Embed Size (px)
Citation preview

DOI: 10.1002/chem.200900654
Synthetic, Mechanistic, and Theoretical Studies on the Generation of IridiumHydride Alkylidene and Iridium Hydride Alkene Isomers
Patricia Lara,[a] Margarita Paneque,[a] Manuel L. Poveda,[a] Laura L. Santos,[a]
Jos� E. V. Valpuesta,[a] Ver�nica Salazar,[c] Ernesto Carmona,*[a] Salvador Moncho,[b]
Gregori Ujaque,[b] Agust� Lled�s,*[b] Celia Maya,[a] and Kurt Mereiter[d]
In memoriam of Professor Raffaello Romeo
Introduction
Transition-metal compounds that contain alkyl, alkylidene,or alkene ligands are important organometallic species thatparticipate directly, or as reactive intermediates, in manychemical transformations.[1,2] As is well known, the late de-velopment of transition-metal alkyls was due largely to ki-netic reasons, for these compounds can undergo severalfacile decomposition reactions. Two fundamental, very fa-miliar rearrangements, that are central to the work hereindescribed, are the a- and the b-H eliminations, that lead toisomeric hydride alkylidene and hydride alkene structures,respectively (Scheme 1). The two reactions are reversibleand it is widely assumed that b-H elimination is more facilethan a-H elimination. However, over the years evidence hasbeen gained showing that in many systems these two hydro-gen migrations may compete. In particular, metal alkyls inwhich the M–Ca–Cb–Hb group of atoms cannot adopt the re-quired syn-coplanar spatial distribution[1] may undergo very
Abstract: Experimental and theoreticalstudies on equilibria between iridiumhydride alkylidene structures,[(TpMe2)Ir(H){=C ACHTUNGTRENNUNG(CH2R)ArO}](TpMe2 =hydrotris(3,5-dimethylpyrazo-lyl)borate; R=H, Me; Ar = substitutedC6H4 group), and their correspondinghydride olefin isomers,[(TpMe2)Ir(H){R(H)C=C(H)OAr}],have been carried out. Compounds ofthese types are obtained either by reac-tion of the unsaturated fragment[(TpMe2)Ir ACHTUNGTRENNUNG(C6H5)2] with o-C6H4(OH)CH2R, or with the substitut-ed anisoles 2,6-Me2C6H3OMe, 2,4,6-Me3C6H2OMe, and 4-Br-2,6-
Me2C6H2OMe. The reactions with thesubstituted anisoles require not onlymultiple C�H bond activation but alsocleavage of the Me�OAr bond and thereversible formation of a C�C bond (asrevealed by 13C labeling studies). Equi-libria between the two tautomericstructures of these complexes wereachieved by prolonged heating at tem-peratures between 100 and 140 8C, withinterconversion of isomeric complexes
requiring inversion of the metal config-uration, as well as the expected migra-tory insertion and hydrogen-elimina-tion reactions. This proposal is support-ed by a detailed computational explo-ration of the mechanism at the quan-tum mechanics (QM) level in the realsystem. For all compounds investigat-ed, the equilibria favor the alkylidenestructure over the olefinic isomer by afactor of between approximately 1 and25. Calculations demonstrate that themain reason for this preference is thestrong Ir–carbene interactions in thecarbene isomers, rather than steric de-stabilization of the olefinic tautomers.
Keywords: alkenes · carbenes ·C�H activation · density functionalcalculations · iridium
[a] P. Lara, Prof. Dr. M. Paneque, Prof. Dr. M. L. Poveda,Dr. L. L. Santos, J. E. V. Valpuesta, Prof. Dr. E. Carmona,Dr. C. MayaInstituto de Investigaciones Qu�micas andDepartamento de Qu�mica Inorg�nicaConsejo Superior de Investigaciones Cient�ficas (CSIC)and Universidad de SevillaAv. Am�rico Vespucio 49, Isla de la Cartuja, 41092 Sevilla (Spain)Fax: (+34) 954460565E-mail : [email protected]
[b] S. Moncho, Dr. G. Ujaque, Prof. Dr. A. Lled�sDepartament de Qu�mica, Universitat Aut�noma de Barcelona08193 Bellaterra (Spain)Fax: (+34) 935812920E-mail : [email protected]
[c] Dr. V. SalazarCIQ, Universidad Aut�noma del Estado de HidalgoPachuca (M�xico)
[d] Prof. Dr. K. MereiterDepartment of Chemistry, Vienna University of TechnologyGetreidemarkt 9/164, 1060 Vienna (Austria)
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.200900654.
� 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 9046 – 90579046

slow b-H elimination. As discussed previously,[3,4] when botha- and b-hydrogen atoms are present in sterically hinderedmetal environments, a-agostic interactions[4] and a-H elimi-nations[3] may become more favorable than their b counter-parts. In agreement with this line of reasoning, an equilibri-um between the two tautomeric structures shown inScheme 1 could be attained, despite the large energy differ-ence that exists between the free alkene and alkylidene mol-ecules. The isomerization of free ethene to ethylidene is en-dothermic by about 80 kcal mol�1, but the ethylidene modelcomplex [Ru ACHTUNGTRENNUNG(=CHCH3)Cl(H) ACHTUNGTRENNUNG(PH3)2] is only ca.16 kcal mol�1 less stable than its olefinic isomer, [Ru-ACHTUNGTRENNUNG(C2H4)Cl(H) ACHTUNGTRENNUNG(PH3)2].[5a] These calculations suggest that asuitable choice of the metal complex may allow accomplish-ment of this goal, a hypothesis that also finds some experi-mental support. Thus, although the conversion of a metal al-kylidene into the corresponding metal alkene is a generalreaction of electrophilic alkylidenes,[1b, 5b–e] and the oppositetransformation, namely, the alkene-to-alkylidene rearrange-ment is a rarely observed process,[6] in recent years new ex-amples of equilibria between metal–alkene and –alkylideneisomers have been found, which add to the very few alreadyknown.[7] Gusev and co-workers have shown that olefin andcarbene isomers form in the reactions of [{RuCl2 ACHTUNGTRENNUNG(p-cymene)}2] with bulky pincer ligands, as a result of C�Hbond activation and a- and b-H eliminations from thepincer-ligand framework.[8a] Also very recently Caulton et al.found analogous equilibria between the p-olefin and car-bene isomers of norbornene in the reaction of this alkenewith some rhenium complexes of pincer ligands of type[ReH4ACHTUNGTRENNUNG(PNPR)] (PNPR =N(SiMe2CH2PR2)2).[8b]
As discussed in the preceding paper in this issue,[9] wehave observed that the reaction of the complex [(TpMe2)Ir-ACHTUNGTRENNUNG(C6H5)2(N2)], 1·N2 (TpMe2 =hydrotris(3,5-dimethylpyrazolyl)-borate) with 2-ethylphenol occurs in the way represented inScheme 2, to give the hydride alkylidene complex 2 c as themajor product accompanied by minor amounts (ca. 5 %) of
its hydride alkene tautomer, 2 o (c and o will be employedthroughout this paper as abbreviations for carbene andolefin, respectively). As reported herein, 2-propylphenolreacts similarly with 1·N2 to give the corresponding hydridealkylidene, 3 c, as the major reaction product with smallamounts of isomer 3 o. Somewhat unexpectedly, the iridiumalkene compounds 4 o–6 o, structurally akin to 2 o, form, re-spectively, in the reactions of 2,6-dimethylanisole, 2,4,6-tri-methylanisole and 4-bromo-2,6-dimethylanisole, with the un-saturated species [(TpMe2)Ir ACHTUNGTRENNUNG(C6H5)2] (1), generated from 1·N2
by dinitrogen dissociation, or by heating [(TpMe2)Ir ACHTUNGTRENNUNG(C2H4)2]in C6H6.
[10] For these and related systems, equilibria betweentautomeric alkylidene and alkene structures have been ob-served and are reported in this paper, together with mecha-nistic studies aimed at understanding the formation of 4 o–6 o through complex reaction pathways that require thecleavage of three C ACHTUNGTRENNUNG(sp3)�H bonds plus one C ACHTUNGTRENNUNG(sp3)�O bond,along with the formation of a C�C bond. We also report acomputational analysis that has been performed with the ob-jectives of 1) providing support to the mechanism proposedto explain interconversion of isomeric alkene and alkylidenestructures; and 2) analyzing the factors that permit stabiliza-tion of the alkylidene tautomers relative to their alkenecounterparts. Parts of this work have already been brieflycommunicated.[11]
Results and Discussion
It has been shown in the preceding paper[9] that the reactionof 1·N2 and 2-ethylphenol at 60 8C gives a kinetic mixture ofthe alkylidene and alkene isomers, 2 c/2 o, of approximately20:1 (Scheme 2). However, if the reaction is performed athigher temperatures (�100 8C), equilibrium between thetwo tautomers is achieved. It is characterized by Keq =9 infavor of alkylidene 2 c and it is insensitive to temperature inthe range 90–130 8C. Naturally, the equilibrium may be ap-proached starting from pure samples of either 2 c or 2 o.
The analogous reaction of 1·N2 and 2-propylphenol at60 8C proceeds similarly (Scheme 3) to give complex 3 c, ac-
companied by minor amounts (ca. 2–3 %) of a compoundproposed to be the isomeric hydride alkene 3 o. Carbene 3 cis a green solid that when heated in solution at temperaturesaround 100 8C, equilibrates with alkene 3 o, with Keq�1.
In the IR spectrum of 3 c a band is recorded at 2140 cm�1
due to n ACHTUNGTRENNUNG(Ir�H), while in the 1H NMR spectrum, a signal atd=�20.47 ppm is assigned to the hydride ligand. The iridi-
Scheme 1. Equilibrium of alkylidene and alkene structures by competinga- and b-H eliminations.
Scheme 2. Formation of the carbene and olefin isomers, 2c and 2o, re-spectively, in the reaction of 1·N2 with 2-ethylphenol. In this and follow-ing schemes, [Ir] stands for TpMe2Ir.
Scheme 3. Reaction of 1·N2 and 2-propylphenol.
Chem. Eur. J. 2009, 15, 9046 – 9057 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 9047
FULL PAPER
um-bound alkylidene carbon atom is responsible for a13C{1H} resonance at d= 278.9 ppm. For olefin isomer 3 o,the structure represented in Scheme 3 is based on the spec-troscopic data obtained and, in particular, the trans stereo-chemistry of the alkene moiety is clearly inferred from theNOESY spectrum.
Taking into consideration that the reaction of 1·N2 withanisole and phenetole gives rise to two different hydride car-bene products,[9] the formation of which requires aromaticortho-C�H bond activation, we have investigated the analo-gous reactions of the 2,6-dimethyl-substituted anisolesshown in Scheme 4. Quite unexpectedly, the hydride olefin
complexes 4 o–6 o are formed in high spectroscopic yields(�90 %) at 60 8C. The three compounds feature a 1H NMRspectroscopic resonance around d=�17 ppm due to the hy-dride ligand, and other 1H and 13C{1H} signals for their aryloxide moiety that are similar to those found for 2 o.[9] In par-ticular, the CH proton of the �CH=CH2 terminus of 4 o ap-pears at d=6.83 ppm as a doublet of doublets (J=11.1 and8.2 Hz), whereas the =CH2 nuclei provide doublets at d=
3.24 (3J ACHTUNGTRENNUNG(H,H)=8.2 Hz) and 4.23 ppm (3J ACHTUNGTRENNUNG(H,H)=11.1 Hz).The reactivity of the hydride and olefin functionalities of
4 o–6 o toward migratory insertion has been tested by carry-ing out the reaction of 4 o with acetonitrile. Scheme 5 shows
that the reaction is reversible: heating at 60 8C in neat aceto-nitrile promotes insertion to give adduct 7, whereas heatinga solution of the latter compound in cyclohexane cleanly re-verses the process by acetonitrile dissociation and b-H elimi-nation. Compound 7 has been characterized by microanaly-sis and spectroscopy (see the Experimental Section) andalso by X-ray crystallography. One of the olefin derivatives,5 o, has been studied by X-ray diffraction, too.
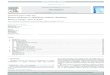
Figure 1 shows ORTEP perspective views of the mole-cules of the two derivatives. The existence in 7 of a chelatingaryloxide alkyl ligand reveals unmistakably that the reaction
of 1·N2 and 2,6-Me2C6H3OMe proceeds with rupture of theArO�Me bond of the ether, followed by C�C coupling ofthe resulting one-carbon unit with one of the aryl ortho-methyl substituents, which prior to the latter event has suf-fered a C�H bond-activation reaction. The Ir�CH2 bond(Ir1�C23 in Figure 1) has a length of 2.04(1) �, comparableto values found for related IrIII�C sigma bonds,[10,12] and itshigher trans influence, in comparison with the aryl oxideand NCMe groups, causes the Ir1�N1 bond trans to the
Scheme 4. The activation of 2,6-dimethyl-substituted anisoles by 1.
Scheme 5. Reversible 1,2-H shift in compound 4 o induced by NCMe.Figure 1. ORTEP representation of the molecular structures of hydridealkene 5o (top) and the NCMe adduct of the insertion product 7(bottom).
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 9046 – 90579048
E. Carmona, A. Lled�s et al.

TpMe2 ligand to be significantly longer than the other two(2.143(1) vs. 2.01(1) �; the latter is the average for Ir1�N3and Ir1�N5). In the hydride alkene compound 5 o it is natu-rally the hydride ligand that exhibits the largest trans influ-ence, its trans Ir�N bond having a length of 2.191(5) �,whilst the average of the other two amounts is 2.080(5) �.The metrical parameters of the Ir�alkene linkage in 5 oreveal relatively weak binding, as expected for an IrIII�alkene interaction. Thus, the Ir�Colefin (2.170(5) �, av.) andC=C bond lengths (1.407(9) �) are longer and shorter, re-spectively, than corresponding bonds in IrI�C2H4 com-plexes.[13] However, they are practically identical to corre-sponding parameters in IrIII�C2H4 complexes,[14] which indi-cates no destabilization of the Ir�alkene bonding in 5 o dueto the steric hindrance created by its alkene terminus.
The structural analogy that exists between compounds4 o–6 o and 2 o, suggests that, as for the latter, equilibria be-tween the hydride alkene and hydride alkylidene isomers ofcompounds 4–6 could be attained in solution. As represent-ed in Scheme 6, prolonged heating of hydride alkenes 4 o–
6 o in benzene or cyclohexane at temperatures between 100and 140 8C (in thick-walled vessels) produces the expectedequilibrium mixtures that are characterized by the indicatedKeq values. Similarly to equilibria based on compounds 2and 3, the hydride alkylidene structures are favored over thetautomeric hydride alkene, but corresponding Keq values arenot very sensitive to the ring substituents and span a rangeof approximately 1–25 for all of the investigated com-pounds.
The rearrangement of the 2,6-dimethyl-substituted ani-soles presented in Scheme 4 is a very unusual reaction. C�Obond cleavage in allylic and vinylic systems is a known reac-tion that is usually facilitated by coordination of the C=Cbond.[12b, 15] However, alkyl and aryl ethers are less reactiveand in the absence of organometal reagents, cleavage oftheir C�O bonds requires heating with concentrated HBr orHI.[16] Many organo-alkali-metal compounds also decom-pose ethers by a variety of mechanisms acting simultaneous-ly,[17] but transition-metal-promoted C�O bond rupture reac-tions, although known, are still somewhat limited.[12a, 18–20]
However, rearrangement of alkyl aryl ethers, ROAr, toalkyl-substituted phenols is an important reaction in organicsynthesis that may occur in the presence of Lewis acid cata-lysts or in their absence.[21] In general, ortho-substituted phe-nols are obtained, as in the Claisen rearrangement of allylic
ethers,[21] but blocking the o-positions with aliphatic groups(e.g., CH3) usually yields the p-substituted phenols, insteadof those resulting from C�C coupling of the R group of theether with one of the o-substituents. Accordingly, observa-tion of the rearrangement of the 2,6-dimethyl-substitutedanisoles shown in Scheme 4 prompted us to convert the che-lating aryloxide alkene ligands of 4 o and 5 o into the corre-sponding 2-ethyl-6-methylphenols (9 a and 9 b in Scheme 7).
This has been achieved by the two-step synthesis shown inScheme 7, which implies hydrogenation of 4 o and 5 o in thepresence of HSiEt3, to give the known IrV complex[(TpMe2)Ir(H)3ACHTUNGTRENNUNG(SiEt3)][22] plus the silyl ethers 8 a and 8 b,which are then readily hydrolyzed to the desired phenols.
To gain information about the C�H bond-activation stepsinvolved in the mechanistic path that leads to compounds4 o and 5 o, the corresponding reactions have been moni-tored by 1H NMR and 13C{1H} NMR spectroscopy, by em-ploying the 13C-enriched anisole ArO13CH3 (Ar=2,6-dime-thylphenyl, ca. 45 % enriched). The unsaturated complex 1was generated from [(TpMe2)Ir ACHTUNGTRENNUNG(C2H4)2] and C6H6 at 50 8C inthe presence of the anisole. Under these conditions, andafter 3 h, a mixture of two hydride carbene intermediates, Aand B was generated. The two compounds were separated
from one another by a combination of column and thin-layer chromatographic procedures (see the ExperimentalSection). Intermediate A was obtained in approximately90 % purity, whereas for B the purity was about 95 %. Inter-mediate A features 1H NMR spectroscopic signals at d=
�14.89 (Ir�H), 15.44 (Ir=CH), and 2.15 ppm (s, 6 H; 2 o-Me), among others, whereas for B the Ir�H and Ir=CH res-onances are found at d=�17.04 and 15.70 ppm, respectively(in the carbenic resonances of A and B the corresponding
Scheme 6. Thermal equilibration of hydride alkenes 4o–6 o with their hy-dride alkylidene tautomers, 4c–6c. Keq = 16 (R=H), 24 (R=Me), and 12(R= Br).
Scheme 7. Synthesis of silyl ethers 8 and phenols 9.
Chem. Eur. J. 2009, 15, 9046 – 9057 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 9049
FULL PAPERIridium Hydride Isomers

13C satellites are also observed). Most informative is the ob-servation of a singlet (relative intensity 3 H) at d= 2.31 ppmfor the unique ortho-methyl group of B, and of two doubletsat d= 4.17 and 2.48 ppm (2J ACHTUNGTRENNUNG(H,H)=13.1 Hz) associated withthe iridium-bound diastereotopic CH2 protons. In the13C{1H} NMR spectrum the label appears exclusively at theircarbene carbon atoms, which resonate at d=259.2 (A) and256.3 ppm (B) and exhibit 1J ACHTUNGTRENNUNG(C,H) couplings of 154 and159 Hz, respectively.
These data reveal unequivocally that the reaction startswith methyl C�H bond activation within the MeO� unit ofthe ether, likely facilitated by coordination of the oxygenatom,[9] to yield an unsaturated [(TpMe2)Ir ACHTUNGTRENNUNG(C6H5) ACHTUNGTRENNUNG(CH2OAr)]species that gives A by a-H elimination. This 1,2-H shift isreversible and eventually the above species undergoes asecond C�H bond activation, which now involves one of theortho-methyl groups, and then an a-H elimination to pro-duce B. By similarity with the related C�H bond activationsof anisole described in the preceding paper,[9] it is probablethat the C�H bond reactions take place through a s-com-plex-assisted metathesis (s-CAM) mechanism.[23] Furtherheating of the reaction mixture results in the disappearanceof the signals associated with the carbene intermediates Aand B : A cleanly transforms into B, which then producesthe final hydride olefin complex 4 o without detection of anyother intermediate. Interestingly, the 13C label distributesunequally between the two olefinic sites of 4 o, favoring theterminal alkene carbon by approximately 70 %, irrespectiveof the reaction time. For the case of 2,4,6-trimethylanisole,45 % 13C-enriched in OMe, this ratio was found to be 55 %in the resulting 5 o. A detailed theoretical study of this com-plex rearrangement is outside the scope of this work, but itis tempting to suggest that species C, resulting from a 1,2-Hshift from iridium to the alkylidene ligand of B, could equili-brate partially with a cyclohexadienone intermediate, D(Scheme 8), prior to experiencing an irreversible a-aryloxide
elimination, subsequent C�C coupling involving the result-ing methylene unit, and b-H elimination (see Scheme 3 inref. [11b]). It should be noted that species of type D are or-ganometallic cyclohexadienones. The free organic moleculesare intermediates or products of some important rearrange-ments of phenols, aromatic ethers, and aromatic esters, likethe photo-Fries rearrangement, the Birch reduction, theWessely oxidation, or the aromatic Claisen rearrange-ment.[16,21]
Interconversion of hydride alkene and hydride alkylidenetautomers—Computational study of the reaction mecha-nism : To understand the mechanism that allows interconver-sion of the hydride carbenes 2 c–6 c with their hydride olefinisomers, 2 o–6 o, both the metal-free and metal-mediated re-distribution pathways have been computed. Firstly, the car-bene to olefin interconversion mechanism for the free or-ganic anion [2-(C2H3)C6H4O]� was analyzed. Figure 2 shows
optimized geometries for the species involved and the corre-sponding energy profile. The reaction can be described as a1,2-H migration. In this mechanism an H atom moves fromthe carbene methyl substituent to the carbene carbon atom,giving rise to Alkene1 through transition state TSL, whichlies 23.7 kcal mol�1 above the free carbene. Alkene2, result-ing from C�C rotation in Alkene1, has practically identicalenergy.
Next, the same mechanism was calculated for the directH-shift in the iridium-bonded carbene formed by coordina-tion of the above anion to the [(TpMe2)Ir(H)] fragment (Fig-ures 3 and 4).
We have located a transition state, TS1, with an arrange-ment of the HC=CH2 fragment very similar to that foundfor the 1,2-migration in the free ligand (TSL). However theenergy barrier (49.2 kcal mol�1) is considerably higher thanthat for the metal-free interconversion, with the weakeningof the Ir=C bond associated with the H migration appearingas the main reason for the increase in the barrier. Thisbond, of length 1.941 � in the alkylidene structure, length-ens to 2.086 � in TS1. There is no metal assistance in this1,2-migration, the migrating proton being 2.187 � awayfrom the iridium center in TS1. All attempts to locate atransition state for the 1,2-H shift with the hydrogen closerto the metal ended up in the same transition state (TS1).
The energy profile for the 1,2-H-migration mechanism inthe [(TpMe2)Ir(H) ACHTUNGTRENNUNG{OC6H4 ACHTUNGTRENNUNG(C2H3)}] system is shown in
Scheme 8. A possible mechanism for the scrambling of the 13C labelacross the olefinic sites of complex 4 o.
Figure 2. Potential-energy profile of metal-free carbene–alkene intercon-version (energies in kcal mol�1).
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 9046 – 90579050
E. Carmona, A. Lled�s et al.
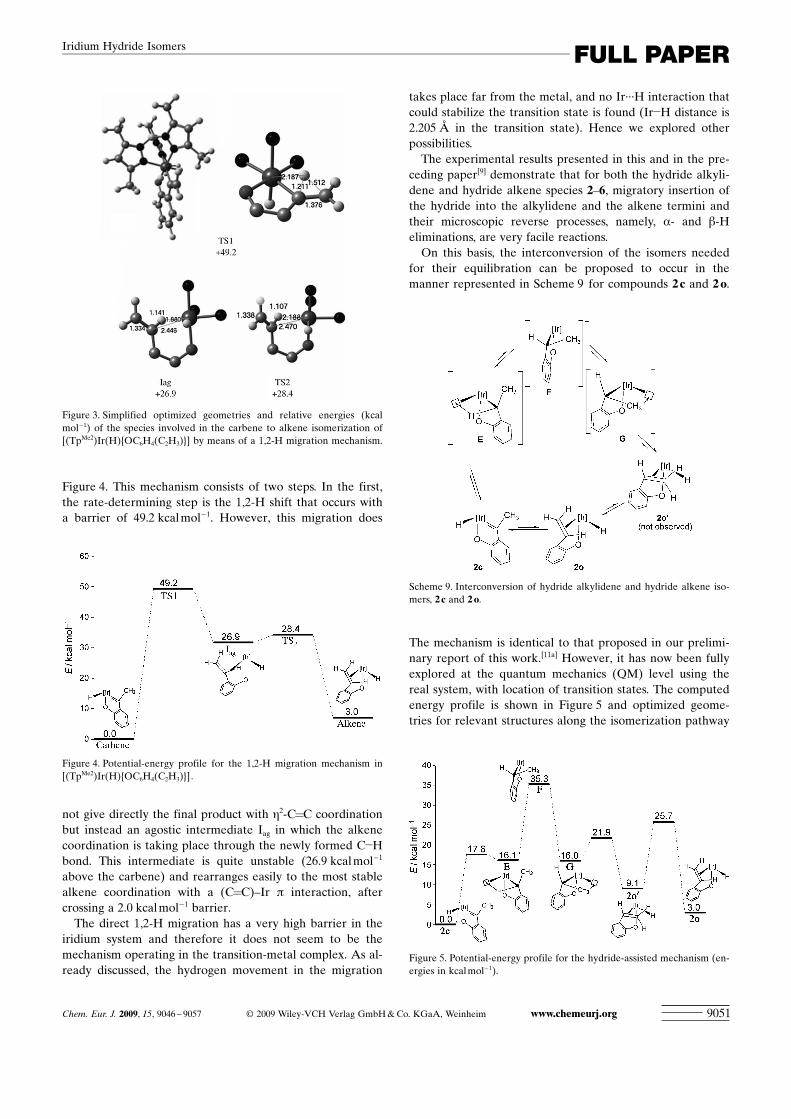
Figure 4. This mechanism consists of two steps. In the first,the rate-determining step is the 1,2-H shift that occurs witha barrier of 49.2 kcal mol�1. However, this migration does
not give directly the final product with h2-C=C coordinationbut instead an agostic intermediate Iag in which the alkenecoordination is taking place through the newly formed C�Hbond. This intermediate is quite unstable (26.9 kcal mol�1
above the carbene) and rearranges easily to the most stablealkene coordination with a (C=C)–Ir p interaction, aftercrossing a 2.0 kcal mol�1 barrier.
The direct 1,2-H migration has a very high barrier in theiridium system and therefore it does not seem to be themechanism operating in the transition-metal complex. As al-ready discussed, the hydrogen movement in the migration
takes place far from the metal, and no Ir···H interaction thatcould stabilize the transition state is found (Ir�H distance is2.205 � in the transition state). Hence we explored otherpossibilities.
The experimental results presented in this and in the pre-ceding paper[9] demonstrate that for both the hydride alkyli-dene and hydride alkene species 2–6, migratory insertion ofthe hydride into the alkylidene and the alkene termini andtheir microscopic reverse processes, namely, a- and b-Heliminations, are very facile reactions.
On this basis, the interconversion of the isomers neededfor their equilibration can be proposed to occur in themanner represented in Scheme 9 for compounds 2 c and 2 o.
The mechanism is identical to that proposed in our prelimi-nary report of this work.[11a] However, it has now been fullyexplored at the quantum mechanics (QM) level using thereal system, with location of transition states. The computedenergy profile is shown in Figure 5 and optimized geome-tries for relevant structures along the isomerization pathway
Figure 3. Simplified optimized geometries and relative energies (kcalmol�1) of the species involved in the carbene to alkene isomerization of[(TpMe2)Ir(H) ACHTUNGTRENNUNG{OC6H4 ACHTUNGTRENNUNG(C2H3)}] by means of a 1,2-H migration mechanism.
Figure 4. Potential-energy profile for the 1,2-H migration mechanism in[(TpMe2)Ir(H) ACHTUNGTRENNUNG{OC6H4 ACHTUNGTRENNUNG(C2H3)}].
Scheme 9. Interconversion of hydride alkylidene and hydride alkene iso-mers, 2 c and 2 o.
Figure 5. Potential-energy profile for the hydride-assisted mechanism (en-ergies in kcal mol�1).
Chem. Eur. J. 2009, 15, 9046 – 9057 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 9051
FULL PAPERIridium Hydride Isomers

are presented in Figure 6. The rate-determining step is ametal inversion through a five-coordinate transition-state F,with energy 35.3 kcal mol�1 higher than the hydride carbene
complex 2 c. This is considerably lower than the 49.2 kcalmol�1 barrier calculated for the direct 1,2-H-migrationmechanism considered above. Agostic interactions stabilizeboth E (a-agostic) and G (b-agostic) intermediates. A struc-ture related to intermediate G, without b-agostic interaction,can also be obtained as a minimum in the potential-energysurface, but it lies 5.5 kcal mol�1 above G and does not par-ticipate in this mechanism. Both E and G may be viewed as
five-coordinate species with a vacant position filled by anagostic interaction and geometry close to square pyramidal.Their interconversion, which actually permits equilibrationof carbene and olefin structures, also occurs through a five-coordinate transition-state, F, in which the iridium atom fea-tures a trigonal bipyramidal geometry. A putative mecha-nism implying carbon inversion (rather than metal inver-sion) at the iridium-bound carbon atom of intermediate Ehas also been computed. Nonetheless, no transition state hasbeen found and a preliminary exploration suggests a muchhigher energy barrier than for metal inversion.
Factors governing the relative stabilities of carbene andolefin isomers : A further point that remains to be addressedin connection with the carbene/olefin equilibria observedfor compounds 2–6 relates to the factors that determine thenearly thermoneutral character of the equilibria, in otherwords, the comparable thermodynamic stability of the car-bene and olefin isomers. As a first approximation to thisproblem, the anionic group [2-(C2H3)C6H4O]� has been calcu-lated in its carbene (H) andolefin (I) forms, with the resultthat I is about 58 kcal mol�1
more stable than H. This data iscomparable to reported valuesfor related pairs of isomers, forethylene is more stable thanmethyl carbene by approximately 80 kcal mol�1, and dimeth-yl carbene, ethyl methyl carbene, and cyclohexylidene areendothermic with respect to their olefinic isomers by ap-proximately 70 kcal mol�1.[5a, 24]
Computation shows that coordination of H and I to[(TpMe2)Ir(H)]+ yields compounds 2 c and 2 o, respectively,with an energy difference of approximately 3 kcal mol�1 infavor of 2 c. Thus, coordination to the metal center overridesthe large energy difference in preference of the free olefin.The optimized geometries of 2 c and 2 o are shown inFigure 7.
Insights into this rather intriguing result and the reasonsthat justify it can be gained from the energy-decompositionanalysis summarized in Scheme 10. DEb represents thebonding energy that results in the formation of 2 c and 2 ofrom [(TpMe2)Ir(H)]+ and the anionic ligand fragments Hand I, respectively, in their free isolated geometries (L� inScheme 10), and has been found to arise from the combina-tion of two terms. One is the distortion of [(TpMe2)Ir(H)]+
and L� from their free geometries to those they adopt in thecomplexes (DEdist) and the other is the interaction energythat stems from the combination of two fragments with op-posite charges to give the real complexes (DEinter). A similarapproach has been used by some of us to discern betweendifferent coordination modes of an ylidic ligand in its com-plex with PdCl2.
[25]
Table 1 summarizes the results of these calculations andcollects also the carbene-minus-olefin values for each of thecalculated magnitudes (DDE). As can be seen, DDEdist of
Figure 6. Optimized geometries and relative energies (kcal mol�1) ofsome species involved in the carbene to alkene isomerization of[(TpMe2)Ir(H) ACHTUNGTRENNUNG{OC6H4 ACHTUNGTRENNUNG(C2H3)}] by means of a hydride-assisted mechanism.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 9046 – 90579052
E. Carmona, A. Lled�s et al.

the ligand favors the carbene over the olefin for the ligandfragment (by 10.9 kcal mol�1) but distortion in the metalfragment favors the olefin by 3.5 kcal mol�1. In total, DDEdist
favors the carbene over the olefin for both fragments by atotal of 7.4 kcal mol�1. However, the main contribution tothe alkylidene stability comes from the interaction term.
DDEinter strongly encourages coordination of L� in the car-bene form by approximately �53.9 kcal mol�1. The additionof distortion and interaction contributions more than com-pensate for the higher stability of the free alkene I, in com-parison with alkylidene H (about 58 kcal mol�1), so that theequilibrium lies on the side of carbene 2 c, by a difference ofabout 3 kcal mol�1. Hence, it is the strength of the Ir�car-bene bond that becomes the main contributor to the stabili-zation of the alkylidene structures of 2–6 over their corre-sponding olefinic isomers.
Conclusion
Equilibria between iridium hydride carbene and hydrideolefin structures may be established from the reactions ofthe unsaturated fragment [(TpMe2)Ir ACHTUNGTRENNUNG(C6H5)2] (1) with eitherthe 2-alkyl-substituted phenols 2-R-C6H4OH (R=C2H5,nC3H7), or the substituted anisoles, 2,6-dimethylanisole,2,4,6-trimethylanisole, and 4-bromo-2,6-dimethylanisole. Thekinetic products of the former reactions are the hydride car-bene complexes 2 c and 3 c, the formation of which requirescleavage of the O�H bond of the phenol groups and activa-tion of two aliphatic C�H bonds. In contrast, the kineticproducts of the reactions involving the substituted anisolesare the hydride olefin compounds 4 o–6 o, which are generat-ed through a complex reaction pathway that involves cleav-age of the O�Me bond of the ether as well as multiple C�Hbond activation and formation of a C�C bond.
A computational study of these equilibria supports thetheory that isomer interconversion occurs by means of car-bene and olefin migratory insertions into the Ir�H bond ofthe corresponding complexes and their respective micro-scopic reverse processes, namely, a- and b-H eliminations.However, due to the chelating nature of the aryloxidealkene and aryloxide alkylidene ligands of these compounds,metal inversion is also needed. In all cases, thermodynamicsfavor the hydride carbene tautomers 2 c–6 c. Whilst, as ex-pected, the alkene structure becomes comparatively destabi-lized by steric hindrance, the main reason behind the obser-vation of these equilibria is the stabilization of the carbenestructure due to the strong TpMe2Ir–alkylidene electronic in-teraction in these complexes.
Experimental Section
General procedures : Microanalyses were made by the MicroanalyticalService of the Instituto de Investigaciones Qu�micas (Sevilla). Infraredspectra were obtained using Perkin–Elmer spectrometers, models 577and 684. The NMR spectroscopic instruments consisted of Bruker DRX-500, DRX-400, and DPX-300 spectrometers. Spectra were referenced toexternal SiMe4 (d =0 ppm) using the residual protio solvent peaks as in-ternal standards (1H NMR spectroscopy experiments) or the characteris-tic resonances of the solvent nuclei (13C NMR spectroscopy experiments).Spectral assignments were made by means of routine one- and two-di-mensional NMR spectroscopy experiments where appropriate. Manipula-tions were performed under argon or oxygen-free dinitrogen, followingconventional Schlenk techniques.
Figure 7. Optimized geometries and relative energies (kcal mol�1) of thehydride alkylidene (2c) and hydride alkene (2o) isomers of[(TpMe2)Ir(H) ACHTUNGTRENNUNG{OC6H4 ACHTUNGTRENNUNG(C2H3)}].
Scheme 10. Energy-decomposition scheme for the formation of com-pounds 2c and 2o (see text). L� represents structures H and I.
Table 1. Energy-decomposition analysis for the formation of compounds2c and 2o (see text). L� represents structures H and I (see Scheme 10for the definition of terms). Energies are given in kcal mol�1.
DEcarbene DEolefin DDE[a]
DEdist [(TpMe2)Ir(H)]+ 15.9 12.4 3.5DEdist (L�) 12.2 23.1 �10.9DEinter �270.2 �216.4 �53.9DEb �242.1 �180.8 �61.3
[a] DDE =DEcarbene�DEolefin.
Chem. Eur. J. 2009, 15, 9046 – 9057 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 9053
FULL PAPERIridium Hydride Isomers

Complexes A and B : The IrI complex [(TpMe2)IrACHTUNGTRENNUNG(C2H4)2] (0.30 g,0.55 mmol) dissolved in C6H6 (3 mL) was reacted with a slight excess of2,6-dimethylanisole (0.090 mL, 0.66 mmol) at 50 8C for 3 h. After removalof the solvent in vacuo, 1H NMR spectroscopy revealed formation of A(ca. 26%) and B (ca. 5 %) together with 4 o (ca. 5%). Two-column chro-matographic separations, the first with Et2O and the second with pentaneas eluents, followed by another two thin-layer chromatography proce-dures, both employing hexane as eluent, allowed separation of the de-sired compounds (A : 30 mg, ca. 90% purity; B : 25 mg, ca. 96 % purity),each made slightly impure by the other. During the chromatography ex-periments partial conversion of A into B took place. Studies employingthe 13C-enriched (ca. 45 %) anisoles ArO13CH3 revealed that the label ap-pears exclusively at the carbene carbon of A and B (1J ACHTUNGTRENNUNG(C,H) values of154 (3 a) and 159 Hz (B) for their Ir=CH� functionalities).
Data for A : 1H NMR (CDCl3, �10 8C): d=15.44 (s, 1 H; Ir=CH); 7.83,6.99, 6.87, 6.67, 6.50 (d, t, t, t, d, 3J ACHTUNGTRENNUNG(H,H)�7.5 Hz, 1 H each; o-, m-, p-,m’-, o’-CHACHTUNGTRENNUNG(Ir-Ph)); 7.02 (s, 3H; CH ACHTUNGTRENNUNG(O-Ar)); 5.80, 5.76, 5.68 (s, 1 H each;3CHpz); 2.48, 2.40, 2.38, 2.19, 1.57, 1.46 (s, 3 H each; 6Mepz); 2.15 (s, 6 H;2Me ACHTUNGTRENNUNG(O-Ar)); �14.89 ppm (s, 1H; IrH); 13C{1H} NMR (CDCl3, �10 8C):d=259.2 (Ir=CH); 158.9 (C1); 151.7, 150.1, 148.6, 143.5, 143.4, 143.1(Cqpz); 142.6, 139.8, 126.5, 126.0, 120.8 (o-, o’-, m-, m’-, p-CH ACHTUNGTRENNUNG(Ir-Ph));136.2 (Cq ACHTUNGTRENNUNG(Ir-Ph)); 129.3, 125.8 (2:1; CH ACHTUNGTRENNUNG(O-Ar)); 106.2, 106.0, 105.4(CHpz); 16.8 (Mear); 15.8, 14.9, 14.5, 13.1, 12.9, 12.9 ppm (Mepz); IR(Nujol): n =2140 cm�1 (Ir-H).
Data for B : 1H NMR (CDCl3, �10 8C): d =15.70 (s, 1H; Ir=CH); 7.15,7.03 (d, m, 1:2, 3J ACHTUNGTRENNUNG(H,H) =7.00 Hz; 3 CHar); 5.83, 5.83, 5.73 (s, 1H each;3CHpz); 4.17, 2.48 (d, 2J ACHTUNGTRENNUNG(H,H) =13.1 Hz, 1H each; IrCH2); 2.51, 2.43,2.38, 2.36, 2.20, 1.93 (s, 3 H each; 6 Mepz); 2.31 (s, 3H; Ar-Me);�17.04 ppm (s, 1H; IrH); 13C{1H} NMR (CDCl3, �10 8C): d=256.3(1J ACHTUNGTRENNUNG(C,H)=159 Hz; Ir=CH); 157.5 (Cq-O); 152.4, 150.0, 148.8, 144.6, 143.9,143.3 (Cqpz); 140.9, 126.8 (Cq-Me); 126.4, 124.6, 124.6 (CHar); 106.9, 106.6,105.4 (CHpz); 16.9, 15.0, 15.0, 14.1, 12.9, 12.8, 12.8 (Mepz+Ar-Me);�13.0 ppm (1J ACHTUNGTRENNUNG(C,H)=127 Hz; Ir-CH2); IR (Nujol): n =2160 cm�1 (Ir-H).
Complex 3c : Compound [(TpMe2)Ir ACHTUNGTRENNUNG(C6H5)2(N2)] (0.30 g, 0.447 mmol) wasdissolved in C6H6 (5 mL) and 2-propylphenol (93 mL, 0.66 mmol) wasadded. The solution was stirred at 60 8C for 14 h, after which time the sol-vent was evaporated under vacuum. NMR spectroscopic monitoring ofthe crude product revealed the formation of 3 in almost quantitativeyield (>95 %). Compound 3c was purified by column chromatographyon silica gel, using a 20:1 mixture of hexane/Et2O as eluent. 1H NMR(CDCl3, 25 8C): d=7.48, 7.24, 7.16, 6.53 (br d, 3J ACHTUNGTRENNUNG(H,H)�7.5 Hz, 1H each;CHar); 5.85, 5.83, 5.55 (s, 1H each; 3CHpz); 3.49 (q, 3J ACHTUNGTRENNUNG(H,H)�7 Hz, 2 H;CH2Me); 2.53, 2.44, 2.37, 2.35, 2.04, 1.34 (s, 3 H each; 6Mepz); 0.94 (t,3J ACHTUNGTRENNUNG(H,H)�7 Hz, 3H; CH3alif), �20.47 ppm (s, 1H; IrH); 13C{1H} NMR(CDCl3, 25 8C): d =278.9 (Ir=C); 190.2 (Carq-O); 152.4, 152.3, 151.7, 150.7,144.4, 143.9, 143.1 (Carq+6Cpzq); 137.0, 122.9, 117.6, 113.6 (4 C-Har); 106.5,106.1, 106.1 (3 CHpz); 40.1 (-CH2-C= ); 16.8, 15.3, 13.1, 12.9, 12.5, 12.3,12.0 ppm (7 Me); IR (Nujol): n =2180 cm�1 (Ir-H); HMRS (FAB): m/z :calcd for [M]+ : 624.2360; found: 624.2344; elemental analysis calcd (%)for C18H20BIrN6O: C 46.2, H 5.2, N 13.5; found: C 46.2, H 5.2, N 13.5.
Complex 3 o : A solution of 3 c in C6H12 (0.030 g, 0.045 mmol; 1 mL) washeated at 100 8C for 48 h. After this time, the volatiles were removed invacuo and from the resulting 3c/3 o mixture (ca. 1:1), and 3o was separat-ed by column chromatography on silica gel using a 20:1 mixture ofhexane/Et2O as eluent. 1H NMR (C6D12, 25 8C): d=6.93, 6.61, 6.26, 6.23(d, t, t, d, 3J ACHTUNGTRENNUNG(H,H)�7.3 Hz, 1H each, CHar); 6.23 (d, 3J ACHTUNGTRENNUNG(H,H)�10.6 Hz,1H; HA, CHar); 5.76, 5.61, 5.53 (s, 1H each; 3CHpz); 4.73 (m, 1H; HB);2.42, 2.34, 2.33, 2.25, 2.18, 1.30 (s, 3H each; 6 Mepz); 0.95 (d, 3J ACHTUNGTRENNUNG(H,H)�6.4 Hz, 3 H; CH3olef), �17.65 ppm (s, 1 H; IrH); 13C{1H} NMR (C6D12,25 8C): d=178.4 (Car-O); 158.4, 156.6, 156.4, 149.4, 148.6, 147.8 (6 Cpzq);138.2 (Cqar); 132.7, 130.0, 120.4, 119.4 (4 C-Har); 113.6, 112.3, 111.6(3 CHpz); 83.0 (CHA); 67.9 (CHB); 21.9 (CH3olef) ; 20.7, 18.4, 18.4, 17.9,17.6, 17.4 ppm (6 Mepz); HMRS (FAB): m/z : calcd for [M]+ : 624.2360;found: 624.2412.
Complex 4 c : A solution of 4 o in C6H12 (0.027 g, 0.043 mmol; 6 mL) washeated at 90 8C for 52 h. After this time, the volatiles were removed invacuo and the resulting 4o/4c mixture (ca. 6:94) was subjected to chro-matography on silica gel (15:1!6:1 hexane/Et2O) to obtain compound
4c in 82 % yield (0.022 g). 1H NMR (CDCl3, 25 8C): d= 7.36, 7.10, 6.49(d, d, dd, 3J ACHTUNGTRENNUNG(H,H)�7.5 Hz, 1 H each; 3 CHar), 5.90, 5.87, 5.58 (s, 1H each;3CHpz); 2.90 (s, 3 H; Ir=CCH3); 2.62, 2.44, 2.38, 2.36, 2.35, 2.08, 1.25 (s,3H each; 6 Mepz+Cq-Me); �20.53 ppm (s, 1H; IrH); 13C{1H} NMR(CDCl3, 25 8C): d=272.8 (Ir=C); 189.1, 151.4, 127.0 (Cq-O, Cq-C =H andCq-Me, resp.); 153.0, 152.0, 152.4, 144.6, 144.3, 143.4 (Cqpz); 136.7, 120.8,114.2 (CHar); 106.9, 106.7, 106.7 (CHpz); 35.1 (1J ACHTUNGTRENNUNG(C,H) =126 Hz; Ir=CCH3); 16.9, 16.3, 14.5, 13.4, 12.6, 12.5, 11.8 ppm (Mepz+C3-Me); IR(Nujol): n =2130 cm�1 (Ir-H); elemental analysis calcd (%) forC24H32BN6OIr: C 46.2, H 5.1, N 13.5; found: C 46.3, H 5.0, N 12.9.
Complex 4 o : Compound [(TpMe2)Ir ACHTUNGTRENNUNG(C2H4)2] (0.30 g, 0.55 mmol) was dis-solved in C6H6 (15 mL) and 2,6-dimethylanisole (0.4 mL, 2.75 mmol) wasadded. The solution was stirred at 60 8C for 12 h and after this time, thesolvent was evaporated under vacuum. NMR spectroscopic monitoring ofthe crude product revealed the formation of 4o in almost quantitativeyield (>90%). Compound 4o was purified by column chromatographyon silica gel, using a 5:1 mixture of hexane/CH2Cl2 as eluent. Yield:49%. It can be crystallized from pentane/Et2O mixtures (2:1) at �20 8C.The use of the corresponding anisole ArO13CH3, with approximately45% 13C-enrichment demonstrated that the 13C label was scrambled be-tween the two olefinic sites. The distribution favors the terminal alkenecarbon by approximately 70 %. 1H NMR (CDCl3, 25 8C): d=7.11, 6.76,6.45 (d, d, t, 3J ACHTUNGTRENNUNG(H,H)�7.5 Hz, 1 H each; 3 CHar); 6.89 (dd, 3Jtrans =11.5 Hz,3Jcis =8.2 Hz, 1H; HC(Ar)= ); 5.95, 5.90, 5.68 (s, 1H each; 3CHpz); 4.26,3.28 (d, 1 H each; H2C= ); 2.08 (s, 3H; Cq-Me); 2.49, 2.43, 2.43, 2.40,2.30, 2.00 (s, 3H each; 6Mepz); �17.47 ppm (s, 1H; IrH); 13C{1H} NMR(CDCl3, 25 8C): d=171.7 (Cq-O); 152.6, 151.0, 150.9, 144.1, 144.0 (Cqpz);130.3 (Cq-Colef.) ; 128.4, 123.6, 114.0 (CHar); 125.2 (C3); 108.2, 107.3, 106.8(CHpz); 76.7 (1J ACHTUNGTRENNUNG(C,H)= 164 Hz; =CH(Ar)); 46.2 (1J ACHTUNGTRENNUNG(C,H)=161 Hz; CH2);16.3 (Cq-Me); 14.2, 13.7, 13.4, 12.9, 12.5 ppm (1:1:1:1:2, Mepz); IR (Nujol):n= 2180 cm�1 (Ir-H); elemental analysis calcd (%) for C24H32BIrN6O: C46.2, H 5.1, N 13.5; found: C 45.8, H 5.1, N 13.1.
Complex 5 c : Compound [(TpMe2)Ir ACHTUNGTRENNUNG(C2H4)2] (0.30 g, 0.55 mmol) was dis-solved in C6H6 (10 mL) and 2,4,6-trimethylanisole (0.17 mL, 1.1 mmol)was added. The solution was stirred at 100 8C for 20 h, after which timethe solvent was evaporated under vacuum. NMR spectroscopic monitor-ing of the crude product revealed the formation of 5c in approximately58% yield. Compound 5c was purified by column chromatography onsilica gel using a 20:1 mixture of hexane/Et2O as eluent. Yield: 33 %.1H NMR (CDCl3, 25 8C): d= 7.12, 7.02 (s, 1 H each; 2 CHar); 5.90, 5.87,5.59 (s, 1H each; 3 CHpz); 2.91 (s, 3H; Ir=CMe); 2.62, 2.45, 2.39, 2.38,2.09, 1.28 (s, 3H each; 6Mepz); 2.35 (s, 3H; C3-Me); 2.23 (s, 3 H; C4-Me);�20.59 ppm (s, 1H; IrH); 13C{1H} NMR (CDCl3, 25 8C): d= 270.1 (Ir=C);187.7 (Cq-O); 152.7, 152.0, 151.7, 144.3, 144.1, 143.1 (Cqpz); 150.8 (Cq-Cq =
Ir); 138.4, 119.4 (CHar); 126.1, 122.8 (Cq-Me); 106.6, 106.4, 106.4 (CHpz);34.5 (1J ACHTUNGTRENNUNG(C,H)= 126 Hz; Ir=CMe); 20.5, 15.9 (Cq-Me); 16.6, 14.2, 13.1,12.3, 12.2, 11.5 ppm (Mepz); IR (Nujol): n =2145 cm�1 (Ir-H); HMRS(FAB): m/z : calcd for [M]+ : 638.2517; found: 638.2512; elemental analy-sis calcd (%) for C25H34BIrN6O·CH2Cl2: C 43.2, H 5.0, N 11.6; found: C42.3, H 5.3, N 11.2.
Complex 5 o : Compound [(TpMe2)Ir ACHTUNGTRENNUNG(C2H4)2] (0.20 g, 0.37 mmol) was dis-solved in C6H6 (8 mL) and 2,4,6-trimethylanisole (0.28 mL, 1.83 mmol)was added. The solution was stirred at 60 8C for 12 h. After this time, thesolvent was distilled off under reduced pressure and NMR spectroscopicmonitoring of the crude product revealed the formation of 5o in almostquantitative yield (>90%). Compound 5o was purified by column chro-matography on silica gel using a (20:1!5:1) mixture of hexane/Et2O aseluent. Yield: 65%. It can be crystallized from pentane/Et2O mixtures(2:1) at 25 8C. The use of the corresponding anisole ArO13CH3 with ap-proximately 45% 13C-enrichment demonstrated that the 13C label wasscrambled between the two olefinic sites of compounds 5o. The distribu-tion favors the terminal alkene carbon by approximately 55 %. 1H NMR(CDCl3, 25 8C): d=6.89, 6.58 (s, s, 1H each; 2CHar); 6.83 (m, 1H; CH-ACHTUNGTRENNUNG(Car)= ); 5.91, 5.86, 5.64 (s, 1H each; 3 CHpz); 4.23, 3.24 (d, 3Jtrans =
11.1 Hz, 3Jcis =8.2 Hz, 1H each; CH2 = ); 2.45, 2.40, 2.39, 2.36, 2.29, 1.96(s, 3 H each; 6Mepz); 2.25 (s, 3H; C4-Me); 2.02 (s, 3 H; C3-Me);�17.44 ppm (s, 1 H; IrH); 13C{1H} NMR (CDCl3, 25 8C): d=169.3 (Cq-O);152.6, 151.0, 150.8, 144.0, 143.9, 143.6 (Cqpz); 130.1 (Cq-Colef) ; 129.3, 123.8
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 9046 – 90579054
E. Carmona, A. Lled�s et al.

(CHar); 124.6, 122.8 (Cq-Me); 108.2, 107.2, 106.8 (CHpz); 76.6 (1J ACHTUNGTRENNUNG(C,H)=
170 Hz; CH ACHTUNGTRENNUNG(Car)= ); 46.1 (1J ACHTUNGTRENNUNG(C,H)=161 Hz; CH2 = ); 20.3, 16.2 (Cq-Me);14.2, 13.7, 13.4, 12.8, 12.5 ppm (1:1:1:1:2; Mepz); IR (Nujol): n= 2189 cm�1
(Ir-H); elemental analysis calcd (%) for C25H34BIrN6O: C 47.1, H 5.3, N13.2; found: C 46.6, H 5.2, N 12.9.
Crystal data of 5o : C25H34BirN6O; Mr =637.59; monoclinic; space groupP21/c ; a=13.28550(5), b=12.69550(5), c =15.04990(5) �; a= 90, b=
95.7984(10), g =908 ; V=2525.420(16) �3; Z=4; T =120(2) K; 1calcd =
1.677 gcm�3 ; m=10.462 mm�1; R1 =0.0344 and wR2 =0.998 (all 4088unique reflections). A crystal of 5o was covered with perfluoropolyetheroil (FOMBLIN, Aldrich) and mounted in a fiber loop. X-ray data (T=
120(2) K) were obtained using a Bruker–Nonius KappaCCD2000 single-crystal diffractometer equipped with a normal-focus sealed-tube X-raysource with graded mirror monochromated CuKa radiation (l=1.5418 �),operating at 45 kV and 70 mA. The crystal-to-detector distance was45 mm. Data collection and data reduction were carried out using HKLDenzo and Scalepack.[26] Unit-cell parameters were determined from aphi scan of 108 using the autoindexing option in Denzo. The structurewas solved by Patterson and difference Fourier synthesis and refined byfull-matrix least-squares procedures, with anisotropic thermal parametersin the last cycles of refinement for all non-hydrogen atoms. The hydrogenatoms were introduced into the geometrically calculated positions and re-fined riding on the corresponding parent atoms, except H1, H2, H22,H23A, and H23B; their positions were determined by difference Fouriersynthesis and refined, together with their isotropic thermal parameter, byleast-squares procedures. Weighted R factors (wR) and all goodness-of-fit(S) are based on F2, conventional R factors (R) are based on F. TheSHELXTL software package[27] (based on SHELXS and SHELXL) wasused for structure solution and refinement. A summary of the fundamen-tal crystal and refinement data, atomic coordinates, anisotropic displace-ment parameters, and bond lengths and angles are given in the Support-ing Information.
Complex 6 c : Compound [(TpMe2)Ir ACHTUNGTRENNUNG(C2H4)2] (0.30 g, 0.55 mmol) was dis-solved in C6H6 (10 mL) and 4-bromo-2,6-dimethylanisole (0.23 mL,1.1 mmol) was added. The solution was stirred at 100 8C for 20 h, andafter this time the solvent was evaporated under vacuum. NMR spectro-scopic monitoring of the crude product revealed the formation of 6 c inapproximately 69% yield. Compound 6c was purified by column chro-matography on silica gel using a 20:1 mixture of hexane/Et2O as eluent.Yield: 30 %. 1H NMR (CDCl3, 25 8C): d=7.50, 6.73 (s, s, 1 H each;2CHar); 5.91, 5.87, 5.60 (s, 1 H each; 3CHpz); 2.84 (s, 3 H; Ir=CMe); 2.59,2.44, 2.38, 2.36, 2.06, 1.23 (s, 3H each; 6Mepz); 2.33 (s, 3 H; C3-Me);�20.27 ppm (s, 1H; IrH); 13C{1H} NMR (CDCl3, 25 8C): d= 272.5 (Ir=C);187.3 (Cq-O); 152.7, 151.9, 151.6, 144.5, 144.4, 144.4 (Cqpz); 152.2 (Cq-C=
Ir); 137.9, 122.5 (CHar); 128.0, 106.8 (Cq-Me); 106.7, 106.6, 106.5 (CHpz);35.4 (1J ACHTUNGTRENNUNG(C,H) =126 Hz; Ir=CMe); 16.7, 14.3, 13.2, 12.4, 12.4, 11.7 (Mepz);16.1 ppm (Cq-Me); IR (Nujol): n=2140 cm�1 (Ir-H); HMRS (FAB): m/z :calcd for [M]+ : 702.1465; found: 702.1478; elemental analysis calcd (%)for C24H31BIrN6OBr·CH2Cl2: C 38.1, H 4.2, N 10.7; found: C 38.9, H 4.2,N 11.5.
Complex 6 o : Compound [(TpMe2)Ir ACHTUNGTRENNUNG(C2H4)2] (0.20 g, 0.37 mmol) was dis-solved in C6H6 (8 mL) and 4-bromo-2,6-dimethylanisole (0.39 mL,1.83 mmol) was added. The solution was stirred at 60 8C for 12 h, andafter this time the solvent was evaporated under vacuum. NMR spectro-scopic monitoring of the crude product revealed the formation of 6o inalmost quantitative yield (>90%). Compound 6o was purified bycolumn chromatography on silica gel using a 20:1!5:1 mixture ofhexane/Et2O as eluent. Yield: 47 %. 1H NMR (CDCl3, 25 8C): d =7.17,6.85 (s, 1 H each; 2CHar); 6.77 (m, 1H; HC(Ar)= ); 5.93, 5.88, 5.66 (s,1H each; 3CHpz); 4.21, 3.24 (d, 3Jtrans =11.1 Hz, 3Jcis =8.3 Hz, 1H each;CH2 = ); 2.47, 2.40, 2.38, 2.38, 2.25, 1.96 (s, 3 H each; 6 Mepz), 2.00 (s, 3H;C3-Me), �17.39 ppm (s, 1H; IrH); 13C{1H} NMR (CDCl3, 25 8C): d=
170.9 (C1); 152.5, 150.9, 150.8, 144.2, 144.1, 143.8 (Cqpz); 132.3 (C2); 130.5,126.0 (CHar); 126.8 (C3); 108.3, 107.3, 106.8 (CHpz); 104.8 (C4); 74.8(1J ACHTUNGTRENNUNG(C,H)=166 Hz, CHACHTUNGTRENNUNG(Car)); 46.3 (1J ACHTUNGTRENNUNG(C,H) =161 Hz; CH2 = ); 16.1 (Cq-Me); 14.2, 13.6, 13.3, 12.9, 12.5, 12.4 ppm (Mepz); IR (Nujol): n=
2185 cm�1 (Ir-H); elemental analysis calcd (%) for C24H31BIrN6OBr: C
41.0, H 4.6, N 12.0; found: C 42.1, H 4.6, N 12.0; HMRS (FAB): m/z :calcd for [M +Na]+ : 726.1441; found: 726.1458.
Complex 7: A solution of 4 o (0.040 g, 0.064 mmol) in NCMe (3 mL) wasstirred at 60 8C for 14 h, and after this period of time the solvent was dis-tilled off under reduced pressure. At this stage, 1H NMR spectroscopicmonitoring demonstrated quantitative formation of compound 7, whichwas isolated as a pure crystalline solid (white needles) by crystallizationfrom pentane/CH2Cl2 (3:1) at �20 8C (0.025 g, isolated yield 60 %). 1HMNR (CDCl3, 25 8C): d= 6.95, 6.85, 6.35 (d, d, t, 3J ACHTUNGTRENNUNG(H,H)�7.5 Hz, 1 Heach; 3 CHar); 5.84, 5.82, 5.67 (s, 1H each; 3 CHpz); 3.23, 2.33 (td, m,2J ACHTUNGTRENNUNG(H,H) =15.4, 3J ACHTUNGTRENNUNG(H,H) = 15.4, 4.4 Hz, 1H each; Ir-CH2CH2); 3.03, 2.73(ddd, ddd, 2J ACHTUNGTRENNUNG(H,H) =11.5 Hz; 3J ACHTUNGTRENNUNG(H,H) =7.7, 4.4 and 15.4, 3.8 Hz, respec-tively; 1H each; Ir-CH2CH2); 2.50, 2.45, 2.42, 2.39, 2.36, 2.13 (s, 3H each;6Mepz); 2.39 (s, 3H; NCCH3); 2.14 ppm (s, 3H; Cq-Me); 13C{1H} NMR(CDCl3, 25 8C): d= 162.9, 128.1, 127.3 (Cq-O, Cq-CH2, and Cq-Me, respec-tively); 151.5, 150.7, 150.2, 143.9, 143.8, 143.4 (Cqpz); 129.6, 127.2, 112.2(CHar); 114.8 (NCCH3); 108.7, 107.8, 106.6 (CHpz); 32.7 (1J ACHTUNGTRENNUNG(C,H)=
123 Hz; Ir-CH2CH2,); 18.8 (Cq-Me); 14.3, 14.1, 13.3, 13.1, 13.0, 12.6(Mepz); 4.2 (1J ACHTUNGTRENNUNG(C,H)=137 Hz; NCCH3); �12.1 ppm (1J ACHTUNGTRENNUNG(C,H) =127 Hz; Ir-CH2CH2); IR (Nujol): n =2289 cm�1 (NC); elemental analysis calcd (%)for C26H35BN7OIr·0.75CH2Cl2: C 44.1, H 5.0, N 13.5; found: C 44.2, H5.0, N 13.5.
Crystal data of 7: C26H35BIrN7O; Mr =664.62; triclinic; space group P1(no. 2); a= 13.290(4), b=13.831(4), c= 18.529(5) �; a =80.716(5), b=
75.807(4), g =79.202(5)8 ; V= 3219.5(15) �3; Z=4; T= 297(2) K; 1calcd =
1.371 gcm�3 ; m=4.17 mm�1; 31148 reflections measured; R1= 0.074 andwR2=0.192 (all 8264 unique reflections); Bruker AXS Smart CCDsystem with MoKa radiation (l =0.71073 �). The structure was solved bydirect methods and refined on F 2 with the SHELXL97 program. Variousrestraints were applied to compensate for the poorly scattering crystal,the solvent content of which was taken into account by the SQUEEZEprocedure of the PLATON program.
Complex 8 a : Compound 4 o (0.1 g, 0.16 mmol) was dissolved in C6H12
(6 mL) and triethylsilane (0.8 mL, 0.16 mmol) was added. The solutionwas placed in a Fischer–Porter vessel, heated, and stirred under 4 atm ofH2 (115 8C for 21 h). After this period of time, the solution turned yellowand the volatiles were removed under vacuum. The compound was puri-fied by column chromatography on silica gel using pentane as eluent.Yield: 60%. The use of the 13C-enriched compound 4o leads to the 13C-enriched silyl ether. The distribution of the 13C label is identical to 4o.1H NMR (CDCl3, 25 8C): d=7.00 (m, 2H; 2 CHar); 6.85 (t, 3J ACHTUNGTRENNUNG(H,H) =
7.5 Hz, 1H); 2.63 (q, 3J ACHTUNGTRENNUNG(H,H) =7.5 Hz, 2H; CqCH2CH3); 2.25 (s, 3 H;CqMe); 1.22 (t, 3J ACHTUNGTRENNUNG(H,H) =7.5 Hz, 3 H; C2CH2CH3); 1.00 (t, 3J ACHTUNGTRENNUNG(H,H) =
8.0 Hz, 9H; Si ACHTUNGTRENNUNG(CH2CH3)3); 0.79 ppm (q, 3J ACHTUNGTRENNUNG(H,H) =8.0 Hz, 6H; Si-ACHTUNGTRENNUNG(CH2CH3)3); 13C{1H} NMR (CDCl3, 25 8C): d= 152.4 (Cq-O); 134.3 (Cq-Et); 128.5, 126.5, 121.3 (CHar); 128.6 (Cq-Me); 23.8 (1J ACHTUNGTRENNUNG(C,H)=126 Hz;C2CH2CH3); 17.8 (1J ACHTUNGTRENNUNG(C,H)=126 Hz; C3Me); 14.3 (1J ACHTUNGTRENNUNG(C,H)=126 Hz;C2CH2CH3); 6.9 (1J ACHTUNGTRENNUNG(C,H)= 125 Hz; SiACHTUNGTRENNUNG(CH2CH3)3); 5.8 ppm (1J ACHTUNGTRENNUNG(C,H)=
117 Hz; Si ACHTUNGTRENNUNG(CH2CH3)3); HMRS (FAB): m/z : calcd for [M]+ : 250.175294;found: 250.175488.
Complex 8 b : Compound 5o (0.20 g, 0.31 mmol) was dissolved in C6H12
(10 mL) and triethylsilane (0.16 mL, 0.32 mmol) was added. The solutionwas placed in a Fischer–Porter vessel, heated, and stirred under 4 atm ofH2 (115 8C for 21 h). After this period of time, the solution turned yellowand the volatiles were removed under vacuum. The compound was puri-fied by column chromatography on silica gel using pentane as eluent.Yield: 60%. The use of the 13C-enriched compound 5o leads to the 13C-enriched silyl ether. The distribution of the 13C label is identical to 5o.1H NMR (CDCl3, 25 8C): d= 6.76, 6.74 (s, 1 H each; 2 CHar); 2.54 (q, 3J-ACHTUNGTRENNUNG(H,H) =7.5 Hz, 2H; C2CH2CH3); 2.21, 2.17 (s, 3H each; CqMe); 1.16 (t,3J ACHTUNGTRENNUNG(H,H) =7.5 Hz, 3H; C2CH2CH3); 0.96 (t, 3J ACHTUNGTRENNUNG(H,H) =8.0 Hz, 9H; Si-ACHTUNGTRENNUNG(CH2CH3)3); 0.73 ppm (q, 3J ACHTUNGTRENNUNG(H,H) = 8.0 Hz, 6H; SiACHTUNGTRENNUNG(CH2CH3)3);13C{1H} NMR (CDCl3, 25 8C): d =150.0 (Cq-O); 133.9 (Cq-Et); 129.1,127.1 (CHar); 130.3, 127.9 (Cq-Me); 23.7 (C2CH2CH3); 20.6, 17.7(1J ACHTUNGTRENNUNG(C,H)=126 Hz; CqMe); 14.3 (1J ACHTUNGTRENNUNG(C,H) =126 Hz; C2CH2CH3); 6.8(1J ACHTUNGTRENNUNG(C,H)=126 Hz; Si ACHTUNGTRENNUNG(CH2CH3)3); 5.8 ppm (1J ACHTUNGTRENNUNG(C,H) =118 Hz; Si-ACHTUNGTRENNUNG(CH2CH3)3); HMRS (FAB): m/z : calcd for [M]+ : 264.190944; found:264.190159.
Chem. Eur. J. 2009, 15, 9046 – 9057 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 9055
FULL PAPERIridium Hydride Isomers

Complex 9 a : Compound 8a (0.06 g, 0.24 mmol) was dissolved in THF(3 mL) and tetrabutylammonium fluoride (1 m in THF; 0.26 mL,0.260 mmol) was added. The solution was stirred for 2 h at room temper-ature. After this period of time, a saturated solution of NH4Cl (3 mL)was added and compound 9a was extracted with ethyl acetate (3 3 mL).The combined organic solvents were dried over Na2SO4 and evaporated.1H NMR (CDCl3, 25 8C): d=6.95, 6.94, 6.74 (d, d, t, 3J ACHTUNGTRENNUNG(H,H) = 7.5 Hz, 1 Heach; 3 CHar); 5.00 (s, 1 H; -OH); 2.62 (q, 3J ACHTUNGTRENNUNG(H,H) =7.5 Hz, 2H;-CH2CH3); 2.24 (s, 3H; Mear); 1.21 ppm (t, 3J ACHTUNGTRENNUNG(H,H) =7.5 Hz, 3H;-CH2CH3); 13C{1H} NMR (CDCl3, 25 8C): d =151.9, 129.4, 123.2 (Cqar);128.4, 126.8, 120.2 (1J ACHTUNGTRENNUNG(C,H)=155 Hz; CHar); 23.1 (1J ACHTUNGTRENNUNG(C,H) =126 Hz;-CH2CH3); 16.1 (Mear); 14.0 ppm (1J ACHTUNGTRENNUNG(C,H)=126 Hz; -CH2CH3). The char-acterization of this compound has been carried out by comparison withliterature data.[28]
Complex 9b : Compound 8b (0.05 g, 0.19 mmol) was dissolved in THF(3 mL) and tetrabutylammonium fluoride (1 m in THF; 0.20 mL,0.204 mmol) was added. The solution was stirred for 2 h at room temper-ature. After this period of time, a saturated solution of NH4Cl (3 mL)was added and compound 9b was extracted with ethyl acetate (3 3 mL).The combined organic solvents were dried over Na2SO4 and evaporated.1H NMR (CDCl3, 25 8C): d=6.75 (s, 2H; 2 CHar); 4.87 (s, 1 H; -OH); 2.57(q, 3J ACHTUNGTRENNUNG(H,H) =7.5 Hz, 2H; C2CH2CH3); 2.20 (s, 6 H; 2 Mear); 1.19 ppm (t,3J ACHTUNGTRENNUNG(H,H) =7.5 Hz, 3H; -CH2CH3); 13C{1H} NMR (CDCl3, 25 8C): d=149.6,129.3, 129.2, 123.1 (Cqar); 129.0, 127.3 (1J ACHTUNGTRENNUNG(C,H)=155 Hz; CHar); 23.2(1J ACHTUNGTRENNUNG(C,H)=128 Hz; -CH2CH3); 20.5, 16.0 (Mear); 14.2 ppm (-CH2CH3);HMRS (FAB): m/z : calcd for [M]+ : 150.1039; found: 150.1045.
Computational details : Preliminary calculations on reactivity alternativeswere performed using Gaussian 03[29] software and the ONIOM schemeapproach using the density functional theory (DFT) with the BHandHfunctional for the QM part and the universal force field (UFF) for theMM part. The reliability of this functional for dealing with the studiedsystems was checked in the previous paper.[11] Partition of the complexwas carried out, including in the MM part the atoms of the Tp ligandthat are not bonded to the metal. With this method the structures of allthe minima and transition states were found and characterized by thenumber of imaginary vibrational frequencies. These calculations wereused as the starting point for the full QM optimizations reported in thispaper. These full QM calculations on the complete system were per-formed using BHandH as implemented in the NWChem software pack-age.[30] No simplifications were introduced in the system, with calculationsbeing performed in the real experimental system. For the Ir atom, theLANL2DZ effective core potential was used to describe the inner elec-trons along with their associated double-z basis set for the remainingelectrons. The 6-31G(d) basis set was used for all the other atoms. Allthe energies given in this report are gas-phase potential energies of fullQM calculations on the complete system expressed in kcal mol�1.
CCDC-722193 (5o) and CCDC-722642 (7) contain the supplementarycrystallographic data for this paper. These data can be obtained free ofcharge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
Acknowledgements
Financial support from the Spanish Ministerio de Ciencia e Innovaci�n(projects CTQ2007-62814, CTQ2008-06866-C02-01/BQU, and ConsoliderIngenio-2010 CSD2007-00006), from the Junta de Andaluc�a (projectFQM672), and from the cooperation project CSIC-CONACyT(2005MX0001) is gratefully acknowledged (FEDER support). S.M. andJ.E.V.V. thank the MICINN and MEC for research grants. The use of thecomputational facilities of the “Centre de Supercomputaci� de Catalu-nya” is greatly appreciated. V.S. thanks the CONACYT for financial sup-port (grants 025424 and 84453).
[1] a) C. Elschenbroich in Organometallics Wiley-VCH, Weinheim,2006 ; b) R. H. Crabtree in The Organometallic Chemistry of the
Transition Metals, Wiley, New York, 2005 ; c) J. P. Collman, J. R.Norton L. S. Hegedus, R. G. Finke in Principles and Applications ofOrganotransition Metal Chemistry, University Science Books, MillValley, 1987.
[2] a) J. A. Moulijin, P. W. N. M. van Leeuwen, R. A. van Santen inCatalysis: An Integrated Approach to Homogeneous, Heterogeneous,and Industrial Catalysis, Elsevier, Amsterdam, 1993 ; b) J. M.Thomas, W. J. Thomas in Principles and Practice of HeterogeneousCatalysis, VCH, Weinheim, 1997; c) P. W. N. M. van Leeuwen inHomogeneous Catalysis, Kluwer Academic, Doderecht, 2004 ;d) Comprehensive Organometallic Chemistry III, Vol. 10–11, Elsevi-er, Amsterdam, 2006.
[3] E. Carmona, M. Paneque, M. L. Poveda, Dalton Trans. 2003, 4022 –4029.
[4] a) J. Jaffart, M. L. Cole, M. Etienne, M. Reinhold, J. E. McGrady, F.Maseras, Dalton Trans. 2003, 4057 –4064; b) J. Jaffart, M. Etienne, F.Maseras, J. E. McGrady, O. Eisenstein, J. Am. Chem. Soc. 2001, 123,6000 – 6013; c) M. D. Fryzuk, S. A. Johnson, S. J. Rettig, J. Am.Chem. Soc. 2001, 123, 1602 – 1612; d) X. Li, L. N. Appelhans, J. W.Faller, R. H. Crabtree, Organometallics 2004, 23, 3378 –3387.
[5] a) J. N. Coalter III, J. C. Bollinger, J. C. Huffman, U. Werner-Zwan-ziger, K. G. Caulton, E. R. Davidson, H. Gerard, E. Clot, O. Eisen-stein, New J. Chem. 2000, 24, 9– 26; b) M. Brookhart, J. R. Tucker,G. R. Husk, J. Am. Chem. Soc. 1981, 103, 979 – 981; c) C. P. Casey,W. H. Miles, H. Tukada, J. Am. Chem. Soc. 1985, 107, 2924 –2931;d) C. Roger, G. S. Bodner, W. G. Hatton, J. A. Gladysz, Organome-tallics 1991, 10, 3266 –3274; e) F. M. Al�as, M. L. Poveda, M. Sellin,E. Carmona, J. Am. Chem. Soc. 1998, 120, 5816 –5817.
[6] a) O. V. Ozerov, L. A. Watson, M. Pink, K. G. Caulton, J. Am.Chem. Soc. 2003, 125, 9604 –9605; b) K. F. Hirsekorn, A. S. Veige,M. P. Marshak, Y. Koldobskaya, P. T. Wolczanski, T. R. Culdari,E. B. Lobcovski, J. Am. Chem. Soc. 2005, 127, 4809 – 4830; c) L.Giannini, G. Guillemot, E. Solari, C. Floriani, N. Re, A. Chiesi-Villa, C. Rizzoli, J. Am. Chem. Soc. 1999, 121, 2797 – 2807; d) R. P.Hughes, S. M. Maddock, A. L. Rheingold, I. A. Guzei, Polyhedron1998, 17, 1037 – 1043; e) J. S. Freundlich, R. R. Schrock, W. M. Davis,J. Am. Chem. Soc. 1996, 118, 3643 –3655; f) G. A. Miller, N. J.Cooper, J. Am. Chem. Soc. 1985, 107, 709 – 711.
[7] a) J. D. Fellmann, R. R. Schrock, D. D. Traficante, Organometallics1982, 1, 481 – 484; b) G. Parkin, E. Bunel, B. J. Burger, M. S. Trim-mer, A. van Asselt, J. E. Bercaw, J. Mol. Catal. 1987, 41, 21–39.
[8] a) V. F. Kuznetsov, K. Abdur-Rashid, A. J. Lough, D. G. Gusev, J.Am. Chem. Soc. 2006, 128, 14388 – 14396; b) O. V. Ozerov, L. A.Watson, M. Pink, K. G. Caulton, J. Am. Chem. Soc. 2007, 129, 6003 –6016.
[9] The preceding paper in this issue: P. Lara, M. Paneque, M. L.Poveda, L. L. Santos, J. E. V. Valpuesta, E. Carmona, S. Moncho, G.Ujaque, A. Lled�s, E. lvarez, K. Mereiter, Chem. Eur. J. 2009,DOI: 10.1002/chem.200900646.
[10] E. Guti�rrez-Puebla, A. Monge, M. C. Nicasio, P. J. P�rez, M. L.Poveda, E. Carmona, Chem. Eur. J. 1998, 4, 2225 –2236.
[11] a) M. Paneque, M. L. Poveda, L. L. Santos, E. Carmona, A. Lled�s,G. Ujaque, K. Mereiter, Angew. Chem. 2004, 116, 3794 –3797;Angew. Chem. Int. Ed. 2004, 43, 3708 –3711; b) P. Lara, M. Paneque,M. L. Poveda, V. Salazar, L. L. Santos, E. Carmona, J. Am. Chem.Soc. 2006, 128, 3512 –3513.
[12] a) E. lvarez, M. Paneque, A. G. Petronilho, M. L. Poveda, L. L.Santos, E. Carmona, K. Mereiter, Organometallics 2007, 26, 1231 –1240; b) M. Paneque, M. L. Poveda, L. L. Santos, E. Carmona, K.Mereiter, Organometallics 2008, 27, 6353 –6359.
[13] See for example: a) Y. Alvarado, O. Boutry, E. Guti�rrez, A.Monge, M. C. Nicasio, M. L. Poveda, P. J. P�rez, C. Ru�z, C. Bianchi-ni, E. Carmona, Chem. Eur. J. 1997, 3, 860 – 873; b) E. Gutierrez-Puebla, A. Monge, M. C. Nicasio, P. J. Perez, M. L. Poveda, L. Rey,C. Ruiz, E. Carmona, Inorg. Chem. 1998, 37, 4538 –4546.
[14] P. Lara, PhD Thesis, University of Sevilla (Spain), 2009.[15] a) Y.-S. Lin, A. Yamamoto in Activation of Unreactive Bonds and
Organic Synthesis (Ed.: S. Murai), Springer, Berlin, 1999 ; pp. 161 –192; b) A. Yamamoto, Adv. Organomet. Chem. 1992, 34, 111 –147;
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 9046 – 90579056
E. Carmona, A. Lled�s et al.

c) J. A. L�pez, K. Mereiter, M. Paneque, M. L. Poveda, O. Serrano,S. Trofimenko, E. Carmona, Chem. Commun. 2006, 3921 –3923.
[16] M. B. Smith, J. March in Advanced Organic Chemistry, 5th ed.,Wiley, New York, 2001, Chapter 18.
[17] A. Maercker, Angew. Chem. 1987, 99, 1002 –1019; Angew. Chem.Int. Ed. Engl. 1987, 26, 972 –989.
[18] a) M. van der Boom, S.-Y. Liou, Y. Ben-David, L. J. W. Shimon, D.Milstein, J. Am. Chem. Soc. 1998, 120, 6531 –6541; b) N. Chatani, H.Tatamidani, Y. Ie, F. Kakiuchi, S. Murai, J. Am. Chem. Soc. 2001,123, 4849 –4850; c) D. B. Grotjahn, H. C. Lo, Organometallics 1996,15, 2860 –2862; d) J. B. Bonanno, T. P. Henry, D. R. Neithamer, P. T.Wolczanski, E. B. Lobkovsky, J. Am. Chem. Soc. 1996, 118, 5132 –5133; e) J. M. Mayer, Polyhedron 1995, 14, 3273 – 3292.
[19] a) J. Arnold, T. D. Tilley, J. Am. Chem. Soc. 1985, 107, 6409 –6410;b) R. Baumann, R. Stumpf, W. M. Davis, L. C. Liang, R. R. Schrock,J. Am. Chem. Soc. 1999, 121, 7822 –7836; c) C. A. Tolman, S. D.Ittel, A. D. English, J. P. Jesson, J. Am. Chem. Soc. 1979, 101, 1742 –1751; d) Y. Yamamoto, R. Sato, F. Matsuo, C. Sudoh, T. Igoshi,Inorg. Chem. 1996, 35, 2329 –2336; e) H. D. Empsall, E. M. Hyde,C. E. Jones, B. L. Shaw, J. Chem. Soc. Dalton Trans. 1974, 1980 –1985.
[20] a) C. A. Bradley, L. F. Veiros, D. Pun, E. Lobkovsky, I. Keresztes, P.Chirik, J. Am. Chem. Soc. 2006, 128, 16600 –16612; b) M. Hirano, H.Sato, N. Kurata, N. Komine, S. Komiya, Organometallics 2007, 26,2005 – 2016; c) M. Tobisu, T. Shimasaki, N. Chatani, Angew. Chem.2008, 120, 4944 –4947; Angew. Chem. Int. Ed. 2008, 47, 4866 –4869.
[21] a) D. A. Whiting in Comprehensive Organic Chemistry, Vol. 1 (Eds.:D. Barton, W. D. Ollis), Pergamon Press, Oxford, 1979, Chapters 4.2;D. A. Whiting in Comprehensive Organic Chemistry, Vol. 1 (Eds.: D.Barton, W. D. Ollis), Pergamon Press, Oxford, 1979, Chapters 4.5;b) K.-F. Wedemeyer in Methoden der Organischen Chemiel, Vol. VI/1c/1 (Eds.: E. M�ller, O. Bayer), Thieme, Stuttgart, 1976, pp. 492 –560.
[22] E. Gutierrez-Puebla, A. Monge, M. Paneque, M. L. Poveda, S. Ta-boada, M. Trujillo, E. Carmona, J. Am. Chem. Soc. 1999, 121, 346 –354.
[23] R. N. Perutz, S. Sabo-Etienne, Angew. Chem. 2007, 119, 2630 –2645;Angew. Chem. Int. Ed. 2007, 46, 2578 –2592.
[24] a) F. Ford, T. Yuzawa, M. S. Platz, S. Matzinger, M. Fulscher, J. Am.Chem. Soc. 1998, 120, 4430 –4438; b) O. V. Ozerov, L. A. Watson,M. Pink, K. G. Caulton, J. Am. Chem. Soc. 2003, 125, 9604 –9605.
[25] L. R. Falvello, J. C. Gines, J. J. Carbo, A. Lledos, R. Navarro, T.Soler, E. P. Urriolabeitia, Inorg. Chem. 2006, 45, 6803 – 6815.
[26] Z. Otwinowski, W. Minor, Methods Enzymol. 1997, 276, 307 – 326.[27] Shelxtl Bruker Axs Inc., 5465 East Cheryl Parkway, Madison.[28] P. G. Gassman, D. R. Amick, J. Am. Chem. Soc. 1978, 100, 7611 –
7619.[29] Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schle-
gel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomer-y, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyen-gar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N.Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K.Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda,O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian,J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E.Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W.Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J.Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C.Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari,J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cio-slowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaro-mi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng,A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W.Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian, Inc., Wall-ingford CT, 2004.
[30] a) NWChem, A Computational Chemistry Package for ParallelComputers, Version 5.1, E. J. Bylaska, W. A. de Jong, N. Govind, K.Kowalski, T. P. Straatsma, M. Valiev, D. Wang, E. Apra, T. L.Windus, J. Hammond, P. Nichols, S. Hirata, M. T. Hackler, Y. Zhao,P.-D. Fan, R. J. Harrison, M. Dupuis, D. M. A. Smith, J. Nieplocha,V. Tipparaju, M. Krishnan, Q. Wu, T. Van Voorhis, A. A. Auer, M.Nooijen, E. Brown, G. Cisneros, G. I. Fann, H. Fruchtl, J. Garza, K.Hirao, R. Kendall, J. A. Nichols, K. Tsemekhman, K. Wolinski, J.Anchell, D. Bernholdt, P. Borowski, T. Clark, D. Clerc, H. Dachsel,M. Deegan, K. Dyall, D. Elwood, E. Glendening, M. Gutowski, A.Hess, J. Jaffe, B. Johnson, J. Ju, R. Kobayashi, R. Kutteh, Z. Lin, R.Littlefield, X. Long, B. Meng, T. Nakajima, S. Niu, L. Pollack, M.Rosing, G. Sandrone, M. Stave, H. Taylor, G. Thomas, J. van Lenthe,A. Wong, Z. Zhang, Pacific Northwest National Laboratory, Rich-land, 2007; b) R. A. Kendall, E. Apra, D. E. Bernholdt, E. J. Bylas-ka, M. Dupuis, G. I. Fann, R. J. Harrison, J. Ju, J. A. Nichols, J. Nie-plocha, T. P. Straatsma, T. L. Windus, A. T. Wong, Comput. Phys.Commun. 2000, 128, 260 –283.
Received: March 12, 2009Published online: August 7, 2009
Chem. Eur. J. 2009, 15, 9046 – 9057 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 9057
FULL PAPERIridium Hydride Isomers








![· SHIM SACD Dire Straits rLove Over Gold] (Private Investigations) ' Clear Cygnus SACD ' , IRIDIUM , IRIDIUM , IRIDIUM 11.5 AWG , , PFA 3455R IRIDIUM Clear Cygnus , 5 Trigon Exxpert](https://img.pdfslide.net/doc/110x75/60d04de1d6909b691a4f38e7/shim-sacd-dire-straits-rlove-over-gold-private-investigations-clear-cygnus.jpg)