Embed Size (px)
Citation preview

doi.org/10.26434/chemrxiv.11860941.v1
The Chemical Bond Across the Periodic Table: Part 1 – First Row andSimple MetalsGabriel Freire Sanzovo Fernandes, Leonardo dos Anjos Cunha, Francisco Bolivar Correto Machado, LuizFerrão
Submitted date: 17/02/2020 • Posted date: 24/02/2020Licence: CC BY-NC-ND 4.0Citation information: Fernandes, Gabriel Freire Sanzovo; Cunha, Leonardo dos Anjos; Machado, FranciscoBolivar Correto; Ferrão, Luiz (2020): The Chemical Bond Across the Periodic Table: Part 1 – First Row andSimple Metals. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.11860941.v1
Chemical bond plays a central role in the description of the physicochemical properties of molecules andsolids and it is essential to several fields in science and engineering, governing the material’s mechanical,electrical, catalytic and optoelectronic properties, among others. Due to this indisputable importance, a properdescription of chemical bond is needed, commonly obtained through solving the Schrödinger equation of thesystem with either molecular orbital theory (molecules) or band theory (solids). However, connecting theseseemingly different concepts is not a straightforward task for students and there is a gap in the availabletextbooks concerning this subject. This work presents a chemical content to be added in the physicalchemistry undergraduate courses, in which the framework of molecular orbitals was used to qualitativelyexplain the standard state of the chemical elements and some properties of the resulting material, such as gasor crystalline solids. Here in Part 1, we were able to show the transition from Van der Waals clusters to metalin alkali and alkaline earth systems. In Part 2 and 3 of this three-part work, the present framework is applied tomain group elements and transition metals. The original content discussed here can be adapted andincorporated in undergraduate and graduate physical chemistry and/or materials science textbooks and alsoserves as a conceptual guide to subsequent disciplines such as quantum chemistry, quantum mechanics andsolid-state physics.
File list (4)
download fileview on ChemRxivThe Chemical Bond across the Periodic Table Part 1_Fir... (731.81 KiB)
download fileview on ChemRxivsupplementary_The Chemical Bond across the Periodic ... (184.69 KiB)
download fileview on ChemRxivThe chemical bond across the periodic table Part 2_mai... (459.25 KiB)
download fileview on ChemRxivThe chemical bond across the periodic table Part 3_tran... (331.06 KiB)
1
The Chemical Bond across the Periodic Table: Part 1 – First Row and Simple Metals
Gabriel F.S. Fernandes1, Leonardo A. Cunha1, Francisco B.C. Machado1, Luiz F. A.
Ferrão1*
1Departamento de Química, Instituto Tecnológico de Aeronáutica, São José dos
Campos, SP 12228-900, Brasil.
__________________________________
*Corresponding author. E-mail address: [email protected]

2
Abstract
Chemical bond plays a central role in the description of the physicochemical properties
of molecules and solids and it is essential to several fields in science and engineering,
governing the material’s mechanical, electrical, catalytic and optoelectronic properties,
among others. Due to this indisputable importance, a proper description of chemical
bond is needed, commonly obtained through solving the Schrödinger equation of the
system with either molecular orbital theory (molecules) or band theory (solids).
However, connecting these seemingly different concepts is not a straightforward task
for students and there is a gap in the available textbooks concerning this subject. This
work presents a chemical content to be added in the physical chemistry undergraduate
courses, in which the framework of molecular orbitals was used to qualitatively explain
the standard state of the chemical elements and some properties of the resulting
material, such as gas or crystalline solids. Here in Part 1, we were able to show the
transition from Van der Waals clusters to metal in alkali and alkaline earth systems. In
Part 2 and 3 of this three-part work, the present framework is applied to main group
elements and transition metals. The original content discussed here can be adapted and
incorporated in undergraduate and graduate physical chemistry and/or materials
science textbooks and also serves as a conceptual guide to subsequent disciplines such
as quantum chemistry, quantum mechanics and solid-state physics.
KEYWORDS: Upper-division Undergraduate, Graduate Education, Physical Chemistry,
Communication / Writing, Textbooks / Reference Books, Atomic Properties / Structure,
Material Science, MO theory, Periodicity / Periodic Table, Quantum Chemistry.

3
I. Introduction
The usual first approach in general chemistry classes for undergraduate students
is to depict the concept of chemical bonding through the electron-localized model
proposed by Lewis1, in which valence electrons, represented by localized dots for each
atom, can be shared to form a covalent bond between two atomic centers. Despite its
simplicity, other important chemical concepts can be built upon the Lewis model, such
as ionic, covalent bonds, intermolecular interactions, the polarity of molecules, stability,
octet rule and chemical reactions.
Going beyond Lewis structures and the octet rule, students are taught concepts
associated with electronic configuration/distribution into levels and sublevels with
different energies, Hund’s rules, Pauli repulsion and ultimately the quantum numbers
that label each one of these electronic states for atoms. Commonly, these ideas are
presented in an ad-hoc manner due to their quantum mechanical origin that requires a
more robust mathematical treatment. Eventually, students learn about the
wavefunction machinery associated with the Schrödinger equation and more abstract
concepts in introductory physical chemistry courses, such as the molecular orbital
theory that explains the chemical bond in molecules and band theory to describe the
electronic behavior in crystalline solid.
The molecular orbital theory arises by solving the Schrödinger equation of the
hydrogen molecule ion (H2+), providing an understanding of the chemical bond between
two protons and one electron from the quantum-mechanical point of view. Considering
a crystalline solid as a simple and big molecule, one can expand the molecular orbital
theory and derive the band theory. This relation is superficially demonstrated in
Physical-Chemistry and Solid-State textbooks2–7, however, only for alkali metals and
earth alkaline metals, using sodium and magnesium as examples. In most of the cases,
there are no discussions concerning their properties, other than electrical properties
(insulating, semiconductor and conductor), based on the occupation of the energy
band.
Understanding general trends that arise from the progression of the discrete
energy levels represented by the molecular orbitals to continuum band regions allows

4
students to comprehend bond formation, crystalline structure, material properties in
solids as well as the standard state for several compounds. For instance, how weakly
bounded agglomerates, such as lithium and beryllium, become conductive metals? Or,
in what way the elements in the carbon group progress from presenting insulator
(carbon) to semiconductor (silicon, germanium) and finally metallic (tin and lead)
properties as we move down across the carbon group in the periodic table? Or, why
nitrogen is a gas at the standard state (298K, 1bar) and the other substances of its
group (phosphorus, arsenic, and antimony) are solid at the standard state (298K, 1bar)?
These chemical notions of the atomic/molecular properties among the elements of the
main group by the molecular orbital theory are important for undergraduate students in
chemistry, physics and materials science/engineering majors.
In the first and second semesters, the engineering students at the Instituto
Tecnológico de Aeronáutica (ITA), in Brazil, are introduced to physical-chemistry
undergraduate introductory courses which focuses in the concepts of chemical bonds
as described above, but directed to demonstrate the formation of energy bands
through molecular orbital in a simple and qualitative manner, explaining the properties
and trends across the periodic table using two main ingredients: 1) the available energy
levels and their occupation based on the electronic structure of the hydrogen atom; and
2) the energy splitting of energy levels due to spatial overlap of electronic densities.
The subjects taught in the referred courses were divided into three-part. In this
first part, the discussion is organized as follows: Section II is dedicated to contextualizing
the proposed content within the data available in the literature, especially in textbooks
covering molecular orbitals (usually in chapters dedicated to molecules) and properties
of solids (usually in chapters dedicated to condensed matter or solids). From that, we
pinpoint the missing information that would benefit from the construction of a bridge
between these subjects (molecules and solids) and the potential broadening in the
scope of the molecular orbital machinery. In section III, the formation of bands and the
state of the elements for the hydrogen, helium and simple metals are discussed.
In the next followed papers, the framework developed here is applied to the
main group elements of the periodic table (In Part 2) and to the transition metals (in
Part 3).

5
II. Molecular orbitals and formation of solids and bands in textbooks
Chemical bonding, molecular orbital, and band theories are common topics in
physical chemistry, solid-state chemistry/physics and electronic structure textbooks.
However, the connection between the molecular orbital and band theories is
superficially discussed without delving into the relationship between energy levels and
their occupation to describe the chemical bond in solids and their physicochemical
properties, the main idea of the present work. Independently of the approach taken,
two main approximations are usually employed to study the electronic structure of
molecules and solids. First, based on the difference between the masses of electrons
and nuclei and consequently the different time scales that govern their motion, the
Born-Oppenheimer approximation2,5,7 is used. This allows us to separate the electronic
degrees of freedom and parametrize the energy as a function of nuclear coordinates.
Therefore, finding the true ground-state of the system corresponds to finding the
nuclear configuration that minimizes the electronic energy. The second approximation
commonly employed is known as the mean-field (or self-consistent field) approach.2,5,7
The electronic problem itself is still too complicated to be solved, due to its many-body
nature. Hence, one searches for the solution of the Schrödinger equation that allows
the treatment of the electrons moving in an average potential/field created by the
others (i.e., independent electrons). The use of these two approximations simplifies the
problem: now the goal is to solve a set of effective Schrödinger-like equations, each one
depending on only one electron coordinate, giving rise to the idea of molecular orbitals.
As previously mentioned, the approach outlined above works for both molecular
and solid-state systems. However, for crystalline solids, which contains on the order of
Avogadro’s number of atoms (~1023), one can simplify the problem even further using
the appropriate physical symmetries of the problem. Given the periodic arrangement of
the atoms in the crystal lattice, the potential felt by the electrons is also periodic. This
allows us to use crystal momentum (k) as a good quantum number to label the
electronic states. Hence, the idea is to find a dispersion relation between the electronic
energy and the crystal momentum, leading to the well-known formation of electronic
bands.

6
Band theory is commonly presented in textbooks2,3,7 to explain the metallic
bonding in solids. The description of this theory, in the textbooks2,3,7, is frequently
based on the nearly free-electron model, wherein in this model the electron is treated
as a free particle, initially, in one-dimensional box then extrapolated to three-
dimensional box, in which the crystalline structure of the solid is taken as the box
parameters. Another way to construct the band theory presented in textbooks2–6 is by
using the linear combination of atomic orbital (CLOA) to describe the one-electron
wavefunction of the molecule, the molecular orbitals. Then, the molecular orbitals
spatial overlap of a large number of atoms/molecules leads to energy bands. Kaxiras6
introduces this topic using the closest approach to the present work, exploring the band
theory from the perspective of quantum chemistry more than most, if not all,
textbooks2–7. In his textbook, the discussion is divided into the topics: noble gas, simple
metals (ns1 and ns2) and main group elements (nsxnpy). The noble gas solid formation is
discussed as happening through the coordination number maximization to compensate
the low cohesion energy, due to the weak attractive character of the Van der Waals
interaction. In the simple metals, the high atomic radius and atomic orbital with no
direction (ns) are used to describe the high spatial overlap between the atomic orbitals,
forming a “sea” of valence electrons. For the main group elements, it is shown that the
valence electrons have directions (px, py and pz), wherein the combination of the s and p
atomic orbitals forming the covalent bond, hybrid orbitals. Some discussion regarding
trends in the main group is discussed, such as the tin increasing its coordination number
to form metallic bonds than hybridize and make chemical bonds (diamond character),
the nitrogen family making three chemical bonds and the ns2 atomic orbital are non-
bonding and the oxygen family forming molecular rings in the solid-state.
However, to the best of the authors' knowledge, there is no previous textbook
that creates a connection between molecular orbitals and band theory topics by
employing the available energy levels and their occupation (from molecular orbitals) to
describe the chemical bond in solids and explain some of their basic physicochemical
properties. This novel content presented herein is represented in the diagram shown in
Figure 1, which contextualizes this content within the available literature data.

7
Figure 1. Representative diagram of the available literature concerning molecular
orbital, band theory and physicochemical properties, focusing on the superposition of
these topics. The author names refer to the following textbooks: Moore - Solid-state
chemistry3; Atkins - Physical chemistry2; Levine - Physical chemistry5; Kaxiras - Atomic
and Electronic structure of solids6; Harrison - Electronic Structure and the Properties of
Solids: The Physics of the Chemical Bond4.
In previous articles8–10, the concepts and the formalism of molecular orbital,
bond order and band theories were individually discussed in more detail. However, the
relation among these concepts could be more explored in a simpler and qualitative
manner, which is the focus of the present work.
III. The formation of bands and the state of the elements
This section is dedicated to present original content in the topic of electronic structure,
in which the chemical bond of a system is analyzed as the system grows, using the
molecular orbital as a simplified tool. Within this approach, one is able to formulate a
global vision of the chemical bonding across the periodic table, including the expected
physical state and some properties of the elements in their standard state. The analysis

8
showed here is focused, but not restricted to, homonuclear systems. The main text
includes essential concepts and vital examples, while the supporting information
presents data in more detail and covers other examples. Namely, the supplementary
material gathers information related to some concepts as molecular orbital, bond order
and atomic orbitals along the second row.
III.A. First row
Before beginning our analysis, we can discuss the representation of the atomic orbitals,
which can be viewed as spatial distributions of the electronic charge. One can use a
simplified representation, as shown in Figure 2, in which the electronic charge
distribution is represented by a line (in 2d representation) or a surface (in 3d
representation), passing through, for example, the region with maximum electronic
density.
Figure 2. An atomic orbital (hydrogen 1s) depicted as the electronic spatial distribution
(on the left) with its corresponding radial distribution (top), highlighting the most
probable radius (rmp). The simplified model (in the right) uses only an isoelectronic
surface to represent the electron spatial distribution.
Within this context, one should keep in mind that the spatial distribution of the
orbitals determines the energy levels of a given system. When overlapping, these

9
orbitals will change their spatial distribution, and therefore, the energy levels will also
change.
II.A. Hydrogen
Hydrogen atom will be used as a metric for the other chemical elements and as
an example of how atomic or molecular orbitals interact to compose a band structure.
Based on these ideas, one can also understand why the hydrogen, at the standard state
(298K, 1bar), is in gaseous state and agglomerate as diatomic molecules. In the
following demonstrations, combinations were considered in a two-by-two way, for
example, two hydrogen atoms forming molecular hydrogen (H2), two molecular
hydrogens composing H4, and so on.
Initially, consider two hydrogen atoms and their 1s atomic orbital to form
molecular hydrogen (H2). According to the LCAO-MO approximation, two atomic
orbitals must produce two molecular orbitals, a bonding molecular orbital, and an anti-
bonding molecular orbital, called and * respectively. This nomenclature for the
molecular orbitals is related to the number of nodal planes containing the bonding
axis2,5. Sigma orbitals () do not have a nodal plane, while pi orbitals (π) present one
nodal plane, delta orbitals (δ), which are rarer, have 2 nodal planes and so on Figure 3.
Figure 3. Interaction of two atomic orbitals forming a bonding and an anti-bonding
(left) and (right) molecular orbitals. The red and blue color refers to the wavefunction
phase, negative and positive, respectively.

10
We shall begin our analysis by noticing that, for each element, a reference
energy level indicated by a red dashed line (Figure 4) represents the highest occupied
atomic orbital, therefore it will change depending on the analyzed chemical element.
Electrons occupying energy levels lower than this reference line will be bonding orbitals,
while electrons in higher energy levels from the reference line are anti-bonding orbitals,
independently of the system size. The energy difference among the atomic orbitals (ns,
np and (n-1)d) was loosely based on the spectroscopy data gathered on NIST Atomic
Spectra Database Levels11. Thus, there was no concern in making the atomic energy
levels accurate, so the student can focus on the modifications in the bonding pattern
that arises from the balance between the occupation of electrons and the available
energy levels, instead of the energy of the initial atomic orbitals. This allows us to use
the same diagram for the complete set of elements considered in the present study and
in the following Part 2 and Part 3.
The two 1s electrons at molecular orbital stabilize the H2 molecule, as shown
in Figure 5. For the H4 molecule, two H2 molecules are combined, therefore, the and
* of one hydrogen molecule will be combined with and * of the other hydrogen
molecule, producing two and two * molecular orbitals. However, it should be noted
that the energy variation for the bonding and anti-bonding molecular orbitals in the H4
molecule is smaller than the H2 molecule. This is a consequence of the surface/volume
ratio, which decreases as the system size increases. As the surface is the responsible for
the overlap and consequently, the possibility to form new bonds connected to the
whole system, this means that bonding energy (molecular orbitals energy variation) for
each aggregation step is expected to monotonically decrease, reaching a limit for the
“infinite size” (solid or strongly bonded liquid) system. This idea is presented in Figure 4,
depicting the formation of hypothetical orbitals in a system of hydrogen atoms.

11
Figure 4. Hypothetical orbitals of a system of hydrogen atoms. The first line shows
noninteracting orbitals for H2 and H4. The second line shows the resulting orbitals
combinations, which can be organized as those with electron depletion of the
internuclear region and those with electron enrichment on the internuclear region. The
third line shows the energy levels corresponding to the formation of the orbital, in
which the arrows represent the energy variations relative to the predecessor system,
highlighting that the energy variations of those orbitals become sequentially smaller.
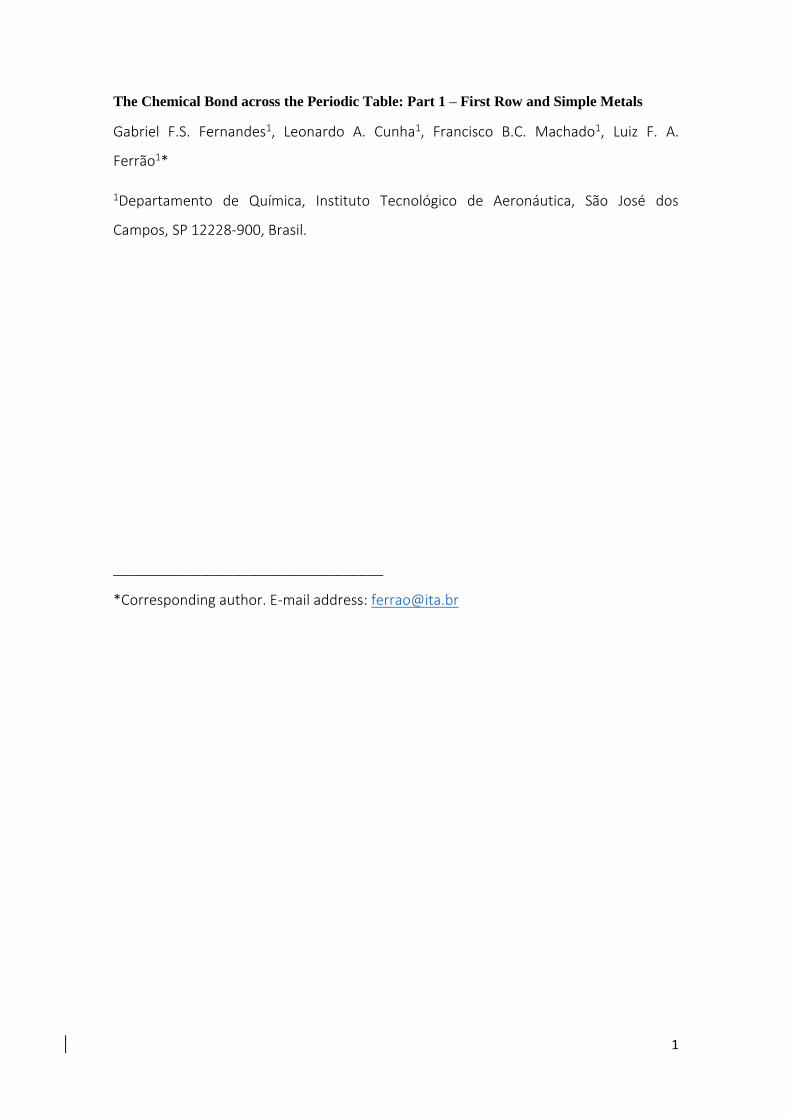
12
Figure 5. Hydrogen atom band structure formation through molecular orbital machinery. The red dashed line refers to the highest occupied atomic orbital.
One could keep overlapping the orbitals in such system, not stopping at H4 or H8,
but conjecturing the electronic structure of a system with up to one mol of hydrogen
atoms. Figure 5 shows the expected energy levels of such system. The dashed red line in
Figure 5 and subsequent Figures represents the highest atomic orbital occupied in the
isolated atoms and will be called hereinafter the reference line. The highest bonding
molecular orbital (energy lower than the reference line), turns into the top of the
valence band, whereas the lowest anti-bonding molecular orbital turns into the bottom
of the conduction band (above the reference line). The transition from molecular
orbitals to energy bands is justified by the energy difference between two consecutive
molecular orbitals (E) for the system containing one mol of atoms (HNA). This can be
estimated by the energy range of the whole s band (valence + conduction) divided by
the number of molecular orbitals in it. Since each atom contributes with one molecular
orbital, then the number of orbitals for HNA is equal to NA. Therefore, we only need to
estimate the energy range of the s band. Since the formation of the diatomic molecule
carries a significant portion of the energy variations involved in this band, one can have

13
an educated guess for the s energy band based solely on the energy variations from two
hydrogen atoms to form H2. This can be obtained from the H2 dissociation energy, equal
to 4.74 eV12,13. Compared to the total energy of the “initially” isolated atoms (E(2H) = -
27.2 eV), this energy variation roughly corresponds to 20% of the system’s total energy.
Now we consider that all other bonds up to the “one mol of atoms” system will further
decrease the lowest energy level by 50% of the energy involved in the formation of the
diatomic. This estimative considers that all bonding external to a diatomic molecule,
due to overlap, does not recover the same binding energy of a single molecule. This is,
up to a point, similar to the bonding energy in an ionic solid (per mol), which is around
1.5 times the binding energy of an ionic pair (Madelung constant). Therefore, the
energy variation of all bonds subsequent to the diatomic can be estimated to be 10%
compared to the isolated atoms total energy. Therefore, we have an estimate for the
valence band energy range equal to 0.3*E(2H).
The energy range of the conduction band can be estimated to be of the same
magnitude of the valence one. However, it should be noticed that the energy difference
between antibonding orbitals (1*) with the reference orbitals are further (in energy)
than the bonding orbitals (1) with the reference ones. In a pictorial way, it is easier to
weaken a bond by depleting the electronic density between the nuclei (antibonding
orbital) than to strengthen a chemical bond by enriching the density between the nuclei
(bonding orbital). In practical terms, we will consider that the H2 antibonding orbital 1*
is 50% further to the reference line than 1, i.e., the energy range of the conduction
band would be equal to 0.3*E(2H) from the diatomic plus 0.1*E(2H) from all other
bonds up to the molar atoms system. Finally, the (valence + conduction bands) energy
range can be estimated to be equal to 0.7*E(2H).
In this sense, we can estimate the energy separation between discrete
molecular orbitals (Equation 2).
∆𝐸 = 𝑎𝑡𝑜𝑚𝑖𝑐 𝑜𝑟𝑏𝑖𝑡𝑎𝑙 𝑒𝑛𝑒𝑟𝑔𝑦∗𝑒𝑛𝑒𝑟𝑔𝑦 𝑣𝑎𝑟𝑖𝑎𝑡𝑖𝑜𝑛
𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑚𝑜𝑙𝑒𝑐𝑢𝑙𝑎𝑟 𝑜𝑟𝑏𝑖𝑡𝑎𝑙𝑠=
=0.7∗(−13.6)
1023 = −10−22 𝑒𝑉
𝑠𝑡𝑎𝑡𝑒 (2)

14
This energy difference between adjacent orbitals is so close (10-22 eV) that we
can effectively treat the bands as formed by continuum states rather than discrete
ones. Take as an example an extremely low-frequency photon (thus very long
wavelength) such as those related to Schumann resonances14. This phenomenon
happens when electric events occur in the atmosphere forming standing waves in a
closed Earth-ionosphere cavity14: the fundamental Schumman resonance is at
approximately 7.83 Hz, with a wavelength equal to Earth’s circumference (l=40.075.000
m). The energy of this extremely low frequency photon (Earth-size wavelength) is in the
order of 10-15 eV, which is very low, but still 10 million times higher than the energy
difference between the orbitals in a band system. Since the slightest thermal energy is
more than sufficient to transfer energy between these states, the band systems can be
treated as continuous.
Thus, for a “one mol of atoms” system the electronic structure description
changes its character from discrete (molecular orbital) to continuum (band structure).
In the situation presented in Figure 5, the hydrogen solid would present a metallic
behavior because of the delocalized electrons and zero band gap between the valence
and conduction bands. However, one can ask why the standard state of hydrogen is a
gas and not a metallic solid? Therefore, in order to infer the standard state of the
elements and answer this question, the bond order analysis should be employed as the
system grows to predict if the system tends to form new bonds.

15
Figure 6. Standard state analysis of the Hydrogen element through Bond Order (B.O.)
and energy criterion. One can see that the H2 possesses a positive B.O. value (B.O.=1)
and stabilization energy gain through the chemical bond, indicating that the diatomic
molecule formation is energetically favorable. However, this is not observed on the H4
molecule, wherein, the B.O. is zero and the occupation of anti-bonding orbitals
destabilizes the molecule, leading to dissociation. Therefore, it is energetically favorable
to maintain the H2 electronic structure (B.O.=0) and interact with other H2 molecule
through intermolecular forces (Van der Waals) rather than make a new chemical bond
(2 molecular orbital).
We should recall that the analysis described in this study is based on the
reference energy level of the “unbound system”, i.e., the reference energy is not always
the isolated atoms, but instead, it is given by the energy levels of the predecessor
system. Consider, for example, the hydrogen system with four atoms (H4 or the 2H2).
The reference, in this case, is given by the 12 levels of the H2 molecule. On H4
molecule, the 12 and 22 orbitals represent the bonding and anti-bonding orbitals,
respectively, and since both orbitals are occupied, the B.O. value is zero (Figure 6). The
bond order has the same value for the 2H2 system, since there is no interaction
between the molecular orbitals of the hydrogen molecules, only non-bonding orbitals.
Furthermore, analyzing the molecular orbitals energy, the 2 molecular orbital in H4
molecule, with respect to the reference level of the H2 system, acts as anti-bonding

16
molecular orbital, therefore destabilizes the molecule. Then, energetically, is favorable
for the hydrogen molecule (H2) to interact with other H2 molecule only through
intermolecular forces (Van der Waals) rather than make a new chemical bond (2
molecular orbital), since H4 would not represent a gain in stability. Here three aspects
should be pointed out: First, even though hydrogen molecules will interact through
intermolecular forces, this is expected to be quite weak, since Van der Waals depends
on the spatial distribution of the electrons and, in the case of hydrogen, this distribution
is very compact, reducing the attractive forces. This is reflected in the very low boiling
point of H2, equal to 20.28 K15. Second, one could imagine if other atomic orbitals
besides 1s could participate in the formation of bonds. However, the next available
orbitals lie on a different level (2s or 2p) which are 10 eV above 1s, thus, these cannot
effectively participate creating bonding orbitals with energetic levels below the atomic
reference line. Third, as the valence and conduction bands of the liquid H2 at low
temperatures are essentially the bonding and antibonding orbital of H2 (low spatial
overlap will lead to low energy splitting), then the energy gap of liquid H2 is related to
the gap between a bonding and an antibonding orbital of H2, which is expected to be
higher than the energy of visible light. Thus, this model allows to predict liquid H2 to be
transparent to visible light, which is experimentally verified.
This indicates that the most stable state for Hydrogen, instead of a crystalline
solid, is in the form of diatomic molecules, which can interact among themselves by
weak van der Waals interactions. However, the discussions based on the information in
Figure 5 leading to the formation of a metallic band essentially relied on the splitting of
the orbitals energy based on their spatial overlap. Even though we showed that this
overlap is not energetically favorable to conglomerate the molecules and propagate the
bonding, a natural question would be: in what cases it would be possible to spatially
overlap the H2 molecules and force it to form the energy levels showed in Figure 4?
Wigner and Huntington16 discussed that in high-pressure conditions (> 220 GPa)17,
hydrogen would exhibit metallic behavior. This very high pressure constitutes the
condition in which the hydrogen orbitals would have a forced spatial overlap. Later on,
it was argued that the nucleus of gas giants like Jupiter, Saturn and some exoplanets18
may attain the extreme pressure conditions and thus, could be constituted of liquid

17
metallic hydrogen. The challenges imposed to create an experimental setup to generate
metallic hydrogen under laboratory conditions make the experimental verification of
this substance to be considered the holy grail of high-pressure physics17,19. In this first
example, we showed that one can have insights into these possible behaviors of
hydrogen by exploring a simple qualitative tool, the formation of molecular orbitals.
II.B. Helium
We shall continue to organize and rationalize the properties of the other
elements using this approach. As an exercise for molecular systems with bond order
zero, consider the Helium element (Figure 7.
Figure 7. Standard state analysis of the Helium element through Bond Order and energy
criterion. As seen for the H2 molecule, the formation of a bigger molecule (He to He2)
with B.O. = 0 with electrons occupying anti-bonding molecular orbitals will lead to
dissociation of the molecule (He to 2 He). In this way, it is energetically preferable to
keep the previous chemical system (He) and interact only by intermolecular forces.
Employing Bond order and molecular orbital energy analysis, one can explain
why it is very unlikely to find, under normal conditions, diatomic Helium. The Helium
atom presents behavior similar to hydrogen (dimer to tetramer in Figure 6), but in this
case, it already occurs in the formation of diatomic, which as discussed before, is

18
responsible for the largest energy variations (Figure 7). The bond order is zero, but
besides that, as the antibonding orbital presents a much higher energy variation than
the bonding one, the occupation of an anti-bonding molecular orbital would destabilize
the molecule, and this would dissociate in two helium atoms. Certainly, these atoms still
will interact by intermolecular forces (Van der Waals), but even this is very weak, using
the same assumption we made when discussing the hydrogen atom: due to the very
compact spatial distribution of the electronic density. Therefore, while helium dimers
can be detected, their interaction energy is very low (95 eV)20 and have a very large
bond length (52 ± 4 Å)20. Therefore, in the scenario of same-element bonding from the
perspective of molecular orbitals, helium is the “worst-case scenario”, since the valence
has the most antibonding population, the energy involved in the antibonding orbitals is
very high and electronic distribution is very compact for intermolecular interactions. It is
interesting that this low interaction reflects in the boiling point of Helium, the lowest in
the periodic table, equal to 4.2 K21. Also, Helium is the only element that, at
atmospheric pressure, is not solid when the temperature tends to 0 K.
III. Second row
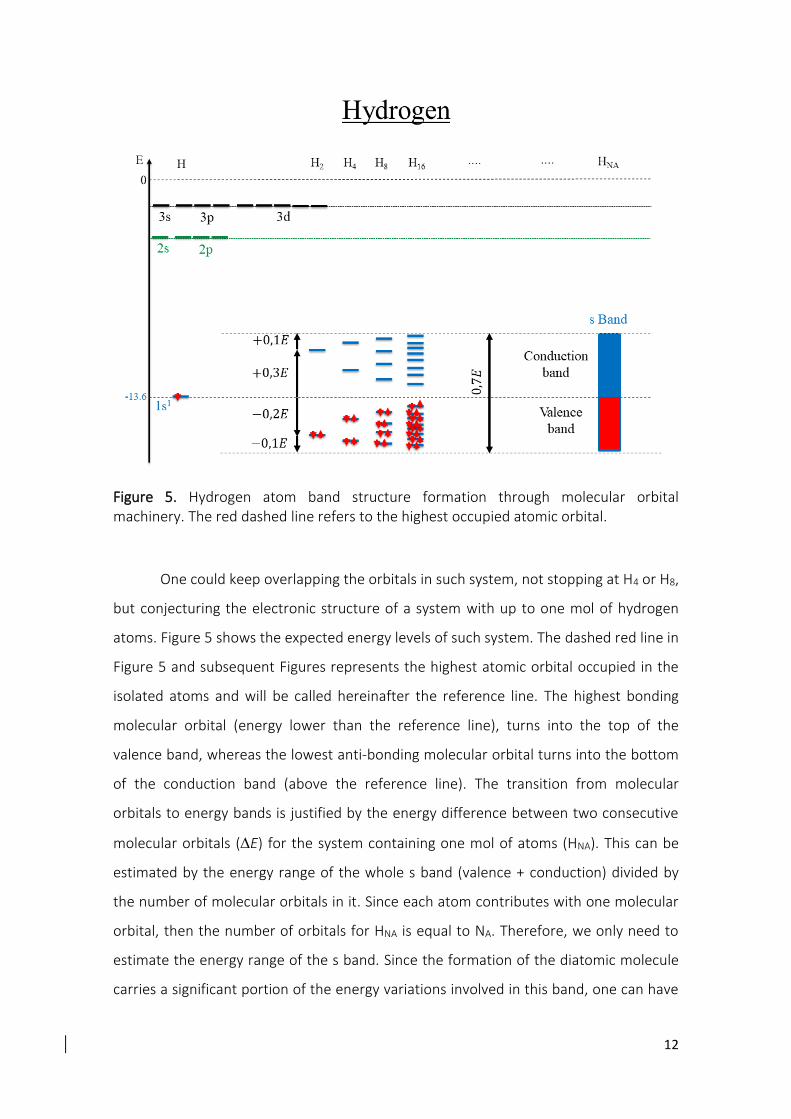
III.A. Simple metals: Lithium and Beryllium elements
In the case of Lithium and Beryllium elements, the occupied 2s and unoccupied
2p atomic orbitals are close in energy (Erro! Fonte de referência não encontrada.), which
suggests a possible competition between the molecular orbitals formed by these atomic
orbitals, as shown in Figure 8.
19
Figure 8. Lithium and beryllium band structures formation through molecular orbital
machinery. The energy proximity between the 2s and 2p atomic orbitals induces an
energy competition among the among the σ2s∗ and the p or p, since the * orbitals
energies increase while the p and p orbitals energies decrease. In this way, rather
than occupying anti-bonding orbitals (σ2s∗ ) becoming weakly bonded, the electrons are
transferred to bonding orbitals (p and p) allowing the formation of the solid.
As the alkali and alkaline earth clusters grow, their atomic orbitals combine
forming 𝜎2𝑠 , 𝜎2𝑠∗ , 𝜎2𝑝, 𝜎2𝑝
∗ ( orbitals formed from 2s and 2p, respectively), p and 𝜋2𝑝∗
orbitals (Figure 8). As expected, the energy competition among the 𝜎2𝑠∗ and the p or
p orbitals arises from the proximity of the 2s and 2p atomic orbitals, since the *
orbitals energies increase while the p and p orbitals energies decrease. In this way,
rather than becoming weaker, the bonds get stronger as the systems grow and the
electrons occupy the bonding regions of s and p bands (s, p, and p), below the
reference line, forming the sp band. This sp band has a high number of available states
coming from the p band (three times larger than in the s band), which allows to
accommodate all electrons that, otherwise, would have to occupy antibonding orbitals
(𝜎2𝑠∗ ) and would lead to the possible dissociation of the system, based on the same
arguments presented for the hydrogen and helium cases. Naturally, this extra bonding

20
energy from the occupation of bonding levels in the p band should be much larger for
the alkaline earth metal, since its extra electron, which would occupy the whole s band,
can migrate to bonding regions of the p band, generating more binding energy per mol.
Experimentally, the binding energy of solid lithium and beryllium, taken from the
enthalpy of sublimation of the solids, are equal to 1.58 eV (153 kJ.mol-1)22 and 3.32 eV
(320 kJ.mol-1)5, respectively. Another consequence of the unfilled bonding band of the
alkali metals is related to its elevated reactivity. These empty orbitals provide sites to
create new chemical bonds with other elements or substances, making this system
prompt to react, given that the energy barrier associated with the binding energy must
be overcome. The differences between the alkali and alkaline earth metals are also
reflected in their crystalline structure. Alkali metals tend to crystallize in body-centered
cubic structure (lower coordination number), while the earth metals form face-centered
cubic or hexagonal lattices (higher coordination number) and exhibit better mechanical
properties such as hardness23–25, tensile strength23–25and Young’s modulus23–25.
In the previous paragraphs, we have been discussing the case of clusters
containing several atoms and those tending to the infinite system (molar), but the
situation is quite different for small-size systems, such as the diatomic and nanoclusters
containing few atoms. For the later, the competition between 𝜎2𝑠∗ and p/p does not
yield a stronger bond, since the bonding orbitals from 2p are expected to lie (slightly)
above the reference line, which is positioned at the 2s orbital energy (last occupied
orbital in the atom). Therefore, from the perspective of energy variation, these bonding
orbitals do not change the bond order of very small alkali and alkaline earth clusters,
thus, they do not aid in the chemical bond. For small systems, the extra electrons in the
𝜎2𝑠∗ orbital decreases the bond strength in the alkaline earth clusters, being
characterized as Van der Waals clusters. Take as an example the lithium and beryllium
diatomic: from the molecular orbital model, the bond order of Li2 and Be2 are equal to 1
and 0, respectively. Experimentally, the dissociation energy of Li2 is around 1.0 eV26,
while in Be2 this value is tenfold lower27. Therefore, this simple model gives a picture of
how the bond in alkaline earth metals change from weak Van der Waals (small systems)
to strongly bonded metals (large clusters and “infinite” systems).

21
VI. Conclusions
In this three-part work (here in Part 1), the molecular orbital concept was
applied as a framework to study the band formation and the standard state of the
elements. The main idea was to analyze changes in bonding patterns given the balance
between electronic configurations/occupation and the available energy levels. The
Hydrogen atom was used as a metric for the other chemical elements and as an
example of how atomic or molecular orbitals interact to compose a band structure.
Within this framework, we were able to show the formation of metallic hydrogen
observed at the nucleus of gas giant planets like Jupiter, Saturn, and some exoplanets.
On the other hand, one can also explain the standard state of the hydrogen as a
diatomic gas and some of its physicochemical properties, such as low boiling point and
transparency to visible light at gas and liquid phase. Based on the Hydrogen structure,
we discussed why it is very unlikely to find, under normal conditions, diatomic Helium.
Through the molecular orbitals diagram, it was shown the transition from weakly
bonded clusters (Van der Waals clusters) to bonded solid (metal) seen in alkaline earth
metals. Moreover, it was discussed the origin of some differences between the
physicochemical properties of the simple metals (alkali and alkaline earth), such as
enthalpy of sublimation, reactivity, crystalline structure, hardness, tensile strength and
Young’s modulus. Those essentially arises due to the one-electron difference in their
atoms, while the clusters/solid possess the same available energy levels. In the
following Parts 2 and 3, the present framework is applied to main group elements and
transition metals, demonstrating some trends in the periodic table and band gap and
light-matter interaction.
Acknowledgements
This work has been supported by Brazilian agencies Fundação de Amparo à
Pesquisa do Estado de São Paulo (FAPESP) under grants 2019/03729-8, 2018/22669-3
and 2017/07707-3, and Conselho Nacional de Desenvolvimento Científico e Tecnológico

22
(CNPq) under grants 309051/2016-9, 406107/2016-5, 307052/2016-8 and
404337/2016-3.
References
1 G. N. Lewis, J. Am. Chem. Soc., 1916, 38, 762–785.
2 P. Atkins and J. De Paula, Physical chemistry, Great Britain by Oxford University Press, Eighth Edi., 2006.
3 E. A. Smart, Lesley E. Moore, Solid state chemistry : an introduction, CRC Press Tayler & Francis Group, New York, Third., 2005.
4 W. A. Harrison, Electronic Structure and the Properties of Solids, Dover Publications, 1989.
5 I. Levine, Physical Chemistry, The McGraw-Hill Companies, New York, 6th editio., 2008.
6 E. Kaxiras, Atomic and Electronic Structure of Solids, 2003.
7 Charles Kittel, Introduction to Solid State Physics, 2010.
8 J. Autschbach, J. Chem. Educ., 2012, 89, 1032–1040.
9 a B. Sannigrahi and T. Kar, J. Chem. Educ., 1988, 65, 674.
10 P. F. Lang, J. Chem. Educ., 2018, ACS.jchemed.8b00239.
11 J. and N. A. T. Kramida, A., Ralchenko, Yu., Reader, 2018.
12 G. Herzberg and A. Monfils, J. Mol. Spectrosc., 1960, 5, 482–498.
13 A. Balakrishnan, V. Smith and B. P. Stoicheff, Phys. Rev. Lett., 1992, 68, 6–9.
14 E. R. Williams, Science (80-. )., 1992, 256, 1184–1187.
15 F. D. Rossim, N. Dame and N. Dame, Int. UNION PURE Appl. Chem., 1968.
16 E. Wigner and H. B. Huntington, J. Chem. Phys., 1935, 3, 764–770.
17 X. Zhang, E. Wang and X. Li, 2018, 134110, 1–11.
18 R. E. Johnson, R. W. Carlson, J. F. Cooper, C. Paranicas, M. H. Moore, M. C. Wong, F. Bagenal, T. Dowling, W. B. McKinnon and others, Cambridge University Press Cambridge, UK, 2004.
19 J. M. Mcmahon, M. A. Morales, C. Pierleoni and D. M. Ceperley, 2012, 84.
20 R. E. Grisenti, W. Schöllkopf and J. P. Toennies, 2000, 1, 4–7.
21 G. Schmidt and W. H. Keesom, Physica, 1937, 4, 963–970.
22 C. Herring, Phys. Rev., 1951, 82, 282–283.
23 J. C. William D. and D. G. Rethwisch, Materials Science and Engineering: An Introduction, John Wiley & Sons, Ltd, Seventh., 2007.

23
24 M. F. Ashby, Materials Selection in Mechanical Design, Elsevier Ltd, Fourth., 2011, vol. 1.
25 J. C. William D. and D. G. Rethwisch, Fundamentals of Materials Science and Engineering: an integrated approach, John Wiley & Sons, Inc., Third., 2008.
26 R. VELASCO, C. OTTINGER and R. N. ZARE, 2009, 5522.
27 A. V Mitin, in AIP Conference Proceedings, 2007, vol. 963, pp. 231–234.

download fileview on ChemRxivThe Chemical Bond across the Periodic Table Part 1_Fir... (731.81 KiB)

Supplementary material
The Chemical Bond across the Periodic Table: Part 1 – First Row and Simple Metals
Gabriel F.S. Fernandes1, Leonardo A. Cunha1, Luiz F. A. Ferrão1*
1Departamento de Química, Instituto Tecnológico de Aeronáutica, São José dos Campos,
SP 12228-900, Brasil.
In the supplementary material, we provide discussion about: molecular orbital, bond
order and atomic orbitals along the second row
__________________________________
*Corresponding author. E-mail address: [email protected]

I.A. Theoretical concepts
I.A.1. Molecular orbital
Classically, two waves can interfere, and the pattern obtained by such
phenomenon depends on the relative phase between them. If the relative phase is equal
to 0°, the interference is constructive and the amplitude of the two stationary waves is
added. On the other hand, if the relative phase is equal to 180°, the interference is
destructive, and the waves amplitudes are subtracted.
This classical idea can be extrapolated to quantum mechanics, in which its effects
arise from the fact that matter also presents wave-like behavior. Electron diffraction9 is
just one of many examples in which one can observe wave-like features for matter. The
non-relativistic Schrödinger equation mathematically describes such properties for a
given system and, through some approximations, many-body wavefunctions may be
expanded as combinations of singe-particle entities known as orbitals (either atomic or
molecular). Due to this wave-matter duality proposed by De Broglie10,11 some
combinations will lead to constructive interference patterns, giving rise to bonding
orbitals. On the other hand, we can also observe destructive interference for some
specific combinations. These will be denoted as anti-bonding orbitals. It is also worth
noticing that such combinations preserve the number of orbitals, i.e. starting from a given
set of N orbitals a new set will also present N different orbitals, built by combinations of
orbitals from the original set.
The functions obtained by this procedure can be treated as building blocks to
describe molecular systems into two categories: the one-electron wavefunctions
centered on the component atoms of the molecule and the one-electron functions that
span over two or more atomic centers. We shall denote the former group as atomic
orbitals, whereas the second group is known as molecular orbitals. The most common
approach in quantum chemistry is to use a linear combination of atomic orbitals to obtain
the molecular orbitals (LCAO-MO)12 that will be part of the molecule’s wavefunction.
In the LCAO-MO procedure, the molecule’s one-particle wave function (molecular
orbital) is built by linear combination (adding or subtracting) atom’s one-particle wave
function (atomic orbital). Disregarding the phase of the wavefunction, the addition of two

atomic orbitals can be classically seen as a constructive interference, in which the
electronic density is located between the atoms (shared or bonding region), stabilizing
the molecule. On the other hand, the subtraction could be interpreted as destructive
interference, in which the molecular orbital formed presents a nodal plane perpendicular
to the chemical bonding axis. The electronic density is dislocated out of the bonding
region, ultimately pulling the nuclei away from each other and weakening the chemical
bond. In this way, the bonding molecular orbital has lower energy than the reference
atomic orbitals used, while the anti-bonding molecular orbital possess higher energy than
the reference atomic orbitals.
I.A.2. Bond order
Bond order is a useful tool commonly used in molecular orbital diagrams to
predict the chemical bond and stability of the molecules. It is defined as the difference
between the number of electrons in bonding molecular orbital and anti-bonding
molecular orbital in relation to the reference system, described in the following Equation
(1).
𝐵𝑂 = 𝑛𝑒−(𝑀𝑂) − 𝑛𝑒−(𝑀𝑂)∗
2 (1)
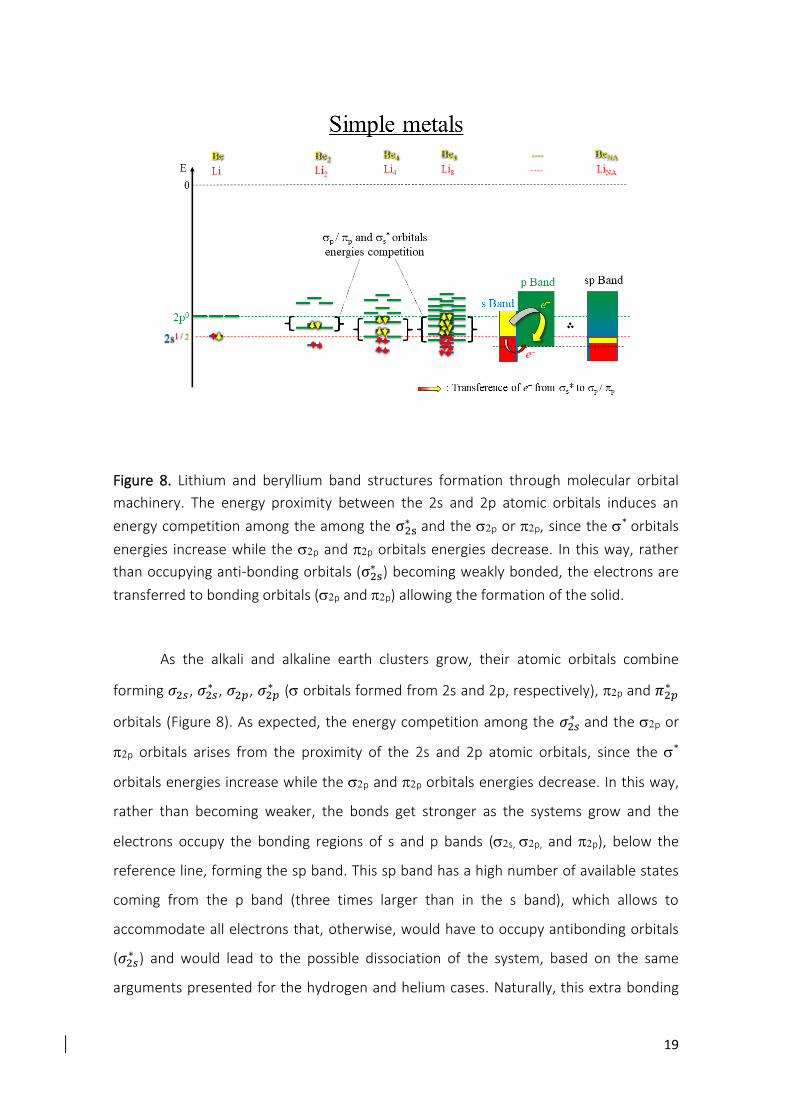
I.A.3. Atomic orbitals along the second row
Before starting the molecular orbital discussion for other elements of the periodic
table, we shall notice the energy gap between the 2s and 2p atomic orbital along the
second row will be discussed (Figure S1), since it plays an important role on the following
discussions.
Figure S1. Atomic orbitals energy diagram for the second row of the main group obtained by the HF/6-311++G(3df,3pd) methodology.
In Figure S1, one can see that the energy gap between the 2s and 2p atomic
orbitals increases from the lithium atom to the fluorine atom, in the same direction of
the effective charge and opposite of the atomic radius. As the atomic nucleus (positive
charge) increases, the effective charge acting on the furthest electrons also increases,
attracting them to the nucleus. Thus, these attracted electrons are more bonded to the
nucleus, reflecting in a lower atomic orbital energy and in a smaller atomic radius. One of
the consequences of this energy gap between orbitals with the same quantum number
is the energetic competition between the and molecular orbitals, which explains why
alkali and earth alkaline atoms form metal solids, the atomic hybridization on boron and
carbon elements and the gaseous standard state of the nitrogen, oxygen and fluorine
elements.
Comentado [LF1]: Incluir He

download fileview on ChemRxivsupplementary_The Chemical Bond across the Periodic ... (184.69 KiB)

The chemical bond across the periodic table: Part 2 – main group elements and trends in
the periodic table
Gabriel F.S. Fernandes1, Leonardo A. Cunha1, Francisco B.C. Machado1, Luiz F. A. Ferrão1*
1Departamento de Química, Instituto Tecnológico de Aeronáutica, São José dos Campos,
SP 12228-900, Brasil.
__________________________________
*Corresponding author. E-mail address: [email protected]

Abstract
Chemical bond plays a central role in the description of the physicochemical properties
of molecules and solids and it is essential to several fields in science and engineering,
governing the material’s mechanical, electrical, catalytic and optoelectronic properties,
among others. Due to this indisputable importance, a proper description of chemical
bond is needed, commonly obtained through solving the Schrödinger equation of the
system with either molecular orbital theory (molecules) or band theory (solids). However,
connecting these seemingly different concepts is not a straightforward task for students
and there is a gap in the available textbooks concerning this subject. This work presents
a chemical content to be added in the physical chemistry undergraduate courses, in
which the framework of molecular orbitals was used to qualitatively explain the standard
state of the chemical elements and some properties of the resulting material, such as gas
or crystalline solids. Here in Part 2, we were able to show the competition between the
hybrid and metallic configuration on main group elements; the change on the electronic
properties from insulator to metal on the carbon family; among some other trends in the
periodic table. The original content discussed here can be adapted and incorporated in
undergraduate and graduate physical-chemistry and/or materials science textbooks and
also serves as a conceptual guide to subsequent disciplines such as quantum chemistry,
quantum mechanics and solid-state physics.
KEYWORDS: Upper-division Undergraduate, Graduate Education, Physical Chemistry,
Communication / Writing, Textbooks / Reference Books, Atomic Properties / Structure,
Material Science, MO theory, Periodicity / Periodic Table, Quantum Chemistry.

I. Introduction
In the previous paper “The chemical bond across the periodic table: Part 1 – first
row and simple metals”, we demonstrated the band formation and the standard state of
the elements through the molecular orbital concept, wherein, our interest lied on a
qualitative picture to help students understand general bonding patterns and how
material properties arise naturally from information related to atomic and molecular
energy levels and the bond order. The observed bonding patterns were analyzed based
on the balance between electronic configurations/occupation and the available energy
levels.
From a bibliographical research in physicochemical and physical chemistry solid
state textbooks2–7, it was noticed that, to the best of the authors knowledge, there is no
previous textbook that creates a connection between molecular orbitals and band theory
topics by employing the available energy levels and their occupation (from molecular
orbitals), to describe the chemical bond in solids and explain some of their basic
physicochemical properties, as discussed previously in Part 1.
Understanding general trends that arise from the progression of the discrete
energy levels represented by the molecular orbitals to continuum band regions allows
students to comprehend bond formation, crystalline structure, material properties in
solids as well as the standard state for several compounds. In this sense, the present
paper focuses on demonstrating the formation of energy bands through molecular orbital
in a simple and qualitative manner, explaining the properties and trends across the
periodic table using three main ingredients: 1) the electronic structure of hydrogen atom;
2) molecular orbitals and energy splitting from spatial overlap and; 3) trends in atomic
radius along the periodic table.
The discussion is organized as follows: Section II is dedicated to explaining the
band formation of the Boron and Carbon elements and why the Nitrogen, Oxygen and
Fluorine are gas at standard conditions. In section III, a brief discussion with respect to
the third-period elements, explaining some electronic structure changes based on the
larger atomic radius of the elements when compared to same-family atoms of the second

row. In section IV, some tendencies of the periodic table are explored, and the carbon
group is used as an example.
II.B. Main group elements
As presented in the supplementary material, the energy gap between 2s and 2p
atomic orbitals (Figure S1) increases at the Nitrogen element, decreasing the energy
competition between the * and molecular orbitals. This atomic energy gap makes the
produced * molecular orbital energetically unfavorable to be occupied due to their high
energy. And, as there are not empty bonding molecular orbitals available, the
propagation of the chemical bond is interrupted at the diatomic level as showed on the
Hydrogen and Helium cases, where the diatomic molecules interact by Van der Waals
intermolecular forces. However, at low temperatures (40K9–14), theses Van der Waals
intermolecular forces are stronger than the molecular vibrations energies, in this way,
the diatomic molecules (N29,10, O2
11 and F212–14) are attracted to each other forming a
molecular solid with electronic structure described by band theory (Erro! Fonte de
referência não encontrada. and S3).
II.B.1. Boron
We now discuss the main group elements of the second row. The main difference
from the simple metal is the occupancy, now the p orbitals are occupied, and the
reference energy lies in this orbital. The consequences of occupying the p atomic orbitals
from boron to fluorine are two-fold. First, the chemical bond presents directionality due
to the angular momentum of the p atomic orbital. Second and related to the first one,
these orbitals with angular momentum/direction can combine in certain symmetries to
allow bonds between two atoms involving multiple orbitals, i.e. and orbitals. While
orbitals are completely symmetric with respect to the internuclear axis, orbitals
present a nodal plane passing through this axis, as shown in Part1 of this three-part paper.
It is worth noting that the relative strength of and orbitals depend on the
internuclear distance. For small distances, the binding energy from orbitals can be

competitive or surpass orbitals. For larger distances, the overlap between two p atomic
orbitals (which symmetries are orthogonal to the internuclear axis, as shown in Part1)
rapidly decreases, lowering the energy splitting in bonding and antibonding orbitals and
consequently, diminishes the tendency in forming this type of orbital to create bonds.
Since the elements in the second row present the shortest atomic radius among the main
group elements, it is expected that these orbitals () are competitive to make the
chemical bond.
Figure 1. Boron band structure formation through molecular orbital machinery. As the
system grows, populating bonding molecular orbitals increases the chemical bond,
favoring the formation of new bonds. On the other hand, the existence of empty bonding
orbitals turns the system prompt to react, while it does not maximize the chemical bonds.
In the presence of other atoms, the boron ground state orbitals can rearrange to
maximize the formation of bonding orbitals with directionality characteristics. Although
in less extension, empty bonding orbitals still occurs in boron after the hybridization.
Approaching two Boron atoms, their 2p1 atomic orbitals interacts producing x,
y, x*, y
*, 𝜎𝑝𝑧 and 𝜎𝑝𝑧
∗ molecular orbitals. The 2p1 electrons occupy the bonding
orbitals (x1 and y
1) in the B2 molecule (Figure 1), and are unpaired, providing reactivity
and paramagnetic features to the molecule. When the boron system achieves a four-
atom size, one can expect x and y orbitals to be doubly occupied (x2 and y
2), or
another occupancy from combinations of and orbitals Either way, there will be

empty bonding orbitals, suggesting that the molecular system can form more stable
systems through more chemical bonds. Also, as discussed previously in Part1 for the alkali
metals, this bond configuration will lead, at any size, to reactive boron systems. As nature
follows minimization of energy, maybe there is another simple way to combine orbitals
and form stronger bonds, maximizing the bond and minimizing the energy. In quantum
mechanics (in fact, any set of linear differential equations), if a set of functions is a
solution to the system, then linear combinations of these functions are also solutions.
Since both s and p orbitals are occupied in boron, the system can combine its atomic
orbitals to form a new set, the so-called hybrid orbitals. The system will try to minimize
the energy by combining the directionality of the p orbitals with the population of the s
orbitals, forming orbitals with one electron each in several “directions”. These orbitals do
not correspond to the lowest energy configuration for a single atom, but in the presence
of other elements, allow the formation of several strong bonds. Therefore, the boron
atom can change its electronic configuration from the ground state (B: 2s22p1) to an
excited state (B*: (sp2)3p1), enabling to make one extra chemical bond. The resulting sp2
hybrid orbitals make directional, localized and strong 𝜎𝑠𝑝2 chemical bonds, in which a
central atom makes a single bond with each other atom, conferring different properties
when compared to alkali and alkaline earth. For example, these bonds involving just
adjacent atoms will have little spatial superposition with further atoms, reducing the
splitting of energy in the orbitals as the system grows. As a result, the 𝜎𝑠𝑝2 valence band
is expected to be energetically very narrow when compared to a metallic valence band,
which involves the spatial superposition of several atoms. The first electronic transitions
in the solid will essentially involve electrons from a strongly bonding orbital (𝜎𝑠𝑝2 valence
band) to a weakly bonding orbital (2p band). This will lead to a semiconductor character
(band gap with energies expected to be within visible light energy), higher vaporization
enthalpy than simple metals and also different mechanical properties and crystalline
structures, since the bonds have “directionality” by directly involving p orbitals in all
bonds. Here it is interesting to recall that this hybrid configuration is not yet completely
satisfactory for boron atoms, since the system still has empty bonding orbitals (2p). Both
simple bond models discussed, one based on the ground state electronic structure
orbitals leading to a "metal", and the other based on excited or hybrid orbitals leading to
“localized” bonds, present empty bond orbitals. Nevertheless, this issue is expected to be
less significant in the latter, which should be closer to the electronic structure
experimentally observed of Boron. As a matter of fact, boron presents several allotropes,
most of them with a semiconductor character and a band gap within visible light15,16. This
non-conformity in simple models is also interesting since it partially explains the exotic
bonds formed by boron, related to its imperfect balance between electrons and orbitals.
This is a situation very different from the next element, carbon.
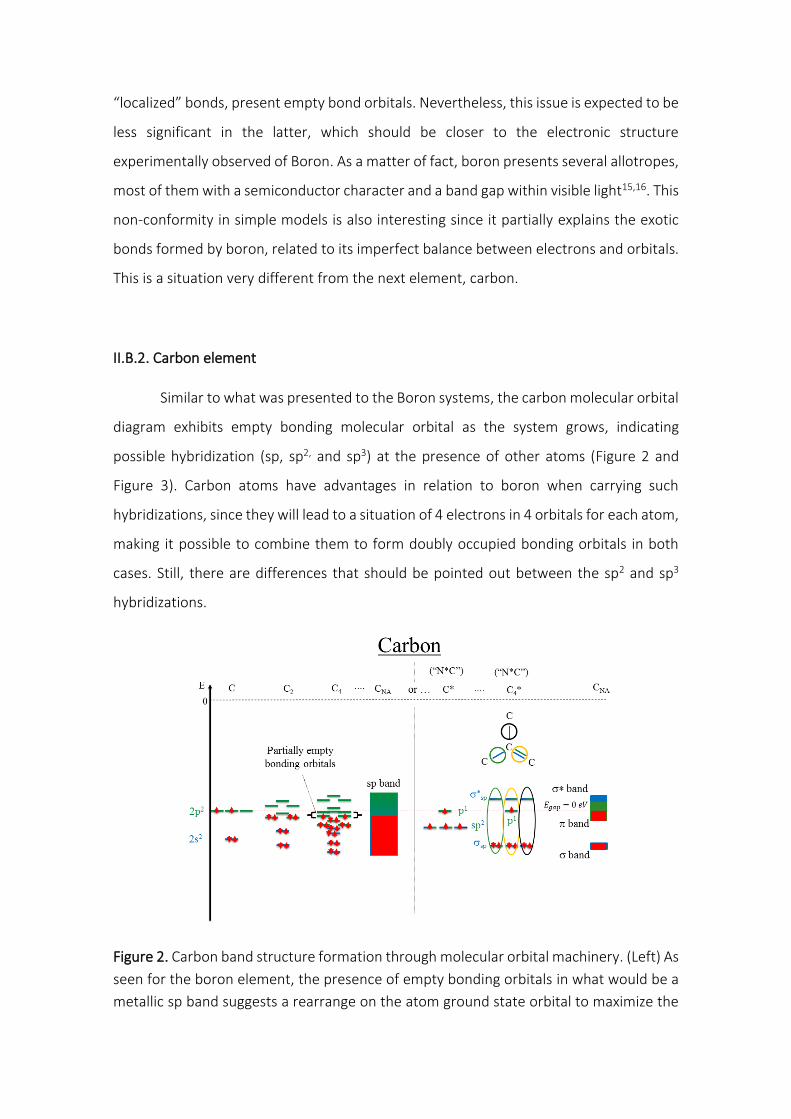
II.B.2. Carbon element
Similar to what was presented to the Boron systems, the carbon molecular orbital
diagram exhibits empty bonding molecular orbital as the system grows, indicating
possible hybridization (sp, sp2, and sp3) at the presence of other atoms (Figure 2 and
Figure 3). Carbon atoms have advantages in relation to boron when carrying such
hybridizations, since they will lead to a situation of 4 electrons in 4 orbitals for each atom,
making it possible to combine them to form doubly occupied bonding orbitals in both
cases. Still, there are differences that should be pointed out between the sp2 and sp3
hybridizations.
Figure 2. Carbon band structure formation through molecular orbital machinery. (Left) As
seen for the boron element, the presence of empty bonding orbitals in what would be a
metallic sp band suggests a rearrange on the atom ground state orbital to maximize the

number of chemical bonds and increase the system binding energy. (Right) The 4
electrons and 4 orbitals of carbon offer two hybridization scenarios to reach one electron
per orbital (ideal for overlap). In this case, the sp2 which leads to a zero band gap.
Figure 3. Carbon band structure formation through molecular orbital machinery. The sp3 carbon (diamond) shows an insulant character since the band gap involves a transition from a bonding orbital to an antibonding orbital, which is expected to be very high in energy, i.e., higher than the visible light energy (Egap > 3 eV).
At first glance, it would be expected that the sp2 hybridization in the carbon
element also presents a semiconductor behavior, as seen in the boron atom.
Nevertheless, the extra electron on carbon electronic configuration (1s2 2s2 2p2) fills the
initially empty band on the boron case, conferring a conductor behavior to the sp2
carbon17.
The sp2 and sp3 hybridizations differ on the number of and molecular orbital
produced after the chemical bond, which will result in different properties and crystalline
structures. From the molecular orbital diagrams (Figure 2 and Figure 3), the electronic
property is easily observed. The sp3 carbon (diamond) shows an insulator character since
the band gap involves a transition from a bonding orbital to an antibonding orbital, which
is expected to be very high in energy (Egap > 3 eV). Experimentally, the band gap is higher
than 5 eV2,18–20. Also, the number of charge carriers (electrons) is smaller, because of the
localized chemical bond (), as discussed in boron. In contrast, the sp2 carbon (graphene)
presents a zero-band gap between the valence () and conduction bands (p*),

conferring a conductor-like behavior. It is worth noting that the orbitals have some
spatial superposition between non-adjacent atoms, even though they present
directionality. One can infer that these orbitals will split in energy as the system grows,
like in metals, but preserving differences, such as a limited spatial superposition (small
width in band energy when compared to the sp band in simple metals) and directionality,
meaning that the bond and physical properties in the direction of the plane containing
the nuclei are very different from out-of-plane properties. Also, as planes (graphene
sheets) interact, the orbitals will further split changing the system properties and hinder
the spatial distribution of the orbitals. Therefore, in single layer systems (graphene), the
charge carriers (electrons) are free to move in the conduction band due to the delocalized
p1 electrons on the band. Since graphene combines localized orbitals with the
delocalized orbitals in an atomic thin layer, it presents very strong mechanical
resistance and high electrical conductivity. In graphite (several layers) these
characteristics are attenuated since the bulk properties are anisotropic and the electron
mobility of orbitals is hindered. These differences in the bonding of diamond (sp3) and
graphene (sp2) result in different crystalline structures, thermal and mechanical
properties such as thermal21 and electrical conductivity22, hardness23 and Young’s
modulus22,23. The diamond carbon crystallizes in face-centered cubic containing four
atoms in one unitary cell, while graphene crystallizes in a hexagonal structure composed
by two graphitic planes formed by two atoms in the unitary cell.
II.B.3. Nitrogen, Oxygen and Fluorine elements
We now move on to nitrogen, oxygen, and fluorine, wherein the supplementary
material is found in the discussion of the oxygen and fluorine elements. On the contrary
to the previously discussed main group elements of the second row, these chemical
elements are found in the gas phase at the standard condition of temperature and
pressure (298K and 1bar). Can we use the same tools to understand how this happens?
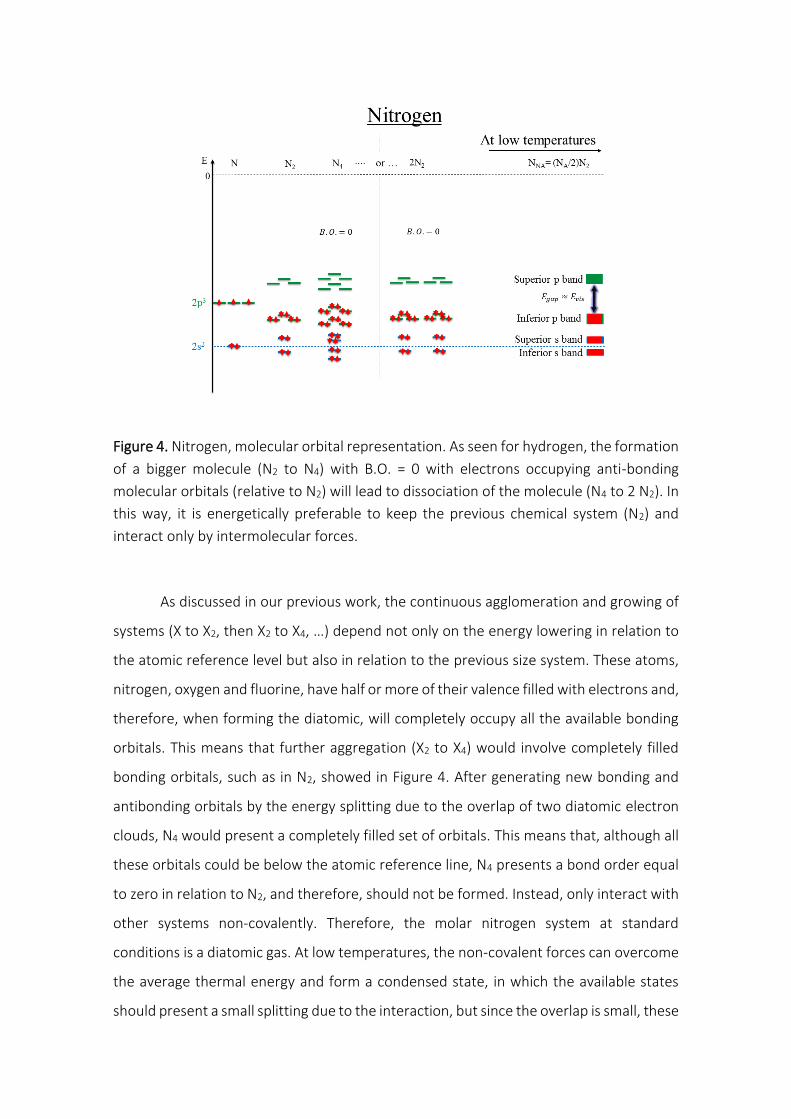
Figure 4. Nitrogen, molecular orbital representation. As seen for hydrogen, the formation
of a bigger molecule (N2 to N4) with B.O. = 0 with electrons occupying anti-bonding
molecular orbitals (relative to N2) will lead to dissociation of the molecule (N4 to 2 N2). In
this way, it is energetically preferable to keep the previous chemical system (N2) and
interact only by intermolecular forces.
As discussed in our previous work, the continuous agglomeration and growing of
systems (X to X2, then X2 to X4, …) depend not only on the energy lowering in relation to
the atomic reference level but also in relation to the previous size system. These atoms,
nitrogen, oxygen and fluorine, have half or more of their valence filled with electrons and,
therefore, when forming the diatomic, will completely occupy all the available bonding
orbitals. This means that further aggregation (X2 to X4) would involve completely filled
bonding orbitals, such as in N2, showed in Figure 4. After generating new bonding and
antibonding orbitals by the energy splitting due to the overlap of two diatomic electron
clouds, N4 would present a completely filled set of orbitals. This means that, although all
these orbitals could be below the atomic reference line, N4 presents a bond order equal
to zero in relation to N2, and therefore, should not be formed. Instead, only interact with
other systems non-covalently. Therefore, the molar nitrogen system at standard
conditions is a diatomic gas. At low temperatures, the non-covalent forces can overcome
the average thermal energy and form a condensed state, in which the available states
should present a small splitting due to the interaction, but since the overlap is small, these

states (or bands) are essentially the states of the diatomic molecules. On the contrary of
hydrogen and helium, this condensation temperature should be considerably higher (77
K) since we now have a second-period element, which has a considerably larger spatial
extension and, therefore, a stronger non-covalent interaction (higher polarizability). We
now concern the interaction between the light with this “low temperature condensed
molar nitrogen system” (liquid N2). The first excitation (lowest possible energy) occurs
from the inferior p band to the superior p band, as shown in Figure 4. This is essentially
the energy involved from bonding to an antibonding orbital, so, it is expected that this
photon energy to be very high when compared to visible light. In fact, liquid nitrogen is
transparent. Since solid nitrogen (63 K) presents very similar interactions and thus, energy
levels, the same should be valid for this system.
As discussed above, nitrogen does not agglomerate to form a solid. However, it is
interesting to conjecture what would be some of large nitrogen cluster properties if it
could be formed. It is not easily attainable, since it involves breaking very strong triple
bonds to form single bonds between several atoms, a condition not favorable for main
group elements of the second period. Although, if the system is subjected to very high
pressures24 or external forces, the N2 molecular orbitals will suffer energy separation due
to the forced spatial overlap, which could lead to a metallic band such as discussed for
hydrogen. If this system could form an sp band such as the simple metals, this band would
be completely filled up to the reference line (regard that the reference line is now on the
p orbital, instead of the s as in simple metals). This means that all available bonding
energy would be used to form the system and thus a very high energy density would be
compressed in this system, which also would be metallic since there is no energy gap
between the valence and conduction band. Also, since all energy levels are occupied up
to the reference line it would be expected that this system to be very reactive under
normal pressure conditions, since it would not be the lowest energetic state. This
situation is analogous to the metallic hydrogen. In fact, it is argued that metallic nitrogen
would be the second non-nuclear highest energetic material, behind only of metallic
hydrogen24. Although it is somewhat far from being the molar nitrogen cluster (NNa),
C2N14 is probably the best substance ever synthesized for comparison: it is a highly
sensitive and energetic azidotetrazole25, whose properties are quite analogous of what

one would expect from the metallic nitrogen, exploding under most stimulus such as the
radiation used to characterize the substance.
lll. Third row
We have been discussing the chemical bond and formation (or not) of condensed
substances from the perspective of the three components, namely, 1) the electronic
structure of hydrogen atom; 2) molecular orbitals and energy splitting from spatial
overlap and; 3) trends in atomic radius across the periodic table. Within this framework,
the main difference in the third-period elements lies in this third aspect: the larger atomic
radius of the elements when compared to same-family atoms of the second row. If one
considers the valence of a given family (one for alkali, three for boron family and so on),
this means that the same electric charge is more diffuse in space as one move down in
the periodic table (at least for simple metals and main group elements, transition metals
will be discussed later). As a consequence, the second aspect (spatial overlap and energy
splitting) is also affected, lowering the energy variation when forming bonding orbitals
and changing the energetic balance between and bonds. In the third period elements,
the binding energy involved in orbitals along the internuclear axis of two atoms (s, pz)
becomes substantially stronger than those involving orbitals orthogonal to the
internuclear axis (px, py). Therefore, bonds are favored over bonds if the system is
allowed to do so. This means that, in the presence of several other atoms, these elements
will tend to break “multiple” bonds with one element to form “single” bonds with several
atoms (or maximize the spatial overlap). What should be the consequences for the simple
metals and main group elements? Since the latter can rely on bonds due to their
occupied p atomic orbitals, these elements should be more affected than simple metals.
In both groups, it is expected a lowering in the binding energy from each bonding orbital,
individually. This means that both third-period simple metals and main group should be
weaker bonded than their second-period counterparts, but we expect possible
qualitative differences only in the latter. We shall now discuss some representative cases
in the main group elements.

lV. Periodic Table tendencies for lower periods
According to the electronic structure description for the second and third rows of
the periodic table, some tendencies can be extrapolated to other periods. First, the
strong and localized chemical bond is unfavorable due to the small atomic/molecular
orbitals overlap caused by increased diffuseness of electronic distribution of the atomic
orbitals with high principal quantum number, producing weak chemical bonds. In this
way, there is a tendency to move from directional and localized bonds to make
delocalized chemical bonds with higher coordination number in order to maintain a
strong total bonding. As discussed in previous examples, this leads to an electronic
structure with metallic behavior. Second, the radius of the internal orbitals is increased,
leading to a higher cross-section among the electrons and nuclei, resulting in a decreasing
of the electrical conductivity in metals. Third, the increase in atomic radius and lower
binding energy have opposite consequences for the simple metals and main group
elements. In the former, this leads to higher reactivity of the solids (alkali and alkaline
earth metals) due to the lower effective charge on the ns1 and ns2 electrons, less
attracted to the nucleus, which also is weakly bonded to other atoms and prompt to
react. In the main group, the weaker bonds lead to lower reactivity due to lower capacity
to form bonding orbitals, and thus, lower electronegativity. This also means that, as a
general trend, all substances tend to present metallic behavior for lower periods,
depending on the balance between available electrons and orbitals. This leads to the
ladder-like line that separates metals and non-metals in the main group.
The next subsections will focus on the discussion of a representative case of the
main group elements, the carbon family, as well as the remaining undiscussed set of
elements, the transition metals.
lV.A. Carbon Group
As mentioned before, one of the periodic table tendencies when moving down
the period along a group is the increase of the metallic character. Within the framework
presented here, this can be attributed to a lowering in the binding energy, and therefore,

in the energy difference between occupied bonding orbitals and unoccupied antibonding
orbitals, which in turn lowers the band gap in the solid.
This behavior is clearly observable on the carbon family because of the variation
of the electrical behavior from insulator (𝐸𝑔 > 3 𝑒𝑉) to semiconductor (0 < 𝐸𝑔 ≤ 3 𝑒𝑉)
and then to metallic (𝐸𝑔 = 0). In terms of electronic structure, the atoms in this family
present the best balance of electrons and available orbitals among the main group family.
This means that by transforming its ground state orbitals to hybrid sp2 or sp3, these atoms
can generate a set of four orbitals with one electron in each. From the perspective of two
overlapping orbitals, this means that two approaching atoms with this configuration will
form a bonding orbital which will be doubly occupied (and an empty antibonding orbital).
The directionality of these hybrid orbitals limits the interaction (overlap) to one orbital
per atom. Another limitation of these directional hybrid orbitals is the low overlap
between non-adjacent atoms, meaning that the aggregation of new atoms does not lead
to significant further energy splitting of the already formed bonding and antibonding
orbitals. Therefore, the valence and conduction bands in solids should be closely related
to the bonding (𝜎𝑠𝑝3) and antibonding (𝜎𝑠𝑝3∗ ) orbitals of each atom pair. As the binding
energy lowers when moving down the period in the periodic table, one expects that this
band gap gets lower for each period. Also, as π bonds are weakened from the third period
on, only the sp3 hybridization is energetically competitive to the metallic band formed
from the ground state orbitals for these elements. One can infer that at a given period,
the bonding energy from the hybrid orbitals would mostly only counterbalance the
hybridization energy cost, consequently, the energy gap would tend to zero. At this point,
the metallic band (using the ground state orbitals) is competitive since, although it also
does not produce highly bonding orbitals, it does not impose a hybridization energy cost.
If that is the case, the system’s geometry changes from the tetrahedral to a geometry
that maximizes the number of adjacent atoms. Experimentally, it is observed that carbon,
silicon and germanium present a face-centered diamond-cubic crystal structure
(tetrahedral) with decreasing band gaps (5.4, 1.1 and 0.7 eV, respectively, as shown in
Figure 5). Interestingly, tin presents two allotropes at atmospheric pressure: below 13.2
ºC (286.2 K) it presents a tetrahedral structure (called -tin, a nonmetallic gray form) like
C, Si, and Ge; above it, tin presents a body-centered tetragonal crystal structure (called
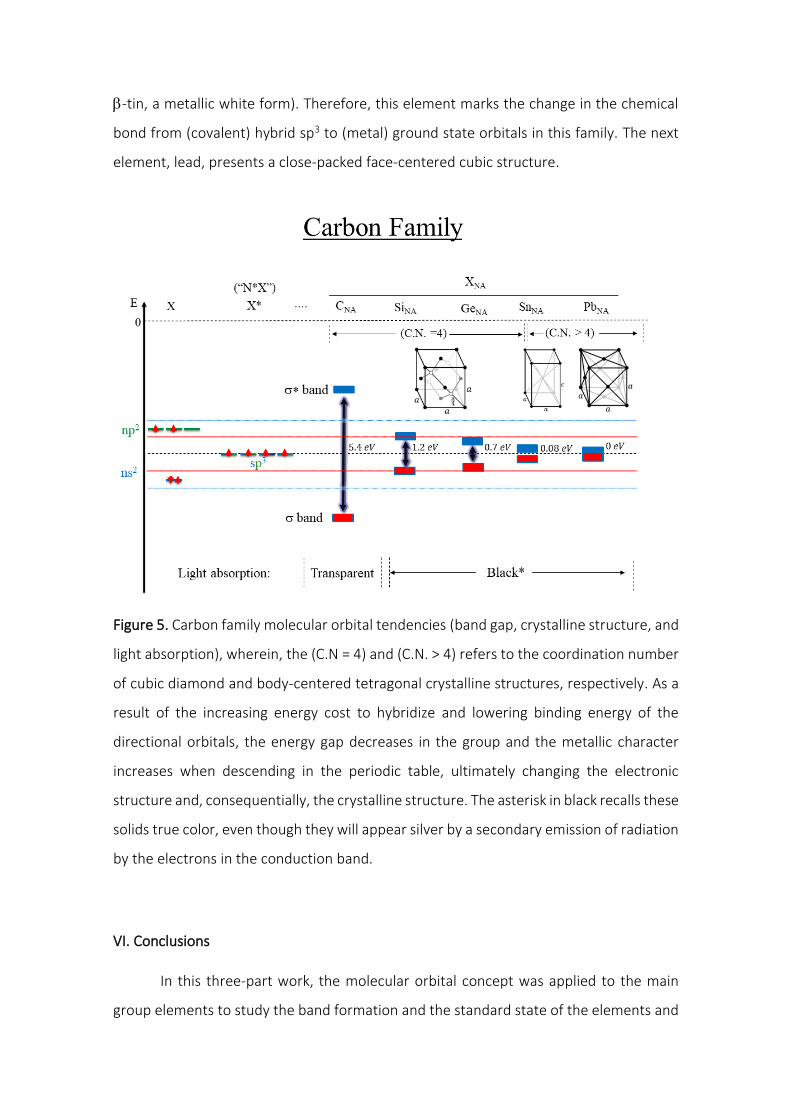
-tin, a metallic white form). Therefore, this element marks the change in the chemical
bond from (covalent) hybrid sp3 to (metal) ground state orbitals in this family. The next
element, lead, presents a close-packed face-centered cubic structure.
Figure 5. Carbon family molecular orbital tendencies (band gap, crystalline structure, and
light absorption), wherein, the (C.N = 4) and (C.N. > 4) refers to the coordination number
of cubic diamond and body-centered tetragonal crystalline structures, respectively. As a
result of the increasing energy cost to hybridize and lowering binding energy of the
directional orbitals, the energy gap decreases in the group and the metallic character
increases when descending in the periodic table, ultimately changing the electronic
structure and, consequentially, the crystalline structure. The asterisk in black recalls these
solids true color, even though they will appear silver by a secondary emission of radiation
by the electrons in the conduction band.
VI. Conclusions
In this three-part work, the molecular orbital concept was applied to the main
group elements to study the band formation and the standard state of the elements and

extrapolated to the lower periods, which allowed us to identify some trends in the
periodic table. On the Boron and Carbon elements, it was seen that the presence of
empty bonding orbitals suggests an electronic configuration able to make more chemical
bonds, composed by hybrid orbitals (sp2 and sp3). The hybrid orbitals are advantageous
when the energy gained through the chemical bonds overcome the necessary energy to
excite the electronic configuration from s2p2 to sp3 or sp2. The sp2 hybridization is present
in both elements, however, in the Boron element, it confers semiconductor behavior and
a tetragonal crystalline structure, while in the Carbon element the sp2 hybridization has
a conductor-like behavior presenting a hexagonal structure. While, the sp3 hybridization
occurs only to the Carbon element, conferring an insulator behavior and different
crystalline structure, face-centered cubic. As seen, it for the Hydrogen and Helium
elements, the molar nitrogen, oxygen and fluorine systems at standard conditions are a
diatomic gas. Since further aggregation of these atoms does not present available energy
levels to propagate the chemical bond. Instead, the chemical bond is interrupted at the
diatomic molecule, interacting with other systems non-covalently.
According to the electronic structure description carried out through this work,
some tendencies could be extrapolated to other periods. First, the strong and localized
chemical bond is unfavorable for the lower periods due to the small atomic/molecular
orbitals overlap. Second, the radius of the internal orbitals is increased, leading to a
higher cross-section among the electrons and nuclei, resulting in a decreasing of the
electrical conductivity in metals. Third, the increase in atomic radius and lower binding
energy have opposite consequences for the simple metals and main group elements. In
the former, this leads to higher reactivity of the solids (alkali and alkaline earth metals)
due to the lower effective charge on the ns1 and ns2 electrons, less attracted to the
nucleus, which also is weakly bonded to other atoms and prompt to react. In the main
group, the weaker bonds lead to lower reactivity due to lower capacity to form bonding
orbitals, and thus, lower electronegativity. This also means that, as a general trend, all
substances tend to present metallic behavior for lower periods, depending on the
balance between available electrons and orbitals.
Acknowledgements

This work has been supported by Brazilian agencies Fundação de Amparo à
Pesquisa do Estado de São Paulo (FAPESP) under grants 2019/03729-8, 2018/22669-3
and 2017/07707-3, and Conselho Nacional de Desenvolvimento Científico e Tecnológico
(CNPq) under grants 309051/2016-9, 406107/2016-5, 307052/2016-8 and
404337/2016-3.
References
1 G. N. Lewis, J. Am. Chem. Soc., 1916, 38, 762–785.
2 E. A. Smart, Lesley E. Moore, Solid state chemistry : an introduction, CRC Press Tayler & Francis Group, New York, Third., 2005.
3 E. Kaxiras, Atomic and Electronic Structure of Solids, 2003.
4 I. Levine, Physical Chemistry, The McGraw-Hill Companies, New York, 6th editio., 2008.
5 P. Atkins and J. De Paula, Physical chemistry, Great Britain by Oxford University Press, Eighth Edi., 2006.
6 J. Autschbach, J. Chem. Educ., 2012, 89, 1032–1040.
7 a B. Sannigrahi and T. Kar, J. Chem. Educ., 1988, 65, 674.
8 P. F. Lang, J. Chem. Educ., 2018, ACS.jchemed.8b00239.
9 W. F. Giauque and J. O. Clayton, J. Am. Chem. Soc., 1933, 55, 4875–4889.
10 A. J. Tursi and E. R. Nixon, J. Chem. Phys., 1970, 52, 1521–1528.
11 Y. A. Freiman and H. J. Jodl, Phys. Rep., 2004, 401, 1–228.
12 S. C. Nyburg, J. Chem. Phys., 1968, 48, 4890–4895.
13 D. E. O’Reilly, E. M. Peterson, D. L. Hogenboom and C. E. Scheie, J. Chem. Phys., 1971, 54, 4194–4199.
14 T. M. Niemczyk, R. R. Getty and G. E. Leroi, J. Chem. Phys., 1973, 59, 5600–5604.
15 O. Madelung, U. Rössler and M. Schulz, Eds., Semiconductors · Non-Tetrahedrally Bonded Elements and Binary Compounds I, Springer Berlin Heidelberg, Berlin, Heidelberg, 2000.
16 A. R. Oganov, J. Chen, C. Gatti, Y. Ma, Y. Ma, C. W. Glass, Z. Liu, T. Yu, O. O. Kurakevych and V. L. Solozhenko, Nature, 2009, 457, 863–867.
17 A. Pisanty, J. Chem. Educ., 1991, 68, 804.
18 Charles Kittel, Introduction to Solid State Physics, 2010.
19 B. D. Fahlman, Materials Chemistry, Springer, 2007.
20 J. C. William D. and D. G. Rethwisch, Materials Science and Engineering: An

Introduction, John Wiley & Sons, Ltd, Seventh., 2007.
21 A. A. Balandin, S. Ghosh, D. L. Nika and E. P. Pokatilov, Fullerenes Nanotub. Carbon Nanostructures, 2010, 18, 474–486.
22 Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924.
23 J. Robertson, Diam. Relat. Mater., 2003, 12, 79–84.
24 S. Jiang, N. Holtgrewe, S. S. Lobanov, F. Su, M. F. Mahmood, R. S. Mcwilliams and A. F. Goncharov, Nat. Commun., 2018, 9, 1–6.
25 T. M. Klapötke, F. A. Martin and J. Stierstorfer, 2011, 403, 4227–4229.

download fileview on ChemRxivThe chemical bond across the periodic table Part 2_mai... (459.25 KiB)

The chemical bond across the periodic table: Part 3 – transition metals and light-matter
interaction
Gabriel F.S. Fernandes1, Leonardo A. Cunha1, Francisco B.C. Machado1, Luiz F. A. Ferrão1*
1Departamento de Química, Instituto Tecnológico de Aeronáutica, São José dos Campos,
SP 12228-900, Brasil.
__________________________________
*Corresponding author. E-mail address: [email protected]

Abstract
Chemical bond plays a central role in the description of the physicochemical properties
of molecules and solids and it is essential to several fields in science and engineering,
governing the material’s mechanical, electrical, catalytic and optoelectronic properties,
among others. Due to this indisputable importance, a proper description of chemical
bond is needed, commonly obtained through solving the Schrödinger equation of the
system with either molecular orbital theory (molecules) or band theory (solids). However,
connecting these seemingly different concepts is not a straightforward task for students
and there is a gap in the available textbooks concerning this subject. This work presents
a chemical content to be added in the physical chemistry undergraduate courses, in
which the framework of molecular orbitals was used to qualitatively explain the standard
state of the chemical elements and some properties of the resulting material, such as gas
or crystalline solids. Here in Part 3, we discuss the basis of the catalytic property of the
transition metals, some trends across the transition metal group, such as the density
increasing, and light-matter interaction. The original content discussed here can be
adapted and incorporated in undergraduate and graduate physical-chemistry and/or
materials science textbooks.
KEYWORDS: Upper-division Undergraduate, Graduate Education, Physical Chemistry,
Communication / Writing, Textbooks / Reference Books, Atomic Properties / Structure,
Material Science, MO theory, Periodicity / Periodic Table, Quantum Chemistry.

I. Introduction
In the proceedings papers of this series, the physicochemical properties, band
formation, standard state of the chemical elements and some periodic table trends were
explained using three main concepts: 1) the available energy levels and their occupation
based on the electronic structure of the hydrogen atom; 2) the energy splitting of energy
levels due to spatial overlap of electronic densities and; 3) trends in atomic radius along
the periodic table. Our interest lied in a qualitative picture to help students understand
general bonding patterns and how the properties of materials arise naturally from
information related to atomic and molecular energy levels and the bond order.
In Part 1, the Hydrogen atom was used as a metric for the other chemical
elements and as an example of how atomic or molecular orbitals interact to compose a
band structure. Within this framework, we were able to show the formation of metallic
hydrogen observed at the nucleus of gas giant planets like Jupiter, Saturn, and some
exoplanets. On the other hand, one can also explain the standard state of the hydrogen
as a diatomic gas and some of its physicochemical properties, such as low boiling point
and transparency to visible light at gas and liquid phase. Based on the Hydrogen structure,
we discussed why it is very unlikely to find, under normal conditions, diatomic Helium.
Through the molecular orbitals diagram, it was shown the transition from weakly bonded
clusters (Van der Waals clusters) to bonded solid (metal) seen in alkali earth metals.
Moreover, differences among the physicochemical properties between the simple metals
that arises due to the one-electron difference in the atomic electronic configuration, such
as enthalpy of sublimation, reactivity, crystalline structure, hardness, tensile strength 1–
3and Young’s modulus.
In Part 2, it was seen that the presence of empty bonding orbitals suggests an
electronic configuration able to make more chemical bonds, composed by hybrid orbitals
(sp2 and sp3). The hybrid orbitals are advantageous when the energy gained through the
chemical bonds overcome the necessary energy to excite the electronic configuration
from s2p2 to sp3 or sp2. The sp2 hybridization is present in both elements, however, in the
Boron element, it confers semiconductor behavior and a tetragonal crystalline structure,
while in the Carbon element the sp2 hybridization has a conductor-like behavior
presenting a hexagonal structure. While, the sp3 hybridization occurs only to the Carbon

element, conferring an insulator behavior and different crystalline structure, face-
centered cubic. As seen, it for the Hydrogen and Helium elements, the molar nitrogen,
oxygen and fluorine systems at standard conditions are a diatomic gas. Since further
aggregation of these atoms does not present available energy levels to propagate the
chemical bond. Instead, the chemical bond is interrupted at the diatomic molecule,
interacting with other systems non-covalently.
According to the electronic structure description carried out through in Part 2,
some tendencies could be extrapolated to other periods. First, the strong and localized
chemical bond is unfavorable for the lower periods due to the small atomic/molecular
orbitals overlap. Second, the radius of the internal orbitals is increased, leading to a
higher cross-section among the electrons and nuclei, resulting in a decreasing of the
electrical conductivity in metals. Third, the increase in atomic radius and lower binding
energy have opposite consequences for the simple metals and main group elements. In
the former, this leads to the higher reactivity of the solids (alkali and alkaline earth metals)
due to the lower effective charge on the ns1 and ns2 electrons, less attracted to the
nucleus, which also is weakly bonded to other atoms and prompt to react. In the main
group, the weaker bonds lead to lower reactivity due to lower capacity to form bonding
orbitals, and thus, lower electronegativity. This also means that, as a general trend, all
substances tend to present metallic behavior for lower periods, depending on the
balance between available electrons and orbitals.
In the present paper, the band formation and physicochemical properties
(electronic and catalytic properties), tendencies to lower periods (density) of the
transition metals group and bandgap/light-matter interaction (mirror effect of metals)
are explained by the molecular orbital framework developed in Part 1. To the best of our
knowledge, the chemical content presented in this paper is not seen in no previous
textbook4–9, whether it is connection between molecular orbitals and band theory topics
by employing the available energy levels and their occupation (from molecular orbitals),
as an electronic structure analysis of the transition metals as the d atomic level is filled
and its consequences in the physicochemical properties.

ll. Periodic Table tendencies for lower periods
ll.A. Transition metals
The main difference in the transition metals is the occupancy of d atomic orbitals
which are valence levels for these atoms. A fair question is: what are the consequences?
These orbitals have angular momentum such as the p orbitals, which translates in
preferential directions of the electronic distribution in space or, simply, directionality.
However, their higher angular momentum (L=2, while in p orbitals L=1) means “more
directions” in space, suggesting that these orbitals could be involved in both covalent and
metallic chemical bonds. Also, since these elements appear only from the fourth period
on, it is expected from the discussions in the previous paper of this series that the energy
splitting from the valence orbitals of these elements should be smaller. Individually, the
d atomic orbital should not make strong chemical bonds as the p atomic orbital (in the
second period) of the main group, however, there are more directions (angular
momentum) which allows the atom to make several chemical bonds with different atoms.
The multiple directionality characteristic of the d orbital combined with their relatively
low binding energy creates spatial regions around transition metal atoms in which other
(adsorbed) species can be submitted to weak binding fields in different orientations,
promoting chemical reactions within the adsorbed molecules that usually would not be
possible if those species were free. If the resulting substance presents lower interaction
with the transition metal (by forming a higher stability product with a “harder” electronic
density or change of polarity), it can be desorbed without directly involving modifications
in the transition metal atoms. This process is one of the bases of the catalyst properties
of the transition metals.
How should the electronic structure of these clusters be? In general, the
electronic structure of the transition metal elements is ns2(n-1)dx, with x=1−10. From the
perspective of the electronic energy diagram we have been using along with this paper
series, this means that the energy reference line lies in the dx orbital as shown in Figure
1. As atoms interact, the energy levels from the valence orbitals (the ones which overlap
significantly) will split. In a simple picture, one can imagine that this procedure, when
repeated to the bulk limit (molar atoms system), could form an sp band and a d band.
The occupancy of these “separate” bands certainly depends on the number of electrons
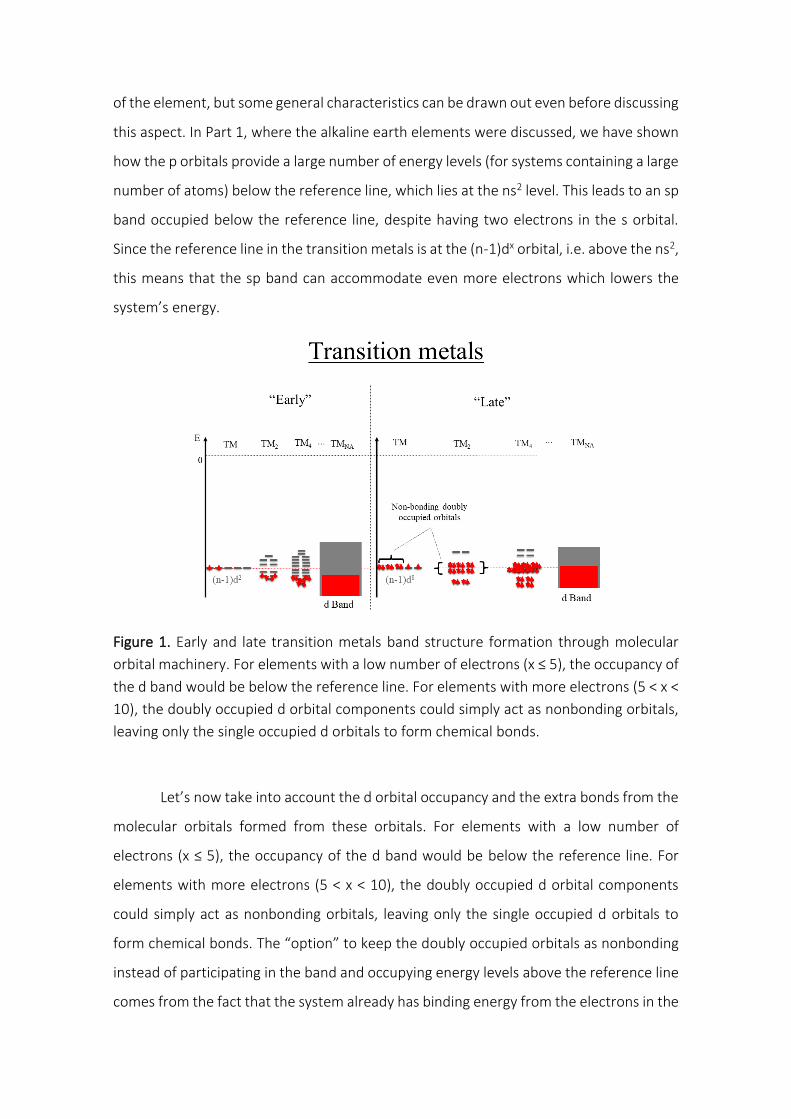
of the element, but some general characteristics can be drawn out even before discussing
this aspect. In Part 1, where the alkaline earth elements were discussed, we have shown
how the p orbitals provide a large number of energy levels (for systems containing a large
number of atoms) below the reference line, which lies at the ns2 level. This leads to an sp
band occupied below the reference line, despite having two electrons in the s orbital.
Since the reference line in the transition metals is at the (n-1)dx orbital, i.e. above the ns2,
this means that the sp band can accommodate even more electrons which lowers the
system’s energy.
Figure 1. Early and late transition metals band structure formation through molecular
orbital machinery. For elements with a low number of electrons (x ≤ 5), the occupancy of
the d band would be below the reference line. For elements with more electrons (5 < x <
10), the doubly occupied d orbital components could simply act as nonbonding orbitals,
leaving only the single occupied d orbitals to form chemical bonds.
Let’s now take into account the d orbital occupancy and the extra bonds from the
molecular orbitals formed from these orbitals. For elements with a low number of
electrons (x ≤ 5), the occupancy of the d band would be below the reference line. For
elements with more electrons (5 < x < 10), the doubly occupied d orbital components
could simply act as nonbonding orbitals, leaving only the single occupied d orbitals to
form chemical bonds. The “option” to keep the doubly occupied orbitals as nonbonding
instead of participating in the band and occupying energy levels above the reference line
comes from the fact that the system already has binding energy from the electrons in the

sp band and does not rely on the bonds from the d orbital to achieve stability. As a
consequence, the energy range of the d band should be broader for the early transition
metals, since more orbitals overlap significantly and therefore, have their energy more
split, while late transition metals will have a narrower d band energy range (Figure 1). In
any case, this means that the sp band always has available levels below the reference line
for all transition metal elements.
The “early” and “late” transition metals may show similar physicochemical
properties due to their similar band structure composed of an sp band plus the unpaired
electrons on the d orbitals. From the perspective of the electronic structure of the early
and late transition metals, it is interesting to discuss limiting cases that should mark
qualitative changes in these systems. There are two families in the late group that should
present some distinct characteristics due to their almost or completely filled d orbitals,
i.e. the coinage (copper) and zinc families.
The elements of the coinage family have an electronic configuration given by (n-
1)d10ns1 and are said to have irregular electronic structure (with respect to the electronic
structure and orbital ordering based on the hydrogen atom). This means that, for this
family, the reference line lies in the s orbital while the d orbital should be lower in energy
and narrow since all d components are doubly occupied (as shown in Figure 1). Given that
picture, these atoms should present a metallic band similar to that of an alkali metal when
forming a solid, i.e., each atom contributes with one electron to the chemical bond, which
is constructed with s and p orbitals and thus forms an sp band with relatively low
occupancy. The d orbitals of each atom are completely filled and, therefore, should not
directly participate in the bonding, only providing certain freedom to the external
electron in the s orbital. This would lead to a metal which should have a relatively high
electron conductivity, but also very reactive since the binding energy involves essentially
one electron per atom.
Obviously, this is not an accurate description of copper, silver and gold solids
which have characteristics of noble metals and are among the most conducting metals.
Then what went wrong in the analysis we used? Maybe it is too simple to capture the
qualitative behavior of the chemical bond or, maybe, it is missing some information. At
least, in this case, the analysis is missing a very important piece of information: the
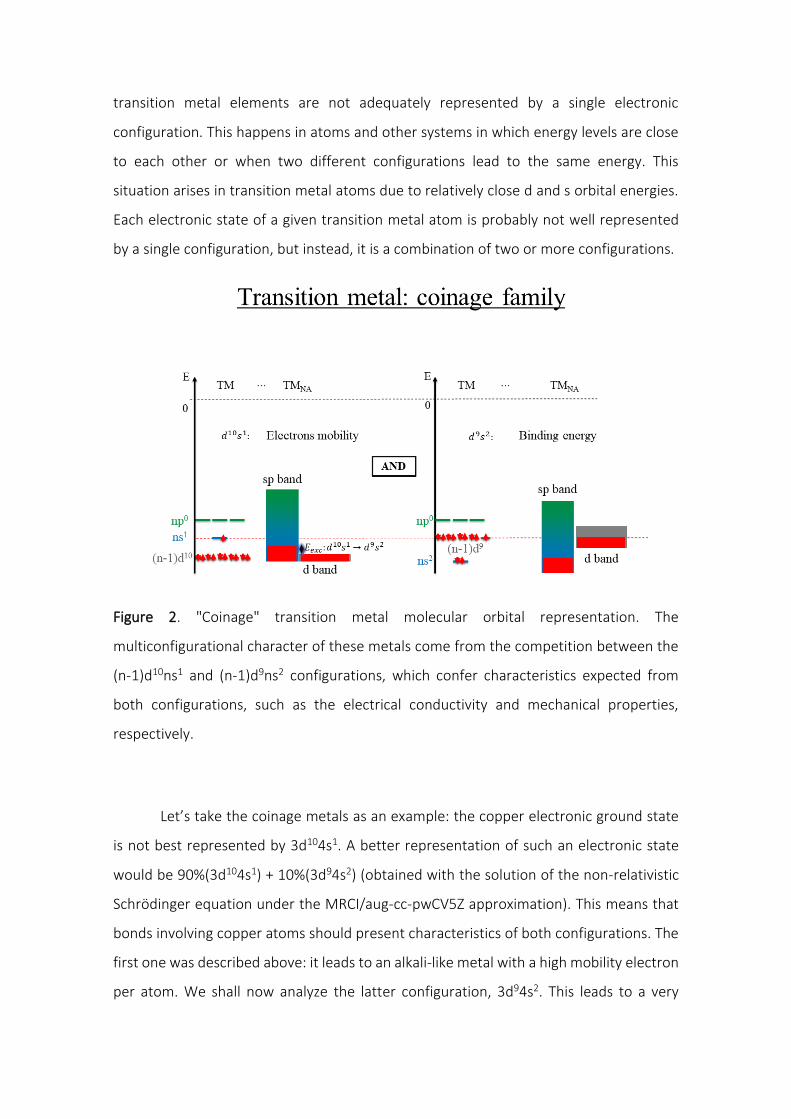
transition metal elements are not adequately represented by a single electronic
configuration. This happens in atoms and other systems in which energy levels are close
to each other or when two different configurations lead to the same energy. This
situation arises in transition metal atoms due to relatively close d and s orbital energies.
Each electronic state of a given transition metal atom is probably not well represented
by a single configuration, but instead, it is a combination of two or more configurations.
Figure 2. "Coinage" transition metal molecular orbital representation. The
multiconfigurational character of these metals come from the competition between the
(n-1)d10ns1 and (n-1)d9ns2 configurations, which confer characteristics expected from
both configurations, such as the electrical conductivity and mechanical properties,
respectively.
Let’s take the coinage metals as an example: the copper electronic ground state
is not best represented by 3d104s1. A better representation of such an electronic state
would be 90%(3d104s1) + 10%(3d94s2) (obtained with the solution of the non-relativistic
Schrödinger equation under the MRCI/aug-cc-pwCV5Z approximation). This means that
bonds involving copper atoms should present characteristics of both configurations. The
first one was described above: it leads to an alkali-like metal with a high mobility electron
per atom. We shall now analyze the latter configuration, 3d94s2. This leads to a very

different bond scenario: the reference line is at the 3d orbital, i.e. above the 4s and thus
the sp band can accommodate more electrons. Besides that, one of the d orbital
components participates directly in the bonding, creating a slightly broader d band which
would be occupied below the reference line (one unpaired d electron per atom). If the
former configuration can be related to the alkali metal, the latter is to some extent
related to the alkaline earth metal, since it has two electrons in an s orbital that will
populate an sp band. However, the binding electrons from the d band can flow to the sp
band and increase the binding energy. In this case, the electrons in both the sp and d
bands would enhance the chemical bond and also the number of electrons available for
conduction. This means that the solid formed by this configuration would have much
more binding energy than an alkaline earth metal (considering the same period of the
periodic table) with a comparable or higher conductivity due to its three electrons per
atom. Combining the solids that would result from both 3d104s1 and 3d94s2 configurations
isolated, one has a more complete picture of the characteristics of copper: a high
conducting metal (from 3d104s1) with high binding energy (from 3d94s2). Since the latter
contributes only with 10% of the atom behavior, the characteristics of this configuration
are attenuate in the resulting solid, making it only regular in its binding energy and
mechanical properties when compared to other transition metals, but sufficient to
reduce its reactivity that would result from a low binding energy per atom. Another effect
that would increase the reactivity is the excess of binding sites unoccupied, but this is not
an issue for this system.
It is worth mentioning another interesting characteristic resulting from the
electronic structure of the coinage metal solids: their interaction with light. When a
photon interacts with the metal, it can excite electrons from the valence to conduction
band with minimal energy. These photons can also change the population of d and sp
bands depending on: the occupation of these bands, the photon energies and other
characteristics of the band structure not discussed here. For most transition metals this
is not a problem since electrons in the solid can “flow” from a given configuration to
another through different bands occupations, and consequently energy changes in a
continuous way. However, in coinage metals, this continuous population changes in d
band are not possible since this band is filled (resulting from (n-1)d10ns1 configuration).

In order to change the d band occupancy, an electronic transition must occur from the d
band to the conduction band (see Figure 2). This transition requires a certain amount of
energy that depends on the relative energies of sp and d bands, and consequently, it
depends on the relative energy of s and d orbitals in the atom. In the case of copper, this
energy separation is in the region of red light. In silver, it occurs in the ultraviolet region,
and in gold, this occurs in the region of yellow light. This property confers the
characteristic color of copper and gold solids, while silver has the common “silver” color
of all other metals since this transition does not occur in the visible light region.
We shall now discuss the zinc family, the latest elements in the transition metal
group. On the contrary to the complexity discussed for the coinage metals, now the
electronic structure of these elements should not lead to further intricacies. The
configuration (n-1)d10ns2 does not have any competing or degenerate energy levels since
both orbitals in “valence” are filled. With s and all d orbital components doubly occupied,
the binding energy of the electrons in this atom is maximized when compared to all other
transition metals (within the same period of the periodic table), which can be seen by
comparing their ionization energies. However, this high electron binding energy in the
atomic system should hinder the capacity of these atoms to make chemical bonds, such
as, up to a point, happens in the noble gases. In this analogy, the main difference between
them is the existence of other available orbitals in zinc family (np0) within the same main
quantum number, in a similar way of how alkali elements differ from hydrogen. So, what
is the expected electronic structure for the system formed as zinc family elements
agglomerate?
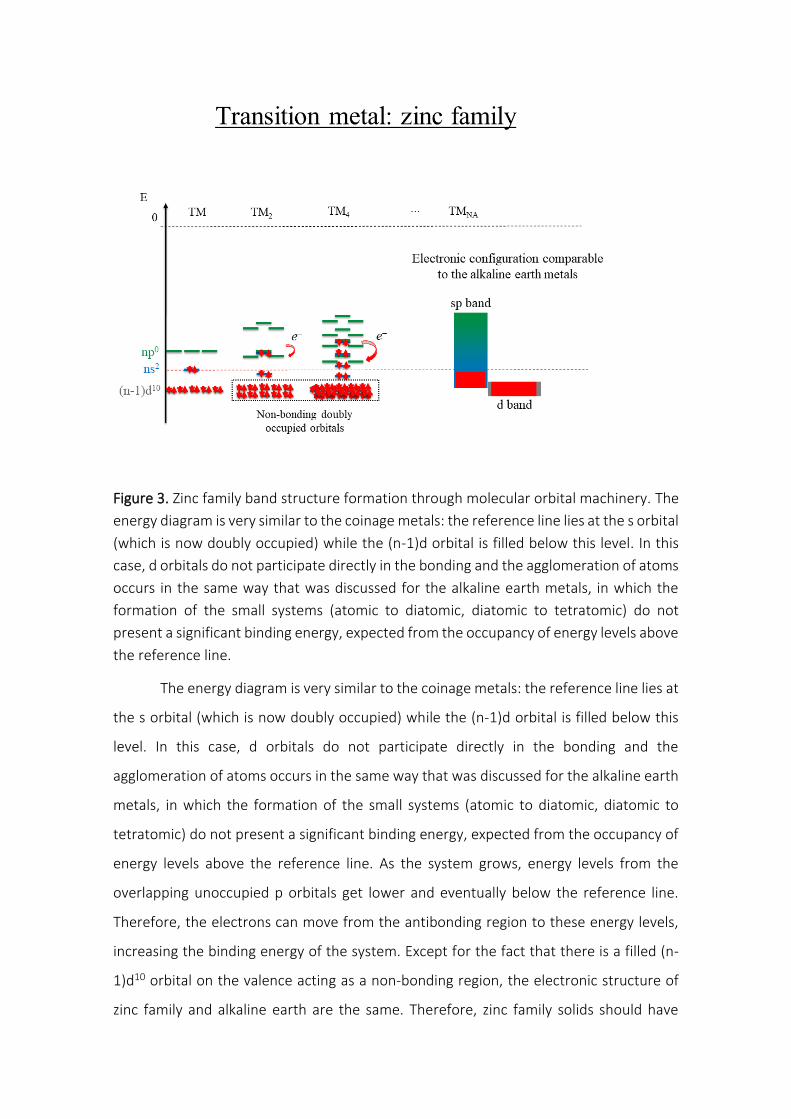
Figure 3. Zinc family band structure formation through molecular orbital machinery. The
energy diagram is very similar to the coinage metals: the reference line lies at the s orbital
(which is now doubly occupied) while the (n-1)d orbital is filled below this level. In this
case, d orbitals do not participate directly in the bonding and the agglomeration of atoms
occurs in the same way that was discussed for the alkaline earth metals, in which the
formation of the small systems (atomic to diatomic, diatomic to tetratomic) do not
present a significant binding energy, expected from the occupancy of energy levels above
the reference line.
The energy diagram is very similar to the coinage metals: the reference line lies at
the s orbital (which is now doubly occupied) while the (n-1)d orbital is filled below this
level. In this case, d orbitals do not participate directly in the bonding and the
agglomeration of atoms occurs in the same way that was discussed for the alkaline earth
metals, in which the formation of the small systems (atomic to diatomic, diatomic to
tetratomic) do not present a significant binding energy, expected from the occupancy of
energy levels above the reference line. As the system grows, energy levels from the
overlapping unoccupied p orbitals get lower and eventually below the reference line.
Therefore, the electrons can move from the antibonding region to these energy levels,
increasing the binding energy of the system. Except for the fact that there is a filled (n-
1)d10 orbital on the valence acting as a non-bonding region, the electronic structure of
zinc family and alkaline earth are the same. Therefore, zinc family solids should have

some characteristics similar to those of the alkaline earth metals (beryllium and
magnesium, mostly)10.
Experimentally, zinc dimer presents atomization energy (dissociation energy)
equal to 3 kJ.mol-1 11,12 while the atomization energy of the solid (sublimation enthalpy)13
is equal to 130 kj.mol-1, the same trend presented by alkaline earth metals.
Descending in the periodic table under the transition metal group we have the
same tendencies as discussed for the simple metals and main group elements: larger
cores and lower binding energies for individual orbitals (due to a lower spatial overlap of
electronic densities). However, a new effect not discussed before should be taken into
account: the relativistic contraction of the s orbitals. Relativistic effects occur in all atoms
and molecules, but their effects scale with the square of the atomic number Z14. From
the perspective of low-resolution spectroscopy and thermochemistry, these effects
become non-negligible in the third period of the periodic table and, by the fifth period,
they become one of the dominant aspects of the wavefunction governing these systems.
The contraction of the s orbitals is one of these effects. In atoms with a high atomic
number, the electrons in the 1s orbital are subjected to a very high potential energy from
the nucleus, since the Coulomb potential that drives the electron-nucleus interaction
depends directly on the charges and are inversely proportional to the “mean” electron-
nucleus distance (also, there is no “shielding” from other orbitals). Therefore, 1s
electrons also have a very high mean kinetic energy (since there is a balance between the
kinetic and potential energies)14. In a classic approach, this would lead to electrons mean
velocities relatively close to the speed of light. The system responds with a contraction of
the spatial distribution of the 1s orbital, which causes a contraction in the spatial
distribution of all other s orbitals in the system, including the s orbital in the valence (or
the outer s orbital)14. While this affects all atoms, when it comes to chemical bond it is
for the transition metals that this plays a central role.
The bond in simple metals depends mostly on the overlapping of the occupied s
orbitals (no matter if they are contracted or not). In the main group elements, the
bonding depends both of s and p orbitals and the contraction of the s orbitals certainly
impact the bonding, but since these orbitals have relatively similar spatial distributions in
lighter molecules, the contraction of s orbitals does not qualitatively change the bonding

in such systems. Take for example the radii of the principal maxima of the outer orbitals
radial distribution of the carbon family elements. For carbon, the 2s and 2p maxima occur
at 0.62 and 0.60 Å, respectively, while for lead, the 6s and 6p maxima occur at 1.01 and
1.22 Å, respectively15. This means that, while there is a change in which orbital is the
outermost one, the s/p maxima ratio “only” changed from 103% to 84%. A similar ratio
occurs for other main group elements. For these elements, the most relevant aspect is
the increment in the s-p orbitals energy gap, increasing the energetic cost of the
hybridization when descending in the periodic table.
For transition metals, on the other hand, there is a dramatic change in the relative
spatial distributions of s and d orbitals, which are the occupied ones in the valence and,
therefore, will directly influence the chemical bonds. Take for example elements of the
coinage family. In copper, the 4s and 3d maxima occur at 1.19 and 0.31 Å, respectively,
while in gold 6s and 5d maxima occur at 1.19 and 0.62 Å, respectively. This means an s/d
ratio changing from 384% in copper to 192% in gold 15. Similar maxima s/d ratios are
observed for other transition metal families. In perspective, the 4s orbitals are roughly
four times larger than 3d, while the 6s it is only two times larger than the 5d. Therefore,
it is expected that the role of the d orbital in the bonding involving 5d transition metal
elements to be increased in comparison to 3d metals (due to larger overlap with other
orbitals). This can attenuate or even invert the trend of lowering the bonding when
descending on the periodic table, at least for the transition metal families with various
singly occupied d orbitals, since those can form bonding energy levels which will be
doubly occupied, increasing the binding energy. Experimentally, the enthalpy of
sublimation of chromium, molybdenum and tungsten, (which have electronic
configuration (n-1)d5ns1) is 395, 657 and 857 kJ.mol-1, respectively16,17.
A counterexample is the zinc family which has the electronic configuration (n-
1)d10ns2. With all d components doubly occupied, this orbital has low participation in the
chemical bond and the solid formed by zinc mostly relies on the binding energy from the
sp band, as discussed before. Descending on the periodic table should lower or difficult
the spatial overlap of s orbitals, and therefore, should lower the binding energy of these
systems. Experimentally, the enthalpy of sublimation (which is directly related to the
binding energy) of zinc, cadmium and mercury is equal to 130, 112 and 62 kJ.mol-1,

respectively13,18. The binding energy in Mercury is low enough that this molar system is
metal (from its sp band) and liquid (low binding energy) at room temperature and at
atmospheric pressure.
Overall, this trend in the binding energy as one descends in the transition metal
families depending on the number of singly occupied d orbitals also explains the trends
of density across the periodic table. Since the binding energy is increased in the direction
of singly occupied d orbitals, this means that the atomic distances decrease in this same
direction. Considering solids with the same crystal structure (close-packed structures),
this means that the solids formed by the elements around the center of the transition
metal and lower in the periodic table should be closer to each other, increasing the
density. Combined to the fact that the atomic number, thus the mass of such systems
increases in the direction of the lower diagonal of the periodic table, that means that the
largest densities should be maximized when approaching the center of the transition
metal block from the right, which is experimentally observed and osmium has the highest
specific mass in the periodic table.
ll.B. Band gap and light-matter interaction
One last aspect involving the light-matter interaction should be discussed: the
color of crystals. The absorption of electromagnetic radiation within visible light depends
on the band gap energy of the system. In the case of the carbon family solids, diamond
has a band gap (5.4 eV) higher than the visible light (1.65 − 3.20 𝑒𝑉), therefore it is
transparent to it. The solids formed by all other carbon family members have band gaps
below 1.65 eV, and therefore, absorb all visible light, meaning that they are black. Then
why are those solids, like most of the other metals, silvery? This happens due to a
secondary effect of the radiation acting upon the solid. In these systems, a large number
of electrons are injected into the conduction band and have high mobility, since there are
plenty of energy levels with different spatial distributions available (this depends also on
the orbitals overlap; orbitals involving the combination of several atoms will promote this
mobility). Since those electrons in the conduction band are still submitted to the
upcoming radiation, they are also affected by it. This weak interaction can be described

in a classical way, i.e., “free” electrons affected by an electromagnetic wave. In this
scenario, the electrons are accelerated by the upcoming radiation tending to move in
resonance with the radiation frequencies. As the electrons accelerate, they also emit an
electromagnetic wave, and as these tend to emit in resonance with the upcoming
radiation, they act as a secondary source of radiation. If the surface of the solid has large
planar or quasi-planar sections (it is polished) this effect is largely enforced since the
accelerating electrons occur in the same directions and thus do not “cancel” each other
(in other words, the inelastic scattering is reduced). Therefore, semiconductors and
metals are black. They look silvery by a secondary effect of a weak interaction of electrons
with the primary source of radiation, acting as a secondary source and mirroring the
primary source, like an antenna of visible light. If this is a representative model for the
experimental observations, another important consequence should occur. To act as an
antenna, the “wave of electrons” in the conduction band needs a minimum spatial extent
in order to order to oscillate in resonance with the primary source, in a certain way
related to the wavelength of this source. As the visible light wavelengths are within the
hundreds of nanometers (400 − 700 nm), it is expected that if the system is significantly
smaller than this, it will probably not be able to respond to the primary source. In fact, it
is experimentally observed that, for example, metallic nanoparticles of aluminum with
spherical morphology and mean diameters of around 100 nm are black. It is worth noting
that these particles are still large enough that their electronic structure is very close to
that of an “infinite size system”. To reinforce this, one can also recall that several
properties of aluminum clusters converge to the bulk limit cubically with the system size
(number of atoms in the cluster)19, and clusters with more than 40 atoms already show
properties that converge to the bulk20. One can estimate that in a much smaller solid
particle, with 10 nm3, there are around 600 atoms, a considerable number which should
already present an electronic structure similar to the bulk.
VI. Conclusions
Within the molecular orbital framework described in Part 1, it was seen that the
basis of the catalytic property of the transition metals relies on the combination of
multiple chemical bonds with low binding energy, in which spatial regions are created

around transition metal atoms in which other (adsorbed) species can be submitted to
weak binding fields in different orientations, promoting chemical reactions within the
adsorbed molecules that usually would not be possible if those species were free. Also, it
was observed that the “early” and “late” transition metals may show similar
physicochemical properties due to their similar band structure composed of an sp band
plus the unpaired electrons on the d orbitals. The coinage (copper) metals possess
qualitative changes in the electronic structure. Their electronic ground state is better
represented by two electronic configurations: (n-1)d10ns1 and (n-1)d9ns2. This means that
bonds involving coinage metals should present characteristics of both configurations. The
first one leads to an alkali-like metal with a high mobility electron per atom. While the
second one to some extent related to the alkaline earth metal, since it has two electrons
in an s orbital that will populate an sp band. However, due to the higher number of
electrons that can populate the sp band, the coinage metals have much more binding
energy than an alkaline earth metal (considering the same period of the periodic table).
Descending in the periodic table under the transition metal group, we observed
the same tendencies discussed for the simple metals (Part 1) and main group elements
(Part 2): larger cores and lower binding energies for individual orbitals (due to a lower
spatial overlap of electronic densities). However, a new effect not discussed before
should be considered: the relativistic contraction of the s orbitals. For transition metals,
there is a dramatic change in the relative spatial distributions of s and d orbitals, this
effect can attenuate or even invert the trend of lowering the bonding when descending
on the transition metal group. Since the binding energy is increased in the direction of
singly occupied d orbitals, this means that the atomic distances decrease in this same
direction. Considering solids with the same crystal structure (close-packed structures),
this means that the solids formed by the elements around the center of the transition
metal and lower in the periodic table should be closer to each other, increasing the
density. Combining this with the fact that the atomic number, thus the mass of such
systems increases in the direction of the lower diagonal of the periodic table, that means
that the largest densities should be maximized when approaching the center of the
transition metal block from the right, which is experimentally observed and osmium has
the highest specific mass in the periodic table.

Also, it was observed that the color of crystals is related to the absorption of
electromagnetic radiation within visible light, which depends on the band gap energy of
the system. Wherein, solids with bandgap higher than the visible light (1.65 - 3.20 eV))
are transparent to it and those solids that have band gaps below 1.65 eV, and therefore,
absorb all visible light, are black. Semiconductors and metals are black, however, they
look silvery by a secondary effect of a weak interaction of electrons with the primary
source of radiation, acting as a secondary source and mirroring the primary source, like
an antenna of visible light.
We hope that the original content discussed along this paper series can be
adapted and incorporated in undergraduate and graduate physical-chemistry and/or
materials science textbooks and also serves as a conceptual guide to subsequent
disciplines such as quantum chemistry, quantum mechanics and solid-state physics.
Acknowledgements
This work has been supported by Brazilian agencies Fundação de Amparo à
Pesquisa do Estado de São Paulo (FAPESP) under grants 2019/03729-8, 2018/22669-3
and 2017/07707-3, and Conselho Nacional de Desenvolvimento Científico e Tecnológico
(CNPq) under grants 309051/2016-9, 406107/2016-5, 307052/2016-8 and
404337/2016-3.
References
1 J. C. William D. and D. G. Rethwisch, Materials Science and Engineering: An Introduction, John Wiley & Sons, Ltd, Seventh., 2007.
2 M. F. Ashby, Materials Selection in Mechanical Design, Elsevier Ltd, Fourth., 2011, vol. 1.
3 J. C. William D. and D. G. Rethwisch, Fundamentals of Materials Science and Engineering: an integrated approach, John Wiley & Sons, Inc., Third., 2008.
4 P. Atkins and J. De Paula, Physical chemistry, Great Britain by Oxford University Press, Eighth Edi., 2006.
5 E. A. Smart, Lesley E. Moore, Solid state chemistry : an introduction, CRC Press Tayler & Francis Group, New York, Third., 2005.

6 W. A. Harrison, Electronic Structure and the Properties of Solids, Dover Publications, 1989.
7 I. Levine, Physical Chemistry, The McGraw-Hill Companies, New York, 6th editio., 2008.
8 E. Kaxiras, Atomic and Electronic Structure of Solids, 2003.
9 Charles Kittel, Introduction to Solid State Physics, 2010.
10 W. B. Jensen, J. Chem. Educ., 2003, 80, 952–961.
11 E. Czuchaj, F. Rebentrost, H. Stoll and H. Preuss, Chem. Phys., 1996, 255, 203–209.
12 M. Czajkowski, R. Bobkowski and L. Krause, Phys. Rev. A, 1990, 41, 277–282.
13 J. R. McCreary and R. J. Thorn, J. Chem. Phys., 1969, 50, 3725–3733.
14 N. Zettili, Quantum Mechanics Concepts and Applications, John Wiley & Sons, Ltd, Second., 2009.
15 J. T. Waber and D. T. Cromer, J. Chem. Phys., 1965, 42, 4116–4123.
16 R. Szwarc, E. R. Plante and J. J. Diamond, J. Res. Notional Bur. Stand. A. Phys. a nd Chem., 1965, 417–421.
17 K. Thurnay, Thermal Properties of Transition Metals, 1998.
18 E. U. Franck, Berichte der Bunsengesellschaft für Phys. Chemie, 1990, 94, 93–93.
19 F. Duque and A. Ma, Eur. Phys. J. D, 1999, 9, 223–227.
20 P. Milani, W. De Heer and A. Cmtelain, Atoms, Mol. Clust., 1991, 135, 133–135.

download fileview on ChemRxivThe chemical bond across the periodic table Part 3_tran... (331.06 KiB)





























