere array (CuO–
S34) appearance, depending on the topology of the host.
The hexagonal feature was inferred based on the small
angle diffraction data. The SAXS patterns exhibit an
intense low angle diffraction peak and other two, lower
intensity peaks which are assigned to the (100), (110) and
(200) planes of the 2D-hexagonal space group P6mm.
The two results were corroborated, and the conclusion
was that the Co3O4@OMS composite appears as
hexagonal nanowires. SEM images of Co3O4–S12
(Fig. 12a, b) show isolated rods 300–600 nm.
We see the influence the NaCl addition has on the
final morphology of the OMS: using a 0.08:1 molar
ratio NaCl:TEOS in the initial solution mixture leads
to nanowire formation (S12), while increasing this
ratio to 0.24:1, nanosphere arrays (S34) are obtained
instead.
This morphology control was, therefore, possible due
to the salt assisted templating process promoted by
NaCl. Nanocasting using Co2? or Cu2? precursors leads
to decomposition of the initial acetate or nitrate
Fig. 10 TEM images of
Co3O4–S12 nanocomposite
J Nanopart Res (2014) 16:2323 Page 15 of 21 2323
123

precursors to Co3O4 or CuO into the silica host. Lower
[NaCl] leads to nanowire formation, while higher
[NaCl] allows nanosphere arrays to form, which span
multiple hundreds of nanometers.The CuO–S34 sample
(Fig. 13a, b) shows agglomerates with the appearance of
chains (Fig. 13a, black arrows) expanding to multiple
micrometers. The nanochannels are interconnected by
metal particles that have a random distribution, inherited
from the mesoporous SBA-15 based silica which
possesses scattered micropores in the nanochannel
walls.(Scheme 1) This nanocasting procedure afforded
reproducible patterned metal oxides and mesoporous
oxides–mesoporous silica mesocomposites.
Catalytic activity of SBA-15-supported MxOy
(Co3O4@S12, CuO@S34) and Co3O4-meso
CH4 oxidation catalyzed by Co3O4(meso) and Co3O4-
SBA-15 has been investigated. The reaction rate
corresponding to the total oxidation of methane is
actually complex as the mechanism implies a couple
of temperature-dependent, competing adsorption
equilibria of CO, CO2, O2, CH4, and H2O on active
centers. For the total oxidation reaction, one may write
the following half-empirical formula:
r ¼ ks Tð Þð Þ pO1=22 þ kl Tð Þð Þ pCH4 ð2Þ
where ks(T) is the reaction constant of the surface-
chemosorbed oxygen and kl(T) represents the reaction
constant of the network oxygen, pO2 and pCH4
represent the partial pressures of oxygen and methane,
respectively. Equation (2) may be simplified applying
the assumption of fast oxygen diffusion from the
gaseous phase onto the catalyst’s surface in order to
replace the oxygen consumed from the network,
together with the negligible effects of CO2 and H2O
(dry gases, low methane concentration, temperatures
higher than 400 �C and conversions lower than 75 %).
Given these conditions, the methane oxidation can be
illustrated by:
Fig. 11 TEM images of
nanocomposite CuO–S34
2323 Page 16 of 21 J Nanopart Res (2014) 16:2323
123
r ¼ kapCH4 ð3Þ
where ka is the apparent reaction constant. The
reaction rate r (mol CH4 s-1 kg-1) can be computed
as:
r ¼ F � Dc CH4ð Þ=mcat ð4Þ
where F is the feed of the reactor, DcCH4is the volume
of reacted methane, and mcat is the weight of the
catalyst used. The reaction constants were computed at
different temperatures in the 250–550 �C range.
Arrhenius equation (5) was used in the logarithmic
form, and ln (k) was plotted as function of the
reciprocal absolute temperature 1/T (K-1) (Fig. 14,
right)
ln k ¼ ln A�Ea=RT ð5ÞThis approach allowed analytical determination of
Ea (the activation energy) and the pre-exponential
factor corresponding to methane oxidation on two
different catalysts, Co3O4@S12 and Co3O4-meso.
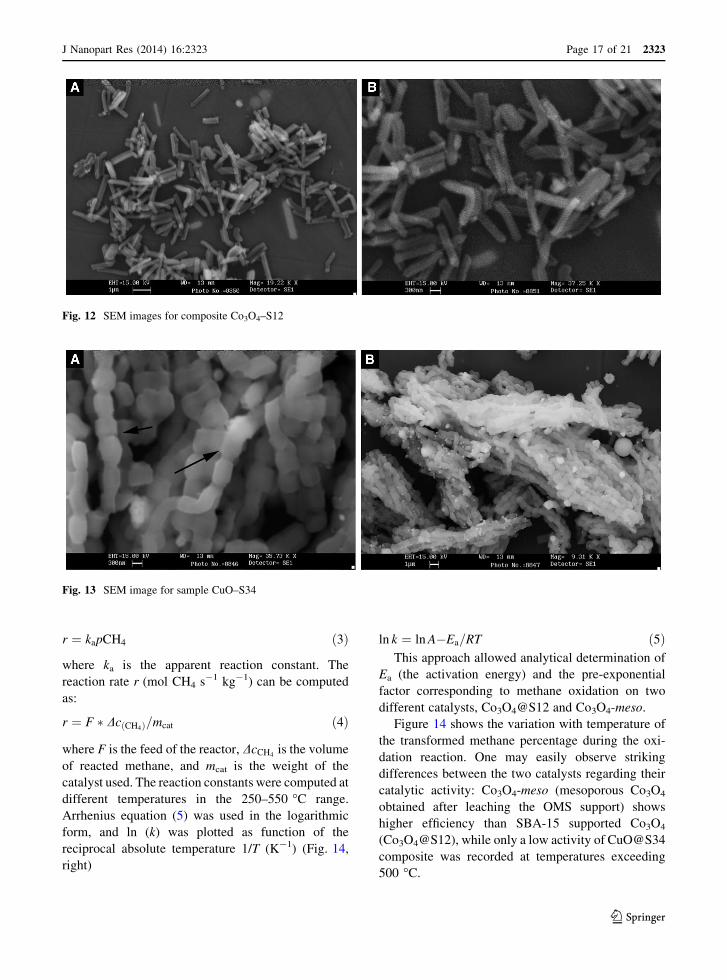
Figure 14 shows the variation with temperature of
the transformed methane percentage during the oxi-
dation reaction. One may easily observe striking
differences between the two catalysts regarding their
catalytic activity: Co3O4-meso (mesoporous Co3O4
obtained after leaching the OMS support) shows
higher efficiency than SBA-15 supported Co3O4
(Co3O4@S12), while only a low activity of CuO@S34
composite was recorded at temperatures exceeding
500 �C.
Fig. 12 SEM images for composite Co3O4–S12
Fig. 13 SEM image for sample CuO–S34
J Nanopart Res (2014) 16:2323 Page 17 of 21 2323
123
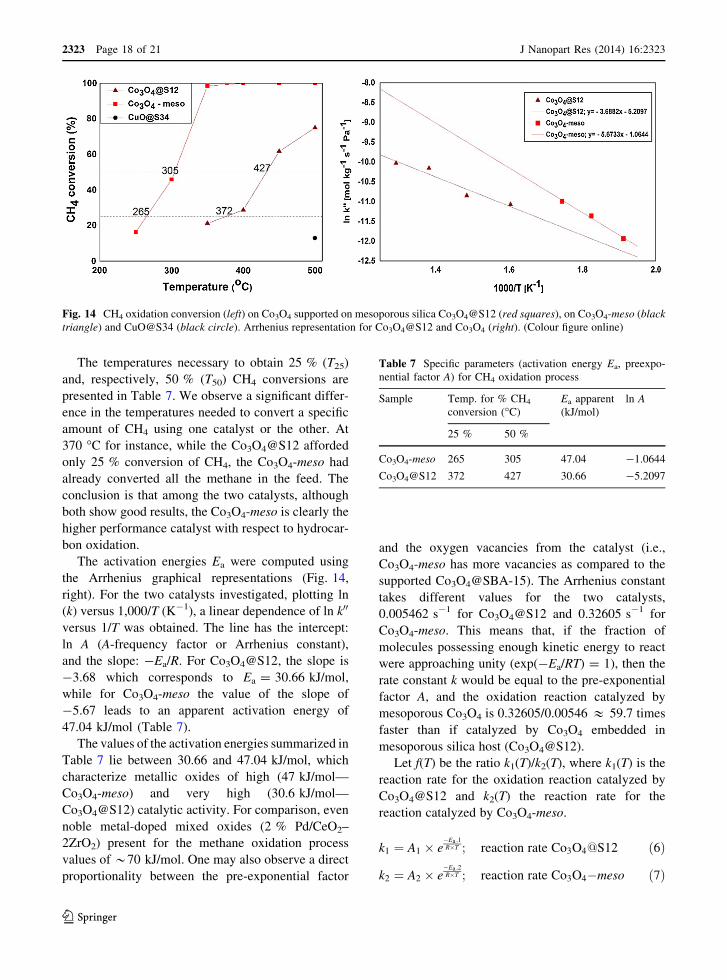
The temperatures necessary to obtain 25 % (T25)
and, respectively, 50 % (T50) CH4 conversions are
presented in Table 7. We observe a significant differ-
ence in the temperatures needed to convert a specific
amount of CH4 using one catalyst or the other. At
370 �C for instance, while the Co3O4@S12 afforded
only 25 % conversion of CH4, the Co3O4-meso had
already converted all the methane in the feed. The
conclusion is that among the two catalysts, although
both show good results, the Co3O4-meso is clearly the
higher performance catalyst with respect to hydrocar-
bon oxidation.
The activation energies Ea were computed using
the Arrhenius graphical representations (Fig. 14,
right). For the two catalysts investigated, plotting ln
(k) versus 1,000/T (K-1), a linear dependence of ln k00
versus 1/T was obtained. The line has the intercept:
ln A (A-frequency factor or Arrhenius constant),
and the slope: -Ea/R. For Co3O4@S12, the slope is
-3.68 which corresponds to Ea = 30.66 kJ/mol,
while for Co3O4-meso the value of the slope of
-5.67 leads to an apparent activation energy of
47.04 kJ/mol (Table 7).
The values of the activation energies summarized in
Table 7 lie between 30.66 and 47.04 kJ/mol, which
characterize metallic oxides of high (47 kJ/mol—
Co3O4-meso) and very high (30.6 kJ/mol—
Co3O4@S12) catalytic activity. For comparison, even
noble metal-doped mixed oxides (2 % Pd/CeO2–
2ZrO2) present for the methane oxidation process
values of *70 kJ/mol. One may also observe a direct
proportionality between the pre-exponential factor
and the oxygen vacancies from the catalyst (i.e.,
Co3O4-meso has more vacancies as compared to the
supported Co3O4@SBA-15). The Arrhenius constant
takes different values for the two catalysts,
0.005462 s-1 for Co3O4@S12 and 0.32605 s-1 for
Co3O4-meso. This means that, if the fraction of
molecules possessing enough kinetic energy to react
were approaching unity (exp(-Ea/RT) = 1), then the
rate constant k would be equal to the pre-exponential
factor A, and the oxidation reaction catalyzed by
mesoporous Co3O4 is 0.32605/0.00546 & 59.7 times
faster than if catalyzed by Co3O4 embedded in
mesoporous silica host (Co3O4@S12).
Let f(T) be the ratio k1(T)/k2(T), where k1(T) is the
reaction rate for the oxidation reaction catalyzed by
Co3O4@S12 and k2(T) the reaction rate for the
reaction catalyzed by Co3O4-meso.
k1 ¼ A1 � e�Ea ;1R�T ; reaction rate Co3O4@S12 ð6Þ
k2 ¼ A2 � e�Ea ;2R�T ; reaction rate Co3O4�meso ð7Þ
Fig. 14 CH4 oxidation conversion (left) on Co3O4 supported on mesoporous silica Co3O4@S12 (red squares), on Co3O4-meso (black
triangle) and CuO@S34 (black circle). Arrhenius representation for Co3O4@S12 and Co3O4 (right). (Colour figure online)
Table 7 Specific parameters (activation energy Ea, preexpo-
nential factor A) for CH4 oxidation process
Sample Temp. for % CH4
conversion (�C)
Ea apparent
(kJ/mol)
ln A
25 % 50 %
Co3O4-meso 265 305 47.04 -1.0644
Co3O4@S12 372 427 30.66 -5.2097
2323 Page 18 of 21 J Nanopart Res (2014) 16:2323
123

f Tð Þ ¼ k1 Tð Þk2 Tð Þ ¼
A1
A2
� e�ðEa ;2�Ea ;1Þ
R�T ¼ 0:01675� e�1;970
T
ð8Þ
f 0 Tð Þ ¼ 33:0003� e�1;970
T � T�2 [ 0; 8T [ 0 ð9Þ
0\f Tð Þ ¼ k1 Tð Þk2 Tð Þ\1; 8T [ 273 K ð10Þ
The reaction rate is actually always faster for
Co3O4(meso) regardless of the temperature. We observe
that f(T) is a continuous and increasing function for any
temperature T (Eq. 8), since the first derivative f0(T) is
always positive (Eq. 9). f(T) is also always lower than
unity for any T [ 273 K (Eq. 10). To put it in words,
this means that the reaction rate will always be higher for
Co3O4-meso than for Co3O4@S12. This result can be
interpreted also in the following way: nanoconfinement
of the mesoporous oxide into the silica host reduces the
activation energy from 47.04 kJ/mol (Co3O4-meso) to
30.66 kJ/mol (Co3O4@S12), while a reverse trend is
observed for the preexponential factor, which is higher
(0.3261 s-1) for Co3O4-meso and lower for confined
Co3O4@S12 (0.0054 s-1). This reflects the lower
probability of collision between the methane and O2
molecules over confined Co3O4@S12 (which contains
12 % cobalt oxide) than over pure mesoporous Co3O4.
The activity depends on the sample textural prop-
erties, the best performance being achieved for the
material with the highest surface area and the most
open pore system. The relatively low temperatures
required for complete oxidation of CH4 also helps
circumventing structural degradation of the catalyst. A
control experiment shows that mesoporous silica
template samples prepared here (S12, S31, S34) have
no catalytic activity at temperatures up to 500 �C,
which confirms that the nanoconfined metal oxide is
responsible for the observed catalytic effect.
Moreover, the metallic oxide reactivity greatly
depends on the coordinative unsaturation degree of the
embedded metal cations. The presence of oxygen
vacancies exposes the unsaturated metal cations effi-
ciently. The two oxidation states of cobalt in Co3O4
allow its participation in reactions involving modifica-
tion of the OS (oxidation state). It has been reported that
the redox properties of the system Co2?/Co3? can
facilitate the catalytic behavior in oxidation of cyclo-
hexanol (Taghavimoghaddam et al. 2012), while also
enhancing the oxygen mobility in Co3O4@OMS mes-
ocomposite. Due to coordinative unsaturation, the
oxygen ions from the superficial layer are generally
less bounded than those from the bulk. This explains the
superficial oxygen ions participating in oxidation reac-
tions, while the mixed valence state of cobalt in Co3O4
(which can be regarded as a 1:1 mixture of Co2O3:CoO,
so Co3?/Co2?=1/1) facilitates the oxidation, Co2?/
Co3? redox couple mediating the oxidation. Substitu-
tion of Co3? with Co2? leads to an increase in
coordinative unsaturation. Among the factors that can
influence the catalytic activity are the above-mentioned
mobility of oxygen vacancies, acid–base properties,
electron affinity of cobalt ions, structural characteristics
of the support and dispersion of active catalyst species
onto the support. The high dispersion of cobalt oxide and
copper oxide in the mesoporous silica significantly
contribute to improved catalytic performance as com-
pared to bulk metal oxides. A key role for this behavior
can be attributed to the small Co3O4 particle size as
depicted before by TEM measurements. Methane
conversion is particularly effective, the conversion
achieved at 350 �C is practically [99 %. CuO meso-
composite on the other hand only showed about 13 %
conversion of methane at 500 �C. This highlights the
mesoporous Co3O4 as a superior catalyst with respect to
alkane oxidation. Mastering the oxidation reaction
mechanism and a thorough knowledge of the inorganic
precursor behavior in mesoporous molecular sieves
(siloxanes), we could gain a new perspective on highly-
efficient catalysts that could accomplish oxidation
reactions at temperatures of about 200–250 �C for
transformations that normally required noble metals or
other expensive plant investments.
Conclusions
In summary, a range of mesocomposites containing
dispersed TM oxides (M=Cu, Co) confined in mesopor-
ous silica OMS have been prepared via the hard-
templating (nanocasting) route and characterized by N2
sorption measurements, SAXS and wide angle XRD
powder diffraction, SEM, and TEM. The mesocompos-
ites obtained exhibited homogeneity, crystallinity con-
firmed by XRD diffraction, and small crystallite sizes
(\10 nm from TEM micrographs) confirmed by the
rather narrow PSD deduced from the N2 sorption
isotherm data. The influence of the silica starting OMS
and the amount of inorganic precursor employed have led
to materials that have been tested and proven to be
J Nanopart Res (2014) 16:2323 Page 19 of 21 2323
123

catalytically active in hydrocarbon oxidation applica-
tions. Choosing a very stable, saturated hydrocarbon in
the alkane series (CnH2n?2, n = 1, methane), we showed
that mesoporous Co3O4 exhibits a high oxidation ability
affording full methane combustion at 350 �C, followed
by the Co3O4@OMS mesocomposite (15.5 % loading)
which converted *50 % of CH4 at 430 �C, while
CuO@OMS composite only afforded a modest 13 %
conversion of methane at 500 �C. This order in catalytic
activity of nanostructured metal oxides mirrors the
previous findings on the bulk materials, which showed
a higher activity for Co3O4 than for CuO or other TM bulk
oxides. The performance of the mesoporous Co3O4
obtained is closely related to that of the very expensive
noble metal based catalysts (Pd@Al2O3), which starts to
convert methane at 300 �C. This Pd-alumina based
catalyst is actually outperformed, since Co3O4@OMS
begins methane conversion below 250 �C, with 18 %
conversion recorded by GC at 250 �C. Further tuning of
the metal precursor quantity can afford a higher activity
for the mesocomposite Co3O4@OMS which will pre-
clude the shortcomings associated with catalyst deacti-
vation in the case of pure mesoporous Co3O4, or
mesoporous framework breakdown during successive
catalytic cycles. Performing as such an active catalyst for
alkane oxidation, we believe that oxidation of other
substrates (hydrocarbons) will proceed under much
milder conditions, which in turn is associated with lower
temperatures, energy consumption and overall ‘‘greener’’
catalysis.
Acknowledgments Support of the Romanian Ministry of
Education and Research through the project PNCDI-2 No.
72-196/2008 ‘‘New complex hydrides for hydrogen storage in
hydride tank suitable for vehicular applications’’—STOHICO
and the financial support of the POSDRU-ID5159 doctoral
fellowship are acknowledged. This work was partially
supported from the Romanian Core Programme (Contract No.
45N/2014). I am grateful to Prof. Cornelia Guran for insightful
discussions. I am in debt to senior researcher Viorica Parvulescu
for catalytic studies and insightful suggestions. I strongly
acknowledge the support received from Prof. Giovanni Principi
regarding training and usage of the research facilities at
Universita Degli Studi di Padova, Italy, where most of this
research was carried out.
References
Armelao L, Barreca D, Gross S, Martucci A, Tieto M, Tondello
E (2001) Cobalt oxide-based films: sol–gel synthesis and
characterization. J Non Cryst 293:477–482
Baldwin TR, Burch R (1990) Remarkable activity enhancement
in the catalytic combustion of methane on supported pal-
ladium catalysts. Catal Lett 6(1):131–138
Benitez MJ, Petracic O, Tuysuz H, Schuth F, Zabel H (2011)
Phys Rev B 83:134424
Brunauer S, Emmett PH, Teller E (1938) Adsorption of gases in
multimolecular layers. J Am Chem Soc 60:309–319
Burch R, Loader PK (1994) Investigation of Pt/Al2O3 and Pd/
Al2O3 catalysts for the combustion of methane at low
concentrations. Appl Catal B 5(1–2):149–164
Carstens JN, Su SC, Bell AT (1998) Factors affecting the cat-
alytic activity of Pd/ZrO2 for the combustion of methane.
J Catal 176:136–142
Comanescu C, Guran C (2011) Influence of NaCl addition on the
synthesis of SBA-15 mesoporous silica. UPB Sci Bull B
73(4):95–104
Cullis CF, Willatt BM (1983) Oxidation of methane over sup-
ported precious metal catalysts. J Catal 83(2):267–285
Dahal N, Ibarra IA, Humphrey SM (2012) High surface area
mesoporous Co3O4 from a direct soft template route.
J Mater Chem 22:12675–12681
Galarneau A, Cambon H, Renzo FD, Fajula F (2001) True
microporosity and surface area of mesoporous SBA-15
silicas as a function of synthesis temperature. Langmuir
17:8328–8335
Hicks RF, Qi H, Young ML, Lee RG (1990) Effect of catalyst
structure on methane oxidation over palladium on alumina.
J Catal 122:295–306
Jain NJ, George A, Bahadur P (1999) Effect of salt on the
micellization of pluronic P65 in aqueous solution. Colloid
Surf A 157:275–283
Kandalkar SG, Gunjakar JL, Lokhande CD (2008) Preparation
of cobalt oxide thin films and its use in supercapacitor
application. Appl Surf Sci 254:5540–5544
Kondo JN, Domen K (2008) Crystallization of mesoporous
metal oxides. Chem Mater 20(3):835–847
Kresge CT, Leonowicz ME, Roth WJ, Vartuli JC, Beck JS (1992)
Ordered mesoporous molecular sieves synthesized by a
liquid crystal template mechanism. Nature 359:710–712
Levy RM (1968) An X-ray study of the participation of the bulk
phase of cobalt oxide in oxidation catalysis. J Phys Chem
72(7):2609–2614
Li Zh, Hoflund GB (1999) Catalytic oxidation of methane over
Pd/Al2O3. React Kinet Catal Lett 66(2):367–374
Li WY, Xu LN, Chen J (2005) Co3O4 nanomaterials in lithium–ion
batteries and gas sensors. Adv Funct Mater 15(5):851–857
Maruyama T, Arai S (1996) Electrochromic properties of cobalt
oxide thin films prepared by chemical vapor deposition.
J Electrochem Soc 143(4):1383–1386
McCarthy JG, Chang YF, Wong VL, Johansson ME (1997)
Kinetics of high temperature methane combustion by metal
oxide catalysts. Div Pet Chem 42:158–162
Milt VG, Lombardo EA, Ulla MA (2002) Stability of cobalt
supported on ZrO2 catalysts for methane combustion. Appl
Catal B 37:63–73
Pena MA, Fierro JLG (2001) Chemical structures and perfor-
mance of perovskite oxides. Chem Rev 101:1981–2018
Pengpanich S, Meeyoo V, Risksomboon T, Bunyakiat K (2002)
Catalytic oxidation of methane over CeO2–ZrO2 mixed
oxide catalysts prepared via sol–gel technique: CO oxida-
tion. Appl Catal A 234:221–233
2323 Page 20 of 21 J Nanopart Res (2014) 16:2323
123

Ribeiro FH, Chow M, Dallabetta RA (1994) Kinetics of the
complete oxidation of methane over supported palladium.
J Catal 146:537–544
Saracco G, Scibilia G, Iannibello A, Baldi G (1996) Methane
combustion on Mg-doped LaCrO3 perovskite catalysts.
Appl Catal B 8:229–244
Sekizawa K, Eguchi K, Widjaja H, Machida M, Arai H (1996)
Property of Pd-supported catalysts for catalytic combus-
tion. Catal Today 28(3):245–250
Shinde VR, Mahadik SB, Gujar TP, Lokhande CD (2006)
Supercapacitive cobalt oxide (Co3O4) thin films by spray
pyrolysis. Appl Surf Sci 252:7487–7492
ShuP, RuanJ, GaoC, Li H,CheS (2009) Formationof mesoporous
Co3O4 replicas of different mesostructures with different pore
sizes. Microporous Mesoporous Mater 123:314–323
Taghavimoghaddam J, Knowles GP, Chaffee AL (2012) Prep-
aration and characterization of mesoporous silica sup-
ported cobalt oxide as a catalyst for the oxidation of
cyclohexanol. J Mol Catal A 358:79–88
Tanev PT, Pinnavaia TJ (1995) A neutral templating route to
mesoporous molecular sieves. Science 267:865–867
Tanev PT, Pinnavaia TJ (1996) Mesoporous silica molecular
sieves prepared by ionic and neutral surfactant templating:
a comparison of physical properties. Chem Mater
8:2068–2079
Wang G, Shen X, Horvat J, Wang B, Liu H, Wexler D, Yao J
(2009) Hydrothermal synthesis and optical, magnetic, and
supercapacitance properties of nanoporous cobalt oxide
nanorods. J Phys Chem C 113:4357–4361
Waters RD, Weimer JJ, Smith JE (1995) An investigation of the
activity of coprecipitated gold catalysts for methane oxi-
dation. Catal Lett 30:181–188
Yang S, Maroto-Valiente A, Benito-Gonzales M, Rodriguez-
Ramos I, Guerrero-Ruiz A (2000) Methane combustion
over supported palladium catalysts: I. Reactivity and active
phases. Appl Catal B 28:223–233
Yisup N, Cao Y, Feng W-L, Dai W-L, Fan K-N (2005) Catalytic
oxidation of methane over novel Ce–Ni–O mixed oxide
catalysts prepared by oxalate gel-coprecipitation. Catal
Lett 99(3–4):207–213
Zamar F, Trovarelli A, Leitenbury CD, Dolcetti G (1995) CeO2-
based solid solution with the fluorite structure as novel and
effective catalysts for methane combustion. Chem Com-
mun 9:965–966
Zavyalova U, Scholz P, Ondruschka B (2007) Influence of
cobalt precursor and fuels on the performance of com-
bustion synthesized Co3O4/c-Al2O3 catalysts for total
oxidation of methane. Appl Catal A 323:226–233
Zhang W, Pauly TR, Pinnavaia TJ (1997) Tailoring the frame-
work and textural mesopores of hms molecular sieves
through an electrically neutral (S�I�) assembly pathway.
Chem Mater 9:2491–2498
Zhao D, Huo Q, Feng J, Chmelka BF, Stucky GD (1998)
Nonionic triblock and star diblock copolymer and oligo-
meric surfactant synthesis of highly ordered, hydrother-
mally stable, mesoporous silica structures. J Am Chem Soc
120:6024–6036
Zhao D, Sun J, Li Q, Stucky GD (2000) Morphological control
of highly ordered mesoporous silica SBA-15. Chem Mater
12(2):275–279
Zhu J, van Ommen JG, Bouwmeester HJM, Lefferts L (2005)
Activation of O2 and CH4 on yttrium-stabilized zirconia for
the partial oxidation of methane to synthesis gas. J Catal
233:434–441
J Nanopart Res (2014) 16:2323 Page 21 of 21 2323
123





























