Embed Size (px)
Citation preview

Constitutive Internalization of Constitutively Active Angiotensin IIAT1A Receptor Mutants Is Blocked by Inverse Agonists*
Received for publication, August 30, 2001, and in revised form, November 20, 2001Published, JBC Papers in Press, November 29, 2001, DOI 10.1074/jbc.M108398200
Stephanie Miserey-Lenkei‡, Charles Parnot‡, Sabine Bardin‡, Pierre Corvol§, and Eric Clauser‡¶
From ‡INSERM EPI 0103, Institut Cochin, 24 rue du Faubourg Saint-Jacques, 75014 Paris, France and §INSERM U36,College de France, 11 place Marcelin Berthelot, 75005 Paris, France
As constitutively active mutants (CAMs) mimic an ac-tive conformation, they can be used to characterize theprocess of G protein-coupled receptor activation. Here,we used CAMs to study the link between activation andinternalization of the angiotensin II AT1A receptor. Thecellular localization of fluorescently tagged N111A,I245T, and L305Q mutants was determined by confocalmicroscopy. In the absence of ligand, CAMs were mostlylocated in intracellular vesicles, whereas the wild-typeAT1A was found at the cell surface. After 2 h incubationwith inverse agonist, losartan, CAMs were translocatedto the plasma membrane. Similar observations weremade in H295, a human adrenocortical cell line whichexpresses physiologically the AT1 receptor. This phe-nomenon, which was not dependent on protein synthe-sis and the pharmacology and kinetics of which weresimilar to the recycling of the wild-type receptor, wascalled “externalization”. After externalization and losar-tan removal, the L305Q CAM underwent rapid ligand-independent endocytocis, with the same kinetics andtemperature sensitivity as the angiotensin II-inducedinternalization of the wild-type AT1A. Moreover, the ad-dition of a second mutation known to block internaliza-tion (�329 truncation) prevented intracellular localiza-tion of the CAM. These data show that AT1A CAMs areconstitutively and permanently internalized and recy-cled. This mechanism is different from the down-regu-lation observed for CAMs of other G protein-coupledreceptors and thus defines a new paradigm for the cel-lular regulation of CAMs.
G protein-coupled receptors (GPCR)1 form one of the largestprotein families, with several hundred members in humans (1).Despite the wide variety of ligands and physiological roles,these receptors are all structurally characterized by seven-transmembrane domains and most of them are thought toshare common activation and desensitization mechanisms.GPCRs are supposed to isomerize spontaneously between an
inactive (R) and an active state (R*), the latter being responsi-ble for G protein coupling and subsequent intracellular signal-ing. This two-state model is probably oversimplified but ishelpful for the interpretation of mutagenesis and pharmacolog-ical data. It is supported by the existence of constitutivelyactive mutants (CAMs) in the GPCR family. These mutantsmimic the active state and therefore present permanent ligand-independent signaling. Ligands able to block this constitutiveactivity are called inverse agonists. In the two-state model, theinverse agonists have preferential affinity for the inactive state(R). Conversely, “regular” agonists preferentially bind the ac-tive state (R*).
Many GPCRs are desensitized after G protein activation, i.e.they become insensitive to agonists. The binding of arrestins tothe receptor is known to play a major role in this process and isfavored by the phosphorylation of the receptor by specificGPCR kinases. The arrestins prevent further interaction withthe G proteins. They also promote internalization via clathrin-coated pit-dependent endocytosis, which results in the disap-pearance of the receptor from the plasma membrane (2). Fi-nally, they participate in the recycling of the receptor, whichcan take a few minutes to a few hours, depending on the typeof GPCR. In some cases, some of the receptors can also bedown-regulated, leading to long-term desensitization. Activa-tion is thus a physiological prerequisite for receptor internal-ization and these two processes are probably highly dependenton each other. As CAMs mimic the active conformation of thereceptor, they should help to elucidate the link between acti-vation and internalization.
In this study, we used the angiotensin II (AngII) type 1receptor (AT1) as a model to address this question. The AT1
receptor regulates the contraction and hypertrophy of vascularsmooth muscle cell contraction and the secretion of aldoster-one. Thus it plays a critical role in the control of blood pressureand sodium homeostasis. This pivotal physiological role makesthe AT1 receptor an important therapeutic target and numer-ous antihypertensive and cardioprotector agents have beendeveloped. As a consequence, it is also one of the most studiedGPCRs. Its cDNA was first cloned in 1991 (3). The AT1 receptor(AT1A and AT1B in rodents) leads to the G�q/11-mediated acti-vation of phospholipase C-�, which generates diacylglyceroland inositol (1,4,5)-trisphosphate. Following the phosphoryla-tion of the intracellular sequences (4, 5), the AT1 receptor israpidly internalized (t1⁄2 � 5 min) (6–10) in clathrin-coated pitsafter interaction with �-arrestin 1 and dynamin 1 and 2 (11).The receptor is then slowly and partly recycled at the plasmamembrane (7, 12, 13). We were able to directly visualize theinternalization process of an EGFP-tagged AT1 receptor usingconfocal microscopy (10).
Only one CAM was identified by site-directed mutagenesis(N111A) (14, 15), but we used a random mutagenesis approachto identify several other CAMs of the AT1A receptor (16). The
* This work was supported by Grant 98126 from Hœscht MarionRoussel and the Institut National Pour la Sante and la RechercheMedicale (INSERM). The costs of publication of this article were de-frayed in part by the payment of page charges. This article musttherefore be hereby marked “advertisement” in accordance with 18U.S.C. Section 1734 solely to indicate this fact.
¶ To whom correspondence should be addressed: INSERM EPI 0103-ICGM, Faculte de Medecine Cochin, Port Royal, 24 rue du Fg StJacques, 75014 Paris, France. Tel.: 33-1-53-73-27-50; Fax: 33-1-53-73-27-51; E-mail: [email protected].
1 The abbreviations used are: GPCR, G protein-coupled receptor;AngII, angiotensin II; AT1, angiotensin II type 1 receptor; IP, inositolphosphate; EGFP, enhanced green fluorescent protein; WT, wild-type;FACS, fluorescence-activated cell sorting; Endo H, endoglycosidase H;AR, adrenergic receptor; FCS, fetal calf serum; CAN, constitutive activemutant.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 277, No. 8, Issue of February 22, pp. 5891–5901, 2002© 2002 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 5891
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

ligand-independent activation of the AT1 signaling pathwayinduced by these mutants is abolished by the inverse agonists,losartan (14, 16) and irbesartan.2 To study the link betweenactivation and internalization/recycling, we analyzed the cellu-lar trafficking of three EGFP-tagged CAMs of the AT1A recep-tor (N111A, I245T, and L305Q).
MATERIALS AND METHODS
Construction of the EGFP-tagged CAM and WT Receptors—TheEGFP-AT1A receptor was constructed in three steps: 1) the nucleotidesequence of the insulin receptor signal peptide (sp) was amplified fromthe pET vector (17) with the following primers: 5� (ACCGGTCGCCAC-CATGGGCACCGGGGG) and 3� (ACCGGTAGGTGGCCCGCGGCGC),which inserted an AgeI site at both ends of the sp. The PCR fragmentwas digested with AgeI and inserted into the AgeI site of pEGFP-C3(CLONTECH), this construct was called ps-EGFP. 2) A linker, consist-ing of six copies of the myc epitope with the following amino acidsequence: Glu-Gln-Lys-Leu-Ile-Ser-Glu-Glu-Asp-Leu-Gly-Arg-Phe(Glu-Gln-Lys-Leu-Ile-Ser-Glu-Glu-Asp-Leu)5, was inserted into theEagI site of pEAT1A� (18). This construct was called 6mycs-AT1A. 3) The6mycs-AT1A vector was digested with HindIII and XmaI and insertedinto the HindIII and XmaI site of ps-EGFP. The construct was calledps-EGFP-6mycs-AT1A and the corresponding receptor was calledEGFP-AT1A.
The EGFP-tagged CAMs were constructed as follows: the BamHIfragment (corresponding to the fragment from amino acid 15 to the stopcodon) of ps-EGFP-6mycs-AT1A cDNA was replaced with the corre-sponding BamHI fragment of pTREC-N111A, pTREC-I245T, orpTREC-L305Q (16). The corresponding receptors were called: EGFP-N111A, EGFP-I245T, and EGFP-L305Q, respectively. The L305Q-EGFP mutant was constructed as follows: after a BstBI digestion,pTREC-L305Q was blunt ended using the large kleenow fragment andthen digested by HindIII. This fragment was inserted into the HindIIIand SmaI sites of pEGFP-N1 (CLONTECH). The truncated mutantswere constructed as follows: for the EGFP-�329, the BamHI fragmentfrom ps-EGFP-6mycs-AT1A cDNA was replaced with the correspondingBamHI fragment from pE�329 (19); for EGFP-L305Q-�329, the SpeI-XbaI fragment of ps-EGFP-6mycs-AT1A�329 (EGFP-�329) was re-placed with the corresponding SpeI-XbaI fragment of pTREC-L305Qcontaining the L305Q mutation.
Cell Culture and Transfection—HEK-293 cells were obtained fromthe ATCC (F-14742, 1573-CRL) and were grown in Dulbecco’s modifiedEagle’s medium supplemented with 7.5% fetal calf serum (FCS), 0.5 mM
glutamine, 100 units/ml penicillin, and 100 �g/ml streptomycin (allfrom Invitrogen). H295 cells were grown in Dulbecco’s modified Eagle’smedium-Ham’s F-12 (Sigma) supplemented with 2% Utroser G, 0.5 mM
glutamine, 50 units/ml penicillin, 50 mg/ml streptomycin (Invitrogen), 5�g/ml insulin, 5 �g/ml transferrin, and 5 ng/ml selenium (Sigma).
For stable expression, HEK-293 cells were transfected with 1 �g/500,000 cells of the plasmid of interest, by use of a liposomal transfec-tion reagent (Dosper, Roche Molecular Diagnosis). Cell lines stablyexpressing the EGFP-AT1A, EGFP-N111A, EGFP-I245T, EGFP-L305Q,EGFP-�329, and EGFP-L305Q-�329 receptors were selected for resist-ance to 750 �g/ml G418 (Invitrogen) and cloned by limiting dilution.
For transient aequorin transfection, the day before transfection,HEK-EGFP-AT1A or HEK-EGFP-L305Q cells were plated in polyal-lylamine (Sigma-Aldrich)-treated white opaque 96-well plates (Cultur-plate, Packard) at a density of 50,000 cells/well. Cells were then trans-fected with 0.1 �g/50,000 cells of the aequorin plasmid (gift from R.Rizzuto, University of Ferrara, Ferrara, Italy, (20)), using a liposomaltransfection reagent (Dosper).
For transient H295 cell transfection, cells were plated in polyal-lylamine-treated 6- or 24-well plates at 50–70% confluence the daybefore transfection. Cells were then transfected with 0.35 �g/well for24-well plates or 0.7 �g/well for 6-well plates of AT1A-EGFP or L305Q-EGFP plasmids, using a liposomal transfection reagent (Lipo-fectAMINE Plus, Invitrogen). FACS analysis indicated that �15% ofthe cells were transfected.
Pharmacological and Signaling Properties of the EGFP-AT1A, EGFP-N111A, EGFP-I245T, EGFP-L305Q, EGFP-�329, and EGFP-L305Q-�329 Receptors in HEK-293 Cells or in H295 Cells—1) Binding exper-iments with [125I]labeled Ang II were performed on intact cells, aspreviously described (21) except that the incubations with [125I]AngII
were performed for 3 h at 4 °C. Binding data were analyzed by linearregression using the Microsoft Excel 5 program.
2) To measure inositol phosphate (IP) production, cells were trans-fected with 0.5 �g/200,000 cells of G�q cDNA (a gift from B. Conklin,Departments of Medicine and Pharmacology, University of California,San Francisco, CA). The cells were metabolically labeled with myo-[3H]inositol as previously described (21) and then the IP content wasdetermined by methanol extraction and separation on a Dowex AG1-X8(Bio-Rad) column. To determine the functional consequences of losar-tan-induced externalization, cells were pretreated with LiCl at 16 °Cinstead of 37 °C.
3) The aequorin assay was performed as previously described (16) onstable HEK-EGFP-AT1A or HEK-EGFP-L305Q cell lines, transientlytransfected with the plasmid encoding the bioluminescent calcium-sensitive protein, aequorin, in 96-well plates. 48 h after transfection,the cells were incubated for 2 h at 37 °C in the presence or absence of 1�M losartan in medium supplemented with 0.5 �M cœlenterazine and1% FCS. The cells were subsequently washed twice with aequorinbuffer and incubated for 30 min at 16 °C in 50 �l of aequorin buffer (16).Cells were stimulated by adding 50 �l of increasing concentrations ofAngII and the luminescence was measured 30 s later in a TopCountcounter (Packard).
Fluorescence-activated Cell Sorting (FACS)—Cells expressing EGFP-tagged receptors were prepared and analyzed by FACS as previouslydescribed (22).
Protein Metabolic Labeling and Immunoprecipitation—Stable HEK-EGFP-AT1A or HEK-EGFP-L305Q cells were starved for 1 h in methi-onine- and cysteine-free Ham’s F-12 (Invitrogen). The starved cellswere labeled for 5 min with 50 �Ci/ml of [35S]Redivue Promix (Amer-sham Pharmacia Biotech) in the same medium. Cells were then rinsedtwice in PBS and incubated for various time in Dulbecco’s modifiedEagle’s medium supplemented with 7.5% FCS at 37 °C. Cells were thenprocessed for immunoprecipitation as previously described (10), exceptthat 1 �g of a monoclonal anti-myc antibody (9E10) was used for theimmunoprecipitation (Santa Cruz Biotechnology). Some immunopre-cipitated samples were treated with Endo H and PNGase F enzymes(both from Biolabs) according to the manufacturers recommendations.
Biochemical Measurement of Internalization and Recycling—Cellswere either pretreated with 10 �M monensin (Sigma) for 1 h at 37 °C orleft untreated. These cells were incubated with 100 nM AngII for 30 minat 4 °C and then incubated at 37 °C for 30 min to allow internalization.At this point an acid wash (0.2 M acetic acid, 0.5 M NaCl in bindingbuffer, 5 min at 4 °C) was performed to remove surface bound AngII.For the recycling studies, cells were additionally incubated in Dulbec-co’s modified Eagle’s medium supplemented with 1% FCS in the pres-ence or absence of 10 �M monensin for various periods of time at 37 °C.To determine the rates of internalization and recycling, binding exper-iments with [125I]labeled AngII were carried out on intact cells for 3 hat 4 °C as previously described (21).
Measurement of Internalization, Externalization, and Re-internaliza-tion by Confocal Microscopy—Confocal microscopy was also used toanalyze receptor trafficking as described previously (10). Cells (50,000cells/well) were seeded on polyallylamine-treated chambered coverglass8 wells (Nunc) and treated for 1 h at 37 °C with 70 �M cycloheximide(Sigma). Cells were then incubated for 30 min at 4 °C with variousligands in Earle’s buffer (10). Internalization was promoted by incubat-ing the cells in Earle’s complete buffer (10) at 37 or at 16 °C for variousperiods of time: 30 min with 100 nM (or 10 nM in Fig. 5C) AngII, 100 nM
[Sar1-Ile8]AngII, 10 �M L162,313, or for 2 h with 1 �M losartan and 10�M irbesartan. For the re-internalization study, after losartan treat-ment, cells were washed for 30 min at 4 °C in Earle’s complete bufferand then incubated at 37 °C or 16 °C. When required cells were treatedwith 10 �M monensin for the entire assay. After the incubation periods,the cells were rinsed in ice-cold Earle’s buffer and fixed by incubatingthem for 10 min in 100% methanol at 4 °C.
Cells were examined with a Leica TCS NT confocal laser scanningmicroscope configured with a Leica DM IRBE inverted microscopeequipped with an argon/helium/neon laser. EGFP fluorescence wasdetected following 100% excitation at 488 nm by use of a spectropho-tometer set with a window between 530 and 600 nm. Images of indi-vidual cells (1024 � 1024 pixels) were obtained by use of a �63 oil-immersion objective. Each image was done on a cross-section throughthe cells.
Quantification of the Subcellular Distribution of the Fluorescence—Digital image analysis using a specific macro software (10, 22) derivedfrom the public domain NIH Image software (developed by the U.S.National Institutes of Health and available on the Internet at rsb.info.nih.gov/nih-image/) was used to measure the subcellular distribution of2 C. Parnot, unpublished results.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants5892
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

the EGFP-tagged receptors. This software allowed to measure the S, C,and N values, which correspond to the mean density of the surface,cytoplasm, and nucleus fluorescence. The background fluorescence (N)was subtracted from the S and C values to give the S� and C� values.The S�/C� ratio provides reliable information about the level of cellsurface expression and the internalization state of the fluorescentreceptor.
Statistics—Results are expressed as mean � S.E. Statistical signif-icance was assessed by the Student’s t test.
RESULTS
Characterization of the EGFP-tagged CAMs of the AngIIAT1A Receptor—The CAM N111A, I245T, L305Q, and the wild-type (WT) AT1A receptors were tagged at the N terminus withEGFP to determine their subcellular localization and traffick-ing. A signal peptide was fused to the N terminus of EGFP toallow the correct exportation of the chimeric protein to theplasma membrane. A spacer, consisting of a tandem repeat ofthe myc epitope (63 amino acids), was inserted between EGFPand the AT1A receptor to prevent steric encumbrance of theAngII-binding site.
The EGFP-tagged CAM and WT receptors were stably trans-fected in HEK-293 cells and their functional properties wereanalyzed (Table I). The Kd of [125I]AngII for the EGFP-AT1A
and the EGFP-L305Q were both similar to the known Kd of thenon-tagged WT receptor (Kd � 0.61 nM for the AT1A receptor(19)). As evaluated by binding of labeled AngII, the EGFP-L305Q receptor presents a lower plasma membrane expressioncompared with the WT EGFP-AT1A receptor (Table I). Wechecked the constitutive activity of the EGFP-L305Q receptorby measuring the agonist-independent production of IP in thestable cell line, after transient transfection of the G�q cDNA toincrease the sensitivity of the assay (16, 23). The results werenormalized with respect to the expression levels of the receptorto allow accurate comparison (Table I). The basal activity of thecell line expressing the EGFP-L305Q receptor was three timesas high as that of the cells expressing the WT EGFP-AT1A,showing that the mutant fused to EGFP retained its constitu-tive activity. Moreover, the production of IP was increased byAngII treatment (Table I). The EGFP-N111A and EGFP-I245Treceptors have similar Kd (0.76 � 0.13 and 0.95 � 0.19 nM,respectively) to that of the WT receptor and the same basalconstitutive activity as the EGFP-L305Q receptor (EGFP-N111A, 770 � 107; EGFP-I245T, 203 � 16). Thus the EGFP-tagged receptors were fully functional in terms of ligand bind-ing and second messenger production.
Cellular Localization of the EGFP-tagged WT and CAM AT1A
Receptors—The EGFP tag enabled the cellular localization ofthe WT and CAM receptors to be determined by confocal mi-croscopy. Interestingly, whereas the WT EGFP-AT1A receptorswere localized at the cell surface, the CAM receptors (N111A,L305Q, and I245T) were mainly located in intracellular vesi-cles in the cytoplasm and some cells expressed low amount ontheir plasma membrane (Fig. 1A).
This constitutive intracellular localization of the CAM re-ceptors was quantified by calculating the ratio of surface and
cytoplasmic fluorescence densities, denoted here as S�/C� (see“Materials and Methods”). For the WT EGFP-AT1A receptor,the S�/C� ratio was typically 1.5–2.0 in the absence of theligand and decreased to 0.5 after AngII-induced internaliza-tion (Table I, Fig. 2C and Ref. 10). The S�/C� ratios of theCAM receptors were dramatically lower than those of the WTreceptor (EGFP-N111A, 0.60 � 0.06; EGFP-I245T, 0.80 �0.09; EGFP-L305Q, 0.66 � 0.04) (Table I and Fig. 2B). TheS�/C� ratios for the WT and L305Q receptors were furtherreduced by incubation with AngII, showing that a non-negligible fraction of the receptors was still at the plasmamembrane and was internalized in the presence of AngII(Fig. 2C). The presence of the EGFP-L305Q receptor at thecell surface was confirmed by binding experiments (Table I).
We assessed whether these differences in basal S�/C� ratiosof WT and CAM receptors were due to differences in the total
FIG. 1. Cellular localization and metabolism of EGFP-AT1A andEGFP-CAMs receptors. A, confocal images of untreated HEK-EGFP-AT1A, EGFP-N111A, EGFP-I245T, and EGFP-L305Q transfected cells.Images are representative of three independent experiments. Scalebar � 5 �m. B, metabolic labeling of the EGFP-AT1A and the EGFP-L305Q receptors. Labeling was carried out with 50 �Ci/ml [35S]methi-onine/cysteine for 5 min and was chased for the indicated time incomplete medium. C, control HEK cells. The receptors are indicated byarrowheads. C, sensitivity of EGFP-AT1A and EGFP-L305Q receptors toEndo H and PNGase F. The glycosylation state was determined bytreating the immunoprecipitated receptors at times 0 and 60 min of thechase with the indicated enzymes. The receptors before and after de-glycosylation are indicated by arrowheads. Results are representativeof three independent experiments.
TABLE IFunctional characterization of the WT EGFP-AT1A and EGFP-L305Q receptors
Results are expressed as mean � S.E. from three independent experiments.
Kd Bmax Basal IP AngII-induced IP production S�/C� ratioTotal
fluorescence(FACS)
nM sites/cell � 103 normalized to105 sites/cell
� stimulation over basal
EGFP-AT1A 0.76 � 0.13 363 � 64 100 � 9 8.80 � 0.40 1.81 � 0.37 6.22EGFP-L305Q 0.57 � 0.05 140 � 25 283 � 13a 1.88 � 0.26b 0.66 � 0.04a 6.06
a p � 0.05 versus EGFP-AT1A.b p � 0.001 versus EGFP-AT1A.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants 5893
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

number of receptors. We used FACS to quantify the total flu-orescence per cell on 10,000 cells stably expressing the EGFP-L305Q and the WT receptors (Table I). The two other CAMspresented comparable total fluorescence (data not shown). Wealso excluded the possibility that plasma membrane targetingwas altered due to the presence of EGFP at the N terminus,because the WT or L305Q receptors with a C-terminal EGFPtag presented the same cellular distribution as their N-termi-nal-tagged counterparts (data not shown).
Metabolic labeling experiments were performed to identifydifferences in the maturation of the WT and L305Q receptors.The WT EGFP-AT1A and the EGFP-L305Q receptors both cor-responded to �80-kDa bands and the maximal intensity wasreached after a 5-min pulse and progressively decreasing aftera 60-min chase (Fig. 1B). The maturation of the proteins wasstudied by their sensitivity to Endo H and PNGase F (Fig. 1C).After a 5-min pulse without chase, both of the receptor types
were sensitive to both enzymes, as indicated by the increase intheir electrophoretic mobility. Surprisingly, after a 60-minchase both receptors were sensitive to Endo H. This migrationprofile, i.e. sensitivity to both enzymes, was also observed on atotal cell extract after deglycosylation. In addition to the �80-kDa band, the WT EGFP-AT1A receptor was also representedby a 65-kDa band, which is not the consequence of EGFP-tagcleavage (data not shown). In conclusion, the WT EGFP-AT1A
and the EGFP-L305Q receptors present similar maturation/degradation and glycosylation profiles and the intracellularlocalization of the CAMs is therefore an intrinsic property ofthe constitutive activity and not due to the intracellular accu-mulation of the receptor during biosynthesis.
These results provide strong evidence for the constitutiveintracellular localization of three different ATA CAMs (N111A,I245T, and L305Q). The CAM receptors remaining at theplasma membrane were fully functional, displaying AngII
FIG. 2. Effect of losartan on the cellular localization of EGFP-tagged WT and CAM receptors. A, cells were examined by confocalmicroscopy after a 2-h incubation at 37 °C with or without 1 �M losartan. Scale bar � 5 �m. B, confocal images were quantified for four cell lines:HEK-EGFP-AT1A, HEK-EGFP-N111A, HEK-EGFP-I245T, and HEK-EGFP-L305Q, and the corresponding S�/C� ratios were calculated. S is themean density of surface fluorescence; C is the mean density of cytoplasmic fluorescence; and N is the mean density of nuclear fluorescenceconsidered to be background. S�/C� � (S-N)/(C-N). n � 10 for each point. Results are expressed as mean � S.E. from three independent experiments., p � 0.01 versus untreated. C, effect of other pharmacological molecules on the cellular localization of EGFP-tagged WT and L305Q receptors.Cells were examined by confocal microscopy after 30 min at 37 °C with 100 nM AngII, 100 nM [Sar1-Ile8]AngII, or 10 �M L162,313, or 2 h at 37 °Cwith 10 �M irbesartan. Confocal images were quantified. n � 10 for each point. Results are expressed as mean � S.E. from three independentexperiments. *, p � 0.05; , p � 0.01; §, p � 0.001 versus untreated.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants5894
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from
binding, as well as AngII-induced signaling and internalizationcomparable to the WT receptor.
Effect of an Inverse Agonist, Losartan, on the Cellular Local-ization of the AT1A CAMs—The inverse agonist, losartan, isknown to inhibit the constitutive signaling activity of the AT1A
CAMs (14, 16). We hypothesized that this compound would alsoaffect the cellular localization of the CAMs. Our results (Fig.2A) confirmed previous findings (8, 10) that losartan does notmodify the membrane localization of the WT EGFP-AT1A re-ceptor. The fluorescence of untreated CAM cells was mainlyintracellular, whereas the treatment with losartan resulted inthe appearance of plasma membrane fluorescence. This wasclearly visible on confocal images (Fig. 2A) and resulted in a50–100% increase in the basal S�/C� ratio of these receptors(Fig. 2B). This plasma membrane translocation resulted in a29% increase in the number of cell surface-binding sites on theL305Q mutant, as measured by [125I]AngII binding after losar-tan wash-away (data not shown). We called this phenomenon“externalization.”
We used the L305Q mutant to determine whether CAMexternalization was specific to inverse agonists or could beobserved with other pharmacological molecules. We tested thepeptide antagonist, [Sar1-Ile8]AngII, the non-peptide agonist,L162,313, and the non-peptide antagonist, irbesartan, whichhas similar inverse agonist properties to losartan (data notshown). [Sar1-Ile8]AngII induced similar levels of internaliza-tion of the WT and L305Q receptors as AngII. L162,313 in-duced a slight internalization of both WT and L305Q receptors(Fig. 2C). Interestingly, irbesartan had a very similar effect tolosartan. It did not modify the localization of the WT receptor,whereas it induced a membrane translocation of the EGFP-L305Q receptors as shown by a �100% increase in the S�/C�ratio (Fig. 2C). Therefore, only the inverse agonists induced theexternalization of the EGFP-L305Q receptor.
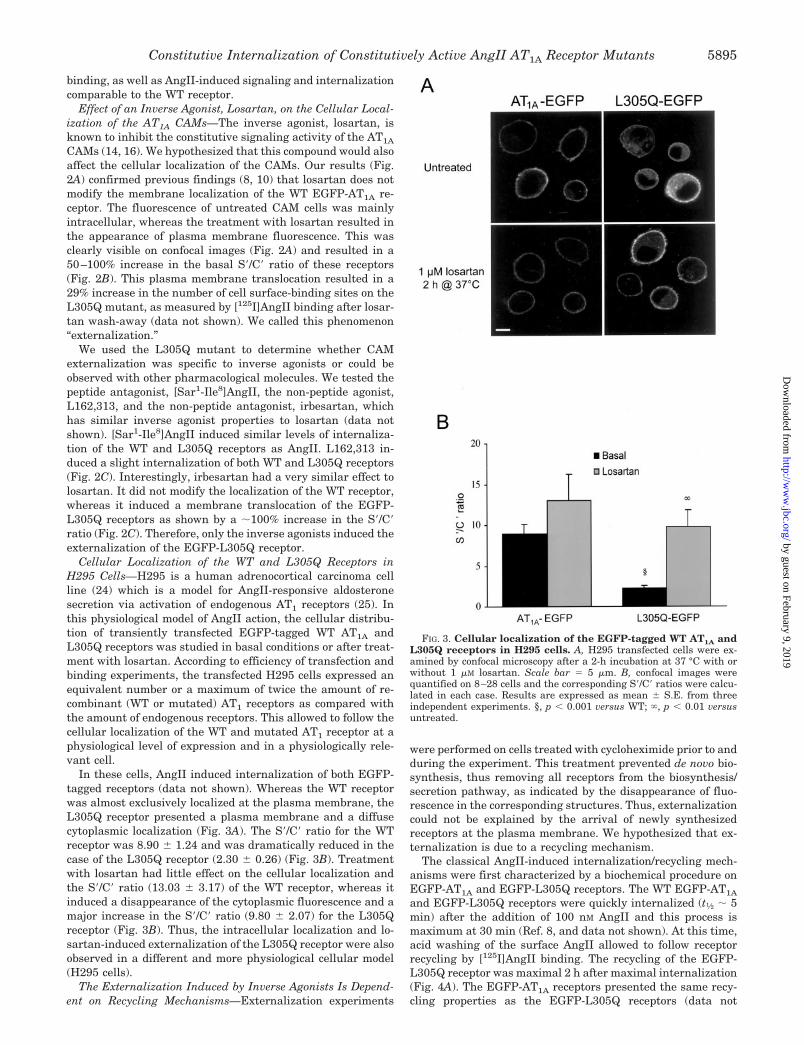
Cellular Localization of the WT and L305Q Receptors inH295 Cells—H295 is a human adrenocortical carcinoma cellline (24) which is a model for AngII-responsive aldosteronesecretion via activation of endogenous AT1 receptors (25). Inthis physiological model of AngII action, the cellular distribu-tion of transiently transfected EGFP-tagged WT AT1A andL305Q receptors was studied in basal conditions or after treat-ment with losartan. According to efficiency of transfection andbinding experiments, the transfected H295 cells expressed anequivalent number or a maximum of twice the amount of re-combinant (WT or mutated) AT1 receptors as compared withthe amount of endogenous receptors. This allowed to follow thecellular localization of the WT and mutated AT1 receptor at aphysiological level of expression and in a physiologically rele-vant cell.
In these cells, AngII induced internalization of both EGFP-tagged receptors (data not shown). Whereas the WT receptorwas almost exclusively localized at the plasma membrane, theL305Q receptor presented a plasma membrane and a diffusecytoplasmic localization (Fig. 3A). The S�/C� ratio for the WTreceptor was 8.90 � 1.24 and was dramatically reduced in thecase of the L305Q receptor (2.30 � 0.26) (Fig. 3B). Treatmentwith losartan had little effect on the cellular localization andthe S�/C� ratio (13.03 � 3.17) of the WT receptor, whereas itinduced a disappearance of the cytoplasmic fluorescence and amajor increase in the S�/C� ratio (9.80 � 2.07) for the L305Qreceptor (Fig. 3B). Thus, the intracellular localization and lo-sartan-induced externalization of the L305Q receptor were alsoobserved in a different and more physiological cellular model(H295 cells).
The Externalization Induced by Inverse Agonists Is Depend-ent on Recycling Mechanisms—Externalization experiments
were performed on cells treated with cycloheximide prior to andduring the experiment. This treatment prevented de novo bio-synthesis, thus removing all receptors from the biosynthesis/secretion pathway, as indicated by the disappearance of fluo-rescence in the corresponding structures. Thus, externalizationcould not be explained by the arrival of newly synthesizedreceptors at the plasma membrane. We hypothesized that ex-ternalization is due to a recycling mechanism.
The classical AngII-induced internalization/recycling mech-anisms were first characterized by a biochemical procedure onEGFP-AT1A and EGFP-L305Q receptors. The WT EGFP-AT1A
and EGFP-L305Q receptors were quickly internalized (t1⁄2 � 5min) after the addition of 100 nM AngII and this process ismaximum at 30 min (Ref. 8, and data not shown). At this time,acid washing of the surface AngII allowed to follow receptorrecycling by [125I]AngII binding. The recycling of the EGFP-L305Q receptor was maximal 2 h after maximal internalization(Fig. 4A). The EGFP-AT1A receptors presented the same recy-cling properties as the EGFP-L305Q receptors (data not
FIG. 3. Cellular localization of the EGFP-tagged WT AT1A andL305Q receptors in H295 cells. A, H295 transfected cells were ex-amined by confocal microscopy after a 2-h incubation at 37 °C with orwithout 1 �M losartan. Scale bar � 5 �m. B, confocal images werequantified on 8–28 cells and the corresponding S�/C� ratios were calcu-lated in each case. Results are expressed as mean � S.E. from threeindependent experiments. §, p � 0.001 versus WT; , p � 0.01 versusuntreated.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants 5895
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

shown). This phenomenon was blocked when the cells weretreated with 10 �M monensin, which is known to inhibit recy-cling (Fig. 4B). These results show that: 1) the EGFP-taggedreceptors recycle equally as well as the untagged receptors, and2) that the L305Q CAM retains normal recycling properties.
Interestingly, the time dependence of losartan-induced ex-ternalization of the L305Q CAM resembled the kinetics of therecycling process. Indeed, after treatment of the HEK-EGFP-L305Q cells with losartan, the S�/C� ratio progressively in-creased until it reached a plateau at 2 h (Fig. 4C). In addition,when EGFP-L305Q cells were pretreated with monensin, lo-sartan was no longer able to promote the externalization of thereceptor, as the S�/C� ratio did not increase compared withuntreated cells and even decreased slightly (Fig. 4D). Thus, theexternalization process observed upon incubation with losartanwas comparable to a recycling mechanism, recycled receptorsbeing blocked at the cell surface by losartan.
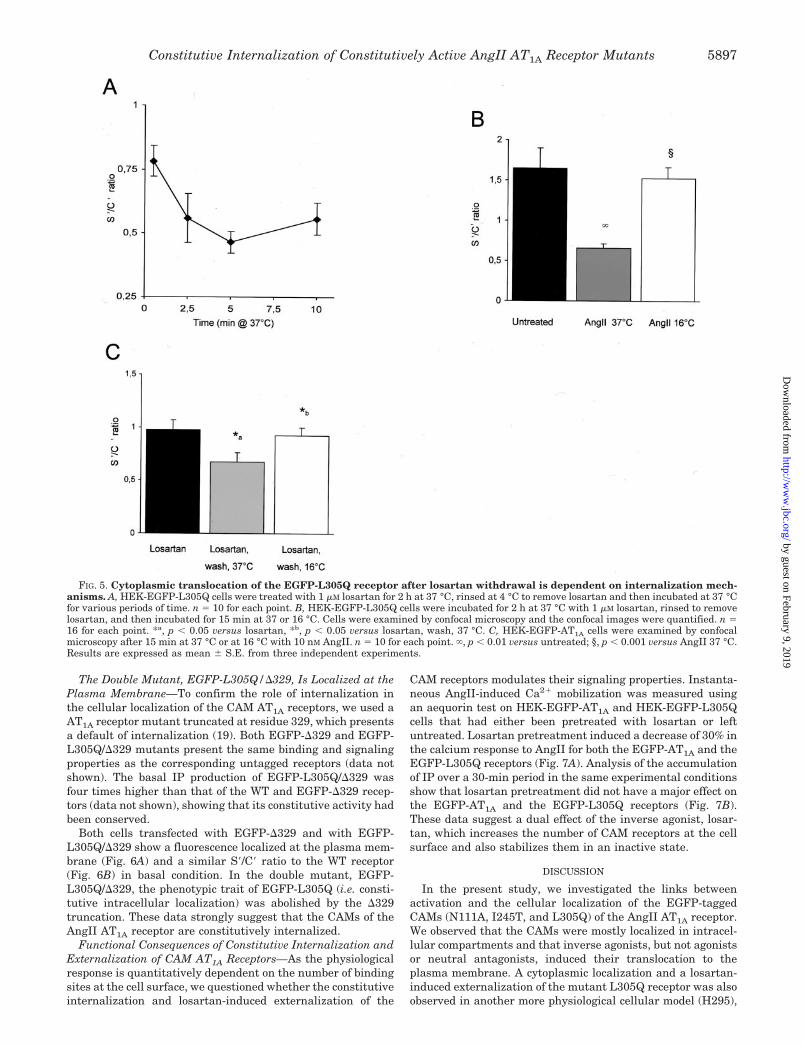
Cytoplasmic Translocation of the EGFP-L305Q Receptor af-ter the Withdrawal of Losartan Is Dependent on InternalizationMechanisms—We questioned what would happen to the exter-nalized receptors after the removal of losartan. Would theypermanently remain at the plasma membrane, suggesting thatthe distribution of CAM receptors is static? Or would they goback into the cells, as would be expected if the observed distri-
bution of CAM receptors is the result of a dynamic cyclingprocess?
To answer these questions, we took advantage of the factthat losartan can be rapidly dissociated from the receptor byrinsing at 4 °C and the sensitivity of the internalization processto temperature. After maximal externalization of the L305Qreceptor with losartan, the inverse agonist was removed byrinsing at 4 °C and the kinetic of reinternalization process wasthen observed at 37 °C, by the microscopy re-internalizationassay (see “Materials and Methods”). The plasma membranefluorescence disappeared from HEK-EGFP-L305Q cells after2.5 min at 37 °C and the effect was maximal at 5 min (Fig. 5A).This was comparable to the kinetics of the WT receptor inter-nalization (t1⁄2 � 2.7 min (10)). In addition, the reinternalizationof the L305Q receptor was abolished when the assay was per-formed at 16 °C (instead of 37 °C) (Fig. 5B), a temperature thatalso blocks the AngII-induced internalization of the WT recep-tor (Fig. 5C) and which blocks the clathrin-dependent internal-ization of other receptors (26).
These results show that losartan-induced externalization isa highly transient process, dependent on the presence of in-verse agonists. Moreover, the removal of losartan results in thecytoplasmic translocation of the EGFP-L305Q receptor depend-ent on internalization mechanisms.
FIG. 4. Losartan-induced externalization is dependent on recycling mechanisms. A, recycling of the EGFP-L305Q receptor afterAngII-induced internalization using the biochemical internalization and recycling assay. B, effect of monensin on recycling of the EGFP-L305Qreceptor. Cell surface receptors were measured by [125I]AngII binding after 30 min internalization and a 3-h recycling period. C, time course oflosartan-induced externalization for the EGFP-L305Q receptor: HEK-EGFP-L305Q cells were examined by confocal microscopy after beingincubated for various periods of time at 37 °C with 1 �M losartan (1 h, n � 27; 1.3 h, n � 15; 1.6 h, n � 17; 2 h, n � 25; 3 h, n � 10). Confocal imageswere quantified on three independent experiments. D, monensin pretreated HEK-EGFP-L305Q cells (n � 22) and untreated cells (n � 10) wereincubated for 2 h at 37 °C with 1 �M losartan with or without monensin. Cells were examined by confocal microscopy and the confocal images werequantified. Results are expressed as mean � S.E. from three independent experiments. *, p � 0.05 versus untreated; , p � 0.01 versus untreated;§, p � 0.001 versus losartan.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants5896
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from
The Double Mutant, EGFP-L305Q/�329, Is Localized at thePlasma Membrane—To confirm the role of internalization inthe cellular localization of the CAM AT1A receptors, we used aAT1A receptor mutant truncated at residue 329, which presentsa default of internalization (19). Both EGFP-�329 and EGFP-L305Q/�329 mutants present the same binding and signalingproperties as the corresponding untagged receptors (data notshown). The basal IP production of EGFP-L305Q/�329 wasfour times higher than that of the WT and EGFP-�329 recep-tors (data not shown), showing that its constitutive activity hadbeen conserved.
Both cells transfected with EGFP-�329 and with EGFP-L305Q/�329 show a fluorescence localized at the plasma mem-brane (Fig. 6A) and a similar S�/C� ratio to the WT receptor(Fig. 6B) in basal condition. In the double mutant, EGFP-L305Q/�329, the phenotypic trait of EGFP-L305Q (i.e. consti-tutive intracellular localization) was abolished by the �329truncation. These data strongly suggest that the CAMs of theAngII AT1A receptor are constitutively internalized.
Functional Consequences of Constitutive Internalization andExternalization of CAM AT1A Receptors—As the physiologicalresponse is quantitatively dependent on the number of bindingsites at the cell surface, we questioned whether the constitutiveinternalization and losartan-induced externalization of the
CAM receptors modulates their signaling properties. Instanta-neous AngII-induced Ca2 mobilization was measured usingan aequorin test on HEK-EGFP-AT1A and HEK-EGFP-L305Qcells that had either been pretreated with losartan or leftuntreated. Losartan pretreatment induced a decrease of 30% inthe calcium response to AngII for both the EGFP-AT1A and theEGFP-L305Q receptors (Fig. 7A). Analysis of the accumulationof IP over a 30-min period in the same experimental conditionsshow that losartan pretreatment did not have a major effect onthe EGFP-AT1A and the EGFP-L305Q receptors (Fig. 7B).These data suggest a dual effect of the inverse agonist, losar-tan, which increases the number of CAM receptors at the cellsurface and also stabilizes them in an inactive state.
DISCUSSION
In the present study, we investigated the links betweenactivation and the cellular localization of the EGFP-taggedCAMs (N111A, I245T, and L305Q) of the AngII AT1A receptor.We observed that the CAMs were mostly localized in intracel-lular compartments and that inverse agonists, but not agonistsor neutral antagonists, induced their translocation to theplasma membrane. A cytoplasmic localization and a losartan-induced externalization of the mutant L305Q receptor was alsoobserved in another more physiological cellular model (H295),
FIG. 5. Cytoplasmic translocation of the EGFP-L305Q receptor after losartan withdrawal is dependent on internalization mech-anisms. A, HEK-EGFP-L305Q cells were treated with 1 �M losartan for 2 h at 37 °C, rinsed at 4 °C to remove losartan and then incubated at 37 °Cfor various periods of time. n � 10 for each point. B, HEK-EGFP-L305Q cells were incubated for 2 h at 37 °C with 1 �M losartan, rinsed to removelosartan, and then incubated for 15 min at 37 or 16 °C. Cells were examined by confocal microscopy and the confocal images were quantified. n �16 for each point. *a, p � 0.05 versus losartan, *b, p � 0.05 versus losartan, wash, 37 °C. C, HEK-EGFP-AT1A cells were examined by confocalmicroscopy after 15 min at 37 °C or at 16 °C with 10 nM AngII. n � 10 for each point. , p � 0.01 versus untreated; §, p � 0.001 versus AngII 37 °C.Results are expressed as mean � S.E. from three independent experiments.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants 5897
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

which expresses endogenous AT1 receptors. Its intracellularlocalization is less pronounced than that observed in trans-fected HEK-293 cells, probably due to differences in the kinet-ics and efficiency of internalization and recycling of the AT1
receptor between the two cell types. These observations are notdue to the impaired folding of the CAMs, which would preventthem from exiting the biosynthetic pathway. We showed indeedthat the biosynthesis of the L305Q CAM and WT AT1A areidentical. Imidazole-like inverse agonists cannot cross theplasma membrane and therefore cannot stabilize the unfoldedreceptor intracellularly, as shown for a misfolded V2 vasopres-sin mutant (27). Several pieces of evidence suggest that it ismore likely that the CAMs of the AT1A receptor are perma-nently internalized and recycled: first, the plasma membranetranslocation of the L305Q CAM, or externalization, induced byinverse agonist has the same kinetics and pharmacology as therecycling mechanism. Second, after externalization by the in-verse agonists, L305Q CAM is quickly and spontaneously re-addressed to the cytoplasm upon ligand removal by a mecha-nism comparable to internalization. Third, the double mutant,EGFP-L305Q/�329, which is constitutively active but lacks adomain required for internalization, is localized at the plasmamembrane. Altogether, these data indicate that the CAMs ofthe AngII AT1A receptor are constitutively and permanentlyinternalized and recycled. Inverse agonists block the CAM inan inactive state when the recycled receptor reaches the
plasma membrane, thus preventing the rapid re-internaliza-tion and resulting in the accumulation of the receptor at themembrane (Fig. 8).
Although the spontaneous internalization of the CAMs of theGPCR family has long been considered as a likely possibility,no direct evidence is available yet. Several CAMs are presentwithin intracellular compartments, such as mutants of thePTH receptor (28), �1B adrenergic receptor (AR) (29), and theyeast pheromone receptors, Ste2p and Ste3p (30). Other stud-ies have shown that CAMs can be spontaneously phosphoryl-ated, desensitized, and/or down-regulated, but they have not
FIG. 6. The double mutant, EGFP-L305Q/�329, is localized atthe plasma membrane. A, untreated HEK-EGFP-L305Q, HEK-EGFP-�329, and HEK-EGFP-L305Q �329 cells were examined by con-focal microscopy. Images are representative of three independent ex-periments. Scale bar � 5 �m. B, confocal images were quantified andthe S�/C� ratio was calculated for each cell line. n � 9 for each point.Results are expressed as mean � S.E. from three independent experi-ments. , p � 0.01 versus EGFP-L305Q.
FIG. 7. Coupling state of the EGFP-AT1A and the EGFP-L305Qreceptors after losartan-induced externalization. A, aequorin as-say: HEK-EGFP-AT1A and HEK-EGFP-L305Q cells were transfectedwith 100 ng/96-well of expression plasmid for mt-aequorin, a biolumi-nescent protein sensitive to calcium. Two days after transfection, cellswere preincubated with coelanterazine either with (gray) or without(black) 1 �M losartan for 2 h at 37 °C and rinsed twice in aequorin bufferat 16 °C. AngII (100 nM) was added and the luminescent signal wasmeasured. B, IP accumulation assay: cells that had been pretreatedwith 1 �M losartan (gray) and untreated cells (black) were stimulatedfor 30 min with 100 nM AngII. Results are expressed as mean � S.E.from three independent experiments. *, p � 0.05 versus untreated; ,p � 0.01 versus untreated.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants5898
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

provided evidence for constitutive internalization (31). Basalphosphorylation is enhanced for two CAMs of the human LHreceptor (32, 33) and for the CAMs of the �1B (34) and �2-AR(35). The �2-AR CAM is also constitutively desensitized andconstitutively down-regulated and both phenomena are re-versed by overnight treatment with the inverse agonist, bet-axolol (35). Constitutive down-regulation reversed by inverseagonists was later observed for several other CAMs, includingmutants of the TRH receptor (36) and the �1B AR (29, 37), andalso for the WT histamine receptor H2, which exhibits naturalconstitutive activity (38). In all cases, the effects of the inverseagonists are only observed after a long period of treatment,consistent with the stabilization of newly synthesized recep-tors. For example, the levels of CAM �2-AR are increased4–7-fold after 24 h treatment, through a protein synthesis-de-pendent process that does not change the subcellular distribu-tion of the receptor (39). This is thus believed to result from theconstitutive addressing of the mutants to degradation path-ways. In some cases, inherent instability and/or folding defectsof the CAMs are involved in this down-regulation process. Thisis clearly the case for the CAMs of the yeast receptors, Ste2pand Ste3p, which remain stuck in the biosynthesis pathwaybecause of impaired folding (30). Stabilizing effects explain howboth inverse agonists and agonists can up-regulate the levels ofStep2 and Step3 CAMs, but also of an �2A CAM (40) and a �2
CAM in Sf9 cells (41) or even in vivo (42).Conversely, the phenomenon observed here is not linked to
the down-regulation or instability of the CAMs of the AT1A
receptor. It is independent of protein synthesis, as all assayswere done on cells treated with cycloheximide. It does notresult from the instability of the CAMs, as their metabolismwas similar to the WT receptor. It does not involve proteinstabilization as other peptide or non-peptide ligands, which
all differ from inverse agonists by their ability to induceinternalization, were unable to relocalize the receptor to theplasma membrane. Our results suggest a very differentmechanism and strongly suggest that AT1A CAMs are consti-tutively and permanently internalized and are then recycled.It is not clear whether the same behavior occurs for otherCAMs in the GPCR family, but there are several reasons whythis phenomenon has never been reported before. First, dueto the difficulty in obtaining antibodies and the recent intro-duction of epitope fluorescent tagging, the cellular distribu-tion of GPCR has only been studied morphologically in alimited number of GPCR. Second, the internalization/recycling kinetics of the AT1A receptor differ from those ofother classical GPCR because it is rapidly internalized (with-in minutes) and slowly recycled (within hours), whereasother GPCR are either rapidly internalized and recycled (�2-AR) or not recycled but degraded (LH receptor). The peculiarkinetic of the AT1A receptor may favor the intracellular ac-cumulation of the receptor.
Some examples in the literature are partially reminiscent ofour observations, suggesting that the phenomenon describedhere may be relevant to other CAMs. Although the kinetics aredifferent, two WT GPCRs have been shown to be constitutivelyinternalized: the thrombin receptor (43) and the cholecystoki-nin receptor type A (44). The WT �1D AR is naturally consti-tutively active and mostly localized in intracellular compart-ments, whereas 24 h in the presence of prazosin causesredistribution of the receptor from intracellular sites to cellularperiphery (45). Finally, other examples include a deletion mu-tant of the �-opioid receptor (46), which is constitutively inter-nalized and recycled, and an inactive mutant of the humanvasopressin receptor, which is constitutively sequestered inarrestin-associated intracellular vesicles (47). These mutantsare not constitutively active, but demonstrate that GPCRs canbe constitutively desensitized by mechanisms distinct fromconstitutive down-regulation.
Although they do not rule out the permanent cycling ofCAMs, recent results on the phosphorylation of the N111A andN111G mutants of the AT1A receptor raise the question of themolecular mechanisms regulating this constitutive internaliza-tion. Unexpectedly, the phosphorylation of N111A and N111Gmutants was not elevated in basal conditions and, unlike theWT receptor, it was not increased by AngII treatment (48).However, the CAMs are normally internalized in response toAngII (Fig. 2C and Ref. 48). The phosphorylation status of thetwo other mutants studied here (I245T and L305Q) is notknown, but the study by Thomas et al. (48) suggests that theAngII-induced phosphorylation of the AT1A receptor is notmandatory for internalization and more generally, that phos-phorylation and internalization can be dissociated. This is alsoin agreement with the fact that phosphorylation is not manda-tory for arrestin binding (49, 50).
Another major difference between our study and previousreports on the regulation of CAMs is the functional conse-quence of treatment with inverse agonists. In the case of CAMsof the �1B AR (37), the TRH receptor (36), and the �2 AR (35),the up-regulation of receptor levels induced by inverse agonistswas accompanied by highly enhanced signaling responses. Con-versely, after losartan-induced externalization of the AT1A
CAM, we observed a reduced AngII-induced calcium mobiliza-tion and no significant change in IP turnover. Unfortunately,pretreatment with losartan does not only block the receptor atthe membrane, but also partly desensitizes it, as shown by thecalcium signaling of the WT receptor. Therefore, the un-changed or even reduced signaling efficiency of the CAM afterlosartan treatment is probably due to the small positive effect
FIG. 8. Model of the cellular distribution of AT1A receptorCAMs. In the basal state the wild-type AT1A receptor was mostlylocalized at the cell surface. Inverse agonists (such as losartan andirbesartan) had no effect on the cellular localization, whereas AngIIinduced the rapid internalization (i) of the receptor in intracellularvesicles. The receptor was then slowly and partly recycled (r) at theplasma membrane. In the basal state the CAMs of the AT1A receptorwere mostly localized in intracellular vesicles. Losartan induced theplasma membrane translocation of the CAMs. In the basal state or afterlosartan-induced externalization, AngII induced the internalization ofthe receptor in intracellular vesicles.
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants 5899
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

of the increased number of receptors at the cell surface, whichis counterbalanced by partial inactivation. Another possibleexplanation is that, as previously discussed by Milligan andBond (51), the signaling efficiency is dependent on the levels ofexpression of G proteins and second messenger generatingenzymes. For example, treatment with inverse agonists did notenhance signaling for the �2 CAM when the mutant was ex-pressed in NG108-15 cells (52).
The initial purpose of this study was to gain insight intothe link between the activation and internalization of theAT1A receptor. We wanted to determine whether the receptoris inexorably targeted for internalization by the cell machin-ery when the receptor is in the active conformation andwhether activation necessarily results in internalization. TheC-terminal deletion mutant, �329, couples to the G proteinbut does not become internalized (19, 53), suggesting thatactivation can be dissociated from internalization. However,this is due to the absence of a large domain necessary for theinteraction with internalization machinery rather than a dif-ference in the overall conformation of the receptor. Moreinterestingly, the peptide ligand [Sar1,Ile4,Ile8]AngII hasbeen reported to activate the N111A and N111G mutantswithout inducing their internalization (data not shown inRef. 48). In contrast, the results presented here suggest thatthe active conformation of the AT1A receptor is an “internal-ization-sensitive” conformation. The three CAMs studiedcarry mutations at different positions in the transmembranedomains but present similar patterns of cellular distribution.This suggests that the active conformation of the WT receptorcannot avoid internalization.
Conversely, the AT1A receptor can quite easily adopt con-formations that do not activate signaling but are recognizedfor internalization. Non-signaling mutants of the AT1A recep-tor (54, 55) are internalized in response to AngII to the samedegree as the WT receptor (8, 55). AngII peptide antagonistsare able to induce internalization of the receptor (8, 10). Thisactivation-independent internalization also takes place forother GPCRs, and is also supported by the fact that mutantsof the �-opioid and vasopressin receptors are constitutivelyinternalized without being constitutively active (46, 47).These results suggest that the activation of the signalingpathways by a GPCR requires a much more specific confor-mation that the conformation required to trigger internaliza-tion. As arrestin binding is mandatory and is the first step forthe internalization of these GPCRs, arrestin probably recog-nizes a broader spectrum of conformations that the Gproteins.
In conclusion, this study shows that the CAMs of the AngIIAT1A receptor are constitutively and permanently internalizedand recycled. The externalization phenomenon described hereshould define a new paradigm for agonist-independent CAMregulation, distinct from the strong down-regulation observedfor a number of other CAMs in the GPCR family and firstexemplified for the �2-AR (35). Furthermore, this study pro-vides important insights on the molecular determinants ofactivation and internalization.
Acknowledgments—We are grateful to Colette Auzan for methodolog-ical assistance and Drs. Sophie Conchon, Bruno Goud, Zsolt Lenkei,and Laurent Muller for helpful discussions. We thank Drs. JeromeBertherat and Lionel Groussin for the gift of H295 cells.
REFERENCES
1. Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G.,Smith, H. O., Yandell, M., Evans, C. A., Holt, R. A., Gocayne, J. D.,Amanatides, P., Ballew, R. M., Huson, D. H., Wortman, J. R., Zhang, Q.,Kodira, C. D., Zheng, X. H., Chen, L., Skupski, M., Subramanian, G.,Thomas, P. D., Zhang, J., Gabor Miklos, G. L., Nelson, C., Broder, S., Clark,
A. G., Nadeau, J., McKusick, V. A., Zinder, N., Levine, A. J., Roberts, R. J.,Simon, M., Slayman, C., Hunkapiller, M., Bolanos, R., Delcher, A., Dew, I.,Fasulo, D., Flanigan, M., Florea, L., Halpern, A., Hannenhalli, S., Kravitz,S., Levy, S., Mobarry, C., Reinert, K., Remington, K., Abu-Threideh, J.,Beasley, E., Biddick, K., Bonazzi, V., Brandon, R., Cargill, M., Chan-dramouliswaran, I., Charlab, R., Chaturvedi, K., Deng, Z., Di Francesco, V.,Dunn, P., Eilbeck, K., Evangelista, C., Gabrielian, A. E., Gan, W., Ge, W.,Gong, F., Gu, Z., Guan, P., Heiman, T. J., Higgins, M. E., Ji, R. R., Ke, Z.,Ketchum, K. A., Lai, Z., Lei, Y., Li, Z., Li, J., Liang, Y., Lin, X., Lu, F.,Merkulov, G. V., Milshina, N., Moore, H. M., Naik, A. K., Narayan, V. A.,Neelam, B., Nusskern, D., Rusch, D. B., Salzberg, S., Shao, W., Shue, B.,Sun, J., Wang, Z., Wang, A., Wang, X., Wang, J., Wei, M., Wides, R., Xiao,C., Yan, C., et al. (2001) Science 291, 1304–1351
2. Ferguson, S. S. (2001) Pharmacol. Rev. 53, 1–243. Murphy, T. J., Alexander, R. W., Griendling, K. K., Runge, M. S., and
Bernstein, K. E. (1991) Nature 351, 233–2364. Smith, R. D., Hunyady, L., Olivares-Reyes, J. A., Mihalik, B., Jayadev, S., and
Catt, K. J. (1998) Mol. Pharmacol. 54, 935–9415. Oppermann, M., Freedman, N. J., Alexander, R. W., and Lefkowitz, R. J.
(1996) J. Biol. Chem. 271, 13266–132726. Hunyady, L., Bor, M., Balla, T., and Catt, K. J. (1994) J. Biol. Chem. 269,
31378–313827. Hein, L., Meinel, L., Pratt, R. E., Dzau, V. J., and Kobilka, B. K. (1997) Mol.
Endocrinol. 11, 1266–12778. Conchon, S., Monnot, C., Teutsch, B., Corvol, P., and Clauser, E. (1994) FEBS
Lett. 349, 365–3709. Chaki, S., Guo, D. F., Yamano, Y., Ohyama, K., Tani, M., Mizukoshi, M.,
Shirai, H., and Inagami, T. (1994) Kidney Int. 46, 1492–149510. Miserey-Lenkei, S., Lenkei, Z., Parnot, C., Corvol, P., and Clauser, E. (2001)
Mol. Endocrinol. 15, 294–30711. Gaborik, Z., Szaszak, M., Szidonya, L., Balla, B., Paku, S., Catt, K. J., Clark,
A. J., and Hunyady, L. (2001) Mol. Pharmacol. 59, 239–24712. Boulay, G., Chretien, L., Richard, D. E., and Guillemette, G. (1994) Endocri-
nology 135, 2130–213613. Anborgh, P. H., Seachrist, J. L., Dale, L. B., and Ferguson, S. S. (2000) Mol.
Endocrinol. 14, 2040–205314. Groblewski, T., Maigret, B., Larguier, R., Lombard, C., Bonnafous, J. C., and
Marie, J. (1997) J. Biol. Chem. 272, 1822–182615. Balmforth, A. J., Lee, A. J., Warburton, P., Donnelly, D., and Ball, S. G. (1997)
J. Biol. Chem. 272, 4245–425116. Parnot, C., Bardin, S., Miserey-Lenkei, S., Guedin, D., Corvol, P., and Clauser,
E. (2000) Proc. Natl. Acad. Sci. U. S. A. 97, 7615–762017. Ellis, L., Clauser, E., Morgan, D. O., Edery, M., Roth, R. A., and Rutter, W. J.
(1986) Cell 45, 721–73218. Conchon, S., Monnot, C., Sirieix, M. E., Bihoreau, C., Corvol, P., and Clauser,
E. (1994) Biochem. Biophys. Res. Commun. 199, 1347–135419. Conchon, S., Peltier, N., Corvol, P., and Clauser, E. (1998) Am. J. Physiol. 274,
E336–34520. Rizzuto, R., Simpson, A. W., Brini, M., and Pozzan, T. (1992) Nature 358,
325–32721. Conchon, S., Barrault, M. B., Miserey, S., Corvol, P., and Clauser, E. (1997)
J. Biol. Chem. 272, 25566–2557222. Lenkei, Z., Beaudet, A., Chartrel, N., De Mota, N., Irinopoulou, T., Braun, B.,
Vaudry, H., and Llorens-Cortes, C. (2000) J. Histochem. Cytochem. 48,1553–1564
23. Burstein, E. S., Spalding, T. A., Brauner-Osborne, H., and Brann, M. R. (1995)FEBS Lett. 363, 261–263
24. Gazdar, A. F., Oie, H. K., Shackleton, C. H., Chen, T. R., Triche, T. J., Myers,C. E., Chrousos, G. P., Brennan, M. F., Stein, C. A., and La Rocca, R. V.(1990) Cancer Res. 50, 5488–5496
25. Bird, I. M., Hanley, N. A., Word, R. A., Mathis, J. M., McCarthy, J. L., Mason,J. I., and Rainey, W. E. (1993) Endocrinology 133, 1555–1561
26. Cao, T. T., Mays, R. W., and von Zastrow, M. (1998) J. Biol. Chem. 273,24592–24602
27. Morello, J. P., Salahpour, A., Laperriere, A., Bernier, V., Arthus, M. F.,Lonergan, M., Petaja-Repo, U., Angers, S., Morin, D., Bichet, D. G., andBouvier, M. (2000) J. Clin. Invest. 105, 887–895
28. Ferrari, S. L., and Bisello, A. (2001) Mol. Endocrinol. 15, 149–16329. Stevens, P. A., Bevan, N., Rees, S., and Milligan, G. (2000) Mol. Pharmacol. 58,
438–44830. Stefan, C. J., Overton, M. C., and Blumer, K. J. (1998) Mol. Biol. Cell 9,
885–89931. Leurs, R., Smit, M. J., Alewijnse, A. E., and Timmerman, H. (1998) Trends
Biochem. Sci 23, 418–42232. Min, K. S., Liu, X., Fabritz, J., Jaquette, J., Abell, A. N., and Ascoli, M. (1998)
J. Biol. Chem. 273, 34911–3491933. Min, L., and Ascoli, M. (2000) Mol. Endocrinol. 14, 1797–181034. Mhaouty-Kodja, S., Barak, L. S., Scheer, A., Abuin, L., Diviani, D., Caron,
M. G., and Cotecchia, S. (1999) Mol. Pharmacol. 55, 339–34735. Pei, G., Samama, P., Lohse, M., Wang, M., Codina, J., and Lefkowitz, R. J.
(1994) Proc. Natl. Acad. Sci. U. S. A. 91, 2699–270236. Heinflink, M., Nussenzveig, D. R., Grimberg, H., Lupu-Meiri, M., Oron, Y., and
Gershengorn, M. C. (1995) Mol. Endocrinol. 9, 1455–146037. Lee, T. W., Cotecchia, S., and Milligan, G. (1997) Biochem. J. 325, 733–73938. Smit, M. J., Leurs, R., Alewijnse, A. E., Blauw, J., Van Nieuw Amerongen,
G. P., Van De Vrede, Y., Roovers, E., and Timmerman, H. (1996) Proc. Natl.Acad. Sci. U. S. A. 93, 6802–6807
39. MacEwan, D. J., and Milligan, G. (1996) FEBS Lett. 399, 108–11240. Betuing, S., Valet, P., Lapalu, S., Peyroulan, D., Hickson, G., Daviaud, D.,
Lafontan, M., and Saulnier-Blache, J. S. (1997) Biochem. Biophys. Res.Commun. 235, 765–773
41. Gether, U., Ballesteros, J. A., Seifert, R., Sanders-Bush, E., Weinstein, H., andKobilka, B. K. (1997) J. Biol. Chem. 272, 2587–2590
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants5900
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

42. Samama, P., Bond, R. A., Rockman, H. A., Milano, C. A., and Lefkowitz, R. J.(1997) Proc. Natl. Acad. Sci. U. S. A. 94, 137–141
43. Shapiro, M. J., Trejo, J., Zeng, D., and Coughlin, S. R. (1996) J. Biol. Chem.271, 32874–32880
44. Tarasova, N. I., Stauber, R. H., Choi, J. K., Hudson, E. A., Czerwinski, G.,Miller, J. L., Pavlakis, G. N., Michejda, C. J., and Wank, S. A. (1997) J. Biol.Chem. 272, 14817–14824
45. McCune, D. F., Edelmann, S. E., Olges, J. R., Post, G. R., Waldrop, B. A.,Waugh, D. J., Perez, D. M., and Piascik, M. T. (2000) Mol. Pharmacol. 57,659–666
46. Segredo, V., Burford, N. T., Lameh, J., and Sadee, W. (1997) J. Neurochem. 68,2395–2404
47. Barak, L. S., Oakley, R. H., Laporte, S. A., and Caron, M. G. (2001) Proc. Natl.Acad. Sci. U. S. A. 98, 93–98
48. Thomas, W. G., Qian, H., Chang, C. S., and Karnik, S. (2000) J. Biol. Chem.275, 2893–2900
49. Bouvier, M., Hausdorff, W. P., De Blasi, A., O’Dowd, B. F., Kobilka, B. K.,Caron, M. G., and Lefkowitz, R. J. (1988) Nature 333, 370–373
50. Hausdorff, W. P., Bouvier, M., O’Dowd, B. F., Irons, G. P., Caron, M. G., andLefkowitz, R. J. (1989) J. Biol. Chem. 264, 12657–12665
51. Milligan, G., and Bond, R. A. (1997) Trends Pharmacol. Sci. 18, 468–47452. MacEwan, D. J., and Milligan, G. (1996) Mol. Pharmacol. 50, 1479–148653. Thomas, W. G., Thekkumkara, T. J., Motel, T. J., and Baker, K. M. (1995)
J. Biol. Chem. 270, 207–21354. Bihoreau, C., Monnot, C., Davies, E., Teutsch, B., Bernstein, K. E., Corvol, P.,
and Clauser, E. (1993) Proc. Natl. Acad. Sci. U. S. A. 90, 5133–513755. Hunyady, L., Baukal, A. J., Balla, T., and Catt, K. J. (1994) J. Biol. Chem. 269,
24798–24804
Constitutive Internalization of Constitutively Active AngII AT1A Receptor Mutants 5901
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from

Stéphanie Miserey-Lenkei, Charles Parnot, Sabine Bardin, Pierre Corvol and Eric ClauserMutants Is Blocked by Inverse Agonists
Receptor1AConstitutive Internalization of Constitutively Active Angiotensin II AT
doi: 10.1074/jbc.M108398200 originally published online November 29, 20012002, 277:5891-5901.J. Biol. Chem.
10.1074/jbc.M108398200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/277/8/5891.full.html#ref-list-1
This article cites 55 references, 31 of which can be accessed free at
by guest on February 9, 2019http://w
ww
.jbc.org/D
ownloaded from





























