Embed Size (px)
Citation preview

Structure
Article
TheArchitectureofCopA fromArcheaoglobus fulgidusStudied by Cryo-Electron Microscopyand Computational DockingGregory S. Allen,1 Chen-Chou Wu,1 Tim Cardozo,3 and David L. Stokes1,2,4,*1Skirball Institute2Department of Cell Biology3Department of PharmacologyNew York University School of Medicine, 540 First Avenue, New York, NY 10016, USA4New York Structural Biology Center, 89 Convent Avenue, New York, NY 10027, USA
*Correspondence: [email protected]
DOI 10.1016/j.str.2011.05.014
SUMMARY
CopA uses ATP to pumpCu+ across cell membranes.X-ray crystallography has defined atomic structuresof several related P-type ATPases. We have deter-mined a structure of CopA at 10 A resolution bycryo-electron microscopy of a new crystal form andused computational molecular docking to study theinteractions between the N-terminal metal-bindingdomain (NMBD) and other elements of the molecule.We found that the shorter-chain lipids used toproduce these crystals are associated with move-ments of the cytoplasmic domains, with a noveldimer interface and with disordering of the NMBD,thus offering evidence for the transience of its inter-action with the other cytoplasmic domains. Dockingidentified a binding site that matched the location ofthe NMBD in our previous structure by cryo-electronmicroscopy, allowing a more detailed view of itsbinding configuration and further support for itsrole in autoinhibition.
INTRODUCTION
Copper is essential to cells as a cofactor for a wide variety of
enzymes such as superoxide dismutase and cytochrome
oxidase. If not carefully controlled, however, Cu can be toxic,
due to its redox potential and its ability to produce free radicals
(Rae et al., 1999). As a result, a system of pumps, transporters,
and metallochaperones has evolved to control the delivery and
distribution of Cu. In general, intracellular Cu scavengers, such
as metallothioneins, ensure that there is a vanishingly low
concentration of free Cu in the cytoplasm (Rae et al., 1999),
and metallochaperones, such as Atox1, CCS, and Sco1/2, are
used to carry the Cu through the cytoplasm and deliver it to
specific targets (Lutsenko et al., 2007). In mammals, Ctr1 is
a secondary transporter on the cell surface that facilitates entry
into the cell (Lee et al., 2001), and two transmembrane
ATPases, ATP7A and ATP7B, pump Cu across the endoplasmic
reticulum and plasma membrane, respectively (Lutsenko et al.,
Structure 19, 1219–12
2007). Mutations in ATP7A give rise to Menkes disease, resulting
from insufficient delivery of Cu in brain and other tissues,
whereas mutations to ATP7B are responsible for Wilson’s
disease, where Cu overload is responsible for liver and brain
dysfunction (Barry et al., 2010; Veldhuis et al., 2009a).
ATP7A andATP7B are homologous to CopA frombacteria and
belong to the family of P-type ATPases (Lutsenko and Kaplan,
1995). This family comprises ATP-dependent, transmembrane
ion pumps that share a common reaction mechanism, mem-
brane topology, domain organization, as well as conserved
sequence motifs implicated in ATP hydrolysis and in formation
of their hallmark phosphoenzyme intermediate (Kuhlbrandt,
2004; Møller et al., 1996). Phylogenetic analysis of P-type
ATPases delineates five subgroups, denoted PI–PV (Axelsen
and Palmgren, 1998). CopA, ATP7A, and ATP7B are in the PIB
subfamily together with transporters of a diverse array of
transition and heavy metal ions such as Cu+, Cu2+, Zn2+, Cd2+,
Co2+, and Pb2+.
PII ATPases are themost thoroughly studied and have become
paradigms for the family. They includeCa2+-ATPase andNa+/K+-
ATPase, which have ten transmembrane helices (M1–10) and two
main cytoplasmic loops inserted between M2/M3 and M4/M5.
The larger cytoplasmic loop (between M4/M5) folds into two
separate domains, thus composing the nucleotide-binding (N)
domain and phosphorylation (P) domain, whereas the smaller
loop (between M2/M3) composes the so-called actuator (A)
domain.Eachdomain ischaracterizedbysequencemotifs related
to their individual roles in the reaction cycle, namely DKTGTLT at
the phosphorylation site of the P domain, isolated residues
surrounding the ATP binding site in the N domain, and the
conserved TGE in the A domain (Olesen et al., 2007). The well-
characterized reactioncyclealternatesbetweenE1andE2states,
in which transmembrane ion binding sites are oriented toward the
intracellular or extracellular milieu, respectively. Switching
between these two states is controlled by phosphorylation of
the catalytic aspartate in the conserved DKTGTLT sequence,
which instigatescoordinatedmovementsofN,P, andAand trans-
membrane domains. By coordinating changes in the affinity and
accessibility of the ion binding sites, ions are transported against
a concentration gradient: that is, E1 binds cytoplasmic ions with
high affinity, whereas E2 binds extracellular (or luminal) ions with
much lower affinity (Jørgensen and Andersen, 1988).
32, September 7, 2011 ª2011 Elsevier Ltd All rights reserved 1219

Structure
Cryo-EM and Computational Docking of CopA
PIB ATPases such asCopA, although functionally and structur-
ally related to PII ATPases, are distinguished by having only eight
transmembrane helices and by bearing one or more N-terminal
metal-binding domains (NMBDs). These NMBDs are homolo-
gous to soluble metallochaperones that carry copper through
the cytoplasm (e.g., Atox1). Both NMBDs and metallochaper-
ones bind Cu+ with high affinity via CxxC sequence motifs.
Although it has been postulated that NMBDs mediate transfer
of Cu+ from the metallochaperones to transport sites, there is
increasing evidence that these domains are instead involved in
autoregulation and, in the case of ATP7A, in targeting the mole-
cule to the basolateral membrane (see reviews by Arguello et al.,
2007; Lutsenko et al., 2007). The structural basis for this autore-
gulation remains to be determined.
X-ray structures have been solved for several intact PII
ATPases, including Ca2+-ATPase from sarcoplasmic reticulum
(SERCA1) (Toyoshima et al., 2000), Na+/K+-ATPase (Morth
et al., 2007), andH+-ATPase (Pedersen et al., 2007). These struc-
tures have demonstrated the nature of the E1 and E2 states as
well as the role of conformational changes in coordinating the
multiple steps of the enzymatic cycle (Olesen et al., 2007).
X-ray structures of PIB-type ATPases have so far only involved
isolated cytoplasmic domains of CopA from Archeaoglobus
fulgidus: specifically, a construct containing the N and P
domains (Sazinsky et al., 2006b; Tsuda and Toyoshima, 2009)
and another construct of the A domain alone (Sazinsky et al.,
2006a). NMR has revealed the binding of ATP to the isolated N
domain (Banci et al., 2010) and has characterized the structural
homology between various NMBDs and the soluble metallocha-
perones (e.g., Banci et al., 2006). Our previous reconstruction of
CopA using cryo-electron microscopy (cryo-EM) is the only
structure of an intact PIB-type ATPase. Although the resolution
was limited (�17 A), this reconstruction served as a template
for building a CopA homology model that defined the location
of the NMBD relative to the other cytoplasmic domains and sug-
gested that the NMBD could regulate CopA activity by holding
the enzyme in an inactive state in the absence of copper and
ATP (Wu et al., 2008). A different conclusion was reached by
a more recent study employing chemical crosslinking, which
suggests that the NMBD binds to the opposite side of the mole-
cule and represents a static structural element of the cyto-
plasmic domain (Lubben et al., 2009).
To reevaluate the location and role of the NMBD, we have
determined a higher-resolution structure using cryo-EM and
helical reconstruction of tubular crystals of CopA from
A. fulgidus. This 10 A resolution structure reveals well-defined
cytoplasmic domains that are readily fit with the A, N, and P
domains from X-ray crystallography. Although the transmem-
brane domain is less distinct, the new map leads us to propose
a novel location for the extra two N-terminal helices that charac-
terize the PIB subfamily. Surprisingly, the NMBD is disordered
in these crystals, presumably due to dramatic changes in the
molecular packing of CopA, thus supporting the idea that the
NMBD is not a static structural element of the cytoplasmic
domain. Based on an all-atom homology model for the cyto-
plasmic domains of CopA, we used computational methods
to dock the NMBD, thus identifying the most favorable site
of interaction. The results are consistent with our previous
structure from cryo-EM and with the proposed autoinhibitory
1220 Structure 19, 1219–1232, September 7, 2011 ª2011 Elsevier Lt
role for NMBD, which would act by retaining CopA in the E2
conformation.
RESULTS
New Crystal Form for CopAOne of our primary goals was to obtain a higher-resolution struc-
ture of CopA in order to understand better the architecture of the
various domains that compose the molecule. As in earlier work,
we used Escherichia coli to express DC-CopA from A. fulgidus
that carried the functionally important N-terminal MBD, which
is common to all PIB ATPases, but not the C-terminal MBD,
which is a unique feature of this particular CopA homolog.
Expression levels were quite high, producing a final yield of
6–9 mg of detergent-purified protein from a 6 l expression with
purity >98% (see Figure S1 available online).
By altering the lipid species and the temperature used for
crystallization, we obtained wider tubular crystals with stronger
diffraction compared to our previous work. Previously, tubular
crystals of DC-CopA (Wu et al., 2008) were produced at 50�Cwith a reconstituted membrane of DOPC (1,2-dioleoyl-sn-glyc-
ero-3-phosphocholine) and in a solution containing the Cu+
chelator BCDS (bathocuproinedisulfonic acid) at pH 6.1. These
tubes were 35 nm in diameter and the corresponding structure
resembled that of SERCA in the E2 enzymatic state, consistent
with the absence of Cu+ in the crystallization media. For the
current work, the lipid was changed to a mixture of DMPC/
DOPE (1,2-dimyristoyl-sn-glycero-3-phosphocholine/1,2-dio-
leoyl-sn-glycero-3-phosphoethanolamine) (4:1 weight ratio),
which produced substantially wider (60 nm diameter) tubular
crystals at 30�C under otherwise identical conditions (Figure 1).
Layer line data from these wider crystals were substantially
stronger and were consistent with the presence of several
related helical symmetries, characterized by systematic differ-
ences in diameter and by the Bessel orders of principal layer
lines: (n1,0, n0,1) equal to either (12,�8), (14,�8), or (14,�10).
Among tubes with (12,�8) symmetry, the variability of unit cell
dimensions for the underlying 2D lattice was small (SD <0.5 A
and <0.4� for the included angle) (Table 1), making the Fourier-
Bessel approach to helical reconstruction viable (Diaz et al.,
2010). Based on two-fold related phase residuals, the final
map from an average of 12 tubes was judged to have 10 A reso-
lution, compared to 17 A from a similar number of tubes in DOPC
(Figure 2C). The visibility of several individual a helices in themap
(discussed below) is consistent with this estimate of resolution.
Packing of DC-CopA in Tubular CrystalsThe reconstruction shows that the orientation of DC-CopA in
DMPC/DOPE tubes is inverted relative to DOPC tubes (Figure 2;
Figure S2). Similar to helical crystals of Ca2+-ATPase (Xu et al.,
2002) and nicotinic acetylcholine receptor (Toyoshima and Un-
win, 1988), DC-CopA molecules in DOPC tubes were oriented
with their cytoplasmic domains facing the outside of the highly
curved bilayer. In these cases, the positive curvature is presum-
ably induced by the larger linear dimension required to accom-
modate the much larger cytoplasmic domains relative to the
transmembrane and luminal domains (Young et al., 1997).
Surprisingly, the large cytoplasmic domains of DC-CopA face
the inside of tubes produced from DMPC/DOPE. This inverted
d All rights reserved

Figure 1. Images of Tubular Crystals of DC-CopA
(A–C) Tubes grown in a 4:1 mixture of DMPC/DOPE at 30�C.(D–F) Tubes grown in DOPC at 45�C. Except for the differences in lipid composition and temperature, the crystallization conditions were otherwise identical.
Low-magnification images of negatively stained crystals are shown in (A) and (D), where the scale bars represent 200 nm. Higher-magnification images of
frozen-hydrated tubes used for helical reconstruction are shown in (B) and (E), where the scale bars represent 50 nm. Computed Fourier transforms in (C) and (F)
reveal layer lines characteristic of helical symmetry, which are indexed according to their Miller index and Bessel order in the right margin (h,k;n). The edge of
these transforms is at �20 A resolution.
See also Figure S1.
Structure
Cryo-EM and Computational Docking of CopA
orientation was also observed for tubular crystals of Na+/K+-
ATPase grown in native membranes from the duck supraorbital
salt gland, although in this case the b subunit added substantial
mass to the outside of these tubes (Rice et al., 2001).
Table 1. Comparison of Unit Cell Dimensions for Tubular Crystals
of DC-CopA
Cytoplasmic Domaina Membraneb
DMPC/DOPE DOPC DMPC/DOPE DOPC
Radius (A) 136 120 200 75
a (A) 63.9 ± 0.24 97.5 ± 1.38 82.9 ± 0.29 78.7 ± 0.92
b (A) 69.8 ± 0.40 61.1 ± 0.76 78.5 ± 0.42 48.7 ± 0.47
g (�) 78.6 ± 0.38 101.0 ± 0.66 99.0 ± 0.40 74.4 ± 0.65
Area (A2) 4370 ± 30.3 5851 ± 69.9 6427 ± 44.6 3656 ± 43.7aUnit cell dimensions measured at a radius corresponding to the middle
of the A domain, where major intermolecular contacts occur.bUnit cell dimensions in the middle of the membrane, that is, halfway
between the density peaks in the mean radial density profile that corre-
spond to the lipid phosphate head groups (see Figure 3).
Structure 19, 1219–12
The packing of DC-CopA molecules is also substantially
different in DMPC/DOPE tubes. In both crystal forms, CopA
forms dimers that are stabilized by reciprocal interactions
between N and A domains. In DOPC tubes, the dimers pack
into ‘‘dimer ribbons’’ reminiscent of Ca2+-ATPase crystals (Tay-
lor et al., 1986), in which contacts between A and N domains
form a continuous ribbon of molecules related by two-fold
symmetry, with another ribbon running in the opposite direction
along the b axis of the unit cell (Figure 2B). In contrast, DMPC/
DOPE tubes lack dimer ribbons; instead, the lattice is character-
ized by a more isotropic set of contacts between neighboring
dimers. As will be discussed in more detail below, the geometry
of the dimer itself is also quite different, with a 50� angle between
the molecules. As a result of these differences, the density of
cytoplasmic domains in DMPC/DOPE tubes is 33% higher
than in DOPC tubes, based on the area of the unit cell at radii cor-
responding to the middle of these domains (Table 1).
In contrast to the cytoplasmic domains, the transmembrane
domains in DMPC/DOPE tubes are less densely packed, with
a density at the center of the membrane that is <60% relative
to DOPC tubes (Table 1). Another way to look at this difference
32, September 7, 2011 ª2011 Elsevier Ltd All rights reserved 1221

A
B
resolution (Å)
25.00 12.50 8.30 6.25
2-fo
ld p
hase
resi
dual
(deg
)
0
10
20
30
40
50
DMPC/DOPEDOPC
DMPC/DOPE
DOPC
C
ab
a
b
N
NA A
Figure 2. Packing of DC-CopA in the Two Types of Tubular Crystals
(A) Cytoplasmic domains of DC-CopA face the inside of DMPC/DOPE tubes
and are tightly packed with an isotropic set of intermolecular contacts.
(B) Cytoplasmic domains face the outside of DOPC tubes and are packed in
‘‘dimer ribbons’’ that run along the b axis of the array. Both types of crystals are
composed of DC-CopA dimers, and two-fold related molecules have been
colored red and blue in this figure with the unit cell outlined in black.
Structure
Cryo-EM and Computational Docking of CopA
1222 Structure 19, 1219–1232, September 7, 2011 ª2011 Elsevier Lt
is to compare membrane and cytoplasmic domains within each
crystal: the unit cell area within the membrane of DOPC tubes is
considerably smaller (64%) than in the region of the cytoplasmic
domains, as one would expect given the smaller size of themole-
cule within the membrane. In contrast, the inverted nature of
the DMPC/DOPE tubes provides a much larger area to the
membrane domains relative to cytoplasmic domains (150%).
This excess membrane area in DMPC/DOPE tubes may
explain the diffuse density seen on the outer surface of the
reconstruction (Figure 3). When images of individual tubes are
projected down their cylindrical axes, several discrete peaks
are observed in the resulting 1D profile (Figure 3C). At the inner-
most radii, these peaks correspond to cytoplasmic domains,
followed by two peaks from the phosphate head groups of the
lipid bilayer. Unlike previous tubes of CopA and of Ca2+-ATPase,
there is an additional density peak on the outer surface of the
membrane in DMPC/DOPE tubes (arrow in Figure 3C). Helical
reconstruction of these tubes produces a considerable amount
of low, diffuse density in this region. More organized features
with somewhat higher density are visible outside the tubes, but
these features lie directly along symmetry axes, where noise
builds up during the reconstruction process (e.g., compare
sections shown in Figures 3A and 3B). From these observations,
we speculate that there is a population of disordered CopAmole-
cules with their cytoplasmic domains facing the outside of the
DMPC/DOPE tubes. Indeed, a symmetric distribution of mole-
cules across the membrane is expected from reconstitution,
and the inverted geometry of the CopA crystals within DMPC/
DOPE tubes produces excess membrane area that could readily
accommodate extra molecules facing the outside of the tubes.
These extra molecules would be unable to participate in regular
crystal contacts given the long distance between their cyto-
plasmic domains. Furthermore, the presence of disordered
molecules would explain why, in the helical reconstruction, the
protein domains within the membrane have much lower contrast
relative to the cytoplasmic domains on the inside of the tube.
Functional Analysis of CopABecause of the dramatic effect of bilayer composition on the
morphology and molecular packing of the tubular crystals, we
used ATPase assays to assess whether lipid had an effect on
DC-CopA function. In particular, we characterized the depen-
dence of ATPase activity on Cu+ or Ag+ concentrations and on
temperature, reasoning that bilayer composition might affect
either the mobility or the stability of the enzyme. The results (Fig-
ure 4) indicate that DC-CopA has an apparent Kd of 0.44 mM for
Cu+ and 0.26 mM for Ag+ in DMPC/DOPE and corresponding
Vmax of 1.97 and 1.76 mmol/mg/min. For comparison, affinities
in DOPC were 0.23 and 0.22 mM for Cu+ and Ag+ with Vmax of
0.94 and 1.48 mmol/mg/min, respectively. These values are
similar to those previously measured by Rice et al. (2006) from
detergent-solubilized DC-CopA: Kd of 0.11 and 0.2 mM for Cu+
and Ag+, respectively, with Vmax of 1.8 and 2.4 mmol/mg/min.
In all cases, ATPase activity was partially inhibited at high
concentrations of either Cu+ or Ag+, probably reflecting the
(C) Two-fold phase statistics for the two helical reconstructions, documenting
a resolution of 17 and 10 A for DOPC and DMPC/DOPE crystals, respectively.
See also Figure S2.
d All rights reserved
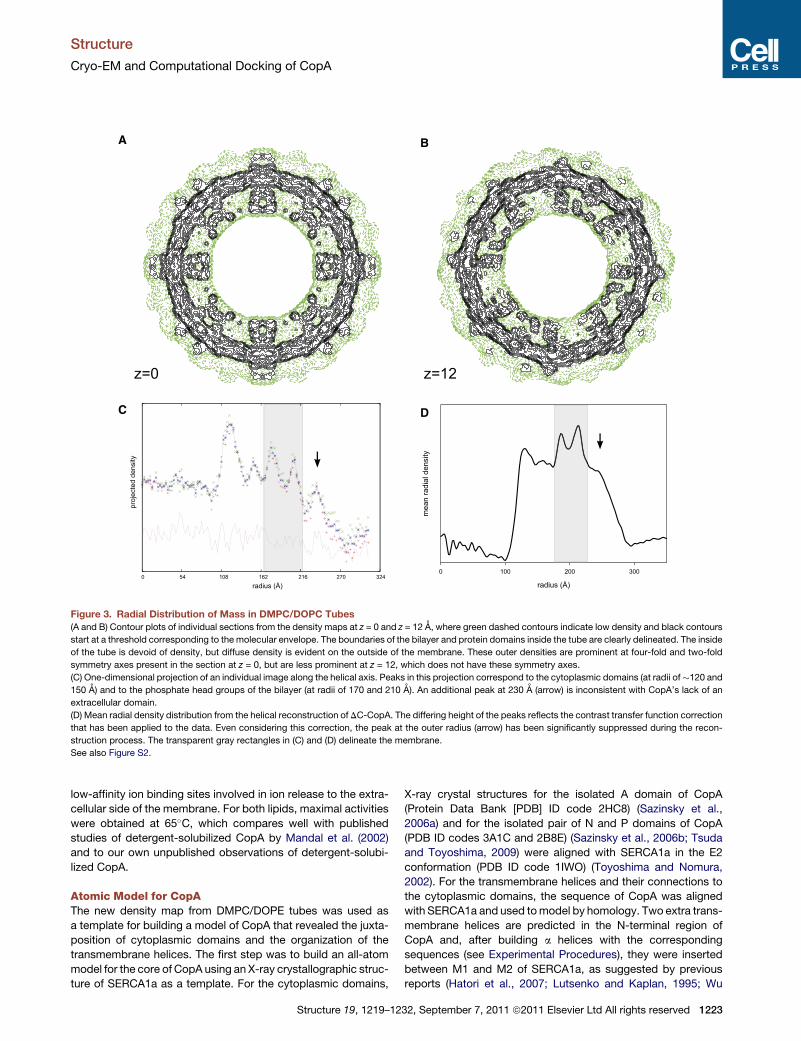
z=0 z=12
0 54 108 162 216 270 324
radius (Å)
proj
ecte
d de
nsity
radius (Å)
0 100 200 300
mea
n ra
dial
den
sity
BA
DC
Figure 3. Radial Distribution of Mass in DMPC/DOPC Tubes
(A and B) Contour plots of individual sections from the density maps at z = 0 and z = 12 A, where green dashed contours indicate low density and black contours
start at a threshold corresponding to the molecular envelope. The boundaries of the bilayer and protein domains inside the tube are clearly delineated. The inside
of the tube is devoid of density, but diffuse density is evident on the outside of the membrane. These outer densities are prominent at four-fold and two-fold
symmetry axes present in the section at z = 0, but are less prominent at z = 12, which does not have these symmetry axes.
(C) One-dimensional projection of an individual image along the helical axis. Peaks in this projection correspond to the cytoplasmic domains (at radii of�120 and
150 A) and to the phosphate head groups of the bilayer (at radii of 170 and 210 A). An additional peak at 230 A (arrow) is inconsistent with CopA’s lack of an
extracellular domain.
(D) Mean radial density distribution from the helical reconstruction of DC-CopA. The differing height of the peaks reflects the contrast transfer function correction
that has been applied to the data. Even considering this correction, the peak at the outer radius (arrow) has been significantly suppressed during the recon-
struction process. The transparent gray rectangles in (C) and (D) delineate the membrane.
See also Figure S2.
Structure
Cryo-EM and Computational Docking of CopA
low-affinity ion binding sites involved in ion release to the extra-
cellular side of the membrane. For both lipids, maximal activities
were obtained at 65�C, which compares well with published
studies of detergent-solubilized CopA by Mandal et al. (2002)
and to our own unpublished observations of detergent-solubi-
lized CopA.
Atomic Model for CopAThe new density map from DMPC/DOPE tubes was used as
a template for building a model of CopA that revealed the juxta-
position of cytoplasmic domains and the organization of the
transmembrane helices. The first step was to build an all-atom
model for the core of CopA using an X-ray crystallographic struc-
ture of SERCA1a as a template. For the cytoplasmic domains,
Structure 19, 1219–12
X-ray crystal structures for the isolated A domain of CopA
(Protein Data Bank [PDB] ID code 2HC8) (Sazinsky et al.,
2006a) and for the isolated pair of N and P domains of CopA
(PDB ID codes 3A1C and 2B8E) (Sazinsky et al., 2006b; Tsuda
and Toyoshima, 2009) were aligned with SERCA1a in the E2
conformation (PDB ID code 1IWO) (Toyoshima and Nomura,
2002). For the transmembrane helices and their connections to
the cytoplasmic domains, the sequence of CopA was aligned
with SERCA1a and used tomodel by homology. Two extra trans-
membrane helices are predicted in the N-terminal region of
CopA and, after building a helices with the corresponding
sequences (see Experimental Procedures), they were inserted
between M1 and M2 of SERCA1a, as suggested by previous
reports (Hatori et al., 2007; Lutsenko and Kaplan, 1995; Wu
32, September 7, 2011 ª2011 Elsevier Ltd All rights reserved 1223

Cu concentration ( M)
0.1 1 10
activ
ity (
mol
es/m
g/m
in)
0.0
0.4
0.8
1.2
1.6
2.0
Ag concentration ( M)
0. 11 100.0
0.4
0.8
1.2
1.6
A B
temperature (°C)0 20 40 60 80 100
activ
ity (
mol
es/m
g/m
in)
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
temperature (°C)0 20 40 60 80 100
0.0
0.5
1.0
1.5
2.0
C D
DMPC/DOPEDOPC
Figure 4. ATPase Activity of Reconstituted DC-CopA
(A and B) Concentration dependence of activity for Cu+- and Ag+-dependent activity. A background level of �0.3 mmol/mg/min has been subtracted from these
data, which have been fitted with the Michaelis–Menten equation for a single binding site. Because there is inhibition at higher ion concentrations, the last two or
three points of each curve have been omitted for this fit. The resulting values for apparent Kd and Vmax are cited in the text.
(C and D) Temperature dependence of activity for Cu+ (C) and Ag+ (D) shows a peak at�65�C, which is consistent with previous work with detergent-solubilized
CopA. In all plots, the open circles represent DC-CopA reconstituted in DMPC/DOPE and the filled circles represent DC-CopA reconstituted in DOPC. Error bars
correspond to the standard error of the mean.
Structure
Cryo-EM and Computational Docking of CopA
et al., 2008). The resulting hybrid homology model lacked the
N-terminal and C-terminal metal-binding domains, which have
no counterpart in the SERCA1 template. This hybrid model
was used for the docking studies described below.
The density map from DMPC/DOPE tubes was used to
reposition the individual domains of this DC-CopA homology
model to produce a fitted model. Specifically, after placing the
homology model roughly into the envelope from the helical
reconstruction, the loops connecting the domains were broken
and the position of each cytoplasmic domain was refined using
cross-correlation (using the Fit-in-Map feature of Chimera). The
density map accounted well for these domains, with evidence
for individual a helices in several locations (Figure 5A; Movie
S1). Surprisingly, the A, N, and P domains of CopA accounted
for all of the densities in this region of the map, indicating that
the NMBD was disordered. In our previous map from DOPC
tubes, the NMBD was seen near the dimer interface, and its
disordering in the current DMPC/DOPE tubes may be due
to changes in this interface. Indeed, Figure 6 shows that there
is an �50� angle between two-fold related molecules in the
1224 Structure 19, 1219–1232, September 7, 2011 ª2011 Elsevier Lt
DMPC/DOPE tubes (defined by axes running from the middle
of the transmembrane helices to catalytic aspartate), whereas
these molecules were almost parallel in DOPC tubes. There is
also a shift in the cytoplasmic domains relative to one another
(Figure 7), which may explain the absence of the NMBD in the
map from DMPC/DOPE tubes.
Protein densities were less distinct within the membrane than
in the extramembranous regions of the map, although the
general outline of the transmembrane domain could be dis-
cerned (Figure 5B). This loss of contrast is common in helical
reconstructions and may be exacerbated in this case by the
extra, disordered molecules hypothesized to face the outside
of the tubes, which would contribute to the background density
within themembrane. Despite the poorer definition of membrane
domains, we were able to use our all-atommodel of DC-CopA to
interpret the transmembrane densities. Specifically, the six
conserved transmembrane helices (M1–M2, M5–M8) fell within
strong density. Similar to maps of SERCA from helical crystals
(Xu et al., 2002; Young et al., 2001; Zhang et al., 1998), a density
was observed at the cytoplasmic surface above M1 (arrowhead
d All rights reserved

Figure 5. Fitting an Atomic Model to the Map from DMPC/DOPE
Tubes
(A) Domains are shown in various shades of red and are labeled accordingly.
Isolated densities for individual a helices are visible in A and P domains, and
other a helices at the periphery of N and P domains fit snugly within the iso-
surface density envelope. The envelope for the transmembrane domain (TM)
shows a distinct bulge at the top right, consistent with the kinked cytoplasmic
end of M1.
(B) Section through the transmembrane region of the densitymap overlaid with
transmembrane helices, which are numbered. This is a view from the extra-
cellular side of the membrane.
See also Movie S1.
Structure
Cryo-EM and Computational Docking of CopA
in Figure 5A) that matches the bent extension of this helix seen in
atomic structures of SERCA, Na+/K+-ATPase, and H+-ATPase.
Additional membrane density was observed surrounding M4,
which we fitted with the extra M2 and M3 helices that are char-
acteristic of PIB ATPases. The location of these helices is slightly
displaced from our previous model from DOPC tubes, with M2
taking the position of M3 in the previous map and M3 falling
Figure 6. Comparison of the DC-CopA Dimer Seen in the Two Helical
(A) Dimer from the DMPC/DOPE tubes viewed parallel to themembrane surface. T
the membrane normal.
(B) The dimer from DMPC/DOPE tubes viewed normal to the membrane surface
(C) Dimer from our previous map from DOPC tubes (PDB ID code 2VOY; Wu et a
transmembrane domain is closely aligned to the membrane normal. The NMBD,
purple for the blue monomer.
(D) Dimer from DOPC tubes viewed normal to the membrane surface. Note that (B
correspond to the membrane.
Structure 19, 1219–12
between M4 and M8. Given the poor contrast within the trans-
membrane domains, the precise location of these two extra
helices remains tentative.
Computational Docking of the NMBDBecause our previous density map from the thinner DOPC tubes
revealed the binding site of the NMBD next to the A domain, we
were disappointed that the current map from DMPC/DOPE
tubes did not provide further information at higher resolution.
Nevertheless, we hypothesized that the NMBD binding site
previously identified next to the A domain, although perturbed,
still represented the most structurally compatible location for
docking of the NMBD. To test this hypothesis, we performed
in silico docking of the NMBD to the other cytoplasmic domains
of CopA models obtained either by homology to the E2 state
of SERCA or by fitting to our map from DMPC/DOPE tubes.
This procedure uses Monte Carlo sampling to identify favor-
able docking sites on the molecular surface and ranks them
energetically.
For our initial docking, we used the hybrid homology model of
DNDC-CopA based on SERCA1 in the E2 conformation as
a template, due to its close resemblance to the structure from
DOPC tubes in which the NMBD was seen. For a search target,
we made a homology model of NMBD based on the structure of
a CopZ metallochaperone from Bacillus subtilis. This particular
template was chosen for its high resolution (1.5 A) and for its
high sequence identity (41%) relative to the CopA NMBD from
A. fulgidus. Although the CopZ template structure had Cu+
bound to its CxxC motif, NMR structures of Cu+-free NMBD
homologs indicated root-mean-square deviations of only �1 A,
suggesting that structural changes induced by Cu+ binding are
minimal. For docking, the homology models for Cu-free NMBD
Crystals
his view illustrates the >30� inclination of the transmembrane domain relative to
.
l., 2008) viewed parallel to the membrane surface showing that in this case the
which is visible in these crystals, is shaded in orange for the red monomer and
) and (D) are at a smaller scale than (A) and (C); the transparent gray rectangles
32, September 7, 2011 ª2011 Elsevier Ltd All rights reserved 1225

Figure 7. Docking of the NMBD to an All-
Atom Model for DC-CopA
(A and B) Two views of the result obtained by
computational docking related by an 180� rotationabout the vertical axis. The NMBD is colored
purple, the A domain is in yellow, the N domain is
in blue, the P domain is in green, and the trans-
membrane helices are in cyan. Key residues
shown are C27 from the GMTCAMC30 Cu-binding
loop of the NMBD, H462 from the ERRSEHP463
ATP-binding loop in the N domain, and the cata-
lytic D424 from the DKTGTLTT431 phosphorylation
loop in the P domain.
(C and D) Equivalent views of an overlay between
the homology model used for docking (same as in
A and B but colored gray and lacking the NMBD)
and the atomic model fitted to the density map
from DMPC/DOPE tubes (colored shades of red).
The P domains are aligned, revealing relative
movements of the N and A domains as well as
a different angle of the transmembrane domain.
See also Figure S3.
Structure
Cryo-EM and Computational Docking of CopA
and CopA were treated as independent, unconstrained mole-
cules free to rotate and translate in all directions in space, and
the ICM docking routine evaluated numerous alternative binding
interfaces, ultimately ranking them according to their predicted
binding energy. Table 2 shows that the lowest-energy solution
was well separated from the other solutions, making it the clearly
preferred location for the NMBD on the molecular surface of
CopA. Significantly, this binding pose is consistent with the loca-
tion of the NMBD in DOPC tubes, despite the lack of physical
constraints or any other information related to the EM structure.
This docking result places the CxxC Cu-binding loop of the
NMBD next to a conserved loop in the N domain that controls
ATP binding and, when bound, the Cu+ ion would lie within
13 A of theMg2+ ion at the catalytic site of the Pdomain (Figure 7).
Unlike the model previously derived directly from the EM struc-
ture, the docking solution packed the two a helices of the NMBD
against the A domain. Specific interactions at this interface also
involve the loops between b strands composing the body of the
A domain (residues 252–256, 270–274, 278–281, 295–298). The
surface area buried in this interaction is 1943 A2, which is divided
between the three domains as follows: 1040 A2 for the A domain,
507 A2 for the P domain, and 374 A2 for the N domain.
For comparison, we also used the fitted model corresponding
to the conformation observed in DMPC/DOPE tubes as a tem-
plate for docking, although in this case the apparent disordering
of the NMBD in the corresponding crystals led us to expect
1226 Structure 19, 1219–1232, September 7, 2011 ª2011 Elsevier Ltd All rights reserved
a lower binding energy. Indeed, the ICM
docking routine determined that the
total energy for this pose was almost
17 kcal/mol higher for the DMPC/DOPE
tube model compared to the SERCA1
homology model (�28.2 versus �44.9).
These energy units are not absolute,
due to inaccuracy in the force field and
the coarseness of the grid on which
potential energies were calculated, but
are indicative of the relative binding
strength. Accordingly, calculations of buried surface area
showed a >40% decrease to 1176 A2. The reason for this
reduced binding energy is evident in Figures 7C and 7D, where
the homology model for CopA is overlaid with the fitted model
for CopA fromDMPC/DOPE tubes. This reveals a significant shift
in the A and N domains relative to the P domain, and the loss of
binding energy is consistent with an alteredmolecular surface for
NMBD docking due to the disrupted interface between A, N, and
P domains. Indeed, intermediate values for binding energy were
obtained in docking studies with other permutations of the
DNDC-CopA model in which the N domain was either moved,
modified, or removed altogether (data not shown). These results
illustrate that all three cytoplasmic domains contribute to the
NMBD binding site and that the binding was strongest when
the conformation resembled the E2 state adopted by SERCA1.
Finally, to complete our model of DC-CopA, we constructed
a linker between the M1 and the NMBD (Figure 7). In SERCA,
twoN-terminal a helices are attached to the front of the A domain
and are connected to the cytoplasmic end of M1 via a flexible
linker. Although the NMBD is associated with the side of the A
domain in our model, the CopA linker sequence is long enough
to wrap under the A domain and to connect to the kinked
extension of M1. Thus, our model of DC-CopA is consistent
with the idea that the physical couplings observed between
SERCA transmembrane helices and the cytoplasmic domains
are conserved in the PIB subfamily of pumps.

Table 2. Results of Computational Docking
RankaEnergy Terms
Totalb vdW Hc vdW Cd H Bonde Electrostaticf Hydrophobicg
1 �44.9 �4.71 �37.83 �4.71 �1.10 �12.32
2 �40.6 �5.76 �32.46 �3.68 �1.31 �11.48
3 �39.4 �5.76 �31.49 �7.75 �2.43 �8.80
4 �36.2 �3.55 �39.56 �2.65 0.42 �13.89
5 �35.9 �4.56 �21.10 �17.63 �4.53 �8.71
6 �35.6 �3.30 �27.97 �7.93 �0.51 �9.68
7 �35.3 0.07 �25.46 �9.20 �1.05 �9.59
8 �35.3 �0.30 �26.78 �3.53 �0.55 �11.70
9 �35.2 �3.05 �28.23 �8.29 �3.38 �11.30
10 �35.2 �7.25 �33.14 �10.48 �2.26 �10.74a Each docking result was ranked according to its total energy. In our application, the lowest-energy solution (rank 1) was well separated from the suc-
ceeding solutions, lending confidence to the result.b Total energy of the conformation. These values are reported as kcal/mol by ICM but should only be taken as relative binding energies due to various
approximations during the calculation.c van der Waals grid potential using a hydrogen probe.d van der Waals grid potential using a carbon probe.eHydrogen-bonding grid potential.f Electrostatic grid potential.g Hydrophobic grid potential.
Structure
Cryo-EM and Computational Docking of CopA
DISCUSSION
Using cryo-EM and image reconstruction, we have produced
a new structure of DC-CopA from A. fulgidus at 10 A resolution
from a new form of helical crystal grown in membranous tubes
composed of shorter-chain lipid (DMPC). Although DC-CopA
forms a dimer as in previous DOPC tubes, there are marked
differences in the intermolecular contacts stabilizing this dimer
and in the packing of this dimer in the unit cell; furthermore,
the transmembrane domain is strongly tilted relative to the
thinner DMPC membrane. At 10 A resolution, X-ray structures
of the three isolated cytoplasmic domains could be fitted
precisely into themap. However, density for theNMBD is absent,
indicating that this domain is disordered. We then used compu-
tational molecular docking to explore the site of NMBD binding
relative to the other CopA cytoplasmic domains and its confor-
mational determinants. Using a homology model based on the
E2 conformation of SERCA1, the lowest-energy pose was
consistent with the site previously identified in tubular crystals
composed of DOPC. Although there were differences with our
previous, manually docked model (i.e., an �180� rotation of the
NMBD relative to the A domain), this new model is nevertheless
consistent with an autoinhibitory interaction between the NMBD
and the N and P domains in the E2 conformation. Comparison of
this CopA homology model with a model fitted to the map from
DMPC/DOPE tubes reveals a distinct shift in N and A domains
that is predicted to reduce the binding energy of the NMBD by
�17 kcal/mol. This result suggests that the affinity of the
NMBD for its preferred site has been lowered to the level of
a weak, Brownian association, explaining why it has become
disordered in the crystal.
Although our previous model of DC-CopA from DOPC tubes is
consistent with the E2 enzymatic state of SERCA1, the new
structure from DMPC/DOPE tubes reveals a significant confor-
Structure 19, 1219–12
mational change. As shown in Figure 7, the A domain has shifted
away from the P domain in the new map, leaving room for the N
domain to swing closer to the catalytic site. Such movement is
generally consistent with conformational changes of SERCA1
during its reaction cycle, which depends on an interplay between
the A and N domains to regulate access to the catalytic aspar-
tate. The overarching idea is that the A domain interacts with
elements of the P domain in E2 states, thus preventing the
approach of the N domain and its associated ATP. In E1 states,
the A domain swings out of the way to enable transfer of
phosphate. Figure S3 compares the position of the A domain
in E2-P and E2 states of SERCA1. In E2-P, the conserved (T/S)
GE278 sequence in the A domain is closest to the catalytic aspar-
tate in the P domain (DKTGTLT430), making direct interactions
with the associated Mg2+ ion. In E2, the (T/S)GE loop has moved
back and interacts with the conserved DG(V/I)ND622 loop at the
periphery of the P domain. In our model fitted to DMPC/DOPE
tubes, this (T/S)GE loop has moved back even more, although
it still appears to interact with residues in the DG(V/I)ND loop.
The latter configuration of cytoplasmic domains has not previ-
ously been seen in structures of P-type ATPases and could
represent a transition from the E2 to the E1 state as the A domain
begins to disengage from the P domain. The increased angle of
the transmembrane domain relative to the P domain (Figures 7C
and 7D) is consistent with this transition, as it eventually results in
a 30� upward rotation of the P domain in the E1 state relative
to the E2 state (Toyoshima and Nomura, 2002). Normally, the
E2-to-E1 transition would be initiated by ions binding to their
transport sites in the membrane. In the current situation, we
speculate that a shearing of the transmembrane domain caused
by its oblique angle relative to the membrane might be respon-
sible for inducing this conformation.
Factors that may contribute to the unusually high tilt of the
membrane domain are (1) the negative curvature of the crystal
32, September 7, 2011 ª2011 Elsevier Ltd All rights reserved 1227

Structure
Cryo-EM and Computational Docking of CopA
contacts between cytoplasmic domains, (2) the intermolecular
contacts that stabilize the dimer, and (3) the thinner bilayer
produced by the shorter aliphatic chains of DMPC, which
composes the bulk of the bilayer. Indeed, the hydrophobic
core thickness of DMPC has been experimentally measured as
23 A compared to 27 A for DOPC (Lewis and Engelman, 1983),
and this difference would be consistent with a tilt of�30� in orderto accommodate a hydrophobic domain with a fixed thickness
(cos�1(23/27)). Given the corresponding difference in the tilt of
the CopA molecule in DMPC/DOPE versus DOPC tubes, it
seems plausible that the thinner DMPC bilayer could be the
driving force for altering the geometry of the dimer and thus
producing the negative curvature of the crystal. Despite the
dramatic effect on crystallization, however, the DMPC/DOPE
bilayer had only modest effects on ATPase activities. It should
be noted that the lipid-to-protein ratio was six times higher for
this assay, where no crystalline arrays were observed to form.
Nevertheless, the approximately 2-fold increase in Kd and Vmax
is comparable to the 2-fold changes in these parameters in
detergent micelles (Rice et al., 2006) and can therefore be
considered to represent the normal range of behavior for CopA
in different hydrophobic environments. Furthermore, this range
is in line with measurements of other P-type ATPases in lipid
and detergent environments (e.g., Cornea and Thomas, 1994;
Warren et al., 1974).
From a structural point of view, a shearing of the transmem-
brane domain would be required tomaintain the upper and lower
boundaries of the transmembrane helices as the domain was
tilted in the bilayer. In other words, one would expect a relative
movement of transmembrane helices along their helical axes.
Such movements have in fact been described for the M1/M2
helical pair of SERCA, both during the transition from E2 to E1
(Toyoshima and Nomura, 2002) and during Ca2+ occlusion by
E1�P (Olesen et al., 2004). In the case of CopA, M2 and M3
are inserted between this M1/M2 pair and placed at the
periphery of the transmembrane domain; accordingly, the entire
M1–M4 bundle would likely slide relative toM5–M8, thus accom-
modating the shear force. By analogywith the structural changes
during the E2-to-E1 transition in SERCA, this movement of
M1–M4would pull the A domain away from the P domain, poten-
tially inducing the conformation seen in DMPC/DOPE tubes. A
corresponding force on the NMBD, which is directly linked to
M1, could also help disengage it from its binding site on the A
domain. According to this hypothesis, M1 would no longer be
tethered to the cytoplasmic domain in the E1 state. This implies
that the more rigid connection between M4 (M2 in SERCA) and
the A domain may be the important element that drives changes
to the transmembrane domain and leads to ion occlusion during
the transition to E1�P.
The site for the NMBD identified by in silico docking is consis-
tent with the corresponding density in our previous map from
DOPC tubes (Wu et al., 2008). The specifics of the interface
between the NMBD and the A domain are different in our pre-
vious model, presumably reflecting our earlier neglect of binding
energy and molecular surface compatibility during manual
assembly and fitting. Nevertheless, in both cases, the NMBD
binds to the side of the A domain with additional interactions
between the GMTCAMC30 Cu-binding loop and the conserved
ERRSEHP463 loop on the N domain. This ERRSEHP463 loop
1228 Structure 19, 1219–1232, September 7, 2011 ª2011 Elsevier Lt
has been shown in NMR and X-ray crystal structures of isolated
domains to be intimately involved in ATP binding (Banci et al.,
2010; Dmitriev et al., 2006). In particular, His462 makes critical
interactions with the a- and b-phosphates and with the adenine
ring of ATP (Sazinsky et al., 2006b; Tsuda and Toyoshima, 2009);
its mutation in ATP7A almost completely prevents ATP binding
(Morgan et al., 2004) and the H462Q mutation is the most
common cause of Wilson’s disease (Thomas et al., 1995).
Interaction between the NMBD and the N domain has been
demonstrated experimentally by Gonzalez-Guerrero et al.
(2009) using pull-down assays. Specifically, they showed that
Cu-free NMBD bound to an N/P-domain construct whereas
Cu-loaded NMBD did not. Furthermore, these investigators
showed that interaction between the Cu-free NMBD and the
N/P-domain construct was disrupted by ADP-Mg2+. Although
our docking result indicates that the majority of binding energy
for NMBD comes from its interface with the A domain, the inter-
face between the NMBD and the N and P domains still accounts
for 891 A2 of buried surface (out of a total of 1934 A2), which
seems sufficient to explain the pull-down results. It is interesting
to note that the altered conformation represented in our DMPC/
DOPE crystals results in a loss of 767 A2 of buried surface (i.e.,
almost the entire interaction with the N and P domains). This
observation, together with the disordering of the NMBD in these
crystals, suggests that the interaction with the A domain alone is
not enough to retain NMDB at its binding site and that its asso-
ciation with the cytoplasmic domains is transient.
This transience is also implied by interaction between the
NMBD andmetallochaperones. Physiologically relevant metallo-
chaperones have been shown to transfer Cu+ to MBDs of both
ATP7A (Walker et al., 2002, 2004) and CopA (Gonzalez-Guerrero
and Arguello, 2008), and NMR structures of the complex
between the HAH1 metallochaperone and related MBDs of
ATP7A indicate that the a-helical face of NMBD interacts with
the corresponding face of the metallochaperone during Cu+
transfer (Achila et al., 2006; Banci et al., 2005a, 2005b). Accord-
ing to our model, these a helices are packed against the A
domain, implying that the NMBD must dissociate prior to inter-
acting with metallochaperones. This dissociation step may be
the kinetic barrier to activation that has been revealed by kinetic
analysis of the transport cycle (Hatori et al., 2009). An alternative
binding site for the NMBD on the opposite side of the A domain,
where it would not have interactions with either the N or P
domain, has been proposed on the basis of chemical crosslink-
ing (Lubben et al., 2009). This alternative is not compatible with
our previous density map or with the results of molecular dock-
ing, and it is difficult to explain how the NMBD would play an
autoinhibitory role from that location.
There is a growing consensus that NMBDs play a regulatory
role for Cu pumps and are not involved in transferring Cu+ to
the transport sites within the transmembrane domain (Arguello
et al., 2007). This conclusion applies both to the human pumps
(ATP7A and ATP7B), which carry six MBDs on their N terminus,
and to CopA from bacteria, which generally have a single
N-terminal MBD. In particular, several studies have shown Cu
transfer from metallochaperones to MBDs (Achila et al., 2006;
Banci et al., 2005a, 2005b; Gonzalez-Guerrero and Arguello,
2008; Larin et al., 1999; Ralle et al., 2004; Singleton et al.,
2009; Strausak et al., 2003) and from metallochaperones to
d All rights reserved

Structure
Cryo-EM and Computational Docking of CopA
transport sites (Gonzalez-Guerrero et al., 2009). However,
disruption of MBDs by either truncation or mutation does not
affect transport activity of bacterial pumps (Bal et al., 2001;
Fan and Rosen, 2002; Mana-Capelli et al., 2003; Mandal and
Arguello, 2003; Mitra and Sharma, 2001; Rice et al., 2006), and
disruption of MBD on human pumps mainly affects enzyme acti-
vation and trafficking (Cater et al., 2007; Huster and Lutsenko,
2003; Strausak et al., 1999; Tsivkovskii et al., 2001; Veldhuis
et al., 2009b; Walker et al., 2002). An enzymatic study of CopA
from Thermotoga maritima concluded that the NMBD was
responsible for kinetic inhibition of the reaction cycle and that
this inhibition was alleviated by its binding of Cu+ (Hatori et al.,
2008). Our structure offers a structural explanation for this auto-
inhibition. Specifically, the NMBD has substantial interactions
with both the A and N domains in our hybrid model of the E2
conformation. Proteolytic digestion patterns of CopA indicate
that these domains undergo large movements during the transi-
tion from the E2 state to the E1�P state, similar to SERCA (Hatori
et al., 2009), and the binding energy of the NMBD would resist
these movements. Cu+ could have a role in disrupting this inter-
action in three ways: (1) by binding to transport sites and
driving a conformational change; (2) by binding directly to the
GMTCAMC30 loop at the interface between the NMBD and the
N domain; and (3) by promoting interaction between the NMBD
and metallochaperones, which would require dissociation of
the NMBD from the A domain in order to form the corresponding
complex.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Plasmids containing the C-terminal truncation of the copA gene (Wu et al.,
2008) behind a pBAD promoter were expressed in E. coli (strain LMG1940)
using LB broth with 0.1 mg/ml ampicillin at 37�C. After allowing 1 l cultures
to reach an OD of 1, expression of CopA was induced with 0.02% arabinose
for 2 hr at 30�C. Cells were then harvested by centrifugation at 10,000 3 g
for 10 min and the pellet was suspended in 50 mM Tris-Cl (pH 7.5), 20% glyc-
erol, 50 mM NaCl, 2 mM b-mercaptoethanol, 0.05 mg/ml DNase I, and
complete protease inhibitor cocktail (Hoffmann-La Roche). Cells were broken
by French press at 20,000 psi and, after removing large debris by centrifuga-
tion at 10,000 3 g for 20 min, membranes were collected by centrifugation at
150,000 3 g for 1 hr and stored at �80�C. Membrane pellets were solubilized
in 10 mg/ml n-dodecyl-b-D-maltoside (DDM; Anatrace), 25 mM Tris-Cl
(pH 7.5), 100 mM NaCl, 20 mM imidazole, 10% glycerol, 2 mM b-mercaptoe-
thanol, and protease inhibitor cocktail at a protein concentration of 10 mg/ml.
After gentle mixing for 30 min, insoluble material was removed by centrifuga-
tion at 150,000 3 g for 30 min and the supernatant was loaded onto a 5 ml
HiTrap Chelating HP column (GE Healthcare), which was charged with 0.1 M
NiCl2 and equilibrated with 25 mM Tris-Cl (pH 7.5), 100 mM NaCl, 20 mM
imidazole, 10% glycerol, 2 mM b-mercaptoethanol, and 0.1% DDM. After
washing, purified DC-CopA was eluted with 25 mM Tris-Cl (pH 7.5), 100 mM
NaCl, 400 mM imidazole, 10% glycerol, 2 mM b-mercaptoethanol, and 0.1%
DDM. Overnight incubation with thrombin (1 U/mg of protein) was used to re-
move the polyhistidine tag. A benzamidine-Sepharose column (Amersham
Biosciences) equilibrated with 25 mM Tris-Cl (pH 7.5), 100 mM NaCl, 10%
glycerol, 2 mM b-mercaptoethanol, and 0.1% DDM was used to remove the
thrombin. Finally, purified DC-CopA protein was dialyzed against 25 mM
Tris-Cl (pH 7.5), 100 mM Na2SO4, 10% glycerol, 2 mM DTT, and 0.1% DDM.
All the steps in this purification were done at 4�C.
ATPase Activity Assays
For measuring ATPase activity, proteoliposomes were made with a 2.5:1 lipid:
protein weight ratio using either DOPC or a 4:1weight ratio of DMPC/DOPE. To
Structure 19, 1219–12
make these proteoliposomes, DC-CopA (solubilized in DDM) and the relevant
lipid were dialyzed against 50 mM Tris-SO4 (pH 7.5), 50 mM Na2SO4, 50 mM
K2SO4, 10 mM MgSO4, and 2 mM b-mercaptoethanol for 5 days at 4�C. Asdescribed in our previous work on CopA (Rice et al., 2006), themalachite green
assay (Kimura et al., 1996) was used to quantitate phosphate production in
50 mM Tris-SO4, 200 mM Na2SO4, 3 mM MgSO4, 5 mM glutathione, 2.4 mM
ATP, 10% glycerol, and 0.025% NaN3. To test dependence on either Cu+ or
Ag+, assays were performed at 70�C; to test temperature dependence, assays
were performed with either 2 mM Cu+ or 1 mM Ag+. In order to prevent buildup
of transport ions inside the vesicles, a small, subsolubilizing amount of deter-
gent (0.01% DDM) was added to make the vesicles leaky. All nonreducing
buffers were pretreated with 100 mg/ml Chelex (Bio-Rad) for at least 1 hr.
Aliquots of DC-CopA proteoliposomes at 0.02 and 0.04 mg/ml were prepared
and stored on ice, where the enzyme is completely inactive. Using a program-
mable PCR thermocycler (Eppendorf North America), duplicate 45 ml aliquots
of the reaction mixture were heated to 70�C for 3, 5, 7, and 9 min and then
cooled to 4�C to stop the reaction. Phosphate was then quantified in 96-well
microtiter plates using a development time of 3 min; absorbance was read
at 630 nm. In order to maintain a pH of 6.3 at a range of different temperatures,
the following buffers were adjusted to the corresponding pHs at room temper-
ature: MES-SO4 (pH 6.2) for 15�C, MES-SO4 (pH 6.3) for 25�C, MES-SO4
(pH 6.4) for 35�C, MES-SO4 (pH 6.5) for 45�C, MES-SO4 (pH 6.6) for 55�C,MOPS-SO4 (pH) 6.9 for 65�C, Tris-SO4 (pH 7.7) for 75�C, Tris-SO4 (pH 8) for
85�C, and Tris-SO4 (pH 8.3) for 95�C. Like in previous work, we observed
a background of 0.3–0.35 mmol/mg/min in the absence of added Cu+ or Ag+;
this background activity was abolished by the Cu chelator BCDS, suggesting
that it arises from trace Cu+ in the solutions. After subtracting the background
activity, the data were fitted with the Michaelis–Menten binding model to
determine the apparent dissociation constant (Kd).
Crystallization and Cryo-Electron Microscopy
DC-CopA tubular crystals were grown with a 4:1 mixture of DMPC/DOPE at
a protein concentration of 1 mg/ml and at a lipid:protein weight ratio of 0.4.
Dialysis was carried out for 5 days in 50 ml dialysis buttons (Hampton Research)
at 30�C against 500ml of 50mMMES (pH 6.1), 25 mMNa2SO4, 25mMK2SO4,
200 mMBCDS, 10 mMMgSO4, and 2 mM b-mercaptoethanol. Stock solutions
of lipid for both crystallization and ATPase assays were made in dodecyl octa-
ethylene glycol ether (C12E8) at 1 mg lipid per 2 mg detergent.
For cryo-EM, a suspension of crystals was deposited on a holey carbon grid
and rapidly frozen in liquid ethane. Samples were imaged at �175�C with a
CT3500 cryoholder (Gatan) at 50,0003 nominal magnification with a CM200
FEG electron microscope (FEI) operating at 200 kV. Electron micrographs
were recorded on Kodak SO-163 film and, after screening by optical diffrac-
tion, suitable images were digitized at 14 mm intervals with a Zeiss SCAI micro-
densitometer (Intergraph).
Helical Reconstruction
Tubular crystals with a diameter of�600 A, displaying symmetric layer lines by
optical diffraction and with defocus values between 1.5 and 2.5 mm, were
selected for helical processing using Fourier-Bessel methods (Diaz et al.,
2010). Layer lines were cataloged with a consistent set of Miller indices, which
reflect the underlying 2D lattice within the membrane. To define the helical
symmetry, Bessel orders were assigned to all of these layer lines (Figure 1).
Initial assignments were based on phase relationships across the meridian,
the radial location of the diffraction maximum, and the self-consistency of
assignments across the transform (e.g., the construction of a plausible n,l plot).
To verify the Bessel order assignments, several alternative indexing schemes
were tested, using EMIP helical reconstruction software (http://cryoem.nysbc.
org/EmIP.html) to compare phase residuals for the determination of out-of-
plane tilt (U). For highly tilted tubes (U�5�), this phase residual was definitive
in identifying the correct indexing. Near-/far-phase residuals and two-fold
phase residuals were also consistently lower with the correct indexing. In
the end, EMIP was used to apply the unbending methods of Beroukhim and
Unwin (1997) to 12 tubes. Correction for the contrast transfer function was
based on an amplitude contrast of 7% (Toyoshima et al., 1993) and the
resolution of the final maps was truncated at 9 A resolution, where the two-
fold related phase residual approached 43� (random data produce a 45�
residual).
32, September 7, 2011 ª2011 Elsevier Ltd All rights reserved 1229

Structure
Cryo-EM and Computational Docking of CopA
Modeling and Docking
All modeling and docking procedures were carried out using the ICM protein
docking software (v3.6-1f; Abagyan and Totrov, 1994; Cardozo et al., 1995)
as previously described (Allen and Stokes, 2011). Briefly, the sequences of
CopA and SERCA1a were aligned by Needleman and Wunsch global align-
ment using zero end-gap penalties (Abagyan and Batalov, 1997). A 3D model
of CopA (omitting N- and C-terminal metal-binding domains) was then con-
structed by threading its sequence onto the X-ray crystallographic structure
for SERCA1a in the E2 conformation (PDB ID code 1IWO) using the alignment
as a guide. For this homology model, the geometries of conserved residues
were fixed, whereas the nonconserved residues were set to their standard
geometries and a local minimization was performed to relieve any clashes.
Structural databases were consulted for probable configurations of the non-
conserved loops or, in the event no reasonable solution was found, the loop
was modeled freely to its lowest-energy conformation (Cardozo et al., 1995).
Existing X-ray crystal structures of CopA cytoplasmic domains (the isolated
P/N-domain pair, PDB ID code 3A1C, and the isolated A domain, PDB ID
code 2HC8) were then aligned with the initial homology model, and this model
was adjusted to exactly match the X-ray structures in overlapping regions.
Thus, the resulting hybrid homology model of CopA inherits X-ray crystallo-
graphic information from both the SERCA1a E2 conformation and the isolated
structures of theCopA cytoplasmic domains. A homologymodel for the NMBD
of CopA based on the X-ray crystallographic structure of the metallochaper-
one CopZ (PDB ID code 2QIF) was built in ICM using the same approach as
above. Finally, we used Coot (Emsley et al., 2010) to make minor adjustments
or refinements to the model and Phyre (Bennett-Lovsey et al., 2008) to build
the extra M2 and M3 helices of CopA, and then several rounds of energy mini-
mization were performed in ICM. Finally, Chimera (Pettersen et al., 2004) was
used to visualize results and to make figures.
After building these atomic models, the NMBD was docked to the hybrid
model of CopA (N, P, and A domains) using ICM protein-protein docking
(Fernandez-Recio et al., 2005). ICM first predicted surface patches on the
cytoplasmic domains that were likely to produce energetically favorable
protein interactions using the optimal docking area method. Next, ICM
computed electrostatic, van der Waals, hydrogen-bond, solvation, and hydro-
phobic potentials on a coarse grid over these surface regions. Interaction
energies were determined for a Monte Carlo sampling of NMBD poses at
each of the surface patches. The 400 most favorable results were then sub-
jected to side-chain energy minimization and ranked according to their overall
binding energy. Buried surface area was determined in CNS (Brunger, 2007)
with a 1.4 A probe radius.
ACCESSION NUMBERS
Coordinates for the two models have been deposited in the Protein Data Bank
under ID codes 3J08 and 3J09, and the map has been deposited in the
Electron Microscopy Databank under accession number 5271.
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and one movie and can be
found with this article online at doi:10.1016/j.str.2011.05.014.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the facilities at the New York Structural
Biology Center and support from NIH grants R01 GM56960 and U54
GM094598 to D.L.S. as well as NIH grant DP2 OD004631 to T.C.
Received: April 4, 2011
Revised: May 9, 2011
Accepted: May 12, 2011
Published online: August 4, 2011
REFERENCES
Abagyan, R.A., and Batalov, S. (1997). Do aligned sequences share the same
fold? J. Mol. Biol. 273, 355–368.
1230 Structure 19, 1219–1232, September 7, 2011 ª2011 Elsevier Lt
Abagyan, R., and Totrov, M. (1994). Biased probability Monte Carlo conforma-
tional searches and electrostatic calculations for peptides and proteins. J.Mol.
Biol. 235, 983–1002.
Achila, D., Banci, L., Bertini, I., Bunce, J., Ciofi-Baffoni, S., and Huffman, D.L.
(2006). Structure of human Wilson protein domains 5 and 6 and their interplay
with domain 4 and the copper chaperone HAH1 in copper uptake. Proc. Natl.
Acad. Sci. USA 103, 5729–5734.
Allen, G.S., and Stokes, D.L. (2011). Modeling, docking, fitting of atomic struc-
tures to 3Dmaps from cryo-electronmicroscopy. MethodsMol. Biol., in press.
Arguello, J.M., Eren, E., and Gonzalez-Guerrero, M. (2007). The structure and
function of heavy metal transport P1B-ATPases. Biometals 20, 233–248.
Axelsen, K.B., and Palmgren, M.G. (1998). Evolution of substrate specificities
in the P-type ATPase superfamily. J. Mol. Evol. 46, 84–101.
Bal, N., Mintz, E., Guillain, F., and Catty, P. (2001). A possible regulatory role for
the metal-binding domain of CadA, the Listeria monocytogenesCd2+-ATPase.
FEBS Lett. 506, 249–252.
Banci, L., Bertini, I., Cantini, F., Chasapis, C.T., Hadjiliadis, N., and Rosato, A.
(2005a). A NMR study of the interaction of a three-domain construct of ATP7A
with copper(I) and copper(I)-HAH1: the interplay of domains. J. Biol. Chem.
280, 38259–38263.
Banci, L., Bertini, I., Ciofi-Baffoni, S., Chasapis, C.T., Hadjiliadis, N., and
Rosato, A. (2005b). An NMR study of the interaction between the human
copper(I) chaperone and the second and fifth metal-binding domains of the
Menkes protein. FEBS J. 272, 865–871.
Banci, L., Bertini, I., Cantini, F., DellaMalva, N., Herrmann, T., Rosato, A., and
Wuthrich, K. (2006). Solution structure and intermolecular interactions of the
third metal-binding domain of ATP7A, the Menkes disease protein. J. Biol.
Chem. 281, 29141–29147.
Banci, L., Bertini, I., Cantini, F., Inagaki, S., Migliardi, M., and Rosato, A. (2010).
The binding mode of ATP revealed by the solution structure of the N-domain of
human ATP7A. J. Biol. Chem. 285, 2537–2544.
Barry, A.N., Shinde, U., and Lutsenko, S. (2010). Structural organization
of human Cu-transporting ATPases: learning from building blocks. J. Biol.
Inorg. Chem. 15, 47–59.
Bennett-Lovsey, R.M., Herbert, A.D., Sternberg, M.J., and Kelley, L.A. (2008).
Exploring the extremes of sequence/structure space with ensemble fold
recognition in the program Phyre. Proteins 70, 611–625.
Beroukhim, R., and Unwin, N. (1997). Distortion correction of tubular crystals:
improvements in the acetylcholine receptor structure. Ultramicroscopy 70,
57–81.
Brunger, A.T. (2007). Version 1.2 of the Crystallography and NMR system. Nat.
Protoc. 2, 2728–2733.
Cardozo, T., Totrov, M., and Abagyan, R. (1995). Homology modeling by the
ICM method. Proteins 23, 403–414.
Cater, M.A., La Fontaine, S., and Mercer, J.F. (2007). Copper binding to the
N-terminal metal-binding sites or the CPC motif is not essential for copper-
induced trafficking of the human Wilson protein (ATP7B). Biochem. J. 401,
143–153.
Cornea, R.L., and Thomas, D.D. (1994). Effects of membrane thickness on the
molecular dynamics and enzymatic activity of reconstituted Ca-ATPase.
Biochemistry 33, 2912–2920.
Diaz, R., Rice, W.J., and Stokes, D.L. (2010). Fourier-Bessel reconstruction of
helical assemblies. Methods Enzymol. 482, 131–165.
Dmitriev, O., Tsivkovskii, R., Abildgaard, F., Morgan, C.T., Markley, J.L., and
Lutsenko, S. (2006). Solution structure of the N-domain of Wilson disease
protein: distinct nucleotide-binding environment and effects of disease muta-
tions. Proc. Natl. Acad. Sci. USA 103, 5302–5307.
Emsley, P., Lohkamp, B., Scott, W.G., and Cowtan, K. (2010). Features and
development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501.
Fan, B., and Rosen, B.P. (2002). Biochemical characterization of CopA, the
Escherichia coli Cu(I)-translocating P-type ATPase. J. Biol. Chem. 277,
46987–46992.
d All rights reserved

Structure
Cryo-EM and Computational Docking of CopA
Fernandez-Recio, J., Totrov, M., Skorodumov, C., and Abagyan, R. (2005).
Optimal docking area: a new method for predicting protein-protein interaction
sites. Proteins 58, 134–143.
Gonzalez-Guerrero, M., and Arguello, J.M. (2008). Mechanism of Cu+-trans-
porting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmem-
brane transport sites. Proc. Natl. Acad. Sci. USA 105, 5992–5997.
Gonzalez-Guerrero, M., Hong, D., and Arguello, J.M. (2009). Chaperone-medi-
ated Cu+ delivery to Cu+ transport ATPases: requirement of nucleotide
binding. J. Biol. Chem. 284, 20804–20811.
Hatori, Y., Majima, E., Tsuda, T., and Toyoshima, C. (2007). Domain organiza-
tion and movements in heavy metal ion pumps: papain digestion of CopA,
a Cu+-transporting ATPase. J. Biol. Chem. 282, 25213–25221.
Hatori, Y., Hirata, A., Toyoshima, C., Lewis, D., Pilankatta, R., and Inesi, G.
(2008). Intermediate phosphorylation reactions in the mechanism of ATP
utilization by the copper ATPase (CopA) of Thermotoga maritima. J. Biol.
Chem. 283, 22541–22549.
Hatori, Y., Lewis, D., Toyoshima, C., and Inesi, G. (2009). Reaction cycle of
Thermotoga maritima copper ATPase and conformational characterization of
catalytically deficient mutants. Biochemistry 48, 4871–4880.
Huster, D., and Lutsenko, S. (2003). The distinct roles of the N-terminal
copper-binding sites in regulation of catalytic activity of the Wilson’s disease
protein. J. Biol. Chem. 278, 32212–32218.
Jørgensen, P.L., and Andersen, J.P. (1988). Structural basis for E1-E2 confor-
mational transitions in Na,K-pump andCa-pump proteins. J.Membr. Biol. 103,
95–120.
Kimura, Y., Kurzydlowski, K., Tada, M., and MacLennan, D.H. (1996).
Phospholamban regulates the Ca2+-ATPase through intramembrane interac-
tions. J. Biol. Chem. 271, 21726–21731.
Kuhlbrandt, W. (2004). Biology, structure and mechanism of P-type ATPases.
Nat. Rev. Mol. Cell Biol. 5, 282–295.
Larin, D., Mekios, C., Das, K., Ross, B., Yang, A.S., and Gilliam, T.C. (1999).
Characterization of the interaction between the Wilson and Menkes disease
proteins and the cytoplasmic copper chaperone, HAH1p. J. Biol. Chem. 274,
28497–28504.
Lee, J., Prohaska, J.R., and Thiele, D.J. (2001). Essential role for mammalian
copper transporter Ctr1 in copper homeostasis and embryonic development.
Proc. Natl. Acad. Sci. USA 98, 6842–6847.
Lewis, B.A., and Engelman, D.M. (1983). Lipid bilayer thickness varies linearly
with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 166,
211–217.
Lubben, M., Portmann, R., Kock, G., Stoll, R., Young, M.M., and Solioz, M.
(2009). Structural model of the CopA copper ATPase of Enterococcus hirae
based on chemical cross-linking. Biometals 22, 363–375.
Lutsenko, S., and Kaplan, J.H. (1995). Organization of P-type ATPases: signif-
icance of structural diversity. Biochemistry 34, 15607–15613.
Lutsenko, S., Barnes, N.L., Bartee, M.Y., and Dmitriev, O.Y. (2007). Function
and regulation of human copper-transporting ATPases. Physiol. Rev. 87,
1011–1046.
Mana-Capelli, S., Mandal, A.K., and Arguello, J.M. (2003). Archaeoglobus
fulgidus CopB is a thermophilic Cu2+-ATPase: functional role of its histidine-
rich N-terminal metal binding domain. J. Biol. Chem. 278, 40534–40541.
Mandal, A.K., and Arguello, J.M. (2003). Functional roles of metal binding
domains of the Archaeoglobus fulgidus Cu(+)-ATPase CopA. Biochemistry
42, 11040–11047.
Mandal, A.K., Cheung, W.D., and Arguello, J.M. (2002). Characterization of a
thermophilic P-type Ag+/Cu+-ATPase from the extremophile Archaeoglobus
fulgidus. J. Biol. Chem. 277, 7201–7208.
Mitra, B., and Sharma, R. (2001). The cysteine-rich amino-terminal domain of
ZntA, a Pb(II)/Zn(II)/Cd(II)-translocating ATPase from Escherichia coli, is not
essential for its function. Biochemistry 40, 7694–7699.
Møller, J.V., Juul, B., and leMaire, M. (1996). Structural organization, ion trans-
port, and energy transduction of P-type ATPases. Biochim. Biophys. Acta
1286, 1–51.
Structure 19, 1219–12
Morgan, C.T., Tsivkovskii, R., Kosinsky, Y.A., Efremov, R.G., and Lutsenko, S.
(2004). The distinct functional properties of the nucleotide-binding domain of
ATP7B, the human copper-transporting ATPase: analysis of the Wilson
disease mutations E1064A, H1069Q, R1151H, and C1104F. J. Biol. Chem.
279, 36363–36371.
Morth, J.P., Pedersen, B.P., Toustrup-Jensen, M.S., Sørensen, T.L., Petersen,
J., Andersen, J.P., Vilsen, B., and Nissen, P. (2007). Crystal structure of the
sodium-potassium pump. Nature 450, 1043–1049.
Olesen, C., Sørensen, T.L., Nielsen, R.C., Møller, J.V., and Nissen, P. (2004).
Dephosphorylation of the calcium pump coupled to counterion occlusion.
Science 306, 2251–2255.
Olesen, C., Picard, M., Winther, A.M., Gyrup, C., Morth, J.P., Oxvig, C., Møller,
J.V., and Nissen, P. (2007). The structural basis of calcium transport by the
calcium pump. Nature 450, 1036–1042.
Pedersen, B.P., Buch-Pedersen, M.J., Morth, J.P., Palmgren, M.G., and
Nissen, P. (2007). Crystal structure of the plasma membrane proton pump.
Nature 450, 1111–1114.
Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M.,
Meng, E.C., and Ferrin, T.E. (2004). UCSF Chimera—a visualization system
for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612.
Rae, T.D., Schmidt, P.J., Pufahl, R.A., Culotta, V.C., and O’Halloran, T.V.
(1999). Undetectable intracellular free copper: the requirement of a copper
chaperone for superoxide dismutase. Science 284, 805–808.
Ralle, M., Lutsenko, S., and Blackburn, N.J. (2004). Copper transfer to the
N-terminal domain of the Wilson disease protein (ATP7B): X-ray absorption
spectroscopy of reconstituted and chaperone-loaded metal binding domains
and their interaction with exogenous ligands. J. Inorg. Biochem. 98, 765–774.
Rice, W.J., Young, H.S., Martin, D.W., Sachs, J.R., and Stokes, D.L. (2001).
Structure of Na+,K+-ATPase at 11-A resolution: comparison with Ca2+-
ATPase in E1 and E2 states. Biophys. J. 80, 2187–2197.
Rice, W.J., Kovalishin, A., and Stokes, D.L. (2006). Role of metal-binding
domains of the copper pump from Archaeoglobus fulgidus. Biochem.
Biophys. Res. Commun. 348, 124–131.
Sazinsky, M.H., Agarwal, S., Arguello, J.M., and Rosenzweig, A.C. (2006a).
Structure of the actuator domain from the Archaeoglobus fulgidus Cu(+)-
ATPase. Biochemistry 45, 9949–9955.
Sazinsky, M.H., Mandal, A.K., Arguello, J.M., and Rosenzweig, A.C. (2006b).
Structure of the ATP binding domain from the Archaeoglobus fulgidus
Cu+-ATPase. J. Biol. Chem. 281, 11161–11166.
Singleton, C., Hearnshaw, S., Zhou, L., Le Brun, N.E., and Hemmings, A.M.
(2009). Mechanistic insights into Cu(I) cluster transfer between the chaperone
CopZ and its cognate Cu(I)-transporting P-type ATPase, CopA. Biochem. J.
424, 347–356.
Strausak, D., La Fontaine, S., Hill, J., Firth, S.D., Lockhart, P.J., and Mercer,
J.F. (1999). The role of GMXCXXC metal binding sites in the copper-induced
redistribution of the Menkes protein. J. Biol. Chem. 274, 11170–11177.
Strausak, D., Howie, M.K., Firth, S.D., Schlicksupp, A., Pipkorn, R., Multhaup,
G., and Mercer, J.F. (2003). Kinetic analysis of the interaction of the copper
chaperone Atox1 with the metal binding sites of the Menkes protein. J. Biol.
Chem. 278, 20821–20827.
Taylor, K.A., Dux, L., and Martonosi, A. (1986). Three-dimensional reconstruc-
tion of negatively stained crystals of the Ca2+-ATPase from muscle sarco-
plasmic reticulum. J. Mol. Biol. 187, 417–427.
Thomas, G.R., Forbes, J.R., Roberts, E.A., Walshe, J.M., and Cox, D.W.
(1995). The Wilson disease gene: spectrum of mutations and their conse-
quences. Nat. Genet. 9, 210–217.
Toyoshima, C., and Nomura, H. (2002). Structural changes in the calcium
pump accompanying the dissociation of calcium. Nature 418, 605–611.
Toyoshima, C., and Unwin, N. (1988). Ion channel of acetylcholine receptor
reconstructed from images of postsynaptic membranes. Nature 336, 247–250.
Toyoshima, C., Yonekura, K., and Sasabe, H. (1993). Contrast transfer for
frozen-hydrated specimens: II. Amplitude contrast at very low frequencies.
Ultramicroscopy 48, 165–176.
32, September 7, 2011 ª2011 Elsevier Ltd All rights reserved 1231

Structure
Cryo-EM and Computational Docking of CopA
Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa, H. (2000). Crystal
structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution.
Nature 405, 647–655.
Tsivkovskii, R., MacArthur, B.C., and Lutsenko, S. (2001). The Lys1010–
Lys1325 fragment of the Wilson’s disease protein binds nucleotides and
interacts with the N-terminal domain of this protein in a copper-dependent
manner. J. Biol. Chem. 276, 2234–2242.
Tsuda, T., and Toyoshima, C. (2009). Nucleotide recognition by CopA, a
Cu+-transporting P-type ATPase. EMBO J. 28, 1782–1791.
Veldhuis, N.A., Gaeth, A.P., Pearson, R.B., Gabriel, K., and Camakaris, J.
(2009a). The multi-layered regulation of copper translocating P-type
ATPases. Biometals 22, 177–190.
Veldhuis, N.A., Valova, V.A., Gaeth, A.P., Palstra, N., Hannan, K.M., Michell,
B.J., Kelly, L.E., Jennings, I., Kemp, B.E., Pearson, R.B., et al. (2009b).
Phosphorylation regulates copper-responsive trafficking of the Menkes
copper transporting P-type ATPase. Int. J. Biochem. Cell Biol. 41, 2403–2412.
Walker, J.M., Tsivkovskii, R., and Lutsenko, S. (2002). Metallochaperone
Atox1 transfers copper to the NH2-terminal domain of the Wilson’s disease
protein and regulates its catalytic activity. J. Biol. Chem. 277, 27953–27959.
1232 Structure 19, 1219–1232, September 7, 2011 ª2011 Elsevier Lt
Walker, J.M., Huster, D., Ralle, M., Morgan, C.T., Blackburn, N.J., and
Lutsenko, S. (2004). The N-terminal metal-binding site 2 of the Wilson’s
disease protein plays a key role in the transfer of copper from Atox1. J. Biol.
Chem. 279, 15376–15384.
Warren, G.B., Toon, P.A., Birdsall, N.J.M., Lee, A.G., andMetcalfe, J.C. (1974).
Reconstitution of a calcium pump using defined membrane components.
Proc. Natl. Acad. Sci. USA 71, 622–626.
Wu, C.C., Rice, W.J., and Stokes, D.L. (2008). Structure of a copper pump
suggests a regulatory role for its metal-binding domain. Structure 16, 976–985.
Xu, C., Rice, W.J., He, W., and Stokes, D.L. (2002). A structural model for the
catalytic cycle of Ca(2+)-ATPase. J. Mol. Biol. 316, 201–211.
Young, H.S., Rigaud, J.-L., Lacapere, J.-J., Reddy, L.G., and Stokes, D.L.
(1997). How to make tubular crystals by reconstitution of detergent-solubilized
Ca2(+)-ATPase. Biophys. J. 72, 2545–2558.
Young, H.S., Xu, C., Zhang, P., and Stokes, D.L. (2001). Locating the thapsi-
gargin-binding site on Ca(2+)-ATPase by cryoelectron microscopy. J. Mol.
Biol. 308, 231–240.
Zhang, P., Toyoshima, C., Yonekura, K., Green, N.M., and Stokes, D.L. (1998).
Structure of the calcium pump from sarcoplasmic reticulum at 8-A resolution.
Nature 392, 835–839.
d All rights reserved





























