Embed Size (px)
Citation preview

Tt
KI
a
ARRAA
KNICHDS(
1
bAidaptgmnmdppm
0h
Vibrational Spectroscopy 70 (2014) 168–186
Contents lists available at ScienceDirect
Vibrational Spectroscopy
jou rn al hom ep age: www .e lsev ier .com/ locate /v ibspec
he effects of conformation and intermolecular hydrogen bonding onhe structural and vibrational spectral data of naproxen molecule
ubilay Balci ∗
stanbul University, Faculty of Science, Department of Physics, Vezneciler, 34134 Istanbul, Turkey
r t i c l e i n f o
rticle history:eceived 9 October 2013eceived in revised form 2 December 2013ccepted 6 December 2013vailable online 16 December 2013
eywords:aproxen
R and Raman spectraonformationydrogen bondingual scale factorscaled quantum mechanical force fieldSQM-FF)
a b s t r a c t
The structural and vibrational properties of naproxen, an inhibitor of cyclooxygenase (COX) enzyme, wereinvestigated by molecular modeling and experimental IR and Raman spectroscopic techniques. Possibleconformers of the molecule were searched via a molecular dynamics simulation carried out with MM2force field. The total energies, equilibrium geometries, force fields, IR and Raman spectral data of the foundstable conformers were determined by means of geometry optimization and harmonic frequency calcula-tions carried out using the B3LYP method and Pople-style basis sets of different size. The stability orderobtained for the lowest-energy conformers was confirmed by high-accuracy thermochemistry calcula-tions performed with G3MP2B3 composite method. Some electronic structure parameters of naproxenand the anharmonicity characters of its vibrational modes were determined by means of natural popula-tion analysis (NPA) and anharmonic frequency calculations at B3LYP/6-31++G(d,p) and B3LYP/6-311++G(d,p)levels of theory. A part of these calculations carried out for free naproxen molecule were repeated alsofor its energetically most favored dimer forms. Two different scaling procedures ((1) “SQM-FF method-
ology” and (2) “Dual scale factors”) were independently applied to the obtained harmonic vibrationalspectral data to fit them to the corresponding experimental data. In the light of the obtained calculationresults, which confirm the remarkable effects of conformation and intermolecular hydrogen bonding onthe structural and vibrational spectral data, in particular, on those associated with the functional groupsin the propanoic acid chain, a reliable assignment of the fundamental bands observed in the experimentalIR and Raman spectra of the molecule was achieved.. Introduction
Naproxen, [S(+)-2-(6-methoxy-2-naphthyl) propionic acid], hasecome one of the most popular and widely used Non-Steroidalnti Inflammatory Drugs (NSAIDs) since approved by U.S. FDA
n December 1991. Unlike the synthetically produced steroidrugs which work by suppressing the immune system, naproxennd other NSAIDs work by preventing the formation of therostaglandins, which are a cluster of bioactive lipids generated inhe human and animal tissues through the activities of cyclooxy-enase isozymes (COX-1 and COX-2), and play important role inany cellular responses and pathophysiologic processes [1]. Today,
aproxen is known as a NSAID compound widely used in the treat-ent of many diseases such as rheumatoid arthritis, osteoarthritis,
egenerative joint disease, ankylosing spondylitis, acute gout and
rimary dismenorrea [2]. It has been also reported that the com-ound is effective in the treatment of traditional and nontraditionaligraine-associated symptoms [3], and it is used in the prevention∗ Tel.: +90 2124555700x15240; fax: +90 2125190834.E-mail addresses: [email protected], [email protected]
924-2031/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.vibspec.2013.12.002
© 2013 Elsevier B.V. All rights reserved.
and treatment of adolescent migraine [4,5] and menstrual migraine[6,7]. According to various epidemiological studies, naproxen andsome other NSAIDs are promising candidates for chemopreventionagainst colon cancer, and some experimental data suggest thatNSAIDs might provide protection against cancers of mammarygland, skin, liver and urinary bladder [8–11]. Besides these, it hasbeen reported that long-term prophylactic use of naproxen canreduce the risk of Alzheimer disease [12–14].
The first structural analysis on naproxen molecule was per-formed by Ravikumar et al. [15], and then this was followed by aseries of structure and conformation studies performed by Wenzeland Buss [16], Bednarek et al. [17], Bachechi et al. [18], Lahmaniet al. [19], King et al. [20], Okulik et al. [21] and Alvarez et al.[22]. Nevertheless, one can reach only three published papers onthe analysis of the vibrational motions of naproxen and associ-ated experimental spectral data. The first of them, based on IRand Raman spectroscopic measurements and molecular model-ing, was reported by Jubert et al. [23]. This was followed by the
other two valuable papers of Luber et al. [24] and Liu et al. [25].Besides this, the effects of solvent–solute interactions, intermolec-ular hydrogen bonding and recrystallization on the morphology ofnaproxen were discussed by Tomasko and Timko [26]. Although
ctrosc
euetcbafaag
2
2
Cfw(pobttsh(utCat1grf3abs
2
iauuiimauvsslbPIcts
K. Balci / Vibrational Spe
ach of the studies mentioned above provides significant contrib-tions to the current literature, none of them has focused on theffects of conformation and intermolecular hydrogen bonding onhe structural and vibrational spectral data of the molecule, specifi-ally. Considering this situation and also some wrong or suspiciousand assignments given in the previous papers, a comprehensivenalysis, the main purpose of which is to reveal the effects of con-ormation and intermolecular hydrogen bonding on the structuralnd vibrational spectral properties of naproxen as well as on its IRnd Raman spectra recorded at room temperature, has been firstiven in this study.
. Methods and calculations
.1. Experimental measurements
Naproxen (CAS number: 22204-53-1; chemical formula:14H14O3; molecular weight: 230.27 g mol−1; purity: 98%, physicalorm: a powder of white color) was provided by FLUKA and usedithout performing any further purification. The FT-IR spectrum
400–4000 cm−1) of the transparent disk sample prepared fromotassium bromide (100 mg) and naproxen (2–3 mg), was recordedn a Jasco 300E model FT-IR spectrometer with resolution of 2 cm−1
y 160 scanning. In addition, the FT-IR spectra of the dilute solu-ions of naproxen prepared with ethanol and methanol solvents (athe concentrations of 0.01–0.1 M) were recorded by using the samepectrometer in the same experimental conditions. On the otherand, the corresponding Raman spectra for the pure compoundin solid phase) and for its ethanol suspension, were recorded bysing a Jasco NRS 3100 model dispersive micro-Raman spectrome-er (equipped with a laser source of 532 nm wavelength, a cooledCD detector of high-sensitivity and a grating of 1200 lines/mm)nd also using a Bruker “Multi Ram” model FT-Raman spec-rometer (source: Nd:YAG Laser of 1064 nm wavelength, power:000 mW; detector: germanium detector cooled by liquid nitro-en). The FT-Raman spectrum (resolution: 1 cm−1) for the spectralegion 4000–50 cm−1, and the dispersive micro-Raman spectrumor the spectral regions 1800–150 cm−1 (resolution: 3.78 cm−1) and400–2700 cm−1 (resolution: 2.89 cm−1) were obtained over 250nd 100 scanning, respectively. After carefully applied baseline andackground corrections, the resultant experimental IR and Ramanpectra reported in this study were obtained.
.2. Molecular modeling calculations
The minimum-energy conformers of naproxen molecule, whichs a naphthalene derivative including the methoxy and propanoiccid substituents (see Fig. 1), were searched by means of a molec-lar dynamics simulation carried out by “Chem3D” software [27]sing Allingers’MM2 force field [28]. The input geometrical data used
n the simulation, where the temperature parameter was graduallyncreased from 0 K to 500 K, were formed from the optimized geo-
etrical parameters reported for monomeric naproxen by Okuliknd Jubert [21]. For each trajectory determined through the sim-lation, the corresponding geometrical parameters were reachedia energy minimization calculations performed by using the sameoftware and force field. These calculations were followed byuccessive geometry optimization and harmonic frequency calcu-ations carried out by Gaussian03 software [29] using B3LYP methodased on the density functional theory (DFT) [30,31] and variousople-style basis sets {6-31G(d), 6-31++G(d,p) and 6-311++G(d,p)}.
n order to examine the stability order for the determined stableonformers, additional high-accuracy thermochemistry calcula-ions were carried out using G3MP2B3 method, which includesome empirical spin–orbit and higher-level energy correctionsopy 70 (2014) 168–186 169
[32–34]; this composite method, which employs the equilibriumgeometrical parameters and zero point energies calculated atB3LYP/6-31G(d) level as well as the energy correction terms cal-culated at CCSD(T,FC)/6-31G(d) and MP2(FC)/G3MP2(Large) levels,is known as the most economic and efficient one among all themethods based on Gaussian-3 theory (G3). For each of the stableconformers, the energies of the frontier molecular orbitals werecalculated at B3LYP/6-31++G(d,p) and B3LYP/6-311++G(d,p) levelsof theory and then these calculation data were used in the defini-tion of the ionization potential (I), electron affinity (A), absoluteelectronegativity (�) and chemical hardness (�) as well as theelectrophilicity index (w) values within the validity of Koopmans’theorem [35], where the energies of the highest occupied molec-ular orbital (HOMO) and the lowest unoccupied molecular orbital(LUMO) are assumed to equal I and A, respectively. In these def-initions realized by using the empirical equations given by Yang,Pearson and Parr [36–42], the absolute electronegativity valueswere taken as −1/2 times the sum of the energies of the frontierorbitals (HOMO and LUMO), while chemical hardness parameterswere equaled to the absolute value of the HOMO–LUMO energygap. On the other hand, for the conformers estimated to havethe largest populations in the molecular medium at room tem-perature, the corresponding natural population analysis (NPA) data{the natural atomic charges, the occupancies and energies of thenatural bonding orbitals (NBOs), the polarization coefficients fornatural hybrid orbitals (NHOs), the electron delocalization ener-gies as well as the Canonical Molecular Orbital (CMO) coefficientsshowing the contributions of the NBOs to the frontier orbitals} werecalculated at the B3LYP/6-311++G(d,p) level of theory by utilizingthe software NBO 5.9 [43]. In addition, the Fukui function values,which show the local reactivity and selectivity of the conform-ers in electrophilic, nucleophlic and radical attacks, were derivedfrom the calculated natural atomic charges using the approachproposed by Parr and Ayers [44,45]. In order to reveal the anhar-monicity characters of the vibrational modes of naproxen, thefrequency calculations were repeated for each stable conformerdominating the room-temperature experimental spectra of themolecule at B3LYP/6-311++G(d,p) level in the anharmonic oscilla-tor approach which includes the cubic and quartic force constantcorrection terms. On the other hand, the overestimations at thecalculated harmonic wavenumbers were corrected by the aid oftwo different types of scaling procedures called “Dual scale fac-tors” [46] and “Scaled quantum mechanical force field (SQM-FF)”methodology [47–49]. In the former procedure, where two differ-ent empirical scale factors are employed to improve the calculatedharmonic wavenumbers, one scale factor {“0.977” for B3LYP/6-31++G(d,p) and “0.980” for B3LYP/6-311++G(d,p)} is employed forthe wavenumbers below 1800 cm−1 while another one {“0.962”for B3LYP/6-31++G(d,p) and “0.967” for B3LYP/6-311++G(d,p)} forthe wavenumbers higher than 1800 cm−1; the scale factors used inthis study were derived from those proposed by Frosch et al. forB3LYP/6-31++G(d,p) level of theory [50] by optimizing them so asto reach the smallest root-mean-square (r.m.s.) error values. Dif-ferently, in the SQM-FF methodology, the scaled wavenumbers arereproduced over the optimized geometry and force field parame-ters obtained from ab initio or DFT calculations. This second scalingprocedure was applied to naproxen molecule by utilizing Collier’ssoftware (“Fcart”, version-06 [51–53]); in the scaling of the forceconstants of the described internal coordinates (see Table S1, sup-porting data), which were derived from the force constants (inCartesian coordinate terms) obtained through the harmonic fre-quency calculations at B3LYP/6-31G(d) level of theory, the original
scale factors proposed by Baker et al. [54] for the same level of the-ory (see Table S2, supporting material) were employed without anymodification. The potential energy distribution (PED) values calcu-lated in the SQM-FF methodology constituted the primary data set
170 K. Balci / Vibrational Spectroscopy 70 (2014) 168–186
en mo
imtRtt[Ionme“c(
Fig. 1. The chemical structure of naprox
n the descriptions of the vibrational normal modes of naproxenolecule. On the other hand, the corresponding Raman intensi-
ies for each normal mode of the molecule were derived from theaman activities calculated at B3LYP/6-31G(d) level of theory andhe scaled wavenumbers obtained in the SQM-FF methodology byhe aid of a set of equations reported by Michalska and Wysokinski55] and Krishnakumar et al. [56] independently from each other.n order to reveal the effects of intermolecular hydrogen bondingn the structural properties and the vibrational spectral data ofaproxen, a part of the calculations carried out for the determinedost stable conformers of the molecule were repeated also for their
nergetically most favored dimer forms by taking into account thecounterpoise corrections” to remove the errors occurred at thealculated energies as a result of the “Basis Set Superposition EffectBSSE)” [57,58].
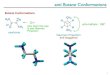
Fig. 2. The illustrations for the possible stab
lecule and the numeration of its atoms.
3. Results and discussion
3.1. Conformation and structural properties
The conformational calculations performed in this study haveindicated that in monomer form, naproxen has at least nine stableconformers (see Fig. 2), six of which were previously reported byLahmani et al. [19]. The stability order given here for the conform-ers is in complete agreement with that reported by these authors.For an easy comparison, all of the determined stable conformersof naproxen can be collected into two different classes so called
here as “cis” and “trans” with respect to the relative positions ofthe carboxyl and methoxy functional groups in the molecule aspreviously proposed by Lahmani et al. [19]; the cis and trans con-formers are marked here with the capitals “C” and “T”, respectively.le conformers of naproxen molecule.

ctroscopy 70 (2014) 168–186 171
T3ogctTclitB(Cltdhlf−0T1cgehag
(wctitatutecbieaoSimvohc
da(acdt8t“
angl
e,
ener
gy
and
elec
tron
ic
stru
ctu
re
par
amet
ers
calc
ula
ted
at
B3LY
P/6-
31++
G(d
,p)
and
B3LY
P/6-
311+
+G(d
,p)
leve
ls
of
theo
ry
for
the
theo
reti
call
y
pos
sibl
e
stab
le
con
form
ers
of
napr
oxen
mol
ecu
le.
l
Con
form
ersa
Dih
edra
l an
gles
(◦ )b
�
(Deb
ye)
Elec
tron
ic
stru
ctu
re
par
amet
ers
(eV
)cEn
ergi
esd
D1
D2
D3
D4
D5
I
A
�
�
w
E tot
al(H
artr
ee)
E rel
ativ
e(k
cal/
mol
)
Conf
. C1
−0.5
−121
.4
88.6
−90.
3
115.
1
1.48
3
5.90
0
1.30
9
4.59
2
3.60
5
1.41
5
−767
.409
1
0.00
0Co
nf. T
1
−0.1
58.3
86.3
92.6
65.1
2.81
2
5.88
0
1.30
6
4.57
4
3.59
3
1.41
1
−767
.400
5
0.26
0Co
nf. C
2
−0.6
−111
.6
−78.
1
101.
6
121.
7
2.95
8
5.90
5
1.29
7
4.60
8
3.60
1
1.40
7
−767
.399
4
0.95
0Co
nf. T
2
0.1
−69.
1
79.6
−100
.0
57.7
2.11
2
5.88
8
1.32
7
4.56
1
3.60
7
1.42
6
−767
.398
9
1.23
4Co
nf. T
3
180.
0
121.
5
−88.
6
90.4
−115
.1
3.28
2 5.
772
1.39
6
4.37
6
3.58
4
1.46
8
−767
.398
6
1.44
1Co
nf. C
3
−179
.9
58.3
87.1
−91.
8
−65.
3
1.42
0 5.
749
1.39
6
4.35
3
3.57
2
1.46
6
−767
.398
2
1.69
9Co
nf. T
4
−179
.9
−111
.3
−77.
7
102.
1
122.
0
2.08
9
5.77
5
1.38
5
4.39
1
3.58
0
1.46
0
−767
.398
2
2.34
3Co
nf. T
5
179.
9
112.
1
78.8
−100
.9
−121
.2
2.09
4
5.77
6
1.38
7
4.38
9
3.58
1
1.46
1
−767
.397
2
2.34
4Co
nf. C
417
9.5
67.5
−80.
4
99.2
−59.
5 3.
363
5.76
0
1.41
8
4.34
2
3.58
9
1.48
3
−767
.396
4
2.73
7
)Co
nf. C
1
−0.5
−121
.3
88.6
−90.
3
115.
2 1.
435
5.95
1
1.35
0
4.60
1
3.65
1
1.44
9
−767
.563
1
0.00
0Co
nf. T
1
−0.1
58.1
86.9
91.6
65.5
2.77
1
5.93
1
1.34
8
4.58
2
3.63
9
1.44
5
−767
.562
7
0.24
8Co
nf. C
2
−0.6
−111
.1
−78.
1
101.
6
122.
1
2.90
1
5.95
5
1.33
7
4.61
8
3.64
6
1.43
9
−767
.561
6
0.93
3Co
nf. T
2
0.1
−69.
2
79.6
−100
.0
57.7
2.05
0
5.93
7
1.36
7
4.57
0
3.65
2
1.45
9
−767
.561
1
1.21
7Co
nf. T
3
179.
9
121.
8
−89.
1
89.8
−114
.7
3.23
3
5.82
3
1.43
8
4.38
5
3.63
0
1.50
3
−767
.560
7
1.49
3Co
nf. C
3
−179
.9
58.7
86.7
−92.
0 −6
4.9
1.39
4
5.80
1
1.44
1
4.35
9
3.62
1
1.50
4
−767
.560
3
1.79
0Co
nf. T
4
−179
.9
−111
.6
−78.
2
101.
4
121.
7
2.03
9
5.82
6
1.42
7
4.39
9
3.62
7
1.49
5
−767
.559
3
2.38
3Co
nf. T
517
9.9
112.
1
78.7
−100
.9
−121
.2
2.07
2
5.82
8
1.45
4
4.37
4
3.64
1
1.51
5
−767
.559
7
2.14
6Co
nf. C
4
179.
5
67.2
−80.
3
99.3
−59.
9
3.29
4
5.80
9
1.46
1
4.34
9
3.63
5
1.51
9
−767
.558
7
2.77
0
rati
ons
for
the
con
form
ers
are
give
n
in
Fig.
2.
The
con
form
ers
are
clas
sifi
ed
as
cis
and
tran
s
wit
h
resp
ect t
o
the
rela
tive
pos
itio
ns
of
met
hox
y
and
carb
oxyl
fun
ctio
nal
grou
ps;
the
con
form
ers
clas
sifi
ed
as
cis
are
mar
ked
thos
e
clas
sifi
ed
as
tran
s
are
mar
ked
by
“T”.
C3
O4
C5;
D2:
C9
C10
C13
C14
;
D3:
C10
C13
C14
O15
;
D4:
C10
C13
C14
O16
;
D5:
C9
C10
C13
C28
. Th
e
nu
mer
atio
n
of
the
atom
s
is
as
give
n
in
Fig.
1.io
n
pot
enti
al
wh
ich
corr
esp
ond
s
to
the
neg
ativ
e of
Hom
o
ener
gy
{−ε H
} in
Koo
pm
ans’
theo
rem
[35]
; A: e
lect
ron
affi
nit
y
wh
ich
corr
esp
ond
s
to
the
neg
ativ
e
of
Lum
o
ener
gy
{−ε L} i
n
Koo
pm
ans’
theo
rem
[35]
; �: c
hem
ical
−
ε H} [
36–4
2];
�:
abso
lute
elec
tron
egat
ivit
y {−
(εH
+
ε L)/
2}
[36–
42];
w:
elec
trop
hil
icit
y
ind
ex
{(�
2/�
)/2}
[42]
.al
ener
gy;
E rel
ativ
e:
rela
tive
ener
gy, d
eriv
ed
from
the
tota
l en
ergy
by
assu
min
g
conf
. C1
as
refe
ren
ce.
K. Balci / Vibrational Spe
he energy values obtained at B3LYP/6-31++G(d,p) and B3LYP/6-11++G(d,p) levels of theory for the determined stable conformersf naproxen are listed in Table 1; as clearly seen, the relative ener-ies of the two conformers labeled here as conf. C1 and conf. T1 areonsiderably lower than those of the others and the “kT” energy ofhe molecular medium (about 0.6 kcal/mol at room temperature).he same energy values indicate that conf. C1 (the methoxy andarboxyl groups locate in “cis” position to each other) is slightlyower in energy than conf. T1 (the same functional groups locaten “trans” position to each other) and accordingly correspondso the most favored conformer in energy for the free molecule.oth the small energy difference between conf. C1 and conf. T10.248 kcal/mol) and the relatively much larger one between conf.1 and conf. C2 (0.933 kcal/mol), predicted at B3LYP/6-311++G(d,p)
evel of theory, can be seen as clear indications that the experimen-al spectra of naproxen be dominated by conf. C1 and conf. T1. Thisetermination was also confirmed by the results obtained from theigh-accuracy thermochemistry calculations performed at G3MP2B3
evel of theory; the energy values obtained at this level of theoryor conf. C1, conf. C2 and conf. T1 are −766.4430, −766.4412 and766.4425 Hartree and indicate that conf. C1 is lower in energy by.314 and 1.106 kcal/mol than conf. T1 and conf. C2, respectively.he difference between the energies of conf. C1 and conf. C2 (about
kcal/mol) yields the energy barrier due to the rotation of thearboxyl group about the C13 C14 bond combining the carboxylroup to the propanoic acid chain. The same is true for the differ-nce between the energies of conf. T1 and conf. T2. On the otherand, the energy difference (about 1.5 kcal/mol) between conf. C1nd conf. T3 yields the energy barrier due to the rotation of methoxyroup about the C3 O4 bond.
For each of the conformers, the corresponding dipole moment�), the electron affinity (A) and ionization potential (I) energies,hich were derived from the energies of the frontier orbitals cal-
ulated at B3LYP/6-31++G(d,p) and B3LYP/6-311++G(d,p) levels ofheory by using the Koopmans’ theorem [35], have been presentedn Table 1. The “global chemical hardness (�)”, “absolute elec-ronegativity (�)” and “electrophilicity indices (w)” [36–42] arelso presented in the same table in comparison. As clearly seen,he dependency of the calculated values to the size of the basis setsed is generally quite weak. Unlike the “total energy” showinghe thermodynamic stability, the “chemical hardness” is a param-ter showing the kinetic stability. From this point of view, one canonclude that conf. C2 is the one exhibiting the highest kinetic sta-ility among all the stable conformers of naproxen, while conf. C1
s thermodynamically the most stable one. As known from the lit-rature [36–42], the chemical hardness and the electronegativityre closely related and both are the indicators of the resistancef an atom or molecule to any change in its electron distribution.ince these two global indicators provide information on the chem-cal potential and reactivity properties of different conformers of a
olecule, considering the chemical hardness and electronegativityalues reported here for the most stable cis and trans conformers,ne can conclude that in the trans structure case, naproxen has aigher chemical potential and a higher reactivity compared to theis structure case.
According to the calculation results, naproxen can form threeifferent types of dimer structures, which are so called heres “Cyclic-cis-dimer”, “Cyclic-trans-dimer” and “Open-chain-dimer”see Fig. 3 for illustrations). The energetically most favored onesmong them are the two “cyclic-cis-dimer” structures formed byonf. C1 and conf. T1; at B3LYP/6-31++G(d,p) level of theory, theifference between their total energies is 0.594 kcal/mol. Besides
his, the considerably higher relative energies (from 1.361 to.952 kcal/mol) as well as much lower stabilization energies (dueo dimerization; see the �E values in Table 2) obtained for thecyclic-trans-dimer” and “open-chain-dimer” structures at the same Table
1Th
e
dih
edra
l
Theo
ry
leve
B3LY
P/6-
31++
G(d
,p)
B3LY
P/6-
311+
+G(d
,p
aTh
e
illu
stby
“C”
wh
ile
bD
1:
C2
cI:
ion
izat
har
dn
ess
{εL
dE t
otal
:
tot

172 K. Balci / Vibrational Spectroscopy 70 (2014) 168–186
F of thef r (conf
lcnCoiC(ct0ea−t
lbN
TTs
o{
�
ig. 3. The possible stable dimer structures determined at B3LYP/6-31++G(d,p) levelorms of the cis conformer (conf. C1) and (b) the dimer forms of the trans conforme
evel of theory should be seen as another clear indication that inondensed phase the experimental room temperature spectra ofaproxen be dominated by the “cyclic-cis-dimer” structures of conf.1 and conf. T1. The effects of intermolecular hydrogen bondingn the electronic structure parameters of naproxen and its stabil-ty can be revealed by a comparison over the data given for conf.1 and conf. T1 (see Table 1) and for their possible dimer structuressee Table 2). Such a comparison indicates that dimerization shouldause remarkable decreases at the values of the ionization poten-ial (I: from 0.032 to 0.738 eV), electronegativity (�: from 0.027 to.072 eV) and chemical hardness (�: from 0.006 to 0.221 eV) param-ters, however, the trend of the changes predicted for the electronffinity (A: from −0.044 to 0.056 eV), electrophilicity index (w: from0.034 to 0.028 eV) and electric dipole moment (�: from −1.500
o 2.804 Debye) parameters depend on the type of dimerization.The effects of dimerization on the frontier orbitals and the
ocalized natural bonding orbitals of naproxen can be revealedy comparisons made over the CMO coefficients and over theBO data which consist of the orbital occupancy and energy
able 2he dipole moment, energy and electronic structure parameters calculated at B3LYP/6-31table cis and trans conformers of naproxen molecule).
Stable dimer structuresa � (Debye) Electronic structure parameters (eV)b
I A � �
Conf. C1 – cyclic-cis-dimer 2.285 5.865 1.291 4.574 3.578
Conf. T1 – cyclic-cis-dimer 1.903 5.830 1.262 4.568 3.546
Conf. C1 – cyclic-trans-dimer 0.928 5.746 1.365 4.381 3.556
Conf. T1 – cyclic-trans-dimer 1.312 5.845 1.279 4.566 3.562
Conf. C1 – open-chain-dimer 1.299 5.771 1.296 4.475 3.533
Conf. T1 – open-chain-dimer 5.616 5.887 1.330 4.556 3.609
a The illustrations for the dimer structures are given in Fig. 3.b I: ionization potential which corresponds to the negative of Homo energy {−εH} in
f Lumo energy {−εL} in Koopmans’ theorem [35]; �: chemical hardness {εL − εH} [36–4(�2/�)/2} [42].
c Etotal: total energy with “counter poise correction”; Erelative: relative energy, derived frEstabilization: stabilization energy due to dimerization {Edimer − 2Emonomer}.
ory for the most stable cis and trans conformers of naproxen molecule. (a) The dimer. T1).
values. The reader can reach these structural data obtained atB3LYP/6-311++G(d,p) level of theory for the monomer and dimerstructures of conf. C1 and conf. T1 in Tables S3 and S4 (sup-porting materials). These comparisons have indicated to a slightchange for the HOMO depending on dimerization but no notableone for the LUMO; unlike the monomer structure case, the�(C1 C8) and �(C6 C7) bonding orbitals are not involved intothe HOMO in the dimer structure. As expected, the effect ofdimerization on the energies of the natural bonding orbitals asso-ciated with the carboxyl group are quite remarkable; in the caseconf. C1, the changes determined for the energies of the non-bonding orbitals n(O15)-1, n(O15)-2, n(O16)-1 and n(O16)-2 are1.308, 0.361, 0.296 and −1.084 eV, while those determined forthe energies of the bonding orbitals �(C14 O15), �(C14 O16)and the antibonding orbital �*(C14 O16) are −0.753, 0.674and −0.399 eV, respectively. Nevertheless, relatively much lower
but again notable increases (between 0.040 and 0.092 eV) havebeen determined for the bonding orbitals �(C7 C8), �(C9 C10),�(C9 C10), �(C11 C12), �(C13 C28), �(O15 H23), �(C19 H21),++G(d,p) level of theory for the stable dimer forms of conf. C1 and conf. T1 (the most
Energiesc
w Etotal (Hartree) Erelative (kcal/mol) �Estabilization (kcal/mol)
1.399 −1534.8231 0.000 −13.3541.377 −1534.8222 0.594 −13.2801.443 −1534.8209 1.361 −11.9931.390 −1534.8214 1.057 −12.8171.395 −1534.8096 8.495 −4.8601.429 −1534.8088 8.952 −4.922
Koopmans’ theorem [35]; A: electron affinity which corresponds to the negative2]; �: absolute electronegativity {−(εH + εL)/2} [36–42]; w: electrophilicity index
om the total energy by assuming the cyclic-cis-dimer form of conf. C1 as reference;

ctrosc
�fcooi
riintnFocascsbtCbat�tab�l−to�t0cfidcdT2��mdtotosaaoa�oTiocho
K. Balci / Vibrational Spe
(C10 C13), �(C13 C14), �(C13 H27). These changes determinedor the energy and occupancy values of the NBOs indicate to aharge transfer from the nonbonding n(O) and bonding �(O H)rbitals centered on the carboxyl group and the �(CC) bondingrbitals centered on the naphthalene ring to the �*(C O) antibond-ng orbital centered on the carboxyl group.
The intramolecular charge transfer between the naphthaleneing and the substituent groups and also its effect on the stabil-ty and reactivity of naproxen molecule can be better understoodf the charge distribution of this molecule is compared to that ofaphthalene molecule, which takes place in naproxen as a consti-utional component. From this point of view, the frontier orbitals,atural atomic charges and electrostatic potential surfaces (seeigs. S1 and S2) calculated at B3LYP/6-311++G(d,p) level of the-ry for naproxen and naphthalene molecules as well as the CMOoefficients, occupancy and energy values calculated for their NBOst the same level of theory constitute a supporting data set. Theums taken over the natural atomic charges of the two moleculesonfirm the charge transfer from the naphthalene ring to theubstituent groups. One can associate the substantial differencesetween the HOMOs of naproxen and naphthalene molecules withhis charge transfer, and considering the frontier orbitals and theMO coefficients reported here (see Table S3), concludes that theonding orbitals �(C2 C3), �(C9 C10), �(C11 C12), �(C1 C8)nd �(C6 C7) on which the HOMO centers are the most suscep-ible ones to electrophilic attacks, while the antibonding orbitals*(C6 C7), �*(C11 C12), �*(C9 C10) and �*(C2 C3) on which
he LUMO centers are the most susceptible ones to the nucleophilicttacks. At B3LYP/6-311++G(d,p) level of theory, the energies of theonding orbitals �(C1 C8), �(C2 C3), �(C6 C7), �(C9 C10) and(C11 C12) which center on the carbon atoms in the naphtha-
ene ring take on values between -6.5 and -7.4 eV (or between150 and −170 kcal/mol), and they are remarkably higher than
hose calculated for the other bonding orbitals of naproxen. On thether hand, the energies of the antibonding �*(C1 C8), �*(C2 C3),*(C6 C7), �*(C9 C10) and �*(C11 C12) orbitals centered on
he carbon atoms in the naphthalene ring take on values about.9 eV (or 20 kcal/mol) and they are much lower than those cal-ulated for the other antibonding orbitals of the molecule. Thesendings clearly explain why the frontier orbitals of naproxen areominated by the �(CC) bonding and �*(CC) antibonding orbitalsentered on the carbon atoms in the naphthalene ring. The NBOata obtained for the monomer forms of conf. C1 and conf. T1 (seeables S4 and S5) have indicated that notable changes (from 1.1 to.2 kcal/mol) should appear at the energies of the bonding orbitals(C10 C13), �(C1 C12), �(C10 C11), �(C9 H21), �(C11 H22),(C7 H19) and �(C28 H29), depending on conformation. Further-ore, these NBO data have confirmed that, naproxen is a highly
elocalized electronic system and there must be strong conjuga-ive interactions between the �(CC) and �*(CC) orbitals centeredn the carbon atoms of the naphthalene ring and also betweenhe p type of nonbonding {n(O4), n(O15)} and the antibondingrbitals �*(C2 C3), �*(C14 O16). It has also been revealed by theame theoretical data that the first order hyperconjugative inter-ctions between the nonbonding orbitals centered on the oxygentoms of the methoxy and carbonyl groups and the antibondingrbitals �*(CC), �*(CO) as well as the sigma–sigma type of inter-ctions (also called “second order hyperconjugation”) between the(CC), �(CH) bonding orbitals and the �*(CC), �*(CO) antibondingrbitals play an important role on the stability of the molecule.he effects of intermolecular hydrogen bonding on these stabiliz-ng interactions between the NBOs can be revealed by a comparison
ver the delocalization energies calculated for the monomer andyclic-cis-dimer forms of conf. C1 (see Table S6); such a comparisonas indicated that, in particular, the delocalization of the electronsccupying the �(C2 C3), �(C9 C10) bonding orbitals and theopy 70 (2014) 168–186 173
n(O15-2), n(O16-2) non-bonding orbitals show strong sensitivity todimerization.
3.2. Chemical reactivity and selectivity
The “Fukui function” values derived from the natural atomiccharges calculated at B3LYP/6-311++G(d,p) level of theory for themonomer and cyclic-cis-dimer forms of conf. C1 and conf. T1 maybe quite useful in the elucidation of the local reactivity and selec-tivity of naproxen in possible nucleophilic, electrophilic and radicalattacks as well as in the prediction of their dependency to con-formation and intermolecular hydrogen bonding; the interestedreader can reach these supporting data from Table S7. Accordingto the “Fukui function” values obtained for the monomer forms ofthe conformers, the hydroxyl-hydrogen atom (H23) and the carbonatoms C9, C12 and C7 of the naphthalene ring are the most favoredcenters for possible nucleophilic attacks, while the C2, C10, C12 car-bon atoms of the naphthalene ring and the methoxy-oxygen (O4)correspond to the most favored centers for electrophilic attacks.On the other hand, the atoms C2, H23, C12, C9 and C7 have beendetermined as the most favored centers for possible radical attacks.Considering these determinations, one can reach to the follow-ing conclusions for naproxen in monomer form; (i) the relativeattractiveness of the atoms is very weakly dependent to the con-formation, (ii) the naphthalene ring is the only active region whichare expected to strongly be subjected to all of the types of attacksmentioned above, (iii) the methoxy group, which is not favoredfor electrophilic attacks, is poorly attractive for both electrophilicand radical attacks, and (iv) the hydroxyl group is quite a favoredregion for both nucleophilic and radical attacks. On the other hand,the Fukui function values obtained for the dimer forms of conf.C1 and conf. T1 indicate that the attractiveness of the hydroxyl-hydrogen atom for possible nucleophilic and radical attacks shouldcompletely disappear when the molecule is in dimer form. Consid-ering the same data, one also concludes that dimerization decreasethe attractiveness of the carbonyl-oxygen atom (O16) for all thepossible molecular attacks, dramatically.
3.3. Geometry and force field
Some selected bond length, bond angle and dihedral angleparameters obtained at B3LYP/6-311++G(d,p) level of theory forconf. C1 and conf. T1 and also for their cyclic-cis-dimer forms are pre-sented in Table 3 in comparison to the corresponding experimentalvalues [18,20], while the complete geometrical data obtained atthe same level of theory for all of the possible stable conformersof naproxen are given in Table S8 in Cartesian coordinate terms asa supporting data. As clearly seen, the theoretical and experimen-tal values in Table 3 well agree with each other. The deviationsbetween the corresponding parameters of the two conformers aregenerally quite small, whereas those determined for the dihedralangles C11 C10 C13 C14 and C11 C10 C13 C28 are of consid-erable amount (63◦ and 48◦, respectively). On the other hand, thesmall differences (ranging from 0.001 to 0.006) between the bondlength values calculated for the corresponding ring CC bonds ofnaproxen (conf. C1 and conf. T1) and naphthalene molecules can beseen as a confirmation for the weak effects of the substituents onthe geometrical parameters associated with the naphthalene ringmoiety of naproxen. The energies calculated for the bonding orbitals�(C3 O4), �(O4 C5), �(C14 O15) and �(C14 O16) of conf. C1and conf. T1 are remarkably lower than all of the other � and �types of bonding orbitals of these two conformers, and accordingly,
the bond lengths calculated for the C3 O4, O4 C5 and C14 O15bonds (respectively, take on the values 1.364, 1.421 and 1.355 A)are considerably shorter than those (about 1.5 A) calculated forthe C10 C13, C13 C14 and C13 C28 bonds of the propanoic acid
174 K. Balci / Vibrational Spectroscopy 70 (2014) 168–186
Table 3The optimized geometrical parameters calculated at B3LYP/6-311++G(d,p) level of theory for the monomer and cyclic-cis-dimer forms of conf. C1 and conf. T1 (the most stablecis and trans conformers of naproxen molecule).
Bond lengths (Å) conf. C1 conf. T1 Bond angles (◦) conf. C1 conf. T1
Parametera* Exp. [20] Mon. Dim. Mon. Dim. Parametera Exp. [20] Mon. Dim. Mon. Dim.
C1 C2 1.429(2) 1.422 1.422 1.422 1.422 C1 C2 C3 119.3 120.2 120.2 120.2 120.1C2 C3 1.375(2) 1.378 1.378 1.378 1.378 C1 C8 C9 119.7 119.3 119.3 119.5 119.5C3 O4 1.371(3) 1.364 1.364 1.364 1.364 C1 C12 C11 120.9 121.2 121.2 121.0 120.9O4 C5 1.428(2) 1.421 1.421 1.421 1.421 C1 C8 C7 118.9 118.4 118.3 118.3 118.3C3 C6 1.419(2) 1.421 1.421 1.422 1.421 C2 C3 O4 125.6 125.2 125.2 125.2 125.2C6 C7 1.366(2) 1.368 1.368 1.368 1.368 C2 C3 C6 120.8 120.3 120.3 120.3 120.3C7 C8 1.419(2) 1.422 1.422 1.423 1.423 C2 C1 C12 121.7 122.1 122.1 122.1 122.2C8 C9 1.420(2) 1.417 1.417 1.415 1.415 C2 C1 C8 119.9 119.7 119.7 119.7 119.7C9 C10 1.375(2) 1.378 1.378 1.380 1.380 C3 O4 C5 117.4 118.4 118.4 118.4 118.4C10 C11 1.420(2) 1.419 1.419 1.416 1.417 C3 C6 C7 120.6 120.3 120.4 120.3 120.4C11 C12 1.373(2) 1.374 1.374 1.376 1.375 O4 C3 C6 113.7 114.5 114.6 114.5 114.5C1 C12 1.429(2) 1.419 1.419 1.418 1.417 C6 C7 C8 120.5 121.2 121.2 121.2 121.2C1 C8 1.419(2) 1.427 1.427 1.428 1.428 C7 C8 C9 121.3 122.4 122.4 122.2 122.2C10 C13 1.529(2) 1.526 1.527 1.529 1.529 C8 C9 C10 120.9 121.7 121.7 121.5 121.5C13 C14 1.510(8) 1.522 1.522 1.522 1.521 C8 C1 C12 118.3 118.2 118.1 118.1 118.1O14 C15 1.324(2) 1.355 1.321 1.355 1.321 C9 C10 C11 117.7 118.8 118.7 118.7 118.7O14 C16 1.212(2) 1.206 1.226 1.206 1.227 C9 C10 C13 119.2 120.6 120.6 121.5 121.6C13 C28 1.530(2) 1.537 1.537 1.537 1.537 C10 C11 C12 120.8 121.0 121.0 121.2 121.2
C10 C13 C14 107.5 109.4 109.1 109.2 109.1C10 C13 C28 112.5 112.6 112.7 112.6 112.7C11 C10 C13 120.6 120.6 120.7 119.8 119.7C13 C14 O15 112.3 112.0 113.5 111.8 113.5C13 C14 O16 124.4 125.8 122.9 125.8 122.9C14 C13 C28 111.4 110.7 111.2 110.9 111.3O15 C14 O16 123.3 122.3 123.6 122.3 123.6– – – – – –
Selected dihedral angles (◦) conf. C1 conf. T1
Parametera Exp. [18] Mon. Dim. Mon. Dim.
C2 C3 O4 C5 9.8(8) −0.5 −0.1 −0.1 −0.3C11 C10 C13 C14 64.9(8) 60.0 58.2 123.2 120.9C11 C10 C13 C28 −59.8(8) −64.5 −65.3 −113.2 −114.9C10 C13 C14 O15 – 88.6 90.9 −87.3 −90.8
assoc
cthntnoowCdabao�Tacnf0C2bOcfi
O15 C14 C13 C28 – −146.8C2 C1 C8 C9 – −179.9
a The numeration of the atoms is as given in Fig. 1; for simplicity, the parameters
hain. The coplanarity of the methoxy plane (C2 C3 O4 C5) withhe naphthalene ring plane should be attributed to the strongyperconjugative interaction between the nonbonding orbital(O4)-2 and the antibonding orbital �*(C2 C3). On the other hand,he hyperconjugative interaction between the nonbonding orbital(O15)-2 and the antibonding orbital �*(C14 O16) as well as thene between the nonbonding orbital n(O16)-2 and the antibondingrbital �*(C14 O15) are the stabilizing intramolecular interactionshich define the planarity of the carboxyl acid moiety. In both conf.1 and conf. T1, the propanoic acid chain is positioned in a perpen-icular position to naphthalene ring and this situation should bettributed not only to the destabilizing repulsive steric interactionsetween the propanoic acid chain and the naphthalene ring butlso to the stabilizing second order hyperconjugative interactionsf the bonding orbital �(C13 H27) with the antibonding orbitals*(C10 C11) and �*(C14 O16). By regarding the data given inable 3, one can estimate that dimerization cause some remark-ble changes on the geometrical parameters associated with thearboxyl group but almost no change on those associated with theaphthalene ring; in the case of conf. C1, the changes estimated
or the bond lengths of O15 H23, O14 C15, O14 C16 are about.030, 0.035 and 0.020 A, while those for the angles C4 O15 H23,13 C14 O15, C13 C14 O16 and O15 C14 O16 are 2.6◦, 1.5◦,.9◦ and 1.2◦, respectively. In particular, the excellent agreement
etween the bond length value (1.321 A) reported here for the14 C15 bonds of conf. C1 and conf. T1 in dimer form and theorresponding experimental value (1.324 A) may be seen as a con-rmation for the reliability of the theoretical estimations.−144.2 148.1 144.3−179.9 179.6 180.0
iated with hydrogen atoms were not included.
The scaled force constants obtained in SQM-FF methodology forthe bond stretching and some selected angle bending coordinates ofconf. C1 and conf. T1 are presented in Table 4 in comparison to thoseof naphthalene molecule. In addition, the complete set of the scaledforce constants obtained for these two conformers can be reachedfrom Table S9 (supporting material). According to these theoreticaldata, the R(C28 H29) and R(C28 H31) bond stretching coordinatesof methyl group are the most sensitive ones to conformation; thedeviations determined for the force constants of these two internalcoordinates of the conformers are 0.105 and 0.111 mdyn/Å, respec-tively. The effects of conformation on the R(C10 C11), R(C11 C12)and R(C13 C14) bond stretching coordinates of the naphthalenering moiety are also notable; the deviations determined for thesethree internal coordinates are 0.064, 0.039 and 0.031 mdyn/Å,respectively. The force constant parameters can be used as reliableindicators for the strengths of the chemical bonds in a molecule. Onthis basis, one can conclude that the C1 C8 bond is considerablyweaker than all the other bonds in the naphthalene ring of naproxen.When the force constants given for the monomer and dimerforms of conf. C1 are compared, it is seen that the R(O14 C16),r(O15 H23) and R(O14 C15) bond stretching coordinates as wellas the ı(O15 C14 O16), ı(C10 C13 C14), ı(C10 C13 C28) andı(C14 O15 H23) angle bending coordinates show the strongestsensitivity to intermolecular hydrogen bonding; very dramatic
changes (−2.795, −2.35 and 0.338 mdyn/Å for the bond stretch-ing coordinates, and −0.205, 0.389, 0.351 and 0.328 mdyn A forthe angle bending coordinates, respectively) were predicted forthe force constants of these seven internal coordinates, depending
K. Balci / Vibrational Spectroscopy 70 (2014) 168–186 175
Table 4The scaled diagonal force constants reproduced in the SQM-FF methodology for some of the bond stretching and angle bending internal coordinates of the monomer andcyclic-cis-dimer forms of conf. C1 and conf. T1 (the most stable cis and trans conformers of naproxen molecule) from the harmonic Cartesian force constants calculated atB3LYP/6-31G(d) level of theory. The corresponding units for the bond stretching and angle bending coordinates are mdyn/Å and mdyn A, respectively.
Internala coordinate Naphthalene Naproxen- conf. C1 Naproxen- conf. T1
Monomer Monomer Dimer Monomer Dimer
R(C1 C2) 4.389 4.392 4.397 4.384 4.387R(C1 C12) 4.401 4.408 4.410 4.430 4.433R(C1 C8) 3.105 3.130 3.132 3.121 3.121R(C2 C3) 5.374 5.208 5.215 5.220 5.222R(C3 O4) – 5.987 5.987 5.988 5.984R(C3 C6) 4.335 4.258 4.257 4.245 4.245R(O4 C5) – 4.954 4.957 4.957 4.962R(C6 C7) 5.373 5.529 5.528 5.541 5.546R(C7 C8) 4.389 4.349 4.349 4.336 4.338R(C8 C9) 4.399 4.449 4.451 4.472 4.478R(C9 C10) 5.362 5.245 5.245 5.165 5.158R(C10 C11) 4.347 4.228 4.222 4.292 4.297R(C10 C13) – 4.057 4.049 4.050 4.046R(C11 C12) 5.358 5.367 5.368 5.328 5.330R(C13 C14) – 3.850 3.883 3.881 3.907R(C13 C28) – 3.958 3.950 3.958 3.960R(O14 C15) – 5.689 6.027 5.683 6.021R(O14 C16) – 12.292 9.498 12.280 9.485R(O15 H23) – 7.225 4.881 7.226 4.871r(C5 H24) – 5.023 5.023 5.023 5.022r(C5 H25) – 4.761 4.761 4.758 4.760r(C5 H26) – 4.759 4.761 4.759 4.759r(C13 H27) – 4.829 4.839 4.815 4.831r(C28 H29) – 4.874 4.871 4.979 4.984r(C28 H30) – 4.924 4.924 4.921 4.919r(C28 H31) – 4.979 4.981 4.868 4.866r(C7 H19) 5.121 5.129 5.133 5.139 5.140r(C6 H20) 5.157 5.216 5.216 5.216 5.218r(C2 H18) 5.119 5.228 5.234 5.232 5.229r(C12 H17) 5.120 5.114 5.118 5.116 5.118r(C9 H21) 5.121 5.083 5.090 5.141 5.141r(C11 H22) 5.158 5.183 5.184 5.121 5.127ı(C13 C14 O15) – 0.726 0.696 0.726 0.697ı(C13 C14 O16) – 0.703 0.686 0.701 0.684ı(O15 C14 O16) – 0.888 0.683 0.888 0.683ı(C10 C13 C14) – 0.699 0.310 0.718 0.321ı(C10 C13 C28) – 0.710 0.359 0.699 0.362ı(C14 C13 C28) – 0.524 0.388 0.524 0.389ı(C3 O4 C5) – 1.449 1.462 1.450 1.462ı(C9 C1O C13) – 0.647 0.655 0.657 0.656ı(C2 C3 C6) 0.659 0.706 0.707 0.705 0.705ı(C1 C8 C7) 0.670 0.662 0.663 0.662 0.663ı(C9 C10 C11) 0.660 0.686 0.685 0.687 0.688ı(C8 C1 C12) 0.670 0.663 0.662 0.663 0.662ı(14 O15 H23) – 0.741 1.069 0.740 1.072ı(O4 C5 H24) – 0.595 0.594 0.595 0.595ı(O4 C5 H25) – 0.582 0.583 0.581 0.584ı(O4 C5 H26) – 0.582 0.582 0.581 0.584ˇ(C6 H20) 0.411 0.398 0.398 0.398 0.398ˇ(C7 H19) 0.427 0.424 0.425 0.424 0.425ˇ(C2 H18) 0.427 0.423 0.425 0.424 0.425ˇ(C9 H21) 0.429 0.433 0.434 0.434 0.434ˇ(C12 H17) 0.429 0.429 0.429 0.428 0.428ˇ(C11 H22) 0.411 0.422 0.419 0.419 0.420ı(C10 C13 H27) – 0.564 0.479 0.564 0.481ı(C13 C28 H29) – 0.484 0.454 0.490 0.455ı(C13 C28 H30) – 0.446 0.447 0.450 0.448
: CH o
ocsnfRirc
ı(C13 C28 H31) – 0.473
a The numeration of the atoms is as given in Fig. 1. R: CC or CO bond stretching; r
n dimerization. Another comparison to be made over the forceonstants of conf. C1 and naphthalene molecule may reveal the sub-tituent effects on the force field parameters associated with theaphthalene ring moiety; such a comparison has shown that the
orce constants of the R(C2 C3), R(C6 C7), R(C7 C8), R(C8 C9),
(C9 C10), R(C10 C11), r(C6 H20) and r(C2 H18) bond stretch-ng coordinates, all of which are related with the naphthaleneing of naproxen, differ from those reported for the correspondingoordinates of naphthalene molecule by values of −0.154, 0.168,
0.454 0.471 0.455
r OH bond stretching; ı: angle bending; ˇ: in-plane bending.
−0.053, 0.073, −0.197, −0.055, 0.059 and 0.113 mdyn/Å, respec-tively.
3.4. Theoretical vibrational spectral data
The anharmonic wavenumbers calculated at B3LYP/6-311++G(d,p) level of theory for the most stable cis and transconformers of naproxen (conf. C1 and conf. T1) and the scaledharmonic wavenumbers obtained at B3LYP/6-31G(d) and

176
K.
Balci /
Vibrational
Spectroscopy 70
(2014) 168–186
Table 5A comparison of the scaled theoretical IR and Raman spectral data obtained for the monomer and cyclic-cis-dimer forms of conf. C1 (the most stable cis conformer of naproxen molecule).
Modesa conf. C1 (monomer) conf. C1 (dimer) Mode descriptionsg
In phase Out of phase
�b (cm−1) �c (cm−1) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra)
1 13 28 33 0 1089 28 1 4277 27 0 1290 (CC)P
2 10 32 35 2 743 47 0 204 44 0 369 (CC)C , (CO)M , (CH3)M
3 40 47 49 2 141 84 1 50 60 1 28 (CC)P + (CC)C
4 93 91 94 2 275 107 0 124 103 9 82 (CO)M , (CH3)M
5 113 117 118 1 31 126 0 50 131 2 4 ˇ(CC)P , (CC)P + (CC)C
6 137 137 140 1 28 158 0 154 140 0 28 �(O· · ·H)C , ı(CO· · ·H)C , R
7 187 180 185 0 28 189 0 10 188 3 1 R + (CH3)M , (CO)M
8 192 193 196 2 57 203 0 75 202 4 43 R + (CH3)P
9 208 210 214 1 130 215 0 153 216 1 76 (CH3)P , ˇ(CO)M
10 225 217 222 0 26 229 0 147 228 2 20 (CH3)P
11 226 226 234 0 37 235 0 41 236 0 22 (CH3)M
12 272 272 279 0 24 318 0 29 330 55 2 �(carboxyl) + ıP(CCC) + ıR
13 282 276 282 3 63 283 0 51 280 0 62 (CO)M
14 383 379 386 3 12 396 12 13 388 0 8 ıR + ˇ(CO)M
15 389 389 393 1 6 399 0 74 397 0 35 R
16 399 395 399 5 94 406 1 161 428 50 1 w(carboxyl) + ıR
17 404 403 402 0 69 402 0 81 401 0 72 R , (CH)R
18 474 469 474 17 16 484 3 29 492 6 11 ı(CCO,CC O)C + ˇ(CO)M + ıR
19 477 474 480 4 1 479 1 8 479 10 6 R
20 522 515 523 6 88 531 1 22 532 4 54 ıR + ı(COC)M
21 528 522 528 1 97 527 2 238 527 3 90 ıR
22 561 554 561 27 13 605 6 11 610 12 1 ı(COC)M , ıR , R , (CH)R
23 592 587 594 19 15 572 2 15 573 9 3 R , (CH)R + (CC)P + (CO)M
24 625 620 632 39 31 671 0 21 682 42 0 scr(carboxyl) + ıR
25 637 633 645 19 24 921 0 2 959 167 13 w(OH)C
26 672 668 669 24 3 656 2 4 658 1 1 R , (CH)R
27 678 674 684 26 10 677 2 15 677 0 2 ıR + scr(carboxyl)28 741 736 738 24 208 737 4 326 739 10 19 ıR , �R + �(CO)M
29 779 750 750 1 34 750 1 62 750 1 19 R + (CH)R
30 785 783 780 23 82 860 2 151 864 12 11 �(CC)P , �(CH3)P
31 792 793 794 8 28 787 3 23 788 17 29 (CC)C + (CH)R
32 810 806 810 10 7 801 3 8 807 13 5 (CH)R
33 823 814 814 6 21 809 23 48 810 15 2 (CH)R
34 859 845 848 5 79 813 4 186 815 3 4 �(CC)P , �R , ıR
35 856 850 855 37 26 856 33 17 853 34 14 (CH)R
36 895 889 887 13 12 886 13 13 886 11 11 (CH)R
37 927 922 924 17 4 925 25 6 925 21 3 �R , ıR + �(CO)M
38 961 955 942 0 1 942 0 1 942 0 1 R(CH)39 971 956 953 0 13 949 7 7 966 10 2 R(CH)40 971 963 963 1 45 964 11 89 963 45 11 �R , ıR , ˇ(CH)R
41 991 986 982 1 46 986 7 56 985 4 60 �(CC)P , �(CH3)P
42 1028 1034 1035 57 7 1036 69 3 1036 36 10 �(CO)M
43 1062 1066 1068 72 6 1072 26 13 1072 25 1 �(CH3)P + �(CC)P
44 1065 1069 1071 20 19 1083 1 37 1084 7 17 �(CC)P , �(CH3)P
45 1130 1121 1124 18 6 1124 18 8 1124 5 7 ˇ(CH)R + �R
46 1128 1135 1144 202 3 1230 1 14 1239 264 12 �(CO)C , �(OH)C + ı(CH)P
47 1143 1145 1151 1 20 1152 1 26 1152 0 12 �(CH3)M
48 1161 1155 1155 3 12 1154 0 4 1154 1 27 ˇ(CH)R , �R
49 1165 1161 1163 36 76 1161 3 95 1161 14 51 ˇ(CH)R , �R + �(CC)P
50 1169 1170 1173 63 83 1175 10 21 1175 92 119 ˇ(CH)R , �R + �(CO)M , �(CC)P , �(OH)C
51 1191 1194 1197 18 41 1198 34 63 1198 3 22 �(CH3)M + �R
52 1224 1228 1227 142 7 1227 81 1 1227 283 7 �R , ˇR(CH) + �(CO)M

K.
Balci /
Vibrational
Spectroscopy 70
(2014) 168–186
177
Table 5 (Continued )
Modesa conf. C1 (monomer) conf. C1 (dimer) Mode descriptionsg
In phase Out of phase
�b (cm−1) �c (cm−1) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra)
53 1249 1256 1261 8 11 1285 9 24 1286 87 46 �R , ˇR(CH) + ı(CH)P
54 1268 1262 1266 81 20 1266 30 1 1266 76 2 ˇ(CH)R , �R , ıR + �(CO)M
55 1270 1267 1274 40 3 1271 120 12 1271 11 4 ˇ(CH)R , �R , ıR + �(CO)M
56 1271 1285 1293 4 21 1462 5 129 1437 61 12 �(OH)C + ı(CH)P
57 1351 1346 1352 4 11 1353 15 29 1353 5 16 ˇ(CH)R , �R
58 1343 1359 1367 10 119 1338 11 22 1335 118 11 �(OH)C , ı(CH)P + �(CC)P , �(CO,C O)C
59 1373 1374 1376 22 624 1370 0 1014 1370 0 395 �(CC)R
60 1382 1384 1380 45 91 1379 6 106 1378 12 31 umb(CH3)P
61 1388 1390 1388 58 134 1388 18 73 1389 112 241 �R , ˇ(CH)R
62 1422 1421 1432 7 154 1432 4 224 1429 71 100 �R , ˇ(CH)R
63 1461 1443 1438 14 66 1440 22 36 1441 43 116 umb(CH3)M + ˇ(CH)R
64 1472 1461 1453 6 82 1454 2 63 1454 9 101 scr(CH3)M
65 1469 1462 1454 7 39 1461 8 30 1461 3 57 scr(CH3)P
66 1479 1470 1468 17 53 1471 10 37 1468 48 81 scr(CH3)P
67 1456 1475 1470 31 69 1469 61 117 1470 1 22 scr(CH3)M
68 1484 1485 1494 28 126 1494 50 193 1494 6 50 ˇ(CH)R , �R
69 1506 1509 1513 31 3 1513 57 5 1513 3 0 �R , ˇ(CH)R
70 1574 1580 1580 5 42 1580 5 52 1580 4 25 �R
71 1609 1612 1613 99 22 1613 164 31 1613 26 17 �R , ˇ(CH)R
72 1627 1634 1632 70 320 1633 123 459 1633 21 155 �R , ˇ(CH)R
73 1769 1768 1765 242 17 1667 12 87 1719 586 9 �(C O)C , �(OH)C
74 2854 2906 2902 53 77 2903 70 146 2903 31 2 �(CH3)M
75 2900 2939 2937 29 101 2938 43 191 2937 22 23 �(CH3)P
76 2893 2963 2958 8 30 2962 6 42 2962 9 17 �(CH)P
77 2933 2968 2959 42 27 2960 6 35 2959 77 15 �(CH3)M
78 2961 3002 3002 32 71 3002 34 124 3002 40 18 �(CH3)P
79 2979 3021 3023 13 18 3023 11 36 3024 23 10 �(CH3)P
80 2997 3032 3029 30 91 3030 41 113 3030 22 70 �(CH3)M
81 2997 3049 3041 14 22 3044 12 21 3044 13 20 �(CH)R
82 3032 3055 3049 10 30 3051 5 23 3051 10 30 �(CH)R
83 3049 3061 3054 13 46 3056 14 45 3056 14 49 �(CH)R
84 3067 3077 3075 7 49 3076 8 47 3076 8 44 �(CH)R
85 3061 3090 3084 13 42 3086 9 129 3086 18 68 �(CH)R
86 3130 3091 3085 11 93 3086 10 51 3086 18 21 �(CH)R
87 3568 3627 3590 48 57 3034 0 543 3125 4370 1 �(OH)C
r.m.s.1 11.1 10.8 9.4 – – 8.3 – – 6.7 –r.m.s.2 16.1 11.9 10.6 – – 10.0 – – 9.0 –
a The corresponding normal modes of the monomer and dimer forms of conf. C1 are given in the same order. The illustrations of the modes are given in Figs. S3–S7.b The anharmonic wavenumbers, calculated at B3LYP/6-311++G(d,p) level of theory.c The scaled wavenumbers, derived from the harmonic wavenumbers calculated at B3LYP/6-311++G(d,p) level of theory using the procedure called “Dual scale factors”.d The scaled wavenumbers, reproduced in the SQM-FF methodology over the geometry and force field data calculated at B3LYP/6-31G(d) level of theory.e The IR intensities, reproduced in the SQM-FF methodology.f The Raman intensities, derived from the Raman activities calculated at B3LYP/6-31G(d) level of theory (see the text).g Based on the PED values given in Table S11 for the dimer form of conf. C1. �: bond stretching; ı: angle bending; ˇ: in-plane bending; : out-of-plane bending; : torsion; �: rocking; w: wagging; scr: scissoring; umb: umbrella.
The subscripts R, P, C, M denote naphthalene ring, propanoic acid chain, carboxyl and methoxy groups”, respectively.r.m.s.1: root-mean-square values, calculated for the spectral region between 50 and 1800 cm−1.r.m.s.2: root-mean-square values, calculated for the spectral region between 50 and 3700 cm−1.

178
K.
Balci /
Vibrational
Spectroscopy 70
(2014) 168–186
Table 6A comparison of the theoretical IR and Raman spectral data obtained for the monomer and cyclic-cis-dimer forms of conf. T1 (the most stable trans conformer of naproxen molecule).
Modesa conf. T1 (monomer) conf. T1 (dimer) Mode descriptionsg
In phase Out of phase
�b (cm−1) �c (cm−1) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra)
1 10 25 27 0 1546 22 0 5123 24 0 2445 (CC)P
2 20 38 37 2 672 60 1 23 48 0 291 (CC)C , (CO)M , (CH3)M
3 39 49 52 2 244 89 2 38 83 0 22 (CC)P + (CC)C
4 95 83 87 2 138 107 2 187 102 5 56 (CO)M , (CH3)M
5 117 114 114 1 44 122 0 18 130 1 3 ˇ(CC)P , (CC)P + (CC)C
6 136 140 144 1 30 162 0 131 150 1 34 �(O· · ·H)C , ı(CO· · ·H)C , R
7 174 172 173 0 106 187 2 126 187 1 2 R + (CH3)M , (CO)M
8 183 181 185 2 26 181 1 26 178 0 5 R + (CH3)P
9 215 216 222 0 45 214 0 27 215 0 5 (CH3)P , ˇ(CO)M
10 226 221 229 1 21 234 3 100 235 0 38 (CH3)P
11 244 235 244 2 7 256 0 13 259 13 12 (CH3)M
12 276 270 277 0 43 325 1 93 342 35 10 �(carboxyl) + ıP(CCC) + ı(COC)M + ıR
13 300 300 304 2 20 288 1 21 282 1 51 (CO)M
14 367 366 369 2 143 380 3 227 374 4 4 ı(Ring)C + ˇ(CO)M
15 389 387 391 2 8 397 0 14 396 1 2 R
16 408 401 403 0 40 413 7 64 432 46 2 w(carboxyl) + ıR
17 404 405 408 3 71 405 0 54 404 1 143 R , (CH)R
18 478 405 478 20 13 490 5 29 490 4 15 ı(CCO,CC O)C + ˇ(CO)M + ıR
19 481 477 479 4 0 478 1 0 478 13 1 R
20 508 503 513 6 35 519 7 89 525 8 1 ıR + ı(COC)M
21 527 521 528 1 128 528 1 155 529 2 73 ıR
22 563 554 562 26 15 606 8 28 609 0 1 ı(COC)M , ıR , R , R(CH)23 593 585 594 21 18 574 2 12 575 4 1 R , (CH)R + (CC)P + (CO)M
24 622 614 628 31 23 685 0 18 695 38 2 Scr(carboxyl) + ıR
25 636 635 652 20 32 921 4 4 962 223 8 w(OH)C
26 666 662 667 26 12 663 3 5 663 1 1 R , (CH)R
27 670 671 673 26 7 653 0 25 658 8 1 ıR + scr(carboxyl)28 739 734 736 29 195 735 6 235 737 12 7 ıR , �R + �(CO)M
29 778 751 751 2 32 750 0 95 751 3 12 R + (CH)R
30 782 777 779 27 131 828 5 25 830 2 5 �(CC)P , �(CH3)P
31 803 796 798 1 14 775 1 309 776 11 37 umb(carboxyl),(CC)C + (CH)R
32 820 809 813 21 20 802 1 14 803 0 10 (CH)R
33 822 820 819 1 30 812 41 22 812 2 26 (CH)R
34 845 842 844 3 54 854 3 112 858 24 2 �(CC)P , �R , ıR
35 867 845 849 37 19 849 6 17 849 53 39 (CH)R
36 901 904 908 14 7 902 17 8 902 13 13 (CH)R
37 925 921 923 22 5 924 12 3 924 38 5 �R , ıR + �(CO)M
38 964 951 938 0 1 938 1 1 938 1 1 R(CH)39 962 956 946 1 2 946 2 2 946 2 1 R(CH)40 976 959 963 1 56 964 13 88 964 3 12 �R , ıR , ˇ(CH)R
41 1003 1005 999 3 43 1001 1 40 1001 5 70 �(CC)P , �(CH3)P
42 1031 1034 1036 50 7 1037 49 6 1037 52 8 �(CO)M
43 1057 1059 1061 53 10 1072 10 6 1071 8 13 �(CH3)P + �(CC)P
44 1077 1078 1082 43 13 1087 16 16 1087 25 20 �(CC)P , �(CH3)P
45 1124 1118 1120 38 9 1123 18 12 1123 10 2 ˇ(CH)R + �R
46 1127 1127 1135 109 8 1240 29 25 1249 202 39 �(CO)C , �(OH)C + ıP(CH)47 1155 1144 1151 1 21 1153 1 20 1153 1 21 �(CH3)M
48 1155 1152 1154 42 8 1149 2 8 1149 23 46 ˇ(CH)R , �R
49 1163 1160 1161 74 69 1159 3 96 1159 1 5 ˇ(CH)R , �R + �(CC)P
50 1169 1166 1171 69 47 1171 32 24 1171 32 33 ˇ(CH)R , �R + �(CO)M , �(CC)P , �(OH)C
51 1196 1192 1196 11 39 1197 2 42 1197 3 18 �(CH3)M + �R
52 1217 1216 1220 147 10 1216 163 13 1215 276 20 �R , ˇR(CH) + �(CO)M

K.
Balci /
Vibrational
Spectroscopy 70
(2014) 168–186
179
Table 6 (Continued )
Modesa conf. T1 (monomer) conf. T1 (dimer) Mode descriptionsg
In phase Out of phase
�b (cm−1) �c (cm−1) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra) �d (cm−1) Int.e (IR) Int.f (Ra)
53 1259 1257 1261 74 7 1290 19 9 1291 24 58 �R , ˇR(CH) + ı(CH)P
54 1269 1265 1272 35 6 1263 97 4 1264 101 2 ˇ(CH)R , �R , ıR + �(CO)M
55 1262 1268 1277 13 19 1273 92 6 1273 20 6 ˇ(CH)R , �R , ıR + �(CO)M
56 1280 1292 1295 3 47 1460 8 125 1439 81 24 �(OH)C + ıP(CH)57 1350 1349 1355 14 10 1359 5 24 1358 1 4 ˇ(CH)R , �R
58 1352 1358 1363 2 299 1339 21 26 1336 122 9 �(OH)C , ı(CH)P + �(CC)P , �(CO,C O)C
59 1368 1369 1377 14 289 1365 0 872 1364 1 176 �(CC)R
60 1373 1385 1382 40 9 1394 5 8 1378 6 14 umb(CH3)P
61 1385 1392 1389 81 247 1389 65 66 1389 100 375 �R , ˇ(CH)R
62 1420 1420 1432 5 174 1430 5 263 1426 66 45 �R , ˇ(CH)R
63 1468 1442 1439 11 61 1440 19 51 1440 16 63 umb(CH3)M + ˇ(CH)R
64 1476 1460 1454 6 81 1455 2 64 1455 9 90 scr(CH3)M
65 1469 1461 1456 5 35 1468 28 42 1462 5 51 scr(CH3)P
66 1476 1469 1467 8 50 1470 49 20 1470 23 81 scr(CH3)P
67 1460 1474 1470 43 79 1485 14 118 1479 9 15 scr(CH3)M
68 1484 1484 1494 34 118 1493 29 180 1495 33 46 ˇ(CH)R , �R
69 1506 1509 1513 29 8 1514 33 14 1513 30 1 �R , ˇ(CH)R
70 1575 1579 1579 3 46 1579 5 60 1579 3 30 �R
71 1607 1610 1611 89 28 1611 134 47 1611 32 10 �R , ˇ(CH)R
72 1627 1634 1632 80 307 1632 89 438 1631 79 155 �R , ˇ(CH)R
73 1770 1766 1765 217 15 1667 10 71 1719 504 9 �(C O)C , �(OH)C
74 2857 2906 2902 54 78 2903 53 108 2903 59 46 �(CH3)M
75 2898 2938 2936 31 101 2937 50 180 2936 18 32 �(CH3)P
76 2896 2962 2954 11 38 2959 8 51 2960 11 21 �(CH)P
77 2925 2967 2959 43 28 2959 31 31 2959 51 20 �(CH3)M
78 2957 3001 3002 33 72 3001 34 124 3001 42 19 �(CH3)P
79 2974 3021 3023 13 17 3024 11 35 3030 23 7 �(CH3)P
80 2994 3032 3030 30 90 3034 33 110 3030 38 67 �(CH3)M
81 3017 3051 3045 7 27 3046 6 29 3046 9 23 �(CH)R
82 3034 3060 3057 2 9 3057 1 9 3057 1 3 �(CH)R
83 3032 3063 3060 16 26 3060 10 30 3057 20 32 �(CH)R
84 3047 3068 3062 19 94 3064 23 145 3064 24 31 �(CH)R
85 3075 3089 3086 13 39 3085 18 49 3085 19 36 �(CH)R
86 3074 3091 3086 11 92 3086 10 112 3087 7 47 �(CH)R
87 3566 3622 3591 47 53 3034 34 469 3127 4015 5 �(OH)C
r.m.s.1 10.9 10.2 8.9 – – 9.2 – – 7.3 – –r.m.s.2 15.6 11.3 10.3 – – 11.0 – – 9.8 – –
a The corresponding normal modes of the monomer and dimer forms of conf. T1 are given in the same order.b The anharmonic wavenumbers, calculated at B3LYP/6-311++G(d,p) level of theory.c The scaled wavenumbers, derived from the harmonic wavenumbers calculated at B3LYP/6-311++G(d,p) level of theory using the scaling procedure called “Dual scale factors”.d The scaled wavenumbers, reproduced in the SQM-FF methodology over the geometry and force field data calculated at B3LYP/6-31G(d) level of theory.e The IR intensities, reproduced in the SQM-FF methodology.f The Raman intensities, derived from the Raman activities calculated at B3LYP/6-31G(d) level of theory (please see the text).g Based on the PED values given in Table S12 for the dimer form of conf. T1. �: bond stretching; ı: angle bending; ˇ: in-plane bending; : out-of-plane bending; : torsion; �: rocking; w: wagging; scr: scissoring; umb: umbrella.
The subscripts R, P, C, M denote naphthalene ring, propanoic acid chain, carboxyl and methoxy groups”, respectively.r.m.s.1: root-mean-square values, calculated for the spectral region between 50 and 1800 cm−1.r.m.s.2: root-mean-square values, calculated for the spectral region between 50 and 3700 cm−1.

180 K. Balci / Vibrational Spectroscopy 70 (2014) 168–186
Table 7The assignments proposed for the fundamental bands observed in the experimental vibrational spectra of naproxen molecule.
Mode1 In solid phase (cm−1)2 In a polar solvent3 Assignments4
This study Refs. [19,23] This study
�(IR)a �(Ra)b �(Ra)c �(IR)d �(Ra)e �(Em)f �(IR)g �(IR)h �(Ra)j
1i – – – – – 33 – – – (CC)P
2o – 57(vs) – – – 51 – – – (CC)R ,(CO)M , (CH3)M
3i – 88(vs) – – – – – – – (CC)P + (CC)C
4i – 119(vs) – – – 113 – – – (CO)M
6i – 147(s) – – – – – – – �(O.H)C , ı(CO.H)C , R
7i,T – 163(s) 166(m) – – 173 – – 167(m) {B3u}: R(Butterfly)9i,C – 207(m) 207(sh) – – 208 – – 207(m) ˇ(CO)M , (CH3)P
10i – 233(m) 230(s) – – 223 – – 232(m) (CH3)P
12i,T – 300(m) 299(m) – 299* – – – 299(m) �(carboxyl), ıP(CCC) + ı(COC)M + ıR
13o – 286(w) 285(sh) – 285* – – – 285(m) (CO)M
14i – – 388(w) 385 – – – – – {B1u}: ıR + ˇ(CO)M
16i – 411(s) 410(s) – 409* – 412(sh) – 410(m) w(carboxyl) + {Ag}: ıR
16o 423(m) – – 424 – – – – 434(m) w(carboxyl) + {Ag}: ıR
17 400(m) 399(m) 400(m) 401* 398 – – – 398(sh) {B1g}: R
18 484(s) 485(m) 492(m) 485 486 – 489(vs) 482(vs) 492(w) ı(CCO,CC O)C , ˇ(CO)M , {B1u}:ıR
19o 473(m) – – 473 – – 475(s) 474(s) – {B3u}: R
20o 523(w) – – 523* – – 520(sh) 523(w) 524(s) {B3g}: ıR , ı(COC)M
21i – 524(vs) 524(s) – 523 521 – – – {Ag}: ıR
23 570(m) 574(m) 573(m) 570 – – 575(m) – 574(w) {B2g}: R
22 600(m) 601(m) 601(m) 601 – – 602(sh) 601(w) 602(w) ı(COC)M + ıR , R , (CH)R
24o,C 665(sh) – – – – – 664(m) 664(sh) – Scr(carboxyl) + {B2u}: ıR
24o,T 674(s) – – – – – 673(m) 674(m) – Scr(carboxyl) + {B2u}: ıR
27i,T – 644(m) 644(m) – 643* – – – 644(m) {B2u}: ıR + scr(carboxyl)25m 643(s) – – 644* – – 640(m) 643(m) – w(OH)C
25o 965(m) – – 966* – – – – – w(OH)C
28 743(m) 742(vs) 742(vs) 743* 741* 741 742(m) 742(m) 742(vs) {Ag}: ıR , �R
29 763(w) 762(s) 761(m) – 761 – 762(w) – 761(m) {B2g}: R
30i,C – 866(s) 866(s) – 865* – – – 868(sh) �(CC)P , �(CH3)P
31 793(s) 795(s) 795(s) 794* 794* 791 793(m) 793(m) 794(m) umb(carboxyl) + {B1g}: (CH)R
32i,T 806(sh) 805(m) 803(sh) – 804* – 806(sh) 805(sh) 805(sh) {Au}: (CH)R
33i 819(vs) – 819 – – 819(s) 819(m) 822(m) {B1g}: (CH)R
34i,C 824(sh) 823(m) 822(m) 825 822 – 825(sh) 824(sh) – �(CC)P + {B1u}: �R , ıR
34i,T – 857(s) 856(sh) – 855 857 – – – �(CC)P + {B1u}: �R , ıR
35o,T 855(vs) – – 856 – – 855(s) 855(s) 856(sh) {B2g}: (CH)R
35o,C 864(vs) – – 865 – – 863(s) 865(s) – {B2g}: (CH)R
36 896(vs) 896(m) 896(m) 897 – – 896(s) 895(s) – {B1g}: (CH)R
37 925(s) 927(m) 926(m) 926* – – 926(m) 926(m) – {B1u}: �R , ıR
40i 961(m) 962(s) 961(s) 962* 960* 966 965(m) 965(m) 961(m) {B3g}: �R , ıR
41 1005(m) 1005(s) 1006(s) 1006* 1004* – – 1002(sh) 1006(m) �(CC)P , �(CH3)P
42 1028(vs) 1031(m) 1030(m) 1029* 1029* 1040 1026(vs) 1030(s) – �(CO)M
43 1072(s) 1072(m) 1072(m) 1073* 1070* – 1069(sh) – 1074(w) �(CH3)P , �(CC)P
44 1091(s) 1093(m) 1092(m) 1092* 1092* – 1090(m) – – �(CC)P , �(CH3)P
45 1120(m) 1122(m) 1123(m) 1120* 1121* – 1120(m) 1118(sh) 1119(sh) {B1u}: ˇ(CH)R
46 – 1236(w) – – – – – – – �(CO)C , �(OH)C + ı(CH)P
48o 1158(vs) – – – – – 1155(m) 1157(s) – {Ag}: ˇ(CH)R
49i – 1155(m) 1156(sh) 1159 – – – – 1157(sh) {B2u}: ˇ(CH)R , �R + �(CC)P
50o 1176(vs) 1176(s) 1175(s) 1179* 1175* – 1175(m) 1176(s) 1175(s) {B2u}: ˇ(CH)R , �R + �(CH3)M
51i 1194(vs) 1196(m) 1195(m) 1195* 1195* – 1193(s) 1194(m) 1195(m) �(CH3)M + {B2u}: �R
52 1228(vs) – 1231 vw 1228 – – 1228(s) 1229(m) – {B3g}: �R , ˇ(CH)R + �(CO)M
53o 1304(m) 1297(m) 1299(m) – – – 1304(m) – 1295(sh) {B1u}: �R , ˇR(CH) + ı(CH)P
54o,T 1264(vs) 1265(m) 1267(m) 1265* – – 1264(vs) 1264(s) – {B3g}: ˇ(CH)R , �R + �(CO)M
55i,C 1277(s) – – 1278* – – 1275(sh) 1274(sh) – {B1u}:ˇ(CH)R ,�R ,ıR + �(CO)M
56o 1436(m) – – 1437* – – 1438(w) 1436(w) – �(OH)C , ı(CH) P
58 1347(m) – 1348(w) 1347* – – 1347(m) 1346(m) – �(OH)C , ı(CH) P
59i 1372(sh) 1370(m) 1372(sh) – 1370* 1377 – – 1376(sh) {Ag}: �(CC)R
60o 1385(sh) 1384(sh) – – – – 1382(sh) – – umb(CH3)P
61o 1395(vs) 1392(vs) 1392(vs) 1397* 1391* 1400 1395(vs) 1393(s) 1390(vs) {B2u}: �R
62 1419(m) 1421(s) 1421(s) 1420 1419 – 1419(s) 1419(m) 1421(s) {B3g}: �R , ˇ(CH)R
63o – 1439(m) 1440(m) 1437* 1439* 1430 – – – umb(CH3)M
64i 1452(s) 1456(s) 1456(s) 1453* 1454* – 1451(vs) 1452(s) 1456(s) scr(CH3)M
65 1462(m) 1462(m) – 1463* 1461* – 1457(sh) – scr(CH3)P
66 1469(m) 1470(m) 1470(sh) – 1469* – 1469(s) 1471(m) – scr(CH3)P
67i 1480(m) 1483(s) – 1481 1483 – 1480(s) 1484(m) – scr(CH3)M
68 1486(m) 1489(s) 1489(s) 1486* 1488* 1490 – – 1487(s) {Ag}: ˇ(CH)R , �R
69 1506(s) 1507(w) 1504(w) 1507* – – 1506(s) 1506(m) – {B2u}: �R , ˇ(CH)R
70 1578(w) 1579(s) 1579(s) – 1579 – 1578(w) 1576(w) 1581(s) {Ag}: �R

K. Balci / Vibrational Spectroscopy 70 (2014) 168–186 181
Table 7 (Continued )
Modea In solid phase (cm−1)b In a polar solventc Assignmentsd
This study Refs. [19,23] This study
�(IR)a �(Ra)b �(Ra)c �(IR)d �(Ra)e �(Em)f �(IR)g �(IR)h �(Ra)j
71i 1605(vs) 1603(w) – 1605 – – 1606(vs) 1607(s) – {B1u}: �R
72 1630(s) 1630(s) 1632s) 1631 1629 – 1631(m) 1636(w) 1632(s) {B3g}: �R
73 1686(vs) 1685(m) 1687(m) 1685* – – 1676(s) 1683(s) – �(C O)C
73o 1728(vs) 1729(m) 1729(m) 1729* – – 1730(s) 1730(s) 1728(m) �(C O)C
73m 1789(m) – 1788(vw) – – – 1788(m) 1789(w) – �(C O)C
74 2881(w)2906(w)
2882(m)2908(w)
2877(m)2908(w)
– 2882 – – – 2878(m) �(CH3)M
75i 2938(s) 2940(s) 2939(s) 2940* 2940* – 2935(s) 2936(s) 2927(s) �(CH3)P
76i – 2964(m) 2962(m) 2965* – – – – – �(CH)P
77 2962(s) – – – 2963* – – – – �(CH3)M
78 2974(s) 2973(s) 2977(s) 2977* 2978* – 2975(s) 2976(s) 2974(s) �(CH3)P
79 2996(m) 2997(m) 2995(sh) – – – – – �(CH3)P
80 3004(m) 3005(s) 3004(s) 3005* 3005* – – – 3002(m) �(CH3)M
81 3029(m) 3031(sh) 3030(sh) – – – 3026(w) – – {B3g}: �(CH)R
83 – 3043(m) 3042(m) – – – – – 3045(w) {B1u}: �(CH)R
87i 3070(m) 3067(s) 3066(s) – 3067* – 3069(w) – 3066(m) �(OH)C
87o 3199(vs) – 3200(w) 3178* – – – – – �(OH)C
1 The numeration of the normal modes is as given in Tables 5 and 6. The superscripts “i” and “o” indicate to the “in-phase” and “out-of-phase” vibrations of naproxen indimer form, while the superscript “m” indicates to the vibrations of naproxen in monomer form. The superscripts “C” and “T” indicate to conf C1 and conf. T1, specifically.
2 The fundamental bands observed in the vibrational spectra of naproxen in solid phase; (a) the wavenumbers obtained from the (KBr) IR spectrum; (b) the wavenumbersobtained from the solid phase FT-Raman spectrum; (c) The wavenumbers obtained from the solid phase dispersive Raman spectrum; (d) the IR spectral data taken from Ref.[23]; (e) the Raman spectral data taken from Ref. [23]; (f) the emission spectral data taken from Ref. [19]. The fundamental bands reassigned in this study are marked withasterisk (*) character.
3 The fundamental bands observed in the vibrational spectra of naproxen in a polar solvent; (g) the wavenumbers obtained from the FT-IR spectrum of naproxen in methanol;( the w
ndings ropan
BfpnahadPisigˇiusdobad{imtioocs1wfis
h) the wavenumbers obtained from the FT-IR spectrum of naproxen in ethanol; (j)
4 Based on the PED values given in Tables S10–S12. �: bond stretching; ı: angle becr: scissoring; umb: umbrella. The subscripts R, P, C, M denote naphthalene ring, p
3LYP/6-311++G(d,p) levels for their monomer and cyclic-cis-dimerorms as well as the associated IR and Raman intensities have beenresented in Tables 5 and 6; in these tables, the correspondingormal modes of the two conformers locate in the same rows andre labeled with the same mode numbers. In addition to these, thearmonic (unscaled) wavenumbers calculated at B3LYP/6-31G(d)nd B3LYP/6-311++G(d,p) levels of theory for the monomer andimer forms of the same structures as well as the correspondingED values produced in the SQM-FF methodology are presentedn Tables S10–S12 as a supporting data set. In these tables, theubscripts “P, C, M, R” which are used in the mode descriptionsndicate the propanoic acid chain, carboxyl, methoxy functionalroups and naphthalene ring moiety, while the characters “�, ı,, , , �, w, umb, scr” denote the bond stretching, angle bending,
n-plane bending, out-of-plane bending, torsion, rocking, wagging,mbrella and scissoring vibrations, respectively. According to thepectral data given for the monomer structures (see Table S10), theependencies of the calculated harmonic wavenumbers on the sizef the basis set used are generally very low for the spectral regionelow 1000 cm−1, but quite high for the region 1000–3200 cm−1
nd, in particular, for the region above 3200 cm−1. The sameata also indicate that an increase in the size of the basis setfrom 6-31G(d) to 6-311++G(d,p)} lead to remarkable down shiftsn the calculated wavenumbers almost for all the vibrational
odes; in the case of conf. C1, the maximum shifts for the abovehree spectral regions are 10, 31 and 72 cm−1, respectively. Themprovements occurred at the wavenumbers after the applicationf two different scaling procedures can be determined by meansf a comparison over the harmonic wavenumbers calculated foronf. C1 and corresponding scaled ones (see Table S10); the “Dualcale factors” procedure has led to improvements ranging from 1 to24 cm−1 which are proportional to the calculated wavenumbers,
hile the SQM-FF procedure has led to improvements rangingrom 1 to 132 cm−1 which are dependent on the types of thenternal coordinates contributing to the normal modes. As can beeen from Tables 5 and 6, the agreement between the anharmonic
avenumbers obtained from the dispersive Raman spectrum of naproxen in ethanol.; ˇ: in-plane bending; : out-of-plane bending; : torsion; �: rocking; w: wagging;oic acid chain, carboxyl and methoxy groups”, respectively.
wavenumbers and the scaled harmonic wavenumbers is quitegood in the low frequency region (below 1800 cm−1), however, thisis not true for the high frequency region (above 2850 cm−1); in thelow frequency region, the corresponding anharmonic wavenum-bers and the scaled harmonic wavenumbers differ from each otherby generally small values below 20 cm−1, while quite large devia-tions (up to 70 cm−1) are seen between them in the high frequencyregion. The dependency of the obtained theoretical wavenumbersto conformation can be revealed by a comparison over the valuesgiven for the monomer structures in Table 5 (for conf. C1) andTable 6 (for conf. T1). Such a comparison has shown that exceptthose reported for the modes 7{R(Butterfly)}, 8{R}, 11{(CH3)M},13{(CO)M}, 14{ıR + ˇ(CO)M}, 20{ı(COC)M + ıR}, 36{(CH)R},41{�(CC)P, �(CH3)P}, 44{�(CC)P, �(CH3)P} and 84{�(CH)R}, thecorresponding scaled wavenumbers of the two conformers differfrom each other by values smaller than 10 cm−1. On the other hand,one can determine the effects of intermolecular hydrogen bondingon the wavenumbers by comparing the corresponding values givenfor the monomer and dimer structures under the columns labeledas “�(d)′′ in the same tables. Such a comparison has indicated thatthe majority of the normal modes dominated by the carboxylgroup vibrations are strongly sensitive to dimerization; the mostprominent ones among them are the modes 12{�(carboxyl)},24{scr(carboxyl)}, 25{w(OH)C}, 46{�(CO)C, �(OH)C}, 56{�(OH)C},58{�(OH)C, ıP(CH)}, 73{�(C O)C, �(OH)C} and 87{�(OH)C}.Through the same comparison, it is also seen that the normalmodes 3{(CC)P}, 30{�(CC)P, �(CH3)P}, 53{ıP(CH)}, 22{ı(COC)M,ıR, R, R(CH)} and 23{R, (CH)R} and 34{(CH)R, R} exhibitrelatively lower but again quite considerable sensitivity to dimer-ization. Comparing the intensity values given for the monomerforms of conf. C1 and conf. T1 (see Tables 5 and 6), one can estimatethat the intensities of some fundamental bands observed in the
experimental IR and Raman spectra of naproxen should be stronglysensitive to conformation; the results from such a comparisonhave indicated that the intensities of the fundamental IR bands dueto the normal modes 16{ıR}, 17{R, (CH)R}, 24{scr(carboxyl)},
182 K. Balci / Vibrational Spectroscopy 70 (2014) 168–186
F (in Km eled h
3��ı8min91�4457stpflam5sf6
ig. 4. The experimental room-temperature FT-IR spectrum of naproxen moleculeonomer and cyclic-cis-dimer forms of its most stable cis and trans conformers, lab
1{(CC)C}, 32, 33{(CH)R}, 43{�(CH3)P, �(CC)P}, 44{�(CC)P,(CH3)P}, 45{ˇ(CH)R, �R}, 46{�(CO)C, �(OH)C}, 48, 49{ˇ(CH)R,R}, 51{�(CH3)M}, 53{ı(CH)P}, 54, 55, 57{ˇ(CH)R, �R}, 58{�(OH)C,(CH)P}, 59{�(CC)R}, 61{�R, ˇ(CH)R}, 66{scr(CH3)P}, 67{scr(CH3)M},1, 82, and 84{�(CH)R} show the strongest sensitivity to confor-ation. Another result obtained from the comparison is that the
ntensities of the fundamental Raman bands associated with theormal modes 1{(CC)P}, 3{(CC)P}, 4{(CO)M, (CH3)M}, 7, 8{R},{(CH3)P, ˇ(CO)M}, 11{(CH3)M}, 12{�(carboxyl)}, 13{(CO)M},4{ıR}, 16{ıR}, 20{ı(COC)M}, 21{ıR}, 26{R, (CH)R}, 30{�(CC)P,(CH3)P}, 31{(CC)C}, 32, 33{(CH)R}, 34{(CH)R, R}, 39{(CH)R},3{�(CH3)P, �(CC)P}, 44{�(CC)P, �(CH3)P}, 46{�(CO)C, �(OH)C},8{ˇ(CH)R, �R}, 50{ˇ(CH)R, �R}, 53{ı(CH)P}, 54, 55{ˇ(CH)R, �R},6{�(OH)C}, 58{�(OH)C, ı(CH)P}, 59{�(CC)R}, 60{umb(CH3)P}, 61,2{�R, ˇ(CH)R} and 82, 83 and 84{�(CH)R} show the strongestensitivity to conformation. On the other hand, the sensitivity ofhe band intensities to intermolecular hydrogen bonding can beredicted by another comparison over the intensity values givenor the monomer and dimer forms of conf. C1 under the columnsabeled as “Int (e)-IR” and “Int (f)-Ra” in Table 5. The results of such
comparison have indicated that the IR intensities of the funda-ental bands associated with the normal modes 12, 16, 25, 43, 46,
2, 53, 55, 56, 58, 61, 62, 71, 72, 73, and 87 should be strongly sen-itivity to dimerization. The sensitivity of the IR intensities of theundamentals due to the normal modes 49, 50, 59, 60, 63, 66, 67,8, 69, 74, and 77 is relatively much weaker but again considerable.
Br) and the scaled theoretical IR spectra obtained in SQM-FF methodology for theere as conf. C1 and conf. T1, respectively.
The comparison results have indicated that dimerization shouldcause very dramatic changes on the intensities of the fundamen-tal Raman bands associated with the normal modes 1–4, 6, 10, 15,16, 21, 28, 30, 34, 56, 58, 59, 61, 62, 68, 72–75, 85, and 87. More-over, considering the same results, one should also expect relativelymuch less but considerable changes for the intensities of the fun-damental Raman bands associated with the normal modes 20, 40,50, 60, 63, 67, 78, and 86 depending on dimerization. The theoreti-cally predicted effects of the conformation and dimerization on theexperimental IR and Raman spectral data of naproxen may be con-firmed by a final comparison over the theoretical and experimentalspectra; such a comparison given in Fig. 4 (for the IR spectra) and inFig. 5 (for the Raman spectra) reveals the better agreement of thetheoretical spectra of the dimer structures with the experimentalones compared to those of the monomers.
3.5. Assignments for the observed fundamental bands
The vibrational spectra of naphthalene which takes place innaproxen molecule as a constitutional fragment have been intenselyinvestigated by Sellers et al. [59], Sipachev [60], Lokshin et al.[61] and Srivastava and Singh [62] using the experimental IR
and Raman spectroscopic techniques as well as the theoreticalmolecular modeling technique based on Hartree-Fock and DFTcalculation methods. In the current literature, there are similarspectroscopic studies also on the other derivatives of naphthalene
K. Balci / Vibrational Spectroscopy 70 (2014) 168–186 183
Fig. 5. The experimental room-temperature FT-Raman spectrum of naproxen (in solid phase) and the scaled theoretical Raman spectra obtained in SQM-FF methodology fort , labele
m[1c[ttatttwTs“stcwvTtTbt
he monomer and cyclic-cis-dimer forms of its most stable cis and trans conformers
olecule such as 1-naphthaldehyde [63], 1-hydroxy naphthalene64], naphthalene acetic acid [65], 1-methoxy naphthalene [66],-naphthalene acetamide [67], 2,3-dimethyl naphthalene [68], 1-hloromethyl-2-methylnaphthalene [69], 1,5-methylnaphthalene70] and 2,3-naphthalenediol [71]. In the light of the improvedheoretical spectral data (see Tables 5 and 6) and also consideringhe previous assignments reported in the above studies, a reliablessignment of the fundamental bands observed in the experimen-al vibrational spectra of naproxen has been given in Table 7; inhe first column of the table where the mode numbers are given,he superscript “C” shows the assignments specifically associatedith conf. C1, and similarly, the assignments associated with conf.
1 are marked with the superscript “T”. In the same column, theuperscripts “i” and “o”, respectively, define the “in-phase” andout-phase” vibrations for the dimer form of naproxen, while theuperscript “m” defines the vibrations for the monomer form ofhe molecule. The reassignments proposed in the fifth and sixtholumns of the same table for some experimental wavenumbers,hich are thought to have been wrongly assigned in the pre-
ious studies on naproxen, are marked by asterisk character (*).he experimental wavenumbers involved into the calculation of
he r.m.s. error values (see Tables 5 and 6) are underlined inable 7. Since the r.m.s. values show the quality of the agreementetween the theory and experiment, one can confidently concludehat the scaled harmonic wavenumbers obtained for the dimerd here as conf. C1 and conf. T1, respectively.
forms of conf. C1 and conf. T1 fit the corresponding experimentalwavenumbers much better than those obtained for their monomerforms. Moreover, the much better agreement of the theoretical IRand Raman intensity data obtained for the dimer forms of the con-formers with the experiment compared to those obtained for theirmonomer forms is also prominent. Accordingly, the assignmentsproposed here for naproxen molecule have been based primarilyon the spectral data obtained for the dimer forms of the two con-formers.
3.5.1. Vibrations of the substituent groupsThe illustrations of the normal modes dominated by the group
vibrations of the substituents in naproxen molecule have beenpresented in Figs. S3–S5 as supporting data. As can be seen alsofrom the mode descriptions given in Tables 5 and 6, the modes1{(CC)P}, 3{(CC)P + (CC)C}, 10{(CH3)P}, 30{�(CC)P, �(CH3)P},41{�(CC)P, �(CH3)P}, 43{�(CH3)P, �(CC)P}, 44{�(CC)P, �(CH3)P},60{umb(CH3)P}, 65, 66{scr(CH3)P}, 75, 76, 78 and 79{�(CH3)P} aredominated by the methyl and ethyl group vibrations (see Fig. S3),and with only few exceptions, the majority of these fourteen modesshow considerable activity either in IR or Raman or in both. It has
been determined that the modes 3, 10, 30, 41 yield strong Ramanbands, while they are weakly active or completely inactive in IR.On the contrary, the modes 43 and 44 yield strong bands in the IRspectra but only medium-intensity bands in the Raman spectra.
184 K. Balci / Vibrational Spectroscopy 70 (2014) 168–186
Table 8A comparison over the corresponding ring vibrations of the various naphthalene derivatives.
Modea Naproxenb Naphthalenec Group-Ad Group-Be Assignmentf
IR1 Ra2 [59] [61] [62] [64] [65] [67] [71] [63] [66] [68]
7 – 163 166 176 – 171 – 183 – – – – {B3u}: R(Butterfly)14 – 388 359 357 357 – – 366 – – 390 370 {B1u}: ıR
17 400 399 389 386 386 428 – 418 410 382 395 395 {B1g}: R
19 473 – 474 476 476 480 488 497 490 488 – 460 {B3u}: R
20 523 – 508 – 506 530 516 530 – 521 525 500 {B3g}: ıR + ı(COC)M
21 – 524 514 510 512 – 514 – – – 526 – {Ag}: ıR
23 570 574 465 463 461 575 550 – 550 538 573 570 {B2g}: R
27 – 644 619 618 618 630 624 661 650 647 – 620 {B2u}: ıR
28 743 742 761 762 758 766 742 – 740 – – – {Ag}: ıR , �R
29 763 762 772 782 770 790 – – 775 – – {B2g}: R
32 806 805 825 841 835 798 800 799 – 849 842 830 {Au}: (CH)R
33 819 – 717 717 717 711 – 730 – 747 714 – {B1g}: (CH)R
35 855 864 – 875 876 876 877 878 851 879 870 880 869 857 880 {B2g}: (CH)R
36 896 896 951 948 943 890 – 891 890 920 – 900 {B1g}: (CH)R
37 925 927 810 846 841 851 – – 840 881 – – {B1u}: �R , ıR
40 961 962 939 938 935 985 – 975 – 1014 1019 – {B3g}: �R , ıR
45 1120 1122 1125 1128 1125 – – 1140 1105 1117 – – {B1u}: ˇ(CH)R
48 1158 – 1163 1158 1158 – 1151 1161 – 1167 – 1160 {Ag}: ˇ(CH)R
49 – 1155 1144 1138 1145 1149 1135 1147 1135 1159 – 1150 {B2u}: ˇ(CH)R , �R
50 1176 1176 1209 1210 1212 1170 1171 1187 1170 1216 1190 1190 {B2u}: ˇ(CH)R , �R
52 1228 – – – – 1180 1189 1210 1190 – 1217 1240 {B3g}: �R , ˇ(CH)R
54 1264 1265 1240 1243 1239 1242 1252 1238 – 1250 1239 1260 {B3g}: ˇ(CH)R , �R
55 1277 – 1265 1270 1269 1271 1265 1302 – 1265 1268 1270 {B1u}:ˇ(CH)R ,�R ,ıR
59 1372 1370 1361 1361 1361 1312 1346 1355 1365 1368 1340 1360 {Ag}: �(CC)R
61 1395 1392 1380 1380 1361 1390 1385 1424 1390 1395 1395 1390 {B2u}: �R
62 1419 1421 1445 1442 – 1445 1410 1440 1450 1452 1448 1400 {B3g}: �R , ˇ(CH)R
68 1486 1489 1460 – 1460 1460 – 1465 1460 1466 – 1470 {Ag}: ˇ(CH)R , �R
69 1506 1507 1509 1506 1504 1519 1509 – 1530 1511 1509 1505 {B2u}: �R , ˇ(CH)R
70 1578 1579 1578 – 1577 1581 1584 1570 – 1579 1581 1570 {Ag}: �R
71 1605 1603 1595 1600 1593 1594 1597 1594 1610 1620 1606 – {B1u}: �R
72 1630 1630 1624 1626 1624 1650 1631 1619 1630 1632 1628 – {B3g}: �R
81 3029 3031 3055 3055 3052 3052 3054 3058 3040 3039 3048 3040 {B3g}: �(CH)R
83 – 3043 3065 3066 3065 3060 3066 3067 3060 3053 3059 3050 {B1u}: �(CH)R
a The numeration of the normal modes is as given in Tables 5–7.b The experimental wavenumbers observed in this study for naproxen. 1 The wavenumbers observed in the (KBr) IR spectrum and 2 the wavenumbers observed in the solid
phase FT-Raman spectrum.c The experimental wavenumbers collected from Refs. [59,61,62] for naphthalene molecule.d The experimental wavenumbers collected from different references for the naphthalene derivatives involved into Group-A {1-hydroxy naphthalene [64], naphthalene
acetic acid [65], 1-naphthalene acetamide [67], 2,3-naphthalenediol [71]}.e The experimental wavenumbers collected from different references for the naphthalene derivatives involved into Group-B {1-naphthaldehyde [63], 1-methoxy naph-
taphth
U7o12ıctbayamb947tmoW4di
halene [66], 2,3-dimethyl naphthalene [68]}.f The assignments proposed in this study for the fundamental bands due to the n
nlike the other modes dominated by the methyl group, the modes5 and 78 yield strong bands both in the IR and Raman spectraf naproxen. The modes 2{(CC)C}, 6{�(O· · ·H)C,ı(CO· · ·H)C,R},2{�(carboxyl) + ı(CCC)P}, 16{w(carboxyl)}, 24{scr(carboxyl)},5{w(OH)C}, 31{umb(carboxyl)}, 46{�(CO)C, �(OH)C}, 56{�(OH)C,(CH)P}, 73{�(C O)C}, 87{�(OH)C} are the ones dominated by thearboxyl group vibrations (see Fig. S4). Of these eleven modes,he modes 2, 6, 12, 56 and 73 yield strong or medium-intensityands in the Raman spectra, however, they are either very weaklyctive or inactive in IR. On the contrary, the modes 24 and 25ield strong IR bands but do not show any activity in Raman. Inll the modes dominated by the carboxyl group vibrations, theodes 16, 31 and 87 are the only ones yielding strong bands
oth in the IR and Raman spectra. The normal modes 4{(CO)M},{ˇ(CO)M}, 11{(CH3)M}, 13{(CO)M}, 22{ı(COC)M}, 42{�(CO)M},7{�(CH3)M}, 51{�(CH3)M}, 63{umb(CH3)M}, 64, 67{Scr (CH3)M},4, 77, 80 {�(CH3)M} are dominated by the methoxy group vibra-ions and the majority of these fourteen modes (see Fig. S5) yield
edium-intensity bands in the Raman spectra, while only the fewf them (the modes 4, 64, 67, 80) exhibit strong activity in Raman.
hen focused on their IR activities, it is seen that the modes2, 51, 64, 77 yield strong bands, while the rest of the modesominated by this functional group are either weakly active or
nactive.
alene ring vibrations of naproxen. These are the same as given in Table 7.
3.6. Vibrations of naphthalene ring
In order to facilitate the discussion, the naphthalene ringvibrations of naproxen, which are illustrated in Figs. S6 andS7 (supporting data), are being associated here with those ofbare naphthalene molecule which belongs to the D2h symmetrypoint group. In this frame, the theoretical intensity data given inTables 5 and 6 lead us to the following conclusions on their IR andRaman activities; (i) the modes 7 (R-butterfly) and 19 (R) yieldbands with strong or medium intensity in the IR and Raman spec-tra of naproxen, while the others dominated by the ring vibrationsof B3u symmetry are completely inactive. (ii) The ring modes 37{�R,ıR}, 55{ˇ(CH)R, �R}, 71{�R} which are of B1u symmetry yield strongbands in the IR spectra, however, they are either weakly active orinactive in Raman. As for the modes 45{ˇ(CH)R}, 53{�R, ˇ(CH)R},83{�(CH)R}, the other three ring modes of the same symmetry, itis seen that the first two of them yield medium-intensity bandsboth in the IR and Raman spectra, while the latest yields medium-intensity bands in the Raman spectra only. (iii) The modes 21{ıR},28{ıR, �R}, 59{�(CC)R}, 68{ˇ(CH)R, �R} and 70{�R} which are of
Ag symmetry yield strong bands in the Raman spectra while theyexhibit either weak activity or inactivity in IR. Unlike them, themode 48{ˇ(CH)R} of the same symmetry yields strong IR bands,however it is completely inactive in Raman as is also true for bare
ctrosc
nwNlosRˇte6atotooom6aRbiTnfisRt
admiRoumia[ltmsim5trdd
4
tnembtaf
[[
[[[[[[
[
K. Balci / Vibrational Spe
aphthalene molecule. (iv) Both of the modes 33 and 36{(CH)R}hich are of B1g symmetry yield very strong bands in the IR spectra.evertheless, while the former is completely Raman inactive, the
ater yields medium-intensity bands in the Raman spectra. On thether hand, the mode 17{R}, which is another ring mode of theame symmetry, yields medium-intensity bands in both the IR andaman spectra. v) The modes 20{ıR, ı(COC)M}, 40{�R, ıR}, 52, 62{�R,(CH)R), 54{ˇ(CH)R, �R}, 72{�R} and 81{�(CH)R} are of B3g symme-
ry, and except the first one, they exhibit generally strong activitiesither in IR or Raman or in both. In this series, the modes 40 and2 are the ones yielding very strong bands in the Raman spectrand medium-intensity bands in the IR spectra. Unlike these two,he modes 52 and 54 yield strong bands in the IR spectra but weakr medium-intensity bands in the Raman spectra. (vi) For naproxen,he vibrational modes of Au symmetry are, in general, weakly activer inactive both in IR and Raman; the mode 32{(CH)R} is the onlyne showing a notable activity among them and yields shoulderr medium-intensity bands in the IR and Raman spectra of theolecule. (vii) The modes 27{ıR}, 49, 50{ˇ(CH)R, �R}, 61{�R} and
9{�R, ˇ(CH)R) are of B2u symmetry. The first two in this seriesre IR inactive, however, they yield medium-intensity bands in theaman spectra. As for the next two, they yield very strong bandsoth in the IR and Raman spectra. Unlike the others, the final mode
n the series yields strong IR bands but weak Raman bands. (viii)he modes 23, 29{R} and 35{(CH)R} are the three modes domi-ated by the ring vibrations of B2g symmetry. Of these modes, therst one yields medium-intensity bands both in the IR and Ramanpectra, while the second one yields medium-intensity bands in theaman spectra only. As for the third one, it yields strong bands inhe IR spectra, however, it does not show any Raman activity.
The effects of the interactions between the substituent groupsnd naphthalene ring moiety on the observed vibrational spectralata of naproxen has to be taken into account if a reliable assign-ent of the fundamental bands associated with its ring vibrations
s aimed. From this point of view, a comparison over the IR andaman bands observed in this study for naproxen and those previ-usly reported for the other naphthalene derivatives may be quiteseful. Such a comparison is given in Table 8 over naphthaleneolecule [59,61,62] and some naphthalene derivatives involved
nto “Group-A {1-hydroxy naphthalene [64], naphthalene aceticcid [65], 1-naphthalene acetamide [67], 2,3-naphthalenediol71]}” and “Group-B {1-naphthaldehyde [63], 1-methoxy naphtha-ene [66], 2,3-dimethyl naphthalene [68]}”; in condensed phase,he molecules in Group-A have a strong potential to form inter-
olecular hydrogen bonds, however, none of those in Group-B hasuch a potential. The results obtained from this comparison havendicated that the effects of the substituent groups on the funda-
ental bands due to the ring modes 14, 23, 27, 32, 33, 36, 37, 50,2, 54, 62 and 68 are much more remarkable than those due tohe other ring vibrations of the molecule. In the light of the sameesults, one can also confirm that dimerization does not cause anyramatic change at the wavenumbers of the fundamental bandsue to the group vibrations of naphthalene ring.
. Conclusion
Molecular dynamics simulations and thermochemistry calcula-ions carried out at different levels of theory have revealed thataproxen in free state should have at least nine stable conform-rs and two of which dominate the experimental spectra of theolecule. The effects of conformation and intermolecular hydrogen
onding on the stabilization of naproxen and also on its struc-ural and vibrational spectral properties were elucidated by theid of the calculations performed for the monomer and dimerorms of these two conformers, labeled here as conf. C1 and conf.
[
[
[
opy 70 (2014) 168–186 185
T1. The calculation results showed that naproxen can form threedifferent dimer structures referred to here as “cyclic-cis-dimer”,“cyclic-trans-dimer” and “open-chain-dimer”, and the first of themcorresponds to the most favored one in energy. A number of com-parisons over the monomer and cyclic-cis-dimer forms of conf. C1and conf. T1 enabled a reliable prediction for the effects of inter-molecular hydrogen bonding and conformation on the geometry,force field and vibrational motions of naproxen; the comparisonresults confirmed that dimerization causes very dramatic changeson the force field and vibrational spectral data associated with thecarboxyl group. A reliable assignment of the fundamental bandsobserved in the experimental IR and Raman spectra of the moleculewas given in the light of the vibrational spectral data calculated inthe harmonic and anharmonic oscillator approaches by taking intoaccount these changes and also those resulting from conformationand substituent effects. In the fitting of the calculated harmonicwavenumbers to the observed wavenumbers, two different scal-ing procedures (“Dual scale factors”, “SQM-FF”) were proceededindependently. Comparisons made over the corresponding exper-imental and theoretical spectral data confirmed not only that theexperimental IR and Raman spectra of naproxen be dominated byits cyclic-cis-dimer form but that the SQM-FF procedure is consid-erably more successful in matching these spectra than both theother scaling procedures used here and the anharmonic oscillatorapproach.
Acknowledgements
This study was supported by the Research Fund of Istanbul Uni-versity; the project number: N-1650/25092007. Dear Prof. WilliamCollier (Oral Roberts University, Department of Chemistry) for pro-viding the Fcart software and Assoc. Prof. Elif Akalin (IstanbulUniversity, Department of Physics) for her valuable support inrecording the FT-Raman spectrum of naproxen deserve the mostspecial thanks for this study.
Appendix A. Supplementary data
Supplementary material related to this article can befound, in the online version, at http://dx.doi.org/10.1016/j.vibspec.2013.12.002.
References
[1] P. Patrignani, Brain Res. Rev. 48 (2) (2005) 352–359.[2] C. Boynton, C. Dick, F. Mayer, J. Clin. Pharmacol. 28 (1988) 512–517.[3] S.D. Silberstein, Neurology 71 (2) (2008) 114–121.[4] D.W. Lewis, M.T. Middlebrook, C. Deline, Ann. Neurol. 36 (1994) 542–543.[5] D.W. Lewis, P. Winner, NeuroRx 3 (2) (2006) 181–191.[6] E.A. MacGregor, Drugs 70 (14) (2010) 1799–1818.[7] E. Sullivan, C. Bushnell, Curr. Pain Headache Rep. 14 (5) (2010) 376–384.[8] R.C. Moon, C.J. Detrisac, C.F. Thomas, G.J. Kelloff, J. Cell. Biochem. Suppl. 161
(1992) 134–138.[9] G.J. Kelloff, C.W. Boone, J.A. Crowell, V.E. Steele, R. Lubet, C.C. Sigman, Cancer
Epidem. Biomar. Prev. 3 (1994) 85–98.10] F.M. Giardiello, G.J. Offerhaus, R.N. DuBois, Eur. J. Cancer 31A (1995) 1071–1076.11] V.E. Steele, C.V. Rao, Y. Zhang, J. Patlolla, D. Boring, L. Kopelovich, M.M. Juliana,
C.J. Grubbs, R.A. Lubet, Cancer Prev. Res. (Phila) 2 (11) (2009) 951–956.12] S.C. Vlad, D.R. Miller, N.W. Kowall, D.T. Felson, Neurology 70 (2008) 1762–1767.13] B.P. Imbimbo, Expert Opin. Invest. Drugs 18 (2009) 1147–1168.14] B.P. Imbimbo, V. Solfrizzi, F. Panza, Front. Aging Neurosci. 2 (2010) 1–14.15] K. Ravikumar, S.S. Rajan, V. Pattabhi, E.J. Gabe, Acta Cryst. C41 (1985) 280–282.16] S. Wenzel, W. Buss, J. Phys. Org. Chem. 5 (1992) 748–754.17] E. Bednarek, W. Bocian, J.Cz. Dobrowolski, L. Kozerski, N. Sadlej-Sosnowska, J.
Sitkowski, J. Mol. Struct. 559 (2001) 369–377.18] F. Bachechi, M. Flieger, M. Sinibaldi, Acta Cryst. C53 (1997) 136–140.
19] F. Lahmani, K. Le Barbu-Debus, N. Seurre, A. Zehnacker-Rentien, Chem. Phys.Lett. 375 (2003) 636–644.20] M.D. King, W.D. Bunchanan, T.M. Korter, Phys. Chem. Chem. Phys. 13 (2011)
4250–4259.21] N. Okulik, A.H. Jubert, J. Mol. Struct. 769 (2006) 135–141.

1 ctrosc
[
[
[[[[[[
[[[
[
[
[[[[[
[[[
[
[[[[[[
[[[[[[[
[[[[[
[[
[[
[
[
[[
86 K. Balci / Vibrational Spe
22] L.A. Alvarez-Valtiera, J.W. Young, D.W. Pratt, Chem. Phys. Lett. 509 (2011)96–101.
23] A. Jubert, M.L. Legarto, N.E. Massa, L.L. Tevez, N.B. Okulik, J. Mol. Struct. 783(2006) 34–51.
24] S. Luber, J. Neugebauer, M. Reiher, J. Chem. Phys. 132 (4) (2010) 44113–44129.25] L. Liu, H. Gao, Spectrochim. Acta Part A 86 (2012) 131–138.26] D.L. Tomasko, M.T. Timko, J. Cryst. Growth 205 (1999) 233–243.27] S.M. Kerwin, J. Am. Chem. Soc. 132 (2010) 2466–2467.28] N.L. Allinger, J. Comput. Chem. 14 (1993) 755–768.29] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheese-
man, J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S.Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A.Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P.Hratchian, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Strat-mann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K.Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich,A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari,J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Ste-fanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith,M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. John-son, W. Chen, M.W. Wong, C. Gonzalez, J.A. Pople, Gaussian 03, Revision E.01,Gaussian, Inc., Wallingford, CT, 2004.
30] A.D. Becke, J. Chem. Phys. 107 (1997) 8554–8560.31] H.L. Schmider, A.D. Becke, J. Chem. Phys. 108 (1998) 9624–9631.32] L.A. Curtiss, K. Raghavachari, P.C. Redfern, V. Rassolov, J.A. Pople, J. Chem. Phys.
109 (1998) 7764–7776.33] L.A. Curtiss, K. Raghavachari, P.C. Redfern, V. Rassolov, J.A. Pople, Chem. Phys.
Lett. 314 (1999) 101–107.34] A.G. Baboul, L.A. Curtiss, P.C. Redfern, K. Raghavachari, J. Chem. Phys. 110 (1999)
7650–7657.35] T. Koopman, Physica 1 (1934) 104–110.36] W. Yang, R.G. Parr, Proc. Natl. Acad. Sci. U.S.A. 82 (1985) 6723–6726.37] R.G. Pearson, Proc. Natl. Acad. Sci. U.S.A. 83 (1986) 8440–8441.38] R.G. Pearson, J. Chem. Sci. 117 (2005) 369–377.
39] R.G. Parr, R.A. Donelly, M. Levy, W.E. Palke, J. Chem. Phys. 68 (1978)3801–3807.40] R.G. Parr, R.G. Pearson, J. Am. Chem. Soc. 105 (1983) 7512–7516.41] R.G. Parr, W. Yang, J. Am. Chem. Soc. 106 (1984) 4049–4050.42] R.G. Parr, L. Szentpaly, S. Liu, J. Am. Chem. Soc. 121 (1999) 1922–1924.
[
[
opy 70 (2014) 168–186
43] F. Weinhold (Ed.), NBO 5.0/5.G Program Manual, Theoretical Chemistry Insti-tute and Department of Chemistry, University of Wisconsin, Madison, 2008.
44] P.W. Ayers, M. Levy, Theor. Chem. Acc. 103 (2000) 353–360.45] P.W. Ayers, R.G. Parr, J. Am. Chem. Soc. 122 (2000) 2010–2018.46] M.D. Halls, J. Velkovski, H.B. Schlegel, Theor. Chem. Acc. 105 (2001) 413–421.47] G. Rauhut, P. Pulay, J. Phys. Chem. 99 (1995) 3093–3100.48] P. Pulay, G. Fogarasi, F. Pang, J.E. Boggs, J. Am. Chem. Soc. 101 (1979) 2550–2560.49] G. Fogarasi, X. Zhou, P.W. Taylor, P. Pulay, J. Am. Chem. Soc. 114 (1992)
8191–8201.50] T. Frosch, M. Schmmit, J. Popp, Anal. Bional. Chem. 387 (2007) 1749–1757.51] W.B. Collier, I. Magdo, T.D. Klots, J. Chem. Phys. 110 (1999) 5710–5720.52] S.E. Lappi, W.B. Collier, S. Franzen, J. Phys. Chem. A 106 (2002) 11446–11455.53] W.B. Collier, QCPE Bull. 13 (1993) 19.54] J. Baker, A.A. Jarzecki, P. Pulay, J. Phys. Chem. A 102 (1998) 1412–1424.55] D. Michalska, R. Wysokinski, Chem. Phys. Lett. 403 (2005) 211–217.56] V. Krishnakumar, G. Keresztury, T. Sundius, R. Ramaswamy, J. Mol. Struct. 702
(2004) 9–21.57] S. Simon, M. Duran, J.J. Dannenberg, J. Chem. Phys. 105 (1996) 11024–11031.58] S.F. Boys, F. Bernardi, Mol. Phys. 19 (1970) 553–566.59] H. Sellers, P. Pulay, J.E. Boggs, J. Am. Chem. Soc. 107 (1985) 6487–6494.60] V.A. Sipachev, J. Mol. Struct. 121 (1985) 143–151.61] B.V. Lokshin, N.E. Borisova, B.M. Senyavin, M.D. Reshetova, Russ. Chem. Bull.
Int. Ed. 51 (9) (2002) 1656–1666.62] A. Srivastava, V.B. Singh, Indian J. Pure Appl. Phys. 45 (2007) 714–720.63] V. Krishnakumar, N. Prabavathi, S. Muthunatesan, Spectrochim. Acta Part A 69
(2008) 528–533.64] V. Krishnakumar, R. Mathammal, J. Raman Spectrosc. 40 (2009) 1599–1604.65] E. Kavitha, N. Sundaraganesan, S. Sebastian, M. Kurt, Spectrochim. Acta Part A
77 (2010) 612–619.66] R.J. Xavier, V. Balachandran, M. Arivazhagan, G. Ilango, Indian J. Pure Appl. Phys.
48 (2010) 245–250.67] C. Ravikumar, L. Padmaja, I. Hubert Joe, Spectrochim. Acta Part A 75 (2010)
859–866.68] T. Prabhu, S. Periandy, S. Mohan, Spectrochim. Acta Part A 78 (2011) 566–574.69] P.B. Nagabalasubramanian, M. Karabacak, S. Periandy, Spectrochim. Acta Part
A 85 (2012) 43–52.70] P.B. Nagabalasubramanian, S. Periandy, Spectrochim. Acta Part A 77 (2010)
1099–1107.71] D. Shoba, S. Periandy, M. Karabacak, S. Ramalingam, Spectrochim. Acta Part A
83 (2011) 540–552.