Embed Size (px)
Citation preview

of May 6, 2018.This information is current as
RecruitmentImmunity via Immune Cell Activation and The Chemokine CCL6 Promotes Innate
KunkelAmos Z. Orlofsky, Cory M. Hogaboam and Steven L. Ana L. Coelho, Matthew A. Schaller, Claudia F. Benjamim,
http://www.jimmunol.org/content/179/8/5474doi: 10.4049/jimmunol.179.8.5474
2007; 179:5474-5482; ;J Immunol
Referenceshttp://www.jimmunol.org/content/179/8/5474.full#ref-list-1
, 19 of which you can access for free at: cites 53 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2007 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Chemokine CCL6 Promotes Innate Immunity via ImmuneCell Activation and Recruitment1
Ana L. Coelho,2* Matthew A. Schaller,* Claudia F. Benjamim,† Amos Z. Orlofsky,‡
Cory M. Hogaboam,* and Steven L. Kunkel*
Septic syndrome is a consequence of innate immune failure. Recent studies showed that the CC chemokine CCL6 enhancedantimicrobial immunity during experimental sepsis through an unknown mechanism. The present study demonstrates that trans-genic CCL6 expression abolishes mortality in a septic peritonitis model via the modulation of resident peritoneal cell activationand, more importantly, through the recruitment of IFN-producing NK cells and killer dendritic cells into the peritoneum. Thus,CCL6 attenuates the immune failure during sepsis, in part, through a protective type 1-cytokine mediated mechanism. TheJournal of Immunology, 2007, 179: 5474–5482.
S evere sepsis, defined as sepsis associated with acute organdysfunction, may result from exuberant innate immuneand procoagulant responses to an infection (1). Despite
important advances in antibiotic development and the improvedeffectiveness of critical care units with advanced ventilator sup-port, the mortality due to sepsis has not dramatically changed overthe past three decades (2). The present limitations for clinical in-tervention in sepsis reflect, in part, our limited understanding of themechanisms involved in the regulation of the innate immune re-sponse during sepsis. It is now appreciated that the immune systemduring sepsis is precariously balanced between pro- and anti-inflammatory mediators. If the patient fails to mount an effectiveinnate response, the clinical outcome could be an overwhelminginfection. Conversely, should the immune system be improperlyregulated, the patient may develop a systemic inflammatory re-sponse syndrome (SIRS),3 characterized by the high expression ofseveral proinflammatory cytokines. SIRS may be just as lethal asan overwhelming infection, thus the immune system initiates acompensatory anti-inflammatory response (CARS), which can leadto a syndrome leaving the patient’s immune system in a state ofimmune suppression. CARS may increase the mortality of pa-tients; this host response can lead to an immunocompromised pa-tient and increase the susceptibility to secondary infection.
Chemokines play important roles in several activities such asangiogenesis/angiostasis, cellular differentiation and activation,
wound healing, lymphocyte homing and development of lymphoidtissue, and influencing the overall type 1/type 2 balance of animmune response (3–5). Clinical studies have identified elevatedlevels of chemokines associated with human sepsis and acute lunginjury (6–9). Because chemokines are essential to the innate im-mune response, they have been considered therapeutic targets dur-ing sepsis.
CCL6 is a CC chemokine initially isolated from mouse bonemarrow (10). This chemokine is mostly a macrophage chemoat-tractant in vitro and in vivo (11, 12), but it can also attract B cells,CD4� lymphocytes and eosinophils (11–13). CCL6 is a murinechemokine with several functional homologues in humans includ-ing macrophage inflammatory protein-1�, CC chemokine F-18,hemofiltrate CC chemokine-1 and hemofiltrate CC-2. These che-mokines have an unusual genomic structure, with a unique secondexon and the ability to bind to and activate CC chemokine receptor1 (14–19). Unlike all other chemokines, CCL6 is IL-4 induciblebut not LPS inducible (13, 20). Several studies have shown thatCCL6 is produced in large quantities during inflammatory and re-modeling disorders, including those that involve alveolar remod-eling, dermal wounding, allergic bronchopulmonary aspergillosis,bleomycin-induced pulmonary fibrosis, experimental demyelinat-ing diseases, and acute and chronic peritonitis (11, 14, 16, 21–24).Recently, it was shown that rat microglia can express CCL6, andthis study shows a possible role of this protein in cell-cell com-munication (22). CCL6 is also up-regulated in the peritoneal fluidof mice following cecal ligation and puncture (CLP) (18, 25, 26).In an experimental model of sepsis, recombinant CCL6 protectedmice from CLP-induced lethality and this protection was associ-ated with increased expression of TNF-�, IL-13, and CCL2 (18).Also, exogenous CCL6 enhanced bacterial clearance and thephagocytic capacity of peritoneal macrophages (18).
In the present study, we investigated the mechanism(s) throughwhich this chemokine protects mice from severe sepsis. To thisend, we used mice transgenically overexpressing CLL6 (CCL6 Tg)in the lung. Our data demonstrate that CCL6 Tg mice are ex-tremely resistant to the lethality following CLP. These mice ex-hibited significantly increased levels of CCL2/MCP-1 at 4 h, andIFN-� at 24 and 72 h, after CLP in the peritoneal lavage fluid.IFN-� was identified as critical cytokine for the survival of CCL6Tg mice, because anti-IFN-� Ab abolished the protective effect ofCCL6. Increased numbers of IFN-producing killer dendritic cells(IKDCs) and NK cells were observed in the peritoneal cavity of
*Department of Pathology, University of Michigan, Ann Arbor, Michigan 48109;†Department of Pharmacology, Universidade Federal do Rio de Janeiro, Rio deJaneiro, Brazil; ‡Albert Einstein College of Medicine, Yeshiva University, Bronx,NY 10461
Received for publication March 30, 2007. Accepted for publication August 7, 2007.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the National Institutes of Health (P50 HL 056402, P01HL 031963, and R01 HL031237).2 Address correspondence and reprint requests to Dr. Ana L. Coelho, Department ofPathology, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan48109. E-mail address: [email protected] Abbreviations used in this paper: SIRS, systemic inflammatory response syndrome;CARS, compensatory anti-inflammatory response; CLP, cecal ligation and puncture;CCL6 Tg, transgenically overexpressing CCL6; IKDC, IFN-producing killer den-dritic cell; WT, wild type; AST, aspartate transaminase; ALT, alanine transaminase;PC, peritoneal cavity; DC, dendritic cell; MHCII, MHC class II.
Copyright © 2007 by The American Association of Immunologists, Inc. 0022-1767/07/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
CLP-CCL6 Tg mice, suggesting these cells were the source of theobserved increases in IFN-�. Thus, these data support the conceptthat CCL6 modulates endogenous macrophages activation possi-bly via CCL2 and, more importantly, facilitated the recruitment ofeffector immune cells with the enhanced ability to generate IFN-�.
Materials and MethodsMice
Specific-pathogen-free, female, C57BL/6 mice (6 to 8 wk of age) wereobtained from Taconic Company, Germantown, NY.
Generation of CCL6 transgenic mice
A murine CCL6 minigene was constructed by PCR using CCL6 cDNA(13) and genomic (27) clones. The resulting minigene consisted of thecomplete CCL6 open reading frame, with intron-3 of the CCL6 gene in itsnatural location, followed by nt 1459–1643 of the vector RJB10A (28),containing the bovine growth hormone polyadenylation signal. The PCRproduct was inserted into a pCRII vector (Invitrogen Life Technologies) inwhich the polylinker orientation was reversed by BstXI cleavage and re-ligation, and the sequence of the resulting construct (pCR-CCL6) was con-firmed. A 2.3-kb segment of the Clara cell 10kd gene promoter was iso-lated as a HindIII fragment from pRtCC10–2300 (29) (a gift from JeffreyWhitsett, Children’s Hospital Research Foundation, Cincinnati, OH) andinserted in the HindIII site of pCR-CCL6. Transient protein expressionfrom this construct was verified by the immunostaining of transfectedH441 lung adenocarcinoma cells. The construct was linearized with NsiIand microinjected into FVB oocytes with the assistance of the Transgenicand ES Cell Core Facility of the Albert Einstein College of Medicine. Sixfounders were backcrossed to C57BL/6 and the hemizygous progeny as-sessed for CCL6 expression by both Western blot of bronchoalveolar la-vage and Northern blot of lung RNA. One positive line was chosen forfurther study. The mice exhibited no gross or histologic abnormality. Allmice were housed five per filter-topped cage and had free access to foodand water during their use in an experiment. The University of MichiganCommittee on Use and Care of Animals approved all animal studies outlinein this manuscript.
CLP experimental model
Animals were subjected to sham or CLP surgery as previously described(30). In brief, mice were anesthetized with an i.p. injection of 2.25 mg
ketamine HCl (Abbot Laboratories) and 150 �g xylazine (Lloyd Labora-tories). Under sterile conditions, a 1–2 cm incision was made on the lowerleft abdomen and the cecum was exposed. The distal portion of the cecumwas partial ligated with 3.0 silk suture and punctured (9 punctures) with a21-gauge needle. The cecum was returned to the peritoneal cavity and theincision was closed with surgical staples. Sham-operated mice were sub-jected to a similar laparotomy without ligation and puncture. Mice wererehydrated with 1 ml saline s.c. and placed on a heating pad until theyrecovered from the anesthetic. Mice in both surgery groups were treatedwith an antibiotic preparation containing Imipenem conjugated with Cila-statin (Primaxin I.V., 10 mg/kg, i.p.; Merck) beginning at 6 h after CLPsurgery and readministered every 12 h thereafter until day 3 postsurgery.Antibiotic therapy increased mouse survival to the 60–70% range, andthese mice were used in the following experiments. Additionally, survivalexperiments were performed without antibiotics, to address the role ofCCL6 in mouse survival without antimicrobial intervention.
In vivo experimental protocol
IFN-� neutralization. Wild-type (WT) and CCL6 Tg mice were treatedi.p. with either control rabbit IgG (500 �g/mouse) or rabbit anti-murineIFN-�-specific IgG (500 �g/mouse) 1 h before CLP surgery. The controlIgG and anti-IFN-� were purified from antiserum using a Protein A column(Pierce). Anti-IFN-� antiserum was raised by immunizing New Zealandwhite rabbits with murine IFN-� (R&D Systems). The polyclonal Abs weretittered by direct ELISA. The Abs did not cross-react with a number ofother murine cytokines, including CXC and CC chemokines, as seen in thefact that IFN-� ELISA established with the Abs did not detect any murinecytokine at a concentration as high as 100 ng/ml. Mouse survival wasmonitored in CLP groups containing ten mice for a total of 7 days. Allmouse survival studies were conducted a maximum of three experiments.NK cells depletion. WT and CCL6 Tg mice were treated i.p. with eitherisotype control (200 �g/mouse) or hamster anti-murine NK 1.1 (200 �g/mouse), provided by Dr. T. Moore (University of Michigan, Ann Arbor,MI), beginning at 24 h prior CLP and readministrated at 48 h after CLPsurgery. Mouse survival was monitored in CLP groups containing ten micefor a total of 7 days. All mouse survival studies were conducted a maxi-mum of three experiments.
Clinical chemistry
Serum was separated from whole blood and the aspartate transaminase(AST) and alanine transaminase (ALT) levels were measured by ClinicalPathology at the University of Michigan Medical School using standard-ized techniques.
Peritoneal cells culture
Peritoneal cells were harvested from sham and CLP mice at 72 h aftersurgery and suspended in RPMI 1640 (BioWhittaker) containing 5% FCS,2 mM L-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin. Cellswere placed in plastic 24-wells plates (1 � 106 cell/well) and incubated at37°C in 5% CO2. After 24 h, supernatants were removed, clarified bycentrifugation, and analyzed by ELISA for IFN-� production.
Determination of CFU
Peritoneal lavages fluid and EDTA-treated blood from 72 h post-CLP wereplaced on ice and serially diluted in sterile saline. A 10-�l aliquot of each
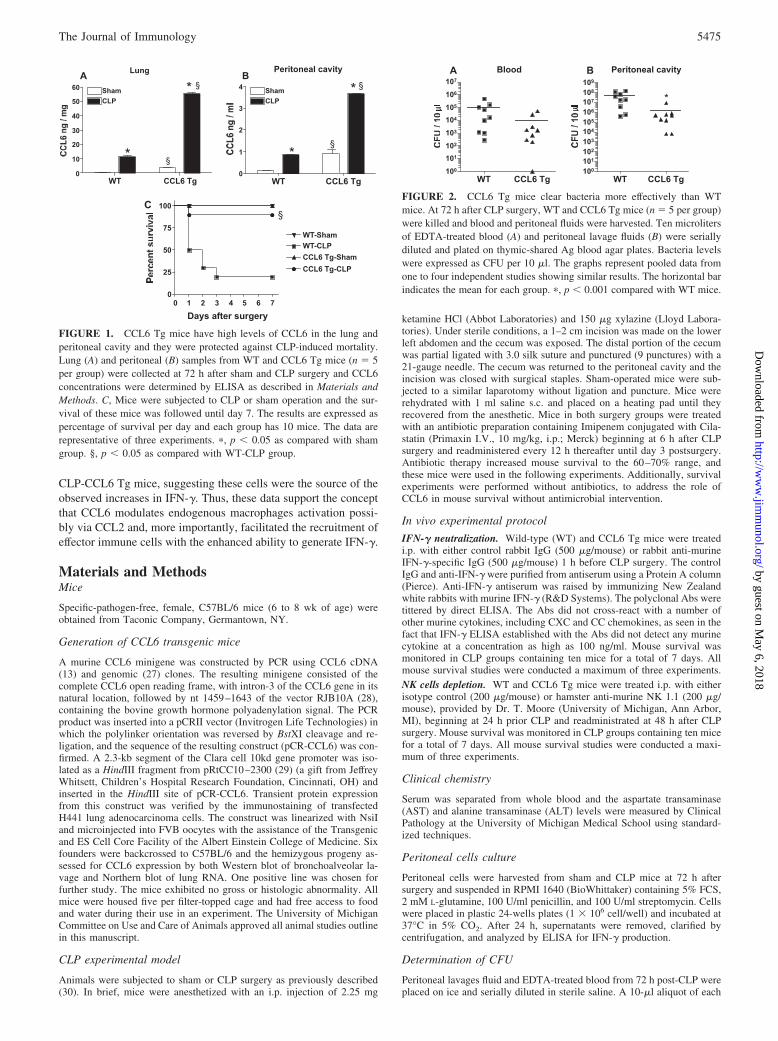
FIGURE 1. CCL6 Tg mice have high levels of CCL6 in the lung andperitoneal cavity and they were protected against CLP-induced mortality.Lung (A) and peritoneal (B) samples from WT and CCL6 Tg mice (n � 5per group) were collected at 72 h after sham and CLP surgery and CCL6concentrations were determined by ELISA as described in Materials andMethods. C, Mice were subjected to CLP or sham operation and the sur-vival of these mice was followed until day 7. The results are expressed aspercentage of survival per day and each group has 10 mice. The data arerepresentative of three experiments. �, p � 0.05 as compared with shamgroup. §, p � 0.05 as compared with WT-CLP group.
FIGURE 2. CCL6 Tg mice clear bacteria more effectively than WTmice. At 72 h after CLP surgery, WT and CCL6 Tg mice (n � 5 per group)were killed and blood and peritoneal fluids were harvested. Ten microlitersof EDTA-treated blood (A) and peritoneal lavage fluids (B) were seriallydiluted and plated on thymic-shared Ag blood agar plates. Bacteria levelswere expressed as CFU per 10 �l. The graphs represent pooled data fromone to four independent studies showing similar results. The horizontal barindicates the mean for each group. �, p � 0.001 compared with WT mice.
5475The Journal of Immunology
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
dilution was spread on thymic-shared Ag agar plates (BD Diagnostic Sys-tems) and incubated at 37°C overnight. Colonies were counted and ex-pressed as CFU/10 �l. Groups contained four to six mice and the experi-ments were repeated on three different occasions. Results were similar foreach experiment and subsequent pooled.
Measurement of cytokines and chemokines by ELISA
Concentrations of murine IFN-�, IL-12 (p70), IL-10, IL-13, TNF-�,CCL2, CCL3, CCL5, and CCL17 were measured in cell-free peritoneallavage fluid and cell culture supernatants using a standardized sandwichELISA previously described (31). In brief, 96-well microtiter plates(Nunc) were coated overnight at 4°C with mAb specific for the murinecytokine and chemokine being measured (R&D System). Wells werewashed with PBS plus 0.05% Tween 20 and nonspecific sites wereblocked with 2% BSA in PBS for 90 min at 37°C. Plates were washedand samples were loaded and incubated at 37°C for 1 h. After washing,a secondary, biotinylated, cytokine/chemokine-specific polyclonal Ab(R&D Systems) was added for 30 min at 37°C. Plates were washedagain and peroxidase-conjugated streptavidin (Bio-Rad) was added.Plates were washed and a chromogenic substrate (Bio-Rad) was addedand incubated at room temperature until fully developed. Reactionswere stopped and read at 490 nm in an ELISA plate reader. Recombi-
nant murine cytokines/chemokines (R&D System) were used to gener-ate standard curves and concentrations were expressed as ng or pg/ml.Experimental groups consisted of triplicate samples in vitro from threeindependent experiments.
Flow cytometry analysis
Peritoneal cells were harvested from sham and CLP groups 72 h aftersurgery. RBC were lysed in lysis buffer and the remaining cells werewashed in PBS and resuspended in PBS containing 1% BSA. Cells wereFc blocked and stained with various Abs. For cells staining, we usedAbs labeled with PE, FITC, PeCy5, or PeCy7 direct against the fol-lowing: CD11b, MHCII, CD19, IgM, CD3, CD4, CD8, NK 1.1 (all fromBD Pharmingen), CD49b (eBioscience), or F4/80 (abCam). For intra-cellular staining, peritoneal cells were collected and placed in plastic24-wells plates (2 � 106 cell/well) and incubated at 37°C in 5% CO2
with protein transporter inhibitor Golgi Plug (BD Pharmingen) to pre-vent the release of IFN-�. After 6 h of incubation, cells were transferredto 5 ml tubes and staining first for the surface markers described aboveand then for IFN-� intracellular (BD Pharmingen). Cells were analyzedwith a Beckman Coulter Cytomics FC500.
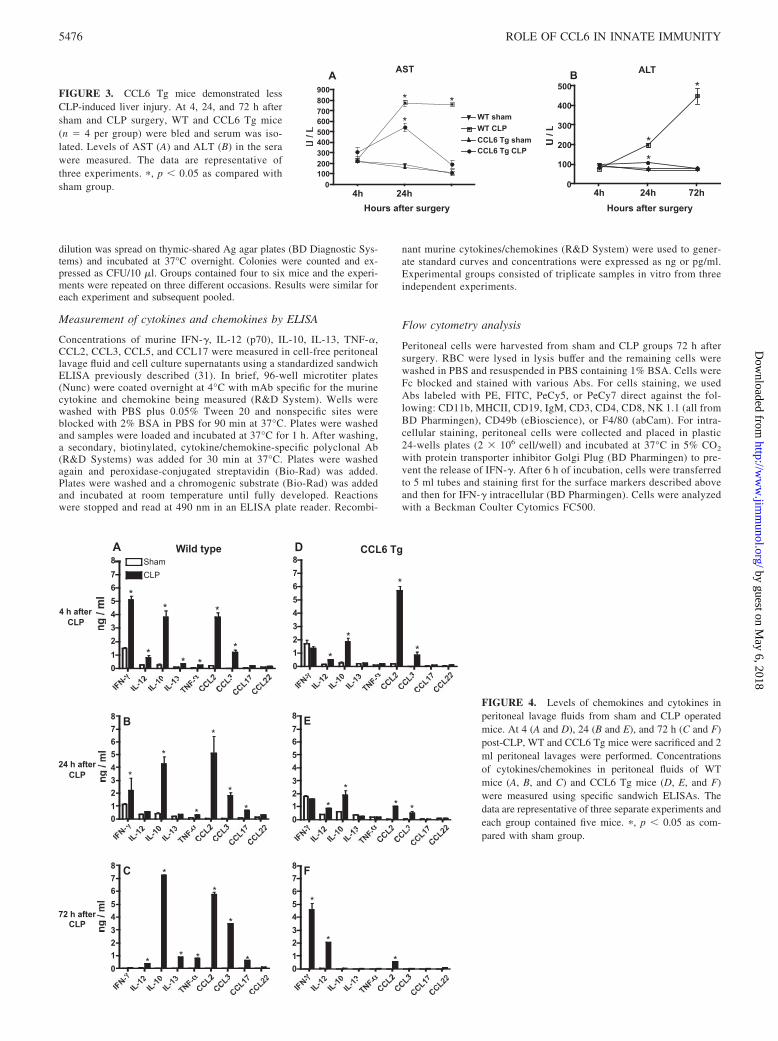
FIGURE 3. CCL6 Tg mice demonstrated lessCLP-induced liver injury. At 4, 24, and 72 h aftersham and CLP surgery, WT and CCL6 Tg mice(n � 4 per group) were bled and serum was iso-lated. Levels of AST (A) and ALT (B) in the serawere measured. The data are representative ofthree experiments. �, p � 0.05 as compared withsham group.
FIGURE 4. Levels of chemokines and cytokines inperitoneal lavage fluids from sham and CLP operatedmice. At 4 (A and D), 24 (B and E), and 72 h (C and F)post-CLP, WT and CCL6 Tg mice were sacrificed and 2ml peritoneal lavages were performed. Concentrationsof cytokines/chemokines in peritoneal fluids of WTmice (A, B, and C) and CCL6 Tg mice (D, E, and F)were measured using specific sandwich ELISAs. Thedata are representative of three separate experiments andeach group contained five mice. �, p � 0.05 as com-pared with sham group.
5476 ROLE OF CCL6 IN INNATE IMMUNITY
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Statistical analysis
All data are shown are means � SE and are representative of three separateexperiments. The means between different treatments were compared byANOVA. When significance was detected, individual differences were an-alyzed using the Bonferroni’s t test for unpaired values. Statistical signif-icance was set at p � 0.05. Survival rates were expressed as percentages,and a log rank test (�2 test) was used to detect differences in mousesurvival.
ResultsExpression and biologic activity of CCL6 during the evolutionof experimental sepsis
CLP-induced sepsis exhibits many of the features of clinical sepsisincluding SIRS and CARS. We previously demonstrated that ex-ogenous CCL6 exerted a protective effect in CLP-induced sepsis,but the mechanism responsible for this protective effect was un-clear. In an attempt to understand the protective role of this che-mokine, CCL6 Tg and WT mice were subjected to experimentalperitonitis. Fig. 1, A and B shows that the baseline and CLP-in-duced CCL6 levels were significantly higher in the CCL6 Tggroup compared with the WT group. At baseline these values were7-fold higher in the lung (Fig. 1A) and 9-fold higher in the peri-toneal cavity (PC) (Fig. 1B). After CLP, these values were 6-foldhigher in the lung (Fig. 1A) and 4-fold higher in the PC (Fig. 1B).To investigate the susceptibility of CCL6 Tg mice to CLP, WT,and CCL6 Tg mice were submitted to sham and CLP withoutantibiotic treatment. Fig. 1C shows mouse survival following shamand CLP. Ninety percent of the CCL6 Tg group was alive at day7 after CLP, whereas only 20% of the WT group was alive at thesame time. Together, these data suggest that increased levels ofCCL6 dramatically protected mice from severe sepsis.
CCL6 Tg mice had enhanced bacterial clearance and reducedbacteremia after CLP
To determine whether the improved survival of CCL6 Tg micewas attributable to improved bacterial clearance, peritoneal, andblood samples were collected to determine local and systemic bac-terial loads, respectively (Fig. 2). At 72 h post-CLP, the bacteria
CFU cultured from plasma (Fig. 2A) and peritoneal wash samples(Fig. 2B) from CCL6 Tg mice were lower compared with WTmice. These results were statistically significant for the PC samples( p � 0.05) but not for the blood samples despite a log differencein the means of these samples.
CCL6 Tg mice have less liver injury induced by CLP
One of the major complications of sepsis is multiple organ failure,which often leads to death (1). To determine the ability of CCL6to prevent CLP-induced liver injury, the levels of liver enzymes inthe serum of sham- and CLP-operated mice were measured in WTand CCL6 Tg groups (Fig. 3). The AST (Fig. 3A) and ALT (Fig.3B) levels of both groups were similar in both sham- and CLP-operated mice at 4 h after surgery. At 24 h post-CLP, the serum ofboth CCL6 Tg and WT mice showed significantly higher levels ofALT and AST compared with sham-operated mice levels, butCLP-CCL6 Tg mice exhibited lower AST and ALT levels (550 �70 and 110 � 10, respectively) than CLP-WT mice (780 � 50 and200 � 32, respectively). At 72 h post-CLP, both AST and ALTreturned to baseline levels in CLP-CCL6 Tg mice, whereas these
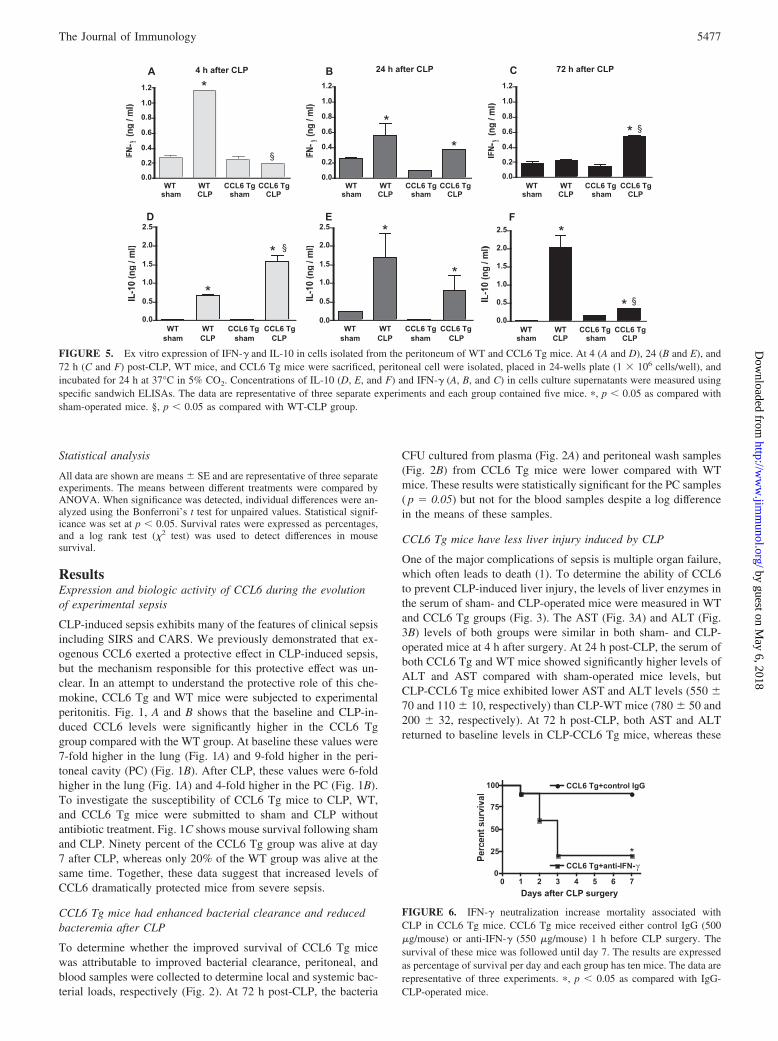
FIGURE 5. Ex vitro expression of IFN-� and IL-10 in cells isolated from the peritoneum of WT and CCL6 Tg mice. At 4 (A and D), 24 (B and E), and72 h (C and F) post-CLP, WT mice, and CCL6 Tg mice were sacrificed, peritoneal cell were isolated, placed in 24-wells plate (1 � 106 cells/well), andincubated for 24 h at 37°C in 5% CO2. Concentrations of IL-10 (D, E, and F) and IFN-� (A, B, and C) in cells culture supernatants were measured usingspecific sandwich ELISAs. The data are representative of three separate experiments and each group contained five mice. �, p � 0.05 as compared withsham-operated mice. §, p � 0.05 as compared with WT-CLP group.
0 1 2 3 4 5 6 70
25
50
75
100
CCL6 Tg+anti-IFN-γ
CCL6 Tg+control IgG
Days after CLP surgery
*
FIGURE 6. IFN-� neutralization increase mortality associated withCLP in CCL6 Tg mice. CCL6 Tg mice received either control IgG (500�g/mouse) or anti-IFN-� (550 �g/mouse) 1 h before CLP surgery. Thesurvival of these mice was followed until day 7. The results are expressedas percentage of survival per day and each group has ten mice. The data arerepresentative of three experiments. �, p � 0.05 as compared with IgG-CLP-operated mice.
5477The Journal of Immunology
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
levels remained elevated in CLP-WT mice. Thus, these data sug-gest that the CCL6 Tg mice recovered faster from the liver injurycaused by CLP.
Time point differences in cytokine and chemokine levels in theperitoneal cavity of WT and CCL6 Tg mice: diminished earlycytokine storm in CCL6 Tg group
Previous studies have shown that the local and systemic levels ofmany cytokines and chemokines are markedly increased, follow-ing CLP surgery (32, 33). To establish the cytokine profile inCCL6 Tg mice, PC fluid from both sham and CLP groups wascollected and analyzed in ELISA (Fig. 4). At 4 h after surgery,higher levels of IFN-� and IL-10 were detected in CLP-WT micecompared with CLP-CCL6 Tg mice (Fig. 4, A and D). At this sametime, only CCL2 levels were significantly increased in the CLP-CCL6 Tg group when compared with WT mice (Fig. 4D). Levelsof CCL3, CCL17, CCL2 and IL-10 were elevated in CLP-WTgroup when compared with CLP-CCL6 Tg group at 24 h post-CLP(Fig. 4, B and E). CCL2, CCL3, CCL17, and particularly, IL-10levels were markedly higher in CLP-WT mice at 72 h after CLP(Fig. 4C). In contrast, at 72 h post-CLP, CLP-CCL6 Tg mice ex-hibited significantly higher levels of IFN-� (0.459 ng/ml vs 0.055ng/ml) and IL-12 (0.225 ng/ml vs 0.054 ng/ml) compared withCLP-WT group at this time after CLP (Fig. 4, C and F).
Cells from the peritoneal cavity of CCL6 Tg mice produce highlevels of IFN-�
Peritoneal cells from WT and CCL6 Tg mice harvested at 4, 24,and 72 h post-sham and post-CLP surgery were cultured in vitro for
24 h and IFN-� and IL-10 levels were measured by ELISA. Con-firming the trends showed in Fig. 4, cells isolated from the peri-toneal cavity of CLP-WT mice generated higher levels of IFN-�
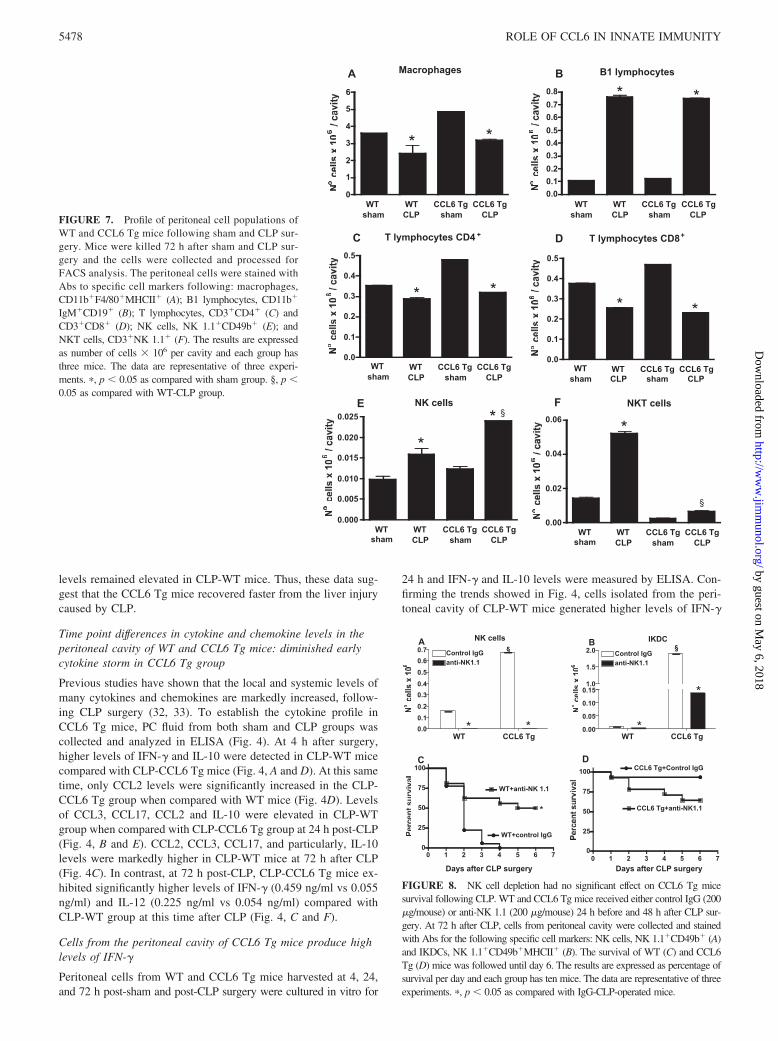
FIGURE 7. Profile of peritoneal cell populations ofWT and CCL6 Tg mice following sham and CLP sur-gery. Mice were killed 72 h after sham and CLP sur-gery and the cells were collected and processed forFACS analysis. The peritoneal cells were stained withAbs to specific cell markers following: macrophages,CD11b�F4/80�MHCII� (A); B1 lymphocytes, CD11b�
IgM�CD19� (B); T lymphocytes, CD3�CD4� (C) andCD3�CD8� (D); NK cells, NK 1.1�CD49b� (E); andNKT cells, CD3�NK 1.1� (F). The results are expressedas number of cells � 106 per cavity and each group hasthree mice. The data are representative of three experi-ments. �, p � 0.05 as compared with sham group. §, p �0.05 as compared with WT-CLP group.
FIGURE 8. NK cell depletion had no significant effect on CCL6 Tg micesurvival following CLP. WT and CCL6 Tg mice received either control IgG (200�g/mouse) or anti-NK 1.1 (200 �g/mouse) 24 h before and 48 h after CLP sur-gery. At 72 h after CLP, cells from peritoneal cavity were collected and stainedwith Abs for the following specific cell markers: NK cells, NK 1.1�CD49b� (A)and IKDCs, NK 1.1�CD49b�MHCII� (B). The survival of WT (C) and CCL6Tg (D) mice was followed until day 6. The results are expressed as percentage ofsurvival per day and each group has ten mice. The data are representative of threeexperiments. �, p � 0.05 as compared with IgG-CLP-operated mice.
5478 ROLE OF CCL6 IN INNATE IMMUNITY
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
4 h after surgery compared with cells from CLP-CCL6 Tg mice(Fig. 5A). However, at later time points IFN-� levels in cells fromCLP-WT mice decreased until 72 h when they were comparable tovalues expressed in cells from sham-WT mice. In contrast, IFN-�expression in cells from CLP-CCL6 Tg mice increased with timeand was significantly higher than in all other experimental groupsat 72 h (Fig. 5, A–C). In contrast, IL-10 expression showed areverse trend (Fig. 5, D–F): In cells from WT-CLP mice, IL-10significantly increased as a function of time between 4 and 72 hwhile significantly decreasing in CLP-CCL6 Tg mice. These datasuggested that IFN-� and IL-10 were expressed in a manner and atlevels conducive for clearance and survival from SIRS.
Neutralization of IFN-� caused CLP-induced lethality in CCL6Tg mice
In an attempt to determine whether the delayed increase in IFN-�levels in the peritoneal cavity of CCL6 Tg mice contributed to aprotective role of CCL6 against sepsis-induced mortality, CCL6Tg mice were pretreated with control rabbit IgG or affinity-purifiedrabbit IgG specific for murine IFN-� 1 h before CLP surgery (Fig.6). When CCL6 Tg mice received anti-IFN-� before CLP, only20% of these mice were alive at day 7 post-CLP compared withmice pretreated with control IgG (90% of survival). These datasuggest a critical role for IFN-� in the protective effect observed inCCL6 Tg mice in our model of severe sepsis.
Peritoneal cell population in WT and CCL6 Tg mice at 72 hafter CLP surgery
To determine the type of cell contributing to increased IFN-�during sepsis, peritoneal cells were isolated 72 h postsurgery fromWT and CCL6 Tg mice subjected to either sham or CLP surgeryand analyzed by flow cytometer. Cells from the peritoneal cavityof sham and CLP mice were stained with Abs to specific mark-ers for macrophages (CD11b�F4/80�MHCII�), B1 lympho-cytes (CD11b�IgM�CD19�), T lymphocytes (CD3�CD4� andCD3�CD8�), NK cells (NK1.1�CD49b�), and NKT cells(CD3�NK1.1�) (Fig. 7). There was no significant difference inthe numbers of macrophages, B1 or T cells in WT and CCL6 Tgmice after CLP surgery (Fig. 7, A–D). In contrast, there was asignificant increase in the number of NK cells in CLP-CCL6 Tgmice compared with CLP-WT mice (Fig. 7E). Additionally, thenumber of NKT cells was significantly lower in both sham- andCLP-operated CCL6 Tg mice compared with WT mice (Fig. 7F).
Depletion of NK cells does not alter CCL6 Tg mice survival
Activated NK cells are a major source of IFN-� during sepsis andare therefore involved in the pathogenesis of sepsis (34, 35). Todetermine whether NK cells were involved in the immunomodu-latory role of CCL6 during severe sepsis, NK cells were depletedbefore CLP surgery. WT and CCL6 Tg mice received control IgGand anti-NK 1.1 Abs 24 h before and 48 h after CLP surgery. Fig.8A shows that the treatment with anti-NK1.1 before CLP com-pletely depleted NK cells in both groups while 97% and 92% ofIKDCs were depleted in WT and CCL6 Tg mice, respectively (Fig.8B). In WT mice (Fig. 8C), the NK cell depletion resulted in sig-nificant protection against CLP, while in CCL6 Tg mice (Fig. 8D)the NK depletion reduced survival, however these differences werenot statistically significant.
IKDC are increased in CLP-CCL6 Tg mice
NK cells and dendritic cells (DC) are central components of innateand adaptive immune responses. NK cells are believed to be themajor producers of IFN-�, which is secreted in response to IL-12(36). Upon stimulation, DCs are also able to produce cytokines,such as IL-12 and TNF-�. Recently, DCs have also been shown tosecrete IFN-� in response to IL-12 alone or in combination withIL-18 (36, 37). Recent studies have characterized a new subset ofDC that share phenotypic and functional properties of both NKcells and DCs. These cells express both NK cell (NK 1.1, CD49b)
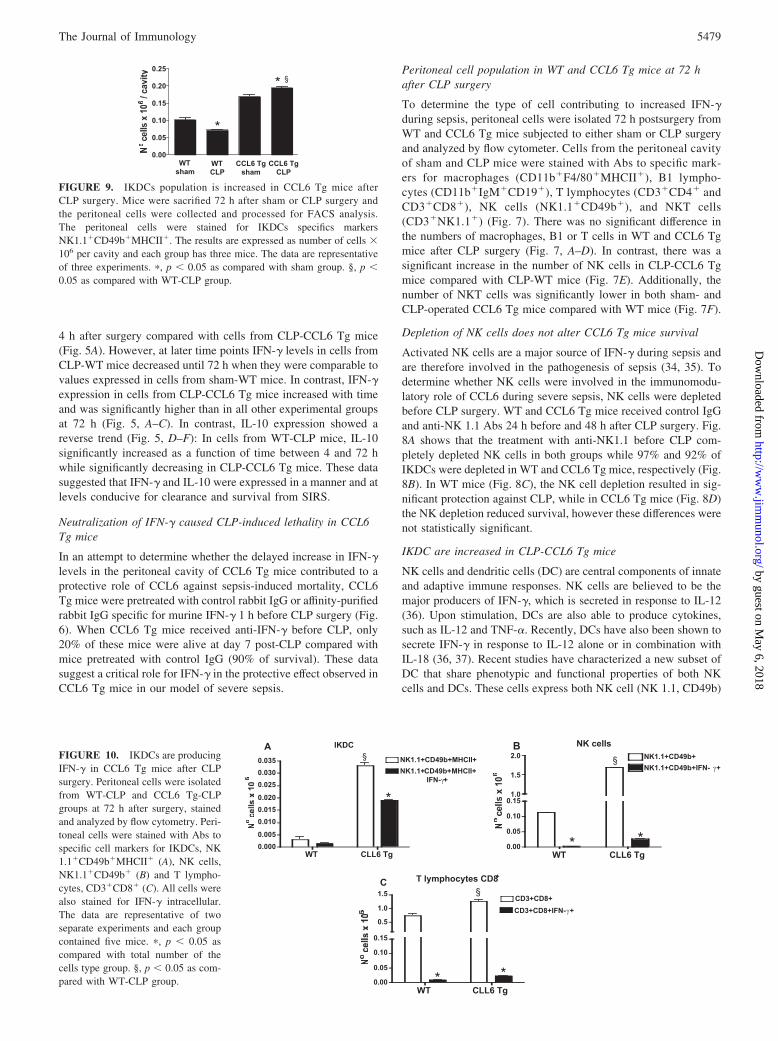
FIGURE 9. IKDCs population is increased in CCL6 Tg mice afterCLP surgery. Mice were sacrified 72 h after sham or CLP surgery andthe peritoneal cells were collected and processed for FACS analysis.The peritoneal cells were stained for IKDCs specifics markersNK1.1�CD49b�MHCII�. The results are expressed as number of cells �106 per cavity and each group has three mice. The data are representativeof three experiments. �, p � 0.05 as compared with sham group. §, p �0.05 as compared with WT-CLP group.
FIGURE 10. IKDCs are producingIFN-� in CCL6 Tg mice after CLPsurgery. Peritoneal cells were isolatedfrom WT-CLP and CCL6 Tg-CLPgroups at 72 h after surgery, stainedand analyzed by flow cytometry. Peri-toneal cells were stained with Abs tospecific cell markers for IKDCs, NK1.1�CD49b�MHCII� (A), NK cells,NK1.1�CD49b� (B) and T lympho-cytes, CD3�CD8� (C). All cells werealso stained for IFN-� intracellular.The data are representative of twoseparate experiments and each groupcontained five mice. �, p � 0.05 ascompared with total number of thecells type group. §, p � 0.05 as com-pared with WT-CLP group.
5479The Journal of Immunology
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

and DC (CD11c, MHC class II (MHCII)) surface markers andproduce significant amounts of IFN-� and IL-12 in response toCpG (38). To determine whether IKDCs were present in theperitoneal cavity of WT and CCL6 Tg sham- and CLP-operatedmice, cells from the peritoneal cavity of sham and CLP micewere stained for NK1.1, CD49b, and MHCII markers and an-alyzed by flow cytometry. The number of IKDCs in CCL6 Tgmice was higher than in WT mice regardless of the surgicalintervention (sham and CLP) (Fig. 9). A 3-fold increase in thenumber of IKDCs was seen when CLP-CCL6 Tg mice werecompared with CLP-WT mice. These data suggest that the pro-tective role of CCL6 during severe sepsis may be related to theeffect of this chemokine on IKDC recruitment into the perito-neal cavity after CLP surgery.
IKDCs are the main cell type producing IFN-� in CCL6 Tgmice
To investigate whether the IKDCs were producing IFN-�, perito-neal cells from WT and CCL6 Tg mice were harvested 72 h afterCLP, stained for NK 1.1, CD49b, and MHCII markers and intra-cellular IFN-� and then analyzed by flow cytometer. Fig. 10Ashows that 63.3% of total IKDCs (NK1.1�CD49b�MHCII�)were positive for IFN-� in CCL6 Tg mice. To investigate whetherother cell types were producing IFN-�, peritoneal cells were alsostained for NK1.1/CD49b (NK cells) and CD3/CD8 (T lympho-cytes) and IFN-�. Only 1.6% of total NK cells and 1.8% of totalCD8� T lymphocytes were positive for IFN-� in CCL6 Tg mice(Fig. 10, B and C). Other cell types such as macrophages, B1 cells,and T lymphocytes CD4� were negative for IFN-� in WT andCCL6 Tg mice at 72 h after CLP (data not shown). These datashow that IKDCs are the key cells producing IFN-� in CCL6 Tgmice at 72 h post-CLP surgery.
DiscussionDysregulation of the immune system occurs during severe sepsis,leading to a rapid death due to the development of multiorganfailure and an increase in complications due to long-term immu-nosupression (39–41). CCL6, a CC chemokine, is present in avariety of inflammatory and remodeling disorders (11, 14, 16, 18,24). We previously showed that the immunoneutralization ofCCL6 enhanced the sepsis-related mortality. In contrast, the ad-ministration of recombinant CCL6 to mice immediately after CLPsurgery significantly improved the survival of these mice (18). Inthis study, we show that mice overexpressing CCL6 are protectedfrom mortality following CLP. In our model of severe sepsis (withno antibiotic treatment), these mice presented an impressive 90%survival rate vs a 20% survival in WT mice. The CCL6 Tg miceexhibited higher levels of CCL6, in the lung and PC, at 24 h post-CLP (data not shown) and the levels remained elevated even at72 h after CLP as shown in Fig. 1. These data agree with otherstudies demonstrating that the production of CCL6 remains ele-vated several days after septic peritonitis (23, 24). In our model,this sustained increase of CCL6 levels seemed to be important inregulating the deleterious effects induced by sepsis, because, inaddition to improved survival, the CLP-CCL6 mice present animproved recovery from liver injury compared with CLP-WTmice. This protective effect is likely caused by an increase inCCL6-mediated cytokine cascades, which provides an apparentcytokine environment for both microorganism clearance and reg-ulate the local inflammatory response.
Previous studies demonstrated that CCL2 has a protective roleduring septic peritonitis (32, 42). Mice treated with CCL2 wereprotected against a lethal dose of LPS or bacteria, and the mech-anism of protection seems to be by way of increased IL-10 pro-
duction concomitant with a decrease of IL-12 and TNF production(43). Several studies demonstrated that CCL2 is produced early inresponse to endotoxin challenge, returning to baselines levels after48 h (18, 32, 42). In our model, CCL2 peaked at 4 h post-CLP inCCL6 Tg mice, which was coincident with low levels of IL-12 andIFN-�. However, at 24 and 72 h, the CCL6 Tg mice presented verylow levels of CCL2. Previous studies demonstrated that CCL6neutralization decreased CCL2 production in the lung (16, 20).However, previous studies showed that the neutralization of CCL6increased CCL2 levels in their model of acute peritonitis (23). InCLP-CCL6 Tg mice, CCL6 levels were elevated at 24 and 72 hpost-CLP and very low levels of CCL2 were found at these timepoints, suggesting that CCL6 may inhibit CCL2 production. Pre-vious studies demonstrated that CCL2 increases the production ofIL-10 in experimental models of sepsis (42, 43). IL-10 has beenidentified as a crucial modulator of the inflammatory response insepsis and it is an important cytokine mediator of sepsis-inducedimmunosupression (44). Prolonged immune suppression is char-acterized by defects in Ag presentation, macrophage paralysis, Tcell anergy, suppressed T cell proliferation, and increased T celland B cell apoptosis, which may be partially attributable to thebiological effects of IL-10. Indeed, in our model, the high andsustained levels of CCL2 and IL-10 in CLP-WT mice correlatedwith poor survival rates. Interestingly, we observed a gradual in-crease of IFN-� and IL-12 in CLP-CCL6 Tg mice with time post-CLP. IFN-� strongly stimulates monocytes/macrophages, increas-ing their microbicidal activity, Ag presentation function, andproduction of proinflammatory cytokines on contact with micro-bial stimuli. Genetics defects in the IFN-� receptor system havebeen described in patients with vaccine-associated bacterial infec-tions, demonstrating the importance of IFN-�-mediated immunityin human host defense against intracellular pathogens (45, 46). Inaddition, the important role of IFN-� in the pathogenesis of LPS-induced shock was confirmed using mice deficient for the IFN-�receptor (47, 48). In this study, we show the crucial role of IFN-�in CCL6-Tg mice during severe sepsis. These mice expressed sig-nificantly elevated levels of IFN-� at 72 h after CLP surgery and90% survival was observed. This increase in IFN-� was importantfor the survival of these mice because, 80% of CCL6 Tg mice diedfollowing anti- IFN-� Ab treatment. In contrast, the neutralizationof IFN-� protected WT mice from sepsis-induced mortality sug-gesting that the rapid increase of IFN-� levels observed at 4 h afterCLP can be deleterious and increase the mortality of these mice(data not shown).
IFN-� is produced primarily by NK cells and a certain subpopu-lation of T lymphocytes (49). In this study, we demonstrated thata NK cell population is increased in CLP-CCL6 Tg mice comparedwith CLP-WT mice. To determine whether NK cells were playinga role in the protective role of CCL6 during severe sepsis, wedepleted NK cells in WT and CCL6 Tg mice. NK cells did notappear to provide a protective role, because we observed only aslight decrease (not statistically significant) in the survival rate ofCCL6 Tg mice. However, the treatment with anti-NK1.1 Ab didnot completely deplete IKDCs population, suggesting that there isstill a small population of IKDCs that may be producing IFN-�.This potentially could explain why there is not a significant dif-ference in the CLP-CCL6 Tg survival rate. In contrast, the treat-ment with anti-NK Ab was protective for WT mice against sepsis-induced mortality. Some studies demonstrated a detrimental rolefor NK cells in experimental models of sepsis (50, 51). ActivatedNK cells are a main source of IFN-� during sepsis and thereforelikely are involved in the pathogenesis of sepsis (52). Indeed, de-pletion of NK cells in septic mice offers protection against cyto-kine- and LPS-induced shock (34, 52).
5480 ROLE OF CCL6 IN INNATE IMMUNITY
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

IKDCs are a new population of murine immune cells that aredistinct from conventional DCs and plasmacytoid DCs becausethey express both NK cell (NK 1.1, CD49b) and DC (CD11c,B220, MHCII) markers (38, 53). It was demonstrated that IKDCsare present in the spleen, lymph node, thymus, and liver of normalmice. Upon activation with CpG, IKDCs secrete high levels ofIFN-� and this secretion depends on autocrine IL-12 (38). Also,these cells are able to directly lyse tumor cells, and present Ag tonaive T cells. In this study, we show that IKDCs were also presentin the peritoneal cavity of both WT and CCL6 Tg mice. CCL6 Tgmice submitted to sham and CLP surgery had higher numbers ofIKDCs in the peritoneal cavity when compared with CLP-WTmice. These data suggest that IKDCs may function in a protectiverole in the CCL6 Tg mice and alter sepsis-induced mortality. Asignificant increase in total numbers and percent of IFN-� positiveIKDC were identified in CCL6 Tg mice post CLP, as comparedwith WT mice. This finding could aid in explaining the increase inIFN-� associated with the CCL6 Tg mice.
Septic mortality is often depicted as being fueled by an imbal-ance in the normal proinflammatory and immunosuppressive ho-meostasis and in severe sepsis this imbalance is exemplified bySIRS and CARS. The present study demonstrated that transgenicCCL6 reduced mortality in a septic peritonitis model, via a mech-anism that involved the recruitment and activation of IFN-� pro-ducing killer DC into the peritoneum. In addition, CCL6 Tg miceexpressed elevated levels of CCL2 early during the septic responseand this chemokine may aid in preventing SIRS and enhancingsurvival. At later time points, when CARS is thought to exert animmunosuppressive environment, the increased levels of IFN-�may have countered the severe immunosuppression and enhancedsurvival in the CCL6 Tg CLP mice. Our studies demonstrate theimportance of CCL6 in attenuating immune failure during severesepsis and future studies will continue to delve into the mecha-nisms and pathways underlying these protective events.
AcknowledgmentsWe thank Dr. Robin Kunkel for her assistance in image processing. Wethank Dr. Judith Connett (Universtity of Michigan, Ann Arbor, MI) for thecritical reading of the manuscript. We thank Holly Evanoff and Pam Lin-coln for their technical assistance.
DisclosuresThe authors have no financial conflict of interest.
References1. Bone, R. C., R. A. Balk, F. B. Cerra, R. P. Dellinger, A. M. Fein, W. A. Knaus,
R. M. Schein, and W. J. Sibbald. 1992. Definitions for sepsis and organ failureand guidelines for the use of innovative therapies in sepsis. The ACCP/SCCMConsensus Conference Committee: American College of Chest Physicians/Soci-ety of Critical Care Medicine. Chest 101: 1644–1655.
2. Bone, R. C., C. J. Grodzin, and R. A. Balk. 1997. Sepsis: a new hypothesis forpathogenesis of the disease process. Chest 112: 235–243.
3. Rossi, D., and A. Zlotnik. 2000. The biology of chemokines and their receptors.Annu. Rev. Immunol. 18: 217–242.
4. D’Ambrosio, D., P. Panina-Bordignon, and F. Sinigaglia. 2003. Chemokine re-ceptors in inflammation: an overview. J. Immunol. Methods 273: 3–13.
5. Rosenkilde, M. M., and T. W. Schwartz. 2004. The chemokine system: a majorregulator of angiogenesis in health and disease. APMIS 112: 481–495.
6. Bossink, A. W., L. Paemen, P. M. Jansen, C. E. Hack, L. G. Thijs, andJ. Van Damme. 1995. Plasma levels of the chemokines monocyte chemotacticproteins-1 and -2 are elevated in human sepsis. Blood 86: 3841–3847.
7. Botha, A. J., F. A. Moore, E. E. Moore, V. M. Peterson, C. C. Silliman, andA. W. Goode. 1996. Sequential systemic platelet-activating factor and interleukin8 primes neutrophils in patients with trauma at risk of multiple organ failure.Br. J. Surg. 83: 1407–1412.
8. Donnelly, S. C., R. M. Strieter, S. L. Kunkel, A. Walz, C. R. Robertson,D. C. Carter, I. S. Grant, A. J. Pollok, and C. Haslett. 1993. Interleukin-8 anddevelopment of adult respiratory distress syndrome in at-risk patient groups.Lancet 341: 643–647.
9. Donnelly, S. C., R. M. Strieter, S. L. Kunkel, A. Walz, D. Steedman, I. S. Grant,A. J. Pollok, D. C. Carter, and C. Haslett. 1994. Chemotactic cytokines in the
established adult respiratory distress syndrome and at-risk patients. Chest 105:98S–99S.
10. Orlofsky, A., M. S. Berger, and M. B. Prystowsky. 1991. Novel expression pat-tern of a new member of the MIP-1 family of cytokine-like genes. Cell Regul. 2:403–412.
11. Asensio, V. C., S. Lassmann, A. Pagenstecher, S. C. Steffensen, S. J. Henriksen,and I. L. Campbell. 1999. C10 is a novel chemokine expressed in experimentalinflammatory demyelinating disorders that promotes recruitment of macrophagesto the central nervous system. Am. J. Pathol. 154: 1181–1191.
12. Berger, M. S., D. D. Taub, A. Orlofsky, T. R. Kleyman, B. Coupaye-Gerard,D. Eisner, and S. A. Cohen. 1996. The chemokine C10: immunological andfunctional analysis of the sequence encoded by the novel second exon. Cytokine8: 439–447.
13. Orlofsky, A., E. Y. Lin, and M. B. Prystowsky. 1994. Selective induction of the� chemokine C10 by IL-4 in mouse macrophages. J. Immunol. 152: 5084–5091.
14. Belperio, J. A., M. Dy, M. D. Burdick, Y. Y. Xue, K. Li, J. A. Elias, andM. P. Keane. 2002. Interaction of IL-13 and C10 in the pathogenesis of bleo-mycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 27: 419–427.
15. Hara, T., K. B. Bacon, L. C. Cho, A. Yoshimura, Y. Morikawa, N. G. Copeland,D. J. Gilbert, N. A. Jenkins, T. J. Schall, and A. Miyajima. 1995. Molecularcloning and functional characterization of a novel member of the C-C chemokinefamily. J. Immunol. 155: 5352–5358.
16. Hogaboam, C. M., C. S. Gallinat, D. D. Taub, R. M. Strieter, S. L. Kunkel, andN. W. Lukacs. 1999. Immunomodulatory role of C10 chemokine in a murinemodel of allergic bronchopulmonary aspergillosis. J. Immunol. 162: 6071–6079.
17. Pardigol, A., U. Forssmann, H. D. Zucht, P. Loetscher, P. Schulz-Knappe,M. Baggiolini, W. G. Forssmann, and H. J. Magert. 1998. HCC-2, a humanchemokine: gene structure, expression pattern, and biological activity. Proc. Natl.Acad. Sci. USA 95: 6308–6313.
18. Steinhauser, M. L., C. M. Hogaboam, A. Matsukawa, N. W. Lukacs,R. M. Strieter, and S. L. Kunkel. 2000. Chemokine C10 promotes disease reso-lution and survival in an experimental model of bacterial sepsis. Infect. Immun.68: 6108–6114.
19. Wang, W., K. B. Bacon, E. R. Oldham, and T. J. Schall. 1998. Molecular cloningand functional characterization of human MIP-1 �, a new C-C chemokine relatedto mouse CCF-18 and C10. J Clin. Immunol. 18: 214–222.
20. Ma, B., Z. Zhu, R. J. Homer, C. Gerard, R. Strieter, and J. A. Elias. 2004. TheC10/CCL6 chemokine and CCR1 play critical roles in the pathogenesis of IL-13-induced inflammation and remodeling. J. Immunol. 172: 1872–1881.
21. Kaesler, S., J. Regenbogen, S. Durka, A. Goppelt, and S. Werner. 2002. Thehealing skin wound: a novel site of action of the chemokine C10. Cytokine 17:157–163.
22. Kanno, M., S. Suzuki, T. Fujiwara, A. Yokoyama, A. Sakamoto, H. Takahashi,Y. Imai, and J. Tanaka. 2005. Functional expression of CCL6 by rat microglia:a possible role of CCL6 in cell-cell communication. J. Neuroimmunol. 167:72–80.
23. LaFleur, A. M., N. W. Lukacs, S. L. Kunkel, and A. Matsukawa. 2004. Role ofCC chemokine CCL6/C10 as a monocyte chemoattractant in a murine acuteperitonitis. Mediators Inflamm. 13: 349–355.
24. Wu, Y., M. B. Prystowsky, and A. Orlofsky. 1999. Sustained high-level produc-tion of murine chemokine C10 during chronic inflammation. Cytokine 11:523–530.
25. Ness, T. L., K. J. Carpenter, J. L. Ewing, C. J. Gerard, C. M. Hogaboam, andS. L. Kunkel. 2004. CCR1 and CC chemokine ligand 5 interactions exacerbateinnate immune responses during sepsis. J. Immunol. 173: 6938–6948.
26. Walley, K. R., N. W. Lukacs, T. J. Standiford, R. M. Strieter, and S. L. Kunkel.1997. Elevated levels of macrophage inflammatory protein 2 in severe murineperitonitis increase neutrophil recruitment and mortality. Infect. Immun. 65:3847–3851.
27. Berger, M. S., C. A. Kozak, A. Gabriel, and M. B. Prystowsky. 1993. The genefor C10, a member of the �-chemokine family, is located on mouse chromosome11 and contains a novel second exon not found in other chemokines. DNA CellBiol. 12: 839–847.
28. Caltabiano, M. M., M. L. Tsang, J. A. Weatherbee, R. Lucas, G. Sathe, J. Sutton,G. D. Johnson, and D. J. Bergsma. 1989. Transient production and secretion ofhuman transforming growth factor TGF-� 2. Gene 85: 479–488.
29. Stripp, B. R., P. L. Sawaya, D. S. Luse, K. A. Wikenheiser, S. E. Wert,J. A. Huffman, D. L. Lattier, G. Singh, S. L. Katyal, and J. A. Whitsett. 1992.cis-acting elements that confer lung epithelial cell expression of the CC10 gene.J. Biol. Chem. 267: 14703–14712.
30. Baker, C. C., I. H. Chaudry, H. O. Gaines, and A. E. Baue. 1983. Evaluation offactors affecting mortality rate after sepsis in a murine cecal ligation and puncturemodel. Surgery 94: 331–335.
31. Evanoff, H., M. Burdick, S. Moore, S. Kunkel, and R. Strieter. 1992. A sensitiveELISA for the detection of human monocyte chemoattractant protein-1 (MCP-1).Immunol. Investig. 21: 39–45.
32. Matsukawa, A., C. M. Hogaboam, N. W. Lukacs, P. M. Lincoln, R. M. Strieter,and S. L. Kunkel. 1999. Endogenous monocyte chemoattractant protein-1(MCP-1) protects mice in a model of acute septic peritonitis: cross-talk betweenMCP-1 and leukotriene B4. J. Immunol. 163: 6148–6154.
33. Walley, K. R., N. W. Lukacs, T. J. Standiford, R. M. Strieter, and S. L. Kunkel.1996. Balance of inflammatory cytokines related to severity and mortality ofmurine sepsis. Infect. Immun. 64: 4733–4738.
34. Carson, W. E., H. Yu, J. Dierksheide, K. Pfeffer, P. Bouchard, R. Clark,J. Durbin, A. S. Baldwin, J. Peschon, P. R. Johnson, et al. 1999. A fatal cytokine-induced systemic inflammatory response reveals a critical role for NK cells.J. Immunol. 162: 4943–4951.
5481The Journal of Immunology
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

35. Goldmann, O., G. S. Chhatwal, and E. Medina. 2005. Contribution of naturalkiller cells to the pathogenesis of septic shock induced by Streptococcus pyogenesin mice. J. Infect. Dis. 191: 1280–1286.
36. Frucht, D. M., T. Fukao, C. Bogdan, H. Schindler, J. J. O’Shea, and S. Koyasu.2001. IFN-� production by antigen-presenting cells: mechanisms emerge. TrendsImmunol. 22:556–560.
37. Stober, D., R. Schirmbeck, and J. Reimann. 2001. IL-12/IL-18-dependent IFN-�release by murine dendritic cells. J. Immunol. 167: 957–965.
38. Pillarisetty, V. G., S. C. Katz, J. I. Bleier, A. B. Shah, and R. P. Dematteo. 2005.Natural killer dendritic cells have both antigen presenting and lytic function andin response to CpG produce IFN-� via autocrine IL-12. J. Immunol. 174:2612–2618.
39. Benjamim, C. F., C. M. Hogaboam, and S. L. Kunkel. 2004. The chronic con-sequences of severe sepsis. J. Leukocyte Biol. 75: 408–412.
40. Cohen. 2002. The immunopathogenesis of sepsis. Nature 420:885–891.41. Riedemann, N. C., R. F. Guo, and P. A. Ward. 2003. The enigma of sepsis.
J. Clin. Invest. 112: 460–467.42. Zisman, D. A., S. L. Kunkel, R. M. Strieter, W. C. Tsai, K. Bucknell,
J. Wilkowski, and T. J. Standiford. 1997. MCP-1 protects mice in lethal endo-toxemia. J. Clin. Invest. 99: 2832–2836.
43. Matsukawa, A., C. M. Hogaboam, N. W. Lukacs, P. M. Lincoln, R. M. Strieter,and S. L. Kunkel. 2000. Endogenous MCP-1 influences systemic cytokine bal-ance in a murine model of acute septic peritonitis. Exp. Mol. Pathol. 68: 77–84.
44. Steinhauser, M. L., C. M. Hogaboam, S. L. Kunkel, N. W. Lukacs, R. M. Strieter,and T. J. Standiford. 1999. IL-10 is a major mediator of sepsis-induced impair-ment in lung antibacterial host defense. J. Immunol. 162: 392–399.
45. Jouanguy, E., F. Altare, S. Lamhamedi, P. Revy, J. F. Emile, M. Newport,M. Levin, S. Blanche, E. Seboun, A. Fischer, and J. L. Casanova. 1996. Inter-feron-�-receptor deficiency in an infant with fatal bacille Calmette-Guerin infec-tion. N. Engl. J. Med. 335: 1956–1961.
46. Jouanguy, E., S. Lamhamedi-Cherradi, F. Altare, M. C. Fondaneche,D. Tuerlinckx, S. Blanche, J. F. Emile, J. L. Gaillard, R. Schreiber, M. Levin, etal. 1997. Partial interferon-� receptor 1 deficiency in a child with tuberculoidbacillus Calmette-Guerin infection and a sibling with clinical tuberculosis.J. Clin. Invest. 100: 2658–2664.
47. Kamijo, R., J. Le, D. Shapiro, E. A. Havell, S. Huang, M. Aguet, M. Bosland, andJ. Vilcek. 1993. Mice that lack the interferon-� receptor have profoundly alteredresponses to infection with Bacillus Calmette-Guerin and subsequent challengewith lipopolysaccharide. J. Exp. Med. 178: 1435–1440.
48. Car, B. D., V. M. Eng, B. Schnyder, L. Ozmen, S. Huang, P. Gallay,D. Heumann, M. Aguet, and B. Ryffel. 1994. Interferon � receptor deficient miceare resistant to endotoxic shock. J. Exp. Med. 179: 1437–1444.
49. Billiau, A. 1996. Interferon-�: biology and role in pathogenesis. Adv. Immunol.62: 61–130.
50. Zeerleder, S., C. E. Hack, C. Caliezi, G. van Mierlo, A. Eerenberg-Belmer,A. Wolbink, and W. A. Wuillenmin. 2005. Activated cytotoxic T cells and NKcells in severe sepsis and septic shock and their role in multiple organ dysfunc-tion. Clin. Immunol. 116: 158–165.
51. Kerr, A. R., L. A. Kirkham, A. Kadioglu, P. W. Andrew, P. Garside,H. Thompson, and T. J. Mitchell. 2005. Identification of a detrimental role for NKcells in pneumococcal pneumonia and sepsis in immunocompromised hosts. Mi-crobes Infect. 7: 845–852.
52. Heremans, H., C. Dillen, J. van Damme, and A. Billiau. 1994. Essential role fornatural killer cells in the lethal lipopolysaccharide-induced Shwartzman-like re-action in mice. Eur. J. Immunol. 24: 1155–1160.
53. Chan, C. W., E. Crafton, H. N. Fan, J. Flook, K. Yoshimura, M. Skarica,D. Brockstedt, T. W. Dubensky, M. F. Stins, L. L. Lanier, et al. 2006. Interferon-producing killer dendritic cells provide a link between innate and adaptive im-munity. Nat. Med. 12: 207–213.
5482 ROLE OF CCL6 IN INNATE IMMUNITY
by guest on May 6, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from













