Embed Size (px)
Citation preview

Cancer Biology and Signal Transduction
Targeting BRG1 Chromatin Remodeler via ItsBromodomain for Enhanced Tumor CellRadiosensitivity In Vitro and In VivoSu-Jung Kwon1, Seul-Ki Lee1, Juri Na2, Shin-Ai Lee1, Han-Sae Lee1, Ji-Hye Park1,June-Key Chung2,3, Hyewon Youn2,3,4, and Jongbum Kwon1
Abstract
Radiotherapy treats cancer by inducing DNA double-strandbreaks (DSB) in tumor cells using ionizing radiation. However,DNA repair in tumor cells often leads to radioresistance andunsuccessful outcome. Inhibition of DNA repair by targetingrepair proteins can increase radiosensitivity of tumor cells. TheBRG1 chromatin remodeling enzyme assists DSB repair by stim-ulating g-H2AX formation and BRG1 binding to acetylated his-tones at DSBs via bromodomain (BRD) is critical for this activity.Here, we show that ectopic expression of BRG1-BRD inhibitedg-H2AX and DSB repair after irradiation and increased the radio-sensitivity in various human cancer cells, including HT29 coloncancer. Dimerization of BRG1-BRD, increasing its chromatinbinding affinity, aggravated the defects in g-H2AX andDSB repairand further enhanced the radiosensitivity. While little affecting
the upstream ATM activation, BRG1-BRD in irradiated HT29 cellsinhibited the recruitment of 53BP1 to damaged chromatin, thedownstream event of g-H2AX, and compromised the G2–Mcheckpoint and increased apoptosis. Importantly, in a xenograftmouse model, BRG1-BRD increased the radiosensitivity of HT29tumors, which was further enhanced by dimerization. These datasuggest that BRG1-BRD radiosensitizes tumor cells by a dominantnegative activity against BRG1, which disrupts g-H2AX and itsdownstream 53BP1 pathways, leading to inefficient DNA repair,G2–M checkpoint defect, and increased apoptosis. This worktherefore identifies BRG1-BRD as a novel tumor radiosensitizerand its action mechanism, providing the first example of chro-matin remodeler as a target for improving cancer radiotherapy.Mol Cancer Ther; 14(2); 597–607. �2014 AACR.
IntroductionRadiotherapy, one of the major approaches of treating cancer,
uses ionizing radiation (IR) to kill tumor cells by inducing DNAdouble-strand breaks (DSB), the most lethal DNA damage. Thecancer killing activity of radiotherapy relies on the sustainedpresence of DSBs that directs the cytotoxic activity on tumor cells.However, DNA repair in tumor cells often leads to radioresistanceand unsuccessful treatment. Blockade of DSB repair through
targeting repair proteins can sensitize tumor cells to IR andincrease radiotherapy efficacy. Therefore, identification of noveltargets to sensitize tumor cells to IR will provide a window for thedevelopment of novel sensitizers for cancer radiotherapy (1–3).
In response to DSBs, cells activate the so-called DNA damageresponse (DDR), a complex signaling pathway that activates theDNA damage checkpoint, halts cell cycle, and directs DNA repairin a highly coordinated fashion. In case DSBs are left unrepaired,cells undergo apoptosis to prevent their damaged genome frombeing transmitted to offspring. DDR is initiated by the activationof the PI3K-like kinase ATM, which leads to recruitment of manydownstream proteins, such as MDC1 and 53BP1, to DSB sitesthrough a DDR signaling cascade. g-H2AX (phosphorylated formof histone H2AX), which is generated by ATM, plays a central rolein recruiting DDR proteins and thereby forming the so-called IR-induced foci or repair foci in the nucleus. The formation of repairfoci following DSB generation is critical for DNA damage check-point activation, damage repair, and cell survival (4–6).
The packaging of the eukaryotic genome into nucleosomes andhigher order chromatin structure presents a barrier to proteinaccess to targetDNAand chromatinmodifications therefore play apivotal role in the DNA-templated nuclear processes such as DNArepair (7, 8). ATP-dependent chromatin remodeling is a prom-inent mechanism for chromatin modification and is mediated bythe multiprotein remodeling complexes that use ATP hydrolysisenergy to slide, evict, or restructure nucleosomes. Recent studieshave shown that the chromatin remodeling complexes such asSWI/SNF and INO80 are involved in DDR (9–14).
We previously have shown that BRG1, the catalytic ATPase ofthe SWI/SNF complex (15), assists DSB repair by stimulating the
1Department of Life Science,TheResearch Center for Cellular Homeo-stasis, EwhaWomans University, Seoul, Korea. 2Department of Nucle-arMedicine andBiomedical Sciences,CancerResearch Institute, SeoulNational University College of Medicine, Seoul, Korea. 3Tumor Micro-environment Global Core Research Center, Cancer Research Institute,Seoul National University College of Medicine, Seoul, Korea. 4CancerImagingCenter, SeoulNationalUniversityHospital, Jongno-Gu, Seoul,Korea.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
S.-J. Kwon, S.-K. Lee, and J. Na contributed equally to this article.
Current address for address: J.-H. Park: Department of Pathology, Beth IsraelDeaconess Medical Center, Harvard Medical School, 330 Brookline Avenue CLS0624, Boston, MA 02115.
Corresponding Authors: Jongbum Kwon, Ewha Womans University, 52Ewhayeodae-gil, Seoul 120-750, Korea. Phone: 82-2-3277-4334; Fax: 82-2-3277-3760; E-mail: [email protected]; and Hyewon Youn, Seoul NationalUniversity College of Medicine, Nuclear Medicine, 28 Yongon-dong, Chongno-gu, Seoul 110-744, Korea. Phone: 82-2-3668-7026; Fax: 82-2-745-7690; E-mail:[email protected]
doi: 10.1158/1535-7163.MCT-14-0372
�2014 American Association for Cancer Research.
MolecularCancerTherapeutics
www.aacrjournals.org 597
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372

formation of g-H2AX and prevents the cell death via apoptosisafter DNA damage (12, 16). The subsequent study has revealedthat the so-called cooperative activation loop mechanism isresponsible for the BRG1 stimulation of g-H2AX (17). Briefly,BRG1 binds to g-H2AX–containing nucleosomes by interactingwith acetylated H3 using bromodomain (BRD), the proteinmodule that recognizes acetyl-Lys moieties (18), and stimulatesATM-mediated g-H2AX formation through nucleosome remodel-ing activity. The g-H2AX then recruits GCN5 histone acetyltrans-ferase to increase H3 acetylation, which leads to further recruit-ment of BRG1 to g-H2AXnucleosomes. This cooperative action ofBRG1, g-H2AX, and acetylated H3 provides an efficient way toaccumulate g-H2AX and acetylated H3 within the sites of DSB.Given the role for BRG1 in DSB repair, we sought to investigatewhether BRG1 could be a target for enhancing the response toradiotherapy in cancer treatment.
Materials and MethodsCell lines and antibodies
The three cancer cells, HT29 (ATCC HTB-38), MDA-MB-231(ATCC HTB-26), and A549 (ATCC CCL-185) were purchasedfrom ATCC and cultured in McCoy's 5A modified medium,DMEM, and F-12K medium, respectively, each supplementedwith 10% FBS. The cell lines were tested and authenticated withDNA fingerprinting using short tandem repeat markers every 50passages. The antibody sources are as follows: anti-BRG1, anti-CHK1, anti-CHK2, anti-p-CHK2(T68), anti-p53, anti-myc (rabbitIgG), anti-a-tubulin, and anti-b-actin antibodies were purchasedfromSantaCruz Biotechnology; anti-g-H2AX, anti-H2A, and anti-p-ATM(S1981) antibodies from EMD Millipore; anti-GAPDHantibody from AbFrontier; anti-ATM and anti-p21 antibodiesfrom EMD Calbiochem; anti-p-CHK1(S345) and anti-p-p53(S15) from Cell Signaling Technology; anti-p16 antibody fromAbcam; and anti-myc antibody (mouse IgG) from Enzo LifeSciences.
Vector constructionThe pCMV/myc/nuc (Invitrogen)-based plasmid vector-expres-
sing BRD(m) has been described previously (17). The expressionvector for BRD(d)was generated by cloning the blunt-end ligationproduct of the PCR-amplified BRG1-BRD sequences into pCMV/myc/nuc. The retroviral vector-expressing BRD(m) and BRD(d)were generated by cloning the PCR products, amplified frompCMV/myc/nuc-BRG1-BRD(m) and pCMV/myc/nuc-BRG1-BRD(d), respectively, into the BamHI-NotI sites of the pMX-purovector such that the encoded proteins contained three copies ofnuclear localization signals,myc and either single or two copies ofBRG1-BRDs. The PCR primer sequences used were 50-GCTCGGA-TCCATGGCAGATCCAAAAAAGAAGAAG-30 and 50-GCTC GCG-GCCGCCTAATCCTTCTCCTCGCCTTC-30.
Transfection and viral infectionPlasmid DNA transfection was performed using the calcium
phosphate method. For retroviral infection, the pMX vectors weretransfected into Plat-E cells with polybrene 10 mg/mL for 48 hoursand the viral supernatants were added to the cell cultures. TheHT29-luc clones harboring empty vector or expressing Myc-BRD(m) or Myc-BRD(d) were established by puromycin selection ofsingle clones following infection with the corresponding pMX-puro viruses.
Counting repair fociThe immunofluorescence microscopy for g-H2AX, p-ATM,
MDC1, and 53BP1 foci measurement was performed as previ-ously described (19, 20). For the sake of convenience and con-sistency, a single optical section of confocal images was capturedinstead of capturing multiple sections, and only foci with certainsize and brightness which were considered to reside on a sameoptical plane were counted. This method gave a consistent andquantitative result that is well correlated with that obtained bywhole foci counting.
Senescence-associated b-galactosidase assayHT29 cells were washed three times with PBS and fixed in 4%
paraformaldehyde for 5minutes. After washwith PBS three times,the cells were incubated with freshly made b-galactosidase stain-ing solution [1 mg/mL of 5-bromo-4-chloro-3-indolyl b-D-galac-topyranoside (X-Gal), 150 mmol/L sodium chloride, 5 mmol/Lpotassium ferricyanide, 5 mmol/L potassium ferrocyanide, 2mmol/L magnesium chloride] for 24 hours at 37�C (withoutCO2). The staining solution was titrated with 0.1 mol/L NaPibuffer to pH 6.0, and the stained cells were observed undermicroscope (Zeiss).
Tumor implantation and IR exposureHT29-luc cells (3� 105) were subcutaneously injected with 40
mL of Matrigel to dorsal part of male BALB/c nu/nu nude mice.After 7days, the grafted tumorswere exposed to9-Gy IRwithhigh-energy X-ray Linear Accelerator (Clinac 4/100, Varian MedicalSystems). All animal experiments described in this study wereperformed under approval from the Seoul National UniversityInstitutional Animal Care and Use Committee (Seoul, Korea).
In vivo bioluminescence imagingIVIS was performed using the IVIS 100 imaging system with a
charge-coupled device camera (Caliper Life Sciences). The micewere kept on the imaging stage under anesthesia with 1.5%isoflurane gas in oxygen at a flow rate of 1.5 L/minute. D-luciferin(Molecular Probes, Invitrogen) was given 150mg/kg bodyweightby intraperitoneal injection and bioluminescence signals werecollected at 10 to 30 minutes with maximum intensity. The micewere positioned prone for in vivo bioluminescence image ofxenograft tumors.
Analysis of in vivo bioluminescence dataThe signal intensities of emitted light from xenograft tumors
were presented as pseudocolor images raging from red (maxi-mum) to blue (minimum) based on their number. Gray-scalephotographs and corresponding pseudocolor images were super-imposed with LIVINGIMAGE version 2.12 (Xenogen) and IGORversion 1.24 (WaveMetrics) image analysis software. Signalsemitted by regions of interest (ROI) were measured and datawere expressed as photon flux (photon/s/cm2/steradian), whichrefers to the photons emitted from a unit solid angle of a sphere.The background signal intensity was subtracted electronically fornormalization both from the images and from themeasurementsof photon flux. Relative intensities of ROI were calculated andplotted with the mean � SD.
Measurement of tumor volume by caliperThe length and width of tumors were measured by a caliper
before bioluminescence imaging analysis and the tumor volumes
Kwon et al.
Mol Cancer Ther; 14(2) February 2015 Molecular Cancer Therapeutics598
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372

were calculated according to the method by Tomayko and Rey-nolds (tumor volume ¼ 1/2(length � width2; ref. 21).
Statistical analysisThe significance of differences between measurements was
evaluated by Student t test using Microsoft Excel. A P value<0.05 was deemed to indicate statistical significance. IC50 wascalculated using SigmaPlot 8.0 software.
Other methodsClonogenic assays, biochemical fractionation experiments,
immunoblot analysis, neutral comet assay, acid extraction andimmunoblot analysis of histones, and IHC of paraffin-embeddedfrozen tissue were performed as previously described (17, 19, 22).Propidium iodide (PI) and Annexin V staining for FACS analysiswas performed as previously described (16).
ResultsBRG1-BRD inhibits DSB repair via blockade of BRG1chromatin binding
Consistent with the cooperative activation loopmodel, ectopicexpression of BRG1-BRD inhibits g-H2AX formation and DSBrepair in 293T human embryonic kidney cells (17). We reasonedthat BRG1-BRD exerts the DNA repair inhibitory activity througha dominant negative function to compete against BRG1 in bind-ing the acetylated histones on g-H2AXnucleosomes (Supplemen-tary Fig. S1). To test this hypothesis, we expressed Myc-taggedBRG1-BRD [designated as BRD(m); m for monomer; Fig. 1A) in293T cells and separated chromatin-bound and -unbound frac-tions by biochemical fractionation. BRD(m) expression (Fig. 1B,lane 2) resulted in a decrease of BRG1 level in the chromatin-bound fraction (Fig. 1C, compare lanes 1 and 2 with lanes 3 and4), showing that BRG1-BRD indeed inhibits BRG1 binding tochromatin.
As dimerization increases chromatin binding affinity of theBRD of CECR2 (19), we tested whether such effect also would beseen for BRG1-BRD. We generated the plasmid vector-expressingtandomly linked dimeric Myc-tagged BRG1-BRD [designated asBRD(d), d for dimer; Fig. 1A). When this vector was transfectedinto 293T cells, BRD(d) was expressed at the similar level as BRD(m; Fig. 1B). BRD(d) bound to chromatin with a higher affinityand inhibited BRG1 chromatin binding more strongly than BRD(m; Fig. 1C, compare lanes 3 and 4 with lanes 5 and 6). Theseresults led us to test whether BRD(d) would have stronger inhib-itory activity against g-H2AX than BRD(m). Indeed, BRD(d)inhibited the g-H2AX induction after irradiation more effectivelythan BRD(m; Fig. 1D–F). In addition, BRD(m) inhibited DSBrepair and BRD(d) showed a stronger effect than BRD(m; Fig. 1GandH). Consistent with these results, the cells expressing BRD(d)showed higher sensitivity to irradiation than the cells expressingBRD(m; Fig. 1I). The results thus far collectively suggest thatBRG1-BRD inhibits DSB repair through direct blockade of BRG1chromatin binding and this activity of BRG1-BRD is enhanced byincreasing its chromatin binding affinity by dimerization.
BRG1-BRD radiosensitizes various human cancer cells,including HT29, by inhibiting g-H2AX and DSB repair
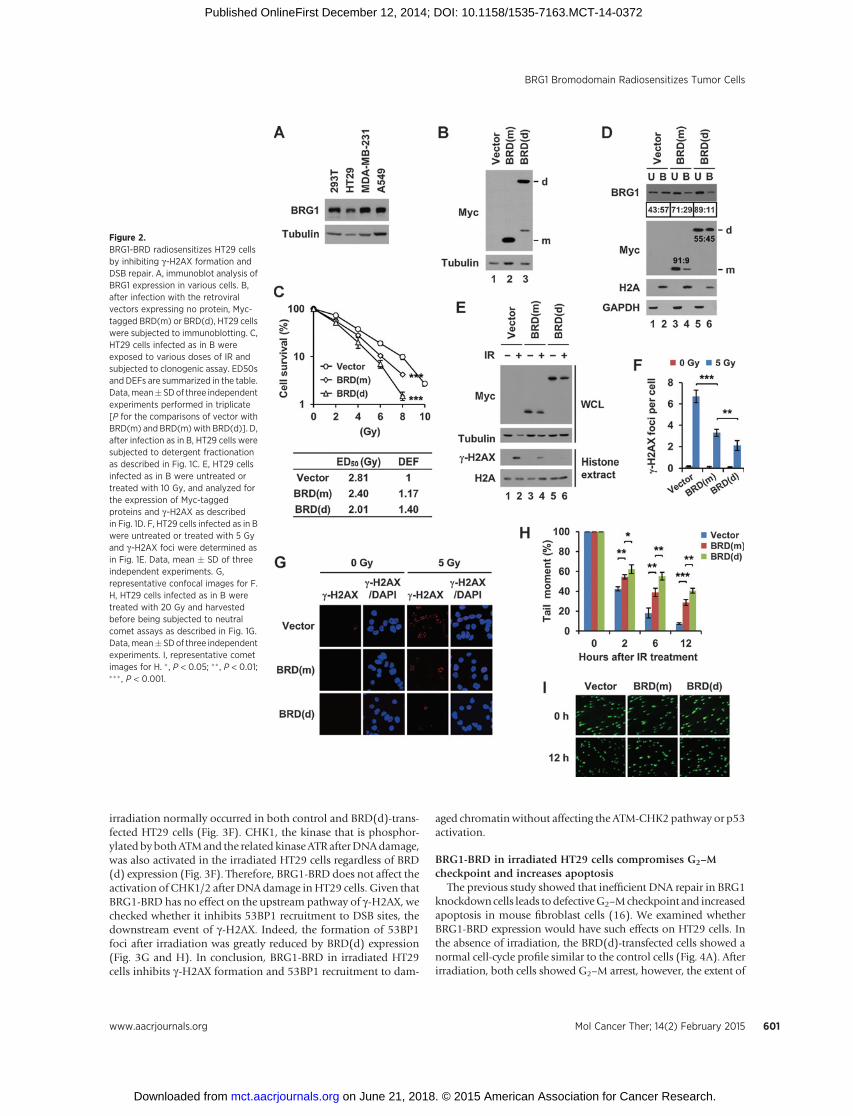
Next, we investigated whether BRG1-BRD radiosensitizeshuman cancer cells. We selected the three cancer cell lines, HT29,MDA-MB-231, and A549, typically used for studying the radio-
resistance problem, as representatives of colon, breast, and lungtumors, respectively. All these cells expressed BRG1 at the com-patible levels with 293T cells (Fig. 2A), making them suitable forour study. When the cancer cells were infected with the retroviralvectors expressing BRD(m) or BRD(d), the two proteins wereexpressed at the similar levels (Fig. 2B andSupplementary Fig. S2Aand B). BRD(m) expression increased the sensitivity of the cancercells to irradiation and the magnitudes of the sensitizing effectswere similar among the three cancer types; the ED50s werereduced by 13% to 15%with the dose enhancement factors (DEF)being 1.15 to 1.17 as compared with empty vector (Fig. 2C andSupplementary Fig. S2C and S2D). Notably, BRD(d) furtherincreased the radiosensitivity of the cancer cells by the significantlevels beyond those obtained by BRD(m); the ED50s werereduced by approximately 22% to 30% with the DEFs being1.28 to 1.44 as compared with empty vector (Fig. 2C and Sup-plementary Fig. S2C and S2D). Therefore, BRG1-BRD has a radio-sensitizing activity on human cancer cells and this activity ofBRG1-BRD is enhanced by dimerization.
To determine whether the increased radiosensitivity of thecancer cells expressing BRG1-BRDwas attributed to defective DSBrepair, we conducted the series of the similar experiments aspreviously described for 293T cells. First, we examined HT29cells. BRG1-BRD inhibited BRG1 binding to chromatin (Fig. 2D,lanes 3 and 4) and this activity of BRG1-BRD was increased bydimerization (Fig. 2D, lanes 5 and 6). The monomeric anddimeric BRG1-BRDs increasingly inhibited the g-H2AX formation(Fig. 2E–G) andDSB repair after irradiation (Fig. 2H and I).Whenwe examined MDA-MB-231 and A549 cells with respect to theeffects of BRD(m) and BRD(d) on g-H2AX and DSB repair, weobtained the similar results (Supplementary Figs. S3A–S3E andS4A–S4E). The viability of 293T and the three cancer cells was notsignificantly affected by the expression of BRD(d) in the absenceof irradiation (Supplementary Fig. S5A and S5B), indicating thatthe radiosensitizing activity of BRG1-BRD is specific to DNAdamage. All these data suggest that BRG1-BRD increases theradiosensitivity of cancer cells by inhibiting DSB repair and theradiosensitizing activity of BRG1-BRD can be enhanced byincreasing its chromatin binding affinity via dimerization.
BRG1-BRD in irradiated HT29 cells inhibits 53BP1 recruitmentto DSB sites without affecting ATM, CHK2, and p53 activations
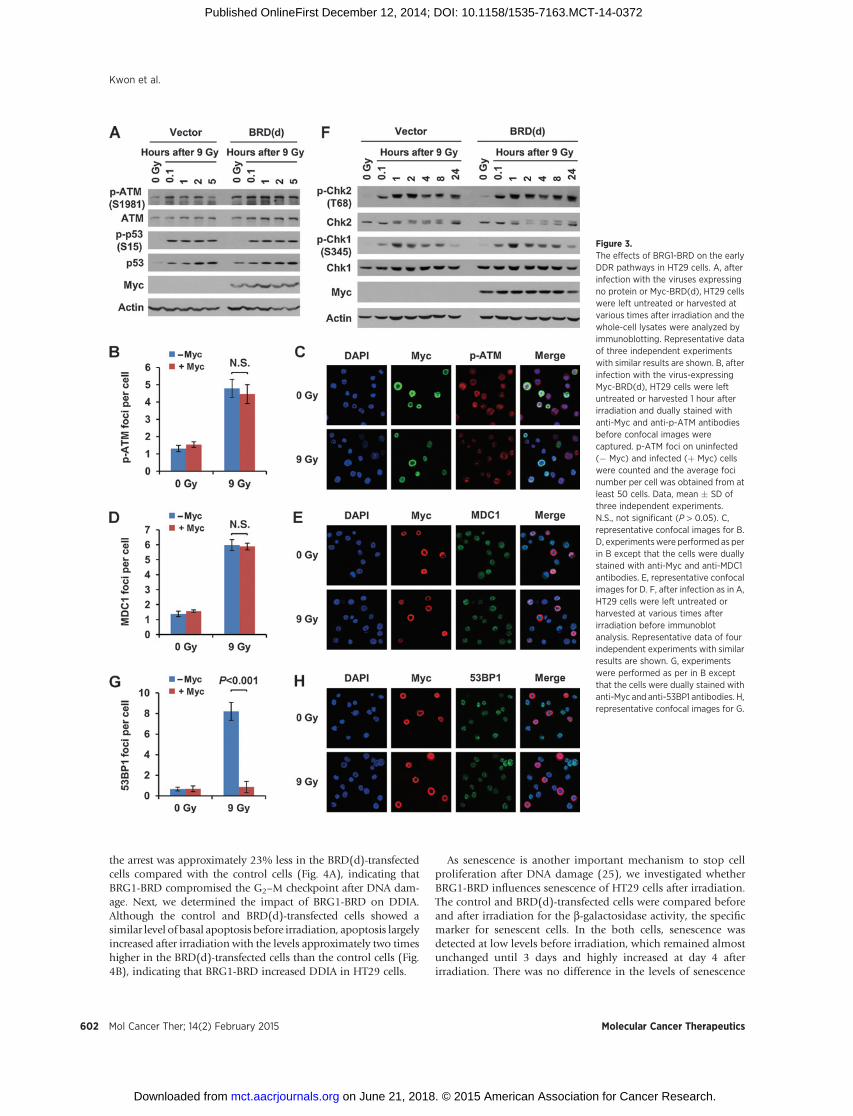
We then investigated what other aspects of DDR than g-H2AXBRG1-BRD affect in cancer cells. We addressed this issue byfocusing on HT29 cells using BRD(d). ATM phosphorylation atSer-1981, indicative of the activation state (23), increased afterirradiation in both control and BRD(d)-transfected cells (Fig. 3A),and phospho-ATM (Fig. 3B and C) and MDC1 foci (Fig. 3D andE), which promote each other in a feedback activation process,normally formed in the irradiated BRD(d)-expressing cells, indi-cating that BRG1-BRD has no effect on ATM activation andrecruitment to damaged chromatin in HT29 cells. Consistentwith these results, ATM-mediated Ser-15 phosphorylation andstabilizationof p53normally occurred in the irradiatedHT29 cellsregardless of BRD(d) expression (Fig. 3A). Unlike in normal cells,the activated p53 in these cells was sustained throughout the timecourse analyzed up to 24 hours (data not shown).
Checkpoint kinase 2 (CHK2) is one of themajor targets of ATMduring DDR and its activation is initiated by phosphorylation atThr-68, leading the G2–Mcell-cycle arrest and in some times DNAdamage-induced apoptosis (DDIA; ref. 24). CHK2 activation after
BRG1 Bromodomain Radiosensitizes Tumor Cells
www.aacrjournals.org Mol Cancer Ther; 14(2) February 2015 599
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372
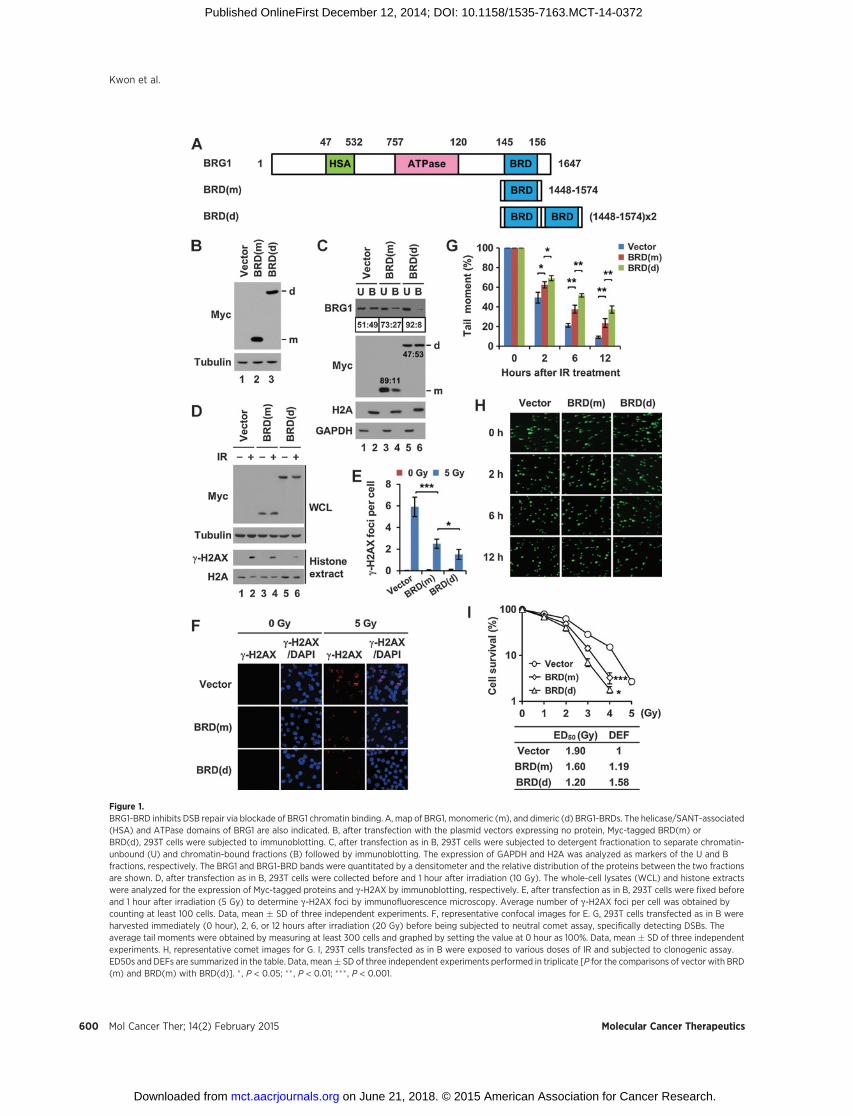
Figure 1.BRG1-BRD inhibits DSB repair via blockade of BRG1 chromatin binding. A, map of BRG1, monomeric (m), and dimeric (d) BRG1-BRDs. The helicase/SANT-associated(HSA) and ATPase domains of BRG1 are also indicated. B, after transfection with the plasmid vectors expressing no protein, Myc-tagged BRD(m) orBRD(d), 293T cells were subjected to immunoblotting. C, after transfection as in B, 293T cells were subjected to detergent fractionation to separate chromatin-unbound (U) and chromatin-bound fractions (B) followed by immunoblotting. The expression of GAPDH and H2A was analyzed as markers of the U and Bfractions, respectively. The BRG1 and BRG1-BRD bands were quantitated by a densitometer and the relative distribution of the proteins between the two fractionsare shown. D, after transfection as in B, 293T cells were collected before and 1 hour after irradiation (10 Gy). The whole-cell lysates (WCL) and histone extractswere analyzed for the expression of Myc-tagged proteins and g-H2AX by immunoblotting, respectively. E, after transfection as in B, 293T cells were fixed beforeand 1 hour after irradiation (5 Gy) to determine g-H2AX foci by immunofluorescence microscopy. Average number of g-H2AX foci per cell was obtained bycounting at least 100 cells. Data, mean � SD of three independent experiments. F, representative confocal images for E. G, 293T cells transfected as in B wereharvested immediately (0 hour), 2, 6, or 12 hours after irradiation (20 Gy) before being subjected to neutral comet assay, specifically detecting DSBs. Theaverage tail moments were obtained by measuring at least 300 cells and graphed by setting the value at 0 hour as 100%. Data, mean � SD of three independentexperiments. H, representative comet images for G. I, 293T cells transfected as in B were exposed to various doses of IR and subjected to clonogenic assay.ED50s and DEFs are summarized in the table. Data, mean� SD of three independent experiments performed in triplicate [P for the comparisons of vector with BRD(m) and BRD(m) with BRD(d)]. � , P < 0.05; �� , P < 0.01; ��� , P < 0.001.
Kwon et al.
Mol Cancer Ther; 14(2) February 2015 Molecular Cancer Therapeutics600
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372
irradiation normally occurred in both control and BRD(d)-trans-fected HT29 cells (Fig. 3F). CHK1, the kinase that is phosphor-ylated bybothATMand the related kinaseATR afterDNAdamage,was also activated in the irradiated HT29 cells regardless of BRD(d) expression (Fig. 3F). Therefore, BRG1-BRD does not affect theactivation of CHK1/2 after DNAdamage inHT29 cells. Given thatBRG1-BRD has no effect on the upstream pathway of g-H2AX, wechecked whether it inhibits 53BP1 recruitment to DSB sites, thedownstream event of g-H2AX. Indeed, the formation of 53BP1foci after irradiation was greatly reduced by BRD(d) expression(Fig. 3G and H). In conclusion, BRG1-BRD in irradiated HT29cells inhibits g-H2AX formation and 53BP1 recruitment to dam-
aged chromatin without affecting the ATM-CHK2 pathway or p53activation.
BRG1-BRD in irradiated HT29 cells compromises G2–Mcheckpoint and increases apoptosis
The previous study showed that inefficient DNA repair in BRG1knockdowncells leads todefectiveG2–Mcheckpoint and increasedapoptosis in mouse fibroblast cells (16). We examined whetherBRG1-BRD expression would have such effects on HT29 cells. Inthe absence of irradiation, the BRD(d)-transfected cells showed anormal cell-cycle profile similar to the control cells (Fig. 4A). Afterirradiation, both cells showed G2–M arrest, however, the extent of
Figure 2.BRG1-BRD radiosensitizes HT29 cellsby inhibiting g-H2AX formation andDSB repair. A, immunoblot analysis ofBRG1 expression in various cells. B,after infection with the retroviralvectors expressing no protein, Myc-tagged BRD(m) or BRD(d), HT29 cellswere subjected to immunoblotting. C,HT29 cells infected as in B wereexposed to various doses of IR andsubjected to clonogenic assay. ED50sand DEFs are summarized in the table.Data,mean�SDof three independentexperiments performed in triplicate[P for the comparisons of vector withBRD(m) and BRD(m) with BRD(d)]. D,after infection as in B, HT29 cells weresubjected to detergent fractionationas described in Fig. 1C. E, HT29 cellsinfected as in B were untreated ortreated with 10 Gy, and analyzed forthe expression of Myc-taggedproteins and g-H2AX as describedin Fig. 1D. F, HT29 cells infected as in Bwere untreated or treated with 5 Gyand g-H2AX foci were determined asin Fig. 1E. Data, mean � SD of threeindependent experiments. G,representative confocal images for F.H, HT29 cells infected as in B weretreated with 20 Gy and harvestedbefore being subjected to neutralcomet assays as described in Fig. 1G.Data,mean�SDof three independentexperiments. I, representative cometimages for H. � , P < 0.05; ��, P < 0.01;��� , P < 0.001.
BRG1 Bromodomain Radiosensitizes Tumor Cells
www.aacrjournals.org Mol Cancer Ther; 14(2) February 2015 601
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372
the arrest was approximately 23% less in the BRD(d)-transfectedcells compared with the control cells (Fig. 4A), indicating thatBRG1-BRD compromised the G2–M checkpoint after DNA dam-age. Next, we determined the impact of BRG1-BRD on DDIA.Although the control and BRD(d)-transfected cells showed asimilar level of basal apoptosis before irradiation, apoptosis largelyincreased after irradiation with the levels approximately two timeshigher in the BRD(d)-transfected cells than the control cells (Fig.4B), indicating that BRG1-BRD increased DDIA in HT29 cells.
As senescence is another important mechanism to stop cellproliferation after DNA damage (25), we investigated whetherBRG1-BRD influences senescence of HT29 cells after irradiation.The control and BRD(d)-transfected cells were compared beforeand after irradiation for the b-galactosidase activity, the specificmarker for senescent cells. In the both cells, senescence wasdetected at low levels before irradiation, which remained almostunchanged until 3 days and highly increased at day 4 afterirradiation. There was no difference in the levels of senescence
Figure 3.The effects of BRG1-BRD on the earlyDDR pathways in HT29 cells. A, afterinfection with the viruses expressingno protein or Myc-BRD(d), HT29 cellswere left untreated or harvested atvarious times after irradiation and thewhole-cell lysates were analyzed byimmunoblotting. Representative dataof three independent experimentswith similar results are shown. B, afterinfection with the virus-expressingMyc-BRD(d), HT29 cells were leftuntreated or harvested 1 hour afterirradiation and dually stained withanti-Myc and anti-p-ATM antibodiesbefore confocal images werecaptured. p-ATM foci on uninfected(� Myc) and infected (þ Myc) cellswere counted and the average focinumber per cell was obtained from atleast 50 cells. Data, mean � SD ofthree independent experiments.N.S., not significant (P > 0.05). C,representative confocal images for B.D, experimentswere performed as perin B except that the cells were duallystained with anti-Myc and anti-MDC1antibodies. E, representative confocalimages for D. F, after infection as in A,HT29 cells were left untreated orharvested at various times afterirradiation before immunoblotanalysis. Representative data of fourindependent experiments with similarresults are shown. G, experimentswere performed as per in B exceptthat the cells were dually stained withanti-Myc and anti-53BP1 antibodies. H,representative confocal images for G.
Kwon et al.
Mol Cancer Ther; 14(2) February 2015 Molecular Cancer Therapeutics602
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372
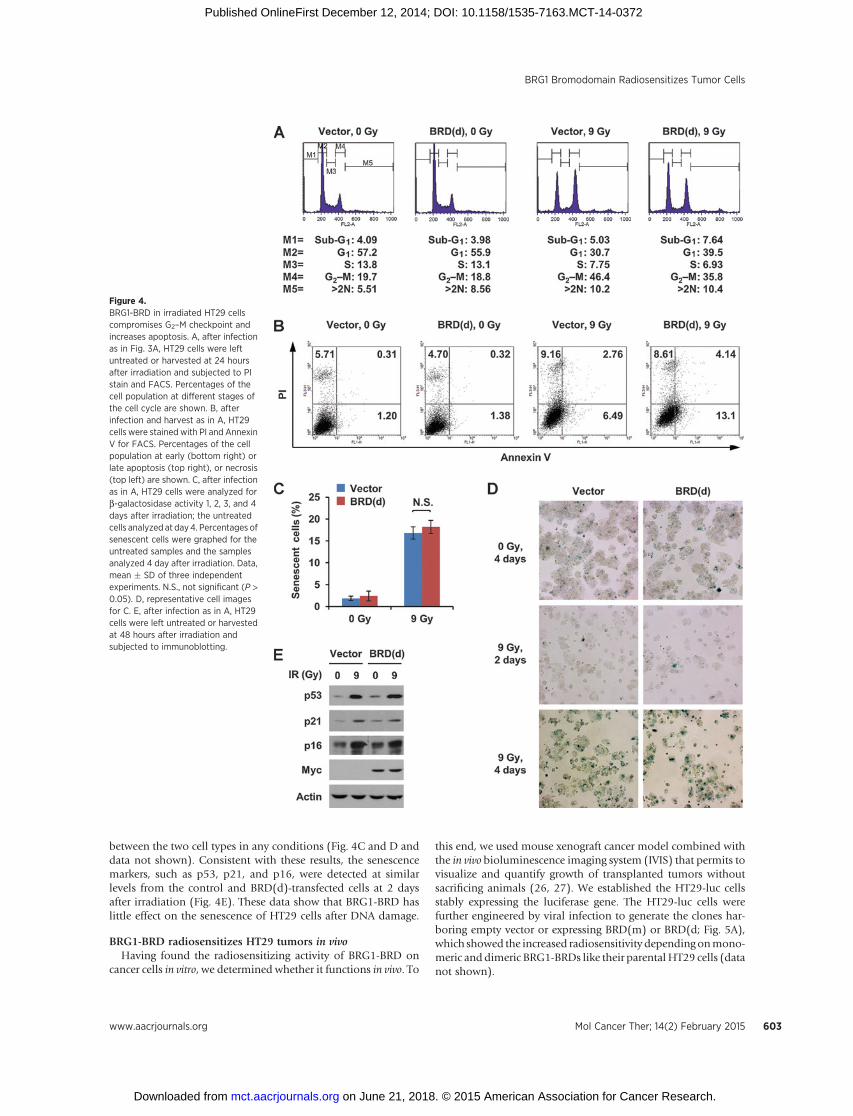
between the two cell types in any conditions (Fig. 4C and D anddata not shown). Consistent with these results, the senescencemarkers, such as p53, p21, and p16, were detected at similarlevels from the control and BRD(d)-transfected cells at 2 daysafter irradiation (Fig. 4E). These data show that BRG1-BRD haslittle effect on the senescence of HT29 cells after DNA damage.
BRG1-BRD radiosensitizes HT29 tumors in vivoHaving found the radiosensitizing activity of BRG1-BRD on
cancer cells in vitro, we determined whether it functions in vivo. To
this end, we used mouse xenograft cancer model combined withthe in vivo bioluminescence imaging system (IVIS) that permits tovisualize and quantify growth of transplanted tumors withoutsacrificing animals (26, 27). We established the HT29-luc cellsstably expressing the luciferase gene. The HT29-luc cells werefurther engineered by viral infection to generate the clones har-boring empty vector or expressing BRD(m) or BRD(d; Fig. 5A),which showed the increased radiosensitivity dependingonmono-meric anddimeric BRG1-BRDs like their parental HT29 cells (datanot shown).
Figure 4.BRG1-BRD in irradiated HT29 cellscompromises G2–M checkpoint andincreases apoptosis. A, after infectionas in Fig. 3A, HT29 cells were leftuntreated or harvested at 24 hoursafter irradiation and subjected to PIstain and FACS. Percentages of thecell population at different stages ofthe cell cycle are shown. B, afterinfection and harvest as in A, HT29cells were stained with PI and AnnexinV for FACS. Percentages of the cellpopulation at early (bottom right) orlate apoptosis (top right), or necrosis(top left) are shown. C, after infectionas in A, HT29 cells were analyzed forb-galactosidase activity 1, 2, 3, and 4days after irradiation; the untreatedcells analyzed at day4. Percentages ofsenescent cells were graphed for theuntreated samples and the samplesanalyzed 4 day after irradiation. Data,mean � SD of three independentexperiments. N.S., not significant (P >0.05). D, representative cell imagesfor C. E, after infection as in A, HT29cells were left untreated or harvestedat 48 hours after irradiation andsubjected to immunoblotting.
BRG1 Bromodomain Radiosensitizes Tumor Cells
www.aacrjournals.org Mol Cancer Ther; 14(2) February 2015 603
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372
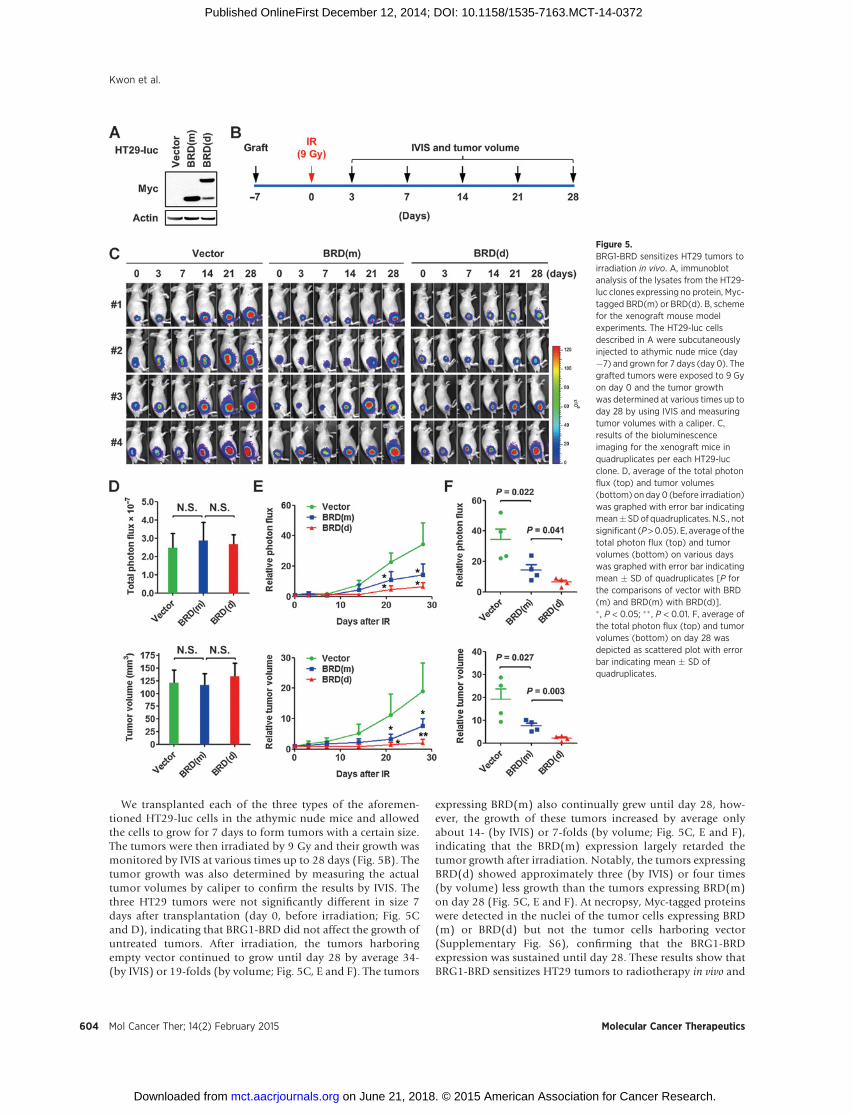
We transplanted each of the three types of the aforemen-tioned HT29-luc cells in the athymic nude mice and allowedthe cells to grow for 7 days to form tumors with a certain size.The tumors were then irradiated by 9 Gy and their growth wasmonitored by IVIS at various times up to 28 days (Fig. 5B). Thetumor growth was also determined by measuring the actualtumor volumes by caliper to confirm the results by IVIS. Thethree HT29 tumors were not significantly different in size 7days after transplantation (day 0, before irradiation; Fig. 5Cand D), indicating that BRG1-BRD did not affect the growth ofuntreated tumors. After irradiation, the tumors harboringempty vector continued to grow until day 28 by average 34-(by IVIS) or 19-folds (by volume; Fig. 5C, E and F). The tumors
expressing BRD(m) also continually grew until day 28, how-ever, the growth of these tumors increased by average onlyabout 14- (by IVIS) or 7-folds (by volume; Fig. 5C, E and F),indicating that the BRD(m) expression largely retarded thetumor growth after irradiation. Notably, the tumors expressingBRD(d) showed approximately three (by IVIS) or four times(by volume) less growth than the tumors expressing BRD(m)on day 28 (Fig. 5C, E and F). At necropsy, Myc-tagged proteinswere detected in the nuclei of the tumor cells expressing BRD(m) or BRD(d) but not the tumor cells harboring vector(Supplementary Fig. S6), confirming that the BRG1-BRDexpression was sustained until day 28. These results show thatBRG1-BRD sensitizes HT29 tumors to radiotherapy in vivo and
Figure 5.BRG1-BRD sensitizes HT29 tumors toirradiation in vivo. A, immunoblotanalysis of the lysates from the HT29-luc clones expressing no protein, Myc-tagged BRD(m) or BRD(d). B, schemefor the xenograft mouse modelexperiments. The HT29-luc cellsdescribed in A were subcutaneouslyinjected to athymic nude mice (day�7) and grown for 7 days (day 0). Thegrafted tumors were exposed to 9 Gyon day 0 and the tumor growthwas determined at various times up today 28 by using IVIS and measuringtumor volumes with a caliper. C,results of the bioluminescenceimaging for the xenograft mice inquadruplicates per each HT29-lucclone. D, average of the total photonflux (top) and tumor volumes(bottom) on day 0 (before irradiation)was graphed with error bar indicatingmean�SDof quadruplicates. N.S., notsignificant (P>0.05). E, average of thetotal photon flux (top) and tumorvolumes (bottom) on various dayswas graphed with error bar indicatingmean � SD of quadruplicates [P forthe comparisons of vector with BRD(m) and BRD(m) with BRD(d)].� , P < 0.05; �� , P < 0.01. F, average ofthe total photon flux (top) and tumorvolumes (bottom) on day 28 wasdepicted as scattered plot with errorbar indicating mean � SD ofquadruplicates.
Kwon et al.
Mol Cancer Ther; 14(2) February 2015 Molecular Cancer Therapeutics604
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372
the tumor radiosensitizing activity of BRG1-BRD is enhancedby dimerization.
BRG1-BRD enhances the sensitivity of HT29 cells to DSB-generating chemotherapeutic drugs
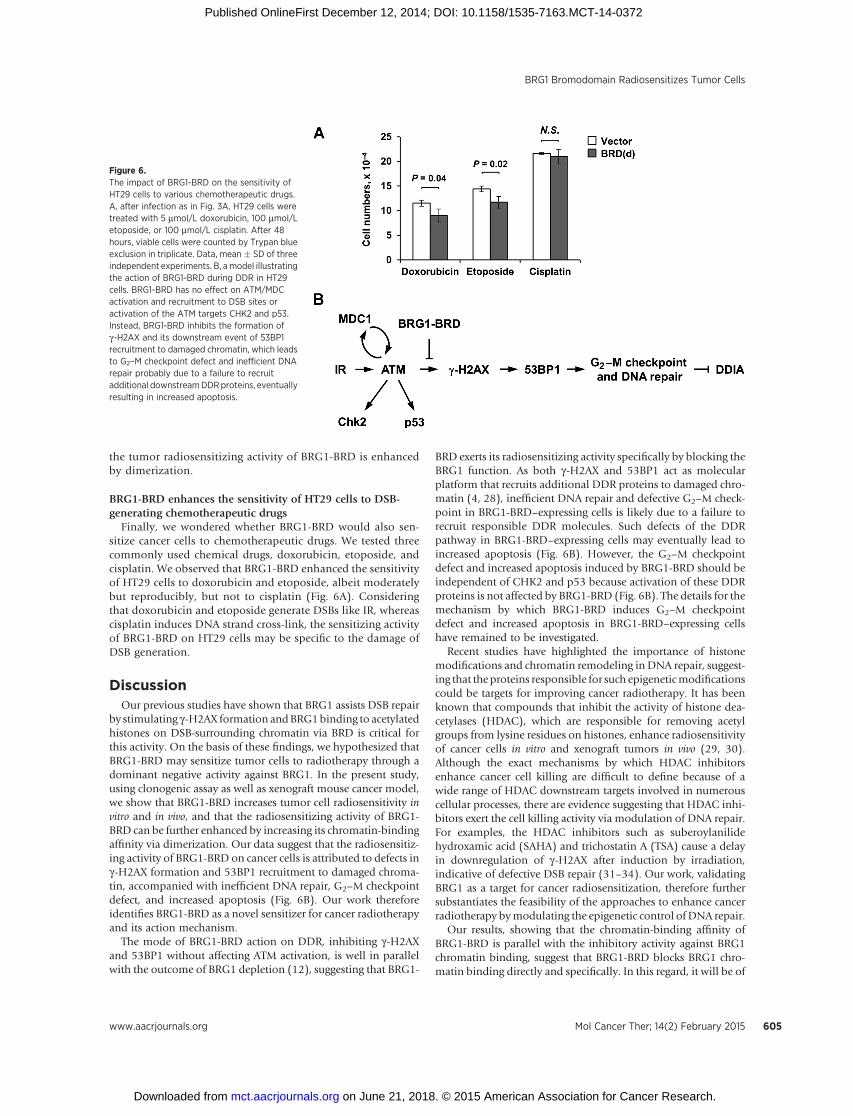
Finally, we wondered whether BRG1-BRD would also sen-sitize cancer cells to chemotherapeutic drugs. We tested threecommonly used chemical drugs, doxorubicin, etoposide, andcisplatin. We observed that BRG1-BRD enhanced the sensitivityof HT29 cells to doxorubicin and etoposide, albeit moderatelybut reproducibly, but not to cisplatin (Fig. 6A). Consideringthat doxorubicin and etoposide generate DSBs like IR, whereascisplatin induces DNA strand cross-link, the sensitizing activityof BRG1-BRD on HT29 cells may be specific to the damage ofDSB generation.
DiscussionOur previous studies have shown that BRG1 assists DSB repair
by stimulating g-H2AX formation andBRG1binding to acetylatedhistones on DSB-surrounding chromatin via BRD is critical forthis activity. On the basis of these findings, we hypothesized thatBRG1-BRD may sensitize tumor cells to radiotherapy through adominant negative activity against BRG1. In the present study,using clonogenic assay as well as xenograft mouse cancer model,we show that BRG1-BRD increases tumor cell radiosensitivity invitro and in vivo, and that the radiosensitizing activity of BRG1-BRD can be further enhanced by increasing its chromatin-bindingaffinity via dimerization. Our data suggest that the radiosensitiz-ing activity of BRG1-BRD on cancer cells is attributed to defects ing-H2AX formation and 53BP1 recruitment to damaged chroma-tin, accompanied with inefficient DNA repair, G2–M checkpointdefect, and increased apoptosis (Fig. 6B). Our work thereforeidentifies BRG1-BRD as a novel sensitizer for cancer radiotherapyand its action mechanism.
The mode of BRG1-BRD action on DDR, inhibiting g-H2AXand 53BP1 without affecting ATM activation, is well in parallelwith the outcome of BRG1 depletion (12), suggesting that BRG1-
BRD exerts its radiosensitizing activity specifically by blocking theBRG1 function. As both g-H2AX and 53BP1 act as molecularplatform that recruits additional DDR proteins to damaged chro-matin (4, 28), inefficient DNA repair and defective G2–M check-point in BRG1-BRD–expressing cells is likely due to a failure torecruit responsible DDR molecules. Such defects of the DDRpathway in BRG1-BRD–expressing cells may eventually lead toincreased apoptosis (Fig. 6B). However, the G2–M checkpointdefect and increased apoptosis induced by BRG1-BRD should beindependent of CHK2 and p53 because activation of these DDRproteins is not affected by BRG1-BRD (Fig. 6B). The details for themechanism by which BRG1-BRD induces G2–M checkpointdefect and increased apoptosis in BRG1-BRD–expressing cellshave remained to be investigated.
Recent studies have highlighted the importance of histonemodifications and chromatin remodeling in DNA repair, suggest-ing that theproteins responsible for such epigeneticmodificationscould be targets for improving cancer radiotherapy. It has beenknown that compounds that inhibit the activity of histone dea-cetylases (HDAC), which are responsible for removing acetylgroups from lysine residues on histones, enhance radiosensitivityof cancer cells in vitro and xenograft tumors in vivo (29, 30).Although the exact mechanisms by which HDAC inhibitorsenhance cancer cell killing are difficult to define because of awide range of HDAC downstream targets involved in numerouscellular processes, there are evidence suggesting that HDAC inhi-bitors exert the cell killing activity via modulation of DNA repair.For examples, the HDAC inhibitors such as suberoylanilidehydroxamic acid (SAHA) and trichostatin A (TSA) cause a delayin downregulation of g-H2AX after induction by irradiation,indicative of defective DSB repair (31–34). Our work, validatingBRG1 as a target for cancer radiosensitization, therefore furthersubstantiates the feasibility of the approaches to enhance cancerradiotherapy bymodulating the epigenetic control ofDNA repair.
Our results, showing that the chromatin-binding affinity ofBRG1-BRD is parallel with the inhibitory activity against BRG1chromatin binding, suggest that BRG1-BRD blocks BRG1 chro-matin binding directly and specifically. In this regard, it will be of
Figure 6.The impact of BRG1-BRD on the sensitivity ofHT29 cells to various chemotherapeutic drugs.A, after infection as in Fig. 3A, HT29 cells weretreated with 5 mmol/L doxorubicin, 100 mmol/Letoposide, or 100 mmol/L cisplatin. After 48hours, viable cells were counted by Trypan blueexclusion in triplicate. Data, mean � SD of threeindependent experiments. B, amodel illustratingthe action of BRG1-BRD during DDR in HT29cells. BRG1-BRD has no effect on ATM/MDCactivation and recruitment to DSB sites oractivation of the ATM targets CHK2 and p53.Instead, BRG1-BRD inhibits the formation ofg-H2AX and its downstream event of 53BP1recruitment to damaged chromatin, which leadsto G2–M checkpoint defect and inefficient DNArepair probably due to a failure to recruitadditional downstreamDDRproteins, eventuallyresulting in increased apoptosis.
BRG1 Bromodomain Radiosensitizes Tumor Cells
www.aacrjournals.org Mol Cancer Ther; 14(2) February 2015 605
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372

interest to test whether higher-order multimerization (more thandimer)would even further enhance the radiosensitizing activity ofBRG1-BRD. These results also emphasize that tumor cell radio-sensitization can be achieved by blocking a specific step of theBRG1-assisted DSB repair process without depleting or inactivat-ing the BRG1 protein itself. This strategymay likely provide betteropportunity to inhibit DNA repair with minimal effects on theBRG10s other cellular functions such as transcription and cell-cycle control (15).
AlthoughBRG1-BRD itself could beused as a radiosensitizer forcancer treatment provided with an appropriate gene deliverysystem, small-molecule drugs that specifically inhibit the inter-action between BRG1-BRD and acetylated histonewould bemoreuseful for clinical applications. It has been known that developingsmall-molecule drugs that target protein–protein interactions isextremely difficult in general because their interacting surfaces areusually wide and shallow (35). However, all the BRD modules,including BRG1-BRD, share a conserved structural feature in thatthey have a central deep and narrow hydrophobic cavity andrecognize acetyl-Lys in a sequence-dependent manner using thispocket (36–38), suggesting the possibility to design small-mol-ecule inhibitors of certain BRDs. Indeed, recent studies havereported the small-molecule compounds called JQ1 and I-BETthat specifically fit into the hydrophobic pocket of the BRD ofbromodomain and extra-terminal (BET) family proteins andfunction as potent inhibitors against the cellular function of theseproteins (39–41). Thus, itmaybe likely possible to develop small-molecule inhibitors that specifically target BRG1-BRD (42, 43). Inthis regard, our work provides the experimental basis for thevalidity of developing specific BRG1-BRD inhibitors as radio-sensitizers for cancer treatment.
In summary, we have established the BRD of BRG1 chromatinremodeling enzyme as a novel radiosensitizer for human tumorcells. To our knowledge, this is the first evidence that ATPasechromatin remodeler can be a target for improving cancer radio-therapy. In addition, our strategy to use the dominant negativityof BRG1-BRD as ameans to radiosensitize tumor cells is based onthe defined molecular mechanisms by which BRG1 assists DSB
repair, which therefore provides an excellent example for themechanism-based approaches to cancer radiotherapeutics. Itshould be noted, however, that, as the radiosensitizing activityof BRG1-BRD on cancer cells is not robust, rational combinationtherapy with commonly used chemotherapeutic drugs wouldpromise more efficient cancer treatment. Further investigationswill allowone to fully understand howBRG1-BRD radiosensitizestumor cells and help to maximize its sensitizing effects, whichmay eventually lead to improvements of the therapeutic outcomefor cancers that are difficult to control due to radioresistance.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: S.-J. Kwon, S.-K. Lee, H.-S. Lee, J.-K. Chung, H. Youn,J. KwonDevelopment of methodology: S.-J. Kwon, S.-K. Lee, J.-H. Park, H. YounAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): S.-J. Kwon, S.-K. Lee, J. Na, S.-A. Lee, H. YounAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): S.-J. Kwon, S.-K. Lee, J. Na, H. Youn, J. KwonWriting, review, and/or revision of the manuscript: J. Na, S.-A. Lee, H. Youn,J. KwonStudy supervision: H. Youn, J. Kwon
Grant SupportS.-J. Kwon, S.-K. Lee, S.-A. Lee, H.-S. Lee, J.-H. Park, and J. Kwon were
supported by the grants of the Korean Health Technology R&D Project(A101709) and the National Research Foundation (NRF) of Korea(2012R1A2A2A01003744). S.-J. Kwon, S.-A. Lee, H.-S. Lee, and J. Kwon werealso supported by NRF-2012R1A5A1048236. J. Na, J.-K. Chung, and H. Younwere supported by NRF-2011-0030680. J-.H. Park was also supported by RPGrant 2009 of Ewha Womans University.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received May 2, 2014; revised November 17, 2014; accepted November 19,2014; published OnlineFirst December 12, 2014.
References1. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with
targeted drugs. Nat Rev Cancer 2011;11:239–53.2. Powell SN, Bindra RS. Targeting the DNA damage response for cancer
therapy. DNA Repair 2009;8:1153–65.3. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling
on cancer chemotherapy response and resistance. Nat Rev Cancer2012;12:587–98.
4. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S,et al. GammaH2AX and cancer. Nat Rev Cancer 2008;8:957–67.
5. Jackson SP, Bartek J. The DNA-damage response in human biology anddisease. Nature 2009;461:1071–8.
6. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play withknives. Mol Cell 2010;40:179–204.
7. Downs JA,NussenzweigMC,Nussenzweig A. Chromatin dynamics and thepreservation of genetic information. Nature 2007;447:951–8.
8. Papamichos-Chronakis M, Peterson CL. Chromatin and the genomeintegrity network. Nat Rev Genet 2012;14:62–75.
9. Chai B, Huang J, Cairns BR, Laurent BC. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand breakrepair. Genes Dev 2005;19:1656–61.
10. van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80complex by H2A phosphorylation links ATP-dependent chromatin remo-deling with DNA double-strand break repair. Cell 2004;119:777–88.
11. Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF,Haber JE, et al. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004;119:767–75.
12. Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN, et al. MammalianSWI/SNF complexes facilitate DNA double-strand break repair by promot-ing gamma-H2AX induction. EMBO J 2006;25:3986–97.
13. Park EJ, Hur SK, Kwon J. Human INO80 chromatin-remodelling complexcontributes to DNA double-strand break repair via the expression ofRad54B and XRCC3 genes. Biochem J 2010;431:179–87.
14. Seeber A, Hauer M, Gasser SM. Nucleosome remodelers in double-strandbreak repair. Curr Opin Genet Dev 2013;23:174–84.
15. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer.Nat Rev Cancer 2011;11:481–92.
16. Park JH, Park EJ, Hur SK, Kim S, Kwon J. Mammalian SWI/SNF chromatinremodeling complexes are required to prevent apoptosis after DNA dam-age. DNA Repair 2009;8:29–39.
17. Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. A cooperative activation loopamong SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J 2010;29:1434–45.
18. Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving theepigenetic landscape through histone readers. Nat Struct Mol Biol 2012;19:1218–27.
Mol Cancer Ther; 14(2) February 2015 Molecular Cancer Therapeutics606
Kwon et al.
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372

19. Lee SK, Park EJ, Lee HS, Lee YS, Kwon J. Genome-wide screen of humanbromodomain-containing proteins identifies Cecr2 as a novel DNA dam-age response protein. Mol Cells 2012;34:85–91.
20. Kwon SJ, Park JH, Park EJ, Lee SA, Lee HS, Kang SW, et al. ATM-mediatedphosphorylation of the chromatin remodeling enzyme BRG1 modulatesDNA double-strand break repair. Oncogene 2014 Jan 13. [Epub ahead ofprint].
21. TomaykoMM, Reynolds CP.Determination of subcutaneous tumor size inathymic (nude) mice. Cancer Chemother Pharmacol 1989;24:148–54.
22. Lee HS, Lee SA, Hur SK, Seo JW, Kwon J. Stabilization and targeting ofINO80 to replication forks by BAP1 during normal DNA synthesis. NatCommun 2014;5:5128.
23. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermo-lecular autophosphorylation and dimer dissociation. Nature 2003;421:499–506.
24. Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer suscepti-bility and cancer therapy - two sides of the same coin? Nat Rev Cancer2007;7:925–36.
25. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008;8:512–22.
26. Kang JH, Chung JK. Molecular-genetic imaging based on reporter geneexpression. J Nucl Med 2008;49 Suppl 2:164S–79S.
27. Choi Y, Jeon YH, Jang JY, Chung JK, Kim CW. Treatment with mANT2shRNA enhances antitumor therapeutic effects induced by MUC1 DNAvaccination. Mol Ther 2011;19:979–89.
28. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus.Nat Rev Mol Cell Biol 2014;15:7–18.
29. Inche AG, La Thangue NB. Chromatin control and cancer-drug discovery:realizing the promise. Drug Discov Today 2006;11:97–109.
30. Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategyfor tumor radiosensitization. J Clin Oncol 2007;25:4051–6.
31. Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone deace-tylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX fociand confines HDAC4 to the cytoplasm in irradiated non-small cell lungcancer. Cancer Res 2006;66:11298–304.
32. Karagiannis TC, Harikrishnan KN, El-Osta A. The histone deacetylaseinhibitor, Trichostatin A, enhances radiation sensitivity and accumulationof gammaH2A.X. Cancer Biol Ther 2005;4:787–93.
33. Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE.Vorinostat, a histone deacetylase inhibitor, enhances the response ofhuman tumor cells to ionizing radiation through prolongation of gam-ma-H2AX foci. Mol Cancer Ther 2006;5:1967–74.
34. Zhang Y, Adachi M, Zou H, Hareyama M, Imai K, Shinomura Y.Histone deacetylase inhibitors enhance phosphorylation of histoneH2AX after ionizing radiation. Int J Radiat Oncol Biol Phys 2006;65:859–66.
35. Wells JA,McClendonCL. Reaching for high-hanging fruit in drug discoveryat protein-protein interfaces. Nature 2007;450:1001–9.
36. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis ofthe human bromodomain family. Cell 2012;149:214–31.
37. Mujtaba S, Zeng L, ZhouMM. Structure and acetyl-lysine recognition of thebromodomain. Oncogene 2007;26:5521–7.
38. ShenW, Xu C, HuangW, Zhang J, Carlson JE, Tu X, et al. Solution structureof humanBrg1bromodomain and its specific binding to acetylatedhistonetails. Biochemistry 2007;46:2100–10.
39. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BETbromodomain inhibition as a therapeutic strategy to target c-Myc. Cell2011;146:904–17.
40. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al.Suppression of inflammation by a synthetic histone mimic. Nature2010;468:1119–23.
41. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al.Selective inhibition of BET bromodomains. Nature 2010;468:1067–73.
42. Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutictargets. Expert Rev Mol Med 2011;13:e29.
43. Hewings DS, Rooney TP, Jennings LE, Hay DA, Schofield CJ, Brennan PE,et al. Progress in the development and application of small moleculeinhibitors of bromodomain-acetyl-lysine interactions. J Med Chem 2012;55:9393–413.
www.aacrjournals.org Mol Cancer Ther; 14(2) February 2015 607
BRG1 Bromodomain Radiosensitizes Tumor Cells
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372

2015;14:597-607. Published OnlineFirst December 12, 2014.Mol Cancer Ther Su-Jung Kwon, Seul-Ki Lee, Juri Na, et al.
In Vivo and In VitroEnhanced Tumor Cell Radiosensitivity Targeting BRG1 Chromatin Remodeler via Its Bromodomain for
Updated version
10.1158/1535-7163.MCT-14-0372doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2014/12/17/1535-7163.MCT-14-0372.DC1
Access the most recent supplemental material at:
Cited articles
http://mct.aacrjournals.org/content/14/2/597.full#ref-list-1
This article cites 42 articles, 7 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/14/2/597.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/14/2/597To request permission to re-use all or part of this article, use this link
on June 21, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst December 12, 2014; DOI: 10.1158/1535-7163.MCT-14-0372





























