Embed Size (px)
Citation preview

UC RiversideUC Riverside Electronic Theses and Dissertations
TitleSynthesis of Isotopically Labeled Co-Enzyme to Probe the Active Site of Tryptophan synthase/ New Synthetic Approach to Tetrahydrocannabinol Analogs
Permalinkhttps://escholarship.org/uc/item/68x2v2nr
AuthorBastin, Baback
Publication Date2015-01-01 Peer reviewed|Thesis/dissertation
eScholarship.org Powered by the California Digital LibraryUniversity of California

UNIVERSITY OF CALIFORNIA RIVERSIDE
Synthesis of Isotopically Labeled Co-Enzyme to Probe the Active Site of Tryptophan synthase/ New Synthetic Approach to Tetrahydrocannabinol Analogs
A Dissertation submitted in partial satisfaction of the requirements for the degree of
Doctor of Philosophy
in
Chemistry
by
Baback Bastin
August 2015
Dissertation Committee:
Dr. Michael Marsella, Chairperson Dr. Richard Hooley
Dr. Thomas Morton

Copyright by Baback Bastin
2015

The Dissertation of Baback Bastin is approved:
Committee Chairperson
University of California, Riverside

iv
Acknowledgements
I am grateful to have been under the guidance of Dr. Michael Marsella. Without his
encouragement, wisdom, and patience, I wouldn’t have grown into the scientist I am
today.
I would also like to thank my parents, Hamid and Fahimeh, and my brother, Bardia, for
their support and love. Hamid and Fahimeh have been a true inspiration to me and always
supportive in all of my endeavors. My mom and dad sacrificed tremendously not only to
raise us but to give us the best education available and I will forever be grateful for their
sacrifices. Bardia has been my best friend and always there for me whenever I needed
him and I am grateful to have a brother as kind hearted and selfless as him.
I am truly thankful in having wonderful lab mates, Mackenzie Alvarez and Aiden
Aceves. They were always around for intellectually stimulating discussions and to help
with experiments in the lab.
Lastly, I would like to dedicate this thesis to the memory of both my grandfathers,
Hassan Bastin and Amir Houshang Aghdaei, who had passed away during my tenure at
UCR.

v
ABSTRACT OF THE DISSERTATION
Synthesis of Isotopically Labeled Co-Enzyme to Probe the Active Site of Tryptophan synthase/ New Synthetic Approach to Tetrahydrocannabinol Analogs
by
Baback Bastin
Doctor of Philosophy, Graduate Program in Chemistry University of California, Riverside, August 2015
Prof. Michael J. Marsella, Chairperson
Identifying enzyme mechanisms at proton level resolution is the ultimate goal of
enzymology. Traditional enzyme mechanistic studies infer protonation states from x-ray
crystal structure and optical spectroscopy. This thesis reports work towards the first
synergistic combination of x-ray crystallography, computational chemistry, synthetic
organic chemistry and solid-state NMR to fully elucidate, at proton level resolution, the
full three-dimensional structure of the catalytic site for Tryptophan synthase during active
catalysis. Specifically, this thesis describes solutions to the synthetic challenges of
introducing site-specific isotopic labels inside the cofactor Pyridoxal-5’-Phosphate (PLP)
and highlights a synthetic route that is consistently more cost-effective and higher
yielding than previous efforts.

vi
The second project presented focuses on efforts towards the synthesis of cannabinoids,
cannabidiol (CBD) and tetrahydrocannabinol (THC). Presently, cannabinoids have
emerged as compounds of interest for a variety of pharmacologic indications. Although
stereochemically simple compounds, economical syntheses of enantiopure cannabinoids
remain elusive. Strategies to address facile syntheses of THC and CBD, as well as their
analogs, will be presented.

vii
Table of Contents
Chapter 1: An Overview of the enzyme Tryptophan synthase: New Methodology to Characterize Each Intermediate at Proton Level Resolution Throughout the Catalytic Cycle
1.1 Introduction ................................................................................................................2 1.2 Tryptophan synthase .................................................................................................2 1.3 Atomic Resolution characterization of E(Q)indoline Intermediate ...............................8 1.4 Characterization of the E(Ain) Intermediate ...........................................................12
References ......................................................................................................................15
Chapter 2: Various Synthetic Approaches Towards Pyridoxal-5’-Phosphate
2.1 Introduction ..............................................................................................................18
2.2 Synthesis Through Pyridone Intermediate ...............................................................18
2.3 Synthesis Through the Kondrat’eva Reaction .........................................................20
2.4 Vollhardt’s Synthesis Through [2+2+2] Cobalt Mediated Cycloaddition ..............21 Reference .......................................................................................................................23 Chapter 3: Current PLP Synthesis and Comparison to Limbach Synthesis 3.1 Introduction ...............................................................................................................25 3.2 Current Published Synthesis of PLP .........................................................................25 3.3 Improvement/Discussion of Synthetic Route to PLP ...............................................27

viii
3.4 Synthesis of Isotopic Analogs of PLP ......................................................................34 3.5 Synthesis of [15N]-2-Aminophenol ...........................................................................36 3.6 Synthetic Approach to [17O]-PLP .............................................................................38 References .......................................................................................................................43 Chapter 4: Synthetic Background/ Synthetic Attempts Towards Cannabinoid Analogs 4.1 Introduction ................................................................................................................45 4.2 Cannabinoid Overview ..............................................................................................45 4.3 Previous Syntheses of Δ1-Tetrahydrocannabinol (Δ1-THC) .....................................48 4.4 Attempts Towards Synthesis of THC ........................................................................52 4.5 Conclusions ................................................................................................................57 References ........................................................................................................................59 Appendix A: Experimental Procedure and Spectroscopic Data ........................................60
Appendix B: 1H-NMR Spectra ..........................................................................................73

ix
List of Figures
Figure 1.1 Crystal structure of Tryptophan synthase ..........................................................3
Figure 1.2 α-site reaction mechanism .................................................................................4
Figure 1.3 β site reaction mechanism .................................................................................5
Figure 1.4 Reaction to form E(Q)Indoline intermediate ..........................................................8
Figure 1.5 Solid state NMR of labeled (red) vs unlabeled (blue) E(Q)Indoline intermediate 9
Figure 1.6 E(Q)indoline complex with with all possible protonation sites circled in green .10
Figure 1.7 Equilibrium and proton exchange of the E(Q)indoline intermediate ...................12
Figure 1.8 E(Ain) intermediate .........................................................................................13
Figure 3.1 Proposed mechanism for the formation of oxazole 2 ......................................29
Figure 3.2 Mechanism of the Diels-Alder reaction that forms compound 3 ....................30
Figure 3.3 Possible intermediates of the Diels-Alder adduct ............................................31
Figure 3.4 Hemiacetal formation of oxidized Pyridoxine in acidic solution ....................32
Figure 3.5 Reaction of 2-Aminophenol (2AP) with the EA-A intermediate ......................38
Figure 3.6 Proposed mechanism for the transformation of 26 to 27 .................................41
Figure 4.1 Left structure shows the monoterpenoid numbering, while the right structure shows the natural occurring (-)-Δ1-THC. Stereochemistry at carbons 3 and 4 are R, R ...49 Figure 4.2 Biosynthetic pathways to THC, CBD and CBC ..............................................54

x
List of Schemes
Scheme 2.1 Conversion of Pyridoxine to PLP a)MnO2, primary amine (R= -p-OEtC6H4, -OH, -t-Butyl), b) H3PO4, P2O5 ..........................................................................................18 Scheme 2.2 Harris, Stiller, and Folkers synthesis a) EtOH/piperidine b)Ac2O, HNO3 c) PCl5, ClC6H5 d) PtO2, H2 e)Pt, H2 f) HCl g) HONO, h)48 % HBr i)H2O, AgCl .............19 Scheme 2.3 Kondrat’eva reaction synthesis to pyridoxine a) HCOOH/EtOH, b) P2O5, DCM c)Diethyl maleate d)Lithium Aluminum Hydride, Et2O .........................................20 Scheme 2.4 Vollhardt synthesis a)n-BuLi, SnMe3Cl b) CH3CN, CpCo(CO2), m-xylenes c) CuI, NaOMe d)1. 48% HBr 2. AgCl, H2O ....................................................................21 Scheme 3.1 Limbach Synthesis a)HCO2H/EtOH, b) P2O5 c) Diethyl maleate d) LAH e) KMnO4, NaHSO3, 50% H2SO4, p-Toluidine f) H3PO4/P2O5 ............................................26 Scheme 3.2 Current published PLP synthesis a) SOCl2/EtOH b) Triethylorthoformate (TEOF) c) P2O5 d) Diethyl Maleate e) LAH f) MnO2/p-Phenetidine g) H3PO4/P2O5 ......28 Scheme 3.3 [15N]-PLP synthesis a) SOCl2/EtOH b) Triethylorthoformate (TEOF) c) P2O5 d) Diethyl Maleate e) LAH f) MnO2/p-Phenetidine g) H3PO4/P2O5 .................................34
Scheme 3.4 [15N,13C]-PLP synthesis a) SOCl2/EtOH b) Triethylorthoformate (TEOF) c) P2O5 d) Diethyl maleate e) LAH f)MnO2/p-Phenetidine g) H3PO4/P2O5 .........................35 Scheme 3.5 Synthesis of [15N]-2AP a) H15NO3(10M), Tetra-n-butylammonium bromide (TBAB), Ethylene Dichloride b)10% Pd/C, NaBH4 ..........................................................36 Scheme 3.6 A new [15N,13C]-PLP synthesis a) SOCl2/EtOH b) triethylorthoformate (TEOF) c) P2O5 d) 2,5-dihydrofuran,hydroquinone, trichloroacetic acid e) MnO2/p-phenetidine f) H3PO4/P2O5 .................................................................................................37 Scheme 3.7 Synthesis of [15N]-2AP a) HN15O3(10M), tetra-n-butylammonium bromide (TBAB), ethylene dichloride b)10% Pd/C, NaBH4 ...........................................................38 Scheme 3.8 Proposed synthesis of [17O]-PLP a) 3.5 M HCl b) Triethylorthoformate/Ac2O then NaHCO3 c) Iminoacetylacetone d) H2SO4 then NaN3 e) H2SO4/EtOH f) LAH g) NaNO2, H2O17 h) MnO2 then p-Phenetidine i) H3PO4/P2O5 ..............................................39
Scheme 4.1 First synthesis of Δ1-THC ..............................................................................50 Scheme 4.2 Mechoulam synthesis .....................................................................................51

xi
Scheme 4.3 Evans stereospecific synthesis of (S,S) Δ1-THC a) 1.Cationic bis(oxazoline)copper (II) catalyst 2.LiOBn b) MeMgBr c) Olivetol d)ZnBr2, MgSO4 .....52
Scheme 4.4 Trost synthesis a) 5 mol % [Mo(CO)3C7H8], sodium dimethyl malonate, 7.5% (R,R)-DACH-pyridyl TROST ligand b) 1. NaOH 2. HCl c) LDA, 4-iodo-2-methylbut-1-ene d) 1. (MeO)2SO2, K2CO3 2. Grubbs II catalyst e) MeLi f) NaSEt g) 1. ZnBr2, MgSO4 2. NaSEt .....................................................................................................53 Scheme 4.5 Olefin Metathesis synthetic route a) K2CO3, MeI b) n-BuLi, TMEDA, DMF c) Ph3P=CH2 d) Grubbs II Generation ...............................................................................55 Scheme 4.6 Diels-Alder route a) K2CO3, MeI b) n-BuLi, TMEDA, DMF c) NaOH, acetone d) Lewis acid catalyst ...........................................................................................56 Scheme 4.7 Biomimetic approach a) K2CO3, MeI b) n-BuLi, TMEDA, DMF c) NaI, AlCl3 d) NaOH, 6-methyl-5-hepten-2-one e) KOH, 6-methyl-5-hepten-2-one ................57 Scheme 4.8 Proposed synthetic pathways to CBD and THC a) 1-10% acid b) MeMgBr c) K-t-pentoxide/NaSEt ..........................................................................................................58

xii
List of Tables
Table 1.1 13C-NMR data from E(Q)indoline intermediate ....................................................11
Table 3.1 Summary of various Diels-Alder reactions .......................................................36
Table 4.1 Chemical structures of phyto-, synthetic and endo-cannabinoids ................. 47-8

1
Chapter 1
An Overview of the enzyme Tryptophan synthase/ New Methodology to Characterize
Each Intermediate, at Proton Level Resolution, Throughout the Catalytic Cycle.

2
1.1 Introduction
Tryptophan is one of the essential amino acids in living organisms. Human beings have
the ability to obtain this amino acid from their diets; however bacteria, yeasts, molds and
some plants use the catalytic ability of Tryptophan synthase to produce L-Tryptophan.1-4
This allows researchers to target this enzyme for the eradication of harmful bacteria or as
a herbicide. Having a clear understanding of the mechanism by which tryptophan
synthase creates L-Tryptophan (L-Trp) allows researchers to create an inhibitor that
would shut down the enzyme and, in turn, destroy the organism without harm to humans
or plants. The following sections will discuss an overview of the enzyme’s catalytic
mechanism, as well how a combination of synthetic, computational, X-ray
crystallography and solid state NMR has allowed for the chemically-detailed three-
dimensional structure of a few of the intermediates of the Tryptophan synthase catalytic
cycle.
1.1 Tryptophan synthase
Tryptophan synthase is a α2β2 bi-enzyme tetramer complex that catalyzes the
transformation of 3-indole-D-glycerol 3’-phosphate (IGP) to L-Trp in bacteria, yeasts,
molds and plants (Figure 1.1).1 The S. typhimurium bi-enzyme complex has been
extensively studied using various techniques1-6 and has been chosen as a model for the
better understanding of substrate channeling and the catalytic mechanism.

3
Figure 1.1 Crystal structure of Tryptophan synthase1
The enzyme is found in bacteria, yeasts, molds and plants. In higher animals, Tryptophan
synthase is non-existent and has become a potential drug target for herbicides and
infectious diseases, i.e. Chlamydia trachomatis and Mycobacterium tuberculosis. 7,8 The
full understanding of the enzymatic catalysis and substrate channeling could prove
helpful in designing potent inhibitors to battle infectious disease and to create herbicides.
The catalytic pathway consists of an α-reaction and a β-reaction. The α-site reaction is
the conversion of 3-indole-D-glycerol 3’-phosphate (IGP) to D-glyceraldehyde 3-
phosphate (G3P) and indole (Figure 1.2). 1-4 For the α-site reaction, the glutamic acid
residue (αGlu49) aids in the retro aldol condensation while the aspartic acid residue
(αAsp60) hydrogen bonds with the amine to further deactivate the ring and allow for
protonation at the β position relative to the amine in the indole ring.

4
Figure 1.2 α-site reaction mechanism2
Once indole has been made, it is channeled down a 25 Å tunnel to the β site where, with
the addition of L-serine (L-Ser), Pyridoxal 5’-Phosphate (PLP), and a monovalent cation
produces the amino acid L-Tryptophan (L-Trp) (Figure 1.3).1-4

5
Figure 1.3 β site reaction mechanism9
Initially, PLP forms an imine with the lysine residue (βLys87) to form an intermediate
internal aldimine, E(Ain). Next, the amino group of L-Ser nucleophilically attacks the
imine in E(Ain), initiating the transamination to form the gem-diamine intermediate,
E(GD1), which then releases the βLys87 residue and forms an external aldimine imine
complex E(Aex1). At this point, the βLys87 deprotonates the α proton and the quinonoid
substrate, E(Q1), is formed. Once the electrons from the pyridine nitrogen resonate back
up and promote elimination of water, the α-aminoacrylate, E(A-A), is formed and Stage 1
of the β site reaction is complete. At this point the α site reaction has been completed and
indole has been shuttled down to react with the E(A-A) intermediate to begin Stage 2 of
the β site reaction. Indole reacts with the E(A-A) through a Michael addition where the

6
enamine moiety of the indole ring attaches to the aforementioned intermediate to form
the new quinonoid structure E(Q2/3). The βLys87 residue then returns and protonates at
the α position giving the E(Aex2). At this point, the uncharged side-chain of the βLys87
residue becomes nucleophilic again and attacks the imine, initiating the second
transamination and giving the second gem-diamine intermediate E(GD2). The last step is
reforming the initial βLys87-PLP imine and freeing L-Trp from the active site.
The overall goal of this project is to define clearly enzymatic catalysis at a
chemical/structural level through a synergistic combination of X-ray crystallography,
computational chemistry, solid state NMR (SSNMR), and synthetic organic chemistry.
In the past, the relationship between protein structure and biological function has been
achieved with the determination of protein structures at a near atomic resolution, genetic
modification of proteins, and bioorganic mechanism experimentation. This has reduced
enzymatic catalysis to a simple set of sequential organic chemical reactions such as
Lewis acid catalysis, Brønsted acid-base catalysis, and nucleophilic/electrophilic
catalysis. This does not account, however, for the enormous rate accelerations achieved
by enzyme active sites. The introduction of computational chemistry has been
indispensible in enzymatic mechanism studies. Most of the current studies have
prioritized the electrostatic field effects at enzyme active sites to stabilizing higher energy
complexes.10-16 In order for a clearer, proton level resolution of enzyme catalysis to
occur, a better insight of the electrostatic microenvironment at the active site is necessary.

7
Though each technique used in the field of enzymology provides insight into enzyme
catalysis, a combination of all the techniques should provide the most detailed
mechanism. For example, a high resolution X-ray crystal structure provides details of the
interaction between enzyme side chains and substrate but at 1.5-2.5 Å resolution it
doesn’t allow for determining protonation states of the acidic/basic functional groups at
the active site. Typically, protonation states are inferred based on solution pKa and
hydrogen bonding pattern intuition. SSNMR allows for proton level resolution under the
same conditions used to solve X-Ray crystal structure and can be used for large
biomolecules, such as the 144 kDa Tryptophan synthase. Work done by McDowell et
al.17 showed that when labeled 13C-Serine was added to the enzyme, SSNMR could easily
identify the E(A-A) intermediate in the crystalline state. Through a combination of
SSNMR, X-Ray crystallography, computational chemistry and synthetic organic
chemistry each individual intermediate of the Tryptophan synthase catalytic cycle can be
resolved at the proton level.

8
1.3 Atomic resolution characterization of the E(Q)indoline Intermediate
The first intermediate fully characterized was a chemical and structural equivalent of the
E(Q2/3) intermediate, E(Q)indoline.5 Through inhibition of the α-reaction (while supplying
indoline instead of indole) the E(A-A) intermediate reacts with indoline to form the
quasi-stable intermediate (Figure 1.4).5
Figure 1.4 Reaction to form E(Q)Indoline intermediate5
The E(Q)indoline intermediate was characterized through solid-state NMR (Figure 1.6), and
distinct 13C resonances are highlighted. With the addition of isotopically labeled serine,
sharp and distinct peaks are present in the spectra (Figure 1.5).5

9
Figure 1.5 Solid state NMR of labeled (red) vs unlabeled (blue) E(Q)Indoline intermediate5
To accurately determine the protonation state of each molecule, a model of the β-subunit
7.0 Å from the substrate was constructed, and the substrate chemical shifts were
calculated. Figure 1.6 shows the possible sites of protonation and several candidate
structures were considered with different protonation sites. Not all the sites can be
simultaneously protonated, so a potential of 28 structures were calculated and compared
with experimental data.5

10
N
O
N
O
N O
O
HPO
OO
EnzNH2
Figure 1.6 E(Q)indoline complex with with all possible protonation sites circled green
According to the experimentally derived and computationally derived chemical shifts,
(Table 1.1), the previously reported protonated Schiff’s base hydrogen bonding to the
phenolic oxygen was found to be incorrect, and a more accurate depiction is fast
exchange between the protonated Schiff’s base and the carboxylic acid in a ratio of 34:66
(but the three-site equilibrium among the protonated Schiff base, the carboxylate, and the
phenolic form in a ratio of 33:58:9 cannot be ruled out) (Figure 1.7). The NMR data
suggest the predominent form is the protonated carboxylic acid hydrogen bonding with
the Schiff’s base, and the X-ray crystal structure also confirms the structure with
distances of the imine nitrogen in hydrogen bonding proximity to the carboxylic acid
proton. Calculations also show a build up of negative charge at Cα in the protonated
carboxylic acid structure, as well. This makes the free lysine from the enzyme more rigid

11
in this structure, as opposed to the free enzyme. The build up of negative charge at Cα not
only helps direct the proton from the lysine residue to Cα position, which is the next step
of the mechanism, but also lowers the energy barrier along the reaction coordinate
through charge stabilization.9 Once Cα is protonated, the lysine residue becomes
nucleophilic, the proton then jumps to the Schiff base making the imine more
electrophilic catalyzing the next step in the catalytic cycle: the transamination reaction.
Exp. Calculated PSB Form
Calculated Acid Form
Two-Site Eq.
Calc. Phenolic
Form
Three-Site Eq.
Cα 103.6 106 101.3 102.9 124.7 104.8 Cβ 54.1 54.0 49.6 51.1 52.7 51.4 C’ 173.0 172.3 169.3 170.3 175.6 170.9 N 296.5 215.1 337.2 295.7 321.7 295.7 C2 50.5 50.1 51.5 51.1 50.2 51.0 C3 28.5 31.9 32.5 32.3 31.8 32.2 N1 83.5 85.0 91.1 89.0 84.5 88.5
Table 1.15 13C-NMR data from E(Q)indoline intermediate

12
Figure 1.7 Equilibrium and proton exchange of the E(Q)indoline intermediate
1.4 Characterization of the E(Ain) Intermediate
By using 15N, 13C, and 31P NMR chemical shift measurements, UV/Vis spectroscopy, X-
ray crystallography and strategic labeling of atoms in the PLP coenzyme the full
protonation state of the E(Ain) was solved. (Figure 1.8).18
N
NN
H
O
O
O
H
OP
O
OO
ENZ
NH3
N
NN
O
O
OO
PO
OO
ENZ
NH3
N
NN
O
OH
O
H
OP
O
OO
ENZ
NH3
N
NN
O
OH
OO
PO
OO
ENZ
NH3
H

13
Figure 1.8 E(Ain) intermediate18
In Figure 1.8, the possible sites of protonation are circled in grey, the side chain residues
are colored blue and the experimentally derived charged states are in red. As
hypothesized, 19 the imine formed with Lys87 and the PLP coenzyme at the C4’ position
should be protonated. The UV/Vis spectroscopy, along with 15N/13C NMR studies,
confirms the protonated Schiff’s base hypothesis. The protonated Schiff’s base (PSB)
makes a more reactive center and allows for a nucleophilic attack at the C4’, as opposed
to the non-protonated tautomer.19-21 Protonation at the imine nitrogen initiates catalysis
and allows for the free serine residue to nucleophilically attack forming the gem-diamine
intermediate while the protonation states of both the phenolic oxygen and the pyridyl
nitrogen serve only in establishing the specificity of the reaction pathway.22-25 Typically,
a absorption maximum between 420-430 nm indicates the E(Ain) complex in PLP

14
dependent enzymes and the 412 nm λmax corresponds to a Schiff base linkage which is in
conjugation with the coenzyme pi-system.26 By viewing the X-ray crystal structure of the
Tryptophan synthase E(Ain) complex, the distance between the Schiff base nitrogen and
the phenolic oxygen is measured around 2.6 Å which correlates to a N−H--O hydrogen
bond. Although UV/Vis and X-ray crystal structure suggest a protonated Schiff base, it
was adamant to probe the active site using solid state NMR to confirm these findings.
These experiments were able to conclude that the phenolic oxygen was deprotonated and
hydrogen bound to the PSB, the pyridine N was deprotonated and hydrogen bound to the
hydroxyl moiety of Ser 377 residue and the phosphoryl group is in the dianionic state.
Having the pyridine nitrogen protonated aids the proton transfer from phenolic oxygen to
the PSB,27,28 which is required for the PSB hypothesis of rate enhancement. The X-ray
crystal structure does show a network of water molecules that keeps a consistent
hydrogen bonding interaction with the phenolic oxygen and this suggests that the
hydrogen bound water is satisfactory for initiating catalysis through a proton transfer to
the Schiff base nitrogen. The measuring of the chemical shifts within the enzyme active
site has allowed a full, proton level, characterization of the E(Ain) intermediate of the
Tryptophan synthase catalytic cycle.

15
REFERENCES:
1. Raboni, S.; Bettati, S.; Mozzarelli., Cell. Mol. Life Sci. 2009, 66, 2391-2403. 2. Dunn, F., M.; Niks, D.; Ngo, H.; Barends, T,R,M.; Schlichting, I,; Trends in Biochem. Sci. 2008, 33, 254-264. 3. Casino, P,. Niks, D., Ngo, H., Pan, P., Brzovic, P., Blumenstein, L., Barens, T.R., Schlichting, I., Dunn, M.F., Biochemistry, 2007, 47, 7713-7727. 5. Weyand, M., Schlichting, I., Biochemistry, 1999, 38, 16469-16480. 6. Pan, P., Woehl, E., Dunn, M.F., Trends in Biochem. Sci. 1997,22, 22-27. 7. Schpatzidis, A., Dealwis, C., Lubetsky, J.B., Liang, P.H., Anderson, K.S., Lolis, E. Biochemistry, 1999, 38, 12665-12674. 8. Finn, J., Langevine, C., Birk, I., Birk, J., Nickerson, K., Rodaway, S., Bioorg. & Med. Chem. Lett. 1999, 9, 2297-2302. 9. Lai, J., Niks, D., Wang, Y., Domratcheva, T., Barends, T. R.M., Schwarz, F., Olsen, R.A., Elliot, D.W., Fatmi, M.Q., Chang, C.A., Schlichtin, I., Dunn, M.F., Mueller, L.J., J. Am. Chem. Soc. 2011, 133, 4-7. 10. Hur, S., Bruice, T.C., Proc. Natl. Acad. Sci. USA. 2003, 100, 12015-12020. 11. Benkovic, S.J., Hammes-Schiffer, S., Science. 2003, 301, 1196-1202. 12. Claeyssens, F.,Harvey, J.N., Manby, F.R., Mata, R.A., Mulholland, A.J., Ranaghan, K.E., Schutz,M., Thiel, S., Thiel, W., Werner, H.J., Angew. Chem. Int. Ed. 2006, 45, 6856-6859. 13. Warshel, A., Sharma, P.K., Kato, M., Xiang, Y., Liu, H., Olsson, M.H., Chem Rev. 2006, 106, 3210-3235. 14. Sigala, P.A., Fafarman, A.T., Bogard, P.E., Boxer, S.G., Herschiag, D., J. Am. Chem. Soc. 2007, 129, 12104-12105. 15. Webb, L.J., Boxer, S.G., Biochem. 2008, 47, 1588-1598. 16. Childs, W., Boxer, S.G., J. Am Chem. Soc. 2010, 132, 6474-6480. 17. McDowell, L.M., Lee, M.S., Schaefer, J., Anderson, K.S. J. Am. Chem. Soc. 1995, 117, 12352-12353.

16
18. Caulkins, B.G., Bastin, B., Yang, C., Neubauer, T.J., Young, R.P., Hilario, E., Huang, Y.M., Chang, C.A., Fan, L., Dunn, M.F., Marsella, M.J., Mueller, L.J., J. Am. Chem. Soc, 2014, 136, 12824-12827. 19. Cordes, E.H., Jencks. W.P., Biochemistry. 1962, 1, 773. 20. Heinert, D., Martell, A.E., J. Am. Chem. Soc. 1963, 85, 188. 21. Metzler, D.E., J. Am. Chem. Soc. 1957, 79, 485. 22. Toney, M.D. Biochim. Biophys. Acta. 2011, 1814, 1407. 23. Crugeiras, J.; Rios, A.; Riveiros, E.; Richard, J. P. J. Am. Chem. Soc. 2011, 133, 3173. 24. Major, D. T.; Gao, J. L. J. Am. Chem. Soc. 2006, 128, 16345.
25. Major, D. T.; Nam, K.; Gao, J. L. J. Am. Chem. Soc. 2006, 128, 8114.
26. Peracchi, A., Bettati, S., Mozzarelli, A., Rossi, G.L., Miles, E.W., Dunn, M.F., Biochemistry. 1996, 35, 1872-1880. 27 Limbach, H. H.; Chan-Huot, M.; Sharif, S.; Tolstoy, P. M.; Shenderovich, I. G.; Denisov, G. S.; Toney, M. D. Biochim. Biophys. Acta. 2011, 1814, 1426. 28 Sharif, S.; Powell, D. R.; Schagen, D.; Steiner, T.; Toney, M. D.; Fogle, E.; Limbach, H. H. Acta Crystallogr., Sect. B 2006, 62, 480.

17
Chapter 2
Various Synthetic Approaches Towards Pyridoxal-5’-Phosphate (PLP)

18
2.1 Introduction Pyridoxine, precursor to PLP (Scheme 2.1), has been synthesized through various
different routes using a variety of different starting materials and synthetic strategies .1-4
The bioactive form of vitamin B6, PLP, was resolved and synthesized in 1944 by
Gunsalus et al.5 Conversion of pyridoxine to the bioactive form, PLP, occurs through two
steps; an oxidation/protection and a phosphorylation/removal of protecting group.
Scheme 2.1 Conversion of Pyridoxine to PLP a) MnO2, primary amine (R= -p-OEtC6H4, -OH, -t-Butyl), b) H3PO4, P2O5
Though numerous syntheses exist, none of them were cost effective, high yielding or able
to directly label any atom throughout the PLP molecule. Herein is a brief overview of the
various syntheses of pyridoxine.
2.2 Synthesis Through Pyridone Intermediate
In 1939, Stiller, Keresztesy and Stevens fully elucidated the strucutre pyridoxine.6 The
complete syntheis of Pyridoxine was accomplished in 1939 (by Harris, Stiller, and
Folkers), and was completely identical, and biologically equivalent in activity, to the
natural vitamin Scheme 2.2.7
N
HOOH
N
HO
NOH
R
N
HO
O
OPO3H2
Pyridoxine Pyridoxal-5'Phosphate
HO
a b

19
Scheme 2.27 Harris, Stiller, and Folkers synthesis a) EtOH/piperidine b)Ac2O, HNO3 c) PCl5, ClC6H5 d) PtO2, H2 e)Pt, H2 f) HCl g) HONO, h)48 % HBr i)H2O, AgCl
The synthesis begins through a condensation between cyanoacetamide and ethyl
acetonoxoalate. The resulting pyridone is nitrated with fuming nitric acid, then converted
to the pyridyl chloride with PCl5. Once isolated, the nitro group is reduced with PtO2 and
H2 to the amine, reductive dechlorination removes the chloride group, and reduces the
cyano group to the primary amine. Refluxing in concentrated hydrochloric acid
protonated both primary amines to make the dihydrochloride. The dihydrochloride was
heated in H2SO4(aq) and to the reaction aqueous solution of sodium nitrite was slowly
added resulting in the 4-ethoxy analog of pyridoxine. The 4-ethoxy analog was refluxed
in 48 % HBr to afford the dibromide species, then refluxed in water and AgCl to yield
O OO
H2N
OCN
NH
CNCH2OC2H5
O
a b
NH
CNCH2OC2H5
O
O2N c
N
CNCH2OC2H5
Cl
O2N
d
N
CNCH2OC2H5
Cl
H2Ne
N
CH2NH2
CH2OC2H5H2Nf
N
CH2NH2
CH2OC2H5ClHH2Ng
N
CH2OC2H5HO
OH
N
CH2BrHO
Br
N
HOOH
HO
h
i
HCl

20
pyridoxine. The overall yield for the reaction was not only very low, but also none of the
starting materials were commercially available isotopically labeled.
2.3 Synthesis Through the Kondrat’eva Reaction
Most of the recent pyridoxine syntheses are centered around the Kondrat’eva approach.8,9
This reaction is an inverse-demand [4+2] cycloaddition between 4-methyl-5-ethoxy
oxazole and a variety of dienophiles and has been shown to form cinchomeronic acids .10
The idea was to place all the necessary functional groups onto the oxazole in order to go
directly to a pyridoxine analog. This new way has shortened the pyridoxine synthesis
tremendously from previous approaches, and is now the most commonly used synthetic
stragey employed. Scheme 2.3 shows the most current synthesis for pyridoxine.
Scheme 2.311 Kondrat’eva reaction synthesis to pyridoxine a) HCOOH/EtOH, b) P2O5, DCM c) Diethyl maleate d) Lithium Aluminum Hydride, Et2O
HO2C NH2
ab
NH
EtO2C
O
N
O OEt
N
HOCO2Et
CO2Et
N
HOOH
c
d
HO

21
The tandem N-formylation/esterfication is performed in a autoclave at high
temperature/pressure with formic acid and ethanol. This ester is then cyclized to the
oxazole The Diels-Alder reaction occurs with diethyl maleate but has been shown to react
with a variety of other dienophiles. The product is reduced with lithium aluminum
hydride to give pyridoxine.
2.4 Vollhardt’s Synthesis Through [2+2+2] Cobalt Mediated Cycloaddition
This particular synthesis uses a [2 + 2 + 2] cycloaddition between α,ω-diynes with nitriles
and α,ω-cyanoalkynes with alkynes Scheme 2.4.4
Scheme 2.4 Vollhardt synthesis a) n-BuLi, SnMe3Cl b) CH3CN, CpCo(CO2), m-xylenes
c) I2, CHCl3 d) CuI, NaOMe e) 1. 48% HBr 2. AgCl, H2O
The first step was the synthesis of the diyne compound by lithiating the bis-2-propynyl
ether, and then reacting with trimethylstannyl chloride. Next, a regioselective cyclization
catalyzed by η5-cyclopentadienyl dicarbonyl cobalt (CpCo(CO2)) was employed with the
OSnMe3
SnMe3 N
OSnMe3
N
OOMe
N
HO
OHHO
Oa
c
e
b
N
OId

22
diyne and acetonitrile refluxed in m-xylenes. The product monodestannylated, upon
chormatographic purfication giving the monostanyl pyridyl compound. Cleavage of the
aryl-tin bond was first initiated with a metal-metal exchange with CuI then, the
methoxide anion was able to nucleophilically displace the resulting aryl-halide bond. The
inner ether was then cleaved by refluxing in HBr and the methoxy group was deprotected
with AgCl.

23
References:
1. Ichiba, A., Emoto, S., Sci. Papers of Inst. Phys. And Chem. Research, 1941, 38, 317-352.
2. Harris, S.A., Folkers, K., J. Am. Chem. Soc., 1939, 61, 3307-3310. 3. Blackwood, R.K., Hess, G.B., Larrabee, C.E., Pilgrim, F.J., J. Am. Chem. Soc. 1958,
80, 6244-6249. 4. Parnell, C.,A., Vollhardt, K.P.C., Tetrahedron, 1985, 41, 5791-5796. 5. Gunsalus, I.C., Bellamy, W.D., Umbreit, W.W., J. Biol. Chem, 1944, 155, 685-686. 6. Stiller, E.T., Keresztesy, J.C., Stevens, J.R., J. Am. Chem. Soc, 1939, 61, 1237-1242. 7. Harris, S.A., Stiller, E.T., Folkers, K., J. Am. Chem. Soc. 1939, 61, 1242-1244. 8. Kondratjeva, G. Y, Khim. Nauka I Prom., 1957, 2, 666. 9. Sharif, S., Schagen, D., Toney, M.D., Limbach, H.H., J. Am. Chem. Soc., 2007, 129,
4440-4455. 10. Firestone, R.A., Harris, E.E., Reuter,W., Tetrahedron, 1967, 23, 943-955. 11. Chan-Huot, M., Niether, C., Sharif, S., Tolstoy, P.M., Toney, M.D., Limbach, H.H., J. Molec. Struc. 2010, 976, 282-289.

24
Chapter 3
Current PLP Synthesis and Comparison to Limbach Synthesis

25
3.1 Introduction
The current synthesis of PLP was published by the Limbach group1, but there were
several issues with their approach. There are several stipulations that were put forth
before we landed on our specific approach. First, we had to design a synthesis that
allowed us to use commercially available isotopically labeled starting material. Second,
the synthesis had to be high yielding and able to recover starting material after each step.
Third, cost analysis played a crucial role in designing the synthesis and this synthesis is
the cheapest route to our desired product. Lastly the synthesis had to be easily
reproducible so that anybody could follow the directions and easily makes isotopic
variants of PLP. We understood there was no one single synthesis to produce all
isotopically labeled PLP analogs, and after reviewing all of the literature available we
determined a modified Kondrat’eva approach satisfied all of our needs for the synthesis
of PLP.

26
3.2 Current Published Synthesis of PLP
Scheme 3.1 details the current published synthesis and this section will detail some of the
issues with the synthesis.
Scheme 3.11 Limbach Synthesis a)HCO2H/EtOH, b) P2O5 c) Diethyl maleate d) LAH e) KMnO4, NaHSO3, 50% H2SO4, p-Toluidine f) H3PO4/P2O5
The first step is a combination of a N-formylation with an esterification of alanine. This
reaction is placed inside of a high-pressure autoclave and heated at 200 °C for 24 hours
yielding 50% of Ethyl-N-formyl-alaninate 1. Next, 1 is cyclized through a 5-exo-trig ring
cyclization which is thermally allowed by Baldwin’s rules,2-4 by dissolving the ethyl-N-
formyl-alaninate in dichloromethane (DCM) then slowly adding P2O5. This reaction yield
was roughly 50% and made 4-methyl-5-ethoxyoxazole 2. Once the oxazole has been
purified it is placed into a small vial, alongside with Diethyl maleate, sealed, and heated
H2N CO2H
a)NH
CO2Et
Ob)
N
O OEt
c)
N
HOCO2Et
CO2Etd)
N
HO
OH
N
HO
N
e)f)
N
HO
O
OPO3H2
OH
OH
1 2
34
56

27
for 3 days. After workup 4,5-ethoxycarbonyl-3-hydroxy-2-methyl-pyridine 3 is obtained
in roughly 60 % yield. Product 3 is reduced to pyridoxine 4 with lithium Aluminum
Hydride (LAH) at 90% turnover. Pyridoxine 4, is then oxidized by an in situ formation of
MnO2 then scavenged from the aqueous mixture with p-Toluidine to give N-
(pyridoxlidene)-tolylamine hydrochloride 5 in 44 % yield. Lastly, the imine free hydroxyl
group is monophosphorylated and the imine is deprotected with a H3PO4/P2O5 mixture,
and the resulting crude product is placed on top of an ion exchange resin and eluted with
water giving 24 % yield of our desired product pyridoxal-5’-phosphate (6). Overall, the
six-step synthesis gives a 1.4 % yield, which corresponds to $9500 for 1 g of 15N-PLP
and ~$93,000 for 1 g of [15N,13C] PLP. This specific synthesis was extremely costly and
some of the steps were questionable in their reproducibility. For step a, placing the
simultaneous N-formylation/esterification returned mainly starting material regardless of
how dry the formic acid and ethanol were. In step c, regardless of how pure the oxazole,
was the yield was not consistent with the literature value and several side products
formed. For step e the allylic oxidation/imine formation did not work at all and the MnO2
was never confirmed to be formed in situ. The aromatic primary amine was too water-
soluble and wouldn’t crystallize out product (5). The following section will discuss the
new method and some changes made to make the synthesis more reproducible, higher
yielding, cheaper and the same number of steps.

28
3.3 Improvements/Discussion of Synthetic Route to PLP
Scheme 3.2 illustrates the modified synthetic route to PLP.
Scheme 3.2 Current published PLP synthesis a) SOCl2/EtOH b) triethylorthoformate (TEOF) c) P2O5 d) diethyl maleate e) LAH f) MnO2/p-phenitidine g) H3PO4/P2O5
The first step of the reaction is an esterification of L-alanine to the ethyl ester5. A solution
of alanine in absolute ethanol is mixed with 4 equivalents of thionyl chloride and heated
gently. The key to this reaction is crystallizing out the alanine ethyl ester 7 through a
layered recrystallization. A pressure recrystallization works by dissolving 7 in a small
amount of absolute ethanol then by gently pipetting an excess of diethyl ether, without
disturbing the ethanol layer, until the heterogeneous solution becomes cloudy. This
mixture is then set aside at room temperature until the formation of crystals begin at
which point the flask is transferred to the freezer and allowed to complete the
H2N CO2H
a)
CO2EtH2Nb)
NH
CO2Et
O
N
O OEt
c)
d)
N
HO
OH
OH
N
HO
N
OEt
OH
e)
f)
N
HO
O
OPO3H2
71
2
4
86

29
crystallizing process overnight. The yield of this reaction was ~100 % and the product 7
continued to the next step without any further purification.
The next step is the N-formylation of compound 76. The solid is reacted with 3 molar
equivalents of triethylorthoformate. Once all of the excess solvent is evaporated, the
crude is sufficiently pure and used in the next step of the synthesis and yielded 95% of
product 1.
The next step of the reaction is the formation of the 4-methyl-5-ethoxy oxazole via a 5-
exo-trig ring cyclization.1 The excess of the dehydrating agent, P2O5, forces the loss of
water instead of the better leaving group, ethanol, to give the desired oxazole 2 in 60 %
yield, Figure 3.1.
Figure 3.1. Proposed mechanism for the formation of oxazole 2, where LA stands for the Lewis acid P2O5
In Figure 3.1, the first step of the proposed mechanism is the 5-exo-trig cyclization,
which is thermally allowed according to Baldwin’s rules.2-4 Next, the oxyanion formed
chelates to the P2O5 and deprotonation at the nitrogen occurs. The phosphorous-oxy
complex is then eliminated through anchimeric assistance (since the non bonding pi

30
electrons on the oxygen are in an anti orientation from the leaving group) and
deprotonation and rearmotization occurs to form the oxazole.
Once the oxazole has been formed, the next step is a Diels-Alder reaction. The electron-
poor dienophile diethyl maleate was used and Figure 3.2 gives a proposed mechanism
according to Firestone, Harris and Reuter.7 In Figure 3.3, the researchers were puzzled by
the mechanism of forming exclusively the 3-hydroxy derivative. The mechanism
suggests unfavorable cis elimination in order to turn the oxo bridgehead into a
hemiacetal. Figure 3.3 demonstrates the 3 possible intermediates formed with the Diels-
Alder reaction of 4-methyl-5-ethoxy oxazole.
Figure 3.2 Mechanism of the Diels-Alder reaction that forms compound 3

31
Figure 3.3 Possible intermediates of the Diels-Alder adduct
Looking at Figure 3.3, intermediate A possesses a carbenium carbon atom at the bridge-
head of the small bicylic. The carbenium ion at the bridgehead is an extremely high-
energy form and therefore was eliminated as a possible intermediate. Intermediate C, as
shown in the mechanism, has a cis-elimination, which is unfavorable. Intermediate B
seemed the likely intermediate. It was proposed that, 7 from the workup, would attack the
carbocation in intermediate B to form a hemiacetal and elimination would occur to form
the aromatic pyridine ring. Unfortunately, if intermediate B were the correct choice, the
3-alkoxy intermediate should have been isolated but never was. Studies also with 18O-
water also never showed any incorporation of the label at the 3-position. As stated earlier,
the 3-hydroxypyridine derivatives was the exclusive product for every dienophile
reaction with the oxazole. The researchers came to two conclusions, either intermediate C
was correct where the oxo-bridgehead spontaneously opens to give a carbocation at the 6
position in the ring, or the cis-elimination is exceptionally easy.
Once 3 has been isolated, the reduction with lithium aluminum hydride afford Pyridoxine
41. Pyridoxine is dissolved into an acidic aqueous solution, with powdered MnO2 added,

32
and stirred at room temperature to initiate the selective allylic oxidation/protection.8 At
this point, it is important to make the p-phenetidine soluble in water by stirring with
water and adding concentrated HCl until a homogenous solution is formed. Once the
reaction has completed, the crude reaction mixture is filtered and the p-phenetidine
solution, alongside with a solution of NaOAc, is added to the filtrate. After allowing the
mixture to sit in the fridge overnight a yellow solid 9, crystallizes out from the aqueous
solution. After filtering and washing with cold water product 9 is isolated, in 95% yield,
and is used through to the next step without any further purification needed. The allylic
oxidation with MnO2 is a well studied reaction9, but here it was necessary to convert the
transformed aldehyde into an imine to protect from hemiacetal formation by the adjacent
primary alcohol Figure 4.3. The hemiacetal is known to form in acidic aqueous solutions
rapidly.
Figure 3.4 Hemi acetal formation of oxidized yridoxine in acidic solution
The final step of the synthesis is the monophosphorylation at the free primary hydroxyl
group and deprotection of the imine group to form pure PLP in its biologically active
N
HO
OOH
N
HOO
HO
H

33
form.10 First, a mixture of 85 % ortho-phosphoric acid and P2O5 are stirred together under
nitrogen gas. Once the exothermic dissolution subsides, 9 is added and the reaction
mixture is gently heated. After heating between 4-5 hours, 0.1 M HCl is added and the
reaction mixture is heated for a few minutes to deprotect the imine and form the
aldehyde.
While the reaction is heating, the cation exchange resin needs to be prepared accordingly.
The resin that works the best is the Dowex® 50WX4 hydrogen form with a 200-400 mesh
size. The resin is then poured into a column and first washed with distilled water,
absolute alcohol and lastly distilled water once more. Next, 1 M HCl is passed through,
followed by distilled water, 1 M NaOH, distilled water, 1 M HCl, and lastly distilled
water until the eluted water is no longer acidic.11
Once the column has been prepared, the crude reaction mixture is cooled to room
temperature and then added to the top of the column and the product is eluted with
distilled water. Typically 1-2 L, sometimes more, of distilled water is passed through to
collect all of the PLP. The water is then evaporated with the rotary evaporator with gentle
heating, never allowing the water bath to exceed 45 °C. All but 5.0 mL of the solution is
evaporated and the aqueous solution contacting PLP is suitable for biological studies.

34
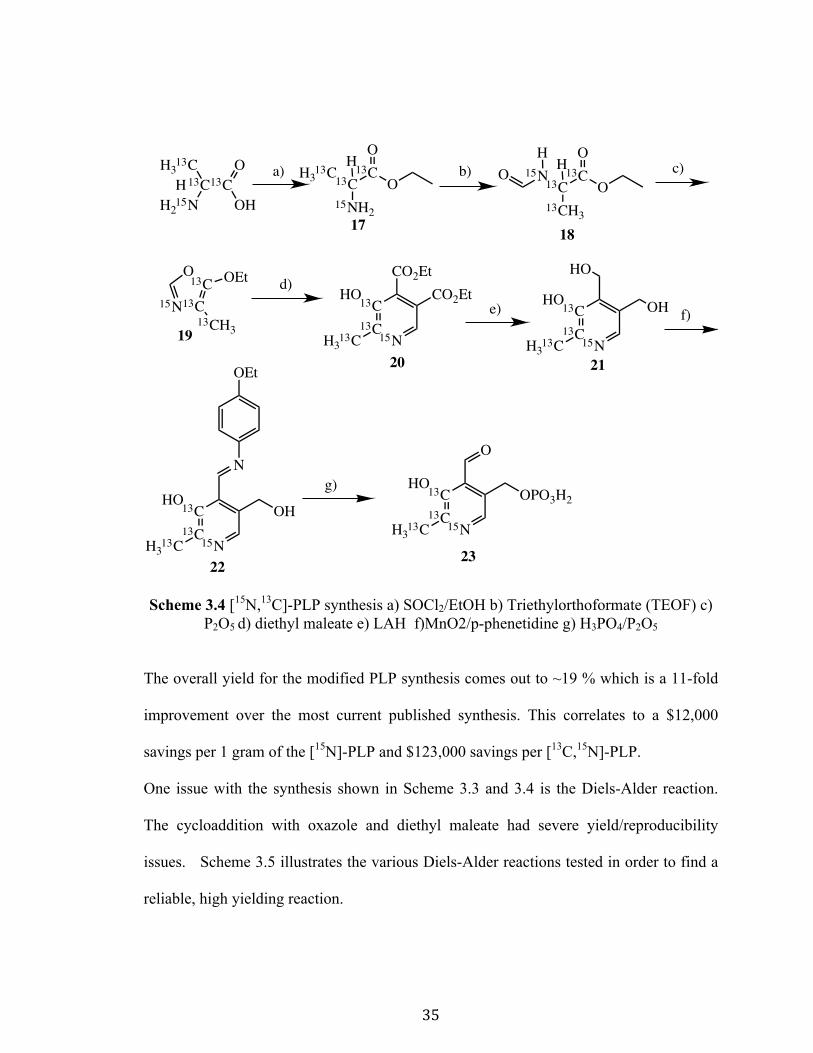
3.4 Synthesis of Isotopic Analogs of PLP
The synthesis discussed in Section 3.3 was utilized to make two isotopic analogs of PLP.
Scheme 3.3 and Scheme 3.4 illustrate synthetic schemes for the synthesis of two
isotopically labeled PLP analogs.
Scheme 3.3 [15N]-PLP synthesis a) SOCl2/EtOH b) triethylorthoformate (TEOF) c) P2O5 d) diethyl maleate e) LAH f) MnO2/p-phenetidine g) H3PO4/P2O5
H215N CO2H
a)
CO2EtH215Nb) CO2Et
15N
O OEt
c)
d)
15N
HO
OH
OH
15N
HO
N
OEt
OH
e)
g)
15N
HO
O
OPO3H2
10 11
1214
15 16
15NH
O
15N
HOCO2Et
CO2Et
13
f)
35
Scheme 3.4 [15N,13C]-PLP synthesis a) SOCl2/EtOH b) Triethylorthoformate (TEOF) c) P2O5 d) diethyl maleate e) LAH f)MnO2/p-phenetidine g) H3PO4/P2O5
The overall yield for the modified PLP synthesis comes out to ~19 % which is a 11-fold
improvement over the most current published synthesis. This correlates to a $12,000
savings per 1 gram of the [15N]-PLP and $123,000 savings per [13C,15N]-PLP.
One issue with the synthesis shown in Scheme 3.3 and 3.4 is the Diels-Alder reaction.
The cycloaddition with oxazole and diethyl maleate had severe yield/reproducibility
issues. Scheme 3.5 illustrates the various Diels-Alder reactions tested in order to find a
reliable, high yielding reaction.
H215N13C13C
H313C H313C13C13C
15NH2
15N13C
13CO
13CH3
OEt13C13C15NH313C
HOCO2Et
CO2Et
13C13C15NH313C
HO
N
OEt
OH13C13C15NH313C
HO
O
OPO3H2
13CH3
13C13C15NOO
OH
O
O
O
Oa) b) c)
d)e)
g)
17 18
19
20 21
23
13C13C15NH313C
HO
HO
OH f)
22
H HH
H

36
Scheme 3.5 Various Diels-Alder reactions attempted
Table 3.1 gives an exhaustive overview of the various reactions attempted and the yields
of pyridoxine isolated.
Table 3.1 Summary of various Diels-Alder reactions
N
O OR
R = -Et, -CO2Et
R1 R1
R1= -CO2Et, -CO2Me, -CH2OH, CH2OAc
OR2 R2
R2= -H, -(O)
O O
R3
R3 = -n-propyl, i-propyl
N
HO
OH
OH
Oxazole Dienophile Catalyst Yield of Pyridoxine
(%) -Et -CO2Et7 None/CaO/Rad.
Inhibitor 27
-CO2Me7 None 30
-CH2OH7 CaO, Hydroquinone < 5
-CH2OAc12 CaO < 5
-H7 Trichloroacetic Acid 60
-C(O)7 None < 15
-n-propyl13 CaO <5
-i-propyl13 CaO <5
-CO2Et -CO2Et14 None 30

37
It became evident that the one dienophile, 2,5-dihydrofuran, was the best choice. Scheme
3.6 now illustrates the new synthetic scheme, which corresponds to a 23 % overall yield.
Scheme 3.6 A new [15N,13C]-PLP synthesis a) SOCl2/EtOH b) triethylorthoformate (TEOF) c) P2O5 d) 2,5-dihydrofuran,hydroquinone, trichloroacetic acid e) MnO2/p-
phenetidine f) H3PO4/P2O5
The synthesis in Scheme 3.6 is essentially the same as the previously reported synthesis
in Schemes 3.3 and 3.4 except for the new Diels-Alder reaction. The new dienophile, 2,5-
dihydrofuran, is used in 20 fold molar excess in relation to the oxazole. It was shown that
a 25 mol % of recrystallized trichloroacetic acid a catalytic amount of hydroquinone
added to the reaction improved yields drastically. The reaction is run in a steel autoclave
heated at 190 °C. for 5 hours.
3.5 Synthesis of [15N]-2-Aminophenol
The inhibitor, 2-Aminophenol (2-AP), works by binding to the E(A-A) intermediate
Figure 3.5.
H215N13C13C
H313C H313C13C13C
15NH2
15N13C
13CO
13CH3
OEt
13C13C15NH313C
HO
N
OEt
OH13C13C15NH313C
HO
O
OPO3H2
13CH3
13C13C15NOO
OH
O
O
O
Oa b c
d f
1718
19
2123
13C13C15NH313C
HO
HO
OH e
22
H HH
H

38
Figure 3.5 Reaction of 2-Aminophenol (2AP) with the EA-A intermediate
This addition was being investigated, and at the time it wasn’t evident if the amine was
attacking the E(A-A) or if it was an electrophilic aromatic substitution reaction. To
determine which case it was, the [15N]-2AP isotope was synthesized in two steps
according to Scheme 3.7.
Scheme 3.7 Synthesis of [15N]-2AP a) HN15O3(10M), tetra-n-butylammonium bromide (TBAB), ethylene dichloride b)10% Pd/C, NaBH4
The nitration of phenol was carried out using [15N]-Nitric Acid and the phase transfer
catalyst TBAB.15 This prohibits the para nitration and limits it to only a mono ortho-
nitrophenol product. The second step is just the reduction of the nitro group to the amine
N
COO
N
HO
H2O3PO
NH2
OH
H
N
COO
N
HO
H2O3PO
H
NH
OH
E(A-A) E(Q)
OH OH15NO2
OH15NH2
a) b)
24 25

39
using Pd/C and sodium borohydride.16 Hydrogen gas is generated, in situ, by adding
concentrated HCl. The overall yield was low, but [15N]-nitric acid is relatively cheap.
3.6 Synthetic Approach to [17O]-PLP
Unfortunately, selectively labeling each individual atom within the PLP molecule
couldn’t be accomplished cheaply, through the modified synthesis. The phenol oxygen
protonation state is still something that needs to be investigated fully. Using the modified
synthesis would mean inserting the 17O label in the second step, making it extremely
costly and an inefficient use of isotopic starting material. Introducing the isotope in the
beginning would cost roughly $80,000 per gram of [17O]-PLP. The goal was to redesign
the synthesis and allow for the 17O label to be inserted towards the end. Scheme 3.8
illustrates a different PLP synthesis, one of the very first, completed by Reuben G. Jones
from Eli Lilly.17
Scheme 3.8 Proposed synthesis of [17O]-PLP a) 3.5 M HCl b) triethylorthoformate/Ac2O then NaHCO3 c) Iminoacetylacetone d) H2SO4 then NaN3 e) H2SO4/EtOH f) LAH g)
NaNO2, H2O17 h) MnO2 then p-phenetidine i) H3PO4/P2O5
EtO2C
OCO2Et
Na
EtO2C
OCO2Et EtO2C
OCO2Et
OH
EtO2C
OCO2Et
NH O
N
H2NCO2H
CO2H
N
H2NCO2Et
CO2Et
N
H17O
HO
OH
N
H17O
O
OPO3H2
a b cd
e f,g h,i
27
29[17O]-Pyridoxine [17O]-PLP
30
26 28

40
In the first step of the synthesis in Scheme 3.8, commercially available enolate of diethyl
oxaloacetate is acidified to transform the enolate anion into the keto form 26.18 Next,
refluxing 26 in triethylorthoformate and acetic anhydride gives the ethoxy analog of 27,
which can then be deprotected to the alcohol with solid sodium bicarbonate. The next
step is just a nucleophillic addition of iminoacetylacetone to 27 at room temperature in
diethyl ether.17 Product 28 is then dissolved in concentrated sulfuric acid and, after an
hour, powdered sodium azide is added slowly.18 Compound 28 initially cyclizes to
diethyl-2-methyl-3-acetyl-4,5-pyridinedicarboxylate. Then upon the addition of sodium
azide a Schmidt reaction occurs and compound 29 is isolated Figure 3.6.

41
Figure 3.6 Proposed mechanism for the transformation of 26 to 27
The reduction of 29 with sodium borohydride and AlCl3 in diglyme has been
unsuccessful so far.18 This particular reduction was chosen to eliminate the formation of
lactones. A possible solution to this might be the esterification of 29 to the ethyl
EtO2CCO2Et
O
NH O
N
O HO CO2EtCO2Et
N
CO2EtCO2Et
O
N3
H
N
N OH CO2EtCO2Et
NN
N
N CO2EtCO2Et
NN
H
H2ON
NCO2Et
CO2Et
H
-N2
OHH
Proton transfer
N
HN
CO2EtCO2Et
-H2O
N
NCO2Et
CO2EtOHH
O
Hydrolysis
N
H2NCO2H
CO2H

42
carboxylate, 30, then reduction using lithium aluminum hydride.19 Once a viable
reduction has been discovered and 3-amino pyridoxine has been isolated, the 17O label
can be inserted using sodium nitrite dissolved in [17O]-H2O. After the insertion of the
isotope, the last two steps are the same as the previous synthesis.

43
References:
1. Chan-Huot, M., Niether, C., Sharif, S., Tolstoy, P.M., Toney, M.D., Limbach, H.H., J. Molec. Struc. 2010, 976, 282-289. 2. Baldwin, J.E., Thomas, R.C., Kruse, L.I., Silberman, L. J. Org. Chem. 1977, 42, 3846-3852. 3. Baldwin, J.E. J. Chem. Soc. Chem. Commun., 1976, 18, 734-736. 4. Baldwin, J.E., Cutting, J., Dupont, W., Kruse, L., Silberman, L., Thomas, R.C. J. Chem. Soc., Chem. Commun., 1976, 18, 736-738. 5. Lee, S.J., Kim, E., Seo, M.L., Do, Y., Lee, Y.A., Lee, S.S., Jung, J.H., Kogiso, M., Shimizu, T. Tetrahedron, 2008, 64, 1301-1308. 6. Dean, A., Ferlin, M.G., Brun, P., Castagliuolo, I., Badocco, D., Pastore, P., Venzo, A., Bombi, G. G., Di Marco, V.B. Dalton Trans., 2008, 7,1689-1697. 7. Firestone, R.A., Harris, E.E., Reuter,W., Tetrahedron, 1967, 23, 943-955. 8. Florentiev, V.L., Ivanov, V.I., Kapreisky, M.Ya., Methods in Enzymology, 1970, 18, 567-598. 9. G. Cahiez, M. Alami, R. J. K. Taylor, M. Reid, J. S. Foot, "Manganese Dioxide", in Paquette, Leo A., Encyclopedia of reagents for organic synthesis, New York: J. Wiley & Sons, 2004. 10. Iwanami, M., Numata, T., Murakami, M., Bull. Chem. Soc. Japan., 1968, 41, 161-165. 11. Armarego, W.L.F., Perrin, D.D., Purification of Laboratory Chemicals. Oxford: Butterworth/Heinemann, 1996. Paperback. 12. Harris, E.E., Zabriskie, J.L., Chamberlin, E.M., Crane, J.P., Peterson, E.R., Reuter, W., JOCS. 1969, 34, 1993-1996. 13. Zou, Y., Shi, X., Zhang, G., Li, Z., Jin, C., Su, W., Org. Process Res. Dev. 2013, 14, 1498-1502. 14. Murakami, M., Iwanami, M,. Bull. Chem. Soc. Jap. 1968, 41, 726-727.

44
15. Joshi, A.V., Baidoosi, M., Mukhopadhyay, S., Sasson, Y., Org. Proc. Research and Develop., 2003, 7, 95-97. 16. Smith, C.J., Ali, A., Chen, L., Hammond, M.J., Anderson, M.S., Chen, Y., Eveland, S.S., Guo, Q., Hyland, S.A., Milot, D.P., Sparrow, C.P., Wright, S.D., Sinclair, P.J. Bioorg. Med. Chem. Lett. 2010, 20, 346-349. 17. Jones, R.G., J. Am. Chem. Soc., 1951, 73, 5244-5247. 18. Jones, R. G. 2-Methyl-3-Acetyl-4,5-Pyridine Dicarboxylic Acid, Lower Alkyl Esters Thereof and Intermediates. USPTO, assignee. Patent 2,724,714. 22 Nov. 1955. Print. 19. Jones, R.G., 2-Methyl-3-Amino-4,5-Di-Hydroxy Methylpyridine And Its Salts and The Preparation Thereof. USPTO, assignee. Patent 2,650,232. 25 Aug. 1953. Print.

45
Chapter 4
Synthetic Background/ Synthetic Attempts Towards Cannabinoid Analogs

46
4.1 Introduction
Cannabinoid analogs have evolved from a recreational to a promising therapeutic agent.
The first isolated cannabinoid analog, Δ1-tetrahydrocannabinol (Δ1-THC), has been used
to treat a wide range of disease (cancer, MS, etc.) along with easing nausea and
stimulating appetite during chemotherapy.1-12 Its efficacy in a wide range of symptom
and disease controls has sparked many synthetic strategies to solve this tricyclic
compound. The following sections will give a background of the previous syntheses of
some cannabinoid analogs and an attempt by our lab to devise a new synthetic route.
4.2 Cannabinoid Overview
Cannabinoids are usually segregated into three distinct classes, phyto-, synthetic, and
endogenous cannabinoids. The phytocannabinoids encompass the over 60 different
compounds that were extracted from the plant Cannabis satvia. Synthetic cannabinoids
are synthetic variants modeled after the phytocannabinoids, and the endogenous
cannabinoids represent those naturally produced within an organism (Table 4.1).

47
Class Name Structure
Phytocannabinoid Δ1-Tetrahydrocannabinol
Cannabidiol
Cannabivarin
Cannabichromene
Synthetic HU-210
Nabilone
CP-55,940
O
OH
OH
HO
O
OH
OH
O
O
OH
OH
O
OH
O
OH
OH
OH
Table 4.1 Chemical structures of phyto-, synthetic and endo- cannabinoids

48
Endocannabinoid Anandamide
2-arachdionoyl glycerol
2-arachidonoyl glycerol
ether
Table 4.1 Chemical structures of phyto-, synthetic and endo- cannabinoids continued.
Most of these compounds act as agonists for either the Cannabinoid receptor 1 (CB1) or
Cannabinoid receptor 2 (CB2) or both. CB1 and CB2 are both G protein-coupled
receptors, with the former being found predominately in the central nervous system,
making it a prime target for CNS disorders.
Δ1-Tetrahydrocannabinol (Δ1-THC) was first isolated and fully characterized in 1964 by
Mechoulam13 and is also responsible for the psychoactive affects during recreational use.
It has been shown to have several therapeutic affects as well. Figure 4.1 illustrates the
numbering scheme of Δ1-THC and the stereochemistry of the naturally occurring
psychoactive and therapeutic analog.
NH
OH
O
O
OOH
OH
OOH
OH

49
Figure 4.1 Left structure shows the monoterpenoid numbering the right structure shows
the natural occurring (-)-Δ1-THC. Stereochemistry at carbons 3 and 4 are R, R.
4.3 Previous Syntheses of Δ1-Tetrahydrocannabinol (Δ1-THC)
The very first synthesis of Δ1-THC was accomplished by Gaoni and Mechoulam in
1965.14 In this synthesis, the lithium derivative of the methoxy protected olivetol, 31,
condenses with citral gave the methoxy protected Cannabidiol (CBD) 32. Heating 32
with methylmagnesium idodie deprotects and converts it to a racemic mixture of CBD.
Upon heating with HCl the CBD is converted to Δ1-THC with an overall yield of 2 %.
(Scheme 4.1)
O
OH1
2
3
45
6
7
O
OHH
H

50
Scheme 4.1 First synthesis of Δ1-THC
Roughly a year later, Taylor and coworkers were able to synthesize a racemic mixture of
Δ6-THC in one step using 10% BF3 as a catalyst in roughly 10-20 % yield.15
Unfortunately this reaction also yielded the cis-Δ1-THC along with the isocannabinoid
33. Mechoulam altered the reaction by using 1 % BF3 and was able to get the trans-Δ1-
THC in roughly 20 % yield along with the cis- Δ1-THC isomer. (Scheme 4.2)
OMe
MeO C5H11
LiOOMe
MeO C5H11
OH
HO C5H11 O
OH
C5H11
H
H
31 32
CBD

51
Scheme 4.2 Mechoulam synthesis
Evans, in 1997, tried making Δ1-THC through the first stereospecific route.16
Unfortunately the inactive (S,S) Δ1-THC was isolated through numerous steps in 21 %
yield. The chiral catalyst, bis(oxazoline) copper (II), was used to catalyze the Diels-Alder
reaction between acrylamide, 34, and 1-acetoxy-3-methylbutadiene 35. The resulting
cycloadduct is then cleaved with LiOBn to form 36, which in turn is reacted with
methylmagnesium bromide to form the Diol 37. The Diol is reacted with olivetol and
cyclized to form (S,S) Δ1-THC. (Scheme 4.3)
(±)-cis-Δ1-THC
(±)-trans-Δ6-THC
(±)-trans-Δ1-THC + cis-Δ1-THC
OH
HO C5H11
OO
OH
C5H11
H
HOH
O C5H11
32
O
OH
C5H11
H
H
10 % BF3
1 % BF3
33

52
Scheme 4.3 Evans stereospecific synthesis of (S,S) Δ1-THC a) 1. Cationic
bis(oxazoline)copper (II) catalyst 2. LiOBn b) MeMgBr c) Olivetol d) ZnBr2, MgSO4
Trost’s synthesis in 2007 was the first stereospecific synthesis, which led to the most
prevalent isomer in Cannabis sativa, (R,R) Δ1-THC, through 12 steps with an overall
yield of 30 %.17 This reaction uses a Molybdenum catalyzed asymmetric allylic alkylation
to set the correct stereochemistry. The first part of the synthesis converts olivetol into the
carbonate 39 in 4 steps. Next, the Mo-catalyzed allylic alkylation of 39 with sodium
dimethyl malonate yields product 40. The product is then decarboxylated, and then
alkylated followed by a ring closing metathesis to form 43. Lastly this product was
isomerized, methylated with MeLi, mono-deprotected using NaSEt, cyclized with ZnBr2,
and lastly fully deprotected, once again with NaSEt, yielding the natural occurring isomer
of Δ1-THC. (Scheme 4.4)
OAc N O
OO
3435
O
HOAc
BnO36 OH
HOH
37
H
HO
OH
C5H11
HO
H
38
O
OH
C5H11
H
H
1-THC(S,S)
ab
c d

53
Scheme 4.4 Trost synthesis a) 5 mol % [Mo(CO)3C7H8], sodium dimethyl malonate, 7.5% (R,R)-DACH-pyridyl TROST ligand b) 1. NaOH 2. HCl c) LDA, 4-iodo-2-
methylbut-1-ene d) 1. (MeO)2SO2, K2CO3 2. Grubbs II catalyst e) MeLi f) NaSEt g) 1. ZnBr2, MgSO4 2. NaSEt
4.4 Attempts towards synthesis of THC
The goal was to design a new synthetic strategy that was shorter and used cheaper
starting material, since olivetol is quite expensive and difficult to synthesize.18 The
following synthetic attempts uses orcinol as the starting material, since we hypothesized
that the shorter carbon chain should reduce the psychotropic side affects but retain the
therapeutic affects and also bind to the CB1 receptor as well as the natural occurring
isomer. The second goal was to design a synthesis that mimicked the biosynthetic route.
Figure 4.1 shows the key intermediate that can be transformed into THC or CBD.
OMe
C5H11 OMe
4 stepsOMe
C5H11 OMe
OCO2CH3
OMe
C5H11 OMe
CO2MeMeO2C
OMe
C5H11 OMe
MeO2COMe
C5H11 OMe
MeO2COMe
C5H11 OMe
MeO2C
O C5H11
OMe
e,f,g
3940
43
44
4142

54
Figure 4.2 Biosynthetic pathways to THC, CBD and CBC
Here, in Figure 4.1, one intermediate is easily converted into three distinct cannabinoids
using three distinct enzymes. The synthetic plan was to functionalize orcinol at the
position ortho to both alcohol groups, with a carbonyl which would allow for either a
Wittig reaction to make a vinylic group, which could potentially react with a terpenoid
through an olefin metathesis or an aldol condensation to create a dienophile to help create
the fused bicylic for THC or the cyclohexane ring for CBD.
CnH2n+1
OH
HOCOOH
O
OH
RCnH2n+1
O
OH
RCnH2n+1
HO
OH
CnH2n+1R
n = 3, 5CBGA
R = COOH (CBCA)R = H (CBC)- CO2
R = COOH (THCA)R = H (THC)- CO2
R = COOH (CBDA)R = H (CBD)- CO2
THCA synthase
CBDAsynthase
CBCA synthase

55
The first synthetic attempt was to utilize the Grubbs catalysis because of its ubiquity and
stability under various conditions. Scheme 4.5 summarizes the synthetic approach.
Scheme 4.5 Olefin Metathesis synthetic route a) K2CO3, MeI b) n-BuLi, TMEDA, DMF c) CH2=PPh3 d) Grubbs II Generation
The first three steps were the same approach that Trost’s synthesis had employed, which
begins by the dimethyl protection of the hydroxyl groups on orcinol.17 Next, 45 was
formylated α to the two hydroxyl groups. Lastly, 46 was reacted with
methyltriphenylphosphonium Bromide, whereby a Wittig reaction takes place to add the
vinylic group and obtain 47. Unfortunately the olefin metathesis between 47 and linalool
would not take place, since neither was able to couple with the Ru center regardless of
which Grubbs catalyst was used.
Next we turned our attention to a slight variation in the synthesis with the key step a
Diels-Alder reaction. (Scheme 4.6)
OH
OH
OMe
OMe
OMe
OMe
O
OMe
OMe
OH
OMe
OMe
OHOH
O
OH
THC CBD
45 46 47
48
a b c
d
OH

56
Scheme 4.6 Diels-Alder route a) K2CO3, MeI b) n-BuLi, TMEDA, DMF c) NaOH, acetone d) Lewis acid catalyst
In this variation of the synthesis steps a and b are identical to the aforementioned route.
Once 46 was obtained an aldol condensation with acetone occurs and 49 is obtained in
high yield. The Diels-Alder step was extensively studied using various dienes and Lewis
acid catalysts. Unfortunately neither of the Diels-Alder reactions gave any appreciable
amount of 50. According to Piermatti et. al.18 this reaction occurs only under extremely
high pressures, (18 kBar), which is not practical or safe, so this synthetic strategy was
deemed useless.
Scheme 4.7 depicts the most up to date attempt (and most promising) synthetic route
towards THC/CBD.
OH
CBD
OH
OH
OMe
OMe
OMe
OMe
O
OMe
OMe
O OTMS
OMe
OTMS
OH
OTHC
OMeO
OH
a b c
d
45 46 49
50

57
Scheme 4.7 Biomimetic approach a) K2CO3, MeI b) n-BuLi, TMEDA, DMF c) NaI, AlCl3 d) KOH, 6-methyl-5-hepten-2-one e) NaOH, 6-methyl-5-hepten-2-one
Once again the first few reactions are very similar as the previous syntheses. In order to
isolate CBD the Aldol condensation of 46 with 6-methyl-5-hepten-2-one gave 52 in
relatively good yields isolated as a solid. The proposed conversion to CBD from 52 is
shown in Scheme 4.8. The monodeprotection of 46 to form 51 was accomplished with
NaI and AlCl3 and was converted to 53 through an Aldol condensation with 6-methyl-5-
hepten-2-one. The proposed conversion of 53 to THC is illustrated in Scheme 4.8 as well.
OH
OH
OH
OMe
OMe
OMe
OMe
O
OH
O
OMe
OMe
OOMe
OH
O
OMe
OH
O
OH
a b c
d e
45 4651
5253
THCCBD

58
A B
Scheme 4.8 Proposed synthetic pathways to CBD and THC a) 1-10% acid b) MeMgBr
c) K-t-pentoxide/NaSEt
In Scheme 4.8 reaction pathway A, 52 could potentially be cyclized using a catalytic
amount of a weak organic acid (for example trifluoroacetic acid). Next, a Grignard
reaction will form the tertiary alcohol. The last steps include a dehydration with a bulky
base and deprotection of the methoxy groups with sodium ethanethiolate, which would
hypothetically give CBD. For reaction pathway B, the tricyclic fusion will occur, since
the one methoxy group had been previously removed. With the cannabinoid skeleton
intact, once again a Grignard reaction with methylmagnesium bromide and
dehydration/deprotection with a bulky base and sodium ethanethiolate, respectively,
should produce Δ1-THC. Though this reaction is not stereospecific (and might give a
mixture of various isomers) it is a good starting point and uses relatively cheap materials.
Preliminary results for the cyclization of 53 indicate a disappearance of the benzylic
alkene in the 1H-NMR spectrum, a disappearance of the –OH functional group in the IR
spectrum, and a m/z peak value which corresponds to the project mass of the product.
OH
OH
O
OMe
OMe
O
OMe
OH
O
OH52
53THC
CBD
OMe
O
OMe
OMe
OMe
OH
OMe
O
O
OMe
O
OH

59
4.5 Conclusion
Various cannabinoids have been previously reported to have potential therapeutic affects
ranging from anti-cancer fighting capabilities to combating various CNS disorders.
Although several syntheses do exist there is still more room for improvement before a
reliable, cheap synthetic drug could be put to market. We had tried several different
routes in order to synthesize CBD and Δ1-THC and found that a biomimetic approach
seemed the most feasible route.

60
References:
1. Pacher, P., Batkai, S., Kunos, G., Pharmcol. Rev., 2006, 58, 389-462. 2. Guindon, J., De Léan, A., Beaulieu, P., Pain, 2006, 121, 85-93. 3. Amar, M., Journal of Ethnopharmacology, 2006, 105, 1-25. 4. Zajicek, J., Fox, P., Sanders, H., Wright, D., Vickery, J., Thompson, A., Lancet, 2003, 362, 1517-1526 5. Compston, A., Coles, A., Lancet, 2008, 372, 1502–17 6. Guzman, M., Sanchez, C., Galve-Roperh, I., J Mol Med, 2001, 78, 613-625. 7. Sanchez, C., Galve-Roperh, I., Canova, C., Brachet, P., Guazman, M., FEBS Lett, 1998, 436, 6-10 8. Sarker, K. P., Obara, S., Nakata, M., Kitajima, I., Maruyama, I., FEBS Lett, 2000, 472, 39-44 9. Chan, G. C-K., Hinds, R. T., Impey, S., Storm, D.R., J. Neurosci, 1998, 18, 5322-5332. 10. Chan, P.C., Sills, R.C., Braun, A.G., Hasaman, J.K., Bucher, J.R., Fundam Appl Toxicol, 1996, 30, 109-117. 11. Petrocellis, L.D., Melck, D., Palmisano, A., Bisogno, T., Laezza, C., Bifulco, M., Di Marzo, V., Proc Natl Acad Sci USA, 1998, 95, 8375-8380. 12. Melck, D., De Petrocellis, L., Orlando, P., Bisogno, T., Laezza, C., Bifulco, M., di Marzo, V., Endocrinology, 2000, 141, 118-126. 13. Mechoulam, R.; Gaoni, Y. J Amer Chem Soc, 1964, 86, 1646. 14. Mechoulam, R.; Gaoni, Y., J Amer Chem Soc, 1965, 87, 3273-3275. 15. Taylor, E.C., Lenard, K., Shvo, Y., J Amer Chem Soc, 1966, 88, 367. 16. Evans, D. A.; Shaughnessy, E. A.; Barnes, D. M. Tetrahedron Letters 1997, 38, 3193. 17. Trost, B. M.; Dogra, K. Organic Letters 2007, 9, 861. 18. Focella, A., Teitel, S., Brossi, A., J. Org. Chem. 1977, 42, 3456-3457.

61
Appendix A Experimental Procedure and Spectroscopic Data
Chemicals and solvents were purchased from Sigma Aldrich and used without further
purification. Unless otherwise noted, reactions were performed using standard synthetic
organic techniques under an atmosphere of nitrogen gas. Nuclear magnetic resonance
(NMR) spectra were acquired using a Varian Inova 300 MHz spectrometer. Chemical
shifts (δ) are reported in parts per million (ppm) and are calibrated to the residual solvent
peak. Coupling constants (J) are reported in Hz. Purifications by column chromatography
were performed using silica gel (40 - 63 µm, 230 - 400 mesh) and appropriate eluent.
Analytical data acquired for all previously reported compounds were fully consistent with
existing spectral data.
Compound 10: A round bottom flask charged with L-15N-Alanine (0.231g, 2.56 mmol) ,
purchased from Sigma-Adlrich, dissolved in 5.0 mL absolute ethanol (1M) is chilled in
an ice-salt bath. Once cooled, SOCl2 (0.65 mL, 8.80 mmol) is added slowly. After the
addition is complete, the mixture is heated to 40 °C for 5 hours. After heating, the solvent
is evaporated and crystallized by dissolving in small amount of absolute ethanol and then
diethyl ether is slowly added on top of the ethanol layer, without disrupting the ethanol
layer. The mixture was then allowed to stand at room temperature until crystals started to
form and then placed into the freezer overnight to complete crystallization. The yield was
0.374 g (95 % yield). 1H NMR (D2O, 400 MHz) δ 1.31 (3H, t, J = 7.2 Hz), δ 1.57 (3H,
dd, J = 2.8, 4.4 Hz), δ 4.18 (1H, qt, J = 7.2 Hz), δ 4.31 (2H, qt, J = 7.2 Hz). 13C-NMR

62
(D2O, 400 MHz) δ 8.40, 10.34, 44.06, 58.75, 166.11. HRMS (m/z): [M+Na] calc for
C5H11(15N)NaO2 141.0657; found 141.0646.
Compound 11: 0.36 g of 10 (2.32 mmol) is dissolved into triethylorthoformate (1.2 mL,
6.96 mmol) and heated at 80 °C for 1.5 hours. The solvent is distilled off in vacuo and 11
was obtained in quantitative yields. 1H NMR (CDCl3, 300 MHz): δ 1.30 (3H t, J = 7.2
Hz), δ 1.45 (3H, dd, J = 2.4, 4.5 Hz), δ 4.23 (2H, qt, J = 7.2 Hz), δ 4.67 (1H, quint, J =
7.2 Hz), δ 6.26 (15NH, dt, J = 92Hz, 5.7 Hz), δ 8.19 (1H, d, J = 16.2 Hz). 13C-NMR
(CDCl3, 400 MHz) δ 14.3, 18.76, 47.07 (d, J = 46 Hz), 61.91, 160.50, 172.80. HRMS
(m/z): [M+H]+ Calc for C6H1215NO3 147.0782; found 147.0800
Compound 12: In a 100 mL round bottom flask 11 (0.53 g, 3.63 mmol) is dissolved in
30 mL dichloromethane. While stirring, 2.08 g of P2O5 is added and the reaction is
refluxed for 24 hrs. The reaction mixture is cooled to room temperature and the reaction
quenched with 20 % aqueous NaOH. The aqueous layer is extracted three times with
dichlormethane, and then the organic phase is washed with water. The organic phase is
then dried with MgSO4 and the solvent removed with a rotary evaporator. The yield of
the reaction was 50 %. 1H NMR (CDCl3, 400 MHz) δ 1.35 (t, 3H, J = 7.2 Hz), δ 2.05 (d,
3H, J = 2 Hz), δ 4.15 (qt, 2H, J = 7.2 Hz), δ 7.37 (d, 1H, J = 12.8 Hz). 13CNMR (CDCl3,
400 MHz) δ 10.2 (d, J = 23.2 Hz), 15.2, 70.3, 104.99, 112.46 (dd, J = 42.8 Hz), 142.31
(d, J = 39.21 Hz). HRMS (m/z): [M+H]+ calc C6H1015NO2 129.0676; found 129.0676.

63
Compound 13: Under N2, 12 (0.681 g, 5.3 mmol) is combined with diethyl maleate (1.7
mL, 10.6 mmol) and 2.5 g SiO2 (1:1 by total mass). The reaction is heated at 120 °C for 6
hrs. Upon the completion of heating, the reaction is cooled to room temp and placed on a
filter and the crude is eluted off with diethyl ether, then dichloromethane and benzene.
The filtrate is evaporated then a small amount of 1.25 M Ethanolic-HCl is added to
dissolve the crude product. To the ethanol solution, diethyl ether is added, gently, on top
in a way to not disrupt the ethanol layer then the bilayer is placed in the freezer overnight
to crystallize out the product. Roughly 0.65 g of 13 was isolated (50 % yield). 1H NMR
(CDCl3, 300 MHz) δ 1.42 (3H, t, J = 7.2 Hz), δ 1.43 (3H, t, J = 7.2 Hz), δ 2.97 (3H, d, J =
3 Hz), δ 4.44 (2H, qt, J = 7.2 Hz), δ 4.54 (2H, qt, J = 7.2 Hz), δ 8.36 (1H, d, J = 3.3 Hz).
HRMS (m/z): [M+H]+ calc for C12H15 (15 N)O5 255.0993; found 255.1026.
Compound 14: Under a flow of N2, lithium aluminum hydride (0.44 g, 13.6 mmol) is
added, very carefully, to 90 mL of anhydrous diethyl ether while stirring at room
temperature. Next, crystalline 13 (0.43 g, 1.7 mmol) is added into the ethereal solution.
The reaction is refluxed overnight in a N2 atmosphere. Once cooled to room temperature,
35 mL of DI H2O is gently added and the mixture is filtered and washed with boiling
water (2x50 mL). The filtrate is then saturated with CO2 for an hour, and then evaporated
to dryness in vacuo. The resulting solid is then extracted with boiling methanol (2 x 40
mL) and the solvent evaporated once again to dryness. A small amount of 3 M
methanolic-HCl is added to dissolve resulting oil and then diethyl ether is gently poured
on top of the methanol layer in a way not to disrupt the layer and lastly the bilayer is

64
placed inside the freezer overnight. Pure 14 (0.26 g) was isolated (90 % yield). 1H NMR
(D2O, 300 MHz) δ 2.50 (3H, d, J = 3.3 Hz), δ 4.68 (2H, s), δ 4.86 (2H, s), δ 7.98 (1H, d, J
= 3.3 Hz). HRMS (m/z): M*+[-H] calc for C8H10 (15N )O3 169.0636; found 169.1360.
Compound 15: Crystalline 14 (0.18 g, 1.05 mmol) was dissolved in 0.3 M H2SO4 (3.5
mL). To the acidic solution, activated MnO2 (95 mg, 1.1 mmol) was added and the
reaction is stirred at room temperature for 3.5 hrs. The reaction is filtered and the filter
cake was rinsed with 1 mL of DI water. At this point the 0.5 M p-phenetidine
hydrochloride solution needs to be made. In a small beaker with 15 mL of DI water, 1.3
mL of p-Phenetidine is added followed by the immediate addition of 1 mL of
concentrated HCl then more DI water is added to make the total volume 20 mL. This
solution is stirred until no solid is present. Once made, 2.8 mL of the p-Phenetidine
hydrochloride solution is added to the filtrate followed immediately by the addition of 4.2
mL of a 2M NaOAc solution. This mixture is placed in the fridge and 15 separates out as
a yellow solid. The solid is filtered off and washed with cold DI water. Roughly 0.29 g of
15 was isolated (95 % yield). 1H NMR (CDCl3, 300 MHz) δ 1.44 (3H, t, J = 6.9 Hz), δ
2.56 (3H, d, J = 2.4 Hz), δ 4.08 (2H, qt, J = 7.2 Hz), δ 4.87 (2H, s), δ 6.96 (2H, d, J = 8.7
Hz), δ 7.34 (2H, d, J = 8.7 Hz), δ 8.06 (1H, d, J = 10.5 Hz), δ 9.15 (1H, s). HRMS (m/z):
[M+H]+ calc for C16H19(15N)NO3 288.1361; found 288.1371.
Compound 16: In a 25 mL round bottom flask, P2O5 (0.75 g, 5.4 mmol) and 85 % o-
H3PO4 (0.41 mL, 7.0 mmol) are stirred until homogenous. Once the reaction is cooled to

65
room temperature, solid 15 (100 mg, 0.35 mmol) is added and stirred at room
temperature until homogeneous. The reaction is then heated at 40 °C for 4 hours, cooled
to room temperature, and then 0.1 M HCl (0.22 mL) is added to the mixture and heated at
60 °C for 15 mins. Before adding the crude product to the ion-exchange resin, it needs to
be prepared as follows. After packing a column with the resin, DI water, ethanol then DI
water are passed through individually and the filtrate discarded. Then the following are
added to the resin in order; 1M HCl, DI water, 1M NaOH, DI water, 1M HCl, and then
once more DI water until eluent is no longer acidic. Once the column is prepared the
crude reaction mixture is added to the column and eluted with a large amount (1 L) of DI
water. The combined filtrate is evaporated on the rotary evaporator to about 5-10 mL. It
is imperative that the water bath temperature not exceed 40 °C. Roughly 700 mg of 16
was isolated (80 % yield). 1H NMR (DMSO-d6, 400 MHz) δ 2.44 (3H, d, J = 2.7 Hz), δ
5.20 (2H, d, J = 7.2 Hz), δ 8.13 (1H, d, J = 10.5 Hz), δ 10.4 (1H, s). HRMS (m/z):
[M+Na]+ calc for C8H10(15N)NaO6P 271.0114; found 271.0103.
Compound 17: See experimental procedure for 10. 1H NMR (DMSO-d6, 300 MHz) δ
1.23 (3H, t, J = 7.2 Hz), δ 1.4 (3H, dt, J = 130, 2.7 Hz) δ 4.02 (1H, m, J = 146 Hz), δ 4.2
(dq, 2H, J = 3.6 Hz), δ 8.50 (15NH, s, br). 13C-NMR δ 13.3, 15.22 (d, J = 136 Hz), 48.96
(ddd, J = 26, 84, 110 Hz), 170.97 (d, J = 246 Hz). HRMS (m/z): [M+Na-H] calc for
13C315NC2H11O2 143.0686; found 143.0677.

66
Compound 18: See experimental procedure for 11. 1H NMR (CDCl3, 300 MHz) δ 1.23
(3H, t, J = 7.2 Hz), δ 1.38 (3H, m, J = 130 Hz), δ 4.15 (2H, dq, J = 3 Hz), δ 4.59 (1H, dsp,
J = 143, 6 Hz), δ 6.25 (15NH, ddd, J = 92, 7.5 Hz), δ 8.12 (1H, ddd, J = 16, 5 Hz). 13C-
NMR (CDCl3, 400 MHz) δ 14.3, 18.78 (d, J = 138 Hz), 47.07 (ddd, J = 46.8, 59, 92 Hz),
61.92, 160.56 (d, J = 53 Hz), 172.81 (d, J = 243 Hz). HRMS (m/z): [M+H]+ Calc for
13C315NC3H11O3 150.0883; found 150.0882.
Compound 19: See experimental procedure for 12. 1H NMR (CDCl3, 400 MHz) δ 1.29
(3H, t, J = 7.2 Hz), δ 1.98 (3H, dsp, J = 2.4, 128 Hz), δ 4.09 (2H, dqt, J = 3.2 Hz), δ 7.31
(1H, m, J = 1.2, 2.8, 4.5 Hz). 13C-NMR (CDCl3, 400 MHz) δ 10.17 (dt, J = 26, 224 Hz),
15.17, 70.32, 112.46 (dd, J = 165, 223 Hz), 142.39, 154.34 (dd, J = 28, 467 Hz). HRMS
(m/z): [M+H]+ calc for 13C315NC3H9O2 132.0777; found 132.0780.
Compound 21: In a steel autoclave, the oxazole 19 (g, mmol) is dissolved in 2,5-
dihydrofuran. Recrystallized trichloroacetic acid (g, mmol) and a small amount of
hydroquinone are added. The reaction is heated in an oven at 190 °C for 5 hours then
allowed to cool to room temperature over night. Excess 2,5-dihydrofuran is evaporated
and the crude is purified through column chromatography to elute with 10:1
DCM/MeOH. The intermediate is then refluxed in 48 % hydrobromic acid (mL, mmol)
for 1 hr then cooled to room temperature. Once cooled, the crude mixture dried in vacuo
and then refluxed in water. Freshly prepared AgCl is added to the refluxing aqueous
solution and the refluxing continued for an additional hour. The reaction is cooled to

67
room temperature then filtered. The filtrate is evaporated to dryness and used without any
further purification. Approximately g of 21 was isolated (%yield). 1H NMR (DMSO-d6,
300 MHz) 13C-NMR (DMSO-d6, 400 MHz) δ 16.32 (J = 136 Hz), 53.46, 56.10, 141.96,
142.10, 142.23, 148.70 (J = 288 Hz), 152.50 (J = 284 Hz). HRMS (m/z): [M+H]+ calc for
13C315NC5H11O3 174.0883; found 174.0908.
Compound 22: See experimental procedure for 15. 1H NMR (CDCl3, 300 MHz) δ 1.38
(3H, t, J = 6.9 Hz), δ 2.51 (3H, d, J= 132 Hz) δ 4.01 (2H, qt, J = 7.2, 5.7 Hz), δ 4.81 (2H,
s), δ 6.89 (2H, d, J = 7.8 Hz), δ 7.28 (2H, d, J = 7.2 Hz), δ 7.88 (1H, m), δ 9.08 (1H, s).
HRMS (m/z): [M+H]+ calc for 13C315NC13H19NO3 291.1461; found 291.1537
Compound 23: See experimental procedure for 16. 1H NMR (D2O, 600 MHz) δ 2.61
(3H, dd, J = 3, 132 Hz), δ 5.10 (2H, d, J = 6 Hz), δ 8.19 (1H, d, J = 6 Hz), δ 10.46 (1H, s).
13C NMR (DMSO-d6, 400 MHz) δ HRMS (m/z): [M+H]+ calc for 13C315NC5H10O6P
252.0389; found 252.0439.
Compound 24: Phenol (0.75 g, 7.8 mmol) was dissolved in ethylene dichloride (16 mL)
and stirred at room temperature. To the mixture tetrabutylammonium bromide (0.25 g,
0.78 mmol), 15N-nitric acid (70 wt%, 1 g), and sufficient water to make up the nitric acid
concentration to 6 wt %. The reaction was stirred overnight at room temperature. Upon
completion of the reaction, the aqueous layer was separated from the organic layer. The
aqueous layer was extracted with pentane, combined with the other organic layer, and the

68
combined organic layers were washed once with DI water. A silica plug eluting with 1:3
EtOAc/Hex gave 0.262 g of 24 (24 % yield). 1H NMR (CDCl3, 300 MHz): δ 7.01 (1H, dt,
J = 1.5, 5.7 Hz), δ 7.18 (1H, dd, J = 1.5, 7.2 Hz), δ 7.60 (1H, dt, J = 1.5, 5.7 Hz), δ 8.13
(1H, dd, J = 1.5, 6.9 Hz), δ 10.6 (1H, s).
Compound 25: In a flame dried 3 neck flask, affixed with reflux condenser, a positive
flow of N2, and a balloon 0.076 g of 10 % Pd/C is dissolved in 7.0 mL of DI water.
sodium borohydride (0.14 g, 3.8 mmol) is then added and the reaction stirred at room
temperature. 24 (0.27 g, 1.9 mmol) is dissolved in 2 M NaOH (10 mL) and added
dropwise. The reaction is stirred at room temperature until the yellow color disappears
and then the reaction mixture was filtered. The filtrate was acidified with 2 M HCl,
filtered once again, and then neutralized with 0.5 M NaOH. The product was extracted
with diethyl ether, and dried over anhydrous magnesium sulfate. Upon removal of the
solvent, 200 mg of 25 was isolated (96 % yield). 1H NMR (CDCl3, 300 MHz) δ 6.66-6.8
(4 H, m) HRMS (m/z): [M+] calc for C6H8NO 110.0498; found 110.0449.
Compound 26/27: To the commercially available sodium salt of oxaloacetate (126 g, 0.6
mol) was added 200 mL of 3.5 N HCl with stirring. The reaction mixture was then
extracted with ether and the organic phase dried over MgSO4. The solvent was
evaporated and the crude ethyl oxaloacetate was heated on a steam bath under vacuum
for 15 minutes. No further purification was necessary, and the crude product was carried
through to the next step. To 26 (126 g, 0.6 mol) was added triethyl orthoformate (140

69
mL, 0.85 mol) and acetic anhydride (130 mL, 1.37 mol) and heated at 140 °C. Heating
continued for 2 hrs, and then the volatiles were distilled off under reduced pressure. To
the resulting crude product was added 150 mL of water, and 50 g of solid NaHCO3 was
gently added with stirring in 1.0 g increments. The aqueous solution was washed twice
with 50 mL portions of ether and then the aqueous solution was gently acidified with an
excess of concentrated hydrochloric acid. The resulting aqueous solution is extracted
three times with ether and then dried over anhydrous MgSO4. The solvent was evaporated
and the crude prodcut was heated on a steam bath, under vacuum, for 15 mins.
Approxamately 100 g of 27 was recovered (70 % yield). 1H NMR (CDCl3, 300 MHz) δ
1.30 (3H, t, J = 7.2 Hz), δ 1.39 (3H, t, J = 7.2 Hz), 4.26 (2H, qt, J = 6.9 Hz), 4.39 (2H, qt,
J = 6.9 Hz), 8.7 (1H, s).
Compound 28: In a round bottom flask, 27 (15.2 g, 70 mmol) was dissolved in 110 mL
diethyl ether. To this solution, iminoacetylacetone (14 g, 88 mmol) was added and the
reaction stirred overnight at room temperature. Once the reaction has commenced the
resulting precipitate was filtered and extracted with warm ethyl acetate. The filtrate was
evaporated to dryness and a 1:1 mixture of ether and petroleum ether added to crystallize
the product. About 9.16 g of 28 was isolated as a yellow crystal (44 % yield). 1H NMR
(CDCl3, 300 MHz) δ 1.29 (3H, t, J = 7.2 Hz), δ 1.36 (3H, t, J = 7.2 Hz), δ 2.19 (3H, s), δ
2.23 (3H, s), δ 4.25 (2H, qt, J = 7.2 Hz), δ 4.34 (2H, qt, J = 7.2 Hz), δ 5.7 (1H, s), δ 8.2
(2H, d, J = 13.8 Hz).

70
Compound 29: In a 500 mL beaker, 28 g of concentrated H2SO4, and 8.0 g of 28 (27
mmol) were stirred at room temperature for 1 hr. After stirring, 1.85 g of powdered NaN3
(28 mmol) was added gently, under ice cooling. The ice bath was removed and the
reaction stirred at room temperature overnight. The solution was then poured over a large
amount of crushed ice and the aqueous solution heated at 70 °C for 10 hrs. Once cooled,
concentrated ammonium hydroxide was added to the reaction mixture until a pH of 2.0
(roughly 36 mL), and the reaction placed into the back of the hood to allow 29 to
crystallize out slowly. Roughly 2.9 g of 29 was isolated (50 % Yield). 1H NMR (DMSO-
d6, 300 MHz) δ 2.58 (3H, s), δ 8.9 (1H, s). HRMS (m/z): [M+H]+ calc for C8H9N2O4
197.0557; found 197.0557.
Compound 30: In a large round bottom flask, 29 (0.55 g, 2.6 mmol) was dissolved in 65
mL of absolute ethanol. To the ethanol solution, 3.3 mL of concentrated H2SO4 was
slowly added into the reaction flask. The reaction was refluxed for 24 hrs, cooled to room
temp., and the solvent removed by rotary evaporation. The crude product was dissolved
in ethyl acetate, washed with NH4OH(aq), and the organic layer dried over MgSO4. After
removal of the solvent, the crude is purified by column chromatography (1:1
EtOAc/DCM). The yield of the diester was 30 0.15 g (23 % yield). 1H NMR (CDCl3,
300 MHz) δ 1.37 (6H, t, 6.6 Hz), δ 2.62 (3H, s), δ 4.38 (4H, qn, J = 7.5 Hz), δ 8.22 (1H,
s).

71
Compound 45: A mixture of orcinol (4.55 g, 36.7 mmol), K2CO3 (25 g, 184 mmol), MeI
(11.4 mL, 184 mmol), and acetone (104 mL) was refluxed for 6 hrs. Once cooled, the
reaction was filtered through celite and the solvent removed. The crude product was
passed through a small silica pad using a mixture of 2 % ethyl acetate in petroleum ether.
Upon removal of the solvent, 4.7 g of 45 was obtained (84 % yield). 1H NMR (CDCl3,
300 MHz) δ 2.32 (3H, s), δ 3.79 (6H, s), δ 6.3 (1H, s), δ 6.35 (2H, s).
Compound 46: Under N2, a mixture of 45 (4.7 g, 31 mmol) in 60 mL of anhydrous
diethyl ether cooled to 0 °C and TMEDA (7.0 mL, 46 mmol), followed by n-butyllithium
(30 mL, 46 mmol) added. The solution was refluxed for 3 hrs, then cooled back down to
0 °C. Once cooled, DMF (7 mL, 92 mmol) was added slowly. The solution is allowed to
warm back up to room temperature slowly and stirred for 2 hrs. At this point the reaction
was quenched with 25 mL of DI water and extracted with ethyl acetate (3 x 125 mL). The
combined organic extracts were dried over anhydrous MgSO4 and the solvent removed in
vacuo. The crude was purified by flash chromatography (60 % diethyl ether in petroleum
ether). Approximately 2.8 g of 46 was isolated (51 % yield). 1H NMR (CDCl3, 300 MHz)
δ 2.38 (3H, s), δ 3.89 (6H, s), δ 6.39 (2H, s), δ 10.45 (1H, s).
Compound 47: Methyl triphenylphosphonium bromide (2.5 g, 7.1 mmol) was stirred in
20 mL of dry THF. To the solution, at room temperature, was added sodium
bis(tirmethylsilyl)amide (8 mL, 8 mmol, 1 M in THF). The solution was stirred for 3 hrs
at room temperature then cooled to -78 °C. To the mixture was added 46 (0.91 g, 5.1

72
mmol) dissolved in 20 mL of dry THF, and the reaction mixture allowed to warm to
room temperature overnight. The reaction was quenched with acetone and water,
extracted with diethyl ether, and the combined organic extracts dried over anhydrous
MgSO4. Silica gel flash chromatography eluting with 5% diethyl ether in petroleum ether
gave 0.52 g of the product (58% yield). 1H NMR (CDCl3, 300 MHz) δ 2.34 (3H, s), δ
3.83 (6H, s), δ 5.38 (1H, dd, J = 2.1, 7.0 Hz), δ 6.01 (1H, dd, J= 2.1, 11.4 Hz), δ 6.38
(2H, s), δ 6.93 (1H, dd, J = 4.2, 9.3 Hz). HRMS (m/z): [M+H]+ calc for C11H14O2
179.1067; found 179.1080.
Compound 49: A solution of 46 (0.56 g, 3.1 mmol) was dissolved in 5.0 mL of acetone
and cooled to 0 °C. To the solution 1 M aq NaOH (5 mL) was added dropwise and the
reaction stirred at room temperature for 3 hrs. At this point, the reaction was cooled once
again to 0 °C and treated with 1 M aq HCl (5 mL) dropwise. DI Water (17 mL) was
added and the mixture extracted with ethyl acetate (3 x 10 mL), and the combined
organic extracts were dried over anhydrous MgSO4. Flash chromatography eluting with
7:3 n-pentane/EtOAc afforded 0.41g of 49 (60 % yield). 1H NMR (CDCl3, 300 MHz) δ
2.37 (6H, s), δ 3.88 (6H, s), δ 6.39 (2H, s), δ 7.13 (1H, d, J = 16.5 Hz), δ 7.97 (1H, d, J =
16.5 Hz). HRMS (m/z): [M+H]+ calc for C13H17O3 221.1172; found 221.1205.
Compound 51: To a solution of 46 (5.4 g, 30 mmol) in 65 mL of acetonitrile and 31 mL
of dichloromethane was slowly added 10.0 g of AlCl3 and 11 g of NaI under ice cooling.
The reaction was stirred at room temperature for 1 hr then 20 mL water is added under
ice cooling. The mixture is extracted with dichloromethane, and the combined organic

73
extracts dried over anhydrous MgSO4. The crude prodcut was purified by flash
chromatography on silica gel eluting with 1:5 EtOAc/Cyclohexane. The reaction gave 4.0
g of 51 (81 % yield). 1H NMR (CDCl3, 300 MHz) δ 2.33 (3H, s), δ 3.88 (3H, s), δ 6.19
(1H, s), δ 6.36 (1H, s), δ 10.26 (1H, s), δ 11.97 (1H, s). HRMS (m/z): [M+H]+ calc for
C9H11O3 167.0703; found 167.0710.
Compound 52: Sodium hydroxide (0.77 g, 19 mmol) was crushed to a powder and
dissolved in 7 mL of absolute ethanol. Once homogenous, 6-methyl-5-hepten-2-one (3.2
mL, 21.7 mmol) was added and the solution is stirred at room temperature for 10
minutes. At this point 51 (2.65 g, 16 mmol) was added and the reaction heated at 40 °C
overnight. The reaction was quenched with dilute aqueous HCl and extracted three times
with ethyl acetate. The combined organic layers were dried over anhydrous MgSO4 and
the solvent removed with rotary evaporation. The crude product was subjected to column
chromatography on silica gel using a 10:1 chloroform/methanol as an eluent. The product
could be further purified through recrystallizing with diethyl ether/hexanes. An amount of
0.36 g of 52 was isolated (28 % yield). 1H NMR (CDCl3, 300 MHz) δ 1.66 (3H, s), δ 1.7
(3H, s), δ 2.31 (3H, s), δ 2.40 (2H, m), δ 2.73 (2H, t, J = 7.8 Hz), δ 3.87 (3H, s), δ 5.18
(1H, t, J = 7.2 Hz), δ 6.30 (1H, s), δ 6.38 (1H, s), δ 7.25 (1H, d, J = 16.5), δ 8.00 (1H, d, J
= 16.5). HRMS (m/z): [M+H]+ calc for C17H23O3 275.1642; found 275.1651
Compound 53: To a 50 mL round bottom flask 1 mL of a 50 % solution of KOH was
added to 10 mL of methanol. While stirring, 6-methyl-5-hepten-2-one (0.15 mL) was

74
added and the reaction stirred at room temperature for 10 minutes. At this point, 46 (0.18
g, 0.99 mmol) was added and the reaction stirred at room temperature for 6 hrs. The
reaction was quenched with dilute aqueous HCl and extracted three times with ethyl
acetate, and the combined organic layers were dried over anhydrous MgSO4. After
removal of the solvent through rotary evaporation, the crude is subjected to flash
chromatography on silica gel eluting with 60 % diethyl ether/petroleum ether solvent
system. Roughly 0.55 g of 53 was isolated (20 % yield). 1H NMR (CDCl3, 300 MHz) δ
1.65 (3H, s), δ 1.70 (3H, s), δ 2.27-2.47 (2H, m), δ 2.36 (3H, s), δ 2.69 (2H, t, J = 7.8
Hz), δ 3.88 (6H,s), δ 5.18 (1H, tt, J = 1.5, 7.2 Hz), δ 6.39 (2H, s), δ 7.14 (1H, d, J = 16.5
Hz), δ 8.00 (1H, d, J = 16.5 Hz). 13C NMR (CDCl3, 300 MHz) δ 17.9, 22.7, 23.6, 25.9,
40.5, 55.9, 104.9, 123.6, 128.6, 132.5, 134.1, 142.6, 160.2, 202.4. HRMS (m/z): [M+] calc
for C18H24O3 288.1725; calc 288.1714.

75
Appendix B: 1H-NMR Spectra
H2 15N
CO2 Et
400 MH
z, D2 O

76
H15N
CO2 Et
O
300 MH
z, CD
Cl3

77
O
15N
OEt
300 MHz, CDCl3

78
15N
HO
CO2 EtCO
2 Et
300 MH
z, CD
Cl3

79
15N
HO
OHOH
300 MH
z, D2 O

80
15N
HO
NOH
OEt
300 MH
z, CD
Cl3

81
15N
HO
O
OPO
3 H2
400 MH
z, DM
SO-d6

82
H2 15N
13CH13C
13CH3O
OEt
400 MH
z, D2 O

83
H15N
13CH13C
13CH3O
OEt
O300 MH
z, CD
Cl3

84
O
15N13C 13C13CH
3 OEt
400 MH
z, CD
Cl3

85
OH
15NH2
300 MH
z, CD
Cl3

86
EtO2 C
OCO
2 Et
OH
300 MH
z, CD
Cl3

87
EtO2 C
OCO
2 Et
NH
O
300 MH
z, CD
Cl3

88
N
H2 N
COOHCO
OH
300 MH
z, DM
SO-d6

89
N
H2 N
CO2 EtCO
2 Et
300 MH
z, CD
Cl3

90
OMe
OMe
300 MHz, CDCl3

91
OMe
OMe
O
300 MHz, CDCl3

92
OMe
OH
O
300 MHz, CDCl3

93
OMe
OMe
300 MH
z, CD
Cl3

94
OMe
OMe
O
300 MH
z, CD
Cl3

95
OMe
OMe
O
300 MH
z, CD
Cl3

96
OMe
OH
O
300 MH
z, CD
Cl3

97
13C13C15N
H3 13C
HO
N
OH
OEt
300 MH
z, CD
Cl3

98
13C13C
15NH3 13C
HO
OO
PO3 H
2
600 MH
z, D2 O





























