Embed Size (px)
Citation preview

Macromol. Chem. Phys. 195, 1973 -1983 (1994) 1973
Synthesis of star-shaped polydimethylsiloxanes containing SiO2 core units
Takuya Ogawa*, Toshio Suzuki, Itaru Mita
Research Center, Dow Corning Asia Ltd., 603 Kishi, Yamakita, Kanagawa 258-01, Japan
(Received September 9, 1993, revised manuscript of October 21, 1993)
SUMMARY Star-shaped polysiloxanes consisting of SiO, cores and polydimethylsiloxane (PDMS)
branches were successfully prepared by three methods using functionalized solvent-soluble silica- tes as a starting material. The first method involved the lithiation of a Si-OH functional silicate with butyllithium, followed by reaction with hexamethylcyclotrisiloxane (D3) and termination with a chlorosilane to afford a star polymer. In this case, the chain ends were substituted with vinyl or Si-OH functional groups. In the second method, a Si-CI functional silicate was used as the source of the SiO, core. A PDMS chain with a lithium silanolate terminal group was reacted with the Si-CI functional silicate to provide a star polymer. In the third method, a PDMS chain with a diethylamino group at one chain end was reacted with the Si-OH functional silicate to form a star polymer. The Mark-Houwink constant a of the star polymer was 0,74, which is very close to that of a linear PDMS.
Introduction
Star-shaped polymers are of great interest due to their unique structures and physical properties, such as their rheological behavior, and as a result several methods for preparing these polymers have been developed. The most convenient and versatile way to introduce the branch point required for star polymer formation is to react a polymer possessing a terminal functional group with a specific compound containing a plurality of reactive sites. For example, the preparation of star polymers via a living polymeriza- tion followed by multifunctional termination using chlorosilanes was demonstrated by Hadjichristidis et al. ’). Dickstein and Lillya prepared telechelic star polydimethylsil- oxanes with a highly defined structure2). According to their method, the reaction product of p-(N,N-bis(trimethylsily1)amino)styrene and sec-butyllithium was used as the initiator, and tetrachlorosilane as the terminator. All polymers showed low poly- dispersity, controlled molecular weights and almost theoretical functionality. Similar approaches to preapare siloxane star polymers are described in various reports 3-6). Some of these describe the preparation of thermotropic rigid rod star-block copoly- mers ’, 6). Polysiloxane star polymers with aluminium or titanium atoms at the branch points were prepared by the reaction of polydimethylsiloxanes having terminal carboxylic acid groups with aluminium isopropoxide or titanium tetraisopropoxide’).
Multi-functional initiators can also be utilized for the preparation of star polymers. Imai et al. prepared siloxane star polymers with different arm lengths using 1,3,5-tris- (1ithiumoxydimethylsilyl)benzene as the initiator *). In addition, stepwise construction of the star polymer branches was achieved to form new types of star-shaped copolymers having three different branches 9).
0 1994, Huthig & Wepf Verlag, Base1 CCC 1022-1 352/94/$08.00

1974 T. Ogawa, T. Suzuki, I. Mita
Dendrimers or starburst polymers, which can be regarded as belonging to the star polymer family, have been extensively investigated. Masamune et al. reported that they obtained multibranched polysiloxanes with terminal Si-H groups starting from an a- hydro-m-hydroxyoligosiloxane and methyltrichlorosilane ’’). Siloxane starburst poly- mers were also prepared by divergent ‘ ‘ 9 ‘’) and convergent methods as reported by Imai et al.
On the other hand, solvent-soluble silicates, sometimes referred to as MQ resins, have also gathered special interest due to their unique inorganic-organic properties 14). The solvent-soluble silicates consist of monofunctional R,SiO,,, units [e. g. (CH,),SiO,,,] and quadrifunctional SiO, units. We hypothesized that attaching PDMS branches to a silicate core may yield star polymers with interesting chemical and physical properties.
In this paper, we describe the preparation of star polymers containing a silicate moiety as the core and PDMS as the branches, as shown in Fig. 1.
Three different preparative methods were attempted each of which yielded essentially the same star polymers. The solution viscosity behavior of the resulting star polymers was also investigated, and the Mark-Houwink constant was evaluated.
Fig. 1. Star-polymers examined in this study (simple scheme)
Results and discussion
Preparation of star polymers using a multi-functional initiator
The first synthetic strategy to produce star polymers with a Si02 core involves the utilization of a Si-OH functional solvent-soluble silicate. The synthetic steps toward the star polymer are illustrated in Scheme 1.
Scheme I:
HCI/H,O (Me,HSi),O + Si(OMe), (Me: CH,)
b [MezHSi01,21 l,5[Siozl
1
Synthesis of star-shaped polydimethylsiloxanes . . . 1975
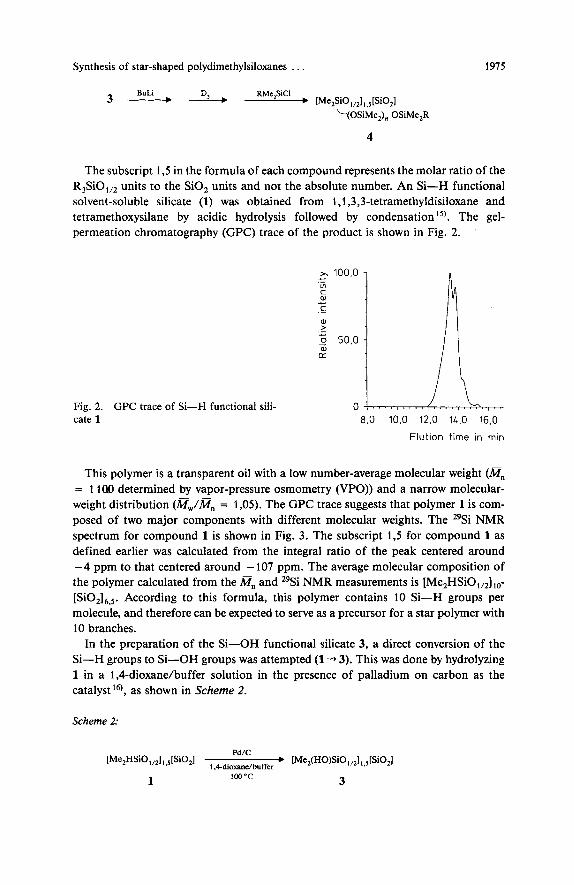
The subscript 1 3 in the formula of each compound represents the molar ratio of the R,SiO,,, units to the SiO, units and not the absolute number. An Si-H functional solvent-soluble silicate (1) was obtained from 1,1,3,3-tetramethyldisiloxane and tetramethoxysilane by acidic hydrolysis followed by condensation 15). The gel- permeation chromatography (GPC) trace of the product is shown in Fig. 2. ‘
Fig. 2. cate 1
GPC trace of Si-H functional sili- 8.0 10.0 12.0 14.0 16.0
Elut ion time in min
This polymer is a transparent oil with a low number-average molecular weight (mn = 1 la0 determined by vapor-pressure osmometry (VPO)) and a narrow molecular- weight distribution (m,.,/Gn = 1,05). The GPC trace suggests that polymer 1 is com- posed of two major components with different molecular weights. The 29Si NMR spectrum for compound 1 is shown in Fig. 3. The subscript 1,5 for compound 1 as defined earlier was calculated from the integral ratio of the peak centered around - 4 ppm to that centered around - 107 ppm. The average molecular composition of the polymer calculated from the a, and 29Si NMR measurements is [Me,HSiO,,,],,- [SiO,],,,. According to this formula, this polymer contains 10 Si-H groups per molecule, and therefore can be expected to serve as a precursor for a star polymer with 10 branches.
In the preparation of the Si-OH functional silicate 3, a direct conversion of the Si-H groups to Si-OH groups was attempted (1 -+ 3). This was done by hydrolyzing 1 in a 1,4-dioxane/buffer solution in the presence of palladium on carbon as the catalyst as shown in Scheme 2.
Scheme 2:
1976 T. Ogawa, T. Suzuki, I. Mita
I
0 -20 -40 -60 -80 -100 6 in ppm
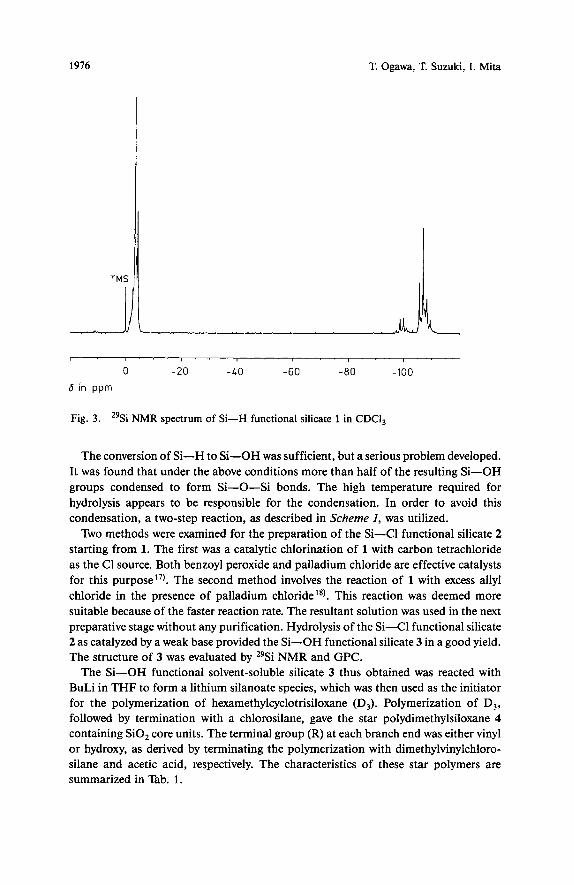
Fig. 3. "Si NMR spectrum of Si-H functional silicate 1 in CDCI,
The conversion of Si-H to Si-OH was sufficient, but a serious problem developed. It was found that under the above conditions more than half of the resulting Si-OH groups condensed to form Si-0-Si bonds. The high temperature required for hydrolysis appears to be responsible for the condensation. In order to avoid this condensation, a two-step reaction, as described in Scheme I, was utilized.
Two methods were examined for the preparation of the Si-CI functional silicate 2 starting from 1. The first was a catalytic chlorination of 1 with carbon tetrachloride as the C1 source. Both benzoyl peroxide and palladium chloride are effective catalysts for this purpose"). The second method involves the reaction of 1 with excess ally1 chloride in the presence of palladium chloridei8). This reaction was deemed more suitable because of the faster reaction rate. The resultant solution was used in the next preparative stage without any purification. Hydrolysis of the Si-Cl functional silicate 2 as catalyzed by a weak base provided the Si-OH functional silicate 3 in a good yield. The structure of 3 was evaluated by 29Si NMR and GPC.
The Si-OH functional solvent-soluble silicate 3 thus obtained was reacted with BuLi in THF to form a lithium silanoate species, which was then used as the initiator for the polymerization of hexamethylcyclotrisiloxane (D,). Polymerization of D,, followed by termination with a chlorosilane, gave the star polydimethylsiloxane 4 containing SiO, core units. The terminal group (R) at each branch end was either vinyl or hydroxy, as derived by terminating the polymerization with dimethylvinylchloro- silane and acetic acid, respectively. The characteristics of these star polymers are summarized in %b. 1.
Synthesis of star-shaped polydimethylsiloxanes . . . 1977
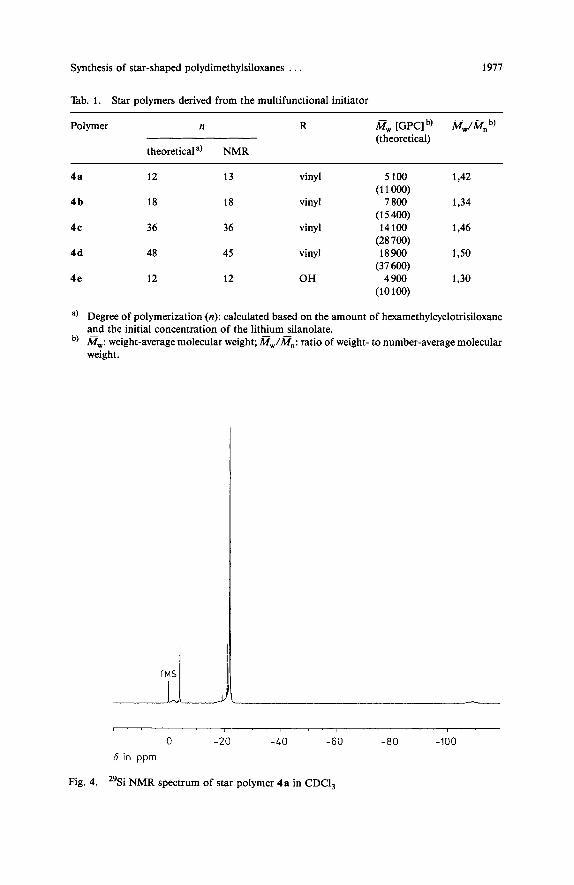
Tab. 1. Star polymers derived from the multifunctional initiator
Polymer n R a, [GPC] b, @,/a,, b, (theoretical)
theoretical a) NMR
4a 12 13 vinyl 5 100 1,42
4 b 18 18 vinyl 7 800 1.34 (1 1
(15400) 4c 36 36 vinyl 14 100
(28 700) 4d 48 45 vinyl 18900
(37 600) 4e 12 12 OH 4 900
(10100)
a) Degree of polymerization (n): calculated based on the amount of hexamethylcyclotrisiloxane and the initial concentration of the lithium silanolate.
b, a,: weight-average molecular weight; M,/M,,: ratio of weight- to number-average molecular weight.
I . , I
0 -20 -40 -60 -80 -100 6 in pprn
Fig. 4. "Si NMR spectrum of star polymer 4 a in CDCI,
1978 T. Ogawa, T. Suzuki, I. Mita
The degree of polymerization, n, in the theoretical column is based upon the amount of D, consumed relative to the initial concentration of lithium silanolate. The values of n calculated from "Si NMR are in good agreement with the theoretical values. The 29 Si NMR spectrum of 4a is shown in Fig. 4. The three major signals at -4,1, -22 and - 108 ppm are assignable to the terminal Me,(CH,=CH)SiO, the Me,SiO groups of the branches and the core SiO,, respectively.
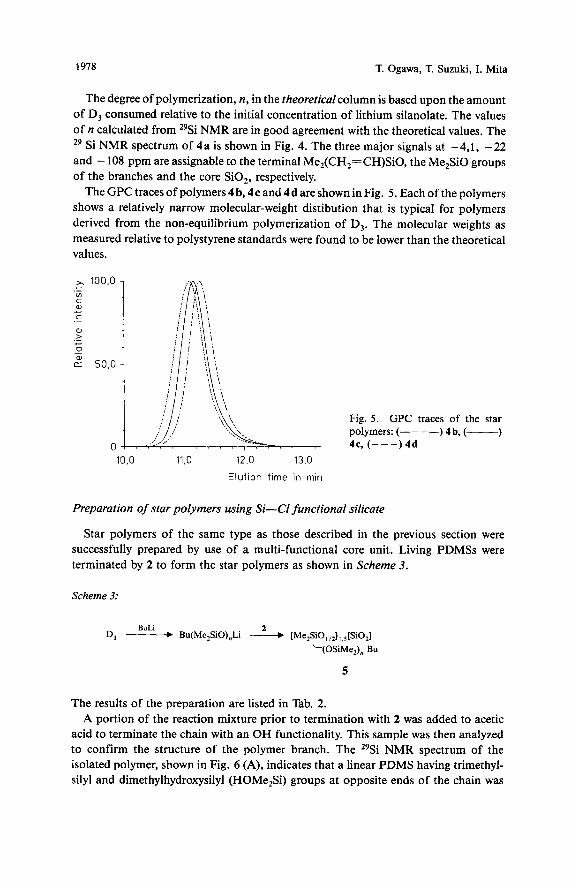
The GPC traces of polymers 4 b, 4c and 4d are shown in Fig. 5 . Each of the polymers shows a relatively narrow molecular-weight distibution that is typical for polymers derived from the non-equilibrium polymerization of D,. The molecular weights as measured relative to polystyrene standards were found to be lower than the theoretical values.
Fig. 5 . GPC traces of the star polymers: (- - - -) 4 b, (-) 4c, ( - - - ) 4 d
10.0 11.0 12.0 13.0
Elution time in rnin
Preparation of star polymers using Si-CI functional silicate
Star polymers of the same type as those described in the previous section were successfully prepared by use of a multi-functional core unit. Living PDMSs were terminated by 2 to form the star polymers as shown in Scheme 3 .
Scheme 3:
BuLi 2 D, ___* Bu(Me,SiO),Li d [Me2Si0,,211,51Si021
i(OSiMe2), Bu
5
The results of the preparation are listed in Tmb. 2. A portion of the reaction mixture prior to termination with 2 was added to acetic
acid to terminate the chain with an OH functionality. This sample was then analyzed to confirm the structure of the polymer branch. The 29Si NMR spectrum of the isolated polymer, shown in Fig. 6 (A), indicates that a linear PDMS having trimethyl- silyl and dimethylhydroxysilyl (HOMe,Si) groups at opposite ends of the chain was
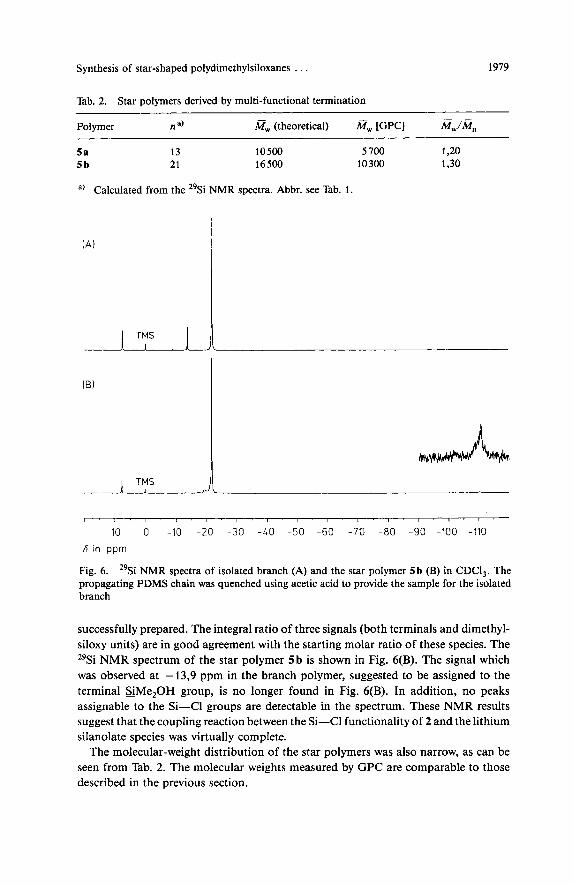
Synthesis of star-shaped polydimethylsiloxanes . . . 1979
Tab. 2. Star polymers derived by multi-functional termination
Polymer n a) a, (theoretical) I%?, [GPC] GJM" ~~~ ~
5 a 13 10500 5b 21 16500
a) Calculated from the 29Si NMR spectra. Abbr. see Tab. 1.
5 700 1,20 10300 1,30
TMS I I
I 1 TMS
. . . . . . . . . . . . . . . . . . . . . . . . . . . .
10 0 -10 -20 -30 -LO -50 -60 -70 -80 -90 -100 -110
6 in ppm
Fig. 6. 29Si NMR spectra of isolated branch (A) and the star polymer 5 b (B) in CDCI,. The propagating PDMS chain was quenched using acetic acid to provide the sample for the isolated branch
successfully prepared. The integral ratio of three signals (both terminals and dimethyl- siloxy units) are in good agreement with the starting molar ratio of these species. The 29Si NMR spectrum of the star polymer 5b is shown in Fig. 6(B). The signal which was observed at - 13,9 ppm in the branch polymer, suggested to be assigned to the terminal sMe,OH group, is no longer found in Fig. 6(B). In addition, no peaks assignable to the Si-Cl groups are detectable in the spectrum. These NMR results suggest that the coupling reaction between the Si-Cl functionality of 2 and the lithium silanolate species was virtually complete.
The molecular-weight distribution of the star polymers was also narrow, as can be seen from Tab. 2. The molecular weights measured by GPC are comparable to those described in the previous section.

1980 T. Ogawa, T. Suzuki, I. Mita
Preparation of star polymers using an Si-NEt, functional PDMS
The third preparative method to generate star polymers containing a SiO, core involves the utilization of aminosilyl functional groups as the reaction site. It is well known that the aminosilyl group easily reacts with a silanol functionality to generate a siloxane linkage19). The synthesis of PDMSs containing an aminosilyl group at one chain end has been recently reportedz0). The synthesis of a star polymer from the aminosilyl functional PDMS was carried out according to the following scheme.
Scheme 4:
Me,ViSiCI b Et,N(SiMe,O),SiMe,Vi BuLi D, Et,NH -
(Et: C,H,; Vi: vinyl) 6
6 + 3 d [Me,SiO,,,],,,[SiO,] L(OSiMe,),OSiMe,Vi
7
The degree of polymerization, n, of the diethylamino-terminated PDMS 6 as calculated from the 29Si NMR analysis was 12, which is in good agreement with the ratio of consumed D, to the initiator. The obtained diethylamino-functional PDMS underwent coupling with the Si-OH functional silicate 3 in the absence of any catalyst to give star polymer 7. The weight-average molecular weight Mw determined by GPC was 5000 (theoretical: 1 1 000), with a polydispersity of 1,3 1. These data are reasonable as compared with those of polymer 4 a in Tab. 1 . The 29Si NMR result was also consistent with the structure of the star polymer.
Solution viscosity of star polymers
The molecular weight-viscosity relationship of the star polymers prepared by the multi-functional initiation method was studied. The intrinsic viscosities of the star polymers as plotted against the molecular weights (Mw from GPC) are shown in Fig. 7. This was also done with linear PDMSs so as to compare the two types of polymers.
As can be seen from the figure, good linearity was obtained for both series of polymers, with the two lines lying almost parallel. The coefficient a of the Mark- Houwink equation (Eq. (1)) for the present star polymers is 0,74,
which is similar to that of the linear PDMSs. It is believed that these star polymers behave as linear flexible molecules due to the high content of dimethylsiloxane branches.

Synthesis of star-shaped polydimethylsiloxanes . . . 1981
Fig. 7. Relationship between the weight- average molecular weights and intrinsic viscosities of the linear and star polymers: (0) star polymers, ( 0 ) linear PDMSs
Experimental part
General
-1.0 -I I
-1 '
-2.0 " ' L 3.0 3.5 L.0 G.5 5.0
log,ofi,
All chemicals are commercially available and were used without further purification. All solvents were reagent grade and distilled under nitrogen from appropriate drying agents prior to use.
Analytical gas chromatography (GC) was performed using a Shimadzu GC-14A gas chromato- graph equipped with a DB-5 capillary column (J & W Scientific). Gel-permeation chromato- graphy (GPC) was carried out with a Tosoh HE-8020 equipped with a series of Tosoh G5000HXL and GMHXGL columns and a refractometer. Toluene was used as the eluent for GPC, and the flow rate was 1 mL/min. Average molecular weights were calculated by use of polystyrene standards. Vapor-pressure osmometry (VPO) was performed in toluene solution using a Corona 117 molecular weight apparatus. 'H and 29Si NMR spectra were recorded with a Bruker ACP300 spectrometer. A1203 sample tubes (Shigemi Standard & Joint Co. Ltd.) and a Si-free probe were used in the 29Si NMR measurements. CDC1, or (CD,),CO were used as the solvent. Chemical shifts were referenced to CHCl, ('H), or tetramethylsilane (29Si). Chromium acetylacetonate [Cr(a~ac)~, 0,02 M] was used as the relaxation agent for 29Si NMR measurements. Intrinsic viscosities were measured by use of a Ubbelohde type capillary viscometer in tetrahydrofuran (THF) at 30°C.
Si-H functional silicate (1)
1,1,3,3-Tetramethyldisiloxane (16,12 g, 0,12 mol), water (9,6 g), conc. HCl (4,8 g) and methanol (9,6 g) were placed in a three-necked flask and cooled with an ice bath. To the mixture was added tetramethoxysilane (24,36 g, 0,16 mol) over a period of 25 min. After the addition was completed, the mixture was stirred for 1 h at room temperature. The mixture separated into two phases.
The upper layer was extracted with hexane and the extract was combined with the lower layer. The resulting organic layer was washed twice with water and was dried with MgSO,. Filtration to remove the drying agent followed by evaporation of the solvent gave a transparent oil. Yield: 21,86 g (85%).
'H NMR (CDC13): 6 = 0,23 &HSi-), 333 (trace, &O-) and 4,73 (Mess&-) . 29Si NMR (CDCl,): 6 = -3,4 (Me2HgO-) and - 107 &03.
Si-CI functional silicate (2)
Si-H functional silicate 1 (1,07 g, 10 mmol for Si-H unit) was dissolved in ally1 chloride (8,2 mL, 0,l mol) and palladium chloride (36 mg, 0,2 mmol) was added. The mixture was

1982 T. Ogawa, T. Suzuki, I. Mita
refluxed for 17 h after which ally1 chloride was removed by evaporation. No Si-H groups were detected in 'H NMR analysis.
'H NMR (CDCI,): 6 = 0,48 &CISi-) and 3,53 (trace, WO-). 29Si NMR (CDCI,): 6 = 6,5 (Me2ClSjO-) and - 109 (530,).
Si-OH functional silicate (3)
An ether solution of the Si-C1 functional silicate 2 (1,29 g) was slowly poured into a mixture of saturated aqueous NaHCO, solution (400 mL) and ether (150 mL) with vigorous stirring. Separation of the organic layer followed by drying and solvent removal gave a slightly yellow oil. Yield: 1,19 g (98%). This polymer required storage as a solution in THF to avoid self- condensation of the Si-OH groups.
29Si NMR ((CD,),CO): 6-= - 11,6 (Me,(HO)SjO-) and - 109 (530,). GPC: a,, = 1250, MJM, = 1,20.
Star polymer (4 a)
To a THF solution of the Si-OH functional silicate 3 (1,15 g of silicate 3,9 mmol of Si-OH) was added butyllithium (hexane solution, 9 mmol) over a period of 5 min with cooling to 0 "C. After stirring for half an hour at O"C, D, (8,O g, 36 mmol) in THF was added to the solution in a single portion. The reaction mixture was allowed to warm to room temperature and the decrease in the D, concentration was monitored by GC analysis. Vinyldimethylchlorosilane (2,5 mL, 18 mmol) was added to the mixture after more than 90% of the D, had been consumed and the resulting mixture was stirred for further 1 h.
The volatile components were removed by vacuum evaporation before dilution with hexane. The resultant solution was washed twice with water and dried using MgSO,. Filtration to remove the drying agent followed by vacuum evaporation gave a colorless oil. Yield: 7,92 g (89%).
,'Si NMR (CDCI,): 6 = -4,l (Me,ViSjO-), -22 (-Me,SiO,-) and -109 (530,).
Star polymer (5 b)
Butyllithium (hexane solution, 16 mmol) was dissolved in 15 mL of THF cooled in an ice bath, and D, (52,7 g, 0,ll mol) in THF was added. The mixture was stirred for 1 h at this temperature, then the cooling bath was removed. The solution was stirred for further 3 h at room temperature before addition of the Si-CI functional silicate 2 (1,71 g, 16 mmol of Si-Cl) in ether (5 mL). The mixture was stirred for an additional 1 h to complete the reaction.
The reaction mixture underwent evaporation and the residue was diluted with hexane. Washing this solution with a saturated aqueous NH,Cl solution followed by drying of the organic layer provided a clear solution. Removal of the solvent followed by centrifugation gave a colorless oil. Yield: 26,9 g (93%).
29Si NMR (CDCI,): 6 = 7,6 (Me,BugO-), -22 (-Me&O-) and - 110 (530,).
Diethylamino-terminated PDMS (6)
Diethylamine (1,M mL, 10 mmol) was dissolved in THF (30 mL) in a three-necked flask with cooling (0 "C). To the solution was added butyllithium (hexane solution, 10 mmol) over a period of 7 min and stirring maintained for 30 min. D, (9,12 g, 41 mmol) in THF was added to the solution in one portion. After stirring for 1 h at 0°C and 2,5 h at room temperature, vinyldi- methylchlorosilane (1,65 mL, 12 mmol) was added. The reaction mixture was stirred for an addi- tional 1 h at room temperature.
Residual solid material was filtered off under an inert atmosphere to yield a pale yellow oil. Yield: 9,52 g (91%). The 29Si NMR and GPC analyses were performed after the sample was quenched in MeOH to give the more stable Si-OMe terminated polysiloxane.

Synthesis of star-shaped polydimethylsiloxanes . . . 1983
29Si NMR (CDCI,): 6 = -4,l (Me2VisO-), -12,3 (MeOMeaO-), -21,l
GPC: M, = 1030, aw/a,, = 1,30. (Me2ViSiOSiMe20-), - 22 (-Meso-) and - 23,3 (MeOMe2SiOaMe20-).
Star polymer (7)
A THF solution of Si-OH functional silicate 3 (0,89 g, 7,2 mmol of Si-OH) was added dropwise to the diethylamino-terminated PDMS 6 (7,56 g, 7,2 mmol), and the mixture was stirred for 1 h at room temperature.
The same work-up as that for the star polymer 5b gave a colorless oil. Yield: 7,OO g (88%). 29Si NMR (CDCI,): 6 = -4,l (Me2Vi$O-), -22 (-MeaO-) and - 110 BO2).
‘1 N. Hadjichristidis, A. Guyot, L. J. Fetters, Macromolecules 11, 889 (1978)
3, L. Wilczek, S. Rubinsztajn, W. W. Fortuniak, J. Chojnowski, I. I. Tverdokhlebova,
4, I. I. Tverdokhlebova, T. A. Larina, I. I. Mamaeva, N. V. Pertsova, L. Wilczek, S. Rubinsztajn,
5 , W. H. Dickstein, C. P. Lillya, Mol. Crysf. Liq. Crysf. 157, 69 (1988) 6, S. K. Bhattacharya, C. A. Smith, W. H. Dickstein, Macromolecules 25, 1373 (1992) ’) H. Kazama, Y. Tezuka, K. Imai, Macromolecules 24, 122 (1991) 8, A. Morikawa, M. Kakimoto, Y. Imai, K. Takano, T. Yamamoto, Polym. P r e p Jpn. 40, 359
9, T. Fujimoto, H. Zhang, T. Kazama, Y. Isono, H. Hasegawa, T. Hashimoto, Polymer 33,2208
lo) H. Uchida, Y. Kabe, K. Yoshino, A. Kawamata, T. Tsumuraya, S. Masamune, J. Am. Chem.
‘ I ) A. Morikawa, M. Kakimoto, Y. Imai, Macromolecules 24, 3469 (1991) 12) A. Morikawa, M. Kakimoto, Y. Imai, Polym. J. (Tokyo) 24, 573 (1992) ‘3 ) A. Morikawa, M. Kakimoto, Y. Imai, Macromolecules 25, 3247 (1992) 1 4 ) W. Noll, “Chemistry and Technology of Silicones”, Academic Press, New York 1968, chapter
10 ‘ V Jpn. Kokai 61-195129 (1986) , Toray Silicone Co. Ltd., inv.: A. Shirahata; Chem. Abstr. 106,
67886~ (1986) 16) G. H. Barnes, Jr., N. E. Daughenbaugh, J. Org. Chem. 31,885 (1966); some kinds of rhodium
catalysts are also effective in the oxidation of Si-H to Si-OH: U. Schubert, C. Egger, K. Rose, C. Alt, J . Mol. Catal. 55, 330 (1989)
”) Jpn. Kokai3-95190,3-95191(1991), KaoCorp., invs.: H. Uchida, Y. Kabe, K. Yoshino; Chem. Abstr. 115, 280276n, 159438~ (1991)
18) Ger. Offen. DE 3518605 (1986), Wacker Chemie, invs.: C. Trieschman, J. Muller, G. Preiner, W. Doskocil; Chem. Abstr. 106, 138941 w (1986)
19) W. Noll, “Chemistry and Technology of Silicones”, Academic Press, New York 1968, chapter 5
20) T. Suzuki, S. Yamada, T. Okawa , Polym. J. (Tokyo) 25, 41 1 (1993)
W. H. Dickstein, C. P. Lillya, Macromolecules 22, 3886 (1989)
R. V. Volkova, Bul/. Po/. Acud. ScL, Chem. 37, 91 (1989)
J. Chojnowski, Vysokomol. Soedin., Ser. B 32, 292 (1990)
(1991)
(1 992)
SOC. 112, 7077 (1990)





























