Embed Size (px)
Citation preview

Terrequinone A biosynthesis through L-tryptophanoxidation, dimerization and bisprenylationCarl J Balibar1,2, Annaleise R Howard-Jones1,2 & Christopher T Walsh1
The antitumor fungal metabolite terrequinone A, identified in extracts of Aspergillus sp., is biosynthesized by the five-genecluster tdiA–tdiE. In this work, we have overproduced all five proteins (TdiA–TdiE) in the bacterial host Escherichia coli, fullyreconstituting the biosynthesis of terrequinone A. This pathway involves aminotransferase activity, head-to-tail dimerizationand bisprenylation of the scaffold to yield the benzoquinone natural product. We have established that TdiD is a pyridoxal-5¢-phosphate–dependent L-tryptophan aminotransferase that generates indolepyruvate for an unusual nonoxidative coupling bythe tridomain nonribosomal peptide synthetase TdiA. TdiC, an NADH-dependent quinone reductase, generates the nucleophilichydroquinone for two distinct rounds of prenylation by the single prenyltransferase TdiB. TdiE is required to shunt thebenzoquinone away from an off-pathway monoprenylated species by an as yet unknown mechanism. Overall, we havebiochemically characterized the complete biosynthetic pathway to terrequinone A, highlighting the nonoxidative dimerizationpathway and the unique asymmetric prenylation involved in its maturation.
Terrequinone A (1) represents a unique member of a family ofbisindolylbenzoquinones commonly known as asterriquinones(Fig. 1). Since the initial discovery of cochliodinol (2)1, an antifungalagent2 from various Chaetomium species3, several new asterriqui-nones, including asterriquinone CT5 (3) and asterriquinone B1 (4),have been isolated from various species of Aspergillus terreus4,5,Humicola and Botryotrichum6. All share a common dihydroxybenzo-quinone core and vary mainly in the pattern of prenylation on theindole substituents. Like terrequinone A, most isolated asterriqui-nones are cytotoxic compounds that have been shown to intercalategenomic DNA, thus predisposing tumor cells to apoptosis7. Structure-activity studies on the asterriquinones have demonstrated that thehydroxyls of the symmetric dihydroxybenzoquinone core are impor-tant for antitumorigenic activity8. However, terrequinone A is the onlyasterriquinone asymmetrical in its quinone core, bearing an isopente-nyl moiety in place of one of the hydroxyls.
Although terrequinone A was originally isolated from Aspergillusterreus9, a gene cluster for this molecule was recently discovered inAspergillus nidulans through a genetic screen involving LaeA, a nuclearmethyltransferase that acts as a global regulator of natural productgene expression in various Aspergillus species10,11. By monitoringgenetic loci that are downregulated in LaeA deletion strains orupregulated in overexpression strains, five contiguous genes,tdiA–tdiE, were found to be involved in secondary metabolite produc-tion. This study constituted the first identification of a biosyntheticpathway for this class of fungal toxins. Bioinformatic analysis of thiscluster predicts that tdiA encodes a single-module nonribosomalpeptide synthetase (NRPS), tdiB an indole prenyltransferase, tdiC anoxidoreductase, tdiD a pyridoxal-5¢-phosphate (PLP; 5)-dependent
aminotransferase and tdiE a gene of unknown function with weakhomology to S-adenosyl-L-methionine (SAM; 6)–dependent methyl-transferases11. Given the predicted functions of the proteins in thecluster, a route to terrequinone A was proposed (Scheme 1).
Unique in this pathway is the presence of a single-module NRPS,TdiA. NRPSs are multifunctional enzymes consisting of semiautono-mous domains that synthesize a myriad of secondary metabolites12.Using an assembly line logic comprising multiple modules, theseenzymes use a thiotemplated mechanism13 to activate, tether andmodify amino acid monomers, sequentially elongating the peptidechain and finally releasing the complete peptide. Absent from theTdiA scaffold is the traditional condensation (C) domain, which isresponsible for formation of an amide bond between amino acidsloaded on sequential modules14,15. Rather, TdiA contains three indi-vidual domains responsible for adenylation, thiolation and thioestercleavage. TdiA’s adenylation (A) domain is responsible for recognitionof substrate and its subsequent activation as an acyl-O-AMP16; thesubstrate is then loaded onto the phosphopantetheine arm of thecognate thiolation (T) domain to form an acylthioester17,18. Thesingle-module TdiA ends with a thioesterase (TE) domain. Such TEdomains traditionally catalyze release of the final product by eitherhydrolysis to the free acid or cyclization to an amide or ester12,19.Notably, there are no amide, ester or free carboxylic acid functional-ities present in terrequinone A, implying a new mechanism of releasefrom this NRPS.
Another notable feature of this pathway involves the prenyltransfer-ase TdiB. The prenylated indole group is a relatively widespread motifthroughout many families of natural products, all of which arethought to be derived biosynthetically from L-tryptophan (7)20. The
Received 16 April; accepted 6 July; published online 12 August 2007; doi:10.1038/nchembio.2007.20
1Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Ave., Boston, Massachusetts 02115, USA. 2These authorscontributed equally to this work. Correspondence should be addressed to C.T.W. ([email protected]).
58 4 VOLUME 3 NUMBER 9 SEPTEMBER 2007 NATURE CHEMICAL BIOLOGY
ART ICL ES

site of attachment of the prenyl group varies widely, with prenylationobserved at all sites around the indole ring. Furthermore, the prenylgroup may be tethered through its C1 position or its C3 position,representing nucleophilic attack of the indole on either end of the allylcation-like transition state of the activated prenyl group21. Biosynthe-tically, prenylated indoles are formed by prenyltransferases (otherwiseknown as dimethylallyl transferases). These enzymes are known to usedimethylallyl diphosphate (DMAPP; 8) as the prenyl source and often,although not always, contain an (N/D)DXXD motif as the signaturefor diphosphate binding. They may be divided into two functionalclassifications: so-called ‘regular’ prenyltransferases catalyze attacks atthe C1 position of DMAPP, whereas ‘reverse’ prenyltransferasesfacilitate attachment of the dimethylallyl group through its C3(ref. 22). Thus, the role of the prenyltransferase is to bind and activateDMAPP and to enable directed capture of the allyl cation at either theC1 or C3 position of DMAPP. Given the presence of two prenylgroups with different connectivities in the terrequinone A scaffold, wefind it intriguing that the Tdi cluster contains only a single geneencoding a prenyltransferase, tdiB.
In this study, we have expressed tdiA, tdiB, tdiC, tdiD and tdiE inE. coli, overproduced and purified the encoded proteins and recon-stituted the full terrequinone A biosynthetic pathway in vitro. Thebiosynthetic route to terrequinone A shows unusual logic, and itscomponent enzymes show many unique functionalities.
RESULTSCloning and expression of Tdi proteinsWe amplified the five genes from the terrequinone A biosyntheticcluster from cDNA derived from Aspergillus nidulans strains A4 orFGSC A26 and cloned the products into C-terminal His6-taggedvectors. After sequencing the five genes, we determined that althoughbioinformatics correctly predicted the sequences of tdiB and tdiC,there were differences in the introns predicted for tdiA, tdiD and tdiE(Supplementary Fig. 1a online). Additionally, we determined that thereported genome sequence spanning tdiD contains an extra base,C464. Expression in E. coli at 30 1C or 15 1C with isopropyl-b-D-
thiogalactoside (IPTG) induction yielded 1.6 mg l–1 TdiA, 2.3 mg l–1
TdiB, 7.5 mg l–1 TdiC, 6.3 mg l–1 TdiD and 16.8 mg l–1 TdiE. Wepurified all proteins to homogeneity using nickel-affinity chromato-graphy and, in the case of TdiA, gel filtration chromatography(Supplementary Fig. 1b).
Characterization of TdiDThe first step in the proposed biosynthetic pathway for terrequinone Ais formation of indole pyruvic acid (IPA; 9) from L-tryptophan usingthe aminotransferase TdiD. PLP-dependent aminotransferases typi-cally contain a covalent PLP cofactor bound as a Schiff base to aconserved lysine in the protein active site. Upon binding of an amine-containing substrate, transimination occurs, yielding an intermediatethat ultimately undergoes hydrolysis to pyridoxamine-5¢-phosphate(PMP; 10), and a keto functionality is installed on the substrate23
(Fig. 2a). Although purified TdiD lacked a bound cofactor, titrationexperiments demonstrated that it was, in fact, able to bind PLP,although weakly. Whereas free PLP has a lmax at 389 nm, the enzyme-bound PLP showed a lmax at 416 nm. TdiD showed linear binding ofPLP, as demonstrated by an increase in absorbance at 416 nm, past10 equivalents (data not shown).
With the ability of TdiD to bind PLP confirmed, we sought to testwhether L-tryptophan could be converted to IPA. Addition ofL-tryptophan to reactions containing TdiD and PLP yielded a decreasein absorbance at 389 nm and a corresponding increase in absorbanceat 323 nm, indicating conversion of PLP to PMP (Fig. 2b). PMPformation was dependent on TdiD (Supplementary Fig. 2a online)and L-tryptophan (Supplementary Fig. 2b). This conversion isindicative of aminotransferase activity and thus IPA formation. Toverify this, we analyzed reactions by HPLC. Over time, L-tryptophanwas consumed, and a new peak that coeluted with authentic IPAevolved (Supplementary Fig. 3 online).
In PLP-dependent aminotransferases, there is generally a secondsubstrate that is responsible for regenerating PLP after PMP forma-tion. This substrate is typically a keto acid that acts as an amineacceptor. We tested three typical a-keto acids (pyruvate (11),a-ketoglutarate (12), and a-keto-g-(methylthio)butyrate (13)) fortheir ability to regenerate PLP, as assayed by either an increase inthe rate of IPA formation or a decrease in the amount of PMP formed.Although none of these compounds acted as a substrate for TdiD,
O
O
HO
HN
NH O
O
HO
HN
NH
OH
O
O
O
HN
NH
O
O
O
HO
HN
NH
OH
1 2
34
2
3 4
1 6
5
4'5'
6'
7'
2'
13' 10'
14'
11'
12'
7
89
11
10
2''
7''
6''5''
4''
Figure 1 Structures of representative asterriquinones. 1, terrequinone A; 2,
cochliodinol; 3, asterriquinone CT5; 4, asterriquinone B1.
NH
CO2H
NH2 TdiD
NH
CO2H
O
NH
HN O
O
OH
HO
TdiA
NH
HN O
O
HO
TdiBTdiCTdiE
9
141
7
Scheme 1 Overall reaction pathway to terrequinone A. 9, IPA; 14,
didemethylasterriquinone D; 1, terrequinone A.
ART ICL ES
NATURE CHEMICAL BIOLOGY VOLUME 3 NUMBER 9 SEPTEMBER 2007 5 8 5
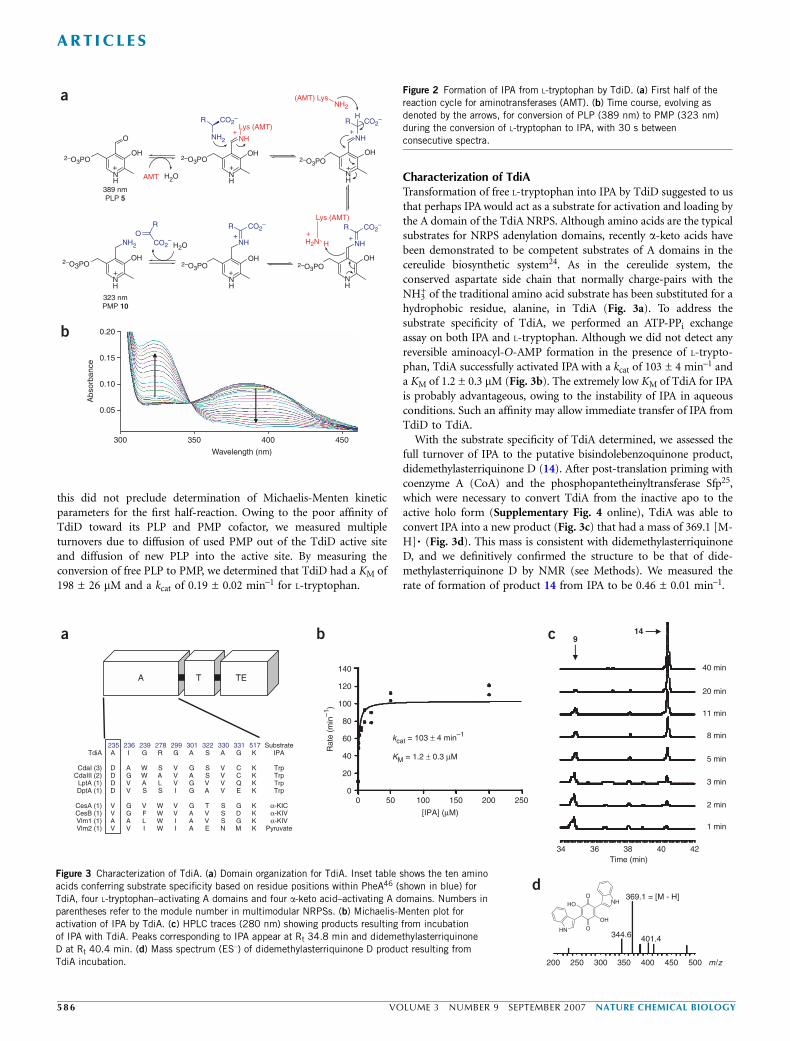
this did not preclude determination of Michaelis-Menten kineticparameters for the first half-reaction. Owing to the poor affinity ofTdiD toward its PLP and PMP cofactor, we measured multipleturnovers due to diffusion of used PMP out of the TdiD active siteand diffusion of new PLP into the active site. By measuring theconversion of free PLP to PMP, we determined that TdiD had a KM of198 ± 26 mM and a kcat of 0.19 ± 0.02 min–1 for L-tryptophan.
Characterization of TdiATransformation of free L-tryptophan into IPA by TdiD suggested to usthat perhaps IPA would act as a substrate for activation and loading bythe A domain of the TdiA NRPS. Although amino acids are the typicalsubstrates for NRPS adenylation domains, recently a-keto acids havebeen demonstrated to be competent substrates of A domains in thecereulide biosynthetic system24. As in the cereulide system, theconserved aspartate side chain that normally charge-pairs with theNH3
+ of the traditional amino acid substrate has been substituted for ahydrophobic residue, alanine, in TdiA (Fig. 3a). To address thesubstrate specificity of TdiA, we performed an ATP-PPi exchangeassay on both IPA and L-tryptophan. Although we did not detect anyreversible aminoacyl-O-AMP formation in the presence of L-trypto-phan, TdiA successfully activated IPA with a kcat of 103 ± 4 min–1 anda KM of 1.2 ± 0.3 mM (Fig. 3b). The extremely low KM of TdiA for IPAis probably advantageous, owing to the instability of IPA in aqueousconditions. Such an affinity may allow immediate transfer of IPA fromTdiD to TdiA.
With the substrate specificity of TdiA determined, we assessed thefull turnover of IPA to the putative bisindolebenzoquinone product,didemethylasterriquinone D (14). After post-translation priming withcoenzyme A (CoA) and the phosphopantetheinyltransferase Sfp25,which were necessary to convert TdiA from the inactive apo to theactive holo form (Supplementary Fig. 4 online), TdiA was able toconvert IPA into a new product (Fig. 3c) that had a mass of 369.1 [M-H]� (Fig. 3d). This mass is consistent with didemethylasterriquinoneD, and we definitively confirmed the structure to be that of dide-methylasterriquinone D by NMR (see Methods). We measured therate of formation of product 14 from IPA to be 0.46 ± 0.01 min–1.
NH
NH
OH
Lys (AMT)NH2
R CO2–
NH
NH+
+
OH2–O3PO
2–O3PO2–O3PO2–O3PO
R CO2– H
(AMT) LysNH2
NH
NH
OH
R CO2–CO2
–
CO2–
RLys (AMT)
+++
++
H2N H
NH
NH
OH
NH
NH2
OH
H2O
OR
323 nmPMP 10
0.20
0.15
0.10
Abs
orba
nce
0.05
300 350 400
Wavelength (nm)
450
NH
O
a
b
OH
+ +
+
2–O3PO 2–O3PO
389 nmPLP 5
H2OAMT
A T TE
235 236 239 278 299 301 322 330 331 517 SubstrateTdiA A I G R G A S A G K IPA
CdaI (3) D A W S V G S V C K TrpCdaIII (2) D G W A V A S V C K Trp
LptA (1) D V A L V G V V Q K TrpDptA (1) D V S S I G A V E K Trp
CesA (1) V G V W V G T S G K α-KICCesB (1) V G F W V A V S D K α-KIVVlm1 (1) A A L W I A V S G K α-KIVVlm2 (1) V V I W I A E N M K Pyruvate
0
20
40
60
80
100
120
140
0 50 100 150 200 250
[IPA] (µM)
Rat
e (m
in–1
)
1 min
2 min
3 min
5 min
8 min
11 min
40 min
Time (min)34 36 38 40 42
20 min
a b c 914
kcat = 103 ± 4 min–1
KM = 1.2 ± 0.3 µM
m/z200 250 300 350 400 450 500
369.1 = [M - H]
344.6 401.4
dNH
HN O
O
OH
HO
Figure 3 Characterization of TdiA. (a) Domain organization for TdiA. Inset table shows the ten amino
acids conferring substrate specificity based on residue positions within PheA46 (shown in blue) for
TdiA, four L-tryptophan–activating A domains and four a-keto acid–activating A domains. Numbers in
parentheses refer to the module number in multimodular NRPSs. (b) Michaelis-Menten plot for
activation of IPA by TdiA. (c) HPLC traces (280 nm) showing products resulting from incubation
of IPA with TdiA. Peaks corresponding to IPA appear at Rt 34.8 min and didemethylasterriquinone
D at Rt 40.4 min. (d) Mass spectrum (ES–) of didemethylasterriquinone D product resulting from
TdiA incubation.
Figure 2 Formation of IPA from L-tryptophan by TdiD. (a) First half of the
reaction cycle for aminotransferases (AMT). (b) Time course, evolving as
denoted by the arrows, for conversion of PLP (389 nm) to PMP (323 nm)
during the conversion of L-tryptophan to IPA, with 30 s between
consecutive spectra.
ART ICL ES
58 6 VOLUME 3 NUMBER 9 SEPTEMBER 2007 NATURE CHEMICAL BIOLOGY

We attempted to form a diaminobenzoquinone by incubating TdiAwith IPA imine generated in situ from coincubation of L-tryptophanand the amino acid oxidase RebO26. Under these conditions, only thepreviously observed dihydroxybenzoquinone core 14 formed (data notshown), probably arising from hydrolysis of the IPA imine to theketone before A domain activation. We presume the inability to formthe diaminobenzoquinone stems from the selectivity of the A domainfor the a-keto acid rather than the a-imino acid.
The symmetric connectivity of the two IPA molecules incorporatedinto the final product is thought to arise by head-to-tail dual Claisencondensations facilitated by the TE domain found at the C terminusof TdiA (Scheme 2). Indeed, when the active site Ser774 containedwithin the conserved GXSXGG motif was mutated to alanine, TdiAwas no longer capable of generating didemethylasterriquinone D fromIPA (Supplementary Fig. 5 online). To our knowledge, this is the firstinstance in which a TE domain has been found to catalyze carbon-carbon bond formation.
Reconstitution of terrequinone AbiosynthesisIn order to investigate the final stages ofterrequinone A biosynthesis, we probed theability of TdiB, TdiC and TdiE to turn overdidemethylasterriquinone D. Given the
homology of TdiC to zinc- and NADH (15)-dependent quinoneoxidoreductases, we included zinc sulfate and NADH in the reactionbuffer. To enable TdiB-mediated prenyltransferase activity, we alsoincluded DMAPP, magnesium chloride and TCEP, and to facilitate thepostulated methyltransferase activity of TdiE, we included S-adeno-sylmethionine (SAM). Under these conditions, didemethylasterriqui-none D was cleanly converted to terrequinone A (Fig. 4a), asconfirmed by NMR and mass spectroscopy (see Methods).
Terrequinone A formation was absolutely dependent on thepresence of DMAPP and NADH (Fig. 4b); however, when wesubstituted NADPH (16) for NADH, we observed an identicalproduct profile (data not shown). We included both divalent zincand magnesium, respectively, in the original reaction mixture to allowfor activity of the oxidoreductase TdiC and the prenyltransferaseTdiB. In the presence of magnesium, zinc was not required forturnover, but we observed a reduction in product formation whenwe omitted both salts from the reaction mixture. This result suggeststhat magnesium can substitute for zinc in the TdiC active site andthat zinc can substitute for magnesium to a certain extent. Theweak magnesium dependence presumably derives from TdiB activity,in line with observations of other reported prenyltransferases in whichmild rate enhancements up to threefold have been observed in thepresence of millimolar concentrations of magnesium or calciumions27–29. The use of magnesium chloride and calcium chloride at5 mM concentrations gave identical product profiles (data notshown); hence, we were able to use these salts interchangeably inassays of the TdiB/TdiC/TdiE enzyme system. Notably, despite thehomology of TdiE to SAM-dependent methyltransferases, TdiE is nota methyltransferase, as SAM was not required for terrequinone Aformation by the TdiB/TdiC/TdiE system (Fig. 4b). We were notentirely surprised by this, for two reasons: TdiE lacks the typical SAMbinding motifs30; and no orphan methyl groups are found in the finalnatural product scaffold.
The greatest yield of terrequinone A required the presence of allthree enzymes: TdiB, TdiC and TdiE (Fig. 4a). None of these inisolation catalyzed turnover of didemethylasterriquinone D. Similarly,the combination of TdiE with either of the other two enzymes gave
A
SH
OO
O
O NH
B
O
O
O
OHN
NH
SO
O
HONH
O
O
HN
B
HO
HN
H
HO
OH
O
OHN
NH
14
T TEA T TE
26
Scheme 2 Mechanism for formation of didemethylasterriquinone D (14)
by TdiA.
a b
Time (min)
11.0 12.0 13.0 14.0 15.0
All
–DMAPP
–NADH
–Mg/Zn
–Mg
–Zn
–SAM
Time (min)
11.0 12.0 13.0 14.0 15.0
BCE
BC
BE
CE
B
C
E
14 1 14
1
Figure 4 Characterization of TdiB/TdiC/TdiE
trienzyme system. HPLC traces (280 nm)
showing products resulting from incubation of
didemethylasterriquinone D with TdiB (5 mM),
TdiC (5 mM) and/or TdiE (5 mM) in the presence
of DMAPP (5 mM), magnesium chloride (5 mM),
zinc sulfate (200 mM), NADH (5 mM), SAM
(250 mM), TCEP (2.5 mM) and BSA (1 mg ml–1)
in 75 mM Tris (pH 8.0) and 10% (vol/vol)
DMSO. (a) Traces resulting from different
TdiB/TdiC/TdiE enzyme combinations.(b) Traces resulting from omission of various
cofactors. Peaks corresponding to starting
material 14 appear at Rt 11.4 min,
O-isopentenyldidemethylasterriquinone D at
Rt 11.7 min, ochrindole D at Rt 13.5 min and
terrequinone A at Rt 14.3 min.
ART ICL ES
NATURE CHEMICAL BIOLOGY VOLUME 3 NUMBER 9 SEPTEMBER 2007 5 8 7

no turnover at all. TdiB and TdiC in combination gave someterrequinone A, but product formation was vastly reduced relativeto the three-enzyme system.
Over the time course of the TdiB/TdiC/TdiE reaction, we observedan intermediate (at retention time (Rt) 13.5 min) forming and thendisappearing from the assay mixture, with apparent conversion toterrequinone A (Fig. 5a). This species proved to be isolable and wasidentified as compound 17 by NMR and mass spectroscopy. Thiscompound corresponds to ochrindole D, which had previously beenisolated from the sclerotia of Aspergillus ochraceus31, further demon-strating that these prenylated bisindolylquinone natural products arewidespread within Aspergilli. Ochrindole D (17) could in turn beconverted to terrequinone A by the action of TdiB alone; TdiC andTdiE were not required for this second prenylation reaction (Fig. 5b).
In the absence of TdiE, bisindolylquinone 14 was converted to amixture of products by TdiB and TdiC (Fig. 5c). The major two peakscorresponded to terrequinone A (Rt 14.3 min) and O-isopentenyldi-demethylasterriquinone D (18) (Rt 11.7 min), as identified by NMRand mass spectroscopy. After isolating 18, we demonstrated that thiscompound was not a substrate for any combination of TdiB, TdiC andTdiE (Fig. 5d). Compound 18 therefore represents a dead-end shuntproduct that cannot be converted to terrequinone A.
Overall, the TdiB/TdiC/TdiE enzyme system robustly convertsdidemethylasterriquinone D to terrequinone A, a transformationinvolving net double prenylation at two distinct sites of the parentscaffold. This activity is dependent on the presence of DMAPP andNAD(P)H and is boosted by divalent metal ions such as magnesium,calcium and/or zinc. In the absence of TdiE, product is formed, butsignificant amounts of a shunt metabolite (8) also form by an off-pathway irreversible O-prenylation event (Scheme 3).
DISCUSSIONTerrequinone A joins a growing family of bisindole alkaloids isolatedfrom a variety of marine and terrestrial sources with a wide spectrum ofpharmacological activities. This class of molecules includes the indolo-carbazoles rebeccamycin (19)32–34 and staurosporine (20)35–37, the
related bacterial pigment violacein (21)38, diketopiperazine-containing fellutarines39,40, piperazine-containing hamacanthins39–42
and dragmacidins39,40, amide-containing coscinamides39, imidizole-or oxozole-containing topsentins39,41,42 and nortopsentins39, imidazo-linone-containing rhopaladins39,40 and pyrimidine-containinghyrtinadines43. Of these, only the gene clusters and biochemicalcharacterization of rebeccamycin, staurosporine32–34,36,37 and viola-cein38 have previously been reported. In all three cases, the bisindolealkaloid is assembled by enzymatic oxidation of L-tryptophan to itsimine, followed by oxidative dimerization to a core that is elaborated byeven further oxidation.
The five-gene terrequinone A locus was recently identified based onthe fact that it is controlled by the A. nidulans global secondarymetabolite regulator LaeA10,11. Here we present in vitro characteriza-tion of the five fungal proteins (TdiA-TdiE) purified from E. coli.Among items of note are (i) the head-to-tail dimerization of tethereda-ketoacyl-NRPS intermediates on TdiA to set up the tetrasubstitutedbenzoquinone nucleus and (ii) bisprenylation by a single enzyme,TdiB. Terrequinone A, and presumably all the related asterriquinones,represent a new biosynthetic logic for nonoxidative dimerization ofL-tryptophan-derived monomers to produce bisindole alkaloids.
The terrequinone A pathway was predicted to start with TdiDacting as an L-tryptophan transaminase. Indeed, although TdiDpurified as the apoprotein from E. coli, it was PLP dependent in itsactivity in converting L-tryptophan to 9. Because the PLP was weaklybound, multiple turnovers occurred without the addition of an a-ketoacid cosubstrate and allowed determination of the kinetic parameters.
The use of a PLP-dependent transaminase as the first oxidative stepin the Tdi pathway is a meaningful divergence point for the flux ofL-tryptophan to 1 relative to the flux of L-tryptophan to rebeccamycin,staurosporine and violacein (Fig. 6), which use a flavoprotein oxi-dase26,32,38. In the latter case, the flavoproteins RebO, StaO and VioAyield the IPA imine as the initial oxidation product. Isomerization tothe eneamine tautomer slows hydrolysis, and one of the a-nitrogens ofthe two starting L-tryptophan molecules is incorporated into thedimerized scaffold32. By contrast, both amino nitrogens are removed
1 min
3 min
5 min
10 min
15 min
20 min
30 min
Time (min)11.0 12.0 13.0 14.0 15.0
1 min
3 min
5 min
10 min
15 min
20 min
30 min
Time (min)11.0 12.0 13.0 14.0 15.012.0 13.0 14.0 15.0
BCE
BC
BE
B
C
E
CE
None
Time (min)
BCE
BC
BE
B
C
E
CE
None *
*
*
*
*
*
*
*
12.0 13.0 14.0 15.0Time (min)
a b c d14 18 17 1 18 17 117 114 17 1
Figure 5 Turnover of intermediates on route to terrequinone A. (a) HPLC traces (280 nm) showing a time course for incubation with TdiB, TdiC and TdiE.
(b) Products resulting from incubation of ochrindole D (17) (Rt 14 min) with TdiB, TdiC and/or TdiE. (c) Time course for incubation with TdiB and TdiC.
(d) Products resulting from incubation of O-isopentenyldidemethylasterriquinone D (18) (Rt 12.2 min) with TdiB, TdiC and/or TdiE. In a and c, peaks
corresponding to starting material 14 appear at Rt 11.4 min, 18 at Rt 11.7 min, 17 at Rt 13.5 min and 1 at Rt 14.3 min. In b and d, peaks corresponding
to 17 appear at Rt 14.0 min, 18 at Rt 12.2 min and 1 at Rt 15.0 min. In d, 17 impurity (Rt 14 min) is converted to 1 (Rt 15 min) in presence of TdiB
(indicated by asterisks).
ART ICL ES
58 8 VOLUME 3 NUMBER 9 SEPTEMBER 2007 NATURE CHEMICAL BIOLOGY
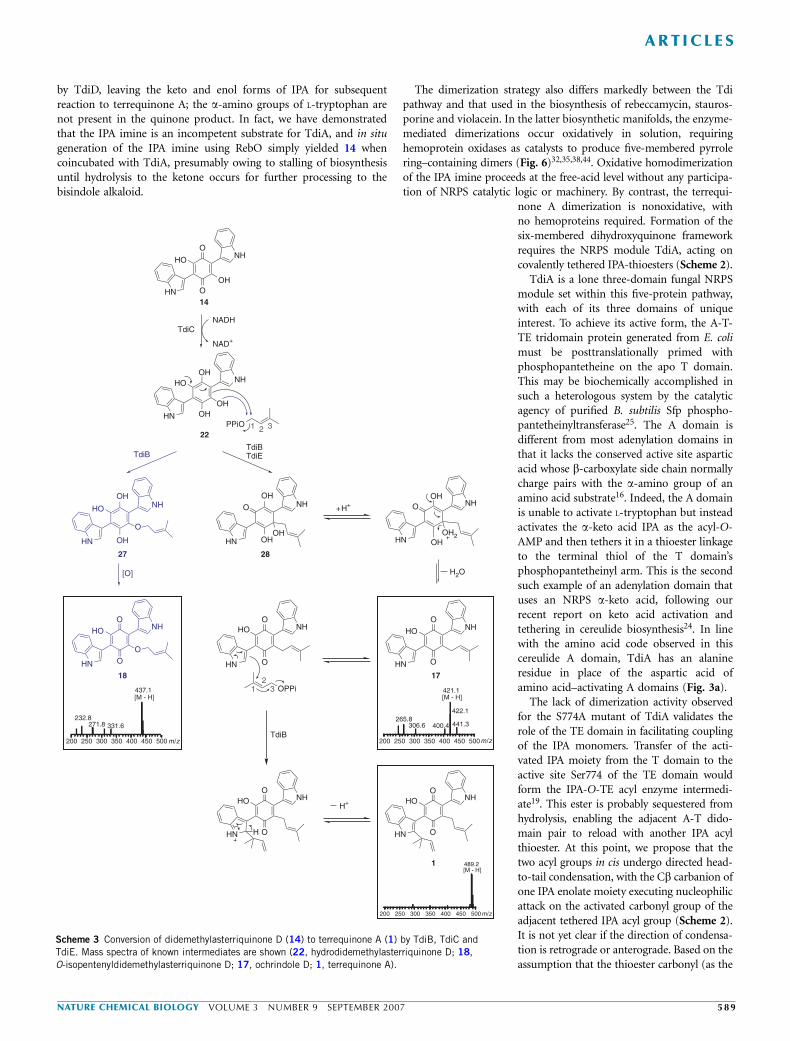
by TdiD, leaving the keto and enol forms of IPA for subsequentreaction to terrequinone A; the a-amino groups of L-tryptophan arenot present in the quinone product. In fact, we have demonstratedthat the IPA imine is an incompetent substrate for TdiA, and in situgeneration of the IPA imine using RebO simply yielded 14 whencoincubated with TdiA, presumably owing to stalling of biosynthesisuntil hydrolysis to the ketone occurs for further processing to thebisindole alkaloid.
The dimerization strategy also differs markedly between the Tdipathway and that used in the biosynthesis of rebeccamycin, stauros-porine and violacein. In the latter biosynthetic manifolds, the enzyme-mediated dimerizations occur oxidatively in solution, requiringhemoprotein oxidases as catalysts to produce five-membered pyrrolering–containing dimers (Fig. 6)32,35,38,44. Oxidative homodimerizationof the IPA imine proceeds at the free-acid level without any participa-tion of NRPS catalytic logic or machinery. By contrast, the terrequi-
none A dimerization is nonoxidative, withno hemoproteins required. Formation of thesix-membered dihydroxyquinone frameworkrequires the NRPS module TdiA, acting oncovalently tethered IPA-thioesters (Scheme 2).
TdiA is a lone three-domain fungal NRPSmodule set within this five-protein pathway,with each of its three domains of uniqueinterest. To achieve its active form, the A-T-TE tridomain protein generated from E. colimust be posttranslationally primed withphosphopantetheine on the apo T domain.This may be biochemically accomplished insuch a heterologous system by the catalyticagency of purified B. subtilis Sfp phospho-pantetheinyltransferase25. The A domain isdifferent from most adenylation domains inthat it lacks the conserved active site asparticacid whose b-carboxylate side chain normallycharge pairs with the a-amino group of anamino acid substrate16. Indeed, the A domainis unable to activate L-tryptophan but insteadactivates the a-keto acid IPA as the acyl-O-AMP and then tethers it in a thioester linkageto the terminal thiol of the T domain’sphosphopantetheinyl arm. This is the secondsuch example of an adenylation domain thatuses an NRPS a-keto acid, following ourrecent report on keto acid activation andtethering in cereulide biosynthesis24. In linewith the amino acid code observed in thiscereulide A domain, TdiA has an alanineresidue in place of the aspartic acid ofamino acid–activating A domains (Fig. 3a).
The lack of dimerization activity observedfor the S774A mutant of TdiA validates therole of the TE domain in facilitating couplingof the IPA monomers. Transfer of the acti-vated IPA moiety from the T domain to theactive site Ser774 of the TE domain wouldform the IPA-O-TE acyl enzyme intermedi-ate19. This ester is probably sequestered fromhydrolysis, enabling the adjacent A-T dido-main pair to reload with another IPA acylthioester. At this point, we propose that thetwo acyl groups in cis undergo directed head-to-tail condensation, with the Cb carbanion ofone IPA enolate moiety executing nucleophilicattack on the activated carbonyl group of theadjacent tethered IPA acyl group (Scheme 2).It is not yet clear if the direction of condensa-tion is retrograde or anterograde. Based on theassumption that the thioester carbonyl (as the
NH
HN O
O
OH
HO
NH
HN O
OHO
14
1
TdiCNADH
NH
HN OH
OH
OH
HO
PPiO22
TdiBTdiBTdiE
NH
HN OH
OH
O
HO
[O]
NH
HN O
O
O
HO
NH
HN OH
OH
O
OH
+H+ NH
HN OH
OHO
OH2
NH
HN O
O
HO
OPPi
NH
HN O
OHO
TdiB
NH
HN O
OHO
H
1718
1 2 3
12
3
NAD+
H+
200 250 300 350 400 450 500
437.1 [M - H]
232.8271.8 331.6
m/z 200 250 300 350 400 450 500
421.1 [M - H]
422.1265.8
441.3306.6 400.4
m/z
200 250 300 350 400 450 500
489.2 [M - H]
m/z
2827
H2O
Scheme 3 Conversion of didemethylasterriquinone D (14) to terrequinone A (1) by TdiB, TdiC and
TdiE. Mass spectra of known intermediates are shown (22, hydrodidemethylasterriquinone D; 18,
O-isopentenyldidemethylasterriquinone D; 17, ochrindole D; 1, terrequinone A).
ART ICL ES
NATURE CHEMICAL BIOLOGY VOLUME 3 NUMBER 9 SEPTEMBER 2007 5 8 9

more thermodynamically activated carbonyl) behaves as the electro-phile, the upstream IPA moiety would be transferred to form a lineartethered dimer in the TE active site. A second, now intramolecular,attack of an IPA enolate on the ester carbonyl will complete thecyclization and release of the dihydroxyquinone 14 from the TE activesite (Scheme 2).
This one NRPS module in TdiA thus activates two keto acids,tethers them sequentially and holds them in cis, one on the T domainas a thioester and one on the TE domain as an oxoester. The TEdomain then directs a double Claisen condensation to generate twocarbon-carbon bonds, fashion the quinone nucleus and catalyze thesequential disconnection of both covalent attachments to the protein.This represents a new disconnection mechanism for an assembly linechain-terminating TE domain. It is probable that all asterriquinonesare generated by such NRPS-mediated dimerization in a deceptivelysimple three-domain, one-module NRPS protein. To our knowledge,this is the first example of a TE domain that is capable of catalyzingcarbon-carbon bond formation.
TdiC, a NADH-dependent oxidoreductase that aligns with knownzinc- and NADH-dependent quinone reductases, acts on the nascentbisindolylquinone scaffold, didemethylasterriquinone D. TdiC-mediated hydride addition to the tetrasubstituted quinone wouldcreate the hydroquinone oxidation state (hydrodidemethylasterriqui-none D; 22) (Scheme 3). This reduction is required to reverse thepolarity of the electrophilic quinone to the nucleophilic hydroquinone22 that acts as a carbanion equivalent for the first prenylation event atC2 of the original quinone nucleus. This occurs by attack on C1 of theallylic cation-like transition state derived from DMAPP in the activesite of the prenyltransferase TdiB. Elimination of water would result innet replacement of a hydroxyl with a prenyl at C2 of the quinone ring.This monoprenylated quinone 17 can be detected and isolated inHPLC assays as an intermediate that builds up and then goes on to thefinal product 1.
In the absence of TdiE, a second monoprenylated species 18 buildsup in the reaction mixture. Compound 18 is prenylated instead on theoxygen at C2 of the dihydroxyquinone. This is an irreversible O-alkylation that is off pathway and competes with the C2 carbonprenylation. This competition between on-pathway C2 prenylation
and off-pathway O-prenylation is where the protein TdiE acts, shunt-ing the flux of the reaction down the productive pathway by an as yetunknown mechanism. TdiE could be a chaperone or partner protein,presenting the bisindolylhydroquinone 22 to the prenyltransferaseTdiB, but that will be the subject of future investigations. We havedemonstrated that TdiE does not act as a methyltransferase, to whichit has weak homology, because SAM is not a required cofactor in theproduction of terrequinone A. This is not entirely surprising, giventhe absence of any orphan methyl groups in the natural product 1.TdiE is reminiscent of VioE in the bisindole alkaloid violaceinpathway, in that VioE is required for shuttling a reactive inter-mediate toward an on-pathway product rather than a dead-endshunt product38.
After isolation of intermediate 17, it can be converted to the finaldoubly prenylated natural product 1 by the action of TdiB andDMAPP alone. Neither TdiC nor TdiE has any detectable role inthis second prenylation step. The monoprenyl benzoquinone nucleusof 17 is again electrophilic and is not a suitable partner for reactionwith another molecule of DMAPP. Instead, the indole ring, a knownnucleophilic site for enzymatic prenylations in other natural products,acts as the reaction partner. In this case, nucleophilic attack occurs viathe indole C2, with DMAPP captured this time at C3 rather than C1,yielding terrequinone A via a reverse prenylation reaction. Thus, TdiBcan carry out two consecutive prenyl transfers to the bisindolylqui-none scaffold of 14: the first prenyl moiety affixes to the hydroquinonenucleus and the second to one of the indole rings directly. Further-more, the versatility of TdiB is emphasized by the observation thatDMAPP can be captured regioselectively at C1 in the first prenylationand then at C3 in the second prenylation. How the single enzyme TdiBcan mediate these two distinct prenylations is worthy of futurestructural and mechanistic study; in particular, the facial selectivityof attack on DMAPP and the concerted or stepwise nature of theseprocesses are of interest.
This is the first case of a prenyltransferase acting iteratively on anatural product scaffold. This is in contrast to the A. fumigatuspathway to fumigaclavine C (23) in which two prenylations, onewith C1 connectivity29 and the other with reverse C3 connectivity45,require two separate prenyltransferases. Although we do not think itsurprising that a single prenyltransferase could be responsible for twoprenylations in other fully symmetrical asterriquinones with identicalprenyl connectivities, the marked asymmetry of 1 emphasizes theversatility of TdiB.
Comparison of the RebD/VioB-mediated oxidative dimerizationroute for two IPA imines with the NRPS-mediated nonoxidativedimerization of IPA-S-enzyme intermediates by TdiA uncovers distinctring sizes and nitrogen contents in the coupled products as well asdiverse orientations of the monomers. The head-to-head heme pro-tein–mediated coupling of the Reb and Vio pathways probably involvesprior one-electron oxidation of each of the IPA eneamine molecules,followed by Cb radical coupling. One of the two nitrogens subse-quently gets eliminated as NH3, while the other acts as a nucleophile ina C-N bond-forming step to give the pyrrole ring. The head-to-tailpathway of the TdiA-mediated coupling gives a six-membered (ratherthan five-membered) ring system. As proposed above, this probablyoccurs by tandem enolate-mediated Claisen condensations on theT- and TE domain-tethered IPA monomers. The TdiA-mediatedcoupling is nonoxidative, in contrast to the oxidative RebD/VioBcouplings, and the subsequent TdiC step is reductive. This reflectsnature’s chemical versatility in its use of distinct enzymes and coen-zymes to control fates and redox states in the dimerization ofL-tryptophan to different natural product scaffolds.
HOO
OHN
NHHN O
NH
HN
O
OH
121
NN
HN O
O
HN
MeO
H
19
NNH
HN OO
Cl Cl
O OH
OH
OMe
HO
20
Figure 6 Biochemically characterized natural products derived fromL-tryptophan dimerization. 19, staurosporine; 20, rebeccamycin; 21,
violacein; 1, terrequinone A. Atoms derived from individual L-tryptophan
monomers are depicted in blue and red.
ART ICL ES
59 0 VOLUME 3 NUMBER 9 SEPTEMBER 2007 NATURE CHEMICAL BIOLOGY

In summary, the Tdi pathway is an intersection of NRPSand isoprenoid biosynthetic machinery, where TdiA and TdiB arenoteworthy catalysts. The bisindolylbenzoquinone framework is con-structed by IPA dimerization on a single NRPS module (TdiA),whereas distinct tandem prenylation regiochemistries are catalyzedby TdiB. In characterizing the biosynthetic pathway to terrequinone A,this work sets the stage for future mechanistic work to uncover moredetails about these enzymes.
METHODSGeneral materials. Bacterial strains, plasmids, materials and instrumentation
are described in Supplementary Methods online.
Cloning and expression of TdiA-TdiE. All fungal DNA isolation and mani-
pulation, as well as cloning and expression of TdiA-TdiE, are described in
Supplementary Methods.
Compound synthesis and characterization. Preparation of dimethylallyl
diphosphate, as well as isolation of 14, 1, 17 and 18 from TdiA, TdiB, TdiC
and TdiE incubations, are described in Supplementary Methods.
Characterization of TdiD. Titration reactions measuring PLP binding by TdiD
contained 40 mM TdiD, 25 mM HEPES (pH 7.75) and 150 mM NaCl. PLP was
titrated in 0.1 equivalents at a time, and the change of absorbance at 416 nm,
which is indicative of the PLP-enzyme complex, was monitored by UV-visible
spectrophotometry.
Reactions to determine the KM of TdiD toward L-tryptophan contained
25 mM HEPES (pH 7.75), 150 mM NaCl, 250 mM PLP, 2 mM TdiD and various
concentrations of L-tryptophan ranging from 1–500 mM. The increase in
absorbance at 323 nm, which is indicative of PMP formation, was monitored
continuously for 20 min by UV-visible spectrophotometry.
Reactions to determine the kcat of TdiD toward L-tryptophan contained
25 mM HEPES (pH 7.75), 150 mM NaCl, 200 mM PLP, 10 mM TdiD and
various concentrations of L-tryptophan ranging from 50–1,000 mM. The
decrease in absorbance at 389 nm, which is indicative of PLP consumption, was
monitored continuously for 12 min by UV-visible spectrophotometry
(e389 ¼ 5,618 M–1 cm–1).
Reactions to determine possible a-keto acid substrates for regenerating PLP
from PMP after reaction with L-tryptophan contained 25 mM HEPES
(pH 7.75), 150 mM NaCl, 250 mM PLP, 500 mM L-tryptophan, 30 mM TdiD
and 500 mM a-keto acid (pyruvate, a-ketoglutarate and a-keto-g-(methylthio)-
butyrate). At various time points ranging between 10 and 180 min, 10 ml was
quenched into 100 ml MeOH and centrifuged at 16,000g to pellet the protein.
The supernatant containing the product was analyzed by HPLC on a Phenom-
enex 250 � 4.6 mm C18 5m Luna column using a gradient of 0%–50%
acetonitrile over 20 min starting in 0.1% TFA in H2O; absorbance at 280 nm
was monitored.
ATP-PPi exchange assay for TdiA A domain substrate specificity. Reactions
(100 ml) contained 75 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM DTT, 5 mM
ATP, 1 mM sodium [32P]pyrophosphate (0.18 mCi) and 0.1 mM TdiA, with
substrate concentrations ranging from 0.1–500 mM for IPA and 0.05–17 mM
for L-tryptophan. Reactions were incubated at 25 1C for 5 min (IPA) or 15 min
(L-tryptophan) and were quenched by addition of 500 ml 1.6% (wt/vol)
activated charcoal, 200 mM tetrasodium pyrophosphate and 3.5% (vol/vol)
perchloric acid in water. The charcoal was pelleted by centrifugation and
washed twice with 500 ml 200 mM tetrasodium pyrophosphate and 3.5%
perchloric acid in water. The radioactivity bound to the charcoal was then
measured by liquid scintillation counting. Note that enzyme concentrations
and reaction times were chosen such that ATP-PPi exchange remained under
10% of equilibrium levels.
Didemethylasterriquinone D formation by TdiA. Reactions to measure
product formation by TdiA contained 50 mM Tris (pH 8.0), 10 mM MgCl2,
200 mM CoA, 5 mM TdiA and 3 mM Sfp. After a 45-min incubation at 25 1C to
prime the T domain, the reaction was initiated by addition of 5 mM ATP and
200 mM IPA. At desired time points ranging from 1–120 min, 100 ml were
quenched into 200 ml MeOH and then centrifuged at 16,000g to pellet protein.
The supernatant containing the product was analyzed by HPLC on a Phenom-
enex 250 � 4.6 mm C18 5m Luna column using a gradient of 0%–100%
acetonitrile over 50 min starting in 0.1% TFA in H2O; absorbance at 280 nm
was monitored.
Reaction of TdiA with imino-IPA. Reactions to measure possible formation of
a diaminobenzoquinone core contained 50 mM Tris (pH 8.0), 10 mM MgCl2,
200 mM CoA, 5 mM TdiA and 3 mM Sfp. After a 45-min incubation at 25 1C to
prime the T domain, the reaction was initiated by addition of 5 mM ATP,
500 mM Trp, 5 mM RebO and 5 mM NH4Cl. After incubation for 5 h at 25 1C,
the 100-ml reaction was quenched into 200 ml MeOH and centrifuged at 16,000g
to pellet protein. The supernatant containing the product, combined with a
50-ml DMSO wash of the pellet, was analyzed by HPLC on a Higgins Analytical
50 � 4.6 mm C18 5m CLIPEUS column using a gradient of 0%–100%
acetonitrile over 15 min starting in 0.1% TFA in H2O, monitoring absorbance
at 280 nm.
Conversion of didemethylasterriquinone D into terrequinone A. Reactions
used to convert didemethylasterriquinone D into terrequinone A contained
100 mM didemethylasterriquinone D, 500 mM DMAPP, 5 mM MgCl2, 75 mM
Tris (pH 8.0), 2.5 mM TCEP, 1 mg ml–1 BSA, 200 mM ZnSO4, 5 mM NADH,
250 mM SAM, 2 mM TdiB, 2 mM TdiC, 2 mM TdiE and 10% (vol/vol) DMSO in
a 100-ml volume. In order to test for necessary cofactors and proteins, reactions
were repeated omitting DMAPP, MgCl2, ZnSO4, NADH, SAM, MgCl2 and
ZnSO4, TdiB, TdiC, TdiE or combinations of the three enzymes. After 2 h,
reactions were quenched with 200 ml methanol and centrifuged at 16,000g to
pellet protein. The supernatant containing the product combined with a 50-ml
DMSO wash of the pellet was analyzed by HPLC on a Higgins Analytical
50 � 4.6 mm C18 5m CLIPEUS column using a gradient of 0%–100%
acetonitrile over 15 min starting in 0.1% TFA in H2O; absorbance at 280 nm
was monitored.
Accession codes. The correct cDNA sequences for tdiA–tdiE have been
deposited in GenBank (tdiA: EF550581; tdiB: EF550582; tdiC: EF550583; tdiD:
EF550584; tdiE: EF550585).
Note: Supplementary information and chemical compound information is available onthe Nature Chemical Biology website.
ACKNOWLEDGMENTSWe gratefully acknowledge the National Institutes of Health grant GM 20011(to C.T.W.) and a Department of Defense National Defense Science andEngineering Graduate Fellowship (to C.J.B.). We thank P.D. Straight fordiscussions and for providing a sample of A. nidulans strain A4, and we thankJ.A. Read for his critical reading of this manuscript.
AUTHOR CONTRIBUTIONSC.J.B. and A.R.H.-J. contributed equally to this work.
COMPETING INTERESTS STATEMENTThe authors declare no competing financial interests.
Published online at http://www.nature.com/naturechemicalbiology
Reprints and permissions information is available online at http://npg.nature.com/
reprintsandpermissions
1. Jerram, W.A. et al. The chemistry of cochliodinol, a metabolite of Chaetomium spp.Can. J. Chem. 53, 727–737 (1975).
2. Meiler, D. & Taylor, A. The effect of cochliodinol, a metabolite of Chaetomiumcochliodes, on the respiration of microsopores of Fusarium oxysporum. Can. J.Microbiol. 17, 83–86 (1971).
3. Brewer, D., Jerram, W.A. & Taylor, A. The production of cochliodinol and a relatedmetabolite by Chaetomium species. Can. J. Microbiol. 14, 861–866 (1968).
4. Yamamoto, Y., Kiriyama, S., Shimizu, S. & Koshimura, S. Antitumor activity ofasterrequinone, a metabolic product from Aspergillus terreus. Gann 67, 623–624(1976).
5. Arai, K. et al. Metabolic products of Aspergillus terreus. IV. Metabolite of the strain IFO8835. (2). The isolation and structure of indolyl benzoquinone pigments. Chem.Pharm. Bull. (Tokyo) 29, 961–969 (1981).
6. Mocek, U. et al. Isolation and structure elucidation of five new asterriquinones fromAspergillus, Humicola and Botryotrichum species. J. Antibiot. (Tokyo) 49, 854–859(1996).
ART ICL ES
NATURE CHEMICAL BIOLOGY VOLUME 3 NUMBER 9 SEPTEMBER 2007 5 9 1

7. Kaji, A., Saito, R., Nomura, M., Miyamoto, K. & Kiriyama, N. Mechanism of thecytotoxicity of asterriquinone, a metabolite of Aspergillus terreus. Anticancer Res. 17,3675–3679 (1997).
8. Shimizu, S., Yamamoto, Y., Inagaki, J. & Koshimura, S. Antitumor effect and structure-activity relationship of asterriquinone analogs. Gann 73, 642–648 (1982).
9. He, J. et al. Cytotoxic and other metabolites of Aspergillus inhabiting the rhizosphere ofSonoran desert plants. J. Nat. Prod. 67, 1985–1991 (2004).
10. Bouhired, S., Weber, M., Kempf-Sontag, A., Keller, N.P. & Hoffmeister, D. Accurateprediction of the Aspergillus nidulans terrequinone gene cluster boundaries using thetranscriptional regulator LaeA. Fungal Genet. Biol. (2007).
11. Bok, J.W. et al. Genomic mining for Aspergillus natural products. Chem. Biol. 13,31–37 (2006).
12. Marahiel, M.A., Stachelhaus, T. & Mootz, H.D. Modular peptide synthetases involved innonribosomal peptide synthesis. Chem. Rev. 97, 2651–2674 (1997).
13. Stein, T. et al. The multiple carrier model of nonribosomal peptide biosynthesis atmodular multienzymatic templates. J. Biol. Chem. 271, 15428–15435 (1996).
14. Belshaw, P.J., Walsh, C.T. & Stachelhaus, T. Aminoacyl-CoAs as probes of condensationdomain selectivity in nonribosomal peptide synthesis. Science 284, 486–489 (1999).
15. Stachelhaus, T., Mootz, H.D., Bergendahl, V. & Marahiel, M.A. Peptide bond formationin nonribosomal peptide biosynthesis. Catalytic role of the condensation domain.J. Biol. Chem. 273, 22773–22781 (1998).
16. Conti, E., Stachelhaus, T., Marahiel, M.A. & Brick, P. Structural basis for the activationof phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16,4174–4183 (1997).
17. Stachelhaus, T., Huser, A. & Marahiel, M.A. Biochemical characterization of peptidylcarrier protein (PCP), the thiolation domain of multifunctional peptide synthetases.Chem. Biol. 3, 913–921 (1996).
18. Weber, T., Baumgartner, R., Renner, C., Marahiel, M.A. & Holak, T.A. Solution structureof PCP, a prototype for the peptidyl carrier domains of modular peptide synthetases.Structure 8, 407–418 (2000).
19. Keating, T.A. & Walsh, C.T. Initiation, elongation, and termination strategies inpolyketide and polypeptide antibiotic biosynthesis. Curr. Opin. Chem. Biol. 3,598–606 (1999).
20. Williams, R.M., Stocking, E.M. & Sanz-Cervera, J.F. Biosynthesis of prenylatedalkaloids derived from tryptophan. Top. Curr. Chem. 209, 97–173 (2000).
21. Dolence, J.M. & Poulter, C.D. in Comprehensive Natural Product Chemistry (ed.Meth-Cohn, O.) 18473–18500 (Elsevier, Oxford, 1999).
22. Grundmann, A. & Li, S.M. Overproduction, purification and characterization ofFtmPT1, a brevianamide F prenyltransferase from Aspergillus fumigatus. Microbiology151, 2199–2207 (2005).
23. Eliot, A.C. & Kirsch, J.F. Pyridoxal phosphate enzymes: mechanistic, structural, andevolutionary considerations. Annu. Rev. Biochem. 73, 383–415 (2004).
24. Magarvey, N.A., Ehling-Schulz, M. & Walsh, C.T. Characterization of the cereulideNRPS alpha-hydroxy acid specifying modules: activation of alpha-keto acids and chiralreduction on the assembly line. J. Am. Chem. Soc. 128, 10698–10699 (2006).
25. Quadri, L.E. et al. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyltransferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry37, 1585–1595 (1998).
26. Nishizawa, T., Aldrich, C.C. & Sherman, D.H. Molecular analysis of the rebeccamycinL-amino acid oxidase from Lechevalieria aerocolonigenes ATCC 39243. J. Bacteriol.187, 2084–2092 (2005).
27. Gebler, J.C. & Poulter, C.D. Purification and characterization of dimethylallyl trypto-phan synthase from Claviceps purpurea. Arch. Biochem. Biophys. 296, 308–313(1992).
28. Lee, S.L., Floss, H.G. & Heinstein, P. Purification and properties of dimethylallylpyro-phosphate:tryptopharm dimethylallyl transferase, the first enzyme of ergot alkaloidbiosynthesis in Claviceps. sp. SD 58. Arch. Biochem. Biophys. 177, 84–94(1976).
29. Unsold, I.A. & Li, S.M. Overproduction, purification and characterization of FgaPT2, adimethylallyltryptophan synthase from Aspergillus fumigatus. Microbiology 151,1499–1505 (2005).
30. Hamahata, A., Takata, Y., Gomi, T. & Fujioka, M. Probing the S-adenosylmethionine-binding site of rat guanidinoacetate methyltransferase. Effect of site-directedmutagenesis of residues that are conserved across mammalian non-nucleic acidmethyltransferases. Biochem. J. 317, 141–145 (1996).
31. de Guzman, F.S. et al. Ochrindoles A-D: new bis-indolyl benzenoids from the sclerotiaof Aspergillus ochraceus NRRL 3519. J. Nat. Prod. 57, 634–639 (1994).
32. Howard-Jones, A.R. & Walsh, C.T. Enzymatic generation of the chromopyrrolic acidscaffold of rebeccamycin by the tandem action of RebO and RebD. Biochemistry 44,15652–15663 (2005).
33. Onaka, H., Taniguchi, S., Igarashi, Y. & Furumai, T. Characterization of the biosyntheticgene cluster of rebeccamycin from Lechevalieria aerocolonigenes ATCC 39243. Biosci.Biotechnol. Biochem. 67, 127–138 (2003).
34. Sanchez, C. et al. The biosynthetic gene cluster for the antitumor rebeccamycin:characterization and generation of indolocarbazole derivatives. Chem. Biol. 9,519–531 (2002).
35. Asamizu, S., Kato, Y., Igarashi, Y., Furumai, T. & Onaka, H. Direct formation ofchromopyrrolic acid from indole-3-pyruvic acid by StaD, a novel hemoprotein inindolocarbazole biosynthesis. Tetrahedr. Lett. 47, 473–475 (2006).
36. Howard-Jones, A.R. & Walsh, C.T. Staurosporine and rebeccamycin aglyconesare assembled by the oxidative action of StaP, StaC, and RebC on chromopyrrolicacid. J. Am. Chem. Soc. 128, 12289–12298 (2006).
37. Onaka, H., Taniguchi, S., Igarashi, Y. & Furumai, T. Cloning of the staurosporinebiosynthetic gene cluster from Streptomyces sp. TP-A0274 and its hetero-logous expression in Streptomyces lividans. J. Antibiot. (Tokyo) 55, 1063–1071(2002).
38. Balibar, C.J. & Walsh, C.T. In vitro biosynthesis of violacein from L-tryptophan by theenzymes VioA-E from Chromobacterium violaceum. Biochemistry 45, 15444–15457(2006).
39. Hibino, S. & Choshi, T. Simple indole alkaloids and those with a nonrearrangedmonoterpenoid unit. Nat. Prod. Rep. 19, 148–180 (2002).
40. Sanchez, C., Mendez, C. & Salas, J.A. Indolocarbazole natural products: occurrence,biosynthesis, and biological activity. Nat. Prod. Rep. 23, 1007–1045 (2006).
41. Bao, B. et al. Bisindole alkaloids of the topsentin and hamacanthin classes from amarine sponge Spongosorites sp. J. Nat. Prod. 70, 2–8 (2007).
42. Oh, K.B. et al. Bis(indole) alkaloids as sortase A inhibitors from the sponge Spongo-sorites sp. Bioorg. Med. Chem. Lett. 15, 4927–4931 (2005).
43. Endo, T., Tsuda, M., Fromont, J. & Kobayashi, J. Hyrtinadine A, a bis-indole alkaloidfrom a marine sponge. J. Nat. Prod. 70, 423–424 (2007).
44. Nishizawa, T., Gruschow, S., Jayamaha, D.H., Nishizawa-Harada, C. & Sherman, D.H.Enzymatic assembly of the bis-indole core of rebeccamycin. J. Am. Chem. Soc. 128,724–725 (2006).
45. Unsold, I.A. & Li, S.M. Reverse prenyltransferase in the biosynthesis of fumigaclavineC in Aspergillus fumigatus: gene expression, purification, and characterization offumigaclavine C synthase FGAPT1. ChemBioChem 7, 158–164 (2006).
46. Stachelhaus, T., Mootz, H.D. & Marahiel, M.A. The specificity-conferring code ofadenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6, 493–505(1999).
ART ICL ES
59 2 VOLUME 3 NUMBER 9 SEPTEMBER 2007 NATURE CHEMICAL BIOLOGY





























