Embed Size (px)
Citation preview

Synthesis of Aromatic Heterocycles by
Palladium(0)-Catalyzed Domino Reactions
and Cyclization of Hydrazone Dianions
Dissertation
zur
Erlangung des Doktorgrades
doctor rerum naturalium (Dr. rer. nat.)
der Mathematisch-Naturwissenschaftlichen Fakultät
der Universität Rostock
vorgelegt von
M.Sc. Thang Ngoc Ngo
geboren am 16 Oktober 1987 in Phu Tho, Vietnam
Rostock, 2016

Die vorliegende Arbeit wurde im Institut für Chemie von April 2012 bis Dezember 2016
angefertigt.
Erster Gutachter Prof. Dr. Peter Langer
Institut für Chemie
Universität Rostock
Zweiter Gutachter Prof. Dr. Bernd Schmidt
Institut für Chemie
Universität Potsdam
Eingereicht am: 10. Oktober 2016
Tag der Verteidigung: 20. Dezember 2016

I
Acknowledgements
First and foremost, I would like to express my sincerest appreciation to Prof. Dr. Dr. h.c.
mult. Peter Langer for giving me the opportunity to be a part of his research group, for giving
me enthusiasm in chemistry, and for supervising me through difficulties during my PhD.
Without his guidance, support, and encouragement, I would never be able to achieve the work
in this dissertation.
I am deeply thankful to Dr. Dang Thanh Tuan and Dr. Tran Quang Hung for providing me
helpful suggestions and advice throughout my PhD. And I wish to express my special gratitude
to Dr. Peter Ehlers for his valuable support in the development of my work and his meticulous
correction of my dissertation.
I would like to express my acknowledgements to Professor Stefan Lochbrunner for his
support in photophysical characterization. And many thanks go to Mr. Wolfgang Breitsprecher
for his help with the absorption and fluorescence measurements. I wish to thank Dr. Jamshed
Iqbal for the biological studies, and his students, Sundas Sarwar, Syeda Abida Ejaz, Syeda
Mahwish Bakht, for conducting the biotests, Syed Jawad Ali Shah for molecular docking study.
My special thanks go to Dr. Dirk Michalik for NMR measurements and valuable
discussion, Dr. Alexander Vilinger for X-Ray measurements, Dr. Holger Feist and Dr. Martin
Hein for their tremendous support and advice, as well as Anna Hallman, Claudia Hahn, Carmen
Esser, and all other members of analytical and technical staffs of the Department of Chemistry,
University of Rostock and LIKAT (Leibniz-Institut für Katalyse).
I am honored to work with Dr. Omer Akrawi, Do Huy Hoang, Pham Ngo Nghia, Elina
Ausekle, and all other members of Professor Peter Langer’s research group in a friendly and
collaborative environment.
Many thanks go to Frank Janert and Stephan Wöhlbrandt for their diligent work during
their bachelor theses.
The financial support by the State of Mecklenburg-Vorpommern is gratefully
acknowledged.
Additionally, I wish to thank Prof. Dr. Dang Nhu Tai, Dr. Nguyen Quyet Chien, Dr. Tran
Thu Thuy, Dr. Pham Duy Nam, and Dr. Vuong Van Truong for their guidance in both chemistry
and life.

II
Finally, I would like to express the deepest thank for the love and encouragement of my
parents and my brother, especially my father, thank you for everything. And I would like to
thank my beloved wife for always being patient, and on my side through every step of life.

III
Declaration
Hereby I declare that this thesis has been written without any assistance from third parties.
Furthermore, I confirm that no sources have been used in the preparation of this thesis other
than those indicated in the thesis itself.
Erklärung
Hiermit erkläre ich, dass ich die vorliegende Arbeit selbstständig angefertigt und ohne
fremde Hilfe verfasst habe, keine außer den von mir angegebenen Hilfsmitteln und Quellen
dazu verwendet habe und die den benutzten Werken inhaltlich und wörtlich entnommenen
Stellen als solche kenntlich gemacht habe.
Rostock, September 2016
Thang N. Ngo

IV
Abstract
This dissertation focuses on the development of new and convenient synthetic approaches
for important or new aromatic heterocycles, which are potential for drug discovery and
advanced materials. Based on selective Pd(0)-catalyzed reactions of easily accessible starting
materials, the precursors with desired active centers were set up for further domino reactions
towards target molecules. This strategy was successfully applied in the development of new
synthetic methods for chromeno[3,4-b]pyrrol-4(3H)-ones, indolo[1,2-f]phenanthridines,
azaindolo[1,2-f]phenanthridines, and naphtho-fused heterocycles. Furthermore, a convenient
synthesis of fluorinated pyrazoles based on one-pot domino reaction of dianinons was
developed.
Zusammenfassung
Diese Dissertation beschäftigt sich mit der Entwicklung neuer und zugleich einfacher
synthetischer Zugänge von wichtigen bzw. neuen aromatischen Heterozyklen, welche von
Interesse für die Entwicklung neuer Wirkstoffe oder Materialien sind. Mittels selektiver Pd(0)
katalysierter Reaktionen von einfach zugänglichen Startmaterialien wurden Vorstufen
synthetisiert, welche im Anschluss durch Domino-Reaktionen zu den entsprechenden
Zielprodukten umgesetzt wurden. Diese Strategie wurde erfolgreich angewendet zur
Darstellung von Chromeno[3,4-b]pyrrol-4(3H)-onen, Indolo[1,2-f]phenanthridinen,
Azaindolo[1,2-f]phenanthridinen und Naphthalin-annellierten Heterozyklen. Außerdem wurde
ein praktischer Zugang zu fluorierten Pyrazolen, basierend auf einer Ein-Topf-Domino-
Reaktion entwickelt.

Table of Contents
Acknowledgements .............................................................................................................................. I
Declaration ......................................................................................................................................... III
Abstract .............................................................................................................................................. IV
List of abbreviations ..............................................................................................................................
1. General introduction ......................................................................................................................1
1.1. The importance of heterocycles ............................................................................................. 1
1.2. Palladium(0) catalytic cycle ................................................................................................... 4
1.2.1. Oxidative addition and reductive elimination .................................................................... 5
1.2.2. Reactions of Pd(II) complexes ........................................................................................... 7
1.2.3. Effects of ligands: Phosphine ligands .............................................................................. 16
1.3. Palladium(0)-catalyzed domino reactions in constructing important heterocycles.............. 18
1.4. The aim of dissertation ......................................................................................................... 21
2. Synthesis of pyrrolocoumarins via palladium(0)-catalyzed domino C-N
coupling/hydroamination reactions ....................................................................................................22
2.1. Introduction .......................................................................................................................... 22
2.2. Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones ................................................................. 24
2.3. Bioactivity and docking study.............................................................................................. 31
2.4. Conclusion ........................................................................................................................... 36
3. Palladium(0)-catalyzed domino C-N coupling/hydroamination/ C-H arylation reactions:
Synthesis of indolo[1,2-f]phenanthridines, azaindolo[1,2-f]phenanthridines .................................37
3.1. Introduction .......................................................................................................................... 37
3.2. Synthesis of indolo[1,2-f]phenanthridines ........................................................................... 39
3.3. Synthesis of azaindolo[1,2-f]phenanthridines ...................................................................... 48
3.4. Absorption and fluorescence properties of azaindolo[1,2-f]phenanthridines ...................... 59
3.5. Unsuccessful results ............................................................................................................. 60
3.6. Conclusion ........................................................................................................................... 63
4. Regioselective synthesis of naphtho-fused heterocycles via palladium(0)-catalyzed tandem
reaction of N-tosylhydrazones .............................................................................................................64
4.1. Introduction .......................................................................................................................... 64
4.2. Synthesis of naphtho-fused heterocycles ............................................................................. 66
4.3. Conclusion ........................................................................................................................... 76
5. Efficient one-pot synthesis of 5-perfluoroalkylpyrazoles by cyclization of hydrazone
dianions .................................................................................................................................................77
5.1. Introduction .......................................................................................................................... 77
5.2. Synthesis of perfluoroalkylated pyrazoles ........................................................................... 80

5.3. Alkaline phosphatase and nucleotide pyrophosphatase activity and SAR ........................... 86
5.4. Conclusion ........................................................................................................................... 88
APPENDIX ............................................................................................................................................. i
Methods for compound characterization and analysis ........................................................................ i
Biological protocols ..............................................................................................................................ii
Molecular docking ............................................................................................................................... vi
General procedure for the synthesis of pyrrolocoumarins ................................................................. vi
General procedure for synthesis of Indolo[1,2-f]phenanthridines ................................................... xvi
General procedure for synthesis of azaindolo[1,2-f]phenanthridines ........................................... xxvii
General procedure for domino reaction of dibromide compounds and N-tosylhydrazones .......xxxvii
General procedure for the synthesis of pyrazoles ......................................................................... xlviii
Crystal data and structure refinement ............................................................................................. lxix
List of tables .................................................................................................................................... lxxv
List of figures .................................................................................................................................. lxxvi
List of schemes .............................................................................................................................. lxxvii
List of References ........................................................................................................................... lxxix

List of abbreviations
°C Degrees Celsius
13C Carbon 13
19F Fluorine 19
1H Hydrogen, proton
Å Angstrom, 10-8 m
Ac Acetyl
AcO Acetate
Ar Aryl
BINAP 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
Bn Benzyl
Boc N-tert-butoxycarbonyl
Calcd Calculated
CataCXium A Di(1-adamantyl)-n-butylphosphine
CI Chemical Ionization
cm-1 Wavenumber
Cy Cyclohexane
DavePhos 2-Dicyclohexylphosphino-2′-(N,N-dimethylamino)biphenyl
dba Dibenzylideneacetone
DMA Dimethylacetamide
DMF N,N-Dimethylformamide
DPEPhos (Oxydi-2,1-phenylene)bis(diphenylphosphine)
Dppe 1,2-Bis(diphenylphosphino)ethane
Dppf 1,1'- Bis(diphenylphosphanyl)ferrocene
EI Electron Ionization
EI-MS Electron Ionization - Mass Spectrometry
Equiv. Equivalent
ESI Electrospray Ionization
Et3N Triethylamine
GC Gas Chromatography
h Hour
HMBC Heteronuclear Multiple-bond Correlation Spectroscopy
HOMO Highest Occupied Molecular Orbital

HSQC Heteronuclear Single Quantum Coherence Spectroscopy
Hz Hertz (S-1)
IR Infrared Spectroscopy
J Coupling Constant
L Ligand
LCD Liquid Crystal Display
LUMO Lowest Unoccupied Molecular Orbital
m/z Mass-to-charge Ratio
MeCN Acetonitrile
mp Melting Point
MS Mass Spectrometry
NMR Nuclear Magnetic Resonance
NOESY Nuclear Overhauser Effect Spectroscopy
Nu Nucleophile
ORTEP Oak Ridge Thermal Ellipsoid Plot
OTf Triflate (Trifluoromethanesulfonate)
Ph Phenyl
PPh3 Triphenylphosphine
ppm Parts per Million
rt Room temperature
RuPhos 2-Dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl
SPhos 2-Dicyclohexylphosphino-2',6'-dimethoxybiphenyl
Tf2O Trifluoromethanesulfonic anhydride
TFA Trifluoroacetic Acid
THF Tetrahydrofuran
TLC Thin Layer Chromatography
TMS Trimethylsilane
UV/Vis Ultraviolet and visible absorption spectroscopy
XantPhos 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene
XPhos 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
λ Wavelength
ϕ Fluorescence quantum yield

General Introduction
1
1. General introduction
1.1. The importance of heterocycles
Heterocyclic molecules could be figuratively compared to jewelry rings ornamented with
gemstones.[1] By introducing one or more heteroatoms to an aromatic carbocyclic system, both
chemical and physical properties change significantly, making the ring system more “precious”.
For example, pyridine, formed by replacing one CH unit in benzene by one nitrogen, could act
as a base or nucleophile while the α-hydrogens could undergo substitution by strong base such
as sodium amide. Another example is pyrrole, an electron-rich five-membered ring heterocycle,
formed by replacing two CH units in benzene by one NH unit, which is prone to electrophilic
reactions, or tend to be oxidized more easily. In addition, heteroatom that possesses one or more
unshared electron pairs, as of pyridine, imidazole, or pyrazole, can take part in weak interactions
such as hydrogen bonds or form complexes with metal ions, playing crucial roles in human
biological systems. Although activities of aromatic systems are improved by incorporation of
heteroatoms, the thermal stability is still retained in molecules of heteroaromatic systems.[1][2]
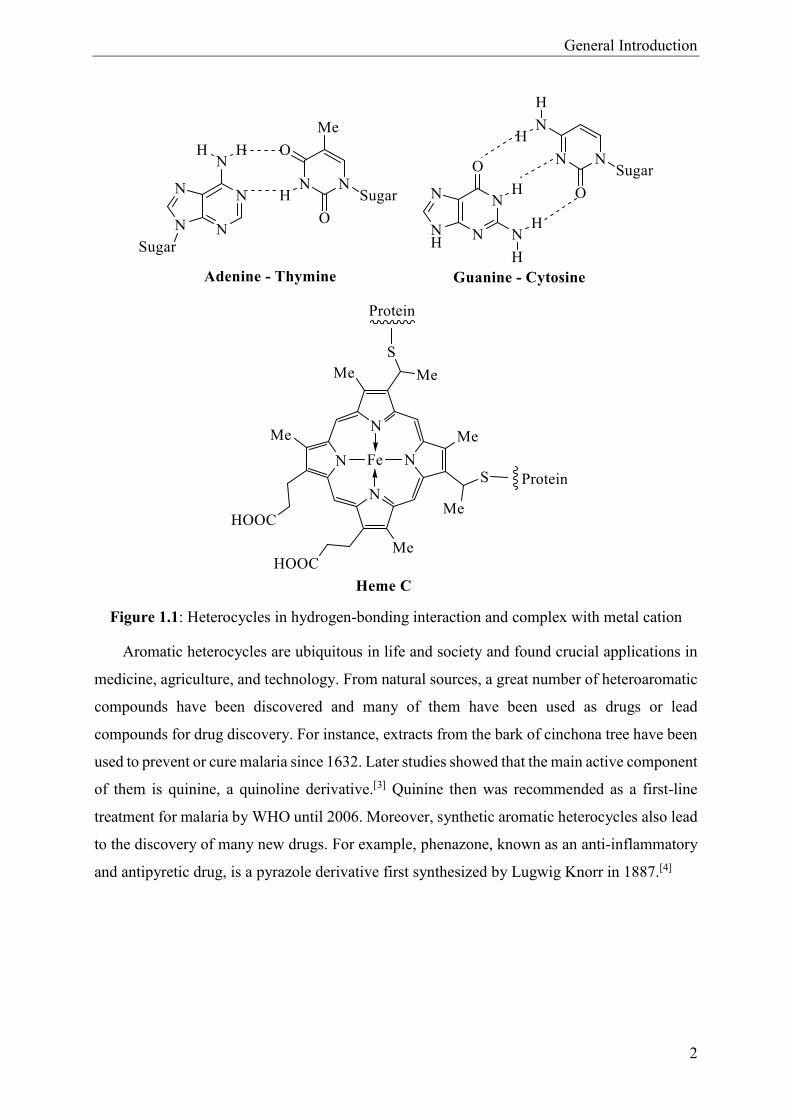
General Introduction
2
Figure 1.1: Heterocycles in hydrogen-bonding interaction and complex with metal cation
Aromatic heterocycles are ubiquitous in life and society and found crucial applications in
medicine, agriculture, and technology. From natural sources, a great number of heteroaromatic
compounds have been discovered and many of them have been used as drugs or lead
compounds for drug discovery. For instance, extracts from the bark of cinchona tree have been
used to prevent or cure malaria since 1632. Later studies showed that the main active component
of them is quinine, a quinoline derivative.[3] Quinine then was recommended as a first-line
treatment for malaria by WHO until 2006. Moreover, synthetic aromatic heterocycles also lead
to the discovery of many new drugs. For example, phenazone, known as an anti-inflammatory
and antipyretic drug, is a pyrazole derivative first synthesized by Lugwig Knorr in 1887.[4]

General Introduction
3
Figure 1.2: Aromatic heterocycles as drugs
Furthermore, synthetic aromatic polyheterocycles are essential components in developing
advanced materials. They are applied in numerous types of light-emitting diodes (OLEDs),
organic photovoltaic devices (OPVs), and organic field effect transistors (OFETs).[5]
Heteroatoms help improving stability, charge mobility, and molecular packing of aromatic
heterocycles over corresponding polycyclic aromatic hydrocarbons (PAHs).[6] For example,
pentacene, a linear PAH consisting of five fused benzene rings, known as an organic
semiconductor with high charge mobility, is vulnerable to oxidation, which slowly degrades
under air and light exposure; therefore, its applications is practically limited.[7] By introducing
heteroatoms to pentacene systems, the stability of obtained derivatives is improved as well as
other properties such as charge mobility, solubility, or molecular packing. Numerous pentacene
derivatives have been discovered by this strategy which possess potential electronic,
photophysical, and optical properties.[8] Moreover, heteroaromatic compounds can form
organometallic chelates with remarkable charge transport and luminescent properties. Among
them, Alq3 (tris(8-hydroxyquinolinato)aluminium), a common component of small molecule
OLEDs, has been used as the emission and electron transport layers.[9] And Ir(ppy)3
(Tris[2-phenylpyridinato-C2,N)iridium(III)), a green emitting complex, is used as the dopant in
phosphorescent organic light-emitting diode (PHOLED).[10]

General Introduction
4
Figure 1.3: Aromatic heterocycles applied in organic materials
Due to their gravity in modern life, extensive effort has been devoted to the development
of synthetic methods and characteristic studies of aromatic heterocycles. Recently, synthetic
methods of heterocyclic and polyheterocyclic compounds have been blooming in the light of
transition metal-catalyzed reactions.[11] Among them, palladium(0) – catalyzed reactions have
been well-studied and found important practical applications.[12–14]
1.2. Palladium(0) catalytic cycle
The chemistry of palladium(0)-catalyzed reaction was incubated in the late 1960s and has
been growing rapidly since then. Because of its practical importance to human life, in 2010, the
Nobel Prize in chemistry was dedicated to professors Heck, Negishi, and Suzuki for their
contribution to the development in the field.[15]
In general, a palladium(0)-catalyzed reaction is a catalytic cycle that includes 3 basic stages:
oxidative addition, reactions of Pd(II) complexes with appropriate nucleophiles, and reductive
elimination. Firstly, the catalytic cycle starts with a Pd(0) species that is oxidized by substrates
to generate a Pd(II) complex, which is called oxidative addition (OA). The new generated Pd(II)
species now behaves as an electrophile. Depending on the nature of nucleophiles, different
processes can take place, such as ligand substitution, transmetalation, or migratory insertion.
Finally, expected products are formed by reductive elimination (RE) of Pd(II) complexes, along

General Introduction
5
with the regeneration of [Pd(0)] catalyst to start a new catalytic cycle. In the case of migratory
insertion, the product and a hydridopalladium(II) halide complex are obtained after β-hydride
elimination, then the hydridopalladium(II) halide complex undergoes reductive elimination to
recreate the [Pd(0)] catalyst.[13,16]
Figure 1.4: A general Pd(0)-catalytic cycle
1.2.1. Oxidative addition and reductive elimination
Oxidative addition (OA) and reductive elimination (RE), which are microscopic reverse,
are involved in all Pd(0)-catalytic cycles.
Scheme 1.1: Oxidative addition and reductive elimination
The process above describes the oxidative addition of R-X to a Pd(0) center. After OA, both
coordination number and oxidative state of metal increase by 2 units. According to the
18-electron rule, Pd(0) with d10 configuration requires 8 more electrons to reach the
configuration of Xe, thus the coordination number of Pd(0) is four. For the oxidative addition
to occur, the metal center must have a lone pair of electrons and a vacant site, that means
16-electron or lower complex is required. For instance, a typical Pd(0) catalyst is Pd(PPh3)4;
before OA can be performed, at least one ligand (PPh3) must leave to activate the catalyst. As
studies have shown, a small amount of Pd(PPh3)2 was found in equilibrium with Pd(PPh3)3. The
two-coordinated species is more reactive than the three-coordinated complex and plays the main

General Introduction
6
role in oxidative addition. The highly active 14-electron Pd(0) species then performs OA with
the substrate, Pd(0) is oxidized to Pd(II) and the coordination number increases by two units,
resulting in a 16-electron square-planar Pd(II) complex, then isomerizes to the more stable trans
complex. Oxidative addition can undergo through 4 types of mechanisms: concerted,
substitution SN2, radical, or ionic mechanisms. In the case of aryl or vinyl halide (pseudohalide),
the mechanism oxidative addition to Pd(0) is widely accepted as concerted pathway for non-
polar substrates or substitution via SNAr for polar substrates.[16,17]
Scheme 1.2: Mechanisms of oxidative addition
Noteworthy, when the substrates contain two or more reactive centers for OA, controlling
the selectivity of OA is very important to obtained desired products. Generally, the rate of OA
for different halogens or pseudohalogens follows this trend: CAr-I > CAr-OTf > CAr- Br >>
CAr-Cl >>> CAr-F. CAr-F bonds, on the other hand, is relatively inert for OA. With a same
halogen, the electron density and steric effect of substituents around C-X decide the selectivity
of the reaction. OA favors less sterically hindered and more electron-positive carbon. For
example, by utilizing chemo-selectivity or site-selectivity on 2,3-dihalogenopyridine, different
products of cross-coupling reactions can be prepared depending on the synthetic strategy. Since
CAr-Br is more reactive than CAr-Cl, the first OA takes place at the 3rd position of
3-bromo-2-chloropyridine. However, with 2,3-dibromopyridine, the first OA takes place at the
2nd position which is more electron-positive.[18] Another demonstration is
5,7-dibromo-8-(trifluoromethylsulfonyloxy)quinolone. In this molecule, there are three
reactive centers for cross-coupling reactions, for example, Suzuki-Miyaura reaction. Evaluating
these three reactive centers, Br5 and Br7 are more reactive than OTf and Br5 is less sterically
hindered than Br7. Therefore, the reactivity order for OA is: C-Br5, C-Br7, and finally C-OTf.
[19] (figure 1.5)

General Introduction
7
Figure 1.5: Selectivity of oxidative addition on substrates with many reactive centers
Reductive elimination is the reverse of oxidative addition to regenerate Pd(0) catalyst from
Pd(II) complex, and in most cases, forms new bond in the product structure. Since RE is the
microscopic reverse of OA, RE can run through those mechanisms as OA, however, the most
important one is concerted pathway. Moreover, cis-coordination of the groups being eliminated
is required for RE. Therefore, in order for RE to perform, the trans Pd(II) complex must
isomerize to cis configuration. If a β-H is present in the molecular, β-hydride elimination may
compete with reductive elimination.
Reductive eliminations which involve H, such as H-H, H-R, H-COR, are particularly fast.
Reductive eliminations involving C(sp2)-C(sp2) bond formation usually take part in
cross-coupling reactions such as Suzuki-Miyaura, Negishi, Stills, Kumada, Hiyama couplings.
Sonogashira coupling, on the other hand, produces C(sp)-C(sp2) bond. Representative examples
for reductive elimination involving C(sp2)-N or C(sp2)-O bond formation are those of
Buchwald-Hartwig reaction.[20]
1.2.2. Reactions of Pd(II) complexes
As mentioned earlier, depending on the nature of nucleophiles interacting with the Pd(II)
complex, the catalytic cycle can run in different directions. Most important are ligand
substitution, demonstrated in Buchwald-Hartwig reaction, transmetalation in cross-coupling
reactions involving an organometallic reagent as the nucleophile, and migratory insertion in
Heck-type reactions or carbene insertion. Those transformations and their demonstration in
reactions utilized in this dissertation will be the main concern of this section.

General Introduction
8
Ligand substitution: Buchwald-Hartwig reaction
Ligand exchange is an important process for cross-coupling reactions involving
C-heteroatom bond formations such as C-N, C-S, C-O, C-P bonds.[21] One of the most important
reactions is Buchwald-Hartwig amination, in which amines are employed as nucleophiles.[22]
Scheme 1.3: Buchwald-Hartwig reaction
As described previously, the four-coordinated Pd(II) complex formed by OA is a 16-electron
square-planar complex. Ligand substitution of Pd(II) complexes can occur via two mechanisms:
associative pathway or dissociative pathway. In associative pathway, nucleophile forms a
penta-coordinated square-pyramidal Pd(II) complex by σ-bonding with empty pz orbital of Pd.
The 18-electron square-pyramidal complex then rearranges the configuration in which the
leaving ligand (usually X) is being on the top of the pyramid via a trigonal bipyramid
intermediate. Finally, the leaving ligand disassociates to form a new square-planar Pd(II)
complex. This process can be considered as an analog of SN2 reaction, in which Pd(II) center
is a soft electrophile. However, when the attack of nucleophile is sterically hindered or the
formation of penta-coordinated square-pyramidal complex is not favored by energy, ligand
exchange prefers via dissociative pathway. In this mechanism, the Pd-X bond is fully broken
to form a T-shaped intermediate, then the nucleophile attacks to the empty site left by X to form
the new square-planar complex. The dissociative pathway is similar to SN1 reaction. There is
no change in oxidation state at the Pd center through ligand substitution.

General Introduction
9
Scheme 1.4: Mechanisms of ligand exchange
Transmetalation: Suzuki and Sonogashira reaction
Scheme 1.5: Transmetalation
Transmetalation is an irreversible process involving the transfer of ligands from one metal
to another. Similar to ligand substitution, there is no change of oxidation state of the metal
centers. In this process, Pd(II) is an electrophilic center while R2-M bond is a nucleophile.
Therefore, increasing the nucleophilicity of R2 and electrophilicity of Pd(II) center accelerate
transmetalation. For transmetalation to proceed, Pd must be more electronegative than M. For
example, Pd(II) complexes participate in transmetalation with organometallic compounds of
metals such as Cu, B, Si, Zn, Al, Sn. Cross-coupling reactions involving transmetalation of
different organometallic reagents with Pd(II) complexes are summarized in figure 1.6.[23]

General Introduction
10
Figure 1.6: Cross-coupling reaction involving transmetalation
Among them, Suzuki-Miyaura cross-coupling reaction is one of the most popular methods
for constructing C(sp2)-C(sp2) bonds.[24] This method employs organoborons as the coupling
partner, which are easily accessible, air and moisture stable, less toxic, and safer for
environment than other organometallic compounds such as organostannane or organozinc.
Since the C-B bond is considered to be highly covalent, R2BX2 is a weak nucleophilic reagent.
Hence, to promote transmetalation, adding a nucleophile or base is often required to increase
the nucleophilicity of B-R2. In fact, many studies show that, without the presence of base,
organoboron compounds do not undergo transmetalation. A nucleophilic base can participate
in the reaction by two ways described in scheme 1.6. Following path A, organoboron compound
is activated by adding one base to the boron center, which is more readily to undergo
transmetalation. In path B, the nucleophilic base replaces X of Pd(II) complex and transforms
it to a new Pd(II) complex that is capable of coordinating to the boron center of organoborane.
The product of transmetalation then arranges to cis configuration for reductive elimination,
affording the product and regenerating Pd(0) catalyst.
Scheme 1.6: Roles of base in Suzuki-Miyaura reaction
While Suzuki-Miyaura reaction is widely used for constructing C(sp2)-C(sp2) bonds,
Sonogashira reaction is the most convenient method for forming C(sp2)-C(sp) bonds.[14,25] The
reaction utilizes organocopper compounds as the coupling partner, generated in situ from

General Introduction
11
terminal alkyne and CuX in the presence of an amine base. In this reaction, CuX is used as
catalytic amount, integrating with Pd cycle as shown in scheme 1.7.
Scheme 1.7: Mechanism of Sonogashira reaction
Insertion and elimination: Heck-type reactions and cross-coupling reactions of
N-tosylhydrazone
In organometallic chemistry, migratory insertion is an inserting process of a ligand to metal
complexes which results in bond formation of it with another ligand on the metal complex. By
that definition, migratory insertion of a ligand to Pd(II) complex can take place commonly in
two main ways: 1,1- and 1,2-migratory insertion. As depicted in scheme 1.8, 1,1-migratory
insertion results in bond formation of R with ligand X-Y at the same position with Pd while
1,2- migratory insertion gives bond at the neighboring atom.
Scheme 1.8: Migratory insertions

General Introduction
12
Ligands that have both donor and acceptor centers at the same atom usually undergo
1,1-migratory insertion. For example, CO gives 1,1-migratory insertion, in which both Pd and
R1 end up attached to carbon of CO.[26]
Scheme 1.9: Migratory insertion of CO
Scheme 1.10: Migratory insertion of carbene
Another important example is the migratory insertion of carbenes R2C, which possess a lone
pair electron that can act as a σ-donor and an empty orbital that can act as an acceptor at carbon
atom. Recently, the coupling reactions of aryl halide and diazo compound to synthesize
substituted olefins, in which carbene is generated in situ, have been attracting a lot of
attention.[27–29] Among them, cross-coupling reactions utilizing N-tosylhydrazone as the
coupling partner have been prove to be efficient to synthesized substituted olefins.[30,31] In this
reaction, under the high temperature and in the presence of a base, such as LiOtBu, a carbene
is generated in situ from N-tosylhydrazones.
Scheme 1.11: Cross-coupling reaction of N-tosylhydrazones involving carbene
On the other hand, 1,2-migratory insertion takes place when ƞ2-ligands, such as alkenes,
react with Pd(II) complexes. A typical example of 1,2-migratory insertion is the insertion of
double bond to Pd(II) complexes, an important step in Heck-Mizoroki cross-coupling
reaction.[32]

General Introduction
13
Scheme 1.12: Heck-Mizoroki reaction
Migratory insertion in Heck-Mizoroki reactions can undergo via three mechanisms: cationic,
neutral, or anionic pathways. Cationic mechanism takes place when Heck reactions of aryl
triflates or aryl halides are catalyzed by palladium-diphosphine in the presence of Ag(I) or Tl(I)
salts. Dissociation of OTf or X anion leaves Pd(II) center a positive charge and a vacant site,
that is attached by the double bond of an alkene. Then migratory insertion of double bond to
PdAr proceeds, forming new Pd(II) complex. During migratory insertion, both Pd-P bonds stay
intact so the enantioselectivity of the product can be achieved by utilizing chiral diphosphine
ligands.
Scheme 1.13: Cationic mechanism of 1,2-migratory insertion
Regularly, without the presence of Ag(I) or Tl(I) salt, migratory insertion of alkenes is
considered undergoing neutral pathway. This mechanism involves the dissociation of one
neutral ligand (phosphine ligand) to create a vacant site for the coordination of double bond
with Pd(II) center, leading to 1,2-migratory insertion of C=C to Ar-Pd.
Scheme 1.14: Neutral mechanism of 1,2-migratory insertion
Recent studies show that the combination of phosphine ligands and Pd(OAc)2 as precatalyst
may generate an anionic species [Pd(L)2OAc]- for oxidative addition of ArX, in which acetate
anion act as a bystander ligand. The product of oxidative addition is believed existing as a
penta-coordinated Pd(II) complex, which is short-live and easy to dissociate X- anion to form
neutral trans-ArPd(OAc)L2 as the key reactive intermediate. The reaction of this Pd(II) species

General Introduction
14
is the rate-determining step of the Heck reactions underwent anionic pathway. Acetate ligand
facilitates the dissociation of one phosphine ligand because of its bidentate nature.
Scheme 1.15: Anionic mechanism of 1,2-migratory insertion
In all three mechanisms, product of insertion is syn-addition of alkene to PdR. Regiochemistry
depends on the mechanism of insertion. Regioselectivity is under the influence of steric factor
when the reaction passes via neutral Pd complexes, which Ar prefer to attach to less hindered
carbon. On the other hand, regioselectivity is governed by electronic factor in mechanisms
involving cationic Pd complexes, which Ar favorably bonds with the less electron-density
carbon.
The reverse process of 1,2-migratory insertion, β-hydride elimination, is a process in which
a metal alkyl is converted to a hydro metal alkene complex. In order for β-hydride elimination
to process, a vacant site on Pd that is cis to the alkyl group is required, and Pd-C-C-H must
arrange on a coplanar conformation to bring β-H atom close enough to Pd to form an agostic
interaction. The Heck reaction is stereoselective for E olefin because the transition state of it is
more favored by energy. In addition, Z-configurated olefin, which is the minor product, can
react with H-Pd to form thermodynamically more stable E-isomer.
Scheme 1.16: β-hydride elimination
β-Hydride elimination produces olefin and hydridopalladium(II) halide complex, which
undergoes reductive elimination to regenerate Pd(0) catalyst.
An analogue of Heck reaction is the direct arylation of aryl halides and arenes without
pre-functionalized, affording biaryl compounds. This reaction has many advantages over
traditional cross-coupling reactions such as utilizing unactivated arenes instead of
pre-functionalized arenes for transmetalation.[33]

General Introduction
15
Scheme 1.17: C-H arylation vs traditional cross-coupling reactions
The reaction of Ar[Pd]X with arenes can be considered as electrophilic aromatic substitution
SEAr, Heck-like, or concerted metalation-deprotonation (CMD). Studies have supported that it
is reasonable to describe direct arylation of electron-rich, π-nucleophilic heteroarenes as SEAr
and that of electron-deficient benzenes as CMD.
Scheme 1.18: Mechanisms C-H arylation
Owning to the fact that there might be more than one C-H site in the structure of arene, more
than one biaryl products could be formed. In general, considering CMD pathway, the more
acidic the C-H bond, the more active it is in C-H activation reaction. On the other hand, the
Heck-like pathway favors the more stable intermediate, in which the positive charge is more
stabilized. Moreover, a strategy to achieve selectivity is to employ directing groups. Normally,
directing groups can coordinate to the Pd center, therefore limit it to a certain geometry, which
can selectively reach to the desired C-H site. For example, ester, amide, carbamate, nitro can
be used as directing groups, in which the ortho C-H to the directing group is usually the reacting
site. After the reaction, directing groups can be removed or transform to other functional
groups.[34]

General Introduction
16
Furthermore, intramolecular direct arylation is often utilized in constructing aromatic
polyheterocyles due to its convenience, which is important in developing synthetic methods in
this dissertation.[35]
Scheme 1.19: Intramolecular direct arylation
1.2.3. Effects of ligands: Phosphine ligands
Ligands are usually used in combination with Pd catalyst to achieve selectivity and
reactivity. In oxidative addition, Pd center of Pd(0) complexes acts as a nucleophile. Strong
σ-donating ligands such as alkyl phosphines and carbenes increase electron density at the Pd(0)
center; hence, facilitate oxidative addition. On the other hand, strong π-acceptor ligands such
as CO, however, slow the process down. Bulky ligands help pushing the equilibrium of Pd(0)
complexes to the two-coordinated 14-electron complex, or in some cases, generating
monoligated 12-electron complexes, which are highly active for OA. Bulky ligands also force
other ligands close together to facilitate reductive elimination. The Pd center of Pd(II)
complexes, on the other hand, is electrophilic and should be sterically accessible for the
incoming nucleophiles. Therefore, controlling the electronic and steric properties of ligands is
crucial for optimizing reaction conditions. In this dissertation, phosphine ligands were mainly
employed because of their availability and many advantages.
Phosphine ligands R3P, known as σ-donating and weak π-accepting ligands (d to σ* of
P-R), are widely used in Pd(0)-catalyzed cross-coupling reaction because they could be
conveniently synthesized in series. Furthermore, their electronic and steric properties could be
modified systematically and predictably by changing R. In addition, phosphines normally exist
in crystal form, are air and thermal stable. Therefore, they are easy to handle and stable under
a wide range of reaction conditions. Notably, in 1998, Fu reported a series of bulky,
electron-rich phosphines, such as P(tBu)3 and PCy3, used in combination with Pd(0)
precatalysts to produce biaryls from unactivated chloroarenes and arylboronic acid in good
yield.[36] At about the same time, Buchwald also reported comparable results with the discovery
of dialkyl biaryl phosphine ligands.[37] Those ligands have been applying widely in
Pd(0)-catalyzed C-C, C-N, C-O bond-forming reactions.[38]

General Introduction
17
Figure 1.7: Phosphine ligands
Moreover, Beller showed that turnover numbers of Suzuki-Miyaura couplings of unactivated
and deactivated chloroarenes could be achieved at 20000 when
di-(1-adamanyl)-n-butylphosphine (cataCXium® A) is used as the ligand.[39] Bulky ligands are
believed to promote the formation of highly active monoligated 12-electron Pd(0) catalyst.[40]
In addition, diphosphines such as Ph2P(CH2)nPPh2, which are bidentate ligands, also play an
important role in Pd-catalyzed reactions. This type of ligands force Pd(II) complexes to the cis
arrangement which accelerate both OA and RE. Bite angle is an important parameter regarding
bidentate ligands and easily modified combining with steric effect of substituents. Many studies
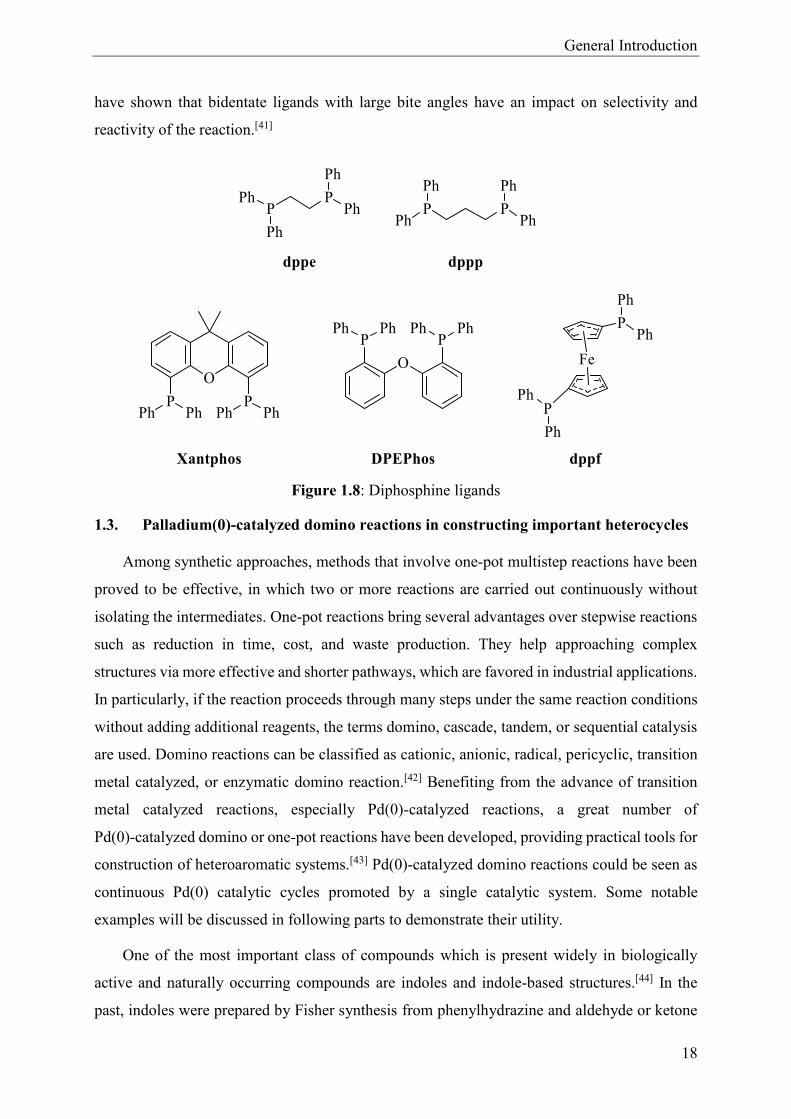
General Introduction
18
have shown that bidentate ligands with large bite angles have an impact on selectivity and
reactivity of the reaction.[41]
Figure 1.8: Diphosphine ligands
1.3. Palladium(0)-catalyzed domino reactions in constructing important heterocycles
Among synthetic approaches, methods that involve one-pot multistep reactions have been
proved to be effective, in which two or more reactions are carried out continuously without
isolating the intermediates. One-pot reactions bring several advantages over stepwise reactions
such as reduction in time, cost, and waste production. They help approaching complex
structures via more effective and shorter pathways, which are favored in industrial applications.
In particularly, if the reaction proceeds through many steps under the same reaction conditions
without adding additional reagents, the terms domino, cascade, tandem, or sequential catalysis
are used. Domino reactions can be classified as cationic, anionic, radical, pericyclic, transition
metal catalyzed, or enzymatic domino reaction.[42] Benefiting from the advance of transition
metal catalyzed reactions, especially Pd(0)-catalyzed reactions, a great number of
Pd(0)-catalyzed domino or one-pot reactions have been developed, providing practical tools for
construction of heteroaromatic systems.[43] Pd(0)-catalyzed domino reactions could be seen as
continuous Pd(0) catalytic cycles promoted by a single catalytic system. Some notable
examples will be discussed in following parts to demonstrate their utility.
One of the most important class of compounds which is present widely in biologically
active and naturally occurring compounds are indoles and indole-based structures.[44] In the
past, indoles were prepared by Fisher synthesis from phenylhydrazine and aldehyde or ketone

General Introduction
19
under acidic conditions. However, phenylhydrazine is very toxic, and the Fisher’s conditions
are somehow harsh. Later a lot of modifications have been studied to improve Fisher
synthesis,[45] such as Buchwald modification, in which involves Pd-catalyzed cross-coupling of
aryl bromide and hydrazones.[46]
Scheme 1.20: Approaches to the synthesis of indoles by Pd(0)-catalyzed domino reactions:
Recently, many convenient methods based on Pd(0)-catalyzed domino reactions have been
developed for the synthesis of indoles.[47] Among them, Larock’s method has proved to be
superior (a, figure 1.9).[48] The domino reaction initiates by oxidative addition of
2-halogenoaniline to form Pd(II) species, followed by regioselective insertion of alkyne to the
Pd(II) center, and subsequent intramolecular palladium displacement by amino group, affording
2,3-disubstituted indoles in good yields. A similar strategy starting
from ortho-alkynyltrifluoroacetanilides and aryl halides was reported by Cacchi (d).[49] The
methodology was well demonstrated in synthesizing the complex structure of
indolo[2,3-a]carbazole in a single step from 1,3-diaceylene precursor.[50]

General Introduction
20
Scheme 1.21: Synthesis of indolo[2,3-a]carbazoles by Cacchi
Starting from ortho-alkynylhalogenoarenes, Ackermann has combined Buchwald-hartwig
amination and intramolecular hydroamination to synthesize 2-substituted indoles with excellent
yields. Moreover, ortho-alkynylhalogenoarenes are easily obtained by Sonogashira reaction of
ortho-dihalogenoarenes and terminal alkynes (c).[51] Later, Ackemann has reported an
improvement that started straightforward from ortho-dihalogenoarenes, terminal alkyenes, and
amines in a one-pot three-component reaction.[52]
Scheme 1.22: Synthesis of indoles by one-pot three-component reaction by Ackemann
In addition, recently methods are also worth mentioning, for example, strategies that employ
aryl and alkenyl C-N bond formation (b)[53] or subsequently C-C and C-N bond formation from
imines and ortho-dihaloarenes (e).[54]
Furthermore, an important class of compounds derived from indole ring is carbazoles,
which are found in many natural alkaloids and some of them possess potential bioactivity.[55]
Carbazoles can be synthesized efficiently by Pd(0)-catalyzed domino reaction, for instance,
two-fold C-N bond formation by Buchwald-Hartwig reaction[56] or C-N and C-C bond
formation by subsequent Buchwald-Hartwig and C-H arylation reactions.[57]
Scheme 1.23: Approaches to the synthesis of carbazoles

General Introduction
21
1.4. The aim of dissertation
Motivated by the importance of aromatic heterocycles, the aim of this dissertation is to
develop new and convenient approaches for important or new aromatic heterocycles, mainly
nitrogen-containing. Compounds targeted in this work are potential for drug discovery and
advanced materials. Regarding biologically active compounds, the structures based on natural
products, which possess highly therapeutic activity, and drugs or novel drug candidates will be
explored. For discovering new organic material, aromatic polyheterocyclic compounds, which
have been proved to be crucial in advanced materials, will be the spotlight.
The synthetic methods developed for desired compounds are aiming to be convenient and
practical. My strategy is based on the selectivity of Pd(0)-catalyzed reactions of easily
accessible starting materials to set up the precursors with desired active centers for further
domino reactions. The precursors then will be converted to target molecules by designed
Pd(0)-catalyzed domino reactions. This strategy will be demonstrated throughout chapter 2 to
chapter 4. Chapter 5 is about domino reactions of dianions for the synthesis of fluorinated
compounds.
In addition, selected synthesized compounds will be submitted to biological studies in
collaboration with the group of Dr. Jamshed Iqbal (Centre for Advanced Drug Research,
COMSATS Institute of Information Technology, Abbottabad, Pakistan). And compounds with
potential application in developing advanced materials will be examined physical properties in
collaboration with the group of Prof. Stefan Lochbrunner (Institut für Physik, Universität
Rostock).

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
22
2. Synthesis of pyrrolocoumarins via palladium(0)-catalyzed
domino C-N coupling/hydroamination reactions
2.1. Introduction
Pyrrolocoumarin is a privileged scaffold formulated from coumarin moiety fused with a
pyrrole unit which widely occurs in biologically active compounds. Notably,
chromeno[3,4-b]pyrrol-4(3H)-one, recognized as the core structure in molecules of marine
alkaloids ningalin B[58] and lamellarin,[59,60] exhibits potent pharmacological properties such as
immunomodulatory activity, cytotoxicity,[60] and HIV-1 integrase inhibition.[61] Some of them
are novel drug candidates or lead compounds for drug discovery. For example, synthetic
modification of lamellarin D leads to a series of Topoisomerase 1 inhibitors.[62]
Figure 2.1: Ningalin B and Lamellarin D derivatives
The diversity of their bioactivities has motivated many researches to develop efficient
synthetic routes for constructing the chromeno[3,4-b]pyrrol-4(3H)-one subunit. Generally,
there are two main strategies for the synthesis of this unique scaffold.[63] Firstly, synthesis of
chromeno[3,4-b]pyrrol-4(3H)-ones can be derived from regioselectively constructing
functionalized pyrrole moiety, following by lactonizing to afford the desired structure. This

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
23
approach is effectively demonstrated in the works of Iwao.[64] The second approach originated
from isoquinolines as starting materials.[65] Recently, synthesis of
chromeno[3,4-b]pyrrol-4(3H)-ones starting from coumarins has attracted a lot of attention. For
example, Langer group has reported a new approach to this scaffold by cyclocondensation
reactions of 1,3-dicarbonyl compounds with 4-chloro-3-nitrocoumarin.[66]
Scheme 2.1: Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones
Moreover, catalytic hydroamination of alkynes has recently become a valuable synthetic
tool in the synthesis of fused nitrogen-containing heterocycles. Several catalytic systems have
been utilized for this type of cyclization such as palladium, gold, mercury, copper, zinc,
rhodium, platinum, indium, and iridium salts.[67] Among them, Pd(II) has been proved its
versatility. For example, Xu and co-workers reported an elegant approach to pyrrolocoumarins
by Pd-catalyzed intramolecular hydroamination of acetylenic aminocoumarins obtained from
4-chloro-3-nitrocoumarin.[68] Interestingly, Ackermann reported that the combination of
Pd(0)-catalyzed Buchwald-Hartwig amination and intramolecular hydroamination of
o-alkynylanilines with amines works harmoniously to form pyrrole-fused systems.[51,69]
Considering this strategy, I believe that it is plausible to obtain the pyrrolocoumarin framework
starting from a ortho-dihalogenated coumarin, following by mono-Sonogashira coupling
reaction to obtain key intermediate o-alkynyl bromocoumarin. The key step then relies on
forming fused-pyrrole ring via palladium catalyzed sequential C-N coupling/intramolecular
hydroamination of o-alkynyl bromocoumarins with amines. After reviewing many reported
synthetic approaches, I found that 3-bromo-4-(trifluoromethanesulfonyloxy)coumarin 2.3

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
24
would be the most suitable educt for synthesizing various analogues of pyrrolocoumarin
(scheme 2.3).
Scheme 2.2: Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones by hydroamination
2.2. Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones
Starting from commercially available 4-hydroxycoumarin 2.1, bromination was carried out
in MeCN at 20 °C by using NBS, affording brominated product 2.2 after 45 minutes with 91%
yield. 3-Bromo-4-hydroxycoumarin 2.2 was then easily transformed into its corresponding
triflate 2.3 by using triflic anhydride in dry DCM. Interestingly, the yield raised up from 55%
to 80% by reducing the temperature to -20 °C compared to 0 °C of the originally published
procedure (Scheme 2.3).[70]
Scheme 2.3: Synthesis of 3-bromo-4-(trifluoromethanesulfonyloxy)coumarin. Conditions: i,
NH4OAc, MeCN, 45 min, 20 °C. ii, 1 (1 equiv.), Tf2O (1.2 equiv.), Et3N (3 equiv.),
CH2Cl2, -20 °C to 20 °C.

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
25
Then, the selective Sonogashira cross-coupling reaction of
3-bromo-4-(trifluoromethanesulfonyloxy)coumarin 2.2 with phenylacetylene was studied as
the model reaction. Since C-OTf is more favored than C-Br in Sonogashira reaction plus the
C4 is more electron-positive than C3, the mono-coupling is expected at C4-OTf. At the first
attempt, the general procedure using Pd(PPh3)2Cl2/CuI in THF at room temperature (20 °C)
was applied, but the result was disappointing with only 7% yield of isolated product yield (entry
1, table 2.1). Only trace of product was detected when Pd(PPh3)4 was used as the catalyst (entry
2 and 3, table 2.1). After studying various conditions, Pd(CH3CN)2Cl2 proved to be the best
catalyst for this reaction so far. The reaction proceeded smoothly in NEt3/DMF (3:2) at room
temperature catalyzed by 5% of Pd(CH3CN)2Cl2 and 10% CuI in 75% yield (entry 7, table 2.1).
DMF must be added to overcome the low solubility of 2.3 in NEt3. Other attempts to raise the
temperature or to change the catalyst resulted in poor yields.
Table 2.1: Optimization for the synthesis of 2.4a
No. Catalyst (5 mol%),
CuI (10%) Base Solvent Time (h) Yield (%)
1 Pd(PPh3)2Cl2 NEt3 THF 4 7
2 Pd(PPh3)3 NEt3 THF 4 trace
3 Pd(PPh3)4 NEt3 CH3CN 4 trace
4 Pd(PPh3)2Cl2 NEt3 CH3CN 4 15
5 Pd(PPh3)2Cl2 NEt3 DMF 4 23
6 Pd(PPh3)2Cl2 DIPEA DMF 4 20
7 Pd(CH3CN)2Cl2 NEt3 DMF 2 75
Conditions: 2.3 (1 equiv.), alkyne (1.2 equiv.), NEt3/DMF (3:2), 20 °C
Applying the optimized conditions, I prepared various alkynylated coumarins 2.4b-g. Both
aromatic and aliphatic alkynes could be employed to achieve desired products (Table 2.2).

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
26
Table 2.2: Synthesis of 2.4
2.4 R1 Yield (%)
b
56
c
22
d
53
e
41
f CH3(CH2)2- 47
g CH3(CH2)3- 45
Conditions: 2.3 (1 equiv.), alkyne (1.2 equiv.), Pd(CH3CN)2Cl2 (5% mol),
CuI (10 % mol), NEt3/DMF (3:2), 20 °C, 2 h.
With the alkynylated coumarins in hand, the reaction of alkynylated coumarins 2.4 with
amines 2.5 affording pyrrolocoumarins 2.6 was investigated. Compound 2.4a and
4-methoxyaniline were firstly used as model substrates for the optimization of the reaction
conditions. During the studies, it became apparent that strong bases, such as NaOtBu or KOtBu,
should be avoided, because of decomposition of the coumarin ring under these conditions.
Therefore, mild inorganic bases, such as Cs2CO3, K2CO3, and K3PO4, were utilized. The
reaction was first studied in toluene as the solvent using Pd(OAc)2 (5%) as the catalytic source
and different phosphine ligands (10%). It is important to note that we could only obtain the
desired product, albeit in only 12% yield when SPhos
(2-Dicyclohexylphosphino-2',6'-dimethoxybiphenyl) was used as the ligand and Cs2CO3 as the
base (entry 4, table 2.3). However, the yield could be improved when DMF or DMA was used
as the solvent. The best result (64% yield) was obtained when the catalyst loading was increased
to 10% and CuI (20%) was used as an additive (entry 14, table 2.3). The role of CuI might be
to promote the hydroamination step. The use of CuI as the catalyst in combination with ligands

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
27
also resulted in the formation of the product as well, however, only in 23% yield (entry 15, table
2.3).
Table 2.3: Optimization of 2.6a
No. Catalyst
(5%) Ligand (10%) Base Solvent
Temp.
(°C)
Time
(h)
Yield
(%)
1 Pd(OAc)2 XantPhosa Cs2CO3 Toluene 110 8 -
2 Pd(OAc)2 (tBu)3P·HBF4 Cs2CO3 Toluene 110 8 -
3 Pd(OAc)2 Cy3P Cs2CO3 Toluene 110 8 -
4 Pd(OAc)2 SPhos Cs2CO3 Toluene 110 8 12
5 Pd(OAc)2 XPhos Cs2CO3 Toluene 110 8 5
6 Pd(OAc)2 SPhos K3PO4 Toluene 110 8 -
7 Pd(OAc)2 SPhos K2CO3 Toluene 110 8 -
8 Pd(OAc)2 XPhos K3PO4 Toluene 110 8 -
9 Pd(OAc)2 XPhos K2CO3 Toluene 110 8 -
10 Pd(OAc)2 SPhos Cs2CO3 DMF 110 2 39
11 Pd(OAc)2 SPhos Cs2CO3 DMA 110 2 35
12 Pd(OAc)2 SPhos Cs2CO3 DMSO 110 8 -
13 Pd(OAc)2c SPhosb Cs2CO3 DMF 80 4 52
14 Pd(OAc)2
c
CuIb SPhosb Cs2CO3 DMF 80 4 64
15 CuIc 1,10-phenantrolineb Cs2CO3 DMF 110 8 23
Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
28
16 CuIc 1H-Benzotriazole-1
-methanolb Cs2CO3 DMF 110 8 15
17 CuIc 1,2-DMEDAb Cs2CO3 DMF 110 8 17
18 CuIc TMEDAb Cs2CO3 DMF 110 8 11
Conditions: 2.4a (0.1 mmol, 1 equiv.), 2.5a (0.12 mmol, 1.2 equiv.), base (0.25 mmol,
2.5 equiv.), solvent 2 mL a5%, b20%, c10%
With the optimized conditions in hand, we extended the scope of the reaction to synthesize
a series of pyrrolocoumarins 2.6b-q using various amines (Table 2.4). The employment of
anilines containing electron donating substituents (more nucleophilic) generally resulted in
better yields than of anilines containing electron withdrawing groups. No product was isolated
from 4-nitroaniline. Aliphatic amines were successfully employed in the reactions, affording
desired products in good yields.
Table 2.4: Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones
Compound R1 R2 Structure Yield
(%)
2.6a
64
2.6b
47

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
29
2.6c
61
2.6d
52
2.6e
41
2.6f
35
2.6g
40
2.6h
82
2.6i
76

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
30
2.6j
61
2.6k
46
2.6l
57
2.6m
47
2.6n
65
2.6o
69
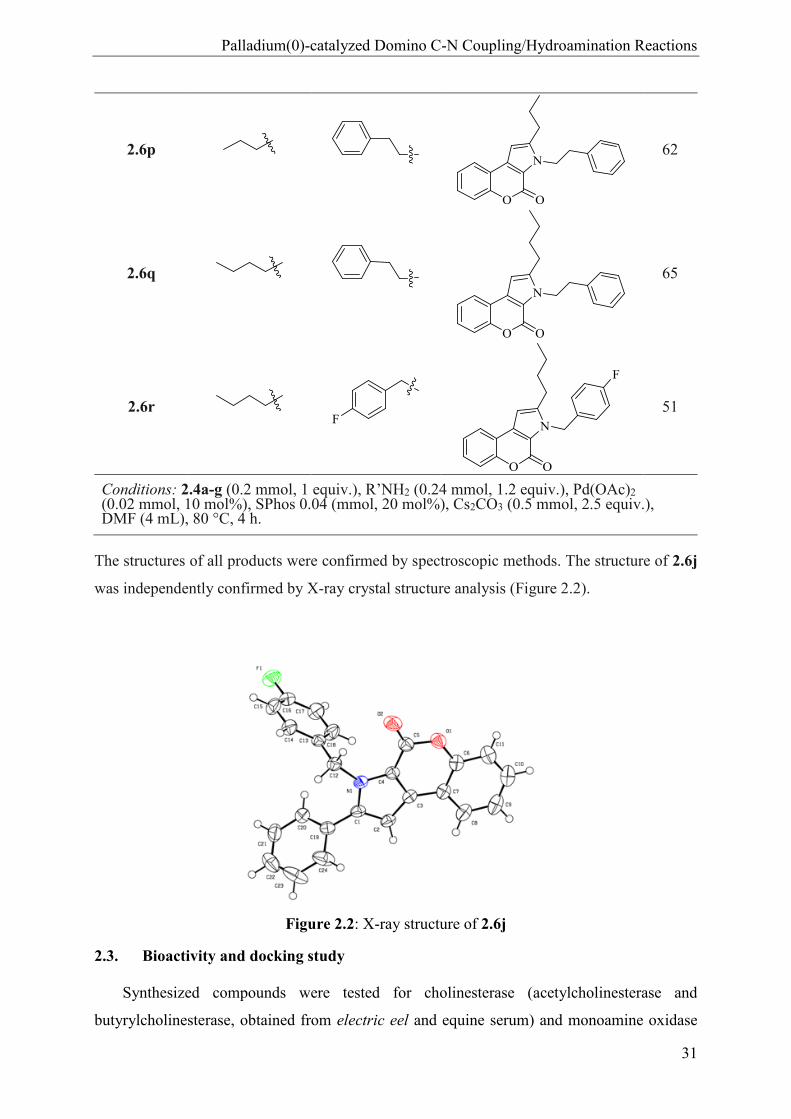
Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
31
2.6p
62
2.6q
65
2.6r
51
Conditions: 2.4a-g (0.2 mmol, 1 equiv.), R’NH2 (0.24 mmol, 1.2 equiv.), Pd(OAc)2 (0.02 mmol, 10 mol%), SPhos 0.04 (mmol, 20 mol%), Cs2CO3 (0.5 mmol, 2.5 equiv.), DMF (4 mL), 80 °C, 4 h.
The structures of all products were confirmed by spectroscopic methods. The structure of 2.6j
was independently confirmed by X-ray crystal structure analysis (Figure 2.2).
Figure 2.2: X-ray structure of 2.6j
2.3. Bioactivity and docking study
Synthesized compounds were tested for cholinesterase (acetylcholinesterase and
butyrylcholinesterase, obtained from electric eel and equine serum) and monoamine oxidase

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
32
(A & B) inhibition. Cholinesterase inhibition is known as one of the most powerful approaches
for the treatment of Alzheimer’s disease.[71] Moreover, monoamine oxidases are considered an
important therapeutic target for neurodegenerative disorders. Among them, selective MAO-A
inhibitors could be used as antidepressants and anxiolytics, and selective MAO-B inhibitors are
used for treatments of Parkinson's disease and Alzheimer's disease.[72] Interestingly, many
coumarin derivatives have been reported as potential inhibitors against both cholinesterases and
monoamine oxidases[73]. Therefore, chromeno[3,4-b]pyrrol-4(3H)-ones could be considered as
a potential approach to cholinesterase and monoamine oxidase inhibitors.
Table 2.5: Anticholinesterase activities of chromeno[3,4-b]pyrrol-4(3H)-ones
AcetylCholinesterase activity ButyrylCholinesterase activity
Compounds IC50±SEM (µM) IC50±SEM (µM)
2.6d 0.47±0.01 57.8±1.23
2.6e 11.8±0.99 82.1±2.45
2.6a 257.6±3.11 127.2±4.21
2.6f 2.87±0.56 159.1±3.11
2.6h 385.1±4.11 51.3±3.11
2.6i 0.47±0.01 208.8±2.56
2.6k 8.01±1.21 39.8±3.11
2.6q 248.7±4.89 14.9±2.16
2.6p 173.5±2.98 9.45±1.56
2.6m 308.7±3.12 34.1±1.78
2.6n 4.04±0.34 57.8±3.12
2.6l 16.8±1.56 18.4±1.89
2.6c 3.59±0.34 43.5±4.78
Donepezil 0.03 ± 0.01 6.41 ± 0.34
Neostigmine 22.2 ± 3.2 49.6 ± 6.11
As the results of anticholinesterase activity study (table 2.5), compounds 2.6d and 2.6i showed
highest inhibitory activity against AChE with the IC50 value of 0.47±0.01 µM. Structurally,
compound 2.6d contains phenyl group at C2 and N1 while 2.6i possesses phenyl group at C2
and phenethyl group at N1. Others modifications of the substituent at C2 and N1 lead to the
devaluation of inhibitory activity against AchE. Notably, the presence of aliphatic substituents,
methoxybenzyl at C2, or benzyl at N1 in the structure of chromeno[3,4-b]pyrrol-4(3H)-ones
significantly decreased the value of IC50 (compounds 2.6a, 2.6h, 2.6m, 2.6p, 2.6q). Compound
2.6i, however, exhibits lowest inhibitory activity against BChE, with the IC50 value of
208.8±2.56 µM. The most active BchE inhibitor was found to be compound 2.6p with the IC50
value of 9.45±1.56 µM. Furthermore, the data suggested that
chromeno[3,4-b]pyrrol-4(3H)-ones displayed selective inhibitory activity against either AChE
or BChE.

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
33
Table 2.6: Monoamine oxidase (A & B) activities of chromeno[3,4-b]pyrrol-4(3H)-ones
Compounds MA0-A
IC 50 µM & SEM VALUE
MAO-B
IC 50 µM & SEM VALUE
2.6d 0.79±0.005 0.33 ±0.06
2.6a 41% 43%
2.6b 0.77±0.003 2.18±0.03
2.6c 2.73±0.08 0.59±0.003
2.6f 1.09±0.01 4.71±0.05
2.6h 1.28±0.04 0.21±0.0005
2.6i 26% 11%
2.6j 0.53±0.001 15%
2.6k 1.21±0.02 40%
2.6q 1.51±0.03 37%
2.6p 5.12±0.07 0.15±0.0001
2.6m 0.69±0.0004 32%
2.6n 1.06±0.06 0.32±0.0002
2.6o 0.52±0.0003 21%
Clorgyline 3.64±0.012 -
Deprenyl - 0.007±0.001
Clorgyline were used as the standard inhibitor for monoamine oxidase A with IC50 value of
3.64±0.012 µM, and deprenyl for monoamine oxidase B with 0.007±0.001 µM. Compound 2.6j
and 2.6o were found to be potential inhibitors against monoamine oxidase A with IC50 value of
0.53±0.001 µM and 0.52±0.0003 µM respectively, which are about 7 times stronger compared
to the standard. Compound 2.6p, with IC50 = 0.15±0.0001 µM, exhibits the most effective
inhibitory activity against monoamine oxidase B among the tested
chromeno[3,4-b]pyrrol-4(3H)-ones. There is no clear trend between structure and monoamine
oxidase activities (table 2.6).
Compounds 2.6d and 2.6i showed similar activities against AChE, therefore these
compounds were chosen for molecular docking study. Both compounds show similar
interactions inside the receptor and form hydrophobic pocket with amino acid residues Trp279,
Ile287, Phe330, Phe33d1, Tyr334, and Gly335. However, the results showed that compound
2.6i established more stable conformation inside the receptor and formed additional hydrogen
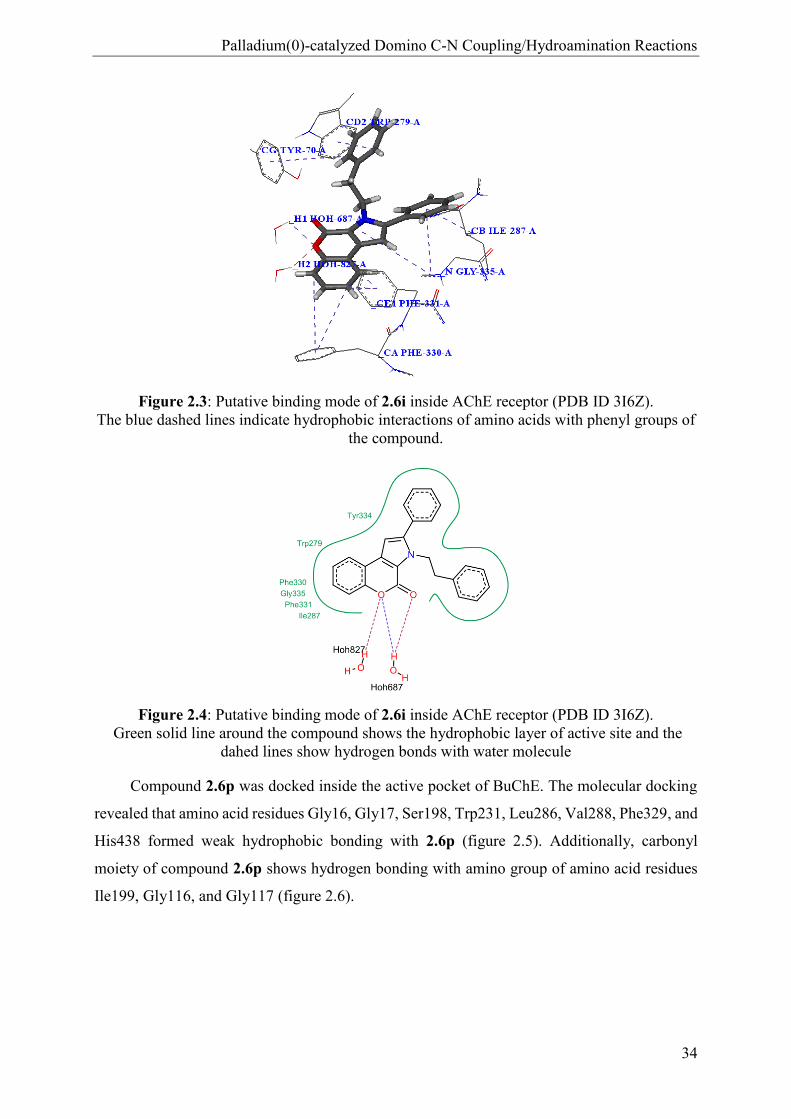
bonds with two water molecules. Figure 2.3 shows the docked pose of 2.6i with AChE receptor
and Figure 2.4 shows interaction in 2D.
Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
34
Figure 2.3: Putative binding mode of 2.6i inside AChE receptor (PDB ID 3I6Z).
The blue dashed lines indicate hydrophobic interactions of amino acids with phenyl groups of
the compound.
Figure 2.4: Putative binding mode of 2.6i inside AChE receptor (PDB ID 3I6Z).
Green solid line around the compound shows the hydrophobic layer of active site and the
dahed lines show hydrogen bonds with water molecule
Compound 2.6p was docked inside the active pocket of BuChE. The molecular docking
revealed that amino acid residues Gly16, Gly17, Ser198, Trp231, Leu286, Val288, Phe329, and
His438 formed weak hydrophobic bonding with 2.6p (figure 2.5). Additionally, carbonyl
moiety of compound 2.6p shows hydrogen bonding with amino group of amino acid residues
Ile199, Gly116, and Gly117 (figure 2.6).

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
35
Figure 2.5: Putative binding mode of 2.6p inside BuChE receptor (PDB ID 1P0I)
The blue dashed lines show the hydrophobic interactions of amino acid residues with phenyl
groups of compound 2.6p and the red dashed lines indicate hydrogen bonds between amino
group of Gly116, Gly117, Ala199 and carbonyl group of compound 2.6p.
Figure 2.6: Putative binding mode of 2.6p inside BuChE receptor (PDB ID 1P0I)
Green solid line around the compound shows the hydrophobic layer of active sites and the
dahed lines depict hydrogen bonds beteween carbonyl group of compound 2.6p with amino
groups of amino acid residues
After docking study, the docked poses were further verified by HYDE assessment. The ΔG
value for the compound 2.6i was calculated as -26 KJmol-1 while that of the 2.6h was only -10
KJmol-1. These results also prove the experimental data, which compound 2.6i is the best
inhibitor against AChE while compound 2.6h exhibit the least active inhibior. Similar results
were found in case of BuChE. The ΔG value for the strongest compound 2.6p is -18 KJmol-1
and for 2.6i is -15 KJmol-1.

Palladium(0)-catalyzed Domino C-N Coupling/Hydroamination Reactions
36
2.4. Conclusion
A convenient method for the synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones was
successfully developed. Moreover, a series of chromeno[3,4-b]pyrrol-4(3H)-ones, which are
difficult to obtained by known methods, were prepared. New synthesized
chromeno[3,4-b]pyrrol-4(3H)-ones were studied regarding their inhibitory activity against
cholinesterases and monoamine oxidases. Among them, compounds 2.6d and 2.6i were found
to be potent selective inhibitors against AChE while compound 2.6p showed the best activity
as selective inhibitor against BuChE. Furthermore, chromeno[3,4-b]pyrrol-4(3H)-ones 2.6j and
2.6o also displayed potent inhibitory activity against monoamine oxidase A.
A part of the results of this chapter were published in:
T. N. Ngo, O. A. Akrawi, T. T. Dang, A. Villinger, P. Langer, Tetrahedron Lett. 2015, 56,
86–88.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
37
3. Palladium(0)-catalyzed domino C-N coupling/hydroamination/
C-H arylation reactions: Synthesis of indolo[1,2-f]phenanthridines,
azaindolo[1,2-f]phenanthridines
3.1. Introduction
Fused phenanthridines are recognized as an important motif among drug-like molecules
and found many applications in medicinal chemistry.[74] For example, nitidine, fagaronine, and
coralyne, which are natural alkaloids containing the benzo[c]phenanthridine moiety, possess
interesting antitumor activity and are important targets for total syntheses and biological
evaluations;[75] ethidium bromide, an intercalating agent, is used as a fluorescent tag;[76] and
pyrrolo[1,2-f]phenanthridines was found capable of inhibiting HIV (figure 3.1).[77]
Figure 3.1: Biologically active phenanthridines
In addition to their biological activities, due to their large π-conjugated electron system and
effects of heteroatom contributing to the conjugated system, fused phenanthridines are essential
components for developing new semiconductors and organic light-emitting diodes (OLEDs).[78]
Particularly, recent studies showed that fused phenanthridines possess interesting optical and
electronic properties, in which phenanthridines fused with N-heterocycles are potential
candidates for the development of new blue-emitting materials.[79] Furthermore, organic dyes
developed from indolo[1,2-f]phenanthridine structure show broad and intense visible
absorptions, which are promising new sensitizers for dye-sensitized solar cells (figure 3.2).[80]

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
38
Figure 3.2: Phenanthridine derivatives with potential physical properties
Although many methods for the synthesis of phenanthridines were developed in recent
years, most of them require many steps and/or harsh conditions, and even more challenging for
fused phenanthridines.[81] Therefore, developing new and efficient methods is important and
necessary for the development of new bioactive molecules and organic materials. In recent
years, the advances in transition metal-catalyzed reactions have facilitated considerably the
approach to complex structures. Among them, palladium-catalyzed domino reactions proved to
be a useful tool for the synthesis of fused heterocyclic compounds with high atom economy. In
2007, Zhang and coworkers published a convenient method to synthesize
indolo[1,2-f]phenanthridines by reaction of arynes with 1-(2-bromophenyl)-1H-indole in the
presence of a Pd catalyst.[82] However, approaching starting materials for this method could be
problematic and requires many synthetic steps. In 2012, Mirua et al. and later in 2013, You et
al. independently reported an interesting Pd-catalyzed domino N–H/C–H arylation for the
regioselective synthesis of N-heterocyclic fused phenanthridines.[83,84] These authors used
readily available 2-arylindoles and 1,2-dibromobenzene as the starting materials, however, the
selectivity of reactions was difficult to control, which may result in mixtures of isomers. A
similar cascade process involving C-H arylations followed by an intramolecular N-arylation
reaction to prepare benzimidazole-fused phenanthridines in moderate to good yields was
published by Peng et al. in 2014.[85] More recently, Wang and Lv reported a one-pot tandem
approach to indolo[1,2-f]phenanthridines employing 2-alkynylanilines and boronic acids via
Cu-catalyzed C-N coupling/hydroamination and Pd-catalyzed C-H arylation.[86]
Retrospectively, I believe that by combining intramolecular C-H arylation with the construction
of indole scaffold, fused phenanthridines could be formed in a one-pot domino reaction. For
constructing indole moiety, sequential Pd-catalyzed C-N coupling and intramolecular
hydroamination of ortho-halogenated phenylacetylene is expected to be most suitable in this
scenario.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
39
Scheme 3.1: Domino reactions for the synthesis of fused-phenanthridines
In this chapter, I wish to describe a new synthetic methodology to efficiently approach
various indolo[1,2-f]phenathridines. The transformations proceed through three sequential
steps in a one-pot reaction: C-N coupling, hydroamination, and C-H arylation reactions with
employment of a single Pd catalyst. The reaction employs commercially available starting
materials: dihalogenated arenes, terminal alkynes, and 2-bromoanilines or anilines.
3.2. Synthesis of indolo[1,2-f]phenanthridines
For studying the reaction, 1-bromo-2-(phenylethynyl)benzene 3.1a and 2-bromoaniline
3.2a were chosen as model substrates. Initially, the reaction was carried out in DMF at 120 °C
using Cs2CO3 as the base in the absence of catalyst and ligand. However, the reaction did not
give the desired product 3.3a. When 10 mol% Pd(OAc)2 and 20 mol% PPh3 were introduced to
the reaction mixture as the catalyst, the product was isolated in 34% yield after 24 h. To improve
the yield, a series of monodentate and bidentate phosphine ligands were examined.
Consequently, XantPhos (10 mol%) and PCy3·HBF4 (20 mol%) were found to be the best
ligands as the reaction resulted in 75% and 74% yields of 3.3a, respectively. For further
investigation, other combinations of various palladium precursors and XantPhos were screened.
No product could be isolated when Pd2(dba)3 was used, while 70% yield of the desired product
was obtained when employing Pd(PPh3)4. It proved to be important to use Cs2CO3 as the base.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
40
Only trace amounts of product were detected when other bases were used, such as K2CO3 and
KOtBu. Variation of solvent and temperature did not lead to improved yields (table 3.1).
Table 3.1: Optimization for the synthesis of 3.3a
Entry Catalyst Ligand Base Yield (%)
1 Pd(OAc)2 PPh3 Cs2CO3 34
2 Pd(OAc)2 BINAP Cs2CO3 34
3 Pd(OAc)2 XantPhos Cs2CO3 75
4 Pd(OAc)2 DPEPhos Cs2CO3 70
5 Pd(OAc)2 DPPE Cs2CO3 38
6 Pd(OAc)2 DPPF Cs2CO3 41
7 Pd(OAc)2 XPhos Cs2CO3 45
8 Pd(OAc)2 SPhos Cs2CO3 34
9 Pd(OAc)2 RuPhos Cs2CO3 38
10 Pd(OAc)2 DavePhos Cs2CO3 64
11 Pd(OAc)2 PCy3·HBF4 Cs2CO3 74
12 Pd(OAc)2 P(tBu)3·HBF4 Cs2CO3 5
13 Pd(PPh3)4 XantPhos Cs2CO3 70
14 Pd2(dba)3 XantPhos Cs2CO3 trace
15 Pd(OAc)2 XantPhos K2CO3 trace
16 Pd(OAc)2 XantPhos KOtBu 5
Conditions: 3.1a (0.1 mmol, 1 equiv.), 3.2a (0.12 mmol, 1.2 equiv.), Pd(OAc)2 (0.01 mmol,
10 mol%), ligand (20 mol% with monodentate ligands, 10 mol% with bidentate ligands),
Cs2CO3 (0.3 mmol, 3 equiv.), DMF (1 mL), 120 °C, 24 h.
With the optimized conditions in hand, I extended the scope of the reaction by modifying
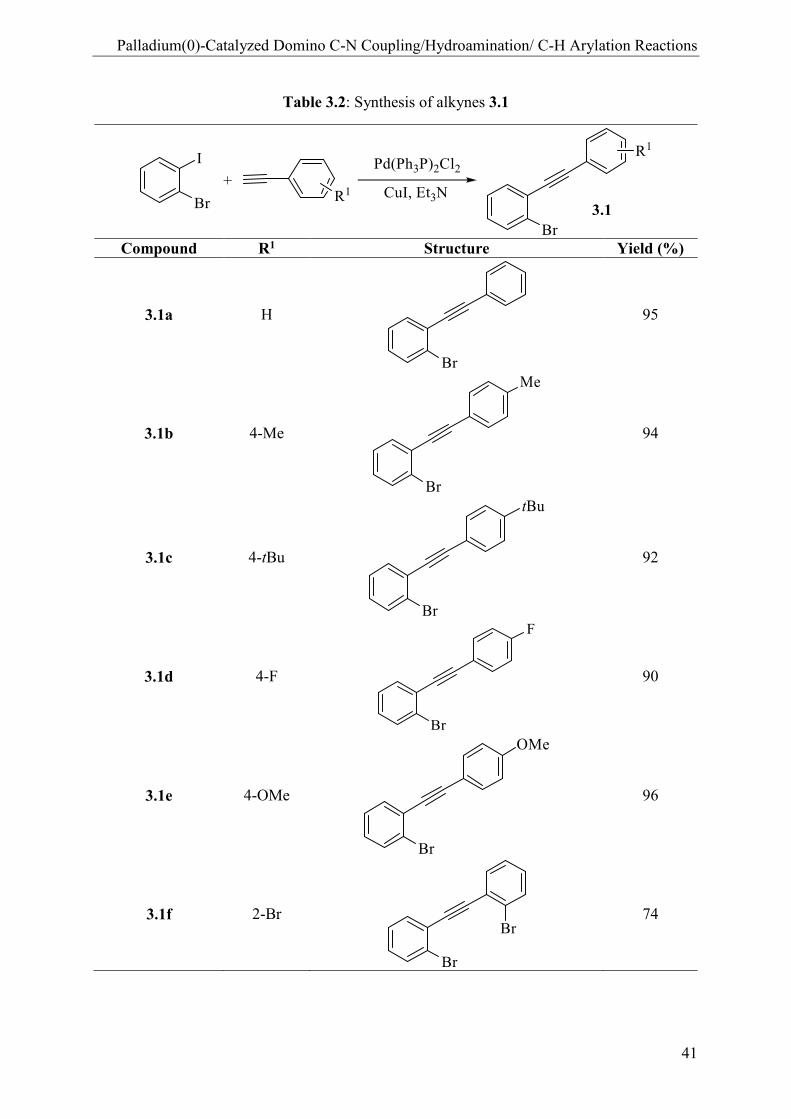
both substrates to prepare a series of indolo[1,2-f]phenanthridines. First, several alkynes 3.1a-f
were synthesized by chemoselective Sonogashira coupling reactions of 2-bromo-iodobenzene
with 1.1 equiv. of various phenylacetylenes, including electron-withdrawing and -donating
substituents. These compounds were obtained in nearly quantitative yield when using reported
procedure.[87]
Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
41
Table 3.2: Synthesis of alkynes 3.1
Compound R1 Structure Yield (%)
3.1a H
95
3.1b 4-Me
94
3.1c 4-tBu
92
3.1d 4-F
90
3.1e 4-OMe
96
3.1f 2-Br
74

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
42
Conditions: 2-bromo-iodobenzene 1 equiv., alkynes 1.1 equiv., Pd(PPh3)2Cl2 2.5 mol%,
CuI 10 mol%, Et3N is used as solvent and base, 25 °C, 4 h.
Then, reactions of 3.1a-e with 2-bromoanilline 3.2a, 2-bromo-4-methylaniline 3.2b, and
2-bromo-4-fluoroaniline 3.2c afforded various indolo[1,2-f]phenathridines 3.3 (Table 3.3). In
general, 1-bromo-2-(phenylethynyl)benzene or 2-bromoaniline derivatives, bearing
electron-donating or -withdrawing groups, afforded the corresponding products in moderate to
good yields under optimized conditions. The presence of substituents located at the aryl group
of the 1-bromo-2-(phenylethynyl)benzene had no pronounced effect on the yield. In contrast,
the structure of the 2-bromoaniline derivative had a greater impact, but did not follow a clear
trend. The best yields were obtained for 3.3h and 3.3j. The structure of 3.3i was unambiguously
confirmed by X-ray crystallography (figure 3.3).
Table 3.3: Synthesis of indolo[1,2-f]phenathridines
Compound R1 R2 Structure
Yield
(%)a
3.3a H (3.1a) H (3.2a)
75
3.3b H (3.1a) Me (3.2b)
66
3.3c Me (3.1b) H (3.2a)
52

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
43
3.3d Me (3.1b) Me (3.2b)
45
3.3e Me (3.1b) F (3.2c)
55
3.3f tBu (3.1c) H (3.2a)
65
3.3g tBu (3.1c) Me (3.2b)
67
3.3h F (3.1d) H (3.2a)
77
3.3i F (3.1d) Me (3.2b)
54
3.3j MeO (3.1e) H (3.2a)
78
3.3k MeO (3.1e) Me (3.2b)
73
Conditions: 3.1 (0.3 mmol, 1 equiv.), 3.2 (0.36 mmol, 1.2 equiv.), Pd(OAc)2 (0.03 mmol,
10 mol%), XantPhos (0.03 mmol, 10 mol%), Cs2CO3 (0.9 mmol, 3 equiv.), DMF (4 mL),
120 °C, 24 h.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
44
Figure 3.3: X-ray crystal structure analysis of 3.3i
Moreover, I conducted additional experiments to study the applicability of less reactive
chlorine substituents in the reaction. I realized that the bromine substituent in acetylenes 3.1
played a crucial role in the reaction, since no conversion of 1-chloro-2-(phenylethynyl)benzene
was observed when reacting with 2-bromoaniline under the optimized conditions.
Scheme 3.2: Reaction of 1-chloro-2-(phenylethynyl)benzene with 2-bromoaniline
A possible mechanistic pathway of the formation of indolo[1,2-f]phenanthridines is proposed
in Scheme 3.3. First, a Buchwald-Hartwig reaction of 3.1b with 2-bromoaniline 3.2a gave
intermediate 3.1i. Intramolecular hydroamination of 3.1i subsequently formed intermediate
3.1ii (which could be isolated and structurally confirmed by NMR spectroscopy). Finally, an
intramolecular C-H activation took place to give indolo[1,2-f]phenanthridine 3.3c.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
45
Scheme 3.3: Proposed pathway for the reaction
Encouraged by the successful isolation of intermediate 3.1ii, I considered the application
of 1,2-bis(2-bromophenyl)ethyne 3.1f as a suitable alternative precursor for the reaction. This
would allow the employment of simple anilines as educts and would widely broaden the scope
of the methodology. To my delight, the reaction of 1,2-bis(2-bromophenyl)ethyne 3.1f with
various anilines proceeded smoothly and produced the desired products 3.5a-j in moderate to
good yields (Table 3.4). Anilines containing a fluoro substituent gave very good results with
72% (3.5g) and 75% (3.5h) isolated yields. When employing an unsymmetrical aniline, a
mixture of two inseparable isomers 3.5e1 and 3.5e2 with 1:3 ratios was formed.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
46
Table 3.4: Reaction of 1,2-bis(2-bromophenyl)ethyne 3.1f with amines
Compound R Products Yield (%)a
3.5a (3.3a) H (3.4a)
52
3.5b (3.3b) 4-Me (3.4b)
55
3.5c 4-tBu (3.4c)
43
3.5d 4-MeO (3.4d)
49
3.5e 3-MeO (3.4e)
43a
3.5f 2-MeO (3.4f)
49

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
47
3.5g 2-F (3.4g)
72
3.5h 4-F (3.4h)
75
3.5i 4-CN (3.4i)
44
3.5j 4-MeS (3.4j)
56
Conditions: 3.1f (0.3 mmol, 1equiv.), 3.4 (0.36 mmol, 1.2 equiv.), Pd(OAc)2 (0.03 mmol,
10 mol%), (PCy3·HBF4 0.06 mmol, 20 mol%), Cs2CO3 (0.9 mmol, 3 equiv.), DMF (4 mL),
120 °C, 24 h. a inseparable mixture (ratio 1:3 determined by NMR)
It is clear that employment of unsymmetrical dibromoacetylenes would result in a
selectivity issue. In order to address this problem, we investigated the reaction of
1-bromo-2-((2-chlorophenyl)ethynyl)benzene 3.1g containing two different halides. The
reaction of 3.1g with p-toluidine afforded, using the optimized conditions, the desired product
3.5b in only 14% yield. As mentioned before, no conversion takes place in the reaction of
1-chloro-2-(phenylethynyl)benzene with 2-bromoaniline under the same conditions. Therefore,
the reactions were assumed to proceed starting from Buchwald Hartwig reaction at the C-Br
bond, followed by hydroamination and intramolecular C-H arylation at the C-Cl bond (scheme
3.4).

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
48
Scheme 3.4: Reaction of 1-bromo-2-((2-chlorophenyl)ethynyl)benzene with p-toluidine
3.3. Synthesis of azaindolo[1,2-f]phenanthridines
As mentioned previously, the structure obtained when phenanthridine fused with
N-heterocycles, such as imidazole, benzoimidazole, indole, and pyrrole, were reported to
possess remarkable optical and electronic properties. For example, phenanthridine fused with
N-heterocycles, such as imidazole, benzoimidazole, indole, and pyrrole, were reported to
possess remarkable optical and electronic properties.[79,83,85] Moreover, azaindoles are also
considered as important core structures and have been studied for decades.[88] However, the
scaffold of phenanthridine fused with azaindole is rarely reported, probably because of
difficulties in synthetic approaches. To my best knowledge, only one work (patented) related
to azaindolo[1,2-f]phenanthridines was published (figure 3.4); authors of the patent claimed
that electroluminescent devices employing these compounds showed improvement in driving
voltage and lifespan in comparison of using Alq3.[89] Therefore, developing an efficient
synthetic method for this scaffold is compelling for further studies of its properties.
Figure 3.4: Azaindolo[1,2-f]phenanthridines applicable in electroluminescent devices
In the reported patent, authors synthesized azaindolo[1,2-f]phenanthridine scaffold starting
from 7-azaindole, following by Ullmann reaction to obtain the key intermediate. This precursor
was transformed to azaindolo[1,2-f]phenanthridine by Pd-catalyzed cascade reaction with
benzyne generated in situ from 2-(trimethylsilyl)phenyl trifluoromethanesulfonate, which was
mentioned in previous part (scheme 3.5).

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
49
Scheme 3.5: Synthesis of azaindolo[1,2-f]phenanthridines via in situ benzyme
Regarding my methodology for the synthesis of fused phenanthridines, the strategy relies
on three sequential steps catalyzed by a single Pd catalyst in a one-pot reaction: C-N coupling,
hydroamination and intramolecular C-H arylation. After contemplation, I realized that this
strategy could be applied efficiently to synthesize phenanthridines-fused azaindole scaffolds
from simple and commercially available dihalogentated pyridines. Especially by employing
chemo- or regioselective of Sonogashira reaction for modifying the alkyne precursors, different
scaffolds, 4- or 7-azaindolo[1,2-f]phenanthridines, could be obtained (scheme 3.6).
Scheme 3.6: Synthesis of azaindolo[1,2-f]phenanthridines
In the following section, the details will be discussed, and furthermore, the optical properties
of selected synthesized compounds will be studied to justify the objective of the proposed idea.
Initially, 3-bromo-2-(phenylethynyl)pyridines 3.6a and 2-bromoaniline were chosen as
model substrates to study the reaction. No desired product was obtained after stirring the
mixture of substrates and cesium carbonate in DMF at 120 °C for 24 h. At the beginning, we
applied the reaction conditions from the previous reactions (table 3.3), which utilizes
Pd(OAc)2/XantPhos as the catalytic system. Interestingly, these conditions produced the
desired product with 39% yield. Furthermore, when the reaction time was increased to 48 h, the
yield of the reaction raised to 64%. Encouraged by this result, we continued to investigate other
combinations of catalytic sources and ligands using the same solvent, base, and temperature

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
50
conditions. To my delight, XantPhos was the best choice of ligand so far. The combination of
Pd(PPh3)4 and XantPhos gave the best result with 68% yield. Other attempts to change the
solvent and base did not lead to higher yields. Notably, decreasing the reaction temperature also
led to a significant decrease of yield. During the reaction, we observed the formation of
intermediate 3.6ii as a potential intermediate of the reaction pathway which is proposed in
Scheme 3.7.
Scheme 3.7: Proposed pathway for the reaction
With the optimized conditions in hand, the scope of the reaction was extended by
modifying the starting alkynes and 2-bromoanilines. Both electron-withdrawing and
electron-donating substituents were introduced into both substrates. Firstly,
3-bromo-2-(alkynyl)pyridines 3.6a-3.6f were synthesized from 2,3-dibromopyridine by
selective Sonogashira reaction at its C2, affording desired compounds with good to excellent
yields (table 3.5).

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
51
Table 3.5: Synthesis of 3-bromo-2-(alkynyl)pyridines
Compound R1 Structure Yield (%)
3.6a H
86
3.6b 4-Me
91
3.6c 4-tBu
95
3.6d 4-F
80
3.6e 4-OMe
91
3.6f 2-Br
77

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
52
Conditions: 2,3-dibromopyridine 1 equiv., alkynes 1.1 equiv., Pd(PPh3)2Cl2 2.5 mol%, CuI
10 mol%, Et3N is used as solvent and base, 25 °C, 4 h.
Then the domino reactions were performed with obtained 3-bromo-2-(alkynyl)pyridines
and several anilines. The reaction proceeded smoothly with various starting materials under
optimized conditions affording the desired products 3.7a-3.7k in moderate to high yields.
However, no apparent effect of the substituents on the yield was observed. Compound 3.7h was
obtained with the highest yield (88%) (Table 3.6). Furthermore, the structure of 3.7j was
independently confirmed by X-ray crystallographic analysis (figure 3.5).
Table 3.6: Synthesis of 4-azaindolo[1,2-f]phenanthridines
Compound R1 R2 Structure Yield (%)a
3.7a H H
68
3.7b H Me
64
3.7c H F
41

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
53
3.7d Me H
61
3.7e Me Me
72
3.7f tBu H
69
3.7g tBu Me
32
3.7h F H
88
3.7i F Me
79

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
54
3.7j MeO H
85
3.7k MeO Me
37
Conditions: 3.6 (0.3 mmol, 1 equiv.), 3.2 (0.36 mmol, 1.2 equiv.), Pd(PPh3)4 (0.03 mmol,
10 mol%), XantPhos (0.03 mmol, 10 mol%), Cs2CO3 (0.9 mmol, 3 equiv.), DMF (4 mL),
120 °C, 24 h.
Figure 3.5: X-ray crystallographic analysis of 3.7j.
Similarly, I assumed that the position of bromine and hydrogen atom participating in the
last step of the reaction could be interchangeable. With this idea in mind, reactions of 3.6f with
various amines were investigated to broaden the scope of this strategy (table 3.7).
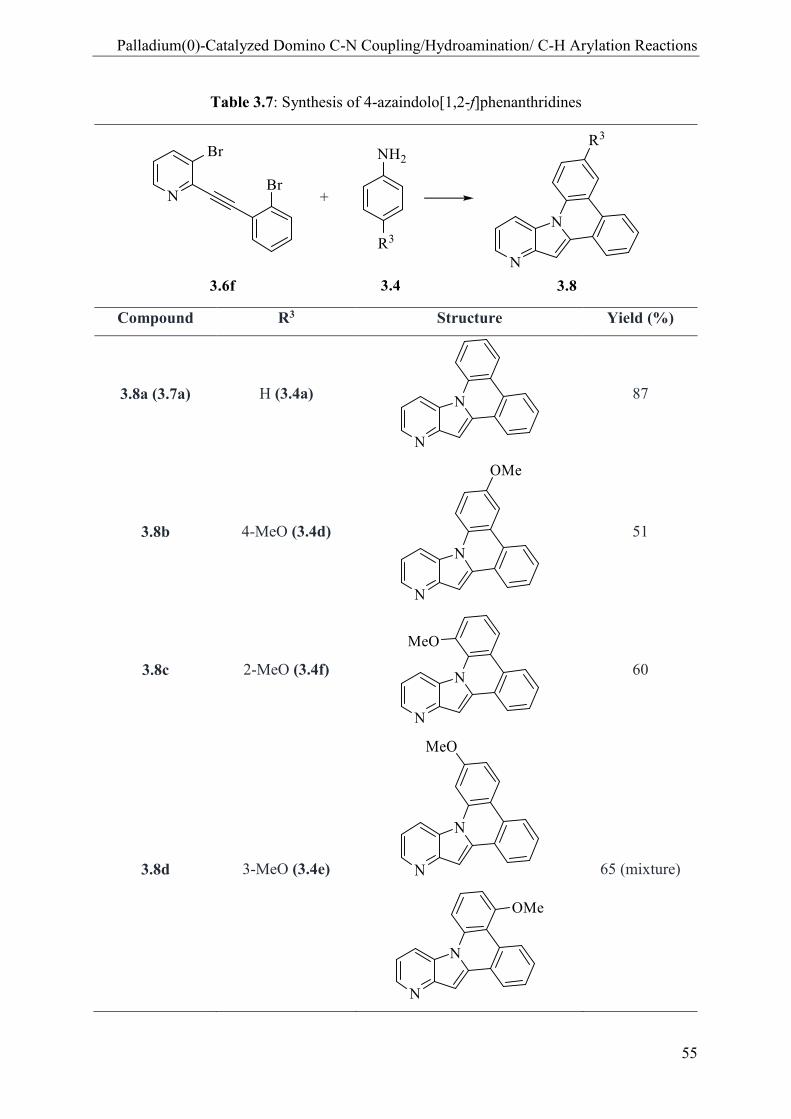
Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
55
Table 3.7: Synthesis of 4-azaindolo[1,2-f]phenanthridines
Compound R3 Structure Yield (%)
3.8a (3.7a) H (3.4a)
87
3.8b 4-MeO (3.4d)
51
3.8c 2-MeO (3.4f)
60
3.8d 3-MeO (3.4e)
65 (mixture)

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
56
3.8e (3.7c) 4-F (3.4g)
76
3.8f (3.7b) 4-CH3 (3.4b)
65
3.8g 4-SMe (3.4j)
81
3.8h
(3.4k)
42
Conditions: 3.6f (0.3 mmol), 3.4 (0.36 mmol), Pd(PPh3)4 (0.03 mmol), XantPhos
(0.03 mmol), Cs2CO3 (0.9 mmol), DMF (4 mL), 120 °C, 24 h.
To my delight, the reaction proceeded without any problem when the same conditions as
in the previous reaction were applied, affording desired products in moderate to high yields.
Interestingly, compounds 3.7a (3.8a), 3.7b (3.8f), 3.7c (3.8e) could be synthesized by this
method with higher yield. Besides, other modifications on the aniline ring gave lower yields
compared to 2-bromoanline. However, the limitation of this method is the selectivity when
meta-substituted amines are used, such as 3-methoxyaniline 3.4e. In this case, using
3-methoxyaniline resulted in a mixture of two products, which were not separable by column
chromatography.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
57
Additionally, the selectivity of the Sonogashira reaction on halogenated pyridines gives us
the possibility to deliver a higher diversity of products by customizing the starting material. To
demonstrate this, the selective Sonogashira mono-coupling reaction of
1-chloro-2-ethynylbenzene on 3-bromo-2-chloropyridine was performed to afford alkyne 3.6g.
The coupling reaction of 3-bromo-2-chloropyridine is controlled by the chemoselectivity of
bromide at position 3 versus the chloride at position 2, compared to 2,3-dibromopyridine in
which reactions at position 2 are more favored. Subsequently, alkyne 3.6g produced various
7-azaindolo[1,2-f]phenanthridines 3.9a-3.9c under optimized conditions as shown in table 3.8.
Noteworthy, the C2-Cl of 3.6g (of pyridine ring) is more reactive than of
1-chloro-2-(phenylethynyl)benzene for the first step of the domino reaction (Buchwald-
Hartwig reaction), as 1-chloro-2-(phenylethynyl)benzene showed no conversion when applied
conditions to perform the domino reaction (scheme 3.2).
Scheme 3.8: Synthesis of 3.6g
Table 3.8: Synthesis of 7-azaindolo[1,2-f]phenanthridines
Compound R Structure Yield (%)a
3.9a 4-MeO (3.4d)
34

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
58
3.9b 4-F (3.4g)
51
3.9c 4-SMe (3.4j)
36
Conditions: 3.6g (0.3 mmol, 1 equiv.), 3.4 (0.36 mmol, 1.2 equiv.), Pd(PPh3)4 (0.03 mmol,
10 mol%), XantPhos (0.03 mmol, 10 mol%), Cs2CO3 (0.9 mmol, 3 equiv.), DMF (4 mL),
120 °C, 24 h. (temperature and reaction time were not optimized)
To explore larger conjugated systems, I considered structure 3.10 (scheme 3.9) as the target
of the domino reaction. Then, 2,3,5,6-tetrabromopyridine was utilized as the starting material,
affording product of selective two-fold Sonogashira reaction at 2 and 6 positions as the set-up
for the domino reaction. Unfortunately, the reaction resulted in an inseparable mixture.
Scheme 3.9: Two-fold domino reaction

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
59
3.4. Absorption and fluorescence properties of azaindolo[1,2-f]phenanthridines
The optical properties of all synthesized compounds were studied by UV/Vis and
fluorescence spectroscopy in CH2Cl2 at 25 °C as summarized in table 3.9. The UV/Vis spectra
show an absorption band in the range of 270-300 nm and several weaker bands in the range of
300-400 nm (figure 3.6). In general, introducing electron-donor groups, such as a methyl or
methoxy group, at the core structure 3.7a causes a slight red shift of the absorption bands. A
stronger redshift was observed for compound 3.8h by extending the conjugated system of the
core structure. Changing the position of the nitrogen atom in the azaindole moiety (compounds
3.9a, 3.9b, 3.9c) caused also a shift to longer wavelengths. The similar trend was also observed
in the emission spectra.
Figure 3.6: Normalized absorption and corrected emission spectra of
azaindolo[1,2-f]phenanthridines in CH2Cl2.
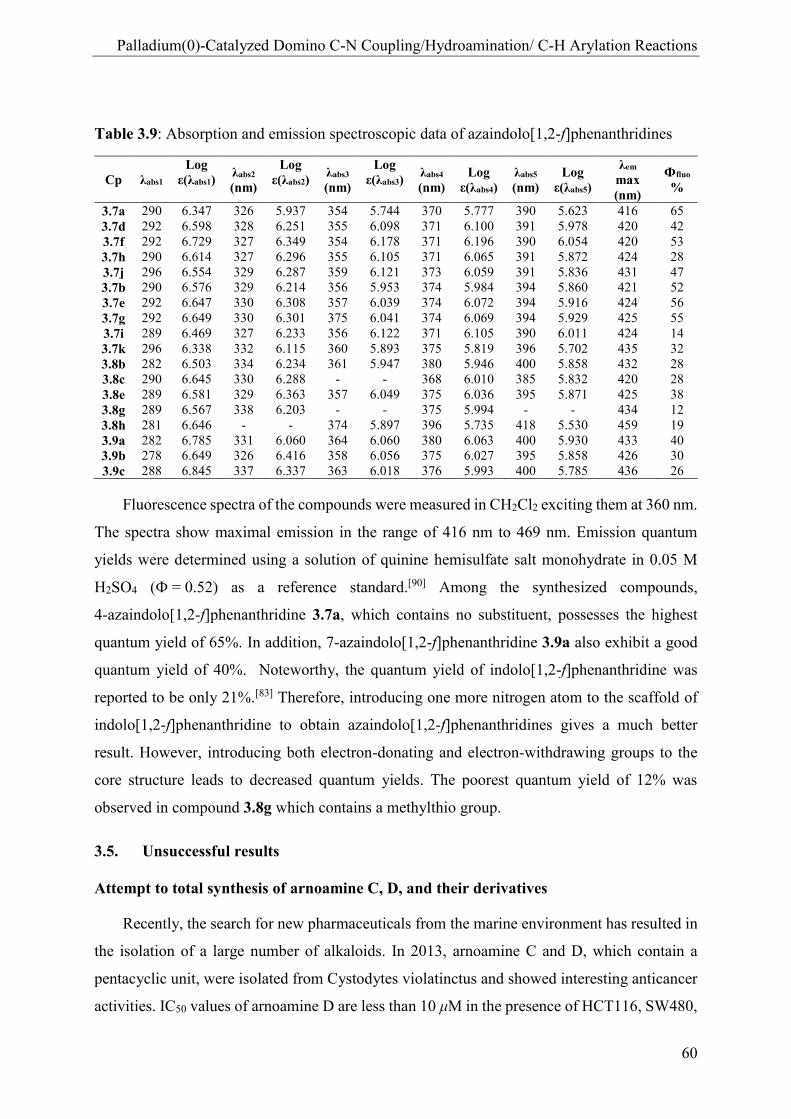
Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
60
Table 3.9: Absorption and emission spectroscopic data of azaindolo[1,2-f]phenanthridines
Cp λabs1
Log
ε(λabs1)
λabs2
(nm)
Log
ε(λabs2)
λabs3
(nm)
Log
ε(λabs3)
λabs4
(nm)
Log
ε(λabs4)
λabs5
(nm)
Log
ε(λabs5)
λem
max
(nm)
Փfluo
%
3.7a 290 6.347 326 5.937 354 5.744 370 5.777 390 5.623 416 65
3.7d 292 6.598 328 6.251 355 6.098 371 6.100 391 5.978 420 42
3.7f 292 6.729 327 6.349 354 6.178 371 6.196 390 6.054 420 53
3.7h 290 6.614 327 6.296 355 6.105 371 6.065 391 5.872 424 28
3.7j 296 6.554 329 6.287 359 6.121 373 6.059 391 5.836 431 47
3.7b 290 6.576 329 6.214 356 5.953 374 5.984 394 5.860 421 52
3.7e 292 6.647 330 6.308 357 6.039 374 6.072 394 5.916 424 56
3.7g 292 6.649 330 6.301 375 6.041 374 6.069 394 5.929 425 55
3.7i 289 6.469 327 6.233 356 6.122 371 6.105 390 6.011 424 14
3.7k 296 6.338 332 6.115 360 5.893 375 5.819 396 5.702 435 32
3.8b 282 6.503 334 6.234 361 5.947 380 5.946 400 5.858 432 28
3.8c 290 6.645 330 6.288 - - 368 6.010 385 5.832 420 28
3.8e 289 6.581 329 6.363 357 6.049 375 6.036 395 5.871 425 38
3.8g 289 6.567 338 6.203 - - 375 5.994 - - 434 12
3.8h 281 6.646 - - 374 5.897 396 5.735 418 5.530 459 19
3.9a 282 6.785 331 6.060 364 6.060 380 6.063 400 5.930 433 40
3.9b 278 6.649 326 6.416 358 6.056 375 6.027 395 5.858 426 30
3.9c 288 6.845 337 6.337 363 6.018 376 5.993 400 5.785 436 26
Fluorescence spectra of the compounds were measured in CH2Cl2 exciting them at 360 nm.
The spectra show maximal emission in the range of 416 nm to 469 nm. Emission quantum
yields were determined using a solution of quinine hemisulfate salt monohydrate in 0.05 M
H2SO4 (Փ = 0.52) as a reference standard.[90] Among the synthesized compounds,
4-azaindolo[1,2-f]phenanthridine 3.7a, which contains no substituent, possesses the highest
quantum yield of 65%. In addition, 7-azaindolo[1,2-f]phenanthridine 3.9a also exhibit a good
quantum yield of 40%. Noteworthy, the quantum yield of indolo[1,2-f]phenanthridine was
reported to be only 21%.[83] Therefore, introducing one more nitrogen atom to the scaffold of
indolo[1,2-f]phenanthridine to obtain azaindolo[1,2-f]phenanthridines gives a much better
result. However, introducing both electron-donating and electron-withdrawing groups to the
core structure leads to decreased quantum yields. The poorest quantum yield of 12% was
observed in compound 3.8g which contains a methylthio group.
3.5. Unsuccessful results
Attempt to total synthesis of arnoamine C, D, and their derivatives
Recently, the search for new pharmaceuticals from the marine environment has resulted in
the isolation of a large number of alkaloids. In 2013, arnoamine C and D, which contain a
pentacyclic unit, were isolated from Cystodytes violatinctus and showed interesting anticancer
activities. IC50 values of arnoamine D are less than 10 μM in the presence of HCT116, SW480,

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
61
and A375 cancer cell lines.[91] Derivatives of Arnoamine C & D would have interesting
bioactivities for applications in medicinal chemistry.
Figure 3.7: Arnoamine C & D
Based on the same strategy described in scheme 3.1, the total synthesis of Arnoamine C &
D and their derivatives could be synthesized using three-step domino reaction (scheme 3.11).
Scheme 3.10: Retrosynthesis analysis of Arnoamines
The starting material 3.11 can be synthesized from 5-chloro-2-methoxyaniline via 3 steps in
scheme 3.11.[92]

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
62
Scheme 3.11: Proposed Synthesis of starting material
However, the precursor 3.11 failed to convert to corresponding desired compound 3.10. I
assumed that chlorine might be not reactive enough for the reaction; therefor, another starting
material containing bromine was employed. For this purpose, 6-hydroxyquinoline was used as
the starting point, following by bromination with NBS and triflation to obtain
5-bromo-6-trifluoromethanesulfonatequinoline 3.12. Selective Sonogashira at the C-OTf of
quinoline 3.12 was performed successfully, affording precursor 3.13 for the domino reaction.
Scheme 3.12: Alternative starting material 3.13
With the initial screening, the formation of desired product was observed together with the
intermediate by GC/MS. But unfortunately, the desired product 3.14 was not separable as the
pure product.

Palladium(0)-Catalyzed Domino C-N Coupling/Hydroamination/ C-H Arylation Reactions
63
Scheme 3.13: Domino reaction of 3.13
3.6. Conclusion
In conclusion, two Pd-catalyzed three-step tandem reactions comprising of the three
sequential reactions: C-N coupling, hydroamination and C-H arylation reaction were
developed. These methods offer a straightforward synthesis of indolo[1,2-f]phenanthridines
under mild conditions with good yields, which are interesting for further applications in the
synthesis of new organic materials and bioactive molecules. In addition, a series of new
azaindolo[1,2-f]phenanthridines were synthesized conveniently by this strategy. The starting
materials were easily accessible by regioselective Sonogashira cross-coupling reaction, which
lead to diverse final products. The absorption and fluorescence properties of all products were
studied. This class of compounds shows promising photophysical properties, in particular, high
quantum yields. Furthermore, other new aromatic polyheterocycles are being explored by the
developed synthetic method.
The results of this chapter were published in:
T. N. Ngo, P. Ehlers, T. T. Dang, A. Villinger, P. Langer, Org. Biomol. Chem. 2015, 13,
3321–3330. Highlighted in Synfacts S001815SF.
and
T. N. Ngo, F. Janert, P. Ehlers, D. H. Hoang, T. T. Dang, A. Villinger, S. Lochbrunner, P.
Langer, Org. Biomol. Chem. 2016, 14, 1293–1301.

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
64
4. Regioselective synthesis of naphtho-fused heterocycles via
palladium(0)-catalyzed tandem reaction of N-tosylhydrazones
4.1. Introduction
Since its first discovery, independently by Mizoroki and Heck, Heck-Mizoroki reaction is
among the most useful synthetic tools due to its high chemoselectivity, mild reaction conditions,
and cheap reaction reagents.[93] Especially, domino processes involving intramolecular
Heck-Mizoroki reaction have been known as one of the most powerful methods to produce
polycarbocyclic or polyheterocyclic compounds with a variety of ring sizes. The active alkyl
intermediate palladium complexes, formed by insertion of double bond to Pd(II) complex, can
participate in sequential intra- or intermolecular processes before the β-hydride elimination
step[94]. For example, the active intermediate alkyl palladium complex could react with another
double bond in the molecule to afford fused bicyclic compounds.[95] Moreover, regioselectivity
in intramolecular Heck reaction has also attracted a lot of attention. Generally, intramolecular
Heck reactions give exo-trig cyclization products, however, several reactions formed
6-endo-trig instead of 5-exo-trig cyclized products.[96] In some cases, the 6-membered ring
might be the result of a sequence of 5-exo-trig, then 3-exo-trig cyclization, finally ring opening
of cyclopropane (Scheme 4.1). If the β-hydride elimination occurs before the ring opening
process, the cyclopropane ring is still retained in the product as the evidence for this
mechanism.[97] Although many studies regarding this process have been achieved, studying the
behavior of palladium on the variety of substrates is still compelling for insightful
understanding.
Scheme 4.1: Pathways of 6-endo-trig and 5-exo-trig cyclizations

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
65
Recently, the utility of easily accessible N-tosylhydrazones as the coupling partner for
metal-catalyzed cross-coupling reactions has attracted growing attention.[28–31,98] As discussed
earlier in chapter 1, the method relies on the insertion of carbene species, generated in situ from
N-tosylhydrazones, to Pd(II) complexes. In 2007, the group of Barluenga developed
palladium-catalyzed cross coupling reactions of N-tosylhydrazones and aryl halides, efficiently
affording polysubstituted olefins.[30] Furthermore, the presence of the double bond in obtained
olefins attracts a sequential intramolecular Heck cyclization. To exemplify, Valdes et. al.
reported a palladium-catalyzed autotandem process which involves cross-coupling of
N-tosylhydrazones with 2,2′-dibromobiphenyls followed by a 5-exo-trig Heck-type cyclization,
final β-hydride elimination step to give the formation of various polycyclic compounds
(Scheme 4.2a).[99]
Scheme 4.2: Pd-catalyzed cyclization of dibrominated compounds with hydrazones
From this perspective, I wish to report a novel tandem process started from the
cross-coupling reaction of N-tosylhydrazones with dibromide compounds, then followed by a
sequence of intramolecular 5-exo-trig, 3-exo-trig cyclization, ring opening, β-hydride
elimination in the presence of a single palladium catalyst to give 6-endo-trig cyclization
products (scheme 4.2b). The obtained fused heterocycles, heterotetracenes and

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
66
heteropentacene, with large π-extended conjugated aromatic system are important in both
materials science and medicinal chemistry.[100] To the best of my knowledge, only few tandem
procedures to access these fused systems were developed to date.[101]
4.2. Synthesis of naphtho-fused heterocycles
For the initial investigation, 3-bromo-2-(2’-chlorophenyl)benzo[b]thiophene (4.1a) and
acetophenone N-tosylhydrazone (4.2a) were chosen as model substrates (Scheme 4.3). Firstly,
I utilized Pd2(dba)3 and XPhos as the catalytic system and LiOtBu as the base, which are known
as standard conditions for the cross-coupling reaction of N-tosylhydrazones and aryl halides.
Two similar reactions at 90 °C in dioxane as the solvent were performed under argon. One
reaction was stopped after 2 hours, and the intermediate as the product of the first coupling was
isolated. The structure of the intermediate 4.1i was confirmed by NMR and mass spectra as the
new terminal double bond was formed and the chlorine still remained in the molecule. The
remaining reaction was continued and the reaction temperature was raised to 100 ºC. After 4 h,
complete conversion of 4.1a and formation of only one product with 72% yield were observed.
Surprisingly, the product was proved to be regioisomer 4.3a by X-ray crystallographic analysis,
which is different from the 6-endo-trig cyclization 4.3a’.
Scheme 4.3: Tandem reaction of 3-bromo-2-(2’-chlorophenyl)benzo[b]thiophene 4.1a with
N-tosylhydrazone of acetophenone 4.2a
Conditions: 4.1a (0.2 mmol, 1 equiv.), 4.2a (0.3 mmol, 1.5 equiv.), Pd2(dba)3 2.5 mol%,
XPhos 10 mol%, LiOtBu (0.8 mmol, 4 equiv.), dioxane (4 mL).

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
67
On the basis of these experimental results in the combination with reported literature on
domino palladium-catalyzed processes as well as Barluenga’s and Valdes’ reports of
N-tosylhydrazones and aryl halides, I postulate that the mechanism of the reaction followed a
sequence of intramolecular 5-exo-trig, 3-exo-trig cyclization, ring opening, and finally
β-hydride elimination. The proposed mechanism is described in Scheme 4.4. The catalytic cycle
is believed to initiate by the first oxidative addition of C-Br bond of 4.1a with palladium catalyst
to form an active Pd-complex (I). This complex reacts with the carbene generated from
N-Tosylhydrazone 4.2a to give new palladium active species (III) via intermediate complex
(II). The reductive elimination of complex (III) regenerates palladium(0) species and an
intermediate (4.4), which was isolated. The second oxidative addition of intermediate (4.4) with
palladium catalyst results in formation of a new active palladium complex (IV). The
intramolecular cyclization of this palladium species (IV) via 5-exo-trig, 3-exo-trig cyclization,
and then ring opening of cyclopropane lead to the more stable palladium complex (VII). A
second reductive elimination of palladium complex (VII) releases the cyclized product (4.3a)
and H[Pd]Cl, which reacts with base to reproduce palladium(0) catalyst (Schem 4.4).

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
68
Scheme 4.4: Proposed mechanism of Pd-catalyzed cyclization of dibrominated compounds
with hydrazones.

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
69
Scheme 4.5: Tandem reaction of 2,2′-dibromobiphenyl 4.1b and
3-bromo-2-(2-bromophenyl)pyridine 4.1c.
Reaction conditions: 1 (0.2 mmol), 4.2a (0.3 mmol, 1.5 equiv.), Pd2(dba)3 2.5 mol%, XPhos
10 mol%, LiOtBu (0.8 mmol, 4 equiv.), dioxane 4 mL, 110 °C 24 h.* inseparable mixture
In addition, the reaction 2,2′-dibromobiphenyl (4.1b) and N-tosylhydrazone of
4’-methylacetophenone (4.2b) was carried out under the same conditions (Scheme 4.5). It is
worth to mention again that the tandem reaction of 2,2′-dibromobiphenyl and N-tosylhydrazone
of cyclic ketones underwent β-hydride elimination after the 5-exo-trig cyclization to afford
5-membered ring spiro compounds. In this work, the absence of β-hydrogens promoted
3-exo-trig cyclization to the double bond of benzothiophene which is not fully conjugated in
the aromatic system. However, in the case of 2,2′-dibromobiphenyl, the alkyl palladium
complex was not able to perform 3-exo-trig cyclization to the stable conjugated aromatic system
of benzene. Therefore, only the uncyclized product was detected after 24 h. The same result
was observed with 3-bromo-2-(2-bromophenyl)pyridine. These results also confirmed the
hypothesis about the mechanism of the reaction.
In order to improve the yield of the desired products, I utilized
3-bromo-2-(2-bromophenyl)benzo[b]thiophene (4.1d) as the starting material instead of
3-bromo-2-(2’-chlorophenyl)benzo[b]thiophene (4.1a). The reaction was performed under the
same conditions at 90 °C. Surprisingly, the reaction completed after 4 h producing only 4.3a
with 83% yield. Then, other combinations of Pd(OAc)2, Pd2(dba)3 and different ligands were
examined, however, no significant improvement of yield was observed. Therefore, the
combination of Pd2(dba)3 and XPhos proved to be the best so far. Increasing the amount of
Pd2(dba)3 to 5 mol% and XPhos to 20% increased the yield to 85%, so it was not reasonable to
use more amount of catalyst. As I reviewed the literature, almost of the publications related to

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
70
the cross-coupling of N-tosylhydrazones employed the conditions with LiOtBu as the base and
dioxane as the solvent, so the combination of LiOtBu and dioxane is believed to be important
for the first cross-coupling of N-tosylhydrazones. Moreover, the reaction was additionally
carried out in toluene but resulted in a complicated mixture, without the possibility to isolate
the pure compound. The reaction was also tested at 60 °C, 90 °C, and 110 °C. At 60 °C, after
4h, I detected mainly the intermediates. At 110 °C, the reaction completed after 2 hours, but
more spots on the TLC were observed, one of them overlapped with the spot of the product,
leading to the difficulty in isolating the pure product, so I found that carrying out the reaction
at 90 °C was most suitable for this purpose. Interestingly, the formation of two intermediates
was observed in this case, because the first coupling of N-tosylhydrazone with 4.1d could
undergo at either C-Br bond (Scheme 4.6). The two intermediates gave the same palladium
complex after the 5-exo-trig cyclization (similar to complex V), thus, only one product was
formed. Besides, double Heck reaction of 4.1d with styrene utilizing above conditions as well
as conditions reported by Blacklock et. al. produced a complicated mixture without the
formation of 4.3a.[102] Therefore, the cross-coupling reaction of N-tosylhydrazone was proved
to be superior in this tandem reaction.
Scheme 4.6: Tandem reaction of 3-bromo-2-(2-bromophenyl)benzo[b]thiophene 4.1d and
N-tosylhydrazones.
Encouraged by these results, I examined the scope and limitation of the tandem reaction
by varying the substrates (table 4.1). Firstly, the tandem reactions of various N-tosylhydrazones
were performed with 3-bromo-2-(2’-bromophenyl)benzo[b]thiophene 4.1d. To my delight, the
optimized conditions were applicable for numerous N-tosylhydrazones of substituted
acetophenones. Both electron-donating and electron-withdrawing groups on N-tosylhydrozones
resulted in only one isomer from good to excellent yields. N-tosylhydrazone of
4’-methoxyacetophenone gave the best yield of 95% (4.3i) while the trifluoromethyl substituent

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
71
on acetophenone gave the poorest yield with 53% (4.3c). There is no predictable effect of
substituents on acetophenones moiety on yields of the reaction. The reaction conditions are
tolerable with several substituents such as unprotected hydroxyl (4.3d) or cyano groups (4.3e).
N-tosylhydrazone derived from hetero-aromatic acetophenone such as 4-acetylpyridine also
gave the desired product under the reaction conditions (4.3h). Interestingly, a heteropentacene
(4.3o) could be successfully prepared with 34% yield in the employment of reaction conditions
with corresponding 3,3'-dibromo-2,2'-bibenzo[b]thiophene. Unfortunately, the reaction of
N-tosylhydrazone of 1,2-diphenylethan-1-one with 4.1d resulted in an inseparable mixture.
Table 4.1: Synthesis of benzo[b]naphtho[2,1-d]thiophenes
Compound Ar Structure Yield (%)
4.3b
90
4.3c
53
4.3d
81
4.3e
68

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
72
4.3f
89
4.3g
75
4.3h
56
4.3i
95
4.3j
80
4.3k
71
4.3l
65
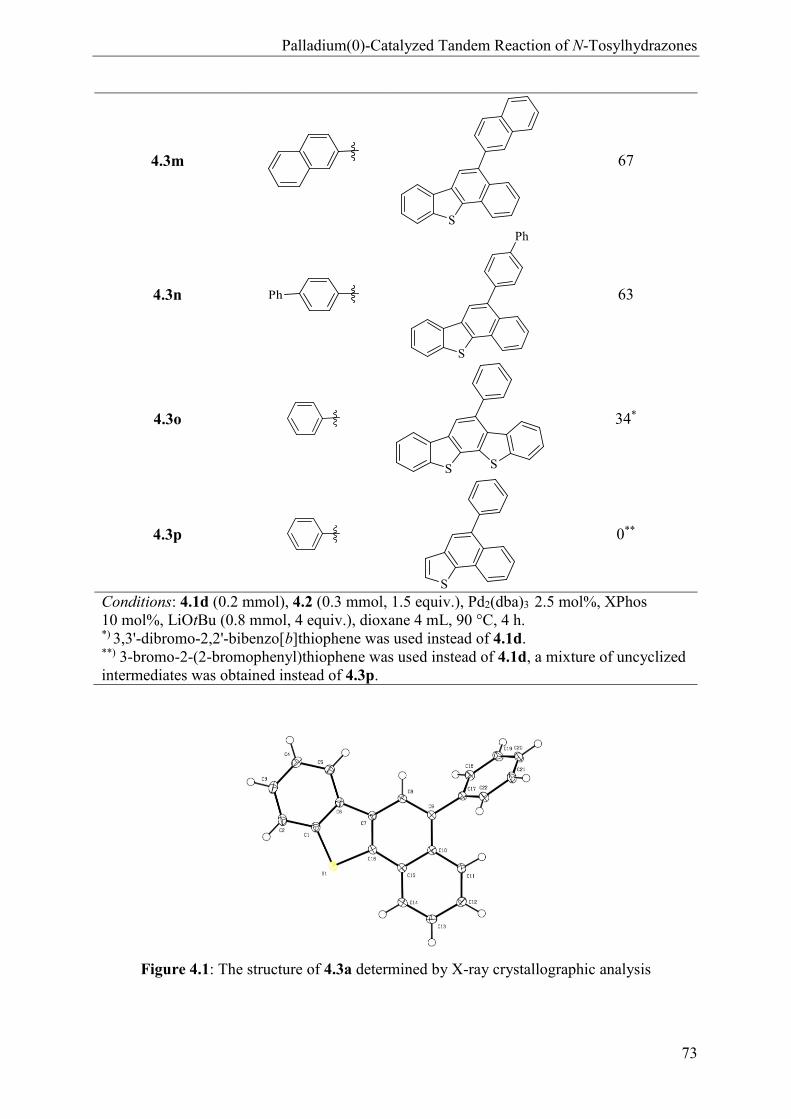
Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
73
4.3m
67
4.3n
63
4.3o
34*
4.3p
0**
Conditions: 4.1d (0.2 mmol), 4.2 (0.3 mmol, 1.5 equiv.), Pd2(dba)3 2.5 mol%, XPhos
10 mol%, LiOtBu (0.8 mmol, 4 equiv.), dioxane 4 mL, 90 °C, 4 h. *) 3,3'-dibromo-2,2'-bibenzo[b]thiophene was used instead of 4.1d. **) 3-bromo-2-(2-bromophenyl)thiophene was used instead of 4.1d, a mixture of uncyclized
intermediates was obtained instead of 4.3p.
Figure 4.1: The structure of 4.3a determined by X-ray crystallographic analysis

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
74
Figure 4.2: The structure of 4.3b determined by X-ray crystallographic analysis
In order to further explore the scope of reaction, I studied the reaction with other dibromide
derivatives from different heterocycles. The reaction gave good yields and regioselectivity
when applied for 3-bromo-2-(2-bromophenyl)benzofuran 4.1e. The reaction was assumed
following the proposed mechanism for 3-bromo-2-(2’-bromophenyl)benzo[b]thiophene,
forming similar isomer (table 4.2). Higher temperature and longer reaction time were required
when 3-bromo-2-(2-bromophenyl)-1-methyl-1H-indole 4.1f was employed as the precursor,
producing benzo[a]carbazole derivatives in relatively lower yields (table 4.3).
Table 4.2: Synthesis of naphtho[1,2-b]benzofurans
Compound Ar Structure Yield (%)
4.5a
60

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
75
4.5b
45
4.5c
77
4.5d
41
Reaction conditions: 4.1e (0.2 mmol), 4.2 (0.3 mmol, 1.5 equiv.), Pd2(dba)3 2.5 mol%,
XPhos 10 mol%, LiOtBu (0.8 mmol, 4 equiv.), dioxane 4 mL, 90 °C, 4 h.
Table 4.3: Synthesis of benzo[a]carbazoles
Compounds Ar Structure Yield
4.6a
31

Palladium(0)-Catalyzed Tandem Reaction of N-Tosylhydrazones
76
4.6b
45
4.6c
39
Reaction conditions: 4.1f (0.2 mmol), 4.2 (0.3 mmol, 1.5 equiv.), Pd2(dba)3 2.5 mol%,
XPhos 10 mol%, LiOtBu (0.8 mmol, 4 equiv.), dioxane 4 mL, 100 °C, 12 h.
4.3. Conclusion
In conclusion, I have developed a tandem reaction for regioselective synthesis of a range
of π-extended polyheterocyclic aromatic compounds. The reaction can be applied to numerous
substrates with the tolerance of various functional groups. Products of the reaction,
heterotetracenes and a heteropentacene, are interesting targets to explore potential applications
in materials science as well as medicinal chemistry. This strategy could be applied in the
synthesis of advanced functional materials containing larger π-extended heteroacene
compounds.
The results of this chapter were published in:
T. N. Ngo, T. T. Dang, A. Villinger, P. Langer, Adv. Synth. Catal. 2016, 358, 1328–1336.

Cyclization of Hydrazone Dianions
77
5. Efficient one-pot synthesis of 5-perfluoroalkylpyrazoles by
cyclization of hydrazone dianions
5.1. Introduction
Dianions, contained two negative charges, are known for versatility in cyclization reactions
with dielectrophiles to form rings with various size. The group of Prof. Langer has developed
several domino or one-pot cyclization reactions based on 1,1-, 1,2-, 1,3-, and 1,4-dianions,
affording important core structures ranging from three- to six-membered rings. These concepts
can apply to synthesize heterocycles, for example, the regioselective synthesis of functionalized
pyrroles by one-pot cyclocondensation of 1,3-dicarbonyl dianions with α-azidoketones. Due to
dianions are highly reactive, reactions with dielectrophiles can result in elimination,
polymerization, decomposition, and formation of open chained products. An important
discovery to overcome the drawback of dianions is the employment of electroneutral dianions,
or masked dianions, which are activated when reacting with Lewis acid. The idea was well
demonstrated by using 1,3-bis-silyl enol ethers as masked dianions in cyclization reactions with
dielectrophiles.[103]
Scheme 5.1: One-pot cyclocondensation of 1,3-dicarbonyl dianions with α-azidoketones
Fluorinated organic compounds have become essential in the development of
agrochemicals, and more importantly, in pharmaceuticals.[104] Recently, the number of
approved drugs or drug candidates containing fluorine is increasing noticeably, contributing
about 25% compounds in pharmaceuticals. In fact, three of ten best-selling drugs contain at
least one fluorine: rosuvastatin used for the treatment of high cholesterol and prevent
cardiovascular disease,[105] sofosbuvir, and ledipasvir used for the treatment of hepatitis C
infection.[106] The high electronegativity combining with its small size makes fluorine

Cyclization of Hydrazone Dianions
78
extremely low polarizability, resulting in drastic changes in physicochemical properties of
fluoroorganic compounds, most importantly enhancing thermal stability and lipophilicity, thus,
increasing bioavailability. In addition, fluorine has the size similar to hydrogen, however, could
act as a hydrogen bond acceptor, benefiting the molecules with similar geometry to those of
hydrogen but completely different interactions.[107]
Moreover, nitrogen-containing heterocycles constitute a large part of biologically active
compounds. Among them, pyrazole moiety is widely recognized, presenting in many leading
drugs,[108] such as Zometapine[109] and Viagra[110] and in agrochemicals, such as Tolfenpyrad
and Fenpyroximate.[111] Recently, the progression of synthetic fluorination methods leads to the
discovery of a pyramid of fluorinated pyrazoles with remarkable biological activities.[112] For
example, many important drugs and agrochemicals, such as Celecoxib (antiarthritic),[113]
Mavacoxib (antiarthritic),[114] Razaxaban (anticoagulant),[115] Fluazolate (herbicide),[116]
Penthiopyrad (fungicide)[117] are derived from trifluoromethylated and perfluoroalkylated
pyrazoles. Therefore, developing new efficient methods for the synthesis of fluorinated
heterocycles, particularly fluorinated pyrazoles, is still in demand.
Figure 5.1: Drugs and agrochemicals containing trifluoromethylated pyrazoles
There are two strategies to introduce florine to a structrue: direct fluorination or synthesis from
pre-fluorinated building blocks. Although direct fluorination is flourishing and gaining
significance with the rapid development of transition metal-catalyzed reactions, the strategy

Cyclization of Hydrazone Dianions
79
involving fluorine-containing intermediates is still efficient in various scenarios. Based on
developed synthetic methods for specific class of heterocycles, which are already proved their
efficiency, fluorine or fluorinated groups, such as trifluoromethyl or perfluoroalkyl, are
embedded in molecules of the precursors.
Particularly, conventional methods for the synthesis of pyrazoles are based on the
cyclocondensation of hydrazine with 1,3-dielectrophiles, such as 1,3-dicarbonyl or
α,β-unsaturated carbonyl compounds.[118] Another approach is 1,3-dipolar cycloadditions of an
alkyne with various 1,3-dipoles, such as diazoalkanes, nitrilimines, or azomethine imines.[119]
Scheme 5.2: Synthesis of pyrazoles
Trifluoromethylated pyrazoles have been prepared by cyclocondensation of
trifluoromethyl-1,3-diketones with phenylhydrazine, however, mixtures of regioisomers were
formed.[120] In order to discover more efficient approaches for the synthesis of
trifluoromethylated pyrazoles, several research groups have focused on the development of new
methods to address the issue of regioselectivity. Frizzo et al. reported a useful method for the
synthesis of 5-trifluoromethylpyrazoles based on the condensation of
4-alkoxy-1,1,1-trifluoro-3-alken-2-ones with phenyl-hydrazine using the ionic liquid
[BMIM][BF4] as the solvent. In general, this method gave very good yields of
5-trifluoromethylpyrazoles, but for some derivatives, regioisomeric mixtures were still
obtained.[121] Recently, some useful methods have been developed to overcome this problem,
e.g., the use of fluorinated alcohols (TFE and HFIP) in the cyclocondensation of
trifluoromethyl-1,3-diketones with phenylhydrazine or the employment of
4-trifluoromethyl-sydnones as starting materials in the cycloaddition reaction with alkynes.[122]
In early 2014, Mykhailiuk and coworkers reported an interesting approach to the synthesis of

Cyclization of Hydrazone Dianions
80
3-trifluoromethylpyrazoles in very good yields based on the [3+2] cycloaddition of CF3CHN2
with alkynes.[123] Very recently, a new method for the synthesis of 3-trifluoromethylpyrazoles
was described via trifluoromethylation/cyclization of α,β-alkynic hydrazone with hypervalent
iodine reagent.[124]
In addition, another interesting approach is the cyclization of hydrazone dianions with
esters. Hauser and co-workers were the first to report the synthesis of 5-substituted pyrazoles
by cyclization of hydrazone 1,4-dianions with esters. The same strategy was applied by the
group of Prof. Langer to prepare pyrazole-5-carboxylates and pyrazole-1,5-dicarboxylates.[125]
I believe that this method could be employed to prepare regioselectively
5-perfluoroalkylpyrazoles (scheme 5.3).
Scheme 5.3: Synthesis of pyrazoles by cyclization of hydrazone 1,4-dianions
In this chapter, a convenient and efficient method for the synthesis 5-trifluoromethyl- and
5-perfluoroalkylpyrazoles by one-pot cyclization of hydrazone 1,4-dianions with fluorinated
esters is being described. In addition, the activity of the pyrazoles prepared as inhibitors of
human tissue-nonspecific alkaline phosphatase (h-TNAP) and human intestinal alkaline
phosphatase (h-IAP) are being discussed. Furthermore, the effects of these molecules were also
tested on two other human ectonucleotidases, ecto-nucleotide pyrophosphatase/
phosphodiesterase-1 (h-NPP1) and h-NPP3.
5.2. Synthesis of perfluoroalkylated pyrazoles
Hydrazones 5.2 were prepared by condensation of ketones 5.1 with hydrazine derivatives.
This reaction proceeded under solvent free (‘green’) conditions at room temperature and is
catalyzed by acetic acid and provided nearly quantitative yields of hydrazones. Then, 5.2 were
converted to their dianions by treatment with 2 equivalents of n-BuLi in THF at -78 °C.
Subsequently, ethyl perfluorocarboxylates 5.3 were added to the reaction mixture. After
warming to ambient temperature, either trifluoroacetic acid (TFA) or p-toluenesulfonic acid

Cyclization of Hydrazone Dianions
81
(PTSA) was added to the reaction mixture to give perfluoroalkylated pyrazoles 5.4 or 5.5,
respectively (Scheme 5.4 & table 5.1). The formation of the product proceeds by the attack of
the carbon of dianion A onto 5.3 to give intermediate B, cyclization by the attack of the nitrogen
atom onto the carbonyl group to give intermediate C, and subsequent acid-mediated
dehydration. Treatment of intermediate C with TFA under reflux in dioxane allowed
5-perfluoromethylated pyrazoles 5.4 in good to excellent isolated yields. On the other hand,
treatment of intermediate C with PTSA (reflux, toluene) gave the N-deprotected
3-perfluoromethylpyrazoles 5.5. This could be explained by the fact that PTSA is a stronger
acid than TFA. The carbamate protecting group was removed when PTSA was used but
remained intact when TFA was used. It is noteworthy that the synthesis of compound 5.4f could
be scaled up to gram quantities. Starting with 10 mmol of 5.1f, product 5.4f was isolated in
88% yield (2.67 g).
Scheme 5.4: Synthesis of pyrazoles 5.4 and 5.5
Conditions: i, neat, acetic acid (catalytic amount, 3 drops), 20 °C; ii, 1) 2.2 equiv. n-BuLi,
THF, -78 °C to 20 °C. 2) 1.5 equiv. 5.3a, -78 °C to 20 °C. 3) TFA, reflux 2h (or PTSA,
reflux, 8h).

Cyclization of Hydrazone Dianions
82
Table 5.1: Synthesis of pyrazoles 5.4 and 5.5
Compound Ar R RF Structure Yield (%)
5.4a
CF3
89
5.4b
CF3
79
5.4c
CF3
81
5.4d
CF3
61
5.4e
CF3
74
5.4f
CF3
87
5.4g
CF3
90
5.4h
CF3
82
5.4i
CF3
78
5.4j
CF3
64

Cyclization of Hydrazone Dianions
83
5.4k
C2F5
95
5.4l
C2F5
91
5.4m
C2F5
93
5.4n
C2F5
85
5.4o
C3F7
83
5.4p
C3F7
76
5.4q
CO2Et CF3
57
5.4r
CO2Et CF3
65
5.5a
H CF3
51
5.5b
H C2F5
56
5.5c
H C3F7
57

Cyclization of Hydrazone Dianions
84
5.5d
H CF3
78
5.5e
H C2F5
86
5.5f
H C3F7
64
5.5g
H CF3
75
5.5h
H C2F5
74
5.5i
H C3F7
67
The cyclization of the dianion of the hydrazone of cyclododecanone 5.2s with ethyl
2,2,2-trifluoroacetate afforded the annulated trifluoromethylated pyrazole 5.4s in 80% yield
(Scheme 5.5). The cyclizations of the hydrazones of cyclohexanone 5.2t, cyclohex-2-en-1-one
5.2u and tetralone 5.2v afforded the corresponding products 5.4t-v.

Cyclization of Hydrazone Dianions
85
Scheme 5.5: Synthesis of 5.4s-v
Conditions:i, neat, acetic acid (catalytic amount, 3 drops), 20 °C; ii, 1) 2.2 equiv. n-BuLi,
THF, -78 °C to 20 °C. 2) 1.5 equiv. 5.3a, -78 °C to 20 °C. 3) TFA, reflux 2h.
Recently, it has been reported that trifluoromethylated indazole D32 represents a highly
selective ligand for the estrogen receptor β. Trifluoromethylated indazole E33 is a useful agent
for the treatment of obesity and diabetes (Figure 5.2). Therefore, it would be interesting to
prepare trifluoromethylated indazoles starting from ring-fused pyrazoles.
Figure 5.2: Bioactive trifluoromethylated indazoles D and E
The dehydrogenation of pyrazoles 5.4s and 5.4t with DDQ afforded the desired indazoles
5.6a and 5.6b in high yields, respectively. These experiments show that this methodology can
be successfully applied also for the synthesis of trifluorinated indazoles (scheme 5.6).

Cyclization of Hydrazone Dianions
86
Scheme 5.6: Synthesis of 5.6a and 5.6b
Conditions:i, 2.0 equiv. DDQ, toluene, reflux, 3 h.
The cyclization of the dianions of oximes 5.7a, b with ethyl trifluoroacetate and subsequent
treatment with TFA (reflux, 8h) afforded 5-trifluoromethylated isoxazoles 5.8a and 5.8b in 57%
and 62% isolated yields, respectively.
Scheme 5.7: Synthesis of isoxazoles 5.8a, b
Conditions:i, 1) 2.2 equiv. n-BuLi, THF, -78 °C to 20 °C. 2) 1.5 equiv. CF3COOEt, -78 °C to
20 °C. 3) TFA, reflux 2h.
5.3. Alkaline phosphatase and nucleotide pyrophosphatase activity and SAR
Intestinal alkaline phosphatase (IAP) is believed to play an important role in detoxification
of bacterial endotoxin, dephosphorylation of triphosphorylated and diphosphorylated
nucleotides, regulation of the intestinal microbiome, and regulation of intestinal lipid
absorption.[126] However, overexpression of IAP might have a connection with some
inflammatory bowel diseases such as Crohn’s disease.[127] On the other hand, tissue

Cyclization of Hydrazone Dianions
87
non-specific alkaline phosphatases (TNAP), which participate in the maintenance of the PPi
level in the body, share significant homology with IAP.[128] Therefore, selective inhibitors
against IAP would be potential therapeutic agents. Interestingly, pyrazole derivatives were
observed having inhibitory activity against alkaline phosphatases.[129] In this context,
embedding fluorine atoms in pyrazole molecules is promising to the discovery of potential
inhibitors for APs. In addition, nucleotide pyrophosphatase/phosphodiesterases (NPPs), which
affect a number of processes like bone mineralization, cell proliferation, motility, and digestion,
are also important therapeutic targets and it is worth to examine the inhibitory activity of
fluorinated pyrazoles against them.[130]
All the fluorinated pyrazole derivatives were tested for human recombinant APs and NPPs
and they were found to be selective inhibitors of APs in comparison to NPPs. Against h-NPP1
and h-NPP3, these compounds exhibited low response of inhibition. The data obtained showed
that all the values were below 50%. Compound 5.4i was found to be the potent inhibitor of
h-TNAP having IC50 value of IC50 ±SEM = 0.45±0.01 µM. It can be suggested that the activity
of this compound might be due to the presence of a phenyl and a trifluoromethyl group at the
parent pyrazole ring. When the activity of this compound was compared with the other
derivatives containing naphthalene ring attached to parent pyrazole it was clearly observed that
compound having phenyl and side chain with less carbon atom, has more impact on activity
against h-TNAP. When the number of carbon and fluorine increased the activity of the
compound was decreased as it was reflected in activity values of 5.4m, 5.5d, 5.5e, and 5.5f.
Levamisole was used as a standard inhibitor against h-TNAP. Other compounds inhibited
h-TNAP with IC50 values in the range of IC50 ±SEM = 0.449±0.001 to 50.3±3.28 µM.
Compound 5.4n exhibited the most potent inhibition of h-IAP with an IC50value of IC50 ±SEM=
0.65±0.04 µM [sic] which is over 120 folds more efficient than the known standard inhibitor
L-phenylalanine. The comprehensive study of the compound structure was justified by
comparing with L-phenylalanine (known reference standard) which contain only one phenyl
ring. This confirmed that the presence of biphenyl group on the pyrazole ring might be
responsible for its high activity against h-IAP. On the other hand, when this compound was
compared with 5.4e containing biphenyl ring with the different carbon side chain it was
observed that with a reduced number of carbon atoms in the side chain, the activity of
compound was decreased against h-IAP. Other pyrazole derivatives displayed h-IAP inhibition
activity in the range IC50 ±SEM = 0.647±0.04 to 7.36±0.25 µM. The above-mentioned data
showed that most pyrazole derivatives were better inhibitors of h-IAP than of h-TNAP.

Cyclization of Hydrazone Dianions
88
Table 5.2: Alkaline phosphatase AP (h-TNAP & h-IAP) and NPP (h-NPP1 & h-NPP3)
inhibition in presence of the synthesized compounds
5.4. Conclusion
In conclusion, I have demonstrated that a series of 5-trifluoromethylated and
5-perfluoroalkylated pyrazoles, including deprotected derivatives, could be efficiently and
selectively synthesized by one-pot cyclization of hydrazone dianions with ethyl
perfluorocarboxylates. In addition, two trifluoromethylated indazoles were prepared from the
corresponding bicyclic hydrazones. The cyclization of oxime dianions afforded
No.
h-TNAP h-IAP h-NPP1 h-NPP3
IC50 a
(µM)±SEM
IC50
a(µM)±SEM (%inhibition)b (%inhibition)b
5.4a 9.52±1.53 1.49±0.38 18.9% 12.5%
5.4b 11.1±1.06 1.22±0.22 23.4% 27.6%
5.4c 10.46±.65 1.63±0.41 21.2% 22.6%
5.4d 26.6±2.56 2.31±0.13 1.65% 4.87%
5.4e 3.23±0.48 1.41±0.05 12.4% 13.5%
5.4f 1.48±0.72 3.52±0.98 7.98% 19.8%
5.4g 2.59±0.38 5.78±0.74 13.8% 6.87%
5.4h 10.1±1.72 1.62±0.23 4.89% 2.76%
5.4i 0.45±0.01 4.46±0.78 23.2% 2.87%
5.4j 3.11±0.84 2.37±0.79 6.98% 1.09%
5.4k 2.11±0.28 5.12±0.84 34.5% 24.8%
5.4l 5.01±0.79 3.71±0.37 2.89% 4.67%
5.4m 1.35±0.06 2.19±0.05 38.4% 14.6%
5.4n
48.6±3.22 0.65±0.04 6.98% 9.87%
5.4o 50.3±3.28 1.35±0.14 28.2% 12.7%
5.4p 25.9±1.38 7.11±0.98 11.5% 17.8%
5.5a 11.6±1.28 7.36±0.25 3.08% 6.08%
5.5b 13.1±0.63 10.5±1.02 14.8% 18.9%
5.5c 4.34±0.03 2.91±0.35 39.1% 34.6%
5.5d 8.09±1.28 1.95±0.26 22.4% 18.7%
5.5e 13.9±1.08 4.47±0.97 6.87% 7.98%
5.5f 12.9±1.38 2.23±0.32 15.9% 10.8%
5.5g 1.62±0.11 2.39±0.36 19.3% 14.7%
5.5h 47.1±3.11 2.57±0.77 25.6% 29.8%
5.5i 2.04±0.17 1.96±0.33 28.2% 31.2%
5.6b 21.2±1.78 6.03±0.75 27.6% 12.6%
5.6a 17.2±0.89 5.84±0.37 12.8% 16.7%
Levamisole 19.21±0.001
L-Phenylalanine ------- 80.21±0.001
Values are expressed as mean ± SEM of n = 3. aThe IC50 is the concentration at which 50%
of the enzyme activity is inhibited. bThe % inhibition of the enzyme activity caused by 0.1
mM of the tested compound.

Cyclization of Hydrazone Dianions
89
trifluoromethyl-substituted isoxazoles. All the compounds were selective inhibitors of APs with
little effect on h-NPP1 and h-NPP3. In addition, the data showed that most of the compounds
presented here inhibited h-IAP more efficiently than h-TNAP. Therefore these compounds
appear as selective inhibitors of h-IAP.The results reported herein are of considerable interest
for further applications in medicinal chemistry.
The results of this chapter were published in:
T. N. Ngo, S. A. Ejaz, T. Q. Hung, T. T. Dang, J. Iqbal, J. Lecka, J. Sevigny, P. Langer, Org.
Biomol. Chem. 2015, 13, 8277-8290.

APPENDIX
i
APPENDIX
Methods for compound characterization and analysis
Melting Points
Micro heating table HMK 67/1825 Kuestner (Büchi apparatus); Melting points are
uncorrected.
Nuclear Magnetic Reasonance Spectroscopy (NMR)
Bruker: AM 250, (62.9 MHz); Bruker: ARX 300, (75.4 MHz), Bruker: ARX 500,
(125 MHz). The chemical shifts are given in parts per million (ppm). Coupling constants are
given in Hz.
References for 1H NMR: TMS (δ = 0.00) or residual deuterated solvent (CDCl3 (δ = 7.26),
C6D6 (δ = 7.16), (CD3)2CO (δ = 2.05), (CD3)2SO (δ = 2.50)), for 13C NMR TMS (δ = 0.00) or
residual deuterated solvent (CDCl3 (δ = 77.16), C6D6 (δ = 128.06), (CD3)2CO (δ = 29.84;
206.26), (CD3)2SO (δ = 39.52)) were taken as internal standard. The splitting pattern was
characterized by s: singlet, d: doublet, t: triplet, q: quartet, quin: quintet, sex: sextet, m:
multiplet. More complicate coupling peaks are represented by combinations of the respective
symbol. For example, dt indicate to doublet of triplet. Distortionless enhancement polarization
transfer (DEPT) spectra were taken to determine the types of carbon signals.
Mass Spectroscopy (MS)
AMD MS40, Varian MAT CH 7, MAT 731 (EI, 70 eV), Intecta AMD 402 (EI, 70 eV, and
CI).
High Resolution Mass Spectroscopy (HRMS)
Finnigan MAT 95 or Varian MAT 311; Bruker FT CIR, AMD 402 (AMD Intectra).
Infrared Spectroscopy (IR)
Bruker IFS 66 (FT IR), Nicolet 205 FT IR; Nicolet Protege 460, Nicolet 360 Smart Orbit
(ATR); KBr, KAP, Nujol, and ATR; Peaks were characterized with abbreviation: w = weak,
m = medium, s = strong, br = broad.

APPENDIX
ii
X-ray Crystal Structure Analysis
Bruker X8Apex diffractometer with CCD camera (Mo Kαradiation and graphite
monochromator, λ=0.71073 Å). The structures were solved by direct methods andrefined by
full-matrix least-squares procedures on F2 with the SHELXTL software package.
UV/Vis spectroscopy
Lambda 5 (Perkin Elmer) and Analytic Jena Specord 50 UV/VIS spectrometer in
acetonitril.
Fluorescence spectroscopy
Fluoromax4P-0759D-0311-FM. The samples were dissolved in dichloromethane. The
quinine hemisulfate salt monohydrate in 0.05M H2SO4 which has a fluorescence yield of 0.52,
was used as the standard for the fluorescent quantum yield determination.
Thin Layer Chromatography (TLC)
Merck Silica 60 F254 on aluminum foil from Macherey-Nagel. Detection under UV light
at 254 nm and/or 365 nm of wavelength and visualize by dipping in TLC stains solution
including conc. H2SO4/vaniline, Cerium-ammonium-molybdate (CAM), ceric sulfate and
Dragendorff reagent.
Column chromatography
Column chromatography was performed over Merck silica gel (63-200 µM) as normal
column and (40-63 µM) as flash column. All the solvent were distilled prior to use.
Biological protocols
The biotests were performed by Sundas Sarwar, Syeda Abida Ejaz, and Syeda Mahwish
Bakht in the group of Dr. Jamshed Iqbal, Centre for Advanced Drug Research, COMSATS
Institute of Information Technology, Abbottabad, Pakistan.
Cell Transfection with Human APs and NPPs
COS-7 cells were transfected with plasmids expressing human APs (TNAP & IAP) or
human NPPs ((NPP-1) or (NPP-3)) in 10-cm plates, by using Lipofectamine. The confluent
cells were incubated for 5 h at 37 °C in DMEM/F-12 in the absence of fetal bovine serum and

APPENDIX
iii
with 6 µg of plasmid DNA and 24 µL of Lipofectamine reagent. The same volume of
DMEM/F-12 containing 20% FBS was added to stop the transfection and cells were harvested
48–72 h later.
Preparation of membrane fractions
The transfected cells were washed three times at 4 °C, with Tris–saline buffer, collected by
scraping in the harvesting buffer (95 mM NaCl, 0.1 mM PMSF, and 45 mM Tris buffer, pH
7.5) and washed twice by centrifugation at 300×g for 5 min at 4 °C. Subsequently, cells were
resuspended in the harvesting buffer containing 10 µg/mL aprotinin and sonicated. Nuclear and
cellular debris were discarded by 10 min centrifugation (300×g at 4 °C). Glycerol was added to
the resulting supernatant at a final concentration of 7.5%.
Samples were kept at -80 °C until used. Protein concentration was estimated using Bradford
microplate assay and bovine serum albumin was used as a standard.
Protocol of Cholinesterase inhibition assay
For the determination of cholinesterase inhibition, electric eel and horse serum were used
as sources of AChE and BChE, respectively. AChE and BChE inhibition was measured in vitro
by the Ellman’s spectrophotometric method with slight modification. The reaction started by
mixing 20 µL assay buffer, 10 µL of test compound and 10 µL of enzymes (0.5 and 3.4 U/mg
of AChE or BChE, respectively). Then the reaction mixture was incubated for 10 min at 25 °C.
At the end of the pre/incubation period, 10 µL of 1 mM acetylthiocholine iodide or
butyrylthiocholine chloride were added to the respective AChE or BChE enzyme solution and
50 µL of 0.5 mM, 5,5’/Dithiobis/2/Nitrobenzoic Acid (DTNB) was added as coloring reagent.
The mixtures were incubated for 15 min at 25 °C. The formation of enzymatic product was
determined by the variation in absorbance measured at 405 nm with a microplate reader
(Bio/Tek ELx800TM, Instruments Inc., Winooski, VT, USA). In this bioactivity assay, the
standard drugs, neostigmine and donepezil were used. The buffer for enzyme dilution
comprised of 50 mM Tris/HCl containing 0.1% (w/v) BSA (pH 8). To remove the effect of
DMSO on enzymes, a blank assay was performed without any enzyme and accounted as
non/enzymatic reaction. The analysis of each concentration was done in triplicate and the IC50
values were calculated with the linear regression parameters. The computer program used for
this purpose is GraphPad Prism 5.0 (San Diego, CA, USA).
Protocol of Monoamine oxidase inhibition assay

APPENDIX
iv
Monoamine oxidase inhibitory activities of the synthesized compounds were evaluated
using standard protocol. Assays were performed in 200 µL final volume in 96 well plate. Rat
liver mitochondria were pretreated for 15 min at room temperature with an aqueous solution of
clorgyline (30 nM) or deprenyl (300 nM) to irreversibly block MAO/A or MAO/B activity,
respectively. Test compounds (2 µL), dissolved in DMSO (100%), were added to 90 µL of
mitochondrial preparation (25.0 µg of protein for rat MAO/A and 5.0 µg protein for rat MAO/B)
and were incubated for 30 min prior to the addition of 90 µL of freshly prepared Amplex Red
fluorogenic substrate. The Amplex Red reagent were used as follows, for a 96 well plate, 1.0 mg
of Amplex Red, dissolved in 200 µL of DMSO (100%) and 100 µL of reconstituted horseradish
peroxidase (HRP 200 U/mL) stock solution (kit vial + 1.0 mL of 50 mM sodium phosphate
buffer) was added to 9700 µL of sodium phosphate buffer (250 mM, pH 7.4). The enzymatic
reaction was started by the addition of 20 µL/well of an aqueous solution of the substrate p/
tyramine (300 µM final concentration). Deprenyl and clorgyline (each in a final concentration
of 1.0 µM) were used to determine non MAO/B and non/MAO/A enzyme activity, respectively.
Fluorescence measurements were performed over 45 min and the concentration response curves
of clorgyline and deprenyl served as positive controls for the rat MAO/A and rat MAO/B assay,
respectively.
Protocol of Alkaline Phosphatase Assay (h-TNAP & h-IAP)
A chemiluminescent substrate, CDP-star, was used for the determination of activity of
h-TNAP and h-IAP. The conditions for the assay were optimized with the slight modifications
in the previously used spectrophotometric method. The assay buffer was composed of 2.5 mM
MgCl2, 0.05 mM ZnCl2 and 8 M DEA (pH 9.8). Initial screening was performed at a
concentration of 0.2 mM of the tested compounds. The total volume of 50 µL contained 10 µL
of tested compound (0.2 mM with final DMSO 1% (v/v)), 20 µL of h-TNAP (46 ng of protein
from COS cell lysate in assay buffer) or of h-IAP (57 ng protein in assay buffer). The mixture
was pre-incubated for 5-7 minutes at 37 °C and luminescence was measured as pre-read using
microplate reader (BioTek FLx800, Instruments, Inc. USA). Then, 20 µL of CDP-star (final
concentration of 110 µM) was added to initiate the reaction and the assay mixture was incubated
for 15 min more at 37 °C. The change in the luminescence was measured as after-read. The
activity of each compound was compared with total activity control (without any inhibitor).
Levamisole (2 mM per well) and L-phenylalanine (4 mM per well) were used as a positive
control for the inhibition of h-TNAP and h-IAP, respectively. For the compounds which
exhibited over 50% inhibition of either h-TNAP activity or h-IAP activity, full concentration

APPENDIX
v
inhibition curves were produced to evaluate IC50 values. For this purpose, 6 to 8 serial dilutions
of each compound were prepared in assay buffer and their dose response curves were obtained
by assaying each inhibitor concentration against both ALPs using the above mentioned reaction
conditions. All experiments were repeated three times in triplicate. The Cheng Prusoff equation
was used to calculate the IC50 values, determined by the non-linear curve fitting program
PRISM 5.0 (GraphPad, San Diego, California, USA).
Protocol of Nucleotide pyrophosphatase (h-NPP-1 & h-NPP-3) activity
The conditions for the assay were optimized with the slight modifications in the previously
used spectrophotometric method. The reaction was carried out in the assay buffer which
contained 5 mM MgCl2, 0.1 mM ZnCl2, 50% glycerol and 50 mM tris-hydrochloride (pH: 9.5).
Initial screening was performed at a concentration of 0.1 mM of the tested compounds. The
total volume of 100 µL contained 70 µL of the assay buffer, 10 µL of tested compound (0.1 mM
with final DMSO 1% (v/v)) and 10 µL of h-NPP-1 (27 ng of protein from COS cell lysate in
assay buffer) or 10 µL of h-NPP-3 (25 mg of protein from COS cell lysate in assay buffer). The
mixture was pre-incubated for 10 minutes at 37 °C and absorbance was measured at 405 nm as
pre-read using microplate reader (BioTek FLx800, Instruments, Inc. USA). The reaction was
then initiated by the addition of 10 µL of p-Nph-5-TMP substrate at a final concentration of
0.5 mM and the reaction mixture was incubated for 30 more min at 37 °C. The change in the
absorbance was measured as after-read. The activity of each compound was compared with the
reaction in absence of synthesized compounds/inhibitors. The compounds which exhibited over
50% inhibition of either the h-NPP-1 activity or h-NPP-3 activity were further evaluated for
determination of IC50 values. For this purpose, their dose response curves were obtained by
assaying each inhibitor concentration against both NPPs using the above mentioned reaction
conditions. All experiments were repeated three times in triplicate. The Cheng Prusoff equation
was used to calculate the IC50 values, determined by the non-linear curve fitting program
PRISM 5.0 (GraphPad, San Diego, California, USA).
Preparation of receptor and ligands
Prior to docking procedures, the receptor crystalline structures were downloaded from
RCSB Protein Data Bank. As no good resolution template was present for eeAChE, the crystal
structure of Torpedo californica AChE (PDB ID 3I6Z) with resolution of 2.19 Å and Human
BuChE (PDB ID 1P0I) of human BuChE with resolution of 2.0 Å were downloaded. Receptors
for docking studies were prepared by using Load or Prepare Utility of LeadIT v2.1.8 from

APPENDIX
vi
BioSolveIT GmbH, Germany. The active site of the receptor was defined by selecting amino
acid residue in 7.5 Å radius around reference ligands i.e. Galantamine (G6X) in AChE receptor
and Butanoic acid (BUA) in BuChE.
Ligands: Chemical structures of the ligands were drawn using ACD/ChemSketch v14.1,
and 3D optimized. Using ANTECHAMBER, Gasteiger charges were added. Ligand structures
were then subjected to energy minimization using default values in Chimera v1.10.04 and saved
as mol2 files.
Molecular docking
Molecular docking was performed by Syed Jawad Ali Shah in the group of Dr. Jamshed
Iqbal, Centre for Advanced Drug Research, COMSATS Institute of Information Technology,
Abbottabad, Pakistan.
Molecular docking was carried out using FlexX utility of LeadIT v2.1.8 from BioSolveIT
GmbH, Germany. The binding site was defined as stated above. Water, amino acid residues and
small molecules were handled automatically by the software. Compounds were protonated as
in aqueous solution. Top ranking 30 poses were kept and further used for HYDE Assessment.
Hyde assessment and visual affinity
Top ranking docked poses were then subjected to HYDE assessment of LeadIT v.2.1.8 to
determine the Binding Free Energy (binding affinity). HYDE is based on two parameters. One
is hydrogen bonding and the second one is dehydration term i.e. ΔGiHYDE = Ʃ ΔGi
Dehydration + Ʃ
ΔGiH-bond
General procedure for the synthesis of pyrrolocoumarins
Compound 2.4 (0.3 mmol), aniline 2.5 (0.36 mmol, 1.2 equiv), Pd(OAc)2 (0.03 mmol,
10% mol), SPhos (0.06 mmol, 20% mol), CuI (20% mol), and Cs2CO3 (2.5 equiv. 0.9 mmol)
were placed in a dried pressure tube equipped with a septum. The reaction was back-filled with
argon three times. Then dry and degassed DMF (4 mL) was added under argon and the septum
was replaced with a Teflon cap. The reaction mixture was allowed to stir at 80 °C for 4 h. Then
the reaction mixture was cooled to room temperature and was filtered through a pad of Celite.
The Celite pad was washed three times with ethyl acetate (3x20 mL). The filtrate was dried
under reduced pressure, and the product 2.6 was obtained after flash chromatography on a silica
gel column.

APPENDIX
vii
3-(4-Methoxyphenyl)-2-phenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6a
White solid, 64% (70.5 mg). M.p.: 197–199 °C.
IR (ATR, cm-1): 3068.7 (w), 2971.0 (w), 2840.9 (w), 1720.8 (s),
1608.6 (m), 1512.0 (s), 1464.4 (s), 1356.6 (m), 1249.1 (m),
1168.0 (s), 1065.4 (s), 973.8 (s), 831.2 (m), 755.7 (s), 693.9 (s),
553.3 (s).
1H NMR (300 MHz, CDCl3) δ 7.89 – 7.79 (m, 1H, CHAr), 7.40 (dd, 3J = 4.9, 4J = 1.4 Hz, 2H,
CHAr), 7.34 – 7.15 (m, 8H, CHAr), 6.95 – 6.84 (m, 3H, CHAr), 3.83 (s, 3H, OCH3).
13C NMR (75 MHz, CDCl3) δ 159.5 (C-OCH3), 154.2 (C=O), 151.6, 145.5, 131.2, 130.5, 130.3
(CAr), 129.3 (2CHAr), 129.0 (2CHAr), 128.2 (3CHAr), 127.9, 123.8, 122.8 (CHAr), 118.3, 117.7
(CAr), 116.9 (CAr), 113.7 (2CHAr), 102.9 (CHAr), 55.3 (OCH3).
MS (EI,70 eV): m/z (%) = 367 (M+, 100), 278 (5), 205 (10), 176 (9), 133 (5), 77 (4).
HRMS (EI, 70 eV): calcd for C24H16O3N1 ([M]+): 367.12029, found: 367.11931.
3-(3-Methoxyphenyl)-2-phenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6b
Yellowish solid, 47% (51.7 mg). M.p.: 182–183 °C.
IR (ATR, cm-1): 3116.2 (w), 3063.7 (w), 2951.1 (w), 2849.3 (w),
1710.4 (s), 1598.8 (m), 1487.0 (m), 1463.4 (s), 1356.2 (m),
1213.5 (s), 1111.8 (m), 1031.5 (s), 989.4 (m), 954.4 (m), 850.0
(m), 762.4 (s), 693.9 (s), 647.1 (m).
1H NMR (300 MHz, CDCl3) δ 7.84 (d, 3J = 7.6 Hz, 1H, CHAr), 7.47 – 7.36 (m, 2H, CHAr),
7.36 – 7.17 (m, 7H, CHAr), 7.06 – 6.74 (m, 4H, CHAr), 3.74 (s, 3H, OCH3).
13C NMR (63 MHz, CDCl3) δ 159.7 (C-OCH3), 154.0 (C=O), 151.6, 145.3, 138.4, 131.1, 130.7
(CAr), 129.3 (CHAr), 129.2 (2 CHAr), 128.6 (CHAr), 128.5 (2 CHAr), 128.3, 124.2, 123.2, 121.1
(CHAr), 118.1, 117.7 (CAr), 117.3, 114.8, 114.5, 103.5 (CHAr), 55.6 (OCH3)
MS (EI,70 eV): m/z (%) = 367 (M+, 100), 278 (5), 205 (7), 176 (9), 92 (4), 77 (4).
HRMS (EI, 70 eV): calcd for C24H16O3N1 ([M]+): 367.12029, found: 367.11972.
3-(2-Methoxyphenyl)-2-phenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6c

APPENDIX
viii
Yellowish solid, 61% (67.2 mg). M.p.: 162–163 °C.
IR (ATR, cm-1): 3067.7 (w), 2923.1 (w), 2843.0 (w), 1717.9 (s), 1598.9
(w), 1503.0 (m), 1465.0 (s), 1279.3 (m), 1220.4 (m), 1162.0 (m), 1108.9
(m), 1060.3 (m), 969.9 (s), 896.1 (m), 756.1 (s), 700.5 (s), 655.3 (m),
549.6 (m).
1H NMR (300 MHz, CDCl3) δ 7.98 – 7.73 (m, 1H, CHAr), 7.43 – 7.35 (m, 3H, CHAr),
7.34 – 7.21 (m, 6H, CHAr), 7.16 (dd, 3J = 7.7, 4J = 1.7 Hz, 1H, CHAr), 7.02 – 6.88 (m, 3H,
CHAr), 3.68 (s, 3H, OCH3).
13C NMR (75 MHz, CDCl3) δ 155.8 (C-OCH3), 154.0 (C=O), 151.6, 145.5, 131.3, 130.4 (CAr),
130.4 (CHAr), 129.8 (CHAr), 128.8 (2CHAr), 128.5 (CHAr ), 128.3 (2 CHAr), 128.0 (CHAr), 126.8
(CAr), 124.0, 123.1, 120.5 (CHAr), 118.4, 117.9 (CAr), 117.2, 112.0, 102.9 (CHAr), 55.8 (OCH3).
MS (EI,70 eV): m/z (%) = 367 (M+, 100), 336 (35), 261 (25), 205 (5), 176 (10), 139 (9), 77
(6).
HRMS (EI, 70 eV): calcd for C24H16O3N1 ([M]+): 367.12029, found: 367.11965.
2,3-Diphenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6d
White solid, 52% (52.6 mg). M.p.: 215–216 °C.
IR (ATR, cm-1): 3051.9 (w), 2922.3 (w), 2849.8 (w), 1714.0 (s),
1590.1 (w), 1495.0 (m), 1468.7 (m), 1390.6 (m), 1352.7 (m), 1224.1
(m), 1165.9 (m), 1110.5 (m), 1063.1 (s), 760.2 (s), 690.5 (s), 558.7
(m).
1H NMR (300 MHz, CDCl3) δ 7.92 – 7.75 (m, 1H, CHAr), 7.51 – 7.13 (m, 13H, CHAr), 6.95 (s,
1H, CHAr).
13C NMR (75 MHz, CDCl3) δ 154.1 (C=O), 151.6, 145.4, 137.5, 131.1, 130.7 (CAr), 129.3
(2CHAr), 128.7 (2 CHAr ), 128.7 (CHAr ), 128.7 (2 CHAr ), 128.5 (CHAr ), 128.5 (2 CHAr ), 128.3,
124.2, 123.12 (CHAr), 118.1, 117.7 (CAr), 117.2, 103.5 (CHAr).
MS (EI,70 eV): m/z (%) = 337 (M+, 100), 307 (5), 291 (13), 278 (5), 205 (12), 176 (10), 146
(8), 77 (11).
HRMS (+ESI, 180 eV): calcd for C23H15O2N1 ([M+H]+): 338.11773, found: 338.11756.
3-(4-Fluorophenyl)-2-phenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6e

APPENDIX
ix
Yellowish solid, 41% (43.7 mg). M.p.: 226–227 °C.
IR (ATR, cm-1): 3412.0 (w), 3096.6 (w), 3069.1 (w), 1710.4 (w),
1609.2 (w), 1507.4 (s), 1467.2 (m), 1355.7 (m), 1220.4 (s), 1117.2
(m), 1064.2 (m), 977.5 (m), 845.3 (m), 756.7 (s), 695.1 (m), 553.3
(s).
1H NMR (300 MHz, CDCl3) δ 7.84 (dt, 3J = 7.6, 4J = 1.1 Hz, 1H, CHAr), 7.41 (dd, 3J = 4.7,
4J = 1.0 Hz, 2H, CHAr), 7.38 – 7.15 (m, 8H, CHAr), 7.13 – 7.02 (m, 2H, CHAr), 6.93 (s, 1H,
CHAr).
19F NMR (282 MHz, CDCl3) δ -112.40.
13C NMR (75 MHz, CDCl3) δ 162.4 (d, 1J = 248.7 Hz, CF), 154.2 (C=O), 151.6, 145.5, 133.5
(d, 4J = 3.3 Hz), 130.9, 130.8 (CAr), 130.4 (d, 3J = 8.8 Hz, 2 CHAr ), 129.3 (2 CHAr ), 128.7
(CHAr ), 128.6 (2 CHAr ), 128.4, 124.3, 123.2 (CHAr), 118.2, 117.6 (CAr), 117.3 (CHAr ), 115.8
(d, 2J = 23.0 Hz, 2 CHAr ), 103.6 (CHAr).
MS (EI,70 eV): m/z (%) = 355 (M+, 100), 309 (11), 224 (6), 205 (8), 176 (9), 155 (5), 95 (7).
77 (2).
2-Phenyl-3-(3-(trifluoromethyl)phenyl)chromeno[3,4-b]pyrrol-4(3H)-one 2.6f
Yellowish solid, 35% (42.5 mg). M.p.: 191–192 °C.
IR (ATR, cm-1): 3059.1 (w), 2922.3 (w), 2851.1 (w), 1713.6 (s),
1612.3 (w), 1498.5 (w), 1463.6 (m), 1401.8 (w), 1330.0 (s), 1251.2
(w), 1166.3 (m), 1125.0 (s), 1064.6 (s), 987.7 (m), 818.9 (m),
765.3 (s), 698.6 (s), 654.4 (m), 550.0 (m).
1H NMR (300 MHz, CDCl3) δ 7.93 – 7.78 (m, 1H, CHAr), 7.70 – 7.60 (m, 1H, CHAr),
7.58 – 7.45 (m, 3H, CHAr), 7.45 – 7.38 (m, 2H, CHAr)), 7.38 – 7.22 (m, 4H, CHAr), 7.22 – 7.12
(m, 2H, CHAr), 6.96 (s, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -62.73.
13C NMR (75 MHz, CDCl3) δ 154.1 (C=O), 151.6, 145.5, 137.8 (CAr), 132.1(CHAr), 131.2
(CAr), 131.2 (q, 2J = 33.1 Hz, CAr-CF3), 130.5 (CAr), 129.4 (2 CHAr ), 129.3 (CHAr ), 128.9
(CHAr), 128.7 (2 CHAr ), 128.6, 125.8 (q, 3J = 3.8 Hz), 125.4 (q, 3J = 3.6 Hz), 124.3, 123.9
(CHAr), 123.58 (q, 1J = 270.2 Hz, CF3), 118.0, 117.4 (CAr), 117.3, 104.1 (CHAr).
MS (EI,70 eV): m/z (%) = 405 (M+, 100), 384 (36), 274 (4), 205 (11), 176 (9), 145 (7), 75 (3).
HRMS (EI, 70 eV): calcd for C24H14F3O2N1 ([M]+): 405.09711, found: 405.09668.

APPENDIX
x
4-(4-Oxo-2-phenylchromeno[3,4-b]pyrrol-3(4H)-yl)benzonitrile 2.6g
Yellow solid, 40% (43.3 mg). M.p.: 233–234 °C.
IR (ATR, cm-1): 3096.1 (w), 3067,6 (w), 3046.2 (w), 2241.8 (w),
2232.5 (w), 1719.4 (m), 1704.5 (s), 1600.6 (w), 1507.9 (m),
1466.4 (m), 1399.7 (m), 1355.2 (w), 1223.5 (w), 1065.2 (m),
977.7 (m), 854.4 (m), 756.5 (s), 696.4 (m), 566.9 (m).
1H NMR (300 MHz, CDCl3) δ 7.89 – 7.78 (m, 1H, CHAr), 7.72 – 7.63 (m, 2H, CHAr),
7.46 – 7.27 (m, 8H, CHAr), 7.16 (dd, 3J = 7.8, 4J = 1.7 Hz, 2H, CHAr), 6.97 (s, 1H, CHAr).
13C NMR (75 MHz, CDCl3) δ 154.1 (C=O), 151.6, 145.3, 141.2 (CAr), 132.6 (2 CHAr), 131.7,
130.3 (CAr), 129.7 (2 CHAr), 129.3 (2 CHAr ), 129.1 (CHAr ), 128.8 (3 CHAr ), 124.5, 123.3
(CHAr), 117.9 (CAr), 117.4 (CHAr), 117.3, 112.5 (CAr), 104.5 (CHAr). (one signal could not be
detected)
MS (EI,70 eV): m/z (%) = 362 (M+, 100), 316 (12), 277 (1), 230 (4), 203 (6), 176 (8), 152 (6),
75 (4).
HRMS (EI, 70 eV): calcd for C24H14O2N2 ([M]+): 362.10498, found: 362.10398.
3-Benzyl-2-phenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6h
Yellowish solid, 82% (86.3 mg). M.p.: 199–201 °C.
IR (ATR, cm-1): 3130.8 (w), 3025.7 (w), 2962.9 (w), 2850.9 (w),
1702.9 (s), 1496.5 (m), 1427.0 (m), 1257.1 (m), 1202.5 (m), 1010.3
(m), 963.0 (m), 892.9 (m), 813.5 (m), 751.8 (s), 727.5 (s), 689.9 (s),
648.6 (m), 584.0 (m).
1H NMR (250 MHz, CDCl3) δ 7.79 (dd, 3J = 7.0, 4J = 1.0 Hz, 1H, CHAr), 7.50 – 7.35 (m, 7H,
CHAr), 7.35 – 7.15 (m, 4H, CHAr), 6.93 (dd, 3J = 7.2, 4J = 2.3 Hz, 2H, CHAr), 6.77 (s, 1H, CHAr),
5.77 (s, 2H, CH2).
13C NMR (63 MHz, CDCl3) δ 154.9 (C=O), 151.4, 145.9, 138.1, 131.2, 130.4 (CAr), 129.5 (2
CHAr), 129.1 (CHAr), 128.7 (2 CHAr), 128.5 (2 CHAr), 127.9 (CHAr), 127.3 (CHAr), 126.4
(2CHAr), 124.1, 122.9 (CHAr), 117.7 (CAr), 117.1, 103.3 (CHAr), 49.1 (CH2) (One signal could
not be detected).
MS (EI,70 eV): m/z (%) = 351 (M+, 76), 334 (4), 260 (4), 232 (7), 203 (5), 176 (6), 157 (3), 91
(100), 65 (10).
HRMS (EI, 70 eV): calcd for C24H17O2N1 ([M]+): 351.12538, found: 351.12528.

APPENDIX
xi
3-Phenethyl-2-phenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6i
Yellowish solid, 76% (83.2 mg). M.p.: 185–186 °C.
IR (ATR, cm-1): 3064.6 (w), 3025.6 (w), 2924.7 (w), 2860.8 (w),
1703.1 (s), 1603.2 (w), 1504.7 (w), 1465.5 (m), 1356.5 (m), 1198.2
(m), 1110.2 (m), 1070.7 (m), 953.6 (m), 757.1 (s), 693.8 (s), 652.7
(m).
1H NMR (300 MHz, CDCl3) δ 7.77 (dd, 3J = 7.7, 4J = 1.3 Hz, 1H, CHAr), 7.53 – 7.33 (m, 5H,
CHAr), 7.34 – 7.20 (m, 3H, CHAr), 7.21 – 7.08 (m, 3H, CHAr), 6.99 – 6.82 (m, 2H, CHAr), 6.62
(s, 1H, CHAr), 4.66 (dd, 3J = 8.2, 3J = 6.8 Hz, 2H, NCH2CH2), 3.10 – 2.96 (m, 2H, NCH2CH2).
13C NMR (63 MHz, CDCl3) δ 155.5 (C=O), 151.8, 146.2, 138.2, 131.6, 130.7 (CAr), 129.8 (2
CHAr , 129.3 (CHAr ), 129.3 (2 CHAr ), 129.0 (2 CHAr ), 128.8 (2 CHAr ), 128.3, 126.9, 124.5,
123.4 (CHAr), 118.2 (CAr), 117.5 (CHAr), 116.9 (CAr), 103.2 (CHAr), 47.9 (NCH2CH2), 38.5
(NCH2CH2).
MS (EI,70 eV): m/z (%) = 365 (M+, 41), 274 (44), 261 (100), 244 (4), 230 (16), 202 (14), 152
(3), 91 (6), 65 (3).
HRMS (EI, 70 eV): calcd for C25H19O2N1 ([M]+): 365.14103, found: 365.14097.
3-(4-Fluorobenzyl)-2-phenylchromeno[3,4-b]pyrrol-4(3H)-one 2.6j
Yellowish solid, 61% (67.5 mg). M.p.: 211–212 °C.
IR (ATR, cm-1): 3114.5 (w), 3054.4 (w), 2943.7 (w), 1703.7 (s),
1604.6 (w), 1506.2 (s), 1465.1 (s), 1381.6 (w), 1295.3 (m), 1208.2
(s), 1113.9 (s), 1040.5 (s), 949.6 (m), 894.5 (m), 765.2 (s), 698.2 (s),
651.5 (m), 538.4 (m).
1H NMR (300 MHz, CDCl3) δ 7.82 – 7.75 (m, 1H, CHAr), 7.49 – 7.43
(m, 3H, CHAr), 7.43 – 7.34 (m, 4H, CHAr), 7.33 – 7.27 (m, 1H, CHAr), 6.90 – 6.86 (m, 4H,
CHAr), 6.76 (s, 1H, CHAr), 5.73 (s, 2H, CH2).
19F NMR (282 MHz, CDCl3) δ -115.07.
13C NMR (63 MHz, CDCl3) δ 162.2 (d, 1J = 245.7 Hz, CF), 155.2 (C=O), 151.6, 146.0, 133.9
(d, 4J = 3.2 Hz), 131.2, 130.7 (CAr), 129.6 (2 CHAr ), 129.4 (CHAr ), 128.9 (2 CHAr ), 128.48 (d,
3J = 8.1 Hz, 2 CHAr ), 128.2, 124.3, 123.1 (CHAr), 117.8 (CAr), 117.3 (CHAr), 117.1 (CAr), 115.6
(d, 2J = 21.6 Hz, 2 CHAr ), 103.6 (CHAr), 48.5 (CH2).
MS (EI,70 eV): m/z (%) = 369 (M+, 70), 260 (4), 232 (6), 203 (4), 176 (5), 109 (100), 83 (7).

APPENDIX
xii
HRMS (EI, 70 eV): calcd for C24H16F1O2N1 ([M]+): 369.11596, found: 369.11563.
2-Phenyl-3-(3-(trifluoromethyl)benzyl)chromeno[3,4-b]pyrrol-4(3H)-one 2.6k
Yellow solid, 46% (57.8 mg). M.p.: 153–154 °C.
IR (ATR, cm-1): 3119.1 (w), 3067.1 (w), 2981.0 (w), 2851.0
(w), 1707.7 (s), 1504.7 (m), 1427.7 (m), 1325.6 (s), 1202.7 (m),
1118.5 (s), 1071.8 (m), 1037.2 (m), 1016.3 (m), 894.9 (m), 817.7
(m), 759.4 (s), 694.5 (s), 653.9 (s), 557.3 (m).
1H NMR (300 MHz, CDCl3) δ 7.86 – 7.70 (m, 1H, CHAr), 7.54 – 7.27 (m, 10H, CHAr), 7.17 (d,3
J = 7.8 Hz, 1H, CHAr), 7.09 (s, 1H, CHAr), 6.78 (s, 1H, CHAr), 5.81 (s, 2H, CH2).
19F NMR (282 MHz, CDCl3) δ -62.70.
13C NMR (63 MHz, CDCl3) δ 155.3 (C=O), 151.6, 146.1, 139.1, 131.0 (CAr), 130.9 (q,
2J = 32.5 Hz, C-CF3), 130.8 (CAr), 130.2 (CHAr ), 129.6 (2 CHAr ), 129.6, 129.3 (CHAr ), 129.1
(2 CHAr), 128.3 (CHAr), 124.5 (q, 3J = 8.1 Hz, CHAr), 124.4 (CHAr), 123.8 (q, 1J = 273 Hz, CF3),
123.7 (q, 3J = 7.7 Hz, CHAr), 123.2 (CHAr), 117.8 (CAr), 117.3 (CHAr), 177.1 (CAr), 103.8
(CHAr), 48.7 (CH2).
MS (EI,70 eV): m/z (%) = 419 (M+, 100), 398 (13), 274 (9), 260 (27), 232 (27), 203 (10), 176
(11), 159 (37), 109 (12), 75 (4).
HRMS (EI, 70 eV): calcd for C25H16F3O2N1 ([M]+): 419.11276, found: 419.11272.
3-(3,4-Dimethoxybenzyl)-2-(4-fluorophenyl)chromeno[3,4-b]pyrrol-4(3H)-one 2.6l
Yellowish solid, 57% (73.4 mg). M.p.: 231–231 °C.
1H NMR (300 MHz, CDCl3) δ 7.80 – 7.73 (m, 1H, CHAr),
7.45 – 7.33 (m, 4H, CHAr), 7.33 – 7.25 (m, 1H, CHAr),
7.23 – 7.09 (m, 2H, CHAr), 6.72 (s, 1H. CHHAr), 6.67 (d,
3J = 8.3 Hz, 1H, CHAr), 6.53 (d, 4J = 2.0 Hz, 1H, CHAr), 6.43
(dd,3J = 8.2, 4J = 2.0 Hz, 1H, CHAr), 5.67 (s, 2H, CH2), 3.79 (s,
3H, OCH3), 3.72 (s, 3H, OCH3).
19F NMR (282 MHz, CDCl3) δ -111.39.
13C NMR (75 MHz, CDCl3) δ 163.3 (d, 1J = 250.0 Hz, CF), 155.2 (C=O), 151.5, 149.0, 148.5,
144.8 (CAr), 131.6 (d, 3J = 8.3 Hz, 2CHAr), 130.6 (CHAr), 130.5 (CAr), 127.5 (d, 4J = 3.5 Hz,
CAr), 124.3, 123.1, 119.2 (CHAr), 117.8 (CAr), 117.2 (CHAr), 117.1 (CAr), 116.1 (d, 2J = 21.7 Hz,
2CHAr), 111.3, 110.3, 103.6 (CHAr), 55.9, 55.9 (OCH3), 48.8 (CH2).

APPENDIX
xiii
MS (EI,70 eV): m/z (%) = 429 (M+, 18), 151 (100), 107 (7), 78 (3).
HRMS (EI, 70 eV): calcd for C26H20F1O4N1 ([M]+): 429.13709, found: 429.13713.
2-(4-(Tert-butyl)phenyl)-3-(4-fluorobenzyl)chromeno[3,4-b]pyrrol-4(3H)-one 2.6m
Yellowish solid, 47% (60.0 mg). M.p.: 187–188 °C.
IR (ATR, cm-1): 3074.1 (w), 2961.9 (w), 2866.9 (w), 1720.6 (s),
1605.8 (m), 1508.8 (m), 1475.7 (m), 1358.2 (m), 1219.9 (m), 1156.6
(m), 1083.2 (m), 1032.4 (m), 970.7 (m), 765.3 (s), 652.1 (m), 575.9
(m), 526.5 (m).
1H NMR (300 MHz, CDCl3) δ 7.87 – 7.70 (m, 1H, CHAr), 7.51 – 7.43
(m, 2H, CHAr), 7.43 – 7.36 (m, 2H, CHAr), 7.36 – 7.27 (m, 3H, CHAr),
6.95 – 6.84 (m, 4H, CHAr), 6.75 (s, 1H, CHHAr), 5.73 (s, 2H, CH2), 1.37 (s, 9H, C(CH3)3).
19F NMR (282 MHz, CDCl3) δ -115.21.
13C NMR (75 MHz, CDCl3) δ 162.1 (d, 1J = 245.6 Hz, CF), 155.1 (C=O), 152.7, 151.6, 146.2,
134.1 (d, 4J = 3.2 Hz, CAr), 130.7 (CAr), 129.3 (2CHAr ), 128.4 (d, 3J = 8.1 Hz, 2CHAr ), 128.2
(CAr), 128.1 (CHAr), 125.9 (2CHAr ), 124.2, 123.1 (CHAr), 117.8 (CAr), 117.2 (CHAr), 116.9
(CAr), 115.51 (d, 2J = 21.5 Hz, 2CHAr), 103.4 (CHAr), 48.6 (CH2), 34.9 (C(CH3)3), 31.4 (3C,
C(CH3)3).
MS (EI,70 eV): m/z (%) = 425 (M+, 94), 410 (21), 301 (26), 273 (6), 109 (100), 83 (6).
HRMS (EI, 70 eV): calcd for C28H24F1O2N1 ([M]+): 425.17856, found: 425.1782.
2-(4-(Tert-butyl)phenyl)-3-phenethylchromeno[3,4-b]pyrrol-4(3H)-one 2.6n
Yellowish solid, 65% (82.1 mg). M.p.: 168–169 °C.
IR (ATR, cm-1): 3030.4 (w), 2962.0 (w), 2921.1 (w), 2847.9 (w),
1704.6 (s), 1514.5 (w), 1490.3 (m), 1452.8 (m), 1398.2 (m),
1284.0 (m), 1070.4 (m), 1014.0 (m), 942.9 (w), 764.3 (s), 700.6
(s), 659.3 (m), 576.7 (m), 532.6 (w).
1H NMR (300 MHz, CDCl3) δ 7.82 – 7.73 (m, 1H, CHAr),
7.50 – 7.33 (m, 4H, CHAr), 7.33 – 7.11 (m, 6H, CHAr), 7.02 – 6.93 (m, 2H, CHAr), 6.62 (s, 1H,
CHHAr), 4.79 – 4.59 (m, 2H, NCH2CH2), 3.12 – 2.76 (m, 2H, NCH2CH2), 1.40 (s, 9H, C(CH3)3).
13C NMR (75 MHz, CDCl3) δ 155.2 (C=O), 152.3, 151.6, 145.9, 138.0, 130.4 (CAr), 129.3
(2CHAr), 129.0 (2CHAr), 128.5 (2CHAr), 128.3 (CAr), 127.9, 126.6 (CHAr), 125.7 (2CHAr),

APPENDIX
xiv
124.1, 123.1 (CHAr), 117.9 (CAr), 117.2 (CHAr), 116.5 (CAr), 102.7 (CHAr), 47.6 (NCH2CH2),
38.3 (NCH2CH2), 34.9 (C(CH3)3), 31.4 (3C, C(CH3)3).
MS (EI,70 eV): m/z (%) = 421 (M+, 75), 317 (77), 302 (84), 274 (100), 230 (7), 202 (8), 105
(9), 77 (9), 57 (36), 41 (10).
HRMS (EI, 70 eV): calcd for C29H27O2N1 ([M]+): 421.20363, found: 421.20335.
2-(4-methoxyphenyl)-3-phenethylchromeno[3,4-b]pyrrol-4(3H)-one 2o
Yellowish solid, 69% (81.8 mg). M.p.: 194–195 °C
1H NMR (300 MHz, CDCl3) δ 7.76 (dd, 3J = 7.7, 4J = 1.2 Hz,
1H, CHAr), 7.48 – 7.33 (m, 2H, CHAr), 7.29 (dd, 3J = 7.6,
4J = 1.7 Hz, 1H, CHAr), 7.22 – 7.10 (m, 5H, CHAr), 7.01 – 6.88
(m, 4H, CHAr), 6.57 (s, 1H, CHAr), 4.84 – 4.35 (m, 2H,
NCH2CH2), 3.88 (s, 3H, OCH3), 3.11 – 2.94 (m, 2H, NCH2CH2).
13C NMR (75 MHz, CDCl3) δ 160.2 (C-OCH3), 155.2 (C=O), 151.6, 145.8, 138.0 (CAr), 130.8
(2CHAr), 130.4 (CAr), 129.1 (2CHAr), 128.6 (2CHAr), 127.9, 126.6, 124.1 (CHAr), 123.5 (CAr),
123.1 (CHAr), 117.9 (CAr), 117.0 (CHAr), 116.4 (CAr), 114.2 (2CHAr), 102.7 (CHAr), 55.5
(OCH3), 47.56 (N CH2CH2), 38.2 (N CH2CH2).
MS (EI,70 eV): m/z (%) = 395 (M+, 57), 304 (56), 291 (100), 276 (24), 217 (12), 190 (6), 91
(6), 77 (8).
HRMS (EI, 70 eV): calcd for C26H21O3N1 ([M]+): 395.15160, found: 395.15141.
3-Phenethyl-2-propylchromeno[3,4-b]pyrrol-4(3H)-one 2.6p
Yellowish oil, 62% (61.6 mg).
IR (ATR, cm-1): 3124.1 (w), 3024.2 (w), 2959.5 (w), 2873.7 (w),
1710.7 (s), 1593.8 (w), 1491.6 (m), 1434.0 (m), 1362.3 (m),
1285.2 (m), 1208.0 (m), 1153.9 (w), 1062.2 (m), 1015.2 (m),
965.1 (m), 895.0 (m), 793.0 (m), 763.1 (s), 748.8 (s), 697.5 (s),
657.1 (m), 575.2 (m), 531.7 (m).
1H NMR (300 MHz, CDCl3) δ 7.83 – 7.64 (m, 1H, CHAr), 7.43 – 7.32 (m, 2H, CHAr),
7.31 – 7.21 (m, 4H, CHAr), 7.15 – 7.08 (m, 2H, CHAr), 6.40 (s, 1H, CHAr), 4.67 – 4.50 (m, 2H),
3.21 – 3.03 (m, 2H), 2.38 – 2.24 (m, 2H), 1.74 – 1.55 (m, 2H, CH2), 0.96 (t, 3J = 7.3 Hz, 3H,
CH3).

APPENDIX
xv
13C NMR (63 MHz, CDCl3) δ 155.0 (C=O), 151.5, 146.3, 138.3, 130.2 (CAr), 129.2 (2CHAr),
128.7 (2CHAr), 127.7, 126.8, 124.0, 123.0 (CHAr), 118.1 (CAr), 117.2 (CHAr), 115.5 (CAr), 100.2
(CHAr), 47.1 (NCH2CH2), 38.2 (NCH2CH2), 28.3, 21.62 (CH2), 14.1 (CH3).
MS (EI,70 eV): m/z (%) = 331 (M+, 74), 240 (100), 227 (42), 212 (17), 198 (39), 181 (5), 115
(9), 91 (12), 77 (12), 65 (5).
HRMS (EI, 70 eV): calcd for C22H21O2N1 ([M]+): 331.15668, found: 331.15666.
2-Butyl-3-phenethylchromeno[3,4-b]pyrrol-4(3H)-one 2.6q
Yellowish oil, 65% (67.2 mg).
IR (ATR, cm-1): 3118.7 (w), 3025.9 (w), 2959.3 (w), 2864.4 (w),
1703.8 (s), 1612.7 (w), 1507.5 (w), 1477.5 (m), 1428.2 (m),
1358.6 (m), 1301.0 (w), 1202.2 (m), 1037.0 (m), 956.3 (w), 829.0
(m), 751.2 (s), 697.4 (m), 570.3 (m).
1H NMR (300 MHz, CDCl3) δ 7.73 (dd, 3J = 7.6, 4J = 1.3 Hz, 1H, CHAr), 7.43 – 7.31 (m, 2H,
CHAr), 7.31 – 7.21 (m, 4H, CHAr), 7.16 – 7.07 (m, 2H, CHAr), 6.39 (s, 1H, CHAr), 4.70 – 4.49
(m, 2H, CH2), 3.10 (t, 3J = 7.3 Hz, 2H, CH2), 2.48 – 2.22 (m, 2H, CH2), 1.73 – 1.48 (m, 2H,
CH2), 1.43 – 1.25 (m, 2H, CH2), 0.93 (t, 3J = 7.3 Hz, 3H, CH3).
13C NMR (75 MHz, CDCl3) δ 154.9 (C=O), 151.4, 146.4, 138.2, 130.1 (CAr), 129.1 (2CHAr),
128.6 (2CHAr), 127.6, 126.7, 123.9, 122.9 (CHAr), 117.9 (CAr), 117.0 (CHAr), 115.3 (CAr), 100.1
(CHAr), 46.9 (NCH2CH2), 38.1 (NCH2CH2), 30.3, 25.8, 22.5 (CH2), 13.9 (CH3).
MS (EI,70 eV): m/z (%) = 345 (M+, 57), 316 (8), 254 (20), 241 (6), 212 (100), 199 (59), 167
(9), 105 (9), 91 (12), 77 (11).
HRMS (EI, 70 eV): calcd for C23H23O2N1 ([M]+): 345.17233, found: 345.17210.
2-Butyl-3-(4-fluorobenzyl)chromeno[3,4-b]pyrrol-4(3H)-one 2.6r
Yellowish oil, 51% (53.4 mg).
IR (ATR, cm-1): 3069.6 (w), 2957.3 (w), 2926.6 (w), 2855.0 (w),
1706.5 (s), 1603.4 (w), 1495.9 (m), 1432.4 (m), 1392.2 (m), 1314.5
(w), 1221.8 (m), 1159.4 (m), 1072.8 (m), 977.5 (m), 895.2 (m), 839.8
(m), 808.7 (m), 767.3 (s), 733.5 (m), 659.8 (m), 551.5 (m).
1H NMR (300 MHz, CDCl3) δ 7.81 – 7.71 (m, 1H, CHAr), 7.43 – 7.32
(m, 2H, CHAr), 7.32 – 7.23 (m, 1H, CHAr), 7.10 – 6.81 (m, 4H, CHAr), 6.54 (s, 1H, CHHAr), 5.73

APPENDIX
xvi
(s, 2H, CH2), 2.67 – 2.55 (m, 2H, CH2), 1.74 – 1.59 (m, 2H, CH2), 1.40 (m, 2H, CH2), 0.92 (t,
3J = 7.3 Hz, 3H, CH3).
19F NMR (282 MHz, CDCl3) δ -115.08.
13C NMR (75 MHz, CDCl3) δ 162.1 (d, 1J = 244.5 Hz, CF), 155.0 (C=O), 151.3, 146.5 (CAr),
133.3 (d, 4J = 3.3 Hz, CAr), 130.2 (CAr), 128.1 (d, 3J = 7.5 Hz, 2CHAr), 127.8 (CAr), 124.0, 122.9
(CHAr), 117.8 (CAr), 117.1, 115.9 (CHAr), 115.7 (d, 2J = 21.7 Hz, 2CHAr), 101.0 (CHAr), 47.6
(NCH2), 30.3, 26.3, 22.4 (CH2), 13.8 (CH3).
MS (EI,70 eV): m/z (%) = 349 (M+, 32), 320 (11), 307 (29), 24 (7), 224 (3), 198 (10), 109
(100), 83 (8), 63 (2).
HRMS (EI, 70 eV): calcd for C22H20F1O2N1 ([M]+): 349.14726, found: 349.14690.
General procedure for synthesis of Indolo[1,2-f]phenanthridines
1-Bromo-2-(phenylethynyl)benzene 3.1 (0.3 mmol), 2-bromoaniline 3.2 (0.33 mmol),
Pd(OAc)2 (0.03 mmol, 10%), XantPhos (0.03 mmol, 10%), and Cs2CO3 (0.9 mmol) were
placed in a dried pressure tube equipped with a septum. The reaction vessel was back-filled
with argon three times. Then dried and degassed DMF (4 mL) was added under argon and the
septum was replaced with a Teflon cap. The reaction mixture was allowed to stir at 120 °C for
24 h. Then the reaction mixture was cooled to room temperature and was filtered through a pad
of Celite. The filtrate was dried under reduced pressure, and the product 3.3 was obtained after
flash chromatography on a silica gel column with heptane.
Indolo[1,2-f]phenanthridine 3.3a:
Yellowish solid, 75% (60.1 mg). M.p.: 150–151 °C.
IR (ATR, cm-1): 3054.8 (w), 1938.2 (w), 1594.9 (w), 1562.1 (w), 1550.6
(w), 1485 (m), 1452.2 (m), 1434.9 (s), 1355.8 (m), 1334.6 (s), 1251.6
(m), 1199.6 (m), 1107.0 (m), 1041.4 (w), 956.6 (w), 788.8 (w), 754.1
(m), 742.5 (s), 711.6 (m), 613.3 (m), 574.7 (m).
1H NMR (300 MHz, CDCl3) δ 8.55 (dd, 3J = 8.5, 4J = 0.8 Hz, 1H, CHAr), 8.45 – 8.35 (m, 1H,
CHAr), 8.32 (dd, 3J = 8.1, 4J = 1.4 Hz, 1H, CHAr), 8.27 – 8.18 (m, 1H, CHAr), 8.18 – 8.09 (m,
1H, CHAr), 7.90 – 7.80 (m, 1H, CHAr), 7.63 – 7.52 (m, 1H, CHAr), 7.53 – 7.44 (m, 2H, CHAr),
7.44 – 7.31 (m, 3H, CHAr), 7.26 (s, 1H, CHAr).

APPENDIX
xvii
13C NMR (63 MHz, CDCl3) δ 136.3, 135.5, 134.2, 130.6 (CAr), 129.0, 128.5, 128.1 (CHAr),
127.1, 126.4 (CAr), 124.4, 124.3, 123.3, 122.7 (CHAr), 122.4 (CAr), 122.3, 122.1, 121.3, 116.6,
114.5, 96.5 (CHAr).
MS (EI,70 eV): m/z (%) = 267 (M+, 100), 239(8), 134(11), 120(4), 106(3).
HRMS (EI, 70 eV): calcd for C20H13N1 ([M]+): 267.10425, found: 267.10431.
6-Methylindolo[1,2-f]phenanthridine 3.3b:
Yellowish solid, 66% (55.6 mg). M.p.: 170–171 °C.
IR (ATR, cm-1): 3120.4 (w), 3043.3 (w), 2914.1 (w), 1930.0 (w), 1593.0
(m), 1562.1 (m), 1550.6 (m), 1488.8 (m), 1446.4 (s), 1355.8 (s), 1332.6
(m), 1251.6 (m), 1195.7 (m), 1112.8 (m), 1043.4 (m), 958.5 (m), 788.8
(s), 754.1 (m), 736.7 (s), 713.6 (m), 615.2 (m), 578.6 (m).
1H NMR (300 MHz, CD2Cl2) δ 8.46 (d, 3J = 8.6 Hz, 1H, CHAr), 8.38 (d, 3J = Hz, 1H, CHAr),
8.32 – 8.24 (m, 1H, CHAr), 8.22 – 8.13 (m, 2H, CHAr), 7.88 – 7.79 (m, 1H, CHAr), 7.57 – 7.48
(m, 2H, CHAr), 7.47 – 7.42 (m, 1H, CHAr), 7.42 – 7.30 (m, 2H, CHAr), 7.29 (d, 4J = 0.7 Hz, 1H,
CHAr), 2.53 (s, 3H, CH3).
13C NMR (63 MHz, CD2Cl2) δ 135.7, 134.4, 134.4, 133.3, 130.8 (CAr), 130.3, 128.8, 128.5
(CHAr), 127.5, 126.7 (CAr), 124.9, 124.7, 123.0, 122.5 (CHAr), 122.5 (CAr), 122.2, 121.5, 116.7,
114.7, 96.4 (CHAr), 21.4 (CH3).
MS (EI,70 eV): m/z (%) = 281 (M+, 100), 252(3), 139(12).
HRMS (+ESI): calcd for C21H16N1 ([M+H]+): 282.12773, found: 282.12775.
3-Methylindolo[1,2-f]phenanthridine 3.3c:
Yellowish solid, 52% (43.8 mg). M.p.: 135–136 °C.
IR (ATR, cm-1): 3114.6 (w), 3043.3 (w), 2912.1 (w), 2854.3 (w),
1907.3 (m), 1598.8 (m), 1564.1 (w), 1556.3 (w), 1494.6 (w), 1440.6
(s), 1351.9 (s), 1340.4 (m), 1253.6 (m), 1199.6 (w), 1112.8 (w),
1035.6 (m), 958.5 (m), 781.1 (m), 757.9 (s), 740.6 (s), 713.6 (s), 615.2 (m), 578.6 (m), 532.3
(s).
1H NMR (300 MHz, CDCl3) δ 8.56 (dd, 3J = 8.5, 4J = 0.9 Hz, 1H, CHAr), 8.43 – 8.30 (m, 2H,
CHAr), 8.08 – 8.01 (m, 2H, CHAr), 7.88 – 7.80 (m, 1H, CHAr), 7.64 – 7.53 (m, 1H, CHAr),
7.44 – 7.30 (m, 4H, CHAr), 7.22 (s, 1H, CHAr), 2.53 (s, 3H, CH3).

APPENDIX
xviii
13C NMR (63 MHz, CDCl3) δ 137.8, 136.3, 135.7, 134.0, 130.7 (CAr), 129.7, 128.8 (CHAr),
127.0 (CAr), 124.3, 124.1 (CHAr), 123.9 (CAr), 123.1, 122.7 (CHAr), 122.3 (CAr), 121.9, 121.9,
121.1, 116.5, 114.3, 95.6 (CHAr), 22.0 (CH3).
MS (EI,70 eV): m/z (%) = 281 (M+, 100), 252 (3), 139 (10), 126 (4).
HRMS (EI, 70 eV): calcd for C21H15N1 ([M]+): 281.11990, found: 281.12033.
3,6-Dimethylindolo[1,2-f]phenanthridine 3.3d:
Yellowish solid, 45% (39.8 mg). M.p.: 185–186 °C.
IR (ATR, cm-1): 3106.9 (w), 3054.8 (w), 2912.1 (w), 2850.4 (w),
2725.1 (w), 1917.0 (w), 1729.9 (w), 1598.8 (m), 1562.1 (m), 1498.5
(w), 1444.5 (s), 1350.0 (s), 1251.6 (m), 1201.5 (s), 1112.8 (m),
1033.7 (m), 960.4 (m), 867.9 (m), 823.5 (s), 784.9 (s), 756.0 (s),
736.7 (s), 713.6 (s), 655.7 (s), 613.3 (m), 574.7 (m), 538.1 (s).
1H NMR (250 MHz, CDCl3) δ 8.40 (d, 3J = 8.6 Hz, 1H, CHAr), 8.37 – 8.29 (m, 1H, CHAr), 8.07
(d, 4J = 1.2 Hz, 1H, CHAr), 8.04 – 7.95 (m, 2H, CHAr), 7.87 – 7.77 (m, 1H, CHAr), 7.42 – 7.24
(m, 4H, CHAr), 7.18 (s, 1H, CHAr), 2.51 (s, 3H, CH3), 2.50 (s, 3H, CH3).
13C NMR (63 MHz, CDCl3) δ 137.7, 135.6, 134.1, 133.8, 132.4, 130.5 (CAr), 129.5, 129.5
(CHAr), 126.9 (CAr), 124.3, 124.3 (CHAr), 123.9 (CAr), 122.6 (CHAr), 122.1 (CAr), 121.7, 121.6,
120.9, 116.3, 114.2, 95.3 (CHAr), 22.0 (CH3), 21.3 (CH3).
MS (EI,70 eV): m/z (%) = 295 (M+, 100), 278 (13), 139 (9).
HRMS (EI, 70 eV): calcd for C22H17N1 ([M]+): 295.13555, found: 295.13567.
6-Fluoro-3-methylindolo[1,2-f]phenanthridine 3.3e:
Yellowish solid, 55% (49.3 mg). M.p.: 165–166°C.
IR (ATR, cm -1): 3106.9 (w), 3049.1 (w), 2917.9 (w), 2856.2 (w),
2736.6 (w), 1917.0 (w), 1729.9 (w), 1606.5 (w), 1567.9 (m), 1496.6
(m), 1448.4 (m), 1429.1 (s), 1353.9 (m), 1276.7 (m), 1249.7 (m),
1203.4 (m), 1172.6 (s), 1116.6 (m), 1068.4 (m), 1041.4 (m), 960.4
(m), 948.9 (m), 867.9 (s), 823.5 (s), 788.8 (s), 754.1 (m), 734.8 (s), 713.6 (m), 655.7 (m), 611.4
(m), 572.8 (m), 536.1 (s).
1H NMR (300 MHz, CDCl3) δ 8.42 (dd, 3J = 9.2, 4J = 4.9 Hz, 1H, CHAr), 8.26 (dd, 3J = 6.5,
4J = 2.6 Hz, 1H, CHAr), 7.96 (d, 3J = 8.1 Hz, 1H, CHAr), 7.89 (dd, 3J = 10.2, 4J = 2.9 Hz, 1H,

APPENDIX
xix
CHAr), 7.86 – 7.73 (m, 2H, CHAr), 7.43 – 7.27 (m, 3H, CHAr), 7.27 – 7.10 (m, 1H, CHAr), 2.49
(s, 3H, CH3) (signal of one H could not be detected).
19F NMR (282 MHz, CDCl3) δ -119.57.
13C NMR (75 MHz, CDCl3) δ 158.7 (d, 1J = 241.8 Hz, CF), 137.9, 135.2, 133.7 (CAr), 132.6 (d,
4J = 2.2 Hz, CAr), 130.4 (CAr), 130.3 (CHAr), 126.1 (d, 4J = 2.5 Hz, CAr), 124.3 (CHAr), 124.2
(d, 3J = 7.6 Hz, CAr), 124.1 (CAr), 122.8, 122.1, 121.9, 121.2 (CHAr), 117.7 (d, 4J = 8.2 Hz),
115.5 (d, 2J = 23.0 Hz, CHAr), 113.8 (CHAr), 110.2 (d, 2J = 23.8 Hz, CHAr), 95.7 (CHAr), 21.9
(CH3).
MS (EI,70 eV): m/z (%) = 299 (M+, 100), 270 (3), 148 (6).
HRMS (EI, 70 eV): calcd for C21H14N1F1 ([M]+): 299.11048, found: 299.11053.
3-(Tert-butyl)indolo[1,2-f]phenanthridine 3.3f:
Yellowish solid, 65% (63.0 mg). M.p.: 151–152 °C.
IR (ATR, cm-1): 3122.3 (w), 3043.3 (w), 2950.7 (w), 2863.9 (w),
2742.4 (w), 2331.6 (w), 1917.0 (w), 1731.8 (w), 1606.5 (w),
1562.1 (w), 1490.8 (w), 1448.4 (s), 1440.6 (s), 1417.5 (s), 1348.1
(s), 1276.7 (m), 1259.4 (m), 1197.6 (m), 1172.6 (m), 1112.8 (m), 1072.3 (m), 1053.0 (m),
1020.2 (m), 958.5 (w), 875.6 (m), 831.2 (m), 788.8 (m), 754.1 (s), 734.8 (s), 723.2 (m), 661.5
(m), 611.4 (m), 588.2 (m), 541.9 (m).
1H NMR (300 MHz, CDCl3) δ 8.54 (d, 3J = 8.4 Hz, 1H, CHAr), 8.41 – 8.31 (m, 2H, CHAr), 8.25
(d, 4J = 1.7 Hz, 1H, CHAr), 8.06 (d, 3J = 8.4 Hz, 1H, CHAr), 7.86 – 7.76 (m, 1H, CHAr),
7.61 – 7.47 (m, 2H, CHAr), 7.41 – 7.28 (m, 3H, CHAr), 1.45 (s, 9H, C(CH3)3). (signal of one H
could not be detected).
13C NMR (63 MHz, CDCl3) δ 136.2, 135.4, 133.9, 130.6, 130.5 (CAr), 128.6 (CHAr), 126.5
(CAr), 126.2, 124.1, 123.9 (CHAr), 123.8 (CAr), 123.0 (CHAr), 122.5 (CAr), 121.8, 121.8, 120.9,
118.6, 116.5, 114.3, 95.7 (CHAr), 35.2 (C(CH3)3), 31.4 (3C, C(CH3)3).
MS (EI,70 eV): m/z (%) = 323 (M+,100), 308 (70), 293 (34), 267 (13), 140 (23).
HRMS (EI, 70 eV): calcd for C24H21N1 ([M]+): 323.16685, found: 323.16687.
3-(Tert-butyl)-6-methylindolo[1,2-f]phenanthridine 3.3g:

APPENDIX
xx
Yellowish solid, 67% (67.7 mg). M.p.: 195–196 °C.
IR (ATR, cm-1): 3124.3 (w), 3049.1 (w), 2950.7 (w), 2863.9 (w),
2732.8 (w), 2331.6 (w), 1913.1 (w), 1731.8, 1606.5 (w), 1564.1
(m), 1488.8 (m), 1446.4 (s), 1421.4 (s), 1351.9 (s), 1280.6 (m),
1261.3 (m), 1195.7 (m), 1114.7 (m), 1066.5 (m), 1045.3 (w),
1024.1 (w), 788.8 (s), 757.9 (s), 736.7 (m), 611.4 (m), 588.2 (m), 555.4 (s).
1H NMR (300 MHz, CD2Cl2) δ 8.46 (d, 3J = 8.5 Hz, 1H, CHAr), 8.38 (d, 3J = 8.2 Hz, 1H, CHAr),
8.30 (d, 4J = 1.7 Hz, 1H, CHAr), 8.22 (s, 1H, CHAr), 8.11 (d, 3J = 8.4 Hz, 1H, CHAr), 7.83 (dd,
3J = 6.8, 4J = 1.9 Hz, 1H, CHAr), 7.61 (dd, 3J = 8.4, 4J = 1.8 Hz, 1H, CHAr), 7.49 – 7.28 (m, 3H,
CHAr), 7.24 (s, 1H, CHAr), 2.55 (s, 3H, CH3), 1.49 (s, 9H, C(CH3)3).
13C NMR (63 MHz, CD2Cl2) δ 151.6, 135.9, 134.5, 134.3, 133.2, 130.9 (CAr), 130.1 (CHAr),
127.0 (CAr), 126.7, 124.7, 124.5 (CHAr), 124.2, 122.8 (CAr), 122.2, 122.1, 121.4, 119.3, 116.8,
114.7, 95.8 (CHAr), 35.6 (C-(CH3)3), 31.7 (3C, C-(CH3)3), 21.4 (CH3).
MS (EI,70 eV): m/z (%) = 337 (M+, 100), 322 (59), 307 (32), 278 (10), 161 (5), 147 (17).
HRMS (EI, 70 eV): calcd for C25H23N1 ([M]+): 337.1825, found: 337.18270.
3-Fluoroindolo[1,2-f]phenanthridine 3.3h:
Yellowish solid, 77% (65.8 mg). M.p.: 129–130 °C.
IR (ATR, cm-1): 3054.8 (w), 2952.6 (w), 2850.4 (w), 2734.7 (w),
2331.6 (w), 1928.6 (w), 1884.2 (w), 1731.8 (w), 1602.6 (m), 1558.3
(m), 1490.8 (m), 1440.6 (s), 1350.0 (s), 1280.6 (m), 1272.9 (m),
1195.7 (m), 1120.5 (m), 887.1 (w), 835.1 (m), 777.2 (m), 754.1 (m), 736.7 (s), 729.0 (s), 651.9
(m), 597.9 (m), 555.4 (m), 534.2 (m).
1H NMR (300 MHz, CDCl3) δ 8.42 – 8.31 (m, 1H, CHAr), 8.26 – 8.17 (m, 1H, CHAr), 8.00 (dd,
3J = 8.1, 4J = 1.3 Hz, 1H, CHAr), 7.90 (dd, 3J = 8.8, 4J = 5.7 Hz, 1H, CHAr), 7.75 – 7.62 (m, 2H,
CHAr), 7.51 – 7.39 (m, 1H, CHAr), 7.33 – 7.13 (m, 3H, CHAr), 7.12 – 7.01 (m, 1H, CHAr), 7.00
(s, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -112.61.
13C NMR (63 MHz, CDCl3) δ 162.5 (d, 1J = 246.4 Hz, CF), 136.2, 134.6, 133.7, 130.3 (CAr),
129.4 (CHAr), 128.9 (d, 3J = 8.2 Hz, CAr), 126.2 (d, 3J = 8.7 Hz, CHAr), 124.1, 123.0 (CHAr),
122.5 (d, 4J = 2.5 Hz, CAr), 122.0, 121.89 (CHAr), 121.2 (d, 4J = 3.1 Hz, CAr), 120.9, 116.3

APPENDIX
xxi
(CHAr), 116.1 (d, 2J = 23.0 Hz, CHAr), 114.2 (CHAr), 108.4 (d, 2J = 23.2 Hz), 95.8 (d,
6J = 1.5 Hz, CHAr).
MS (EI,70 eV): m/z (%) = 285 (M+, 100), 257 (7), 143 (10).
HRMS (EI, 70 eV): calcd for C20H12N1F1 ([M]+): 285.09483, found: 285.09458.
3-Fluoro-6-methylindolo[1,2-f]phenanthridine 3.3i:
Yellowish solid, 54% (48.4 mg). M.p.: 203–204 °C.
IR (ATR, cm-1): 3108.8 (w), 3051.0 (w), 2917.9 (w), 2850.4 (w),
2732.8 (w), 2325.8 (w), 1928.6 (w), 1895.8 (w), 1731.8 (w), 1614.2
(m), 1554.4 (m), 1486.9 (m), 1438.7 (m), 1350.0 (m), 1280.6 (m),
1274.8(m), 1189.9, 1120.5, 1076.1, 1041.4, 1024.1, 960.4, 900.6,
844.7(m), 784.9 (s), 736.7 (s), 653.8 (m), 632.6 (m), 615.2 (m), 607.5 (m), 574.7(m), 530.4 (m).
1H NMR (300 MHz, CDCl3) δ 8.40 – 8.21 (m, 2H, CHAr), 8.01 (dd, 3J = 8.8, 4J = 5.7 Hz, 1H,
CHAr), 7.87 (s, 1H, CHAr), 7.84 – 7.64 (m, 2H, CHAr), 7.43 – 7.28 (m, 3H, CHAr), 7.21 – 6.99
(m, 2H, CHAr), 2.46 (s, 3H, CH3).
19F NMR (282 MHz, CDCl3) δ -112.82.
13C NMR (75 MHz, CDCl3) δ 162.6 (d, 1J = 246.2 Hz, CF), 134.6, 134.2, 133.8, 132.6 (CAr),
130.4 (CHAr), 129.0 (d, 3J = 8.2 Hz, CAr), 126.3 (d, 3J = 8.7 Hz, CHAr), 124.5 (CHAr), 122.7 (d,
4J = 2.4 Hz, CAr), 122.0, 121.8 (CHAr), 121.2 (d, 4J = 3.0 Hz, CAr), 121.1, 116.3 (CHAr), 116.1
(d, 2J = 22.7 Hz, CHAr) (one signal of the doublet is overlapped with the signal at 116.3), 114.2
(CHAr), 108.5 (d, 2J = 23.3 Hz, CHAr), 95.6 (CHAr), 21.2 (CH3) (signal of one CAr could not be
detected).
MS (EI,70 eV): m/z (%) = 299 (M+, 100), 149 (8).
HRMS (EI, 70 eV): calcd for C21H14N1F1 ([M]+): 299.11048, found: 299.10984.
3-Methoxyindolo[1,2-f]phenanthridine 3.3j:
Yellowish solid, 78% (69.5 mg). M.p.: 143–144 °C.
IR (ATR, cm-1): 3108.8 (w), 3043.3 (w), 2919.8 (w), 2850.4 (w),
2732.8 (w), 2323.9 (w), 2057.8 (w), 1918.9 (w), 1891.9 (w),
1731.8 (w), 1610.3 (m), 1558.3 (m), 1492.7 (s), 1427.1 (s), 1348.1
(m), 1286.4 (m), 1199.6 (m), 1120.5 (m), 1078.1 (m), 1037.6 (m), 1024.1 (m), 958.5 (m), 910.3
(m), 835.1 (m), 777.2 (m), 740.6 (s), 653.8 (m), 638.4 (m), 607.5 (m), 565.1 (m), 538.1.

APPENDIX
xxii
1H NMR (300 MHz, CDCl3) δ 8.49 (dd, 3J = 8.5, 4J = 0.9 Hz, 1H, CHAr), 8.37 – 8.28 (m, 1H,
CHAr), 8.18 (dd, 3J = 8.1, 4J = 1.4 Hz, 1H, CHAr), 7.97 (d, 3J = 8.8 Hz, 1H, CHAr), 7.84 – 7.73
(m, 1H, CHAr), 7.60 – 7.49 (m, 2H, CHAr), 7.41 – 7.27 (m, 3H, CHAr), 7.03 (dd, 3J = 8.8,
4J = 2.5 Hz, 1H, CHAr), 3.92 (s, 3H, OCH3). (Signal of one H could not be detected).
13C NMR (63 MHz, CDCl3) δ 159.5 (C-OCH3), 136.3, 135.6, 133.8, 130.8 (CAr), 128.9 (CHAr),
128.4 (CAr), 125.8, 124.0, 122.9 (CHAr), 121.9 (CAr), 121.8, 121.6, 120.8 (CHAr), 119.8 (CAr),
116.4, 116.1, 114.2, 105.8, 94.7 (CHAr), 55.5 (OCH3).
MS (EI, 70 eV): m/z (%) = 297 (M+, 100), 282 (19), 254 (56), 226 (4), 149 (12), 126 (12).
HRMS (EI, 70 eV): calcd for C21H15N1O1 ([M]+): 297.11482, found: 297.11474.
3-Methoxy-6-methylindolo[1,2-f]phenanthridine 3.3k:
Yellowish solid, 73% (68.1 mg). M.p.: 188–189 °C.
IR (ATR, cm-1): 3108.8 (w), 3045.2 (w), 2910.2 (w), 2840.8 (w),
2725.1 (w), 2325.8 (w), 2055.8 (w), 1918.9 (w), 1888.1 (w),
1731.8 (w), 1608.4 (m), 1556.3 (m), 1488.8 (m), 1431.0 (m),
1350.0 (m), 1286.4 (m), 1201.5 (m), 1130.1 (m), 1072.3 (m),
1037.6 (m), 1024.1 (m), 958.5 (m), 919.9 (m), 838.9 (m), 781.1 (s), 736.7 (s), 655.7 (m), 609.4
(m), 565.1 (m), 547.7 (s).
1H NMR (300 MHz, CDCl3) δ 8.41 – 8.21 (m, 2H), 8.06 – 7.91 (m, 2H), 7.86 – 7.72 (m, 1H),
7.57 (d, 4J = 2.3 Hz, 1H), 7.43 – 7.28 (m, 3H), 7.04 (dd, 3J = 8.8, 4J = 2.4 Hz, 2H), 3.94 (s, 3H,
OCH3), 2.47 (s, 3H, CH3).
13C NMR (63 MHz, CDCl3) δ 159.5 (C-OCH3), 135.5, 134.2, 133.7, 132.3, 130.6 (CAr), 129.8
(CHAr), 128.4 (CAr), 125.9, 124.3 (CHAr), 121.8 (CAr), 121.6, 121.4, 120.7 (CHAr), 119.9 (CAr),
116.3, 116.1, 114.2, 105.7, 94.4 (CHAr), 55.6 (OCH3), 21.2 (CH3).
MS (EI, 70 eV): m/z (%) = 311 (M+, 100), 296 (13), 268 (42), 156 (10), 133 (10).
HRMS (EI, 70 eV): calcd for C22H17N1O1 ([M]+): 311.13047, found: 311.13016.
For compounds 3.5c, 3.5d, 3.5h, 3.5i, 3.6j, 3.5g, 3.5f, 20% of PCy3·HBF4 was used instead of
XantPhos.
6-(Tert-butyl)indolo[1,2-f]phenanthridine 3.5c:

APPENDIX
xxiii
Yellowish solid, 43% (41.7 mg). M.p.: 146–168 °C.
IR (ATR, cm-1): 3065.8 (w), 3043.2 (w), 2955.5 (w), 2898.7 (w), 2859.4
(w), 1953.0 (w), 1921.3 (w), 1881.4 (w), 1593.0 (m), 1552.4 (m), 1487.5
(m), 1448.4 (s), 1408.1 (w), 1357.5 (m), 1255.0 (m), 1228.8 (m), 1206.2
(m), 1120.3 (m), 1019.2 (m), 945.7 (m), 877.9 (m), 802.3 (m), 755.3 (s),
732.5 (s), 624.4 (m), 526.5 (m).
1H NMR (250 MHz, Acetone) δ 8.62 (d, 3J = 8.8 Hz, 1H, CHAr), 8.57 – 8.47 (m, 3H, CHAr),
8.33 – 8.24 (m, 1H, CHAr), 7.84 (dd, 3J = 7.1, 4J = 1.5 Hz, 1H, CHAr), 7.76 (dd, 3J = 8.8,
4J = 2.3 Hz, 1H, CHAr), 7.64 – 7.49 (m, 2H, CHAr), 7.48 – 7.25 (m, 3H, CHAr), 1.48 (s, 9H,
C(CH3)3).
13C NMR (63 MHz, Acetone) δ 147.0, 135.9, 134.8, 134.7, 131.4 (CAr), 129.3, 129.1 (CHAr),
128.1 (CAr), 127.5 (CHAr), 127.1 (CAr), 125.3, 123.8, 123.1, 122.7 (CHAr), 122.5 (CAr), 122.0,
121.8, 117.1, 115.2, 97.2 (CHAr), 35.4 (3C, C(CH3)3), 31.8 (C(CH3)3).
MS (EI, 70 eV): m/z (%) = 323 (M+, 100), 308 (90), 293 (28), 267 (16), 239 (3), 140 (22).
HRMS (EI, 70 eV): calcd for C24H21N1 ([M]+): 323.16685, found: 323.16686.
6-Methoxyindolo[1,2-f]phenanthridine 3.5d:
Yellowish solid, 49% (43.7 mg). M.p.: 154–155 °C.
IR (ATR, cm-1): 3105.0 (w), 3043.3 (w), 2910.2 (w), 2836.9 (w), 2325.8
(w), 2053.9 (w), 1917.0 (w), 1890.0 (w), 1731.8 (w), 1620.0 (w), 1562.1
(m), 1488.8 (m), 1438.7 (m), 1357.7(m), 1288.3 (m), 1197.6 (m), 1130.1
(m), 1070.4 (m), 1041.4 (m), 1026.0 (m), 958.5 (m), 919.9 (m), 838.9
(m), 790.7 (S), 734.8 (S), 655.7 (m), 607.5 (m), 563.1(m).
1H NMR (250 MHz, CD2Cl2) δ 8.38 (d, 3J = 9.2 Hz, 1H, CHAr), 8.23 (d, 3J = 8.4 Hz, 1H, CHAr),
8.15 (dd, 3J = 6.2, 4J = 3.1 Hz, 1H, CHAr), 8.10 – 7.93 (m, 1H, CHAr), 7.74 (d, 4J = 2.8 Hz, 1H,
CHAr), 7.70 – 7.57 (m, 1H, CHAr), 7.46 – 7.26 (m, 2H, CHAr), 7.27 – 6.95 (m, 4H, CHAr), 3.80
(s, 3H, OCH3).
13C NMR (63 MHz, CD2Cl2) δ 156.2 (C-OCH3), 135.3, 134.1, 130.7, 130.6 (CAr), 129.0, 128.4
(CHAr), 127.2, 126.8 (CAr), 124.7 (CHAr), 123.8 (CAr), 123.3, 122.5, 121.9, 121.5, 118.0, 115.8,
114.4, 108.9, 96.3 (CHAr), 55.7 (OCH3).
MS (EI,70 eV): m/z (%) = 297 (M+, 100), 282 (25), 254 (53), 226 (5), 148 (11), 127 (14).
HRMS (EI, 70 eV): calcd for C21H15N1O1 ([M]+): 297.13047, found: 297.13000.

APPENDIX
xxiv
6-Fluoroindolo[1,2-f]phenanthridine 3.5h:
Yellowish solid, 75% (64.1 mg). M.p.: 169–170 °C.
IR (ATR, cm-1): 3049.1 (w), 2140.7 (w), 1918.9 (w), 1729.9 (w), 1621.9
(w), 1567.9 (m), 1488.8 (m), 1448.4 (s), 1357.7 (m), 1276.7 (m), 1247.8
(m), 1197.6 (m), 1180.3 (s), 1139.8 (m), 1066.5 (m), 1041.4 (m), 1024.1
(m), 960.4 (m), 921.9 (m), 838.9 (m), 796.5 (s), 734.8 (s), 659.6 (m),
611.4 (m), 565.1(m).
1H NMR (300 MHz, CD2Cl2) δ 8.39 (dd, 3J = 9.2, 4J = 4.9 Hz, 1H, CHAr), 8.29 – 8.18 (m, 1H,
CHAr), 8.13 – 7.97 (m, 2H, CHAr), 7.89 (dd, 3J = 10.3, 4J = 2.9 Hz, 1H, CHAr), 7.85 – 7.75 (m,
1H, CHAr), 7.54 – 7.41 (m, 2H, CHAr), 7.41 – 7.30 (m, 2H, CHAr), 7.30 – 7.20 (m, 1H, CHAr),
7.19 (s, 1H, CHAr).
19F NMR (282 MHz, CD2Cl2) δ -120.10.
13C NMR (63 MHz, CD2Cl2) δ 159.2 (d, 1J = 241.2 Hz, CF), 135.3, 134.2, 132.9 (d,
4J = 2.2 Hz), 130.7 (CAr), 129.4, 128.5 (CHAr), 126.8, 126.5 (d, 4J = 2.5 Hz, CAr), 124.6 (CHAr),
124.5 (d, 3J = 7.7 Hz, CAr), 123.1, 122.8, 122.4, 121.7 (CHAr), 118.2 (d, 3J = 8.2 Hz, CHAr),
116.0 (d, 2J = 23.1 Hz, CHAr), 114.3 (CHAr), 110.6 (d, 2J = 23.9 Hz, CHAr), 96.8 (CHAr).
MS (EI, 70 eV): m/z (%) = 285 (M+, 100), 257 (8), 143 (10), 128 (4).
HRMS (EI, 70 eV): calcd for C20H12N1F1 ([M]+): 285.09483, found: 285.09477.
Indolo[1,2-f]phenanthridine-6-carbonitrile 3.5i:
Yellowish solid, 44% (38.5 mg). M.p.: 217–218 °C.
IR (ATR, cm -1): 3068.3 (w), 2221.7 (m), 1593.0 (m), 1556.3 (m), 1490.8
(m), 1446.4 (s), 1409.8 (m), 1353.9 (m), 1288.3 (m), 1251.6 (m), 1207.3
(m), 1182.2 (w), 1145.6 (w), 1068.4 (w), 1045.3 (w), 1024.1 (w), 958.5
(w), 919.9 (w), 877.5 (m), 833.1 (m), 798.4 (s), 734.8 (s), 657.6 (m),
611.4 (m), 565.1 (m).
1H NMR (300 MHz, CD2Cl2) δ 8.45 (d, 4J = 1.8 Hz, 1H, CHAr), 8.42 (d, 3J = 8.8 Hz, 1H, CHAr),
8.20 (dd, 3J = 6.4, 4J = 2.5 Hz, 1H, CHAr), 8.14 – 7.97 (m, 2H, CHAr), 7.85 – 7.77 (m, 1H,
CHAr), 7.73 (dd, 3J = 8.8, 4J = 1.9 Hz, 1H, CHAr), 7.59 – 7.45 (m, 2H, CHAr), 7.45 – 7.32 (m,
2H, CHAr), 7.22 (s, 1H, CHAr).

APPENDIX
xxv
13C NMR (63 MHz, CD2Cl2) δ 138.9, 135.4, 134.5 (CAr), 132.3 (CHAr), 131.2 (CAr), 129.9,
128.9, 128.8 (CHAr), 126.7, 125.5 (CAr), 124.7, 123.5, 123.3 (CHAr), 123.2 (CAr), 122.9, 121.9
(CHAr), 119.4 (CN), 117.2, 114.8 (CHAr), 106.9 (CAr), 98.4 (CHAr).
MS (EI,70 eV): m/z (%) = 292 (M+, 100), 264 (10), 146 (8), 132 (9), 118 (3).
HRMS (EI, 70 eV): calcd for C21H12N2 ([M]+): 292.0995, found: 292.09960.
6-(Methylthio)indolo[1,2-f]phenanthridine 3.5j:
Yellowish solid, 56% (52.6 mg). M.p.: 143–144 °C.
IR (ATR, cm-1): 3043.3 (w), 2917.9 (w), 1915.1 (w), 1591.1 (w), 1546.7
(m), 1490.8 (m), 1448.4 (s), 1398.2 (m), 1353.9 (m), 1288.3 (m),
1253.6(m), 1203.4 (w), 1188.0 (m), 1164.9 (m), 1114.7 (m), 1024.1 (w),
954.6 (m), 916.1 (w), 864.0 (w), 790.7 (s), 734.8 (s), 613.3 (m), 582.4
(m).
1H NMR (250 MHz, CD2Cl2) δ 8.38 (d, 3J = 8.8 Hz, 1H, CHAr), 8.24 (d, 3J = 8.2 Hz, 1H, CHAr),
8.20 – 8.09 (m, 2H, CHAr), 8.09 – 7.98 (m, 1H, CHAr), 7.79 – 7.63 (m, 1H, CHAr), 7.50 – 7.33
(m, 3H, CHAr), 7.33 – 7.20 (m, 2H, CHAr), 7.18 (s, 1H, CHAr), 2.51 (s, 3H, SCH3).
13C NMR (63 MHz, CD2Cl2) δ 135.3, 134.2, 134.2, 133.3, 130.8 (CAr), 129.0, 128.4, 128.3
(CHAr), 126.7, 126.7 (CAr), 124.6 (CHAr), 123.2 (CAr), 123.1, 123.0, 122.6, 122.2, 121.5, 117.4,
114.6, 96.8 (CHAr), 16.7 (SCH3).
MS (EI,70 eV): m/z (%) = 313 (M+, 100), 298 (47), 265 (12), 254 (25), 156 (9), 132 (5).
HRMS (EI, 70 eV): calcd for C21H15N1S1 ([M]+): 313.09197, found: 313.09196.
8-Fluoroindolo[1,2-f]phenanthridine 3.5g:
Yellowish solid, 72% (61.6 mg). M.p.: 115–116 °C.
IR (ATR, cm-1): 3041.3 (w), 2923.7 (w), 1901.6 (w), 1604.6 (w), 1552.5
(w), 1494.6 (w), 1475.3 (m), 1442.6 (s), 1436.8 (m), 1346.1 (m), 1288.3
(m), 1251.6 (m), 1213.1 (m), 1184.1 (m), 1134.0 (m), 1074.2 (w), 1018.3
(w), 954.6 (w), 912.2 (m), 781.1 (m), 759.9 (s), 736.7 (s), 551.6 (s).
1H NMR (300 MHz, CDCl3) δ 8.23 – 8.02 (m, 3H, CHAr), 8.02 – 7.87 (m, 1H, CHAr),
7.85 – 7.74 (m, 1H, CHAr), 7.59 – 7.41 (m, 2H, CHAr), 7.41 – 7.27 (m, 5H, CHAr).
19F NMR (282 MHz, CDCl3) δ -111.04.

APPENDIX
xxvi
13C NMR (75 MHz, CDCl3) δ 152.3 (d, 1J = 249.6 Hz, CF), 135.6 (d, 4J = 1.2 Hz), 135.5, 130.1
(CAr), 129.0, 127.9 (CHAr), 126.8, 126.5 (d, 4J = 2.7 Hz), 126.2 (d, 4J = 3.0 Hz, CAr), 124.0
(CHAr), 123.9 (d, 3J = 8.5 Hz, CHAr), 123.2 (d, 3J = 11.1 Hz, CAr), 123.0, 122.0 (CHAr), 121.7
(d, 4J = 4.1 Hz, CHAr), 120.5 (CHAr), 119.5 (d, 4J = 2.9 Hz, CHAr), 116.0 (d, 2J = 22.3 Hz,
CHAr), 115.8 (d, 2J = 26.6 Hz, CHAr), 98.0 (CHAr).
MS (EI, 70 eV): m/z (%) = 285 (M+, 100), 264 (12), 142 (9).
HRMS (EI, 70 eV): calcd for C20H12N1F1 ([M]+): 285.09483, found: 285.09470.
8-Methoxyindolo[1,2-f]phenanthridine 3.5f:
Yellowish oil, 49% (43.7 mg).
1H NMR (250 MHz, Acetone) δ 8.39 – 8.31 (m, 1H, CHAr), 8.25 (dd,
3J = 6.4, 4J = 2.6 Hz, 1H, CHAr), 8.05 (d, 3J = 7.9 Hz, 1H, CHAr),
7.80 – 7.68 (m, 2H, CHAr), 7.56 – 7.48 (m, 2H, CHAr), 7.40 (m, 2H,
CHAr), 7.35 – 7.21 (m, 3H, CHAr), 3.95 (d, 3J = 5.1 Hz, 3H).
13C NMR (63 MHz, Acetone) δ 150.7 (C-OCH3), 137.2, 136.6, 130.8 (CAr), 129.6, 128.8
(CHAr), 127.9, 127.6, 126.2 (CAr), 125.6 (CHAr), 125.0 (CAr), 124.7, 124.1, 122.0, 121.4, 120.9,
118.3, 116.9, 112.8, 98.3 (CHAr), 55.8 (OCH3).
MS (EI,70 eV): m/z (%) = 297 (M+,100), 282 (56), 252 (13), 141 (12), 126 (10), 113 (4).
HRMS (EI, 70 eV): calcd for C21H15N1O1 ([M]+): 297.11482, found: 297.11451.
1-(2-Bromophenyl)-2-(p-tolyl)-1H-indole 3.1ii
1H NMR (300 MHz, CDCl3) δ 7.66 – 7.56 (m, 2H, CHAr),
7.30 – 7.03 (m, 7H, CHAr), 6.95 (d, 3J = 8.0 Hz, 2H, CHAr),
6.90 – 6.83 (m, 1H, CHAr), 6.71 (d, 4J = 0.7 Hz, 1H, CHAr), 2.20 (s,
3H, CH3).
13C NMR (75 MHz, CDCl3) δ 141.5, 138.9, 138.4, 137.4 (CAr), 133.8, 131.5, 129.8 (CHAr),
129.7 (CAr), 129.1 (2 CHAr ), 128.5 (CAr), 128.5 (2CHAr ), 128.5 (CHAr), 124.2 (CAr), 122.3,
120.9, 120.6, 111.0, 103.1 (CHAr), 21.3 (CH3).
MS (EI, 70 eV): m/z (%) = 361 (M+,100), 281 (51), 267 (74), 190 (5), 165 (6), 133 (50).
HRMS (EI, 70 eV): calcd for C21H16N1Br1 ([M]+): 361.04606, found: 361.04591.

APPENDIX
xxvii
General procedure for synthesis of azaindolo[1,2-f]phenanthridines
3-bromo-2-(phenylethynyl)pyridine 3.6 (0.3 mmol), 2-bromoaniline 3.2 (1.1 equiv.,
0.33 mmol), Pd(PPh3)4 (10 mol%, 0.03 mmol,), XantPhos (10 mol%, 0.03 mmol), and Cs2CO3
(3 equiv., 0.9 mmol) were placed in a dried pressure tube equipped with a septum. The reaction
was back-filled with argon three times. Then dried and degassed DMF (4 mL) was added under
argon and the septum was replaced with a Teflon cap. The reaction mixture was allowed to stir
at 120 °C for 24 h. Then the reaction mixture was cooled to room temperature and was filtered
through a pad of Celite. The filtrate was dried under reduced pressure, and the product 3.7 was
obtained after flash chromatography on a silica gel column with ethyl acetate.
Pyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7a
Yellowish solid, 86% (69.1 mg). M.p.: 144–145 °C.
IR (ATR, cm-1): = 3123 (w), 3099 (w), 3062 (w), 3034 (w), 1887 (w),
1598 (m), 1580 (w), 1556 (s), 1503 (w), 1488 (w), 1479 (m), 1453 (m),
1440 (s), 1414 (s), 1401 (w), 1378 (m), 1356 (m), 1325 (w), 1311 (w),
1303 (w), 1279 (m), 1236 (m), 1186 (m), 1138 (w), 1127 (w), 1110 (w), 1073 (w), 1051 (w),
1042 (w), 973 (w), 954 (w), 943 (w), 923 (w), 908 (w), 877 (w), 862 (w), 833 (w), 805 (w), 774
(w), 745 (s), 731 (w), 708 (m), 666 (w), 640 (w), 617 (m), 608 (w), 584 (w), 574 (w), 556 (w),
537 (w).
1H NMR (300 MHz, CDCl3) δ = 8.62 (dd, 3J = 4.6, 4J = 1.0 Hz, 1H, CHAr), 8.50 (d, 3J = 8.6 Hz,
1H, CHAr), 8.35 – 8.17 (m, 2H, CHAr), 8.17 – 7.90 (m, 2H, CHAr), 7.53 – 7.43 (m, 3H, CHAr),
7.39 – 7.09 (m, 3H, CHAr).
13C NMR (63 MHz, CDCl3) δ = 147.6 (CAr), 144.9 (CHAr), 138.3, 135.2 (CAr), 129.0, 128.9,
128.5 (CHAr), 127.0, 126.9, 125.1 (CAr), 124.9, 124.3, 123.7, 122.4 (CHAr), 121.9 (CAr), 121.3,
116.2, 116.0, 96.8 (CHAr).
MS (EI, 70 eV): m/z(%) = 268 (M+, 100), 240 (7), 214 (3), 134 (10), 120 (13), 106 (5).
HRMS (EI): Calculated for C19H12N2 (M+): 268.09950, found: 268.09952.
3-Methylpyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7d
Yellowish solid, 61% (51.6 mg). M.p.:162–164 °C.
IR(ATR, cm-1): = 3120 (w), 3093 (w), 3061 (w), 3032 (w), 2917
(w), 2851 (w), 1914 (w), 1883 (w), 1613 (w), 1598 (m), 1575 (w),
1557 (m), 1550 (w), 1493 (w), 1481 (w), 1444 (s), 1414 (s), 1375

APPENDIX
xxviii
(m), 1349 (m), 1306 (m), 1283 (m), 1238 (m), 1208 (w), 1189 (m), 1164 (w), 1149 (w), 1127
(w), 1115 (w), 1079 (m), 1039 (m), 956 (m), 943 (w), 923 (m), 913 (w), 903 (w), 873 (m), 834
(w), 810 (s), 772 (m), 755 (s), 734 (s), 714 (w), 660 (w), 651 (w), 623 (m), 610 (m), 578 (s),
545 (w), 532 (s).
1H NMR (300 MHz, CDCl3) δ = 8.72 – 8.54 (m, 1H, CHAr), 8.50 – 8.36 (m, 1H, CHAr), 8.19
(dd, 3J = 10.5, 3J = 5.7 Hz, 2H, CHAr), 7.95 – 7.78 (m, 2H, CHAr), 7.54 – 7.36 (m, 1H, CHAr),
7.34 – 7.09 (m, 4H, CHAr), 2.45 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3) δ = 147.9 (CAr), 144.8 (CHAr), 138.8, 138.5, 135.3 (CAr), 129.7,
128.8 (CHAr), 126.8 (CAr), 124.8, 124.1, 123.5 (CHAr), 122.6 (CAr), 122.5 (CHAr), 121.9 (CAr),
121.0, 115.9, 115.8, 96.1 (CHAr), 22.0 (CH3). (one signal of C tertiary could not be detected).
MS (EI, 70 eV): m/z(%) = 282 (M+, 100), 266 (4), 140 (10), 128 (2), 126 (5).
HR-MS (EI): calculated for C20H14N2 (M+): 282.11515, found: 282.11477.
3-(Tert-butyl)pyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 8f
Yellowish solid, 69% (67.1 mg). M.p.: 176–178 °C.
IR (ATR, cm-1): = 3060 (w), 3025 (w), 2947 (m), 2902 (w), 2860
(w), 1931 (w), 1884 (w), 1732 (w), 1615 (w), 1598 (m), 1578 (w),
1557 (m), 1504 (w), 1494 (m), 1479 (w), 1463 (w), 1443 (s), 1413
(s), 1392 (w), 1357 (w), 1348 (m), 1303 (w), 1275 (m), 1265 (m), 1242 (w), 1205 (w), 1187
(m), 1162 (w), 28 1151 (w), 1127 (w), 1115 (w), 1097 (w), 1070 (w), 1053 (w), 1039 (w), 1021
(w), 970 (w), 956 (m), 925 (w), 913 (w), 875 (m), 832 (w), 813 (m), 788 (s), 769 (m), 756 (s),
738 (s), 710 (w), 657 (w), 648 (w), 623 (s), 608 (w), 584 (m), 542 (s).
1H NMR (300 MHz, CDCl3) δ = 8.63 (d, 4J = 4.1 Hz, 1H, CHAr), 8.53 (d, 3J = 8.6 Hz, 1H,
CHAr), 8.41 – 8.29 (m, 2H, CHAr), 8.24 (d, 4J = 1.7 Hz, 1H, CHAr), 8.10 (d, 3J = 8.4 Hz, 1H,
CHAr), 7.64 – 7.48 (m, 2H, CHAr), 7.42 – 7.32 (m, 2H, CHAr), 7.22 (dd, 3J = 8.5, 4J = 4.6 Hz,
1H, CHAr), 1.47 (s, 9H, C(CH3)3).
13C NMR (75 MHz, CDCl3) δ = 152.1, 148.0 (CAr), 144.9 (CHAr), 138.5, 135.5 (CAr), 128.9
(CHAr), 127.0, 126.6 (CAr), 126.5, 124.9, 124.2, 123.6 (CHAr), 122.8, 122.4 (CAr), 121.1, 118.7,
116.1, 116.0, 96.3 (CHAr), 35.4 (C(CH3)3), 31.4 (3C, C(CH3)3).
MS (EI, 70 eV): m/z(%) = 324 (M+, 100), 309 (98), 294 (28), 290 (4), 281 (12), 268 (15), 240
(4), 154 (4), 146 (4), 140 (24), 132 (6), 126 (4), 41 (3), 39 (3).
HR-MS (EI): calculated for C23H20N2 (M+): 324.16210, found: 324.16196.

APPENDIX
xxix
3-Fluoropyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7h
Yellowish solid, 88% (75.5 mg). M.p.: 213–215 °C.
IR (ATR, cm-1): = 3059 (w), 3035 (w), 1616 (m), 1603 (m), 1580
(w), 1558 (m), 1551 (m), 1489 (m), 1443 (s), 1416 (s), 1379 (w),
1349 (m), 1331 (w), 1304 (w), 1272 (m), 1245 (w), 1236 (w), 1180
(s), 1135 (w), 1128 (w), 1106 (w), 1072 (w), 1054 (w), 1032 (w), 957 (m), 933 (w), 915 (w),
892 (m), 854 (w), 819 (w), 810 (m), 784 (w), 763 (s), 755 (m), 733 (s), 706 (w), 652 (m), 631
(m), 621 (m), 606 (w), 599 (m), 583 (w), 545 (w), 533 (m).
1H NMR (300 MHz, CDCl3) δ = 8.61 (d, 4J = 3.6 Hz, 1H, CHAr), 8.41 (d, 3J = 8.6 Hz, 1H,
CHAr), 8.23 – 8.11 (m, 1H, CHAr), 8.02 (dd, 3J = 8.1, 4J = 1.3 Hz, 1H, CHAr), 7.95 (dd, 3J = 8.8,
3J = 5.7 Hz, 1H, CHAr), 7.66 (dd, 3J = 10.6, 4J = 2.5 Hz, 1H, CHAr), 7.55 – 7.45 (m, 1H, CHAr),
7.32 – 7.23 (m, 2H, CHAr), 7.23 – 7.08 (m, 3H, CHAr).
19F NMR (63 MHz, CDCl3) δ = 110.77 Hz.
13C NMR (63 MHz, CDCl3) δ = 163.1 (d, 1J = 248.2 Hz, CF), 147.7 (CAr), 145.1 (CHAr), 137.5,
135.5 (CAr), 129.7 (CHAr), 129.1 (d, 3J = 8.4 Hz, CAr), 127.2 (d, 3J = 8.9 Hz, CHAr), 126.8 (CAr),
124.4, 123.7 (CHAr), 121.5 (d, 4J = 2.5 Hz, CAr), 121.1 (CHAr), 121.1 (CAr), 116.5 (d,
2J = 23.2 Hz, CHAr), 116.2, 116.0 (CHAr), 108.5 (d, 2J = 23.4 Hz, CHAr), 96.5 (d, 6J = 1.3 Hz,
CHAr).
MS (EI, 70 eV): m/z(%) = 286 (M+, 100), 258 (7), 232 (3), 195 (2), 143 (10), 129 (11), 115 (3).
HR-MS (EI): calculated for C19H11N2F1 (M+): 286.09008, found: 286.08980.
3-Methoxypyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7j
Yellowish solid, 85% (76.0 mg). M.p.: 200–201 °C.
IR (ATR, cm-1): = 3089 (w), 3032 (w), 2999 (w), 2958 (w), 2930
(w), 2908 (w), 2831 (w), 1609 (s), 1601 (s), 1550 (s), 1493 (s),
1450 (s), 1438 (w), 1428 (w), 1414 (s), 1379 (w), 1347 (m), 1334
(w), 1303 (w), 1278 (s), 1244 (w), 1219 (s), 1190 (m), 1181 (w), 1141 (w), 1130 (w), 1111 (w),
1079 (w), 1073 (w), 1056 (w), 1041 (w), 1033 (w), 1021 (m), 980 (w), 971 (w), 956 (m), 912
(w), 905 (w), 883 (w), 872 (w), 865 (w), 830 (m), 823 (w), 813 (s), 785 (w), 766 (s), 753 (s),
736 (s), 705 (w), 656 (m), 634 (m), 623 (m), 607 (s), 584 (m), 555 (m), 539 (m).
1H NMR (300 MHz, CDCl3) δ = 8.49 (d, 4J = 4.2 Hz, 1H, CHAr), 8.31 (d, 3J = 8.5 Hz, 1H,
CHAr), 8.08 (d, 3J = 8.1 Hz, 1H, CHAr), 7.98 (dd, 3J = 8.1, 4J = 1.0 Hz, 1H, CHAr), 7.79 (d,

APPENDIX
xxx
3J = 8.8 Hz, 1H, CHAr), 7.44 – 7.29 (m, 2H, CHAr), 7.22 – 7.12 (m, 1H, CHAr), 7.07 (dd,
3J = 8.5, 4J = 4.6 Hz, 1H, CHAr), 7.02 (s, 1H, CHAr), 6.90 (dd, 3J = 8.8, 4J = 2.4 Hz, 1H, CHAr),
3.79 (s, 3H, OCH3).
13C NMR (75 MHz, CDCl3) δ = 160.2 (CAr-OCH3), 148.0 (CAr), 144.7 (CHAr), 138.6, 135.5
(CAr), 129.1 (CHAr), 128.5, 126.8 (CAr), 126.6, 124.2, 123.5 (CHAr), 121.7 (CAr), 120.9 (CHAr),
118.6 (CAr), 116.3, 116.0, 115.6, 105.6, 95.2 (CHAr), 55.5 (OCH3).
MS (EI, 70 eV): m/z(%) = 298 (M+, 100), 283 (20), 255 (63), 227 (7), 149 (9), 127 (4), 113 (5),
99 (3). HR-MS (EI): calculated for C20H14N2O1 (M+): 298.11006, found: 298.10998.
6-Methylpyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7b
Yellow solid, 64% (54.1 mg). M.p.: 184–186 °C.
IR (ATR, cm-1): = 3124 (w), 3069 (w), 3036 (w), 2954 (w), 2916 (m),
2850 (m), 2746 (w), 1942 (w), 1900 (w), 1877 (w), 1860 (w), 1823 (w),
1795 (w), 1600 (m), 1577 (w), 1569 (m), 1553 (s), 1524 (w), 1496 (m),
1450 (s), 1416 (s), 1387 (w), 1372 (w), 1354 (m), 1324 (w), 1310 (w),
1300 (w), 1279 (m), 1236 (w), 1193 (w), 1182 (m), 1165 (w), 1145 (w), 1124 (m), 1116 (m),
1066 (w), 1042 (w), 999 (w), 972 (w), 955 (m), 937 (w), 910 (m), 883 (w), 866 (m), 853 (m),
828 (w), 801 (m), 790 (s), 772 (m), 748 (s), 730 (w), 720 (w), 710 (w), 694 (w), 660 (w), 643
(m), 620 (w), 576 (s), 540 (m).
1H NMR (300 MHz, CDCl3) δ = 8.60 (s, 1H, CHAr), 8.39 (d, 3J = 8.5 Hz, 1H, CHAr),
8.13 – 7.95 (m, 3H, CHAr), 7.91 (s, 1H, CHAr), 7.54 – 7.35 (m, 2H, CHAr), 7.30 (s, 1H, CHAr),
7.19 (d, 3J = 7.0 Hz, 2H, CHAr), 2.39 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3) δ = 147.2 (CAr), 144.4 (CHAr), 139.9, 138.3, 133.1, 133.0 (CAr),
129.8, 128.8, 128.3 (CHAr), 127.0, 125.0 (CAr), 125.0, 124.4, 122.4 (CHAr), 121.7 (CAr), 121.3,
116.0, 115.7, 96.2 (CHAr), 21.2 (CH3).
MS (EI, 70 eV): m/z(%) = 282 (M+, 100), 266 (4), 252 (3), 140 (16), 126 (5), 113 (2), 100 (2).
HRMS (EI): calculated for C20H14N2 (M+): 282.11515, found: 282.11484.
3,6-Dimethylpyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7e

APPENDIX
xxxi
Yellowish solid, 72% (63.9 mg). M.p.: 208–210 °C.
IR (ATR, cm-1): = 3154 (w), 3115 (w), 3097 (w), 3056 (w), 3025
(w), 3004 (w), 2952 (w), 2917 (m), 2851 (w), 1920 (w), 1898 (w),
1854 (w), 1613 (w), 1601 (m), 1569 (w), 1549 (s), 1491 (m), 1481
(w), 1434 (w), 1416 (s), 1373 (w), 1352 (w), 1300 (w), 1281 (m),
1262 (w), 1237 (m), 1205 (w), 1192 (m), 1152 (w), 1121 (w), 1099 (w), 1069 (w), 1041 (m),
957 (m), 929 (w), 906 (w), 877 (m), 867 (w), 817 (w), 806 (s), 784 (s), 754 (s), 741 (w), 716
(w), 694 (w), 660 (m), 629 (w), 622 (w), 590 (m), 578 (m), 560 (w), 533 (s).
1H NMR (300 MHz, CDCl3) δ = 8.59 (s, 1H, CHAr), 8.36 (d, 3J = 8.5 Hz, 1H, CHAr), 7.98 (d,
3J = 8.5 Hz, 1H, CHAr), 7.88 (d, 3J = 8.3 Hz, 2H, CHAr), 7.79 (s, 1H, CHAr), 7.35 – 6.87 (m, 4H,
CHAr), 2.45 (s, 3H, CH3), 2.38 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3) δ = 147.7 (CAr), 144.6 (CHAr), 138.6, 138.4, 133.1, 132.8 (CAr),
129.6, 129.5 (CHAr), 126.8, 126.7 (CAr), 124.8, 124.2 (CHAr), 122.6 (CAr), 122.4 (CHAr), 121.7
(CAr), 120.9 (CHAr), 115.6 (2 CHAr ), 95.6 (CHAr), 22.0 (CH3), 21.2 (CH3).
MS (EI, 70 eV): m/z(%) = 296 (M+, 100), 279 (11), 266 (2), 148 (6), 147 (3), 146 (3), 140 (7),
126 (2). HR-MS (EI): calculated for C21H16N2 ([M+1]+1): 297.13862, found: 297.13882.
3-(Tert-butyl)-6-methylpyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7g
Yellow solid, 32% (32.4 mg). M.p. 197–199 °C.
IR (ATR, cm-1): = 3033 (w), 2956 (m), 2914 (w), 2864 (w), 1615
(w), 1600 (w), 1569 (w), 1556 (m), 1548 (m), 1489 (w), 1482 (w),
1460 (w), 1444 (w), 1427 (w), 1414 (s), 1391 (w), 1379 (w), 1357
(m), 1353 (m), 1301 (w), 1280 (m), 1263 (m), 1240 (w), 1202 (w),
1187 (w), 1159 (w), 1131 (w), 1121 (w), 1067 (w), 1045 (w), 958 (m), 941 (w), 925 (w), 904
(w), 880 (m), 865 (w), 830 (m), 811 (w), 786 (s), 774 (s), 757 (s), 736 (w), 722 (m), 666 (w),
656 (w), 634 (w), 620 (w), 611 (w), 578 (m), 552 (s), 533 (w).
1H NMR (300 MHz, CDCl3): δ = 8.67 – 8.56 (m, 1H, CHAr), 8.49 (d, 3J = 8.5 Hz, 1H, CHAr),
8.23 – 8.07 (m, 4H, CHAr), 7.63 – 7.57 (m, 1H, CHAr), 7.36 – 7.29 (m, 2H, CHAr), 7.20 (dd,
3J = 8.5 Hz, 4J = 4.5 Hz, 1H, CHAr), 2.51 (s, 3H, CH3), 1.48 (s, 9H, C(CH3)3).
13C NMR (75 MHz, CDCl3): δ = 152.0, 147.9 (CAr), 144.8 (CHAr), 138.4, 133.4, 133.1 (CAr),
129.8 (CHAr), 126.7 (CAr), 126.3, 125.0, 124.3 (CHAr), 122.9, 122.3 (CAr), 121.0, 118.6, 116.0,
115.8, 96.0 (CHAr), 35.4 (C(CH3)3), 31.5 (CH3), 21.3 (3C, C(CH3)3) (one signal of CAr could
not be detected).

APPENDIX
xxxii
MS (EI, 70 eV): m/z(%) = 338 (M+, 100), 323 (85), 308 (29), 295 (9), 282 (11) , 266 (3), 161
(7), 153 (4), 152 (3), 147 (23), 140 (8), 139 (5), 133 (4), 41 (5), 39 (4). HR-MS (EI): calculated
for C24H22N2 (M+): 338.17775, found: 338.17773.
3-Fluoro-6-methylpyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7i
Yellow solid, 79% (80.1 mg). M.p.: 233–235 °C.
IR (ATR, cm-1): = 3033 (w), 2956 (m), 2914 (w), 2864 (w), 1615
(w), 1600 (w), 1569 (w), 1556 (m), 1548 (m), 1489 (w), 1482 (w),
1460 (w), 1444 (w), 1427 (w), 1414 (s), 1391 (w), 1379 (w), 1357
(m), 1353 (m), 1301 (w), 1280 (m), 1263 (m), 1240 (w), 1202 (w),
1187 (w), 1159 (w), 1131 (w), 1121 (w), 1067 (w), 1045 (w), 958 (m), 941 (w), 925 (w), 904
(w), 880 (m), 865 (w), 830 (m), 811 (w), 786 (s), 774 (s), 757 (s), 736 (w), 722 (m), 666 (w),
656 (w), 634 (w), 620 (w), 611 (w), 578 (m), 552 (s), 533 (w).
1H NMR (300 MHz, CDCl3) δ = 8.68 (s, 1H, CHAr), 8.45 (d, 3J = 8.5 Hz, 1H, CHAr),
8.11 – 8.01 (m, 2H, CHAr), 7.84 (s, 1H, CHAr), 7.76 – 7.69 (m, 1H, CHAr), 7.35 – 7.22 (m, 3H,
CHAr), 7.19 (dd, 3J = 8.4 Hz, 4J = 2.3 Hz, 1H, CHAr), 2.48 (s, 3H, CH3).
19F NMR (63 MHz, CDCl3) δ = 110.92.
13C NMR (63 MHz, CDCl3) δ = 163.1 (d, 1J = 247.9 Hz, CF), 147.5 (CAr), 144.9 (CHAr), 137.5,
133.3, 133.3 (CAr), 130.5 (CHAr), 129.1 (d, 3J = 8.4 Hz, CAr), 127.2 (d, 3J = 8.9 Hz, CHAr), 124.6
(CHAr), 121.6 (d, 4J = 2.4 Hz, CAr), 121.0 (CHAr), 120.9 (d, 4J = 3.0 Hz, CAr), 116.4 (d,
2J = 23.2 Hz, CHAr), 116.0, 115.8 (CHAr), 108.5 (d, 2J = 23.4 Hz, CHAr), 96.1 (CHAr), 21.2
(CH3). (Signal of one CAr could not be detected).
MS (EI, 70 eV): m/z(%) = 338 (M+, 100), 323 (85), 308 (29), 295 (9), 282 (11), 266 (3), 161
(7), 153 (4), 147 (23), 140 (8), 139 (5), 133 (4), 41 (5), 39 (4). HR-MS (EI): calculated for
C20H13F1N2 (M+): 338.17775, found: 338.17773.
3-Methoxy-6-methylpyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7k
Yellow solid, 37% (34.6 mg). M.p.: 183–185 °C.
IR (ATR, cm-1): = 3094 (w), 3029 (w), 3005 (w), 2916 (w), 2839
(w), 2054 (m), 1722 (w), 1610 (s), 1572 (w), 1546 (s), 1493 (s),
1467 (w), 1452 (w), 1432 (w), 1418 (s), 1378 (w), 1353 (w), 1333
(w), 1300 (w), 1282 (s), 1243 (w), 1223 (s), 1194 (m), 1188 (m),
1152 (w), 1125 (w), 1074 (w), 1028 (s), 959 (m), 936 (w), 906 (w), 860 (w), 833 (m), 819 (m),

APPENDIX
xxxiii
803 (w), 782 (s), 773 (m), 751 (s), 710 (w), 682 (w), 668 (w), 633 (m), 621 (w), 587 (w), 579
(w), 545 (s).
1H NMR (300 MHz, CDCl3) δ = 8.57 (s, 1H, CHAr), 8.47 (d, 3J = 8.5 Hz, 1H, CHAr), 8.12 (d,
3J = 8.5 Hz, 1H, CHAr), 7.99 (d, 3J = 8.8 Hz, 1H, CHAr), 7.93 (s, 1H, CHAr), 7.53 (d, 4J = 2.4 Hz,
1H, CHAr), 7.31 (dd, 3J = 8.5, 4J = 1.1 Hz, 1H, CHAr), 7.24 – 7.13 (m, 2H, CHAr), 7.07 (dd,
3J = 8.8, 4J = 2.4 Hz, 1H, CHAr), 3.95 (s, 3H, OCH3), 2.46 (s, 3H, CH3).
13C NMR (63 MHz, CDCl3) δ = 160.5 (C-OCH3), 147.2 (CAr), 143.7 (CHAr), 139.1, 133.3 (CAr),
130.2 (CHAr), 128.8, 127.0 (CAr), 126.9, 124.5 (CHAr), 121.7 (CAr), 121.4 (CHAr), 118.6 (CAr),
116.4, 115.9, 115.4, 105.8, 94.5 (CHAr), 55.7 (OCH3), 21.3 (CH3). (one signal of CAr could not
be detected).
MS (EI, 70 eV): m/z(%) = 312 (M+, 100), 297 (16), 269 (53), 253 (4), 239 (2), 156 (10), 134
(7), 121 (3), 120 (3), 107 (2). HR-MS (EI): calculated for C21H16N2O1 (M+): 312.12571, found:
312.12596.
6-Fluoropyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.7c (3.8e)
Yellow solid, 41% (35.2 mg). M.p.: 220–221 °C.
IR (ATR, cm-1): = 3131.1 (w), 3056.8 (w), 3031.7 (w), 2920.8 (w),
2850.5 (w), 1942.2 (w), 1889.7 (w), 1573.4 (m), 1557.6 (s), 1496.3 (m),
1452.2 (m), 1420.5 (s), 1280.2 (m), 1243.7 (m), 1201.1 (m), 1174.1 (m),
1137.7 (m), 1108.0 (w), 1168.8 (m), 957.1 (m), 909.6 (m), 856.1 (m),
806.8 (m), 782.8 (m), 746.5 (s), 577.9 (m).
1H NMR (300 MHz, CDCl3) δ = 8.65 (d, 4J = 4.0 Hz, 1H, CHAr), 8.44 (d, 3J = 8.6 Hz, 1H,
CHAr), 8.23 (dd, 3J = 9.2, 4J = 4.8 Hz, 1H, CHAr), 8.16 – 8.07 (m, 1H, CHAr), 8.07 – 8.00 (m,
1H, CHAr), 7.90 (dd, 3J = 10.0, 4J = 2.8 Hz, 1H, CHAr), 7.60 – 7.47 (m, 2H, CHAr), 7.36 (s, 1H,
CHAr), 7.28 – 7.16 (m, 2H, CHAr).
19F NMR (282 MHz, CDCl3) δ -118.20.
13C NMR (75 MHz, CDCl3) δ = 159.0 (d, 1J = 243.3 Hz, CF), 147.7 (CAr), 145.3 (CHAr), 137.9,
131.8 (d, 4J = 2.1 Hz, CAr), 129.3, 129.0 (CHAr), 126.8 (CAr), 126.2 (d, 4J = 2.5 Hz, CAr), 125.6
(CAr), 125.1 (CHAr), 124.0 (d, 3J = 7.7 Hz, CAr), 122.7, 120.7 (CHAr), 117.5 (d, 3J = 8.1 Hz,
CHAr), 116.5 (CHAr), 116.1 (d, 2J = 23.2 Hz, CHAr), 110.6 (d, 2J = 23.9 Hz, CHAr), 97.1 (CHAr).
MS (EI, 70 eV): m/z(%) = 286 (M+, 100), 258 (7), 232 (5), 208 (2), 195 (3), 168 (3), 143 (6),
128 (3), 99 (2), 87 (2), 75 (2), 62 (5), 51 (3), 39 (3).

APPENDIX
xxxiv
HR-MS (EI): calculated for C19H11N2F1 (M+): 286.09008, found: 286.09014
6-Methoxypyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.8b
Yellowish solid, 51% (45.6 mg). M.p.: 156–157 °C.
IR (ATR, cm-1): = 3133.5 (w), 3055.6 (w), 2919.6 (w), 2849.1 (w),
1957.7 (w), 1925.7 (w), 1900.2 (w), 1726.6 (w), 1616.8 (m), 1606.1 (m),
1569.7 (m), 1552.5 (s), 1500.3 (m), 1453.4 (m), 1419.4 (s), 1350.4 (m),
1218.9 (m), 1185.5 (m), 1138.4 (m), 1018.3 (m), 956.6 (m), 850.5 (m),
789.6 (m), 755.3 (s), 694.9 (m), 583.2 (m).
1H NMR (300 MHz, CDCl3) δ = 8.61 (d, 4J = 4.0 Hz, 1H, CHAr), 8.41 (d, 3J = 8.6 Hz, 1H,
CHAr), 8.13 (d, 3J = 9.2 Hz, 1H, CHAr), 8.10 – 8.00 (m, 2H, CHAr), 7.64 (d, 4J = 2.8 Hz, 1H,
CHAr), 7.53 – 7.39 (m, 2H, CHAr), 7.32 (s, 1H, CHAr), 7.19 (dd, 3J = 8.6, 4J = 4.6 Hz, 1H, CHAr),
7.03 (dd, 3J = 9.1, 4J = 2.9 Hz, 1H, CHAr), 3.89 (s, 3H, OCH3).
13C NMR (63 MHz, CDCl3) δ = 155.7, 147.4 (CAr), 144.7 (CHAr), 137.9, 129.7 (CAr), 128.8,
128.6 (CHAr), 126.8, 126.7, 125.4 (CAr), 125.1 (CHAr), 123.3(CAr), 122.5, 120.8, 117.1, 116.0,
115.3, 108.4, 96.3 (CHAr), 55.7 (OCH3).
MS (EI, 70 eV): m/z(%) = 298 (M+, 100), 283 (37), 255 (70), 227 (11). 200 (5), 174 (4), 149
(8), 127 (7), 114 (11), 100 (5), 87 (7), 75 (4), 63 (4), 51 (4), 39 (6).
HR-MS (EI): calculated for C20H14N2O1 (M+): 298.11006, found: 298.10977.
8-Methoxypyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.8c
Yellow oil, 60% (53.6 mg).
1H NMR (300 MHz, CDCl3) δ = 8.59 (dd, 3J = 4.5, 4J = 0.9 Hz, 1H,
CHAr), 8.25 – 8.12 (m, 2H, CHAr), 8.10 – 8.03 (m, 1H, CHAr), 7.94 (dd,
3J = 8.1, 5J = 0.9 Hz, 1H, CHAr), 7.65 – 7.45 (m, 3H, CHAr), 7.39 (t,
3J = 8.1 Hz, 1H, CHAr), 7.22 – 7.07 (m, 2H, CHAr), 3.90 (s, 3H, OMe).
13C NMR (63 MHz, CDCl3) δ = 149.5, 146.9 (CAr), 144.4 (CHAr), 139.1, 129.9 (CAr), 128.9,
128.8 (CHAr), 127.3, 125.9, 125.4 (CAr), 124.9 (2CHAr), 124.7 (CHAr), 124.1 (CAr), 123.1, 116.4,
115.0, 112.1, 97.6 (CHAr), 55.9 (OCH3).
MS (EI, 70 eV): m/z(%) = 298 (M+, 100), 283 (83), 253 (13), 227 (7), 201 (4), 175 (2), 142
(10), 127 (6), 114 (7), 100 (7).
HRMS (EI): calculated for C20H14N2O1 (M+): 298.11006, found: 298.11014.

APPENDIX
xxxv
6-(Methylthio)pyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.8g
Yellow solid, 81% (77.2 mg). M.p.: 172–173 °C.
IR (ATR, cm-1): = 3031.8 (w), 3001.0 (w), 2958.0 (w), 2917.4 (w),
2849.2 (w), 1597.0 (w), 1646.3 (m), 1488.3 (w), 1449.2 (m), 1413.4 (s),
1354.0 (m), 1278.7 (m), 1189.0 (m), 1115.3 (w), 955.0 (m), 855.7 (w),
789.8 (s), 750.4 (s), 580.6 (m).
1H NMR (300 MHz, CDCl3) δ = 8.61 (s, 1H, CHAr), 8.42 (d, 3J = 8.5 Hz, 1H, CHAr),
8.17 – 7.97 (m, 4H, CHAr), 7.57 – 7.42 (m, 2H, CHAr), 7.35 (dd, 3J = 8.8, 4J = 2.1 Hz, 1H,
CHAr), 7.31 (s, 1H, CHAr), 7.22 (dd, 3J = 8.5, 4J = 4.6 Hz, 1H, CHAr), 2.57 (s, 3H, SCH3).
13C NMR (75 MHz, CDCl3) δ = 147.2 (CAr), 144.6 (CHAr), 138.2, 133.5, 133.0 (CAr), 129.0,
128.8, 127.9 (CHAr), 126.4, 125.2 (CAr), 125.1, 122.8 (CHAr), 122.6 (CAr), 122.4, 121.3, 116.5,
116.2, 96.7 (CHAr), 16.8 (SCH3). (one signal of CAr could not be detected).
MS (EI, 70 eV): m/z(%) = 314 (M+, 100), 299 (52), 266 (9), 255 (28), 227 (4), 157 (13), 127
(6), 113 (3), 100 (2).
HRMS (EI): calculated for C20H14N2S1 (M+): 314.08722, found: 314.08694.
Benzo[c]pyrido[2',3':4,5]pyrrolo[1,2-f]phenanthridine 3.8h
Yellow solid, 42% (40.1 mg). M.p.: 231–232 °C.
IR (ATR, cm-1): = 3096.6 (w), 2958.7 (w), 2850.5 (w), 1954.5 (w),
1915.3 (w), 1808.3, 1713.7 (w), 1621.8 (w), 1576.9 (m), 1543.0 (m),
1416.8 (s), 1389.4 (m), 1274.8 (m), 1029.8 (m), 807 (m), 752.5 (s), 611.2
(m), 566.6 (m).
1H NMR (300 MHz, CDCl3) δ = 8.64 (d, 4J = 4.0 Hz, 1H, CHAr), 8.34 – 8.11 (m, 4H, CHAr),
7.96 – 7.84 (m, 2H, CHAr), 7.79 (d, 3J = 8.7 Hz, 1H, CHAr), 7.60 – 7.47 (m, 3H, CHAr), 7.45 (d,
5J = 0.6 Hz, 1H, CHAr), 7.43 – 7.34 (m, 1H, CHAr), 7.03 (dd, 3J = 8.5, 4J = 4.5 Hz, 1H, CHAr).
13C NMR (63 MHz, CDCl3) δ = 147.6 (CAr), 145.4 (CHAr), 139.8, 134.1, 129.9, 129.2 (CAr),
129.0, 128.7, 128.3 (CHAr), 127.6 (CAr), 127.2 (CHAr), 125.9 (CAr), 125.2, 124.7, 124.6, 124.3
(CHAr), 123.7 (CAr), 123.0, 122.1 (CHAr), 121.0 (CAr), 120.5, 114.0, 97.7 (CHAr).
MS (EI, 70 eV): m/z(%) = 318 (M+, 100), 291 (12), 237 (2), 159 (41), 144 (23), 131 (9), 105
(2), 87 (1).
HRMS (EI): calculated for C23H14N2 (M+): 318.11515, found: 318.11507.

APPENDIX
xxxvi
6-Methoxypyrido[3',2':4,5]pyrrolo[1,2-f]phenanthridine 3.9a
Yellowish solid, 34% (30.4 mg). M.p.: 186–187 °C.
IR (ATR, cm-1): = 3104.4 (w), 2997.7 (w), 2930.7 (w), 2830.5 (w),
2089.0 (w), 1892.9 (w), 1713.5 (w), 1620.3 (w), 1563.3 (m), 1543.8 (s),
1499.5 (m), 1453.9 (m), 1407.9 (m), 1327 (m), 1293.2 (m), 1215.2 (m),
1075.3 (m), 1042.8 (m), 1016.0 (m), 945.9 (w), 855.2 (m), 792.4 (m),
758.9 (m), 729.0 (m), 607.9 (m), 566.3 (m).
1H NMR (300 MHz, CDCl3) δ = 10.15 (d, 3J = 9.3 Hz, 1H, CHAr), 8.50 (dd, 4J = 4.6,
4J = 1.6 Hz, 1H, CHAr), 8.24 – 7.97 (m, 3H, CHAr), 7.72 (d, 4J = 2.8 Hz, 1H, CHAr), 7.57 – 7.39
(m, 2H, CHAr), 7.34 – 7.16 (m, 2H, CHAr), 7.08 (s, 1H, CHAr), 3.95 (s, 3H, OCH3).
13C NMR (63 MHz, CDCl3) δ = 155.8 (C-OCH3), 146.9 (CAr), 141.8 (CHAr), 134.6, 129.9 (CAr),
128.4, 128.3, 128.2 (CHAr), 127.4, 125.8 (CAr), 124.3, 122.8 (CHAr), 122.7, 122.1 (CAr), 120.4,
117.5, 115.5, 107.5, 92.4 (CHAr), 55.7 (OCH3).
MS (EI, 70 eV): m/z(%) = 298 (M+, 100), 283 (34), 255 (61), 227 (7), 201 (3), 175 (2), 149
(15), 127 (16), 113 (4), 100 (5).
HRMS (EI): calculated for C20H14N2O1 (M+): 298.11066, found: 298.11047
6-Fluoropyrido[3',2':4,5]pyrrolo[1,2-f]phenanthridine 3.9b
Yellowish solid, 51% (43.8 mg). M.p.: 201–202 °C.
IR (ATR, cm-1): = 3122.0 (w), 3054.0 (w), 1920.0 (w), 1884.2 (w),
1798.7 (w), 1661.0 (w), 1620.4 (w), 1567.0 (m), 1497.6 (m), 1454.6 (m),
1409.4 (m), 1329.7 (m), 1267.7 (m), 1174.5 (m), 1141.0 (m), 892.5 (m),
824.2 (m), 752.8 (m), 724.4 (m), 662.7 (w), 593.9 (m), 566.3 (m).
1H NMR (300 MHz, CDCl3) δ = 10.26 (dd, 3J = 9.3, 3J = 5.5 Hz, 1H, CHAr), 8.50 (dd, 4J = 4.5,
4J = 1.3 Hz, 1H, CHAr), 8.25 – 8.03 (m, 3H, CHAr), 7.91 (dd, 3J = 10.3, 4J = 2.7 Hz, 1H, CHAr),
7.52 (dd, 3J = 6.0, 4J = 3.3 Hz, 2H, CHAr), 7.41 – 7.26 (m, 2H, CHAr), 7.11 (s, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ = -118.48.
13C NMR (63 MHz, CDCl3) δ = 159.3 (d, 1J = 242.1 Hz, CF), 142.1 (CHAr), 134.6, 131.9, 130.4
(CAr), 128.9, 128.7, 128.4 (CHAr), 126.8 (d, 4J = 1.9 Hz, CAr), 125.8 (CAr), 124.3 (CHAr), 123.4
(d, 3J = 7.6 Hz, CAr), 122.9 (CHAr), 122.2 (CAr), 121.0 (d, 3J = 7.8 Hz, CHAr), 117.9 (CHAr),
116.4 (d, 2J = 22.4 Hz, CHAr), 109.3 (d, 2J = 24.0 Hz, CHAr), 93.0 (CHAr).
MS (EI, 70 eV): m/z(%) = 286 (M+, 100), 258 (10), 232 (3), 143 (17), 129 (4), 115 (3).

APPENDIX
xxxvii
HRMS (EI): calculated for C19H11N2F1 (M+): 286.09008, found: 286.08990.
6-(Methylthio)pyrido[3',2':4,5]pyrrolo[1,2-f]phenanthridine 3.9c
Yellow solid, 36% (33.9 mg). M.p.: 168–169 °C.
IR (ATR, cm-1): = 3107.2 (w), 3037.2 (w), 2918.0 (w), 2849.6 (w),
2731.2 (w), 2520.1 8w), 2387.1 (w), 2116.4 (w), 1959.0 (w), 1916.1 (w),
1724.9 (w), 1595.9 (m), 1541.6 (m), 1453.3 (s), 1405.6 (s), 1323.5 (m),
1103.3 (m), 955.8 (m), 818.2 (m), 793.9 (s), 758.7 (s), 730.2 (s), 646.5
(m), 585.3 (m).
1H NMR (300 MHz, CDCl3) δ = 10.14 (d, 3J = 8.9 Hz, 1H, CHAr), 8.49 (dd, 4J = 4.6,
4J = 1.7 Hz, 1H, CHAr), 8.27 – 8.11 (m, 2H, CHAr), 8.10 – 7.99 (m, 2H, CHAr), 7.57 – 7.41 (m,
3H, CHAr), 7.26 (dd, 3J = 7.9, 4J = 4.7 Hz, 1H, CHAr), 7.06 (s, 1H, CHAr), 2.61 (s, 3H, SCH3).
13C NMR (75 MHz, CDCl3) δ = 147.1 (CAr), 142.0 (CHAr), 134.7, 133.4, 132.9 (CAr), 128.5 (2
CHAr), 128.4, 128.2 (CHAr), 127.0, 125.7 (CAr), 124.2, 122.7 (CHAr), 122.3 (CAr), 122.2 (CHAr),
122.0 (CAr), 119.8, 117.7, 93.0 (CHAr), 17.2 (SCH3).
MS (EI, 70 eV): m/z(%) = 314 (M+, 100), 299 (59), 266 (7), 255 (29), 227 (3), 201 (1), 157
(14), 133 (5), 113 (2).
HRMS (EI): calculated for C20H14N2S1 (M+): 314.08722, found: 314.08645.
General procedure for domino reaction of dibromide compounds and
N-tosylhydrazones
Dibromide compound 4.1 (0.2 mmol), N-tosylhydrazone 4.2 (1.5 equiv., 0.3 mmol),
Pd2(dba)3 (2.5 mol%, 4.6 mg, 0.005 mmol), XPhos (10 mol%, 9.6 mg, 0.01 mmol), and LiOtBu
(4 equiv. 64 mg, 0.8 mmol) were placed in a dried pressure tube equipped with a septum. The
reaction vessel was back-filled with argon three times. Then dried and degassed dioxane (4 mL)
was added to the reaction mixture under argon and the septum was replaced with a Teflon cap.
The reaction mixture was stirred at 20 °C for 15 mins and then at 90 °C for 4 hours. After
cooling to room temperature, water (10 mL) and ethyl acetate (10 mL) were added and the
organic layer was separated. The aqueous layer was extracted three times with ethyl acetate
(3x10 mL). Combined organic layers were dried over MgSO4 and the solvent was removed
under reduced pressure. The residue was chromatographed (silica gel, heptane) to obtain the
pure product.
5-Phenylbenzo[b]naphtho[2,1-d]thiophene 4.3a

APPENDIX
xxxviii
White solid, 83% (52 mg). M.p.: 196–197 °C.
IR (ATR, cm-1): 3041.6 (w), 1950.5 (w), 1893.8 (w), 1802.3 (w), 1745.3
(w), 1432.7 (w), 1339.2 (w), 1247.1 (w), 1155.1 (w), 1071.7 (w), 873.6
(m), 744.8 (s), 697.5 (s).
1H NMR (250 MHz, CDCl3) δ 8.27 – 8.15 (m, 2H, CHAr), 8.13 (s, 1H, CHAr), 8.06 – 7.92 (m,
2H, CHAr), 7.71 – 7.44 (m, 9H, CHAr).
13C NMR (63 MHz, CDCl3) δ 140.9, 139.4, 138.2, 137.0, 136.8, 132.3, 131.1 (CAr), 130.5 (2
CHAr), 129.3 (CAr), 128.5 (2CHAr), 127.6 (2CHAr), 126.8, 126.5, 126.4, 124.9, 124.8, 123.1,
121.8, 120.7 (CHAr).
MS (EI, 70 eV): m/z (%) = 310 (M+, 100), 276 (4), 154 (28), 131 (7), 118 (4).
HRMS (EI): Calculated for C22H14S1 (M+): 310.08107, found: 310.08151.
5-(p-Tolyl)benzo[b]naphtho[2,1-d]thiophene 4.3b
White solid, 90% (58 mg). M.p.: 143–144 °C.
IR (ATR, cm-1): 3043.5 (w), 2919.8 (w), 2860.6 (w), 2726.5 (w), 1895.5
(w), 1512.0 (w), 1433.0 (w), 1339.7 (w), 1107.1 (w), 875.8 (m), 819.9
(m), 747.0 (s), 607.2 (m).
1H NMR (300 MHz, CDCl3) δ 8.24 – 8.16 (m, 2H, CHAr), 8.13 (s, 1H,
CHAr), 8.07 – 8.02 (m, 1H, CHAr), 8.02 – 7.95 (m, 1H, CHAr), 7.68 – 7.60 (m, 1H, CHAr),
7.56 – 7.46 (m, 5H, CHAr), 7.38 (d, 3J = 7.8 Hz, 2H, CHAr), 2.52 (s, 3H, CH3).
13C NMR (75 MHz, CDCl3) δ 139.4, 138.1, 138.0, 137.3, 136.8, 132.3, 131.2 (CAr), 130.3
(2CHAr), 129.3 (CAr), 129.2 (2CHAr), 127.6, 126.7, 126.4, 126.3, 124.9, 124.7, 123.1, 121.7,
120.7 (CHAr), 21.4 (CH3) (signal of one tertiary carbon is overlapped).
MS (EI, 70 eV): m/z (%) = 324 (M+, 100), 308 (36), 154 (10), 125 (2), 39 (7).
HRMS (EI): Calculated for C23H16S1 (M+): 324.09672, found: 324.09641.
5-(4-(Trifluoromethyl)phenyl)benzo[b]naphtho[2,1-d]thiophene 4.3c
Yellowish solid, 53% (40 mg). M.p.: 167–169 °C.
IR (ATR, cm-1): 3066.7 (w), 2922.7 (w), 2851.0 (w), 1923.3 (w),
1724.8 (w), 1614.7 (m), 1435.9 (m), 1319.7 (s), 1159.9 (s), 1102.4 (s),
1063.7 (s), 841.6 (m), 749.9 (s), 632.4 (m).

APPENDIX
xxxix
1H NMR (300 MHz, CDCl3) δ 8.27 – 8.15 (m, 2H, CHAr), 8.11 – 8.07 (m, 1H, CHAr),
8.04 – 7.95 (m, 1H, CHAr), 7.89 (dd, 3J = 7.2, 3J = 6.8 Hz, 1H, CHAr), 7.85 – 7.77 (m, 2H,
CHAr), 7.75 – 7.61 (m, 3H, CHAr), 7.61 – 7.46 (m, 3H, CHAr).
19F NMR (282 MHz, CDCl3) δ -62.62.
13C NMR (75 MHz, CDCl3) δ 144.7, 139.4, 137.8, 136.6, 136.5, 132.2 (CAr), 130.8 (2CHAr),
130.6 (CAr), 129.8 (q, 2J = 32.5 Hz, C-CF3), 129.3 (CAr), 127.1, 127.0, 126.8, 126.6 (CHAr),
125.5 (q, 3J = 3.8 Hz, 2CHAr), 125.1, 124.9 (CHAr), 124.5 (q, 1J = 272.0 Hz, CF3), 123.2, 121.7,
120.9 (CHAr).
HRMS (EI): Calculated for C23H13F3S1 (M+): 378.06848, found: 378.06817.
4-(Benzo[b]naphtho[2,1-d]thiophen-5-yl)phenol 4.3d
White solid, 81% (53 mg). M.p.: 195–197 °C.
IR (ATR, cm-1): 3534.5 (w), 3163.9 (w, br), 3040.1 (w), 2924.4 (w),
1888.9 (w), 1592.8 (m), 1506.0 (m), 1433.2 (m), 1339.9 (m), 1226.1 (s),
991.2 (m), 832.1 (s), 748.9 (s), 607.3 (m).
1H NMR (300 MHz, CDCl3) δ 8.24 – 8.15 (m, 2H, CHAr), 8.09 (s, 1H,
CHAr), 8.04 – 7.92 (m, 2H, CHAr), 7.69 – 7.58 (m, 1H, CHAr), 7.56 – 7.41 (m, 5H, CHAr),
7.08 – 6.96 (m, 2H, CHAr), 4.90 (s, br, 1H, OH).
13C NMR (63 MHz, CDCl3) δ 155.2, 139.4, 137.7, 136.8, 136.8, 133.5, 132.3 (CAr), 131.7
(2CHAr), 131.4, 129.3 (CAr), 127.6, 126.8, 126.5, 126.4, 125.0, 124.7, 123.1, 121.8, 120.7
(CHAr), 115.4 (2CHAr).
MS (EI, 70 eV): m/z (%) = 326 (M+, 100), 308 (9), 295 (17), 271 (4), 224 (2), 148 (18), 135
(6).
HRMS (EI): Calculated for C22H14O1S1 (M+): 326.07599, found: 326.07548.
4-(Benzo[b]naphtho[2,1-d]thiophen-5-yl)benzonitrile 4.3e
White solid, 68% (46 mg). M.p.: 215–216 °C.
IR (ATR, cm-1): 3405.7 (w), 3046.0 (w), 3014.7 (w), 2223.6 (m), 1938.8
(w), 1602.3 (m), 1508.3 (m), 1341.4 (m), 989.5 (m), 836.7 (m), 747.0
(s), 720.4 (m), 614.1 (m), 573.9 (m).

APPENDIX
xl
1H NMR (300 MHz, CDCl3) δ 8.28 – 8.15 (m, 2H, CHAr), 8.07 (s, 1H, CHAr), 8.03 – 7.94 (m,
1H, CHAr), 7.92 – 7.78 (m, 3H, CHAr), 7.75 – 7.60 (m, 3H, CHAr), 7.60 – 7.45 (m, 3H, CHAr).
13C NMR (75 MHz, CDCl3) δ 145.7, 139.2, 138.0, 136.4, 135.9 (CAr), 132.2 (2CHAr), 132.0
(CAr), 131.0 (2CHAr), 130.1, 129.2 (CAr), 127.1, 126.9, 126.6, 126.5, 125.1, 124.8, 123.0, 121.6,
120.7 (CHAr), 118.9 (CN), 111.3 (CAr).
MS (EI, 70 eV): m/z (%) = 335 (M+, 100), 167 (15), 153 (14), 131 (9).
HRMS (EI): Calculated for C23H13N1S1 (M+): 335.07632, found: 335.07571.
5-(4-Fluorophenyl)benzo[b]naphtho[2,1-d]thiophene 4.3f
Yellowish solid, 89% (58 mg). M.p.: 176–177 °C.
IR (ATR, cm-1): 3063.9 (w), 2926.3 (w), 1889.9 (w), 1667.1 (w), 1597.5
(m), 1504.2 (m), 1434.1 (m), 1215.5 (m), 1153.0 (m), 1089.1 (m), 876.9
(m), 835.6 (m), 749.0 (s), 605.1 (m).
1H NMR (250 MHz, CDCl3) δ 8.28 – 8.14 (m, 2H, CHAr), 8.08 (s, 1H,
CHAr), 8.03 – 7.88 (m, 2H, CHAr), 7.71 – 7.58 (m, 1H, CHAr), 7.58 – 7.46 (m, 5H, CHAr),
7.33 – 7.17 (m, 2H, CHAr).
19F NMR (235 MHz, CDCl3) δ -115.13.
13C NMR (63 MHz, CDCl3) δ 162.5 (d, 1J = 246.5 Hz, CF), 139.4, 137.2, 137.0 (CAr), 136.8 (d,
4J = 3.4 Hz, CAr), 136.7, 132.2 (CAr), 132.0 (d, 3J = 8.0 Hz, 2CHAr), 131.1, 129.3 (CAr), 127.3,
126.9, 126.6, 126.5, 125.0, 124.8, 123.1, 121.7, 120.8, 115.5 (d, 2J = 21.3 Hz, 2CHAr).
MS (EI, 70 eV): m/z (%) = 328 (M+, 100), 163 (23), 154 (10), 140 (8).
HRMS (EI): Calculated for C22H13F1S1 (M+): 328.07165, found: 328.07113.
5-(2-Fluorophenyl)benzo[b]naphtho[2,1-d]thiophene 4.3g
Yellowish solid, 75% (49 mg). M.p.: 193–194 °C.
IR (ATR, cm-1): 3054.6 (w), 1693.9 (w), 1575.4 (w), 1488.7 (m), 1449.4
(w), 1339.4 (w), 1247.6 (w), 1210.9 (w), 1095.3 (w), 887.4 (m), 746.9
(s), 725.7 (m), 611.8 (m).
1H NMR (250 MHz, CDCl3) δ 8.25 – 8.17 (m, 2H, CHAr), 8.15 (s, 1H, CHAr), 8.03 – 7.94 (m,
1H, CHAr), 7.82 – 7.74 (m, 1H, CHAr), 7.69 – 7.60 (m, 1H, CHAr), 7.57 – 7.45 (m, 5H, CHAr),
7.38 – 7.22 (m, 2H, CHAr).
19F NMR (235 MHz, CDCl3) δ -113.51.

APPENDIX
xli
13C NMR (63 MHz, CDCl3) δ 160.5 (d, 1J = 246.7 Hz, CF), 139.3, 137.8, 136.7 (CAr), 132.8 (d,
4J = 3.4 Hz, CHAr), 132.2, 131.7, 131.1 (CAr), 129.8 (d, 3J = 8.0 Hz, CHAr), 129.2, 128.2 (d,
2J = 16.3 Hz, CAr), 127.3 (d, 4J = 1.2 Hz, CHAr), 126.9, 126.7, 126.5, 125.0, 124.8 (CHAr), 124.4
(d, 3J = 3.6 Hz, CHAr), 123.1, 121.8, 121.6, 115.9 (d, 2J = 22.2 Hz, CHAr).
MS (EI, 70 eV): m/z (%) = 328 (M+, 100), 163 (23), 154 (11), 131(5).
HRMS (EI): Calculated for C22H13F1S1 (M+): 328.07165, found: 328.07116.
4-(Benzo[b]naphtho[2,1-d]thiophen-5-yl)pyridine 4.3h
White solid, 56% (35 mg). M.p.: 170–171 °C.
IR (ATR, cm-1): 3066.6 (w), 3022.5 (w), 2922.7 (w), 1935.3 (w), 1693.5
(w), 1591.4 (m), 1402.2 (m), 1340.3 (w), 1062.1 (w), 988.3 (w), 823.4
(m), 749.2 (s), 726.3 (s), 617.4 (m).
1H NMR (300 MHz, CDCl3) δ 8.85 (s, br, 2H, CHAr), 8.26 – 8.16 (m, 2H,
CHAr), 8.10 (s, 1H, CHAr), 8.03 – 7.96 (m, 1H, CHAr), 7.93 (d, 3J = 8.2 Hz, 1H, CHAr),
7.73 – 7.43 (m, 6H, CHAr).
13C NMR (75 MHz, CDCl3) δ 149.6, 139.3, 138.4, 136.5, 135.1, 132.2, 129.9, 129.4 (CAr),
127.3, 127.1, 126.7, 126.7, 125.2, 124.9, 123.2, 121.7, 120.7 (CHAr). (signals of 4CHAr could
not be detected).
MS (EI, 70 eV): m/z (%) = 311 (M+, 100), 282 (12), 155 (23), 141 (14), 107 (4). HRMS (EI):
Calculated for C21H13N1S1 (M+): 311.07632, found: 311.07582.
5-(4-Methoxyphenyl)benzo[b]naphtho[2,1-d]thiophene 4.3i
White solid, 95% (65 mg). M.p.: 178–179 °C.
IR (ATR, cm-1): 3043.2 (w), 2947.3 (w), 2832.6 (w), 1604.6 (m),
1509.9 (m), 1432.5 (m), 1290.1 (w), 1240.1 (m), 1168.9 (m), 1031.0
(m), 875.6 (m), 829.8 (m), 751.5 (s), 723.4 (,), 606.7 (m). 551.1 (m).
1H NMR (250 MHz, CDCl3) δ 8.25 – 8.15 (m, 2H, CHAr), 8.11 (s, 1H,
CHAr), 8.06 – 7.93 (m, 2H, CHAr), 7.69 – 7.58 (m, 1H, CHAr), 7.58 – 7.44 (m, 5H, CHAr),
7.16 – 7.05 (m, 2H, CHAr), 3.94 (s, 3H, OCH3).
13C NMR (63 MHz, CDCl3) δ 159.2, 139.4, 137.8, 136.8, 136.7, 133.2, 132.3 (CAr), 131.5
(2CHAr), 131.4, 129.3 (CAr), 127.6, 126.7, 126.4, 126.4, 124.9, 124.7, 123.1, 121.7, 120.7
(CHAr), 113.9 (2CHAr), 55.5 (OCH3).

APPENDIX
xlii
MS (EI, 70 eV): m/z (%) = 340 (M+, 100), 325 (26), 295 (36), 269 (3), 148 (26), 135 (9).
HRMS (EI): Calculated for C23H16O1S1 (M+): 340.09164, found: 340.09158.
5-(3-Methoxyphenyl)benzo[b]naphtho[2,1-d]thiophene 4.3j
White solid, 80% (55 mg). M.p.: 138–139 °C.
IR (ATR, cm-1): 3047.3 (w), 2960.5 (w), 2831.0 (w), 1705.6 (w), 1582,9
(m), 1485.3 (m), 1459.4 (m), 1255.7 (m), 1214.0 (m), 1077.1 (m), 1034.1
(m), 952,2 (m), 789.8 (s), 748.3 (s), 649.4 (m), 570.3 (m).
1H NMR (250 MHz, CDCl3) δ 8.26 – 8.16 (m, 2H, CHAr), 8.14 (s, 1H,
CHAr), 8.07 – 7.90 (m, 2H, CHAr), 7.68-7.59 (m, 1H, CHAr), 7.58 – 7.41 (m, 4H, CHAr),
7.22 – 7.10 (m, 2H, CHAr), 7.09-7.00 (m, 1H, CHAr), 3.90 (s, 3H, OCH3).
13C NMR (63 MHz, CDCl3) δ 159.7, 142.3, 139.4, 138.0, 137.1, 136.8, 132.2, 131.1 (CAr),
129.5 (CHAr), 129.3 (CAr), 127.6, 126.8, 126.5, 126.4, 124.9, 124.8, 123.1, 123.0, 121.8, 120.6,
116.1, 113.1 (CHAr), 55.5 (OCH3).
MS (EI, 70 eV): m/z (%) = 340 (M+, 100), 325 (3), 308 (10), 295 (33), 148 (22), 135 (8).
HRMS (EI): Calculated for C23H16O1S1 (M+): 340.09164, found: 340.09152.
5-(2-Methoxyphenyl)benzo[b]naphtho[2,1-d]thiophene 4.3k
White solid, 71% (48 mg). M.p.: 216–218 °C.
IR (ATR, cm-1): 3064.5 (w), 2952.0 (w), 2834.7 (w), 1578.4 (w), 1491.1
(m), 1461.4 (m), 1432.8 (m), 1339.4 (w), 1289.0 (m), 1241.1 (m), 1111.2
(m), 1024.2 (m), 875.4 (m), 746.2 (s), 724.4 (m), 614.1 (m), 551.5 (m).
1H NMR (300 MHz, CDCl3) δ 8.23 – 8.14 (m, 2H, CHAr), 8.12 (s, 1H, CHAr), 8.02 – 7.93 (m,
1H, CHAr), 7.69 (d, 3J = 8.4 Hz, 1H, CHAr), 7.64-7.56 (m, 1H, CHAr), 7.55 – 7.43 (m, 4H,
CHAr), 7.40 (dd, 3J = 7.4, 4J = 1.7 Hz, 1H, CHAr), 7.19 – 7.05 (m, 2H, CHAr), 3.72 (s, 3H,
OCH3).
13C NMR (63 MHz, CDCl3) δ 157.6, 139.3, 137.1, 136.9, 134.9, 132.4 (CAr), 132.3 (CHAr),
131.6, 129.7 (CAr), 129.4 (CHAr), 129.0 (CAr), 127.9, 126.6, 126.3, 126.2, 124.8, 124.6, 123.1,
121.8, 121.1, 120.9, 111.2 (CHAr), 55.7 (OCH3).
MS (EI, 70 eV): m/z (%) = 340 (M+, 100), 324 (21), 308 (5), 297 (28), 265 (4), 148 (21), 135
(9).

APPENDIX
xliii
HRMS (EI): Calculated for C23H16O1S1 (M+): 340.09164, found: 340.09157.
5-(Naphthalen-1-yl)benzo[b]naphtho[2,1-d]thiophene 4.3l
White solid, 65% (47 mg). M.p.: 217–218 °C
IR (ATR, cm-1): 3043.8 (w), 2952.5 (w), 2850.6 (w), 1921.1 (w), 1506.2
(w), 1432.7 (m), 1235.3 (w), 1102.5 (w), 1018.6 (m), 879.7 (m), 798.7
(m), 772.9 (s), 748.4 (s), 725.4 (s), 698.6 (m), 600.2 (m), 578.3 (m).
1H NMR (300 MHz, CDCl3) δ 8.28 – 8.23 (m, 1H, CHAr), 8.21 (s, 1H,
CHAr), 8.19 – 8.10 (m, 1H, CHAr), 8.07 – 7.92 (m, 3H, CHAr), 7.70 – 7.56 (m, 3H, CHAr),
7.55 – 7.43 (m, 5H, CHAr), 7.43 – 7.27 (m, 2H, CHAr).
13C NMR (75 MHz, CDCl3) δ 139.4, 138.5, 137.3, 136.8, 136.3, 133.7, 133.2, 132.4, 132.2,
129.1 (CAr), 128.4, 128.3, 128.3, 128.0, 126.9, 126.8, 126.5, 126.5, 126.3, 126.1, 125.6, 124.9,
124.8, 123.2, 121.8, 121.7 (CHAr).
MS (EI, 70 eV): m/z (%) = 360 (M+, 100), 179 (46), 120 (3).
HRMS (EI): Calculated for C26H16S1 (M+): 360.09672, found: 360.09677.
5-(Naphthalen-2-yl)benzo[b]naphtho[2,1-d]thiophene 4.3m
White solid, 67% (48 mg). M.p.: 190–192 °C.
IR (ATR, cm-1): 3036.6 (w), 2919.9 (w), 2849.9 (w), 1920.8 (w),
1704.2 (w), 1596.4 (w), 1434.9 (w), 1240.1 (w), 962.3 (m), 858.5
(m), 815.7 (m), 747.2 (s), 726.9 (s), 669.6 (m), 627.2 (m).
1H NMR (250 MHz, CDCl3) δ 8.27 – 8.16 (m, 3H, CHAr), 8.09 – 7.91
(m, 6H, CHAr), 7.75 – 7.47 (m, 7H, CHAr).
13C NMR (63 MHz, CDCl3) δ 139.4, 138.5, 138.1, 137.2, 136.8, 133.6, 132.8, 132.4, 131.3,
129.4 (CAr), 129.1, 128.8, 128.2, 127.9, 127.9, 127.6, 126.9 (CHAr), 126.6 (2CHAr), 126.5,
126.3, 125.0, 124.8, 123.1, 121.8, 121.1 (CHAr).
MS (EI, 70 eV): m/z (%) = 360 (M+, 100), 179 (43), 143 (4).
HRMS (EI): Calculated for C26H16S1 (M+): 360.09672, found: 360.09575.
5-([1,1'-Biphenyl]-4-yl)benzo[b]naphtho[2,1-d]thiophene 4.3n

APPENDIX
xliv
White solid, 63% (49 mg). M.p.: 190–192 °C.
IR (ATR, cm-1): 3031.2 (w), 1599.4 (w), 1487.9 (w), 1433.1 (w), 1339.8
(w), 1235.3 (w), 990.0 (w), 888.3 (w), 842.8 (m), 747.0 (s), 724.2 (s),
693.0 (m), 645.3 (m), 579.8 (m).
1H NMR (250 MHz, CDCl3) δ 8.27 – 8.18 (m, 2H, CHAr), 8.17 (s, 1H,
CHAr), 8.13 – 8.06 (m, 1H, CHAr), 8.04 – 7.95 (m, 1H, CHAr), 7.84 – 7.60 (m, 7H, CHAr),
7.60 – 7.46 (m, 5H, CHAr), 7.46 – 7.37 (m, 1H, CHAr).
13C NMR (63 MHz, CDCl3) δ 140.9, 140.5, 139.9, 139.4, 137.7, 137.1, 136.8, 132.3, 131.1
(CAr), 130.9 (2CHAr), 129.4 (CAr), 129.0 (2CHAr), 127.6, 127.6 (CHAr), 127.3 (2CHAr), 127.2
(2CHAr), 126.9, 126.6, 126.4, 125.0, 124.8, 123.1, 121.8, 120.8 (CHAr).
MS (EI, 70 eV): m/z (%) = 386 (M+, 100), 308 (20), 193 (10), 154 (6), 77 (5).
HRMS (EI): Calculated for C28H18S1 (M+): 386.11237, found: 386.11180.
6-Phenylbenzo[b]benzo[4,5]thio[3,2-g]benzothiophene 4.3o
Yellow, 34% (25 mg). M.p.: 230–231 °C.
IR (ATR, cm-1): 3054.1 (w), 3025.7 (w), 2920.0 (w), 2849.8 (w),
1942.8 (w), 1905.1 (w), 1820.8 (w), 1788.0 (w), 1750.0 (w), 1693.4
(w), 1600.7 (w), 1435.4 (w), 1405.1 (m), 1330.9 (m), 1231.1(m),
1157.7 (m), 1111.9 (m), 876.9 (m), 755.7 (s), 728.2 (s), 699.6 (s),
646.1 (m), 568.9 (m).
1H NMR (300 MHz, CDCl3) δ 8.21 – 8.12 (m, 1H, CHAr), 8.02 (s, 1H, CHAr), 7.97 – 7.91 (m,
1H, CHAr), 7.91 – 7.86 (m, 1H, CHAr), 7.60 – 7.54 (m, 5H, CHAr), 7.53 – 7.44 (m, 2H, CHAr),
7.42 – 7.33 (m, 1H, CHAr), 7.20 – 7.07 (m, 2H, CHAr).
13C NMR (63 MHz, CDCl3) δ 141.5, 139.7, 139.4, 136.5, 136.2, 136.1, 133.8, 133.7, 132.4,
132.0 (CAr), 129.6 (2CHAr), 128.9 (2CHAr), 128.1, 127.0, 126.3, 125.1, 125.0, 124.4, 123.2,
123.0, 122.2, 120.4 (CHAr).
MS (EI, 70 eV): m/z (%) = 366 (M+, 100), 332 (5), 182 (30), 160 (6), 121 (2).
HR-MS(+ESI): calculated for C24H15S2 (M+H)+: 367.06097, found: 367.06144.
5-Phenylnaphtho[1,2-b]benzofuran 4.5a

APPENDIX
xlv
White solid, 60% (35 mg). M.p.: 119–120 °C.
IR (ATR, cm-1): 3030.2 (w), 2922.0 (w), 2850.8 (w), 1892.7 (w), 1576.1
(w), 1493.1 (w), 1457.9 (m), 1440.5 (m), 1403.5 (m), 1355.6 (m), 1235.5
(m), 1192.5 (m), 1171.9 (m), 1048.3 (m), 883.3 (m), 787.1 (m), 769.4
(m), 741.9 (s), 697.9 (s), 603.0 (m), 582.5 (m).
1H NMR (300 MHz, CDCl3) δ 8.54 (dd, 3J = 8.2, 5J = 0.5 Hz, 1H, CHAr), 8.00 (d, 3J = 8.2 Hz,
2H, CHAr), 7.96 (s, 1H, CHAr), 7.75 (d, 3J = 8.2 Hz, 1H, CHAr), 7.71-7.63 (m, 1H, CHAr),
7.61 – 7.37 (m, 8H, CHAr).
13C NMR (75 MHz, CDCl3) δ 156.3, 151.8, 141.1, 136.2, 131.6 (CAr), 130.5 (2CHAr), 128.5
(2CHAr), 127.4, 127.2, 126.5, 126.5, 126.3 (CHAr), 125.2 (CAr), 123.2 (CHAr), 121.7 (CAr),
121.3, 120.4, 119.5, 118.8 (CAr), 112.0 (CHAr).
MS (EI, 70 eV): m/z (%) = 294 (M+, 100), 263 (15), 239 (4), 146 (8), 132 (15), 119 (10).
HRMS (EI): Calculated for C22H14O1 (M+): 294.10392, found: 294.10358.
5-(p-Tolyl)naphtho[1,2-b]benzofuran 4.5b
White solid, 45% (28 mg). M.p.: 173–174 °C.
IR (ATR, cm-1): 3025.9 (w), 2917.4 (w), 2863.5 (w), 2731.7 (w), 1930.0
(w), 1814.0 (w), 1634.2 (w), 1577.5 (w), 1459.1 (m), 1351.9 (m),
1236.6 (w), 1195.9 (m), 1049.3 (m), 822.6 (m), 766.1 (m), 739.8 (s),
601.2 (m).
1H NMR (250 MHz, CDCl3) δ 8.58 – 8.49 (m, 1H, CHAr), 8.08 – 7.97 (m, 2H, CHAr), 7.95 (s,
1H, CHAr), 7.81 – 7.60 (m, 2H, CHAr), 7.58 – 7.30 (m, 7H, CHAr), 2.51 (s, 3H, CH3).
13C NMR (63 MHz, CDCl3) δ 156.3, 151.7, 138.2, 137.1, 136.1, 131.7 (CAr), 130.4 (2CHAr),
129.2 (2CHAr), 127.3, 126.4, 126.4, 126.2 (CHAr), 125.3 (CAr), 123.1 (CHAr), 121.7 (CAr), 121.2,
120.4, 119.4 (CHAr), 118.8 (CAr), 112.0 (CHAr), 21.4 (CH3).
MS (EI, 70 eV): m/z (%) = 308 (M+, 100), 292 (10), 279 (6), 263 (5), 250 (5), 207 (10), 187
(4), 154 (8), 132 (11).
HRMS (EI): Calculated for C23H16O1 (M+): 308.11957, found: 308.11961.
5-(4-Methoxyphenyl)naphtho[1,2-b]benzofuran 4.5c

APPENDIX
xlvi
White solid, 77% (50 mg). M.p.: 168–169 °C.
IR (ATR, cm-1): 3019.8 (w), 2958.7 (w), 2906.4 (w), 2542.6 (w),
2351.3 (w), 2051.5 (w), 1923.9 (w), 1605.3 (m), 1509.4 (m), 1460.4
(m), 1355.7 (m), 1238.0 (m), 1104.2 (m), 1031.5 (m), 827.2 (m), 768.5
(m), 745.5 (s), 686.4 (m), 601.3 (m), 555.4 (m).
1H NMR (250 MHz, CDCl3) δ 8.58 – 8.47 (m, 1H, CHAr), 8.06 – 7.97 (m, 2H, CHAr), 7.94 (s,
1H), 7.79 – 7.60 (m, 2H, CHAr), 7.60 – 7.34 (m, 5H, CHAr), 7.17 – 7.00 (m, 2H, CHAr), 3.93 (s,
3H, OCH3).
13C NMR (63 MHz, CDCl3) δ 159.1 (C-OCH3), 156.3, 151.7, 135.8, 133.4, 131.8 (CAr), 131.6
(2CHAr), 127.2, 126.4, 126.4, 126.2 (CHAr), 125.3 (CAr), 123.1 (CHAr), 121.7 (CAr), 121.2,
120.4, 119.4 (CHAr), 118.8 (CAr), 113.9 (2CHAr), 112.0 (CHAr), 55.5 (OCH3).
MS (EI, 70 eV): m/z (%) = 324 (M+, 100), 309 (31), 279 (21), 252 (17), 226 (5), 162 (8), 140
(7), 113 (10).
HRMS (EI): Calculated for C23H16O2 (M+): 324.11448, found: 324.11403.
5-([1,1'-Biphenyl]-4-yl)naphtho[1,2-b]benzofuran 4.5d
White solid, 41% (30 mg). M.p.: 187–188 °C.
IR (ATR, cm-1): 3029.6 (w), 2922.4 (w), 2850.5 (w), 1927.1 (w), 1580.5
(w), 1487.1 (m), 1440.6 (m), 1352.2 (m), 1236.5 (w), 1177.3 (m), 1049.4
(m), 838.7 (m), 763.6 (m), 741.9 (s), 692.7 (s), 606.2 (m), 587.4 (m).
1H NMR (300 MHz, CDCl3) δ 8.60 – 8.53 (m, 1H, CHAr), 8.09 (d,
3J = 8.5 Hz, 1H, CHAr), 8.05 – 7.97 (m, 2H, CHAr), 7.82 – 7.70 (m, 5H, CHAr), 7.70 – 7.62 (m,
3H, CHAr), 7.59 – 7.47 (m, 4H, CHAr), 7.47 – 7.36 (m, 2H, CHAr).
13C NMR (75 MHz, CDCl3) δ 156.3, 151.9, 141.0, 140.3, 140.1, 135.7, 131.6 (CAr), 130.9
(2CHAr), 129.0 (2CHAr), 127.5 (CHAr), 127.3 (2CHAr), 127.2 (3CHAr), 126.6, 126.5, 126.3
(CHAr), 125.2 (CAr), 123.2 (CHAr), 121.7 (CAr), 121.3, 120.4, 119.6 (CHAr), 118.8 (CAr), 112.0
(CHAr).
MS (EI, 70 eV): m/z (%) = 370 (M+, 100), 339 (4), 292 (13), 263 (6) CHAr, 185 (8), 169 (7).
HRMS (EI): Calculated for C28H18O1 (M+): 370.13522, found: 370.13465.
11-Methyl-5-phenyl-11H-benzo[a]carbazole 4.6a

APPENDIX
xlvii
Yellowish solid, 31% (19 mg). M. p.: 153–154 °C.
IR (ATR, cm-1): 3079.1 (w), 3055.4 (w), 3026.2 (w), 2918.9 (w), 1597.8
(w), 1518.4 (w), 1448.8 (m), 1334.5 (m), 1263.1 (m), 1133.2 (m), 1017.0
(m), 882.1 (m), 769.2 (m), 735.3 (s), 699.5 (s), 578.3 (s).
1H NMR (250 MHz, CDCl3) δ 8.81 (d, 3J = 8.1 Hz, 1H, CHAr),
8.17 – 8.04 (m, 3H, CHAr), 7.66 – 7.43 (m, 9H, CHAr), 7.35 – 7.27 (m, 1H, CHAr), 4.47 (s, 3H,
NCH3).
13C NMR (63 MHz, CDCl3) δ 142.1, 141.2, 135.3, 132.7, 132.0 (CAr), 130.7 (2CHAr), 128.4
(2CHAr), 127.9, 127.0, 125.2, 125.0, 124.8 (CHAr), 123.2, 123.0 (CAr), 122.4, 120.4, 119.8,
119.8 (CHAr), 118.6 (CAr), 109.2 (CHAr), 34.4 (NCH3).
MS (EI, 70 eV): m/z (%) = 307 (M+, 100), 291 (35), 152 (12), 146 (23), 131 (5), 118 (3).
HRMS (EI): Calculated for C23H17N1 (M+): 307.13555, found: 307.13495.
5-(4-Methoxyphenyl)-11-methyl-11H-benzo[a]carbazole 4.6b
Yellowish solid, 45% (30 mg). M.p.: 154–155 °C.
IR (ATR, cm-1): 3119.2 (w), 3029.2 (w), 2956.9 (w), 2836.1 (w),
1926.1 (w), 1766.2 (w), 1658.8 (w), 1606.6 (m), 1572.5 (m), 1508.6
(s), 1440.4 (s), 1335.6 (m), 1233.7 (s), 1169.8 (s), 1103.7 (m), 1027.6
(s), 884.2 (m), 835.5 (s), 738.4 (s), 690.2 (m), 547.7 (s).
1H NMR (300 MHz, CDCl3) δ 8.79 (d, 3J = 8.1 Hz, 1H, CHAr),
8.19 – 8.05 (m, 3H, CHAr), 7.66 – 7.43 (m, 6H, CHAr), 7.37 – 7.28 (m, 1H, CHAr), 7.12 – 7.04
(m, 2H, CHAr), 4.44 (s, 3H, NCH3), 3.93 (s, 3H, OCH3).
13C NMR (75 MHz, CDCl3) δ 158.9, 141.2, 135.2, 134.4, 132.3, 132.3 (CAr), 131.7 (2CHAr),
127.9, 125.1, 124.9, 124.7 (CHAr), 123.2, 123.0 (CAr), 122.4, 120.4, 119.8, 119.7 (CHAr), 118.6
(CAr), 113.9 (2CHAr), 109.2 (CHAr), 55.5 (OCH3), 34.3 (NCH3).
MS (EI, 70 eV): m/z (%) = 337 (M+, 100), 322 (37), 278 (27), 168 (10), 145 (20).
HRMS (EI): Calculated for C24H19O1N1 (M+): 337.14612, found: 337.14559.
5-([1,1'-Biphenyl]-4-yl)-11-methyl-11H-benzo[a]carbazole 4.6c

APPENDIX
xlviii
White solid, 39% (30 mg). M.p.: 218–219 °C.
IR (ATR, cm-1): 3056.4 (w), 3026.0 (w), 1594.1 (w), 1516.7 (w), 1464.9
(w), 1375.0 (w), 1334.9 (w), 1264.7 (w), 1227.0 (w), 1062.7 (w), 897.7
(m), 845.0 (m), 770.6 (m), 726.0 (s), 693.7 (s), 637.5 (m), 583.0 (m).
1H NMR (300 MHz, CDCl3) δ 8.82 (d, 3J = 8.4 Hz, 1H, CHAr),
8.25 – 8.11 (m, 3H, CHAr), 7.85 – 7.45 (m, 12H, CHAr), 7.45 – 7.29 (m,
2H, CHAr), 4.47 (s, 3H, NCH3).
13C NMR (63 MHz, CDCl3) δ 141.2, 141.2, 141.1, 139.9, 135.4, 132.3, 132.0 (CAr), 131.1
(2CHAr), 129.0 (2CHAr), 127.9, 127.4 (CHAr ), 127.3 (2CHAr), 127.2 (2CHAr), 125.3, 125.1,
124.9 (CHAr), 123.2, 123.1 (CAr), 122.4, 120.5, 119.8, 119.8 (CHAr), 118.6 (CAr), 109.2 (CHAr),
34.4 (NCH3).
MS (EI, 70 eV): m/z (%) = 383 (M+, 100), 367 (14), 341 (1), 291 (10), 184 (9), 152 (4), 77 (4).
HRMS (EI): Calculated for C29H21N1 (M+): 383.16685, found 383.16645.
2-(2-Chlorophenyl)-3-(1-phenylvinyl)benzo[b]thiophene 4.4
Yellowish oil.
IR (ATR, cm-1):3054.8 (w), 3023.3 (w), 1609.9 (m), 1491.6 (m), 1430.9
(m), 1241.0 (w), 1059.9 (m), 1026.7 (w), 904.5 (m), 777.9 (m), 748.6
(s), 732.2 (s), 701.8 (s), 597.7 (m).
1H NMR (300 MHz, CDCl3) δ 8.00 – 7.81 (m, 1H, CHAr), 7.59 – 7.45
(m, 1H, CHAr), 7.45 – 7.34 (m, 5H, CHAr), 7.34 – 7.13 (m, 6H, CHAr), 5.82 (d, 2J = 1.3 Hz, 1H,
C=CH2), 5.34 (d, 2J = 1.3 Hz, 1H, C=CH2).
13C NMR (75 MHz, CDCl3) δ 142.2, 140.1, 139.7, 139.4, 137.8, 135.8, 134.8, 133.4 (CAr),
132.8, 129.8, 129.7 (CHAr), 128.3 (2CHAr), 127.8 (CHAr), 126.9 (2CHAr), 126.4, 124.7, 124.4,
124.2, 122.2 (CHAr), 118.1 (C=CH2).
MS (EI, 70 eV): m/z (%) = 346 (M+, 16), 311 (100), 295 (7), 269 (6), 234 (38), 189 (8), 154
(16), 77 (7).
HRMS (EI): Calculated for C22H1535Cl1S1 (M
+): 346.05775, found: 346.05749.
General procedure for the synthesis of pyrazoles
Acetophenone 5.1 (0.5 mmol, 1 eq) and phenylhydrazine (0.525 mmol, 1.05 eq) were
dissolved in 2 mL of DCM. The solution was stirred for 1 min at room temperature and then

APPENDIX
xlix
the solvent was removed under reduced pressure. The residue was stirred at room temperature
and 3 drops of acetic acid were added. The reaction mixture was stirred for 5 min to complete
the reaction. (In most cases, the product was solid and the reaction mixture became solid as the
reaction completed). The reaction carried out nearly qualitative. Then the crude product was
dissolved in 10 mL of DCM. After that, solvent, water, and acetic acid were removed under
reduced pressure and the product was transferred to the next step without further purification.
To a solution of hyrazone 5.2 (0.5 mmol) in 7 mL of dry THF was added n-butyllithium
(0.44 mL, 2.2 equiv., 2.5 M solution in hexane) slowly at -78 °C under Ar. After adding n-
-butyllithium, the mixture was allowed to warm to 20 °C and stirred at that temperature for 15
min. Then the reaction was cooled to -78 °C again and ethyl perfluorocarboxylate 5.3 (1.5 eq,
solution in 1 mL THF) was added at this temperature. The reaction was allowed to warm to
20 °C and stirred for 30 min, subsequently, 1 mL of TFA was added. The mixture was stirred
under reflux for 2 h. After cooling, a saturated aqueous solution of NaHCO3 was added until no
CO2 evolution was observed. Then THF was removed and the remained water was extracted
with ethyl acetate (10 mL 3x). Combined organic layers were dried over MgSO4 and the solvent
was removed under reduced pressure. The residue was purified by chromatography (silica gel,
n-heptane/DCM).
1,3-Diphenyl-5-(trifluoromethyl)-1H-pyrazole 5.4a
White solid, 89% (128 mg). M.p.: 53–54 °C.
IR (ATR, cm-1): ν = 549 (s), 615 (m), 685 (s), 760 (s), 773 (s), 812 (s),
956 (m), 9872 (s), 1028 (s), 1072 (s), 1118 (s), 1138 (m), 1211 (s), 1232
(s), 1288 (s), 1363 (m), 1444 (s), 1502 (m), 1556 (m), 1593 (m), 3054 (w), 3070 (w), 3139 (w).
1H NMR (300 MHz, CDCl3) δ 7.92 – 7.83 (m, 2H, CHPh), 7.65 – 7.32 (m, 8H, CHPh), 7.12 (s,
1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -57.59.
13C NMR (75 MHz, CDCl3) δ 151.8, 139.3 (CAr), 134.0 (q, 2J = 39.2 Hz, C-CF3), 131.9 (CAr),
129.4 (CHAr), 129.26 (2CHAr), 128.9 (2CHAr), 128.8 (CHAr), 126.0 (2CHAr), 125.9, 125.9
(CHAr), 119.9 (q, 1J = 269.2 Hz, CF3), 106.2 (q, 3J = 2.4 Hz, CHHetAr).
GC-MS (EI, 70 eV): m/z (%) = 288 (100), 267 (37), 219 (9), 77 (20).
HRMS (EI): calcd. for C16H11F3N2 ([M]+): 288.08688, found: 288.08678.
3-(4-Methoxyphenyl)-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 5.4b

APPENDIX
l
Yellow solid, 79% (127 mg). M.p.: 77–78 °C.
IR (ATR, cm-1): ν = 548 (m), 617 (m), 685 (s), 767 (s), 810 (s),
833 (m), 956 (m), 987 (s), 1031 (s), 1080 (s), 1088 (s), 1115 (s),
1151 (s), 1209 (s), 1248 (s), 1290 (s), 1359 (w), 1435 (s), 1450 (s), 1502 (s), 1558 (m), 1595
(m), 1614 (m), 2845 (w), 2943 (w), 2970 (w), 3022 (w), 3068 (w), 3130 (w).
1H NMR (300 MHz, CDCl3) δ 7.86 – 7.73 (m, 2H, CHAr), 7.62 – 7.44 (m, 5H, CHAr), 7.04 (s,
1H, CHAr), 7.02 – 6.90 (m, 2H, CHAr), 3.85 (s, 3H, OCH3).
19F NMR (282 MHz, CDCl3) δ -57.59.
13C NMR (75 MHz, CDCl3) δ 160.2, 151.6, 139.4 (CAr), 133.9 (q, 2J = 39.0 Hz, C-CF3), 129.3
(CHAr), 129.2 (2CHAr), 127.3 (2CHAr), 125.9, 125.9 (CHAr), 124.5 (CAr), 119.9 (q,
1J = 269.1 Hz, CF3), 114.3 (2CHAr), 105.8 (q, 3J = 2.4 Hz, CHHetAr), 55.47 (OCH3).
GC-MS (EI, 70 eV): m/z (%) = 318 (100), 303 (26), 275 (12), 77 (12).
HRMS (EI): calcd. for C17H13F3N2O ([M]+): 318.09745, found: 318.09788.
3-(3-Methoxyphenyl)-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 5.4c
Pale yellow oil, 81% (129 mg).
IR (ATR, cm-1): ν = 544 (m), 627 (m), 688 (s), 766 (s), 816 (m), 847
(m), 916 (w), 987 (s), 1041 (s), 1089 (s), 1124 (s), 1143 (s), 1197 (s),
1224 (s), 1257 (m), 1284 (m), 1354 (m), 1433 (s), 1464 (m), 1500 (s), 1556 (m), 1597 (m), 2835
(w), 2939 (w), 3003 (w), 3063 (w), 3138 (w).
1H NMR (300 MHz, CDCl3) δ 7.63 – 7.47 (m, 5H, CHAr), 7.47 – 7.40 (m, 2H, CHAr), 7.35 (t,
3J = 8.1 Hz, 1H, CHAr), 7.10 (s, 1H, CHAr, ), 6.93 (m, 1H, CHAr), 3.87 (s, 3H, OCH3).
19F NMR (282 MHz, CDCl3) δ -57.60.
13C NMR (75 MHz, CDCl3) δ 160.2, 151.6, 139.3 (CAr), 134.0 (q, 2J = 39.2 Hz, C-CF3), 133.2
(CAr), 130.0, 129.5 (CHAr), 129.3 (2CHAr), 125.9, 125.9 (CHAr), 119.9 (q, 1J = 269.2 Hz, CF3),
118.5, 114.8, 111.1 (CHAr), 106.4 (q, 3J = 2.4 Hz, CHHetAr), 55.5 (OCH3).
GC-MS (EI, 70 eV): m/z (%) = 318 (100), 297 (8), 267 (8), 205 (3), 77 (20). HRMS (EI): calcd.
for C17H13F3N2O ([M]+): 318.09745, found: 318.09718.
3-(2-Methoxyphenyl)-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 5.4d

APPENDIX
li
Pale yellow solid, 61% (109 mg). M.p.: 70–71 °C.
IR (ATR, cm-1): ν = 542 (m), 692 (s), 750 (s), 777 (s), 814 (s), 989 (s),
1028 (s), 1068 (s), 1084 (s), 1113 (s), 1157 (s), 1201 (s), 1230 (s), 1246
(s), 1290 (s), 1354 (m), 1421 (m), 1437 (s), 1456 (m), 1473 (s), 1504 (m), 1556 (m), 1585 (m),
1595 (m), 2841 (w), 2943 (w), 3057 (w), 3180 (w).
1H NMR (300 MHz, CDCl3) δ 8.04 (dd, 3J = 7.6, 4J = 1.7 Hz, 1H, CHAr), 7.64 – 7.42 (m, 5H,
CHAr), 7.41 – 7.30 (m, 2H, CHAr), 7.08 – 6.91 (m, 2H, CHAr), 3.96 (s, 3H, OCH3).
19F NMR (282 MHz, CDCl3) δ -57.38.
13C NMR (75 MHz, CDCl3) δ 156.9, 148.6, 139.4 (CAr), 132.9 (q, 2J = 39.0 Hz, C-CF3), 129.8,
129.1 (CHAr), 129.1 (2CHAr), 128.8, 125.8, 125.8, 120.9 (CHAr), 120.6 (CAr), 120.0 (q,
1J = 269.0 Hz, CF3), 111.3, 110.3 (q, 3J = 2.5 Hz, CHHetAr), 55.5 (OCH3).
GC-MS (EI, 70 eV): m/z (%) = 318 (100), 289 (64), 267 (27), 249 (28), 221 (14), 77 (60).
HRMS (EI): calcd. for C17H13F3N2O ([M]+): 318.09745, found: 318.09699.
3-([1,1'-Biphenyl]-4-yl)-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 5.4e
Pale yellow solid, 74% (134 mg). M.p.: 94–95 °C.
IR (ATR, cm-1): ν = 546 (m), 685 (s), 727 (s), 758 (s), 820 (s), 845
(m), 987 (s), 1086 (s), 1117 (s), 1149 (s), 1159 (s), 1201 (m), 1226
(s), 1290 (s), 1358 (m), 1410 (m), 1443 (s), 1502 (s), 1566 (w), 1595 (m), 2850 (w), 2920 (w),
3032 (w), 3053 (w), 3144 (w).
1H NMR (300 MHz, CDCl3) δ 7.99 – 7.90 (m, 2H, CHAr), 7.72 – 7.63 (m, 4H, CHAr),
7.63 – 7.33 (m, 8H, CHAr), 7.16 (d, 5J = 0.3 Hz, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -57.57 (s).
13C NMR (75 MHz, CDCl3) δ 151.4, 141.5, 140.7, 139.4 (CAr), 134.1 (q, 2J = 39.2 Hz, C-CF3),
130.8 (CAr), 129.5 (CH), 129.3 (2CHAr), 128.9 (2CHAr), 127.6 (CHAr), 127.6 (2CHAr), 127.2
(2CHAr), 126.4 (2CHAr), 125.9, 125.9 (CHAr), 119.9 (q, 1J = 269.1 Hz, CF3), 106.3 (q,
3J = 2.4 Hz, CHHetAr).
GC-MS (EI, 70 eV): m/z (%) = 364 (100), 343 (11), 152 (10), 77(10).
HRMS (EI): calcd. for C22H15F3N2 ([M]+): 364.11818, found: 364.11765.
1-Phenyl-3-(p-tolyl)-5-(trifluoromethyl)-1H-pyrazole 5.4f

APPENDIX
lii
Pale yellow solid, 87% (140 mg). M.p: 71–72 °C.
IR (ATR, cm-1): ν = 544 (m), 623 (m), 687 (s), 768 (s), 798 (s), 827
(m), 989 (s), 1086 (s), 1122 (s), 1155 (s), 1203 (m), 1232 (s), 1290
(m), 1440 (s), 1504 (m), 1556 (m), 1595 (w), 2357 (w), 2860 (w), 2922 (w), 3022 (w), 3061
(w).
1H NMR (250 MHz, CDCl3) δ 7.82 – 7.65 (m, 2H, CHAr), 7.63 – 7.44 (m, 5H, CHAr),
7.33 – 7.18 (m, 2H, CHAr), 7.08 (s, 1H, CHAr), 2.40 (s, 3H, CH3).
19F NMR (282 MHz, CDCl3) δ -57.58.
13C NMR (63 MHz, CDCl3) δ 151.8, 139.4, 138.7 (CAr), 133.9 (q, 2J = 39.2 Hz, C-CF3), 129.6
(2CHAr), 129.4 (CHAr), 129.2 (2CHAr), 129.1 (CAr), 125.9 (4CHAr), 119.9 (q, 1J = 269.2 Hz,
CF3), 106.1 (q, 3J = 2.4 Hz, CHHetAr), 21.4 (CH3).
GC-MS (EI, 70 eV): m/z (%) = 302 (100), 281 (18), 267 (8), 233 (7), 77 (14).
HRMS (ESI): calcd. for C17H13F3N2 ([M+H]+): 303.11036, found: 303.11038.
3-(2-Fluorophenyl)-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 5.4g
Pale yellow solid, 90% (138 mg). M.p. 73–74 °C.
IR (ATR, cm-1): ν = 553 (m), 625 (m), 661 (m), 686 (s), 750 (s), 771 (s),
820 (s), 833 (s), 947 (m), 960 (m), 989 (s), 1028 (m), 1070 (s), 1115 (s),
1165 (s), 1201 (s), 1238 (s), 1261 (m), 1290 (s), 1358 (m), 1423 (m), 1444 (s), 1471 (m), 1500
(m), 1554 (m), 1581 (m), 1587 (m), 1913 (w), 3056 (w), 3076 (w), 3165 (w).
1H NMR (300 MHz, CDCl3) δ 8.19 – 8.07 (m, 1H, CHAr), 7.68 – 7.50 (m, 5H, CHAr),
7.50 – 7.35 (m, 1H, CHAr), 7.35 – 7.15 (m, 3H, CHAr).
19F NMR (282 MHz, CDCl3) δ -57.59, -116.00.
13C NMR (126 MHz, CDCl3) δ 160.5 (d, 1J = 250.0 Hz, CF), 146.5, 139.3 (CAr), 134.3 – 133.3
(m, 1C, CAr), 130.2 (d, 3J = 8.5 Hz, CHAr), 129.5 (CHAr), 129.3 (2CHAr), 128.6 (d, 3J = 11.5 Hz,
CHAr), 125.9, 125.9 (CHAr), 124.6 (d, 4J = 3.5 Hz, CHAr), 119.9 (q, 1J = 269.2 Hz, CF3), 119.9
(d, 2J = 20.8 Hz, CAr), 116.3 (d, 2J = 22.0 Hz, CHAr), 109.9-109.5 (m, 1C, CHHetAr).
GC-MS (EI, 70 eV): m/z (%) = 306 (100), 285 (38), 267 (9), 237 (7), 77 (15).
3-(4-Fluorophenyl)-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 5.4h

APPENDIX
liii
Yellow solid, 82% (125 mg). M.p.: 64–65 °C.
IR (ATR, cm-1): ν = 544 (m), 619 (m), 692 (s), 750 (m), 769 (s), 808
(s), 837 (s), 956 (m), 987 (s), 1028 (m), 1066 (s), 1076 (s), 1086 (s),
1111 (s), 1155 (s), 1207 (s), 1228 (s), 1290 (s), 1440 (s), 1502 (s), 1525 (m), 1558 (m), 1595
(m), 1606 (m), 1888 (w), 3058 (w), 3141 (w).
1H NMR (300 MHz, CDCl3) δ 7.91 – 7.75 (m, 2H, CHAr), 7.63 – 7.44 (m, 5H, CHAr),
7.20 – 7.07 (m, 2H, CHAr), 7.06 (s, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -57.65, -112.97.
13C NMR (75 MHz, CDCl3) δ 163.2 (d, 1J = 247.9 Hz, CF), 150.9, 139.3 (CAr), 134.2 (q,
2J = 39.2 Hz, C-CF3), 129.5 (CHAr), 129.3 (2CHAr), 128.1 (d, 4J = 3.2 Hz, CAr), 127.8 (d,
3J = 8.2 Hz, 2CHAr), 125.8, 125.8 (CHAr), 119.8 (q, 1J = 269.2 Hz, CF3), 115.9 (d, 2J = 21.8 Hz,
2CHAr), 106.0 (q, 3J = 2.4 Hz, CHHetAr).
GC-MS (EI, 70 eV): m/z (%) = 306 (100), 285 (40), 237 (9), 77 (17).
HRMS (EI): calcd. for C16H10F4N2 ([M]+): 306.07746, found: 306.07713.
3-(Naphthalen-2-yl)-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 5.4i
Pale yellow solid, 78% (102 mg). M.p.: 71–71 °C.
IR (ATR, cm-1): ν = 542 (m), 687 (s), 742 (s), 767 (s), 798 (s), 804
(s), 819 (m), 856 (s), 885 (m), 945 (m), 989 (s), 1028 (m), 1057 (s),
1074 (s), 1086 (s), 1113 (s), 1153 (s), 1230 (s), 1246 (s), 1290 (s), 1431 (m), 1485 (m), 1504
(s), 1556 (w), 1595 (m), 3028 (w), 3055 (w).
1H NMR (300 MHz, Acetone) δ 8.50 (s, 1H, CHAr), 8.13 (d, 3J = 8.3 Hz, 1H, CHAr), 8.04 – 7.88
(m, 3H, CHAr), 7.72 – 7.47 (m, 8H, CHAr).
19F NMR (282 MHz, Acetone) δ 119.44.
13C NMR (75 MHz, Acetone) δ 152.5, 140.4, 134.6, 134.6 (CAr), 134.5 (q, 2J = 39.0 Hz,
C-CF3), 130.6 (CAr), 130.3 (CHAr), 130.3 (2CHAr), 129.5, 129.3, 128.8, 127.6, 127.4, 126.9,
126.9, 125.7, 124.6 (CHAr), 121.1 (q, 1J = 268.4 Hz, CF3), 107.6 (q, 4J = 2.5 Hz, CHHetAr).
GC-MS (EI, 70 eV): m/z (%) = 338 (100), 317 (14), 127 (17), 77 (20).
HRMS (EI): calcd. for C20H13F3N2 ([M]+): 338.10253, found: 338.10259.
1-Phenyl-5-(trifluoromethyl)-3-(4-(trifluoromethyl)phenyl)-1H-pyrazole 5.4j

APPENDIX
liv
Yellow solid, 64% (113 mg). M.p.: 50–51 °C.
IR (ATR, cm-1): ν = 552 (m), 592 (s), 625 (m), 687 (s), 766 (s), 804
(s), 845 (s), 991 (s), 1062 (s), 1068 (s), 1089 (s), 1093 (s), 1109 (s),
1132 (s), 1232 (s), 1288 (s), 1323 (s), 1419 (w), 1446 (m), 1504 (m), 1531 (w), 1558 (w), 1595
(m), 1622 (m), 3063 (w), 3145 (w).
1H NMR (300 MHz, CDCl3) δ 7.98 (d, 3J = 8.1 Hz, 2H, CHAr), 7.69 (d, 3J = 8.1 Hz, 2H, CHAr),
7.62 – 7.46 (m, 5H, CHAr), 7.16 (s, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -57.71, -62.63.
13C NMR (75 MHz, CDCl3) δ 150.3, 139.1 (CAr), 135.3 (d, 4J = 1.3 Hz, CAr), 134.5 (q,
2J = 39.6 Hz, C-CF3), 130.7 (q, 2J = 32.5 Hz, CAr), 129.7 (CHAr), 129.4 (2CHAr), 126.17
(2CHAr), 125.9 (q, 3J = 3.8 Hz, 2CHAr), 125.8 (q, 4J = 1.0 Hz, 2CHAr), 124.3 (q, 1J = 272.3 Hz,
CF3), 119.7 (q, 1J = 269.3 Hz, CF3), 106.5 (q, 3J = 2.4 Hz, CHHetAr).
GC-MS (EI, 70 eV): m/z (%) = 356 (100), 335 (51), 287 (10), 267 (10), 77 (27).
HRMS (EI): calcd. for C17H10F6N2 ([M]+): 356.07427, found: 356.07482.
5-(Perfluoroethyl)-1,3-diphenyl-1H-pyrazole 5.4k.
Pale yellow solid, 95% (161 mg). M.p.: 140–142 °C.
IR (ATR, cm-1): ν = 544 (w), 580 (w), 602 (m), 619 (m), 630 (m), 688
(s), 711 (m), 750 (s), 765 (s), 777 (s), 814 (m), 914 (m), 937 (s), 958 (s),
1020 (m), 1041 (s), 1076 (m), 1092 (s), 1134 (s), 1190 (s), 1203 (s), 1223 (s), 1331 (m), 1444
(m), 1498 (m), 1595 (w), 3076 (w), 3151 (w).
1H NMR (300 MHz, CDCl3) δ 7.90 – 7.82 (m, 2H, CHAr), 7.55 – 7.48 (m, 5H, CHAr),
7.48 – 7.32 (m, 3H, CHAr), 7.10 (d, 5J = 0.9 Hz, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -83.38 (t, 3JF-F = 2.9 Hz), -106.28 (q, 3JF-F = 2.9 Hz).
13C NMR (63 MHz, CDCl3) δ 152.1, 139.9 (CAr), 132.0 (t, 2J = 27.6 Hz, C-CF3), 131.7 (CAr),
129.7 (CHAr), 129.1 (2CHAr), 128.9 (2CHAr), 128.9 (CHAr), 126.9 (2CHAr), 126.0 (2CHAr),
107.4 – 107.0 (m, CHAr), signal of CF2CF3 could not be detected.
GC-MS (EI, 70 eV): m/z (%) = 338 (100), 319 (6), 269 (14), 219 (8), 77 (24).
HRMS (EI): calcd. for C17H11F5N2 ([M]+): 388.08369, found: 388.08322.
3-(4-Methoxyphenyl)-5-(perfluoroethyl)-1-phenyl-1H-pyrazole 5.4l

APPENDIX
lv
Pale yellow solid, 91% (168 mg). M.p.: 82–83 °C.
IR (ATR, cm-1): ν = 532 (s), 576 (m), 602 (m), 617 (m), 634 (m),
694 (s), 748 (s), 771 (s), 804 (s), 835 (s), 935 (s), 958 (s), 1026
(s), 1036 (s), 1092 (s), 1134 (s), 1176 (s), 1190 (s), 1215 (s), 1250 (s), 1292 (m), 1331 (m),
1443 (m), 1502 (m), 1552 (w), 1596 (m), 1614 (m), 2839 (w), 2943 (w), 3010 (w), 3139 (w).
1H NMR (300 MHz, CDCl3) δ 7.91 – 7.68 (m, 2H, CHAr), 7.57 – 7.44 (m, 5H, CHAr), 7.02 (s,
1H, CHAr), 7.00 – 6.92 (m, 2H, CHAr), 3.85 (s, 3H, CH3).
19F NMR (282 MHz, CDCl3) δ -83.39 (t, 3JF-F = 2.7 Hz), -106.26 (q, 3JF-F = 2.6 Hz).
13C NMR (63 MHz, CDCl3) δ 160.2, 151.9, 139.9 (CAr), 131.9 (t, 2J = 27.6 Hz, C-CF3), 129.6
(CHAr), 129.0 (2CHAr), 127.3 (2CHAr), 126.9 (2CHAr), 124.5 (CAr), 114.3 (2 CHAr),
106.9 – 106.5 (m, CHAr), 55.5 (OCH3), signal of CF2CF3 could not be detected.
GC-MS (EI, 70 eV): m/z (%) = 368 (100), 353 (20), 77 (14).
HRMS (EI): calcd. for C18H13F5N2O ([M]+): 368.09426, found: 368.09390.
3-(Naphthalen-2-yl)-5-(perfluoroethyl)-1-phenyl-1H-pyrazole 5.4m
Pale brown solid, 93% (181 mg). M.p.: 104–106 °C.
IR (ATR, cm-1): ν = 544 (w), 575 (m), 617 (m), 628 (m), 642 (m),
685 (s), 748 (s), 771 (s), 804 (s), 860 (s), 887 (m), 941 (s), 980 (m),
1020 (s), 1043 (s), 1095 (s), 1134 (s), 1190 (s), 1329 (m), 1431 (m), 1487 (m), 1504 (m),
1594.92 (m), 3043 (w), 3058 (w).
1H NMR (300 MHz, CDCl3) δ 8.33 (s, 1H, CHAr), 8.01 (dd, 3J = 8.6, 4J = 1.7 Hz, 1H, CHAr),
7.95 – 7.79 (m, 3H, CHAr), 7.64 – 7.41 (m, 7H, CHAr), 7.23 (d, 5J = 0.7 Hz, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -83.31 (t, 3JF-F = 2.9 Hz), -106.28 (q, 3JF-F = 2.9 Hz).
13C NMR (75 MHz, CDCl3) δ 152.1, 139.9 (CAr), 133.6 (2CAr), 132.2 (t, 2J = 27.9 Hz,
C-CF3), 129.8 (CHAr), 129.1 (2CHAr), 128.7, 128.4, 127.9 (CHAr), 126.9 (2CHAr), 126.6, 126.5,
125.0, 123.9 (CHAr), 107.6 – 107.3 (m, 1CHAr), signal of one CAr and CF2CF3 could not be
detected.
GC-MS (EI, 70 eV): m/z (%) = 388 (100), 269 (7), 194 (6), 127 (16), 77 (18).
HRMS (EI): calcd. for C21H13F5N2 ([M]+): 388.09934, found: 388.09878.
3-([1,1'-Biphenyl]-4-yl)-5-(perfluoroethyl)-1-phenyl-1H-pyrazole 5.4n

APPENDIX
lvi
Pale yellow solid, 85% (178 mg). M.p.: 140–142 °C.
IR (ATR, cm-1): ν = 694 (s), 727 (s), 746 (s), 765 (s), 819 (s), 846
(m), 937 (s), 958 (s), 1020 (m), 1041 (s), 1072 (m), 1093 (s), 1134
(s), 1186 (s), 1219 (s), 1278 (w), 1331 (m), 1409 (w), 1441 (m), 1502 (m), 1597 (m), 3061 (w),
3132 (w).
1H NMR (300 MHz, CDCl3) δ 7.86 (d, 3J = 8.0 Hz, 2H, CHAr), 7.73 – 7.51 (m, 4H, CHAr),
7.51 – 7.21 (m, 8H, CHAr), 7.04 (s, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -83.33 (t, 3JF-F = 2.5 Hz), -106.24 (q, 3JF-F = 2.5 Hz).
13C NMR (63 MHz, CDCl3) δ 151.7, 141.6, 140.7, 139.9 (CAr), 132.1 (t, 2J = 27.4 Hz, C-CF3),
130.7 (CAr), 129.7 (CHAr), 129.1 (2CHAr), 129.0 (2CHAr), 127.7 (CHAr), 127.6 (2CHAr), 127.2
(2CHAr), 126.9 (2CHAr), 126.4 (2CHAr), 125.4 – 110.9 (m, CF2CF3), 107.4 – 107.1 (m, 1CHAr).
GC-MS (EI, 70 eV): m/z (%) = 414 (100), 395 (3), 345 (5), 295 (4), 207 (3), 152 (8), 116 (1),
77 (8).
HRMS (EI): calcd. for C23H15F5N2 ([M]+): 414.11499, found: 414.11460.
5-(Perfluoropropyl)-1,3-diphenyl-1H-pyrazole 5.4o
White solid, 83% (161 mg). M.p.: 90–91 °C.
IR (ATR, cm-1): ν = 532 (m), 590 (m), 648 (s), 692 (s), 746 (s), 765 (s),
777 (s), 814 (s), 874 (s), 957 (m), 1003 (m), 1026 (m), 1078 (m), 1109
(s), 1138 (s), 1182 (s), 1223 (s), 1344 (s), 1443 (m), 1500 (s), 1595 (w), 3053 (w), 3157 (w).
1H NMR (300 MHz, CDCl3) δ 7.91 – 7.82 (m, 2H, CHAr), 7.50 (s, 5H, CHAr), 7.48 – 7.32 (m,
3H, CHAr), 7.12 (s, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -80.04 (t, 3JF-F = 10.1 Hz), -103.84 – -104.15 (m),
-124.86 – -125.05 (m).
13C NMR (63 MHz, CDCl3) δ 152.1, 139.9 (CAr), 132.3 (t, 2J = 28.7 Hz, C-CF3), 131.7 (CAr),
130.0 (CHAr), 129.3 (2CHAr), 129.2 (2CHAr), 129.1 (CHAr), 127.4 (2CHAr), 126.3 (2CHAr),
125.8 – 92.5 (CF2CF2CF3), 108.0 – 107.6 (m, 1CHAr).
GC-MS (EI, 70 eV): m/z (%) = 388 (100), 269 (39), 77 (18).
HRMS (EI): calcd. for C18H11F7N2 ([M]+): 388.08050, found: 388.07991.
3-(4-Methoxyphenyl)-5-(perfluoropropyl)-1-phenyl-1H-pyrazole (4p):

APPENDIX
lvii
Pale solid, 76% (160 mg). M.p.: 97–98 °C.
IR (ATR, cm-1): ν = 644 (m), 692 (s), 744 (s), 773 (s), 800 (s),
872 (s), 904 (m), 958 (m), 1006 (s), 1030 (s), 1070 (m), 1078
(m), 1109 (s), 1138 (s), 1178 (s), 1184 (s), 1223 (s), 1251 (s), 1344 (m), 1435 (s), 1446 (s),
1502 (s), 1523 (m), 1552 (w), 1596 (w), 1614 (m), 2835 (w), 2939 (w), 2964 (w), 3001 (w),
3066 (w).
1H NMR (300 MHz, CDCl3) δ 7.83 – 7.79 (m, 1H, CHAr), 7.79 – 7.76 (m, 1H, CHAr), 7.49 (s,
5H, CHAr), 7.03 (s, 1H, CHAr), 7.01 – 6.96 (m, 1H, CHAr), 6.96 – 6.92 (m, 1H, CHAr), 3.85 (s,
3H, OCH3).
19F NMR (282 MHz, CDCl3) δ -80.05 (t, 3JF-F = 10.1 Hz), -103.83 – -104.18
(m), -124.90 – -125.02 (m).
13C NMR (63 MHz, CDCl3) δ 160.2, 151.9, 139.9 (CAr), 132.9 – 131.6 (m, 1CAr), 129.6 (CHAr),
128.9 (2CHAr), 127.3 (2CHAr), 127.1 (2CHAr), 124.5 (CAr), 114.3 (2CHAr), 107.2 – 106.9 (m,
1CHHetAr), 55.5 (OCH3). (signals of CF2CF2CF3 could not be detected).
GC-MS (EI, 70 eV): m/z (%) = 418 (100), 375 (4), 299 (12), 77 (9).
HRMS (EI): calcd. for C19H13F7N2O ([M]+): 418.09106, found: 418.09036.
Ethyl 3-(4-methoxyphenyl)-5-(trifluoromethyl)-1H-pyrazole-1-carboxylate 5.4q
White solid, 57% (89 mg). M.p.: 73–74 °C.
IR (ATR, cm-1): ν = 3519.0 (w), 3117.7 (w), 2966.8 (w), 2842.0
(w), 1766.3 (s), 1613.6 (m), 1587.0 (m), 1464.0 (m), 1441.8
(m), 1293.7 (s), 1143.8 (s), 1006.6 (m), 946.1 (m), 834.4 (s), 746.3 (m), 680.4 (m), 554.2 (m).
1H NMR (300 MHz, CDCl3) δ 7.87 – 7.71 (m, 2H, CHAr), 7.10 (s, 1H, CHAr), 7.01 – 6.89 (m,
2H, CHAr), 4.57 (q, 3J = 7.1 Hz, 2H, OCH2CH3), 3.85 (s, 3H, OCH3), 1.49 (t, 3J = 7.1 Hz, 3H,
OCH2CH3).
19F NMR (282 MHz, CDCl3) δ -60.03.
13C NMR (75 MHz, CDCl3) δ 161.1 (C-OCH3), 153.5 (CAr), 148.3 (C=O), 135.8 (q,
2J = 41.3 Hz, C-CF3), 128.0 (2CHAr), 123.1 (CAr), 119.3 (q, 1J = 269.1 Hz, CF3), 114.4 (2CHAr),
110.8 (q, 3J = 3.2 Hz, CHHetAr), 65.5 (OCH2CH3), 55.5 (OCH3), 14.1 (OCH2CH3).
GC-MS (EI, 70 eV): m/z (%) = 314 (47), 270 (12), 255 (17), 242 (100), 227 (58), 213 (17), 199
(36), 170 (12), 151 (22), 120 (5), 101 (5), 75 (6), 63 (5).

APPENDIX
lviii
HRMS (+EI): calcd. for C14H13O3F3N2 ([M]+): 314.08728, found: 314.08732.
Ethyl 3-(2-fluorophenyl)-5-(trifluoromethyl)-1H-pyrazole-1-carboxylate (4r):
White solid, 57% (86 mg). M.p.: 84–85 °C.
IR (ATR, cm-1): ν = 3505.3 (w), 3171.4 (w), 3076.4 (w), 2996.6 (w),
2918.6 (w), 1761.2 (s), 1620.7 (w), 1591.2 (w), 1446.9 (s), 1304.1
(m), 1229.2 (m), 1141.8 (s), 1026.1 (m), 1032.2 (m), 949.0 (m), 840.6 (m), 747.9 (m), 676.2
(m), 552.1 (w).
1H NMR (300 MHz, CDCl3) δ 8.10 (td, 3J = 7.7, 4J = 1.8 Hz, 1H, CHAr), 7.47 – 7.34 (m, 1H,
CHAr), 7.30 (d, 4J = 3.5 Hz, 1H, CHAr), 7.27 – 7.10 (m, 2H, CHAr), 4.59 (q, 3J = 7.1 Hz, 2H,
OCH2CH3), 1.50 (t, 3J = 7.1 Hz, 3H, OCH2CH3).
19F NMR (282 MHz, CDCl3) δ -60.00, -115.67.
13C NMR (63 MHz, CDCl3) δ 160.8 (d, 1J = 251.0 Hz, CF), 148.8 (CAr), 148.1 (C=O), 135.5
(q, 2J = 42.0 Hz C-CF3), 131.5 (d, 3J = 8.6 Hz, CHAr), 129.2 (d, 4J = 2.8 Hz, CHAr), 124.7 (d,
3J = 3.5 Hz, CHAr), 119.3 (q, 1J = 269.2 Hz, CF3), 118.5 (d, 2J = 11.5 Hz, CAr), 116.4 (d,
2J = 21.9 Hz, CHAr), 114.5 – 113.4 (m, CHAr), 65.7 (OCH2CH3), 14.1 (OCH2CH3).
2-Phenyl-3-(trifluoromethyl)-4,5,6,7,8,9,10,11,12,13-decahydro-2H-cyclododeca[c]pyrazo
le 5.4s
White solid, 80% (140 mg). M.p.: 56–57 °C.
IR (ATR, cm-1): ν = 546 (m), 692 (s), 771 (s), 993 (s), 1086 (s), 1111 (s),
1168 (s), 1242 (m), 1309 (m), 1329 (m), 1350 (m), 1452 (m), 1504 (s),
1556 (w), 1595 (m), 2856 (m), 2904 (m), 2933 (m).
1H NMR (250 MHz, CDCl3) δ 7.44 (s, 5H, CHAr), 2.73 – 2.47 (m, 4H, CH2), 1.90 – 1.62 (m,
4H, CH2), 1.59 – 1.30 (m, 12H, CH2).
19F NMR (282 MHz, CDCl3) δ -55.61.
13C NMR (63 MHz, CDCl3) δ 153.2, 140.1 (CAr), 129.0 (2CHAr), 129.0 (q, 2J = 37.2 Hz,
C-CF3), 128.9 (CHAr), 126.2 (2CHAr), 122.9 (q, 3J = 3.2 Hz, CAr), 120.9 (q, 1J = 269.4 Hz, CF3),
28.9, 28.3, 26.0, 25.8, 25.4, 25.2 (CH2), 23.0 (2CH2), 22.9, 20.8(CH2).
GC-MS (EI, 70 eV): m/z (%) = 350 (100), 331 (9), 307 (50), 293 (41), 281 (71), 267 (36), 253
(43), 240 (79), 77 (40).
HRMS (EI): calcd. for C20H25F3N2 ([M]+): 350.19643, found: 350.19626.

APPENDIX
lix
2-Phenyl-3-(trifluoromethyl)-4,5,6,7-tetrahydro-2H-indazole 5.4t
Yellow oil, 82% (109 mg).
IR (ATR, cm-1): ν = 546 (m), 681 (m), 692 (s), 744 (m), 766 (s), 914 (m), 964
(m), 993 (s), 1028 (m), 1074 (s), 1097 (s), 1116 (s), 1149 (s), 1170 (s), 1213
(m), 1296 (m), 1352 (m), 1377 (m), 1464 (m), 1504 (s), 1574 (w), 1598 (m),
2854 (w), 2939 (m).
1H NMR (300 MHz, CDCl3) δ 7.49 – 7.37 (m, 5H, CHAr), 3.07 – 2.53 (m, 4H, CH2), 2.13 – 1.61
(m, 4H, CH2).
19F NMR (282 MHz, CDCl3) δ -55.92.
13C NMR (75 MHz, CDCl3) δ 150.2, 139.8 (CAr), 129.0 (2CHAr), 128.9 (CHAr), 128.1 (q,
2J = 38.2 Hz, C-CF3), 125.9 (2CHAr), 120.9 (q, 1J = 269.4 Hz, CF3), 119.8 (q, 3J = 1.7 Hz, CAr),
23.4, 22.8, 22.8 (CH2), 20.9 (q, 4J= 1.3 Hz, CH2).
GC-MS (EI, 70 eV): m/z (%) = 266 (100), 247 (6), 238 (48), 217 (4), 197 (52), 77(39).
HRMS (EI): calcd. For C14H13F3N2 ([M]+): 266.10253, found: 266.10212.
2-Phenyl-3-(trifluoromethyl)-4,5-dihydro-2H-indazole 5.4u
Brown oil, 66% (87 mg).
IR (ATR, cm-1): ν = 536 (m), 596 (m), 661 (m), 690 (s), 765 (s), 993 (s), 1095
(s), 1117 (s), 1163 (s), 1207 (m), 1304 (m), 1325 (m), 1377 (m), 1462 (m),
1502 (s), 1577 (w), 1597 (m), 1705 (w), 2837 (w), 2898 (w), 2937 (w), 3060
(w).
1H NMR (250 MHz, CDCl3) δ 7.53 – 7.32 (m, 5H, CHAr), 6.60 (dt, 3J = 9.9, 4J = 2.0 Hz, 1H,
CH=CH), 6.14 (dt, 3J = 9.9, 3J = 4.3 Hz, 1H, CH=CH), 3.08 – 2.78 (m, 2H, CH2), 2.55 – 2.36
(m, 2H, CH2).
19F NMR (282 MHz, CDCl3) δ -56.16.
13C NMR (63 MHz, CDCl3) δ 148.6, 139.7 (CAr), 131.5 (CH=CH), 129.1 (2CHAr), 128.9
(CHAr), 127.7 (q, 2J = 38.4 Hz, C-CF3), 125.9 (2C, CHAr), 120.8 (q, 1J = 269.6 Hz, CF3), 119.7
(CHAr). 118.7 (q, 3J = 1.8 Hz, CAr), 23.4 (CH2), 18.5 (q, 4J = 1.3 Hz, CH2).
GC-MS (EI, 70 eV): m/z (%) = 314 (100), 245 (33), 218 (10), 77 (27), 51 (12).
HRMS (EI): calcd. for C14H11F3N2 ([M]+): 264.08688, found: 264.08643.
2-Phenyl-3-(trifluoromethyl)-4,5-dihydro-2H-benzo[g]indazole 5.4v

APPENDIX
lx
Brown solid, 82% (130 mg). M.p.: 78–79 °C.
IR (ATR, cm-1): ν = 549 (m), 648 (m), 696 (s), 731 (s), 781 (s), 897 (m),
945 (m), 987 (s), 1026 (m), 1047 (s), 1103 (s), 1157 (s), 1174 (s), 1226
(s), 1267 (m), 1307 (m), 1331 (m), 1352 (m), 1377 (m), 1446 (s), 1500
(s), 1595 (m), 2848 (w), 2901 (w), 2964 (w).
1H NMR (300 MHz, CDCl3) δ 8.07 – 7.75 (m, 1H, CHAr), 7.67 – 7.35 (m, 5H, CHAr),
7.36 – 7.26 (m, 3H, CHAr), 3.16 – 2.77 (m, 4H, CH2).
19F NMR (282 MHz, CDCl3) δ -56.04.
13C NMR (75 MHz, CDCl3) δ 148.9, 139.8, 136.5 (CAr), 129.2 (CHAr), 129.2 (2CHAr), 128.6
(CAr), 128.5, 128.5 (CHAr), 128.3 (q, 2J = 38.3 Hz, C-CF3), 127.2 (CHAr), 126.2 (2CHAr), 122.8
(CHAr), 120.7 (q, 1J = 269.6 Hz, CF3), 120.1 (q, 3J = 1.8 Hz, CAr), 28.9 (CH2), 19.4 (q,
4J = 1.3 Hz, CH2).
GC-MS (EI, 70 eV): m/z (%) = 314 (100), 245 (33), 218 (10), 142 (13), 115 (10), 77 (27).
HRMS (EI): calcd. For C18H13F3N2 ([M]+): 314.10253, found: 314.10234.
5-(4-Methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazole 5.5a
Compound 5.5a was synthesized following the general procedure using
4-methoxyacetophenone, ethyl carbamate for the first step and ethyl trifluoroacetate for the next
step. After adding ethyl trifluoroacetate, the reaction temperature was allowed to rise to 20 °C
and stirred for 30 min. Then the solvent was removed under reduced pressure. After that, the
residue was dissolved in toluene and 2 mmol (4 eq) of PTSA was added. The reaction mixture
was stirred under reflux for 8 h. After cooling, a saturated aqueous solution of NaHCO3 was
added until no evolution of CO2 was observed. Then THF was removed and the remained water
was extracted with ethyl acetate (10 mLx3). Combined organic layers were dried with MgSO4
and the solvent was removed under reduced pressure. The residue was purified by
chromatography (silica gel, heptane/DCM). The product was isolated as a white solid (51%, 62
mge).
M.p.: 147–148 °C.
IR (ATR, cm-1): ν = 3225.7 (w), 2974.4 (w), 2845.7 (w), 1614.5
(m), 1574.2 (m), 1516.7 (m), 1490.1 (m), 1458.8 (s), 1440.5 (m),
1274.9 (s), 1243.7 (s), 1109.9 (s), 1056.2 (s), 980.9 (m), 836.3 (s), 795.3 (s), 742.4 (m).

APPENDIX
lxi
1H NMR (300 MHz, CDCl3) δ 9.24 (s, br, 1H), 7.95 – 7.31 (m, 2H, CHAr), 7.11 – 6.77 (m, 2H,
CHAr), 6.66 – 6.62 (m, 1H, CHAr), 3.85 (s, 3H, OCH3).
19F NMR (282 MHz, CDCl3) δ -62.17 (s).
13C NMR (75 MHz, CDCl3) δ 160.7, 145.2(CAr), 143.7 (q, 2J = 38.1 Hz, C-CF3), 127.3 (2CHAr),
121.3 (q, 1J = 268.6 Hz, CF3), 120.7 (CAr), 114.8 (2CHAr), 100.5 (q, 3J = 1.8 Hz, CHHetAr), 55.5
(OCH3).
GC-MS (EI, 70 eV): m/z (%) = 242 (100), 227 (41), 223 (10), 199 (41), 169 (3), 151 (24).
HRMS (+EI): calcd. for C11H9O1F3N2 ([M]+): 242.06615, found: 242.0660.
5-(4-Methoxyphenyl)-3-(perfluoroethyl)-1H-pyrazole 5.5b
Compound 5.5b was synthesized following the procedure for
compound 5.5a using 4-methoxyacetophenone for the first step
and ethyl pentafluoropropionate for the next step. The product
was isolated as pale yellow solid (56%, 82 mg). M.p.: 137–138 °C.
IR (ATR, cm-1): ν = 3185.5 (w), 3142.1 (w), 3033.4 (w), 1619.0 (m), 1513.2 (s), 1330.6 (m),
1258.5 (s), 1185.6 (s), 1029.1 (s), 933.0 (s), 830.5 (m), 747.8 (m), 614.4 (m).
1H NMR (300 MHz, MeOD) δ 7.90 – 7.49 (m, 2H, CHAr), 7.16 – 6.94 (m, 2H, CHAr), 6.86 (s,
1H, CHAr), 3.86 (s, 3H, OCH3).
19F NMR (282 MHz, MeOD) δ -86.12, -114.10.
13C NMR (63 MHz, MeOD) δ 161.9 (C-OCH3), 128.3 (2CHAr), 115.6 (2CHAr), 102.2 (CHHetAr),
55.8 (OCH3), (signals of 3CAr and CF2CF3 could not be detected).
GC-MS (EI, 70 eV): m/z (%) = 292 (199), 277 (30), 249 (39), 223 (12), 151 (20), 111 (6).
HRMS (EI): calcd. for C12H9O1N2F5 ([M]+): 292.06296, found: 292.06263.
5-(4-Methoxyphenyl)-3-(perfluoropropyl)-1H-pyrazole 5.5c
Compound 5.5c was synthesized following the procedure for
compound 5.5a using 4-methoxyacetophenone for the first step
and ethyl heptafluorobutyrate for the next step. The product was
isolated as pale yellow solid (57%, 98 mg). M.p.: 127–128 °C.
IR (ATR, cm-1): ν = 3144.1 (w), 3031.2 (w), 2947.1 (w), 1890.6 (w), 1617.6 (m), 1512.5 (s),
1427.8 (m), 1348.6 (m), 1311.7 (m), 1255.9 (m), 1229.3 (s), 1178.2 (s), 1106.6 (s), 1029.6 (m),
1000.1 (m), 874.2 (s), 829.8 (m), 746.3 (s), 652.1 (m), 620.4 (m).

APPENDIX
lxii
1H NMR (300 MHz, MeOD) δ 7.78 – 7.47 (m, 2H, CHAr), 7.22 – 6.91 (m, 2H, CHAr), 6.85 (s,
1H, CHAr), 3.85 (s, 3H).
19F NMR (282 MHz, MeOD) δ -81.15 – -82.64 (m), -111.79 (d, 3J = 9.5 Hz),
-127.00 – -130.06 (m).
13C NMR (63 MHz, MeOD) δ 161.9 (C-OCH3), 146.4, 143.0 (CAr), 128.3 (2CHAr), 122.2 (CAr),
115.6 (2CHAr), 102.4 (CHHetAr), 55.8 (OCH3) (signals of CF2CF2CF3 could not be detected).
GC-MS (EI, 70 eV): m/z (%) = 342 (100), 327 (18), 299 (25), 223 (29), 208 (4), 180 (5), 151
(15), 111 (6). HRMS (EI): calcd. for C13H9O1N2F7 ([M]+): 342.05976, found: 342.05959.
5-(Naphthalen-2-yl)-3-(trifluoromethyl)-1H-pyrazole 5.5d
Compound 5.5d was synthesized following the procedure for
compound 5.5a using 2-acetonaphthone for the first step and ethyl
trifluoroacetate for the next step. The product was isolated as white
solid (78%,102 mg). M.p.: 180–181 °C.
IR (ATR, cm-1): ν = 3227.5 (w), 3043.6 (w), 2923.5 (w), 1631.9 (w), 1608.5 (w), 1582.5 (w),
1567.3 (w), 1500.0 (m), 1474.0 (w), 1255.6 (m), 1150.1 (s), 1120.6 (s), 1103.6 (s), 992.0 (m),
860.5 (m), 803.6 (s), 741.1 (s), 713.1 (m), 679.4 (m), 601.7 (m).
1H NMR (300 MHz, MeOD) δ 8.26 (s, 1H, CHAr), 8.11 – 7.72 (m, 4H, CHAr), 7.56 (dd,
3J = 6.3 Hz, 4J = 3.0 Hz, 2H, CHAr), 7.10 (s, 1H, CHAr).
19F NMR (282 MHz, MeOD) δ -63.58.
13C NMR (75 MHz, Acetone) δ 145.5 (CAr), 144.3 (q, 2J = 37.5 Hz, C-CF3), 134.4, 134.4 (CAr),
129.9, 129.1, 128.7, 127.8, 127.7 (CHAr), 126.7 (CAr), 125.6, 124.3 (CHAr), 122.9 (q,
1J = 267.5 Hz, CF3), 102.1 (CHAr).
GC-MS (EI, 70 eV): m/z (%) = 262 (100), 243 (7), 214 (10), 183 (6), 165 (22), 152 (1), 139 (4),
127 (7), 69 (3).
HRMS (+ESI): calcd. for C14H10F3N2 ([M+H]+): 263.07906, found: 263.07925.
5-(Naphthalen-2-yl)-3-(perfluoroethyl)-1H-pyrazole 5.5e
Compound 5.5e was synthesized following the procedure for
compound 5.5a using 2-acetonaphthone for the first step and ethyl
pentafluoropropionate for the next step. The product was isolated
as pale yellow solid (86%, 134 mg). M.p.: 124–126 °C.

APPENDIX
lxiii
IR (ATR, cm-1): ν = 3147.0 (w), 3114.4 (w), 2933.1 (w), 1955.6 (w), 1906.3 (w), 1785.8 (w),
1679.0 (w), 1583.7 (w), 1566.5 (w), 1513.6 (w), 1431.1 (w), 1332.5 (s), 1214.1 (s), 1183.3 (s),
1131.7 (s), 1069.4 (m), 1028.9 (s), 940.5 (s), 801.5 (m), 745.4 (s), 619.4 (m).
1H NMR (300 MHz, MeOD) δ 8.21 (d, 4J = 4.6 Hz, 1H, CHAr), 8.08 – 7.70 (m, 4H, CHAr),
7.62 – 7.42 (m, 2H, CHAr), 7.07 (s, 1H, CHAr).
19F NMR (282 MHz, MeOD) δ -86.02, -114.00.
13C NMR (63 MHz, MeOD) δ 146.4, 134.8 (CAr), 130.1, 129.3, 128.8 (CHAr), 127.9 (2CHAr),
126.9 (CAr), 125.8, 124.4, 103.3 (CHAr), (signals of 2CAr and CF2CF3 could not be detected).
GC-MS (EI, 70 eV): m/z (%) = 312 (100), 293 (5), 243 (18), 214 (13), 194 (4), 165 (17), 121
(11), 82 (3), 69 (2).
HRMS (+ESI): calcd. for C15H10F5N2 ([M+H]+): 313.07587, found: 313.07617.
3-(Perfluoropropyl)-5-(naphthalen-2-yl)-1H-pyrazole 5.5f
Compound 5.5f was synthesized following the procedure for
compound 5.5a using 2-acetonaphthone for the first step and ethyl
heptafluorobutyrate for the next step. The product was isolated as
pale yellow solid (64%, 116 mg). M.p.: 181–182 °C.
IR (ATR, cm-1): ν = 3183.6 (w), 3067.3 (w), 2877.8 (w), 1565.2 (w), 1512.8 (w), 1415.8 (w),
1349.1 (m), 1224.1 (m), 1176.5 (s), 1103.7 (m), 1000.7 (m), 874.8 (m), 791.3 (m), 744.3 (m),
651.2 (m).
1H NMR (300 MHz, MeOD) δ 8.26 (s, 1H, CHAr), 8.12 – 7.72 (m, 4H, CHAr), 7.68 – 7.44 (m,
2H, CHAr), 7.11 (s, 1H, CHAr).
19F NMR (282 MHz, MeOD) δ -81.78 (t, 3JF-F = 9.5 Hz), -111.71 (d, 3JF-F = 8.6 Hz), -128.28
(s).
13C NMR (75 MHz, Acetone) δ 145.7, 134.4, 134.4 (CAr), 129.9, 129.2, 128.7, 127.8, 127.8,
125.7, 124.3, 103.6 (CHAr), (signals of 2CAr and CF2CF2CF3 could not be detected).
GC-MS (EI, 70 eV): m/z (%) = 362 (100), 343 (9), 243 (34), 214 (17), 194 (4), 165 (14), 122
(13), 83 (4), 69 (4).
HRMS (+ESI): calcd. for C16H10F7N2 ([M+H]+): 363.007267, found: 363.07305.
3-(Trifluoromethyl)-5-(4-(trifluoromethyl)phenyl)-1H-pyrazole 5.5g

APPENDIX
lxiv
Compound 5.5g was synthesized following the procedure for
compound 5.5a using 4-trifluoromethylacetophenone for the first
step and ethyl trifluoroacetate for the next step. The product was
isolated as white solid (75%, 105 mg). M.p.: 135–136 °C.
IR (ATR, cm-1): ν = 3239.9 (w), 3161.7 (w), 3066.3 (w), 2987.4 (w), 1914.8 (w), 1790.5 (w),
1738.4 (w), 1671.9 (w), 1622.3 (w), 1587.6 (w), 1494.7 (w), 1325.4 (m), 1253.0 (m), 1172.6
(m), 1109.0 (s), 1068.0 (m), 982.9 (m), 916.7 (m), 813.6 (m), 747.3 (m), 661.5 (m), 592.3 (m).
1H NMR (300 MHz, MeOD) δ 7.95 (d, 3J = 8.2 Hz, 2H, CHAr), 7.80 (d, 3J = 8.3 Hz, 2H, CHAr),
7.12 (s, 1H, CHAr).
19F NMR (282 MHz, MeOD) δ -63.66, -64.34.
13C NMR (75 MHz, CDCl3) δ 144.2 (CAr), 143.5 (q, 2J = 38.0 Hz, CAr), 131.5 (q, 2J = 33.0 Hz,
C-CF3), 131.2 (CAr), 126.4 (q, 4J = 3.8 Hz, 2CHAr), 125.8 (2CHAr), 123.7 (q, 1J = 272.3 Hz,
CF3), 120.7 (q, 1J = 269.0 Hz, CF3), 102.1 (CHHetAr).
GC-MS (EI, 70 eV): m/z (%) = 280 (100), 261 (23), 231 (7), 211 (20), 201 (5), 182 (15), 164
(4), 145 (8), 133 (49, 87 (2), 69 (6).
HRMS (+ESI): calcd. for C11H7F6N2 ([M+H]+): 281.05579, found: 281.05099.
5-(Perfluoroethyl)-3-(4-(trifluoromethyl)phenyl)-1H-pyrazole 5.5h
Compound 5.5h was synthesized following the procedure for
compound 5.5a using 4-trifluoromethylacetophenone for the first
step and ethyl pentafluoropropionatefor the next step. The product
was isolated as white solid (74%, 122 mg). M.p.: 142–143 °C.
IR (ATR, cm-1): ν = 3191.2 (w), 3032.7 (w), 2887.0 (w), 1623.7 (w), 1590.6 (w), 1465.6 (w),
1429.3 (w), 1325.2 (s), 1225.8 (s), 1194.1 (s), 1125.4 (s), 1063.8 (m), 1030.5 (m), 973,8 (m),
939.2 (m), 841.6 (m), 807.8 (m), 750.1 (m), 693.5 (m), 622.0 (w), 591.5 (w).
1H NMR (300 MHz, MeOD) δ 7.96 (d, 3J = 8.2 Hz, 2H, CHAr), 7.80 (d, 3J = 8.3 Hz, 2H, CHAr),
7.14 (s, 1H, CHAr).
19F NMR (282 MHz, MeOD) δ -64.34, -86.12, -114.07.
13C NMR (75 MHz, CDCl3) δ 144.6 (CAr), 131.7 (q, 2J = 32.9 Hz, C-CF3), 129.2 (CAr), 126.4
(q, 4J = 3.8 Hz, 2CHAr), 125.9 (2CHAr), 123.8 (q, 1J = 272.2 Hz, CF3), 103.9 (CHHetAr), (signals
of 1CAr and CF2CF3 could not be detected).

APPENDIX
lxv
GC-MS (EI, 70 eV): m/z (%) = 330 (81), 311 (21), 261 (100), 232 (4), 213 (9), 182 (6), 164
(18), 145 (5), 121 (5), 105 (5), 69 (6).
HRMS (+ESI): calcd. for C12H7F8N2 ([M+H]+): 331.04760, found: 331.04806.
5-(Perfluoropropyl)-3-(4-(trifluoromethyl)phenyl)-1H-pyrazole 5.5i
Compound 5.5i was synthesized following the procedure for
compound 5.5a using 4-trifluoromethylacetophenone for the first
step and ethyl heptafluorobutyrate for the next step. The product
was isolated as white solid (67%, 127 mg). M.p. 109–110 °C.
IR (ATR, cm-1): ν = 3171.2 (w), 3034.4 (w), 2950.7 (w), 2887.4 (w), 1623.5 (w), 1591.8 (w),
1467.1 (w), 1424.8 (w), 1326.4 (s), 1276.0 (w), 1232.3 (s), 1171.5 (m), 1109.2 (s), 1063.0 (s),
1001.4 (m), 876.1 (m), 842.4 (m), 810.1 (m), 747.9 (m), 652.6 (m), 592.2 (m).
1H NMR (300 MHz, MeOD) δ 7.96 (d, 3J = 8.2 Hz, 2H, CHAr), 7.80 (d, 3J = 8.3 Hz, 2H, CHAr),
7.14 (s, 1H, CHAr).
19F NMR (282 MHz, MeOD) δ -64.35 (s), -81.84 (t, 3J = 9.6 Hz), -111.76 (s), -128.29 – -128.38
(m).
(Due to there are many Fs in the molecule, the signals of carbons are splited and very difficult
to identify.)
GC-MS (EI, 70 eV): m/z (%) = 380 (64), 361 (23), 261 (100), 213 (8), 182 (5), 164 (17), 69 (6).
HRMS (+ESI): calcd. for C13H7F10N2 ([M+H]+): 381.04441, found: 381.04428.
Synthesis of 2-phenyl-3-(trifluoromethyl)-2H-indazole 5.6a
To a solution of 5.4s (100 mg) in toluene (7 mL), DDQ (2 equiv.) was added. The reaction
mixture was stirred under reflux for 3h. Then the reaction mixture was cooled to room
temperature and ethyl acetate (10 mL) and water (10 mL) were added. The organic layer was
separated and washed with water three times. After drying and removal of solvent, the residue
was purified by chromatography (silica gel, n-heptane/DCM). The product was isolated as a
yellow oil (71%, 71 mg).
IR (ATR, cm-1): ν = 534 (m), 567 (m), 627 (m), 640 (m), 692 (s), 743 (s), 768
(s), 829 (m), 914 (m), 933 (m), 989 (s), 1001 (s), 1030 (m), 1074 (s), 1103 (s),
1147 (s), 1174 (s), 1223 (s), 1298 (s), 1381 (w), 1429 (s), 1469 (m), 1500 (s),
1522 (m), 1551 (w), 1597 (m), 2361 (w), 2858 (w), 2929 (w), 3066 (w).

APPENDIX
lxvi
1H NMR (300 MHz, CDCl3) δ 7.90 – 7.76 (m, 2H, CHAr), 7.64 – 7.49 (m, 5H, CHAr),
7.49 – 7.37 (m, 1H, CHAr), 7.37 – 7.27 (m, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -54.47.
13C NMR (75 MHz, CDCl3) δ 148.3, 139.7 (CAr), 130.1 (CHAr), 129.2 (2CHAr), 127.4 (CHAr),
126.3 (2CHAr), 125.2 (CHAr), 123.8 (q, 2J = 39.5 Hz, C-CF3), 121.7 (CAr), 121.1 (q,
1J = 269.0 Hz, CF3), 119.5 (q, 4J = 1.7 Hz, CHAr), 118.6 (CHAr).
GC-MS (EI, 70 eV): m/z (%) = 262 (100), 236 (7), 193 (34), 166 (11), 77 (15), 51 (12).
HRMS (EI): calcd. for C14H9F3N2 ([M]+): 262.07123, found: 262.07106.
Synthesis of 2-phenyl-3-(trifluoromethyl)-2H-benzo[g]indazole 5.6b
To a solution of 5.4t (100 mg) in toluene (7 mL), DDQ (2 equiv.) was added. The reaction
mixture was stirred under reflux for 3h. Then the reaction mixture was cooled to room
temperature and ethyl acetate (10 mL) and water (10 mL) were added. The organic layer was
separated and washed with water three times. After drying and removal of solvent, the residue
was purified by chromatography (silica gel, n-heptane/DCM). The product was isolated as a
pale yellow solid (90%, 90 mg). M.p.: 100–102 °C.
IR (ATR, cm-1): ν = 549 (s), 681 (s), 746 (s), 767 (s), 804 (s), 885 (m),
982 (s), 1045 (s), 1099 (s), 1176 (s), 1217 (s), 1238 (m), 1269 (m), 1309
(m), 1385 (w), 1441 (s), 1473 (m), 1504 (s), 1558 (w), 1597 (m), 3024
(w), 3049 (w).
1H NMR (300 MHz, CDCl3) δ 8.66 (dd, 3J = 5.7, 4J = 3.5 Hz, 1H), 7.93 – 7.80 (m, 1H),
7.76 – 7.52 (m, 9H).
19F NMR (282 MHz, CDCl3) δ -54.74 (s).
13C NMR (63 MHz, CDCl3) δ 146.1, 139.7, 132.3 (CAr), 129.8 (CHAr), 129.1 (2CHAr), 128.5,
127.7, 127.5, 127.3, 126.4, 126.3 (CHAr), 125.0 (CAr), 124.7 (q, 2J = 39.8 Hz, C-CF3), 122.5
(CHAr), 120.9 (q, 1J = 269.2 Hz, CF3), 119.3 (CAr), 116.9 (q, 4J = 1.9 Hz, CHAr).
GC-MS (EI, 70 eV): m/z (%) = 312 (100), 242 (30), 77 (10).
HRMS (EI): calcd. for C18H11F3N2([M]+): 312.08688, found: 312.08667.
3-(4-methoxyphenyl)-5-(trifluoromethyl)isoxazole 5.8a

APPENDIX
lxvii
Compound 5.8a was synthesized following the general procedure using
4-methoxyacetophenone, hydroxylamine for the first step and ethyl trifluoroacetate for the next
step. The product was isolated as a white solid (57%, 69 mg). M.p.
76-78 °C.
IR (ATR, cm-1): ν = 3114.5 (w), 2963.7 (w), 2844.2 (w), 1611.5
(m), 1532.2 (w), 1459.8 (m), 1432.7 (m), 1319.7 (m), 1242.4 (m),
1174.7 (m), 1114.0 (s), 1023.3 (m), 966.8 (m), 915.7 (m), 837.2 (m), 821.7 (m), 747.2 (m),
680.0 (w).
1H NMR (300 MHz, CDCl3) δ 7.82 – 7.68 (m, 2H, CHAr), 7.04 – 6.96 (m, 2H, CHAr), 6.94 (d,
5J = 0.9 Hz, 1H, CHAr), 3.87 (s, 3H, OCH3).
19F NMR (282 MHz, CDCl3) δ -64.24.
13C NMR (75 MHz, CDCl3) δ 162.3, 161.8 (CAr), 159.1 (q, 2J = 42.4 Hz, C-CF3), 128.6
(2CHAr), 119.9 (CAr), 118.1 (q, 1J = 270.3 Hz, CF3), 114.7 (2CHAr), 103.3 (d, 3J = 2.1 Hz,
CHHetAr), 55.5 (OCH3).
GC-MS (EI, 70 eV): m/z (%) = 243 (82), 174 (82), 146 (100), 131 (14), 119 (7), 103 (9), 92
(11), 76 (18), 63 (15), 50 (9).
HRMS (+EI): calcd. for C11H8O2F3N1 ([M]+): 243.05016, found: 243.05028.
3-(naphthalen-2-yl)-5-(trifluoromethyl)isoxazole 5.8b
Compound 5.8b was synthesized following the general procedure using 2-acetonaphthone,
hydroxylamine for the first step and ethyl trifluoroacetate for the next step. The product was
isolated as a white (62%, 82 mg). M.p. 103–104 °C.
IR (ATR, cm-1): ν = 3232.3 (w), 3126.3 (w), 2921.0 (w), 1631.3 (w),
1486.7 (m), 1438.6 (m), 1303.8 (m), 1143.6 (s), 1104.9 (s), 1057.6
(m), 964.1 (m), 901.5 (m), 827.7 (s), 745.5 (m), 633.1 (w).
1H NMR (300 MHz, CDCl3) δ 8.26 (d, 4J = 0.7 Hz, 1H, CHAr), 8.00 – 7.84 (m, 4H, CHAr),
7.64 – 7.48 (m, 2H, CHAr), 7.14 (d, 4J = 0.9 Hz, 1H, CHAr).
19F NMR (282 MHz, CDCl3) δ -64.15.
13C NMR (75 MHz, CDCl3) δ 162.8 (CAr), 159.4 (q, 2J = 42.7 Hz, C-CF3), 134.5, 133.2 (CAr),
129.3, 128.7, 128.1, 127.7, 127.3, 127.2 (CHAr), 124.8 (CAr), 123.7 (CHAr), 118.1 (d,
1J = 270.5 Hz, CF3), 103.7 (d, 3J = 2.1 Hz, CHHetAr).

APPENDIX
lxviii
GC-MS (EI, 70 eV): m/z (%) = 263 (100), 194 (72), 166 (41), 139 (21), 127 (68), 115 (12), 97
(8), 69 (10).
HRMS (+ESI): calcd. for C14H9O1F3N1 ([M+H]+): 264.06307, found: 264.06321.

APPENDIX
lxix
Crystal data and structure refinement
Crystal data and structure refinement for compound 2.6h
is_cm11
Crystal data
Chemical formula C24H16FNO2
Mr 369.38
Crystal system, space group Monoclinic, P21/c
Temperature (K) 173
a, b, c (Å) 7.7148 (5), 20.3158 (13), 11.7110 (8)
β (°) 91.418 (4)
V (Å3) 1834.9 (2)
Z 4
Radiation type Mo Kα
µ (mm−1) 0.09
Crystal size (mm) 0.46 × 0.12 × 0.10
Data collection
Diffractometer Bruker Apex Kappa II-CCD-
diffractometer
Absorption correction Multi-scan
(SADABS; Sheldrick, 2004)
Tmin, Tmax 0.959, 0.991
No. of measured, independent and
observed [I > 2σ(I)] reflections
25493, 4882, 2572
Rint 0.071
(sin θ/λ)max (Å−1) 0.682
Refinement
R[F2 > 2σ(F2)], wR(F2), S 0.049, 0.126, 1.00
No. of reflections 4882
No. of parameters 253
H-atom treatment H-atom parameters constrained
Δρmax, Δρmin (e Å−3) 0.20, −0.21

APPENDIX
lxx
Crystal data and structure refinement for compound 3.3i
is_indolo_d2
Crystal data
Chemical formula C21H14FN
Mr 299.33
Crystal system, space group Monoclinic, P21/c
Temperature (K) 173
a, b, c (Å) 10.5670 (7), 20.0282 (12), 6.8420 (5)
β (°) 95.652 (2)
V (Å3) 1440.99 (17)
Z 4
Radiation type Mo Kα
µ (mm−1) 0.09
Crystal size (mm) 0.33 × 0.27 × 0.22
Data collection
Diffractometer Bruker Apex Kappa II-CCD-
diffractometer
Absorption correction Multi-scan
(SADABS; Sheldrick, 2004)
Tmin, Tmax 0.671, 0.746
No. of measured, independent and
observed [I > 2σ(I)] reflections
19527, 3832, 2993
Rint 0.038
(sin θ/λ)max (Å−1) 0.682
Refinement
R[F2 > 2σ(F2)], wR(F2), S 0.052, 0.129, 1.10
No. of reflections 3832
No. of parameters 279
No. of restraints 68
H-atom treatment H-atom parameters constrained

APPENDIX
lxxi
Δρmax, Δρmin (e Å−3) 0.21, −0.20
Crystal data and structure refinement for compound 3.7j
is_es4
Crystal data
Chemical formula C20H14N2O
Mr 298.33
Crystal system, space group Triclinic, P
Temperature (K) 173
a, b, c (Å) 12.2153 (2), 13.5599 (2), 18.6521 (3)
α, β, γ (°) 95.666 (1), 104.369 (1), 107.679 (1)
V (Å3) 2800.32 (8)
Z 8
Radiation type Mo Kα
µ (mm−1) 0.09
Crystal size (mm) 0.55 × 0.28 × 0.19
Data collection
Diffractometer Bruker-Nonius Apex X8-CCD-
diffractometer
Absorption correction Multi-scan
(SADABS; Sheldrick, 2004)
Tmin, Tmax 0.707, 0.746
No. of measured, independent and
observed [I > 2σ(I)] reflections
93666, 18586, 13060
Rint 0.033
(sin θ/λ)max (Å−1) 0.735
Refinement
R[F2 > 2σ(F2)], wR(F2), S 0.050, 0.147, 1.01
No. of reflections 18586
No. of parameters 833
H-atom treatment H-atom parameters constrained

APPENDIX
lxxii
Δρmax, Δρmin (e Å−3) 0.47, −0.28
Crystal data and structure refinement for compound 4.3a
is_tn_hb
Crystal data
Chemical formula C22H14S
Mr 310.39
Crystal system, space group Triclinic, P
Temperature (K) 123
a, b, c (Å) 6.0433 (2), 10.7998 (3), 12.1236 (3)
α, β, γ (°) 108.612 (1), 92.292 (1), 95.272 (1)
V (Å3) 744.69 (4)
Z 2
Radiation type Mo Kα
µ (mm−1) 0.21
Crystal size (mm) 0.29 × 0.20 × 0.14
Data collection
Diffractometer Bruker-Nonius Apex X8-CCD-
diffractometer
Absorption correction Multi-scan
(SADABS; Sheldrick, 2004)
Tmin, Tmax 0.722, 0.746
No. of measured, independent and
observed [I > 2σ(I)] reflections
16019, 4325, 3897
Rint 0.021
(sin θ/λ)max (Å−1) 0.703
Refinement
R[F2 > 2σ(F2)], wR(F2), S 0.035, 0.096, 1.06
No. of reflections 4325
No. of parameters 208
H-atom treatment H-atom parameters constrained

APPENDIX
lxxiii
Δρmax, Δρmin (e Å−3) 0.45, −0.26
Crystal data and structure refinement for compound 4.3b
is_thang1203
Crystal data
Chemical formula C23H16S
Mr 324.42
Crystal system, space group Triclinic, P
Temperature (K) 123
a, b, c (Å) 5.9864 (2), 11.0375 (4), 13.4379 (5)
α, β, γ (°) 112.071 (2), 96.127 (2), 92.844 (2)
V (Å3) 814.25 (5)
Z 2
Radiation type Mo Kα
µ (mm−1) 0.20
Crystal size (mm) 0.19 × 0.15 × 0.10
Data collection
Diffractometer Bruker-Nonius Apex X8-CCD-
diffractometer
Absorption correction Multi-scan
(SADABS; Sheldrick, 2004)
Tmin, Tmax 0.710, 0.746
No. of measured, independent and
observed [I > 2σ(I)] reflections
27542, 5885, 4610
Rint 0.029
(sin θ/λ)max (Å−1) 0.756
Refinement
R[F2 > 2σ(F2)], wR(F2), S 0.041, 0.112, 1.02
No. of reflections 5885
No. of parameters 218
H-atom treatment H-atom parameters constrained

APPENDIX
lxxiv
Δρmax, Δρmin (e Å−3) 0.46, −0.26

APPENDIX
lxxv
List of tables
Table 2.1: Optimization for the synthesis of 2.4a ................................................................... 25
Table 2.2: Synthesis of 2.4 ...................................................................................................... 26
Table 2.3: Optimization of 2.6a ............................................................................................... 27
Table 2.4: Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones ................................................... 28
Table 2.5: Anticholinesterase activities of chromeno[3,4-b]pyrrol-4(3H)-ones ..................... 32
Table 2.6: Monoamine oxidase (A & B) activities of chromeno[3,4-b]pyrrol-4(3H)-ones .... 33
Table 3.1: Optimization for the synthesis of 3.3a ................................................................... 40
Table 3.2: Synthesis of alkynes 3.1 ......................................................................................... 41
Table 3.3: Synthesis of indolo[1,2-f]phenathridines ............................................................... 42
Table 3.4: Reaction of 1,2-bis(2-bromophenyl)ethyne 3.1f with amines ................................ 46
Table 3.5: Synthesis of 3-bromo-2-(alkynyl)pyridines ........................................................... 51
Table 3.6: Synthesis of 4-azaindolo[1,2-f]phenanthridines ..................................................... 52
Table 3.7: Synthesis of 4-azaindolo[1,2-f]phenanthridines ..................................................... 55
Table 3.8: Synthesis of 7-azaindolo[1,2-f]phenanthridines ..................................................... 57
Table 3.9: Absorption and emission spectroscopic data of azaindolo[1,2-f]phenanthridines . 60
Table 4.1: Synthesis of benzo[b]naphtho[2,1-d]thiophenes .................................................... 71
Table 4.2: Synthesis of naphtho[1,2-b]benzofurans ................................................................ 74
Table 4.3: Synthesis of benzo[a]carbazoles ............................................................................ 75
Table 5.1: Synthesis of pyrazoles 5.4 and 5.5 ......................................................................... 82
Table 5.2: Alkaline phosphatase AP (h-TNAP & h-IAP) and NPP (h-NPP1 & h-NPP3)
inhibition in presence of the synthesized compounds .............................................................. 88

APPENDIX
lxxvi
List of figures
Figure 1.1: Heterocycles in hydrogen-bonding interaction and complex with metal cation..... 2
Figure 1.2: Aromatic heterocycles as drugs .............................................................................. 3
Figure 1.3: Aromatic heterocycles applied in organic materials ............................................... 4
Figure 1.4: A general Pd(0)-catalytic cycle ............................................................................... 5
Figure 1.5: Selectivity of oxidative addition on substrates with many reactive centers ........... 7
Figure 1.6: Cross-coupling reaction involving transmetalation .............................................. 10
Figure 1.7: Phosphine ligands ................................................................................................. 17
Figure 1.8: Diphosphine ligands ............................................................................................. 18
Figure 2.1: Ningalin B and Lamellarin D derivatives ............................................................. 22
Figure 2.2: X-ray structure of 2.6h .......................................................................................... 31
Figure 2.3: Putative binding mode of 2.6i inside AChE receptor (PDB ID 3I6Z). ................. 34
Figure 2.4: Putative binding mode of 2.6i inside AChE receptor (PDB ID 3I6Z). ................. 34
Figure 2.5: Putative binding mode of 2.6p inside BuChE receptor (PDB ID 1P0I) ............... 35
Figure 2.6: Putative binding mode of 2.6p inside BuChE receptor (PDB ID 1P0I) ............... 35
Figure 3.1: Biologically active phenanthridines ...................................................................... 37
Figure 3.2: Phenanthridine derivatives with potential physical properties ............................. 38
Figure 3.3: X-ray crystal structure analysis of 3.3i ................................................................. 44
Figure 3.4: Azaindolo[1,2-f]phenanthridines applicable in electroluminescent devices ........ 48
Figure 3.5: X-ray crystallographic analysis of 3.7j. ................................................................ 54
Figure 3.6: Normalized absorption and corrected emission spectra of
azaindolo[1,2-f]phenanthridines in CH2Cl2. ............................................................................. 59
Figure 3.7: Arnoamine C & D ................................................................................................. 61
Figure 4.1: The structure of 4.3a determined by X-ray crystallographic analysis .................. 73
Figure 4.2: The structure of 4.3b determined by X-ray crystallographic analysis .................. 74
Figure 5.1: Drugs and agrochemicals containing trifluoromethylated pyrazoles.................... 78
Figure 5.2: Bioactive trifluoromethylated indazoles D and E ................................................. 85

APPENDIX
lxxvii
List of schemes
Scheme 1.1: Oxidative addition and reductive elimination ....................................................... 5
Scheme 1.2: Mechanisms of oxidative addition ........................................................................ 6
Scheme 1.3: Buchwald-Hartwig reaction .................................................................................. 8
Scheme 1.4: Mechanisms of ligand exchange ........................................................................... 9
Scheme 1.5: Transmetalation ..................................................................................................... 9
Scheme 1.6: Roles of base in Suzuki-Miyaura reaction .......................................................... 10
Scheme 1.7: Mechanism of Sonogashira reaction ................................................................... 11
Scheme 1.8: Migratory insertions ............................................................................................ 11
Scheme 1.9: Migratory insertion of CO ................................................................................... 12
Scheme 1.10: Migratory insertion of carbene .......................................................................... 12
Scheme 1.11: Cross-coupling reaction of N-tosylhydrazones involving carbene ................... 12
Scheme 1.12: Heck-Mizoroki reaction .................................................................................... 13
Scheme 1.13: Cationic mechanism of 1,2-migratory insertion ............................................... 13
Scheme 1.14: Neutral mechanism of 1,2-migratory insertion ................................................. 13
Scheme 1.15: Anionic mechanism of 1,2-migratory insertion ................................................ 14
Scheme 1.16: β-hydride elimination ........................................................................................ 14
Scheme 1.17: C-H arylation vs traditional cross-coupling reactions ....................................... 15
Scheme 1.18: Mechanisms C-H arylation ............................................................................... 15
Scheme 1.19: Intramolecular direct arylation .......................................................................... 16
Scheme 1.20: Approaches to the synthesis of indoles by Pd(0)-catalyzed domino reactions: 19
Scheme 1.21: Synthesis of indolo[2,3-a]carbazoles by Cacchi ............................................... 20
Scheme 1.22: Synthesis of indoles by one-pot three-component reaction by Ackemann ....... 20
Scheme 1.23: Approaches to the synthesis of carbazoles ........................................................ 20
Scheme 2.1: Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones ................................................ 23
Scheme 2.2: Synthesis of chromeno[3,4-b]pyrrol-4(3H)-ones by hydroamination ................ 24
Scheme 2.3: Synthesis of 3-bromo-4-(trifluoromethanesulfonyloxy)coumarin ...................... 24
Scheme 3.1: Domino reactions for the synthesis of fused-phenanthridines ............................ 39
Scheme 3.2: Reaction of 1-chloro-2-(phenylethynyl)benzene with 2-bromoaniline............... 44
Scheme 3.3: Proposed pathway for the reaction ...................................................................... 45
Scheme 3.4: Reaction of 1-bromo-2-((2-chlorophenyl)ethynyl)benzene with p-toluidine ..... 48
Scheme 3.5: Synthesis of azaindolo[1,2-f]phenanthridines via in situ benzyme ..................... 49
Scheme 3.6: Synthesis of azaindolo[1,2-f]phenanthridines ..................................................... 49
Scheme 3.7: Proposed pathway for the reaction ...................................................................... 50
Scheme 3.8: Synthesis of 3.6g ................................................................................................. 57
Scheme 3.9: Two-fold domino reaction .................................................................................. 58
Scheme 3.10: Retrosynthesis analysis of Arnoamines ............................................................ 61
Scheme 3.11: Proposed Synthesis of starting marterial ........................................................... 62
Scheme 3.12: Alternative starting material 3.13 ...................................................................... 62
Scheme 3.13: Domino reaction of 3.13 ................................................................................... 63
Scheme 4.1: Pathways of 6-endo-trig and 5-exo-trig cyclizations .......................................... 64
Scheme 4.2: Pd-catalyzed cyclization of dibrominated compounds with hydrazones ............ 65
Scheme 4.3: Tandem reaction of 3-bromo-2-(2’-chlorophenyl)benzo[b]thiophene 4.1a with
N-tosylhydrazone of acetophenone 4.2a .................................................................................. 66

APPENDIX
lxxviii
Scheme 4.4: Proposed mechanism of Pd-catalyzed cyclization of dibrominated compounds
with hydrazones. ....................................................................................................................... 68
Scheme 4.5: Tandem reaction of 2,2′-dibromobiphenyl 4.1b and
3-bromo-2-(2-bromophenyl)pyridine 4.1c. .............................................................................. 69
Scheme 4.6: Tandem reaction of 3-bromo-2-(2-bromophenyl)benzo[b]thiophene 4.1d and
N-tosylhydrazones. ................................................................................................................... 70
Scheme 5.1: One-pot cyclocondensation of 1,3-dicarbonyl dianions with α-azidoketones .... 77
Scheme 5.2: Synthesis of pyrazoles ......................................................................................... 79
Scheme 5.3: Synthesis of pyrazoles by cyclization of hydrazone 1,4-dianions ...................... 80
Scheme 5.4: Synthesis of pyrazoles 5.4 and 5.5 ...................................................................... 81
Scheme 5.5: Synthesis of 5.4s-v .............................................................................................. 85
Scheme 5.6: Synthesis of 5.6a and 5.6b .................................................................................. 86
Scheme 5.7: Synthesis of isoxazoles 5.8a, b ........................................................................... 86

APPENDIX
lxxix
List of References
[1] A. F. Pozharskiĭ, A. R. Katritzky, A. T. Soldatenkov, Heterocycles in life and society,
Wiley, Chichester West Sussex, 2011.
[2] J. A. Joule, K. Mills, Heterocyclic chemistry, Wiley, Oxford, 2010.
[3] V. Cechinel-Filho, Plant bioactives and drug discovery, Wiley, Hoboken, N.J., 2012.
[4] a) K. Brune, Acute Pain 1997, 1, 33–40; b) E. Ravina, The evolution of drug discovery,
Wiley-VCH, Weinheim, 2011.
[5] a) W. Jiang, Y. Li, Z. Wang, Chem. Soc. Rev. 2013, 42, 6113–6127; b) W. Wu, Y. Liu, D.
Zhu, Chem. Soc. Rev. 2010, 39, 1489–1502; c) Y. Lin, Y. Li, X. Zhan, Chem. Soc. Rev.
2012, 41, 4245–4272.
[6] K. E. Maly, Cryst. Growth Des. 2011, 11, 5628–5633.
[7] a) G. Horowitz, Adv. Mater. 1998, 10, 365–377; b) J. E. Anthony, J. S. Brooks, D. L. Eaton,
S. R. Parkin, J. Am. Chem. Soc. 2001, 123, 9482–9483.
[8] a) X. Wang, K.-C. Lau, J. Phys. Chem. C 2012, 116, 22749–22758; b) S.-Z. Weng, P.
Shukla, M.-Y. Kuo, Y.-C. Chang, H.-S. Sheu, I. Chao, Y.-T. Tao, ACS Catal. 2009, 1,
2071–2079; c) U. H. F. Bunz, Acc. Chem. Res. 2015, 48, 1676–1686.
[9] a) M. Cölle, J. Gmeiner, W. Milius, H. Hillebrecht, W. Brütting, Adv. Funct. Mater. 2003,
13, 108–112; b) R. Katakura, Y. Koide, Inorg. Chem. 2006, 45, 5730–5732; c) V. A.
Montes, R. Pohl, J. Shinar, P. Anzenbacher, JR, Chem. Eur. J. 2006, 12, 4523–4535.
[10] H. Yersin, Highly efficient OLEDs with phosphorescent materials, Wiley-VCH,
Weinheim, 2007.
[11] A. d. Meijere, S. Bräse, M. Oestreich, Metal-catalyzed cross-coupling reactions and
more, Wiley-VCH, Weinheim, 2014.
[12] J. J. Li, G. W. Gribble, Palladium in Heterocyclic Chemistry, Elsevier Science, 2006.
[13] J. Tsuji, Palladium reagents and catalysts, John Wiley & Sons, Chichester, West
Sussex, Hoboken, N.J., 2004.
[14] C. Torborg, M. Beller, Adv. Synth. Catal. 2009, 351, 3027–3043.
[15] a) P. G. Gildner, T. J. Colacot, Organometallics 2015, 34, 5497–5508; b) X.-F. Wu, P.
Anbarasan, H. Neumann, M. Beller, Angew. Chem. Int. Ed. 2010, 49, 9047–9050.
[16] E.-i. Negishi, Handbook of organopalladium chemistry for organic synthesis, John
Wiley & Sons, Inc, New York, Great Britain, 2002.
[17] a) B. U. W. Maes, S. Verbeeck, T. Verhelst, A. Ekomie, N. von Wolff, G. Lefevre, E.
A. Mitchell, A. Jutand, Chem. Eur. J. 2015, 21, 7858–7865; b) C. Amatore, F. Pfluger,
Organometallics 1990, 9, 2276–2282; c) M. Portnoy, D. Milstein, Organometallics 1993,
12, 1665–1673.
[18] T. N. Ngo, F. Janert, P. Ehlers, D. H. Hoang, T. T. Dang, A. Villinger, S. Lochbrunner,
P. Langer, Org. Biomol. Chem. 2016, 14, 1293–1301.

APPENDIX
lxxx
[19] N. Eleya, A. Mahal, M. Hein, A. Villinger, P. Langer, Adv. Synth. Catal. 2011, 353,
2761–2774.
[20] J. F. Hartwig, Acc. Chem. Res. 1998, 31, 852–860.
[21] a) J. R. Dehli, J. Legros, C. Bolm, Chem. Commun. 2005, 973–986; b) R. Frlan, D.
Kikelj, Synthesis 2006, 2006, 2271–2285; c) B. Schlummer, U. Scholz, Adv. Synth. Catal.
2004, 346, 1599–1626; d) M. A. Fernandez-Rodriguez, Q. Shen, J. F. Hartwig, J. Chem.
Soc. 2006, 128, 2180–2181; e) G. Bastug, S. P. Nolan, J. Org. Chem. 2013, 78, 9303–9308;
f) F. Tappe, V. Trepohl, M. Oestreich, Synthesis 2010, 2010, 3037–3062.
[22] J. P. Wolfe, S. Wagaw, J.-F. Marcoux, S. L. Buchwald, Acc. Chem. Res. 1998, 31, 805–
818.
[23] a) E.-i. Negishi, J. Organomet. Chem. 2002, 653, 34–40; b) K. C. Nicolaou, P. G.
Bulger, D. Sarlah, Angew. Chem. Int. Ed. 2005, 44, 4442–4489; c) S. P. Stanforth,
Tetrahedron 1998, 54, 263–303.
[24] a) S. R. Chemler, D. Trauner, S. J. Danishefsky, Angew. Chem. Int. Ed. 2001, 40, 4544–
4568; b) S. Kotha, K. Lahiri, D. Kashinath, Tetrahedron 2002, 58, 9633–9695; c) A. J. J.
Lennox, G. C. Lloyd-Jones, Angew. Chem. Int. Ed. 2013, 52, 7362–7370; d) H. Li,
Johansson Seechurn, Carin C. C., T. J. Colacot, ACS Catal. 2012, 2, 1147–1164; e) R.
Rossi, F. Bellina, M. Lessi, Adv. Synth. Catal. 2012, 354, 1181–1255; f) M. Sasaki, H.
Fuwa, Synlett 2004, 1851–1874; g) M. Tobisu, N. Chatani, Angew. Chem. Int. Ed. 2009,
48, 3565–3568; h) V. Polshettiwar, A. Decottignies, C. Len, A. Fihri, ChemSusChem 2010,
3, 502–522; i) A. Suzuki, Chem. Commun. 2005, 4759–4763; j) A. Suzuki, Angew. Chem.
Int. Ed. 2011, 50, 6722–6737.
[25] a) R. Chinchilla, C. Najera, Chem. Rev. 2007, 107, 874–922; b) R. Chinchilla, C. Najera,
Chem. Soc. Rev. 2011, 40, 5084–5121; c) R. Chinchilla, C. Najera, Chem. Rev. 2014, 114,
1783–1826; d) K. Sonogashira, J. Organomet. Chem. 2002, 653, 46–49.
[26] X.-F. Wu, H. Neumann, M. Beller, Chem. Rev. 2013, 113, 1–35.
[27] Z. Liu, J. Wang, J. Org. Chem. 2013, 78, 10024–10030.
[28] Z. Shao, H. Zhang, Chem. Soc. Rev. 2012, 41, 560–572.
[29] Q. Xiao, Y. Zhang, J. Wang, Acc. Chem. Res. 2013, 46, 236–247.
[30] J. Barluenga, P. Moriel, C. Valdes, F. Aznar, Angew. Chem. Int. Ed. 2007, 46, 5587–
5590.
[31] J. Barluenga, C. Valdes, Angew. Chem. Int. Ed. 2011, 50, 7486–7500.
[32] a) G. A. Molander, J. P. Wolfe, M. Larhed, Cross coupling and Heck-type reactions,
Thieme, Stuttgart, 2013; b) J. P. Knowles, A. Whiting, Org. Biomol. Chem. 2007, 5, 31–
44.
[33] a) L.-C. Campeau, M. Parisien, A. Jean, K. Fagnou, J. Chem. Soc. 2006, 128, 581–590;
b) D. Alberico, M. E. Scott, M. Lautens, Chem. Rev. 2007, 107, 174–238; c) G. P.
McGlacken, L. M. Bateman, Chem. Soc. Rev. 2009, 38, 2447–2464.
[34] a) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord, F. Glorius, Angew. Chem. Int. Ed.
2012, 51, 10236–10254; b) S. R. Neufeldt, M. S. Sanford, Acc. Chem. Res. 2012, 45, 936–
946.

APPENDIX
lxxxi
[35] L.-C. Campeau, K. Fagnou, Chem. Commun. 2006, 1253–1264.
[36] A. F. Littke, G. C. Fu, Angew. Chem. Int. Ed. 1998, 37, 3387–3388.
[37] D. W. Old, J. P. Wolfe, S. L. Buchwald, J. Am. Chem. Soc. 1998, 120, 9722–9723.
[38] a) R. Martin, S. L. Buchwald, Acc. Chem. Res. 2008, 41, 1461–1473; b) D. S. Surry, S.
L. Buchwald, Angew. Chem. Int. Ed. 2008, 47, 6338–6361; c) D. S. Surry, S. L. Buchwald,
Chem. Sci. 2011, 2, 27–50; d) A. Aranyos, D. W. Old, A. Kiyomori, J. P. Wolfe, J. P.
Sadighi, S. L. Buchwald, J. Am. Chem. Soc. 1999, 121, 4369–4378.
[39] A. Zapf, A. Ehrentraut, M. Beller, Angew. Chem. Int. Ed. 2000, 39, 4153–4155.
[40] a) A. Zapf, M. Beller, Chem. Commun. 2005, 431–440; b) J. P. Stambuli, S. R. Stauffer,
K. H. Shaughnessy, J. F. Hartwig, J. Am. Chem. Soc. 2001, 123, 2677–2678.
[41] a) M.-N. Birkholz, Z. Freixa, van Leeuwen, Piet W N M, Chem. Soc. Rev. 2009, 38,
1099–1118; b) P. Dierkes, van Leeuwen, Piet W. N. M., J. Chem. Soc., Dalton Trans. 1999,
1519–1530; c) M. Kranenburg, van der Burgt, Yuri E. M., P. C. J. Kamer, van Leeuwen,
Piet W. N. M., K. Goubitz, J. Fraanje, Organometallics 1995, 14, 3081–3089; d) L. Qiu, F.
Y. Kwong, J. Wu, W. H. Lam, S. Chan, W.-Y. Yu, Y.-M. Li, R. Guo, Z. Zhou, A. S. C.
Chan, J. Chem. Soc. 2006, 128, 5955–5965; e) van Leeuwen, Piet W. N. M., P. C. J. Kamer,
J. N. H. Reek, P. Dierkes, Chem. Rev. 2000, 100, 2741–2770.
[42] a) L.-F. Tietze, Domino reactions, Wiley-VCH, Weinheim, 2013; b) L.-F. Tietze, G.
Brasche, K. M. Gericke, Domino reactions in organic synthesis, Wiley-VCH, Weinheim,
2006.
[43] L.-Q. Lu, J.-R. Chen, W.-J. Xiao, Acc. Chem. Res. 2012, 45, 1278–1293.
[44] a) M. Shiri, Chem. Rev. 2012, 112, 3508–3549; b) A. J. Kochanowska-Karamyan, M.
T. Hamann, Chem. Rev. 2010, 110, 4489–4497.
[45] G. W. Gribble, J. Chem. Soc., Perkin Trans. 1 2000, 1045–1075.
[46] S. Wagaw, B. H. Yang, S. L. Buchwald, J. Am. Chem. Soc. 1998, 120, 6621–6622.
[47] a) J. Barluenga, F. Rodriguez, F. J. Fananas, Chem. Asian. J. 2009, 4, 1036–1048; b) S.
Cacchi, G. Fabrizi, Chem. Rev. 2005, 105, 2873–2920; c) G. R. Humphrey, J. T. Kuethe,
Chem. Rev. 2006, 106, 2875–2911; d) M. Inman, C. J. Moody, Chem. Sci. 2013, 4, 29–41;
e) K. Krüger, A. Tillack, M. Beller, Adv. Synth. Catal. 2008, 350, 2153–2167.
[48] R. C. Larock, E. K. Yum, J. Am. Chem. Soc. 1991, 113, 6689–6690.
[49] A. Antonio, C. Sandro, M. Fabio, Tetrahedron Lett. 1992, 33, 3915–3918.
[50] M. G. Saulnier, D. B. Frennesson, M. S. Deshpande, D. M. Vyas, Tetrahedron Lett.
1995, 36, 7841–7844.
[51] L. Ackermann, Org. Lett. 2005, 7, 439–442.
[52] L. T. Kaspar, L. Ackermann, Tetrahedron 2005, 61, 11311–11316.
[53] M. C. Willis, G. N. Brace, I. P. Holmes, Angew. Chem. Int. Ed. 2005, 44, 403–406.
[54] J. Barluenga, A. Jimenez-Aquino, F. Aznar, C. Valdes, J. Chem. Soc. 2009, 131, 4031–
4041.
[55] A. W. Schmidt, K. R. Reddy, H.-J. Knolker, Chem. Rev. 2012, 112, 3193–3328.

APPENDIX
lxxxii
[56] a) K. Nozaki, K. Takahashi, K. Nakano, T. Hiyama, H.-Z. Tang, M. Fujiki, S.
Yamaguchi, K. Tamao, Angew. Chem. Int. Ed. 2003, 42, 2051–2053; b) Y. Zhou, J. G.
Verkade, Adv. Synth. Catal. 2010, 352, 616–620.
[57] a) L. Ackermann, A. Althammer, Angew. Chem. Int. Ed. 2007, 46, 1627–1629; b) L.
Ackermann, A. Althammer, P. Mayer, Synthesis 2009, 2009, 3493–3503.
[58] H. Kang, W. Fenical, J. Org. Chem. 1997, 62, 3254–3262.
[59] a) R. J. Andersen, D. J. Faulkner, C. H. He, G. D. van Duyne, J. Clardy, J. Am. Chem.
Soc. 1985, 107, 5492–5495; b) N. Lindquist, W. Fenical, G. D. van Duyne, J. Clardy, J.
Org. Chem. 1988, 53, 4570–4574.
[60] H. Fan, J. Peng, M. T. Hamann, J.-F. Hu, Chem. Rev. 2008, 108, 264–287.
[61] a) H. Kamiyama, Y. Kubo, H. Sato, N. Yamamoto, T. Fukuda, F. Ishibashi, M. Iwao,
Bioorg. Med. Chem. 2011, 19, 7541–7550; b) M. V. Reddy, M. R. Rao, D. Rhodes, M. S.
Hansen, K. Rubins, F. D. Bushman, Y. Venkateswarlu, D. J. Faulkner, J. Med. Chem. 1999,
42, 1901–1907; c) C. Ridley, Bioorg. Med. Chem. 2002, 10, 3285–3290.
[62] a) T. Ohta, T. Fukuda, F. Ishibashi, M. Iwao, J. Org. Chem. 2009, 74, 8143–8153; b) C.
Tardy, M. Facompre, W. Laine, B. Baldeyrou, D. Garcia-Gravalos, A. Francesch, C.
Mateo, A. Pastor, J. A. Jimenez, I. Manzanares et al., Bioorg. Med. Chem. 2004, 12, 1697–
1712.
[63] M. Iwao, T. Fukuda, F. Ishibashi, Heterocycles 2011, 83, 491.
[64] M. Komatsubara, T. Umeki, T. Fukuda, M. Iwao, J. Org. Chem. 2014, 79, 529–537.
[65] a) P. Cironi, I. Manzanares, F. Albericio, M. Alvarez, Org. Lett. 2003, 5, 2959–2962;
b) F. Ishibashi, S. Tanabe, T. Oda, M. Iwao, J. Nat. Prod. 2002, 65, 500–504.
[66] a) M. Zeeshan, V. O. Iaroshenko, S. Dudkin, D. M. Volochnyuk, P. Langer,
Tetrahedron Lett. 2010, 51, 3897–3898; b) O. Fatunsin, V. Iaroshenko, S. Dudkin, M.
Shkoor, D. Volochnyuk, A. Gevorgyan, P. Langer, Synlett 2010, 2010, 1533–1535.
[67] a) K. Cariou, B. Ronan, S. Mignani, L. Fensterbank, M. Malacria, Angew. Chem. Int.
Ed. 2007, 46, 1881–1884; b) K. Hiroya, S. Itoh, M. Ozawa, Y. Kanamori, T. Sakamoto,
Tetrahedron Lett. 2002, 43, 1277–1280; c) T. Kurisaki, T. Naniwa, H. Yamamoto, H.
Imagawa, M. Nishizawa, Tetrahedron Lett. 2007, 48, 1871–1874; d) X. Li, A. R. Chianese,
T. Vogel, R. H. Crabtree, Org. Lett. 2005, 7, 5437–5440; e) N. Sakai, K. Annaka, T.
Konakahara, Tetrahedron Lett. 2006, 47, 631–634; f) B. M. Trost, A. McClory, Angew.
Chem. Int. Ed. 2007, 46, 2074–2077; g) Y. Yin, Z. Chai, W.-Y. Ma, G. Zhao, Synthesis
2008, 2008, 4036–4040; h) Y. Zhang, J. P. Donahue, C.-J. Li, Org. Lett. 2007, 9, 627–630.
[68] L. Chen, M.-H. Xu, Adv. Synth. Catal. 2009, 351, 2005–2012.
[69] Z.-Y. Tang, Q.-S. Hu, Adv. Synth. Catal. 2006, 348, 846–850.
[70] L. Zhang, T. Meng, R. Fan, J. Wu, J. Org. Chem. 2007, 72, 7279–7286.
[71] a) M. Pohanka, Expert Opin. Ther. Pat. 2012, 22, 871–886; b) M. Weinstock, CNS
Drugs 1999, 12, 307–323.
[72] a) M. B. H. Youdim, D. Edmondson, K. F. Tipton, Nat. Rev. Neurosci. 2006, 7, 295–
309; b) M. Bortolato, K. Chen, J. C. Shih, Adv. Drug. Deliv. Rev. 2008, 60, 1527–1533.

APPENDIX
lxxxiii
[73] a) X. Zhou, X.-B. Wang, T. Wang, L.-Y. Kong, Bioorg. Med. Chem. 2008, 16, 8011–
8021; b) A. Saeed, S. Zaib, S. Ashraf, J. Iftikhar, M. Muddassar, K. Y. J. Zhang, J. Iqbal,
Bioorg. Chem. 2015, 63, 58–63; c) X. He, Y.-Y. Chen, J.-B. Shi, W.-J. Tang, Z.-X. Pan,
Z.-Q. Dong, B.-A. Song, J. Li, X.-H. Liu, Bioorg. Med. Chem. 2014, 22, 3732–3738; d) M.
Huang, S.-S. Xie, N. Jiang, J.-S. Lan, L.-Y. Kong, X.-B. Wang, Bioorg. Med. Chem. Lett.
2015, 25, 508–513; e) I. Orhan, H. Gulcan, Curr. Top. Med. Chem. 2015, 15, 1673–1682;
f) L. Pisani, R. Farina, M. Catto, R. M. Iacobazzi, O. Nicolotti, S. Cellamare, G. F.
Mangiatordi, N. Denora, R. Soto-Otero, L. Siragusa et al., J. Med. Chem. 2016, 59, 6791–
6806; g) A. Prasopthum, P. Pouyfung, S. Sarapusit, E. Srisook, P. Rongnoparut, Drug
Metab. Pharmacokinet. 2015, 30, 174–181; h) B.-F. Ruan, H.-J. Cheng, J. Ren, H.-L. Li,
L.-L. Guo, X.-X. Zhang, C. Liao, Eur. J. Med. Chem. 2015, 103, 185–190; i) P. O. Patil, S.
B. Bari, S. D. Firke, P. K. Deshmukh, S. T. Donda, D. A. Patil, Bioorg. Med. Chem. 2013,
21, 2434–2450.
[74] a) T. Ishikawa, Med. Res. Rev. 2001, 21, 61–72; b) T. Ishikawa, H. Ishii, Heterocycles
1999, 50, 627; c) V. Simánek, Z. Dvorák, V. Kubán, B. Klejdus, J. Vicar, J. Ulrichová, J.
Hlavac, Heterocycles 2006, 68, 2403.
[75] S. D. Phillips, R. N. Castle, J. Heterocycl. Chem. 1981, 18, 223–232.
[76] D. Glick (Ed.) Methods of Biochemical Analysis, John Wiley & Sons, Inc, Hoboken,
NJ, USA, 1971.
[77] A. M. Almerico, F. Mingoia, P. Diana, P. Barraja, A. Montalbano, A. Lauria, R. Loddo,
L. Sanna, D. Delpiano, M. G. Setzu et al., Eur. J. Med. Chem. 2002, 37, 3–10.
[78] a) V. Abet, A. Nunez, F. Mendicuti, C. Burgos, J. Alvarez-Builla, J. Org. Chem. 2008,
73, 8800–8807; b) E. Ahmed, A. L. Briseno, Y. Xia, S. A. Jenekhe, J. Chem. Soc. 2008,
130, 1118–1119; c) S. L. Bondarev, V. N. Knyukshto, S. A. Tikhomirov, A. N. Pyrko, Opt.
Spectrosc. 2006, 100, 386–393; d) A. Mishra, P. Bauerle, Angew. Chem. Int. Ed. 2012, 51,
2020–2067.
[79] C. Ritchie, G. J. T. Cooper, Y.-F. Song, C. Streb, H. Yin, A. D. C. Parenty, D. A.
MacLaren, L. Cronin, Nat. Chem. 2009, 1, 47–52.
[80] C. Baik, D. Kim, M.-S. Kang, K. Song, S. O. Kang, J. Ko, Tetrahedron 2009, 65, 5302–
5307.
[81] a) N. P. Buu-Hoï, P. Jacquignon, C. T. Long, J. Chem. Soc. 1957, 0, 505–509; b) D. Li,
B. Zhao, E. J. LaVoie, J. Org. Chem. 2000, 65, 2802–2805; c) M. Lysén, J. L. Kristensen,
P. Vedsø, M. Begtrup, Org. Lett. 2002, 4, 257–259; d) J. Pawlas, M. Begtrup, Org. Lett.
2002, 4, 2687–2690.
[82] C. Xie, Y. Zhang, Z. Huang, P. Xu, J. Org. Chem. 2007, 72, 5431–5434.
[83] L. Yan, D. Zhao, J. Lan, Y. Cheng, Q. Guo, X. Li, N. Wu, J. You, Org. Biomol. Chem.
2013, 11, 7966–7977.
[84] T. Satoh, M. Miura, D. Takeda, K. Hirano, Heterocycles 2012, 86, 487.
[85] C. Chen, G. Shang, J. Zhou, Y. Yu, B. Li, J. Peng, Org. Lett. 2014, 16, 1872–1875.
[86] J. Gao, Y. Shao, J. Zhu, J. Zhu, H. Mao, X. Wang, X. Lv, J. Org. Chem. 2014, 79, 9000–
9008.

APPENDIX
lxxxiv
[87] a) P. M. Byers, J. I. Rashid, R. K. Mohamed, I. V. Alabugin, Org. Lett. 2012, 14, 6032–
6035; b) T. Ishida, S. Kikuchi, T. Tsubo, T. Yamada, Org. Lett. 2013, 15, 848–851; c) M.
Kuhn, F. C. Falk, J. Paradies, Org. Lett. 2011, 13, 4100–4103; d) K. Yoshida, T. Furuyama,
C. Wang, A. Muranaka, D. Hashizume, S. Yasuike, M. Uchiyama, J. Org. Chem. 2012, 77,
729–732.
[88] a) T. Wang, Z. Yin, Z. Zhang, J. A. Bender, Z. Yang, G. Johnson, Z. Yang, L. M.
Zadjura, C. J. D'Arienzo, D. DiGiugno Parker et al., J. Med. Chem. 2009, 52, 7778–7787;
b) T. Wang, Z. Yang, Z. Zhang, Y.-F. Gong, K. A. Riccardi, P.-F. Lin, D. D. Parker, S.
Rahematpura, M. Mathew, M. Zheng et al., Bioorg. Med. Chem. Lett. 2013, 23, 213–217;
c) S. P. Tanis, M. B. Plewe, T. W. Johnson, S. L. Butler, D. Dalvie, D. DeLisle, K. R. Dress,
Q. Hu, B. Huang, J. E. Kuehler et al., Bioorg. Med. Chem. Lett. 2010, 20, 7429–7434; d)
M. B. Plewe, S. L. Butler, K. R. Dress, Q. Hu, T. W. Johnson, J. E. Kuehler, A. Kuki, H.
Lam, W. Liu, D. Nowlin et al., J. Med. Chem. 2009, 52, 7211–7219.
[89] S. J. Jung, G. Y. Kim, J. U. Lee, S. J. Eum, J. D. Lee, KR20150027659.
[90] S. R. Meech, D. Phillips, J. Photochem. 1983, 23, 193–217.
[91] N. Bontemps, F. Gattacceca, C. Long, O. P. Thomas, B. Banaigs, J. Nat. Prod. 2013,
76, 1801–1805.
[92] K. Venkateswarlu, K. Suneel, B. Das, K. N. Reddy, T. S. Reddy, Synth. Commun. 2008,
39, 215–219.
[93] a) R. F. Heck, Acc. Chem. Res. 1979, 12, 146–151; b) T. Mizoroki, K. Mori, A. Ozaki,
Bull. Chem. Soc. Jpn. 1971, 44, 581; c) L. Yin, J. Liebscher, Chem. Rev. 2007, 107, 133–
173; d) A. Biffis, M. Zecca, M. Basato, J. Mol. Catal. A: Chem. 2001, 173, 249–274; e) W.
Cabri, I. Candiani, Acc. Chem. Res. 1995, 28, 2–7.
[94] a) I. P. Beletskaya, A. V. Cheprakov, Chem. Rev. 2000, 100, 3009–3066; b) A. B.
Dounay, L. E. Overman, Chem. Rev. 2003, 103, 2945–2964; c) S. E. Gibson, R. J.
Middleton, Contemp. Org. Synth. 1996, 3, 447–471; d) G. A. Molander, J. P. Wolfe, M.
Larhed, Cross coupling and Heck-type reactions, Thieme, Stuttgart, 2013.
[95] G. Poli, G. Giambastiani, A. Heumann, Tetrahedron 2000, 56, 5959–5989.
[96] a) S. Y. Lau, N. G. Andersen, B. A. Keay, Org. Lett. 2001, 3, 181–184; b) S. P.
Maddaford, N. G. Andersen, W. A. Cristofoli, B. A. Keay, J. Am. Chem. Soc. 1996, 118,
10766–10773; c) Z. Owczarczyk, F. Lamaty, E. J. Vawter, E. Negishi, J. Am. Chem. Soc.
1992, 114, 10091–10092; d) A. C. Albéniz, P. Espinet, Y.-S. Lin, J. Am. Chem. Soc. 1996,
118, 7145–7152; e) D. Balcells, F. Maseras, B. A. Keay, T. Ziegler, Organometallics 2004,
23, 2784–2796.
[97] a) F. E. Meyer, P. J. Parsons, A. de Meijere, J. Org. Chem. 1991, 56, 6487–6488; b) D.
Soorukram, P. Knochel, Org. Lett. 2007, 9, 1021–1023.
[98] a) J. Barluenga, L. Florentino, F. Aznar, C. Valdes, Org. Lett. 2011, 13, 510–513; b) J.
R. Fulton, V. K. Aggarwal, J. de Vicente, Eur. J. Org. Chem. 2005, 2005, 1479–1492.
[99] a) R. Barroso, M.-P. Cabal, R. Badía-Laiño, C. Valdés, Chem. Eur. J. 2015, 21, 16463–
16473; b) R. Barroso, R. A. Valencia, M.-P. Cabal, C. Valdés, Org. Lett. 2014, 16, 2264–
2267.

APPENDIX
lxxxv
[100] a) J. E. Anthony, Chem. Rev. 2006, 106, 5028–5048; b) T. A. Grese, L. D. Pennington,
J. P. Sluka, M. D. Adrian, H. W. Cole, T. R. Fuson, D. E. Magee, D. L. Phillips, E. R.
Rowley, P. K. Shetler et al., J. Med. Chem. 1998, 41, 1272–1283; c) H.-J. Knölker, K. R.
Reddy, Chem. Rev. 2002, 102, 4303–4428; d) M. J. Rospondek, L. Marynowski, M. Góra,
Org. Geochem. 2007, 38, 1729–1756; e) D. R. Boyd, N. D. Sharma, F. Hempenstall, M. A.
Kennedy, J. F. Malone, C. C. R. Allen, S. M. Resnick, D. T. Gibson, J. Org. Chem. 1999,
64, 4005–4011.
[101] a) R. Che, Z. Wu, Z. Li, H. Xiang, X. Zhou, Chem. Eur. J. 2014, 20, 7258–7261; b)
C.-C. Chen, C.-M. Chen, M.-J. Wu, J. Org. Chem. 2014, 79, 4704–4711; c) G. Ferrara, T.
Jin, M. Akhtaruzzaman, A. Islam, L. Han, H. Jiang, Y. Yamamoto, Tetrahedron Lett. 2012,
53, 1946–1950; d) G. Ferrara, T. Jin, K. Oniwa, J. Zhao, A. M. Asiri, Y. Yamamoto,
Tetrahedron Lett. 2012, 53, 914–918; e) K. Hirano, Y. Inaba, N. Takahashi, M. Shimano,
S. Oishi, N. Fujii, H. Ohno, J. Org. Chem. 2011, 76, 1212–1227.
[102] M. Prashad, Y. Liu, X. Y. Mak, D. Har, O. Repič, T. J. Blacklock, Tetrahedron Lett.
2002, 43, 8559–8562.
[103] a) T. Dang, N. Kelzhanova, Z. Abilov, M. Turmukhanova, P. Langer, Synlett 2012, 23,
1283–1290; b) P. Langer, Chem. Eur. J. 2001, 7, 3858–3866; c) P. Langer, Synthesis 2002,
2002, 441–459; d) P. Langer, M. Döring, Eur. J. Org. Chem. 2002, 2002, 221–234; e) P.
Langer, W. Freiberg, Chem. Rev. 2004, 104, 4125–4150.
[104] a) D. Barnes-Seeman, J. Beck, C. Springer, Curr. Top. Med. Chem. 2014, 14, 855–864;
b) H.-J. Bohm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Muller, U. Obst-Sander, M.
Stahl, Chembiochem 2004, 5, 637–643; c) K. L. Kirk, Org. Process Res. Dev. 2008, 12,
305–321; d) S. Purser, P. R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 2008,
37, 320–330.
[105] P. M. Ridker, E. Danielson, F. A. H. Fonseca, J. Genest, A. M. Gotto, JR, J. J. P.
Kastelein, W. Koenig, P. Libby, A. J. Lorenzatti, J. G. MacFadyen et al., N. Engl. J. Med.
2008, 359, 2195–2207.
[106] a) N. Afdhal, K. R. Reddy, D. R. Nelson, E. Lawitz, S. C. Gordon, E. Schiff, R. Nahass,
R. Ghalib, N. Gitlin, R. Herring et al., N. Engl. J. Med. 2014, 370, 1483–1493; b) N. Afdhal,
S. Zeuzem, P. Kwo, M. Chojkier, N. Gitlin, M. Puoti, M. Romero-Gomez, J.-P. Zarski, K.
Agarwal, P. Buggisch et al., N. Engl. J. Med. 2014, 370, 1889–1898; c) E. J. Gane, C. A.
Stedman, R. H. Hyland, X. Ding, E. Svarovskaia, W. T. Symonds, R. G. Hindes, M. M.
Berrey, N. Engl. J. Med. 2013, 368, 34–44; d) E. Lawitz, A. Mangia, D. Wyles, M.
Rodriguez-Torres, T. Hassanein, S. C. Gordon, M. Schultz, M. N. Davis, Z. Kayali, K. R.
Reddy et al., N. Engl. J. Med. 2013, 368, 1878–1887.
[107] a) J. C. Biffinger, H. W. Kim, S. G. DiMagno, Chembiochem 2004, 5, 622–627; b) P.
Kirsch, Modern fluoroorganic chemistry, Wiley-VCH, Weinheim, Great Britain, 2013.
[108] a) S. Fustero, M. Sánchez-Roselló, P. Barrio, A. Simón-Fuentes, Chem. Rev. 2011, 111,
6984–7034; b) J.-Y. Yoon, S.-g. Lee, H. Shin, Curr. Org. Chem. 2011, 15, 657–674.
[109] H. A. DeWald, S. Lobbestael, B. P. H. Poschel, J. Med. Chem. 1981, 24, 982–987.
[110] N. K. Terrett, A. S. Bell, D. Brown, P. Ellis, Bioorg. Med. Chem. Lett. 1996, 6, 1819–
1824.
[111] M. Kim, C. Sim, D. Shin, E. Suh, K. Cho, Crop Prot. 2006, 25, 542–548.

APPENDIX
lxxxvi
[112] a) T. Furuya, A. S. Kamlet, T. Ritter, Nature 2011, 473, 470–477; b) P. Jeschke,
Chembiochem 2004, 5, 571–589; c) K. Muller, C. Faeh, F. Diederich, Science 2007, 317,
1881–1886; d) D. A. Nagib, D. W. C. MacMillan, Nature 2011, 480, 224–228; e) O. A.
Tomashenko, V. V. Grushin, Chem. Rev. 2011, 111, 4475–4521.
[113] M. A. Chowdhury, K. R. A. Abdellatif, Y. Dong, D. Das, M. R. Suresh, E. E. Knaus, J.
Med. Chem. 2009, 52, 1525–1529.
[114] S. R. Cox, S. Liao, M. Payne-Johnson, R. J. Zielinski, M. R. Stegemann, J. Vet.
Pharmacol. Ther. 2011, 34, 1–11.
[115] D. Zhang, N. Raghavan, S.-Y. Chen, H. Zhang, M. Quan, L. Lecureux, L. M. Patrone,
P. Y. S. Lam, S. J. Bonacorsi, R. M. Knabb et al., Drug Metab. Dispos. 2008, 36, 303–315.
[116] B. D. Maxwell, J. Labelled Cpd. Radiopharm. 2000, 43, 645–654.
[117] A. K. Culbreath, T. B. Brenneman, R. C. Kemerait, JR, G. G. Hammes, Pest. Manag.
Sci. 2009, 65, 66–73.
[118] a) S. Hernández, I. Moreno, R. SanMartin, G. Gómez, M. T. Herrero, E. Domínguez, J.
Org. Chem. 2010, 75, 434–441; b) P. S. Humphries, J. M. Finefield, Tetrahedron Lett.
2006, 47, 2443–2446; c) R. R. Ranatunge, M. Augustyniak, U. K. Bandarage, R. A. Earl,
J. L. Ellis, D. S. Garvey, D. R. Janero, L. G. Letts, A. M. Martino, M. G. Murty et al., J.
Med. Chem. 2004, 47, 2180–2193; d) S. Fustero, R. Román, J. F. Sanz-Cervera, A.
Simón-Fuentes, J. Bueno, S. Villanova, J. Org. Chem. 2008, 73, 8545–8552.
[119] T. Eicher, S. Hauptmann, A. Speicher, The Chemistry of Heterocycles, Wiley-VCH,
Weinheim, FRG, 2003.
[120] J. C. Sloop, C. L. Bumgardner, W. Loehle, J. Fluor. Chem. 2002, 118, 135–147.
[121] L. Buriol, C. P. Frizzo, L. D. T. Prola, D. N. Moreira, M. R. B. Marzari, E. Scapin, N.
Zanatta, H. G. Bonacorso, M. A. P. Martins, Catal. Lett. 2011, 141, 1130–1135.
[122] S. Fustero, R. Román, J. F. Sanz-Cervera, A. Simón-Fuentes, A. C. Cuñat, S. Villanova,
M. Murguía, J. Org. Chem. 2008, 73, 3523–3529.
[123] E. Y. Slobodyanyuk, O. S. Artamonov, O. V. Shishkin, P. K. Mykhailiuk, Eur. J. Org.
Chem. 2014, 2014, 2487–2495.
[124] G. Ji, X. Wang, S. Zhang, Y. Xu, Y. Ye, M. Li, Y. Zhang, J. Wang, Chem. Commun.
2014, 50, 4361–4363.
[125] a) R. S. Foote, C. F. Beam, C. R. Hauser, J. Heterocycl. Chem. 1970, 7, 589–592; b) N.
Matsumura, A. Kunugihara, S. Yoneda, Tetrahedron Lett. 1983, 24, 3239–3242; c) T. T.
Dang, T. T. Dang, C. Fischer, H. Görls, P. Langer, Tetrahedron 2008, 64, 2207–2215; d)
T. T. Dang, T. T. Dang, P. Langer, Tetrahedron Lett. 2007, 48, 3591–3593; e) T. T. Dang,
T. T. Dang, P. Langer, Synlett 2011, 2633–2642.
[126] a) J. M. Bates, J. Akerlund, E. Mittge, K. Guillemin, Cell Host Microbe 2007, 2, 371–
382; b) J. Fawley, D. M. Gourlay, J. Surg. Res. 2016, 202, 225–234; c) J.-P. Lalles, Nutr.
Rev. 2010, 68, 323–332.
[127] M. I. Torres, P. Lorite, M. A. Lopez-Casado, A. Rios, Pathol. Res. Pract. 2007, 203,
485–487.

APPENDIX
lxxxvii
[128] a) H. Orimo, J. Nippon Med. Sch. 2010, 77, 4–12; b) M. P. Whyte, Endocr. Rev. 1994,
15, 439–461; c) J. E. Coleman, Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 441–483.
[129] S. Sidique, R. Ardecky, Y. Su, S. Narisawa, B. Brown, J. L. Millan, E. Sergienko, N.
D. P. Cosford, Bioorg. Med. Chem. Lett. 2009, 19, 222–225.
[130] a) M. al-Rashida, J. Iqbal, Med. Res. Rev. 2014, 34, 703–743; b) Y. Baqi, Mini-Rev.
Med. Chem. 2015, 15, 21–33; c) M. Rashida, J. Iqbal, Mini-Rev. Med. Chem. 2015, 15, 41–
51.

Publication list
1. T. N. Ngo, T. T. Dang, A. Villinger, P. Langer, Adv. Synth. Catal. 2016, 358, 1328–1336.
2. H. Do, H. Tran, L. Ohlendorf, T. N. Ngo, T. Dang, P. Ehlers, A. Villinger, P. Langer,
Synlett 2015, 26, 2429–2433.
3. T. Q. Hung, T. N. Ngo, D. H. Hoang, T. T. Dang, K. Ayub, A. Villinger, S. Lochbrunner,
G.-U. Flechsig, P. Langer, Eur. J. Org. Chem. 2015, 2015, 1007–1019.
4. T. N. Ngo, O. A. Akrawi, T. T. Dang, A. Villinger, P. Langer, Tetrahedron Lett. 2015, 56,
86–88.
5. T. N. Ngo, F. Janert, P. Ehlers, D. H. Hoang, T. T. Dang, A. Villinger, S. Lochbrunner, P.
Langer, Org. Biomol. Chem. 2016, 14, 1293–1301.
6. T. N. Ngo, P. Ehlers, T. T. Dang, A. Villinger, P. Langer, Org. Biomol. Chem. 2015, 13,
3321–3330.
7. T. N. Ngo, S. A. Ejaz, T. Q. Hung, T. T. Dang, J. Iqbal, J. Lecka, J. Sevigny, P. Langer,
Org. Biomol. Chem. 2015, 13, 8277–8290.
8. N. N. Pham, T. T. Dang, T. N. Ngo, A. Villinger, P. Ehlers, P. Langer, Org. Biomol. Chem.
2015, 13, 6047–6058.
9. T. Q. Hung, D. H. Hoang, T. N. Ngo, T. T. Dang, K. Ayub, A. Villinger, A. Friedrich, S.
Lochbrunner, G.-U. Flechsig, P. Langer, Org. Biomol. Chem. 2014, 12, 6151–6166.
10. T. Q. Hung, T. N. Ngo, D. H. Hoang, T. T. Dang, A. Villinger, P. Langer, Org. Biomol.
Chem. 2014, 12, 2596–2605.


![m i s try & Modern Chemistry & Applications · aromatic heterocyclic ring system [5]. Heterocycles have enormous potential as the most promising scaffolds as lead structures for the](https://img.pdfslide.net/doc/110x75/5f32fa6dc6b2f5053d3cae9f/m-i-s-try-modern-chemistry-applications-aromatic-heterocyclic-ring-system.jpg)




![Studies on the Syntheses of Heterocycles from 3-Arylsydnone-4- carbohydroximic Acid Chlorides with N-Arylmaleimides, [1,4]Naphthoquinone and Aromatic Amines](https://img.pdfslide.net/doc/110x75/56649e725503460f94b71384/studies-on-the-syntheses-of-heterocycles-from-3-arylsydnone-4-carbohydroximic.jpg)





















