Embed Size (px)
Citation preview

Characterization of GATA3 Mutations in the Hypoparathyroidism,Deafness, and Renal Dysplasia (HDR) Syndrome*
Received for publication, February 18, 2004Published, JBC Papers in Press, February 24, 2004, DOI 10.1074/jbc.M401797200
M. Andrew Nesbit,a,b Michael R. Bowl,a,b,c Brian Harding,a Asif Ali,a,d Alejandro Ayala,e
Carol Crowe,f Angus Dobbie,g Geeta Hampson,h Ian Holdaway,i Michael A. Levine,j
Robert McWilliams,k Susan Rigden,l Julian Sampson,m Andrew J. Williams,n
and Rajesh V. Thakkera,o
From the aAcademic Endocrine Unit, Nuffield Department of Medicine, University of Oxford, Oxford Centre for Diabetes,Endocrinology and Metabolism, Churchill Hospital, Oxford OX3 7LJ, United Kingdom, the ePediatric and ReproductiveEndocrinology Branch, National Institutes of Health, Bethesda, Maryland 20892-1284, the fDivision of Genetics,Department of Pediatrics, MetroHealth Medical Center, Cleveland, Ohio 44109, the gDepartment of Clinical Genetics, TheChurchill Hospital, Oxford OX3 7L3, United Kingdom, the hDepartment of Chemical Pathology, The Guy’s, King’s Collegeand St Thomas’ Hospitals Medical and Dental School, St Thomas’ Hospital, London SE1 7EH, United Kingdom, theiDepartment of Endocrinology, Auckland Hospital, Park Rd., Auckland 1, New Zealand, the jDepartment of PediatricEndocrinology, The Children’s Hospital at The Cleveland Clinic, Cleveland, Ohio 44195-0001, the kDivisions ofHematology and Oncology, Mayo Clinic, Rochester, Minnesota 55905, the lPaediatric Renal Unit, Guy’s Hospital, StThomas Street, London SE1 9RT, United Kingdom, the mInstitute of Medical Genetics, University of Wales College ofMedicine, Cardiff CF14 4XN, United Kingdom, and the nDepartment of Nephrology, Morriston Hospital,Swansea SA6 6NL, United Kingdom
The hypoparathyroidism, deafness, and renal dyspla-sia (HDR) syndrome is an autosomal dominant disordercaused by mutations of the dual zinc finger transcrip-tion factor, GATA3. The C-terminal zinc finger (ZnF2)binds DNA, whereas the N-terminal finger (ZnF1) stabi-lizes this DNA binding and interacts with other zincfinger proteins, such as the Friends of GATA (FOG). Wehave investigated seven HDR probands and their fami-lies for GATA3 abnormalities and have identified twononsense mutations (Glu-228 3 Stop and Arg-367 3Stop); two intragenic deletions that result in frameshiftsfrom codons 201 and 355 with premature terminationsat codons 205 and 370, respectively; one acceptor splicesite mutation that leads to a frameshift from codon 351and a premature termination at codon 367; and two mis-sense mutations (Cys-3183 Arg and Asn-3203 Lys). Thefunctional effects of these mutations, together with apreviously reported GATA3 ZnF1 mutation and sevenother engineered ZnF1 mutations, were assessed byelectrophoretic mobility shift, dissociation, yeast two-hybrid and glutathione S-transferase pull-down assays.Mutations involving GATA3 ZnF2 or adjacent basicamino acids resulted in a loss of DNA binding, but thoseof ZnF1 either lead to a loss of interaction with specificFOG2 ZnFs or altered DNA-binding affinity. These find-ings are consistent with the proposed three-dimensionalmodel of ZnF1, which has separate DNA and proteinbinding surfaces. Thus, our results, which expand thespectrum of HDR-associated GATA3 mutations and re-port the first acceptor splice site mutation, help to elu-cidate the molecular mechanisms that alter the functionof this zinc finger transcription factor and its role incausing this developmental anomaly.
GATA3 belongs to a family of zinc finger transcription fac-tors that are involved in vertebrate embryonic development(1–3). The six mammalian GATA proteins (GATA-1 to -6) sharerelated Cys-X2-Cys-X17-Cys-X2-Cys (where X represents anyamino acid residue) zinc finger DNA-binding domains (see Fig.1) and bind to the consensus motif 5�-(A/T)GATA(A/G)-3� (4).The C-terminal finger (ZnF2)1 is essential for DNA binding,whereas the N-terminal finger (ZnF1) appears to stabilize thisbinding and to physically interact with other multitype zincfinger proteins, such as the Friends of GATA (FOG) (5–7).Thus, FOG-1 and FOG-2 have been shown, in mammals, tomodulate the biological activities of GATA1 and GATA4, re-spectively (5–7). Furthermore, the importance of these interac-tions of GATA and FOG family members are underscored bytheir evolutionary conservation, because it has been shownthat the Drosophila GATA factor, Pannier, interacts with aFOG-like protein referred to as U-shaped (8, 9). The mamma-lian GATA factors can be subdivided into two families based ontheir structures and patterns of expression (10, 11). Thus, thestructurally related proteins GATA4, -5, and -6 are expressedin overlapping patterns in the heart, gut, urogenital system,and smooth muscle cell lineages, whereas GATA1, -2, and -3are expressed in the hematopoietic cell lineages in which theycontrol development of the erythroid, hematopoietic stem celland T cell lineages, respectively (10, 11). In addition, GATA3 isalso expressed in the developing parathyroids, inner ear, andkidneys (12, 13). These expression patterns are consistent withthe disease phenotypes that have been reported in the fewpatients with genetic abnormalities involving three of theGATA members. Thus, GATA1 mutations lead to dyserythro-poietic anemia, thrombocytopenia (14), and the megakaryo-blastic leukemia of Down’s syndrome (15); GATA3 haploinsuf-
* The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
b Both authors contributed equally to this work.c A Medical Research Council (MRC) Ph.D. student.d An MRC Clinical Training Fellow.o To whom correspondence should be addressed. Tel.: 44-1865-857-
501; Fax: 44-1865-857-502; E-mail: [email protected].
1 The abbreviations used are: ZnF, zinc finger; HDR, hypoparathy-roidism, deafness, and renal dysplasia; FOG, Friend of GATA; GST,glutathione S-transferase; EMSA, electrophoretic mobility shift assay;WT, wild-type; PTH, parathyroid hormone; TA, transactivating; CAPS,3-(cyclohexylamino)-1-propanesulphonic acid; GFP, green fluorescentprotein; PBS, phosphate-buffered saline; DAPI, 4�,6-diamidino-2-phe-nylindole; RT, reverse transcription; BD, binding domain; AD, activa-tion domain; DDO, double drop-out; QDO, quaternary drop-out.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 21, Issue of May 21, pp. 22624–22634, 2004Printed in U.S.A.
This paper is available on line at http://www.jbc.org22624
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

ficiency is associated with the hypoparathyroidism, deafness,and renal dysplasia (HDR) syndrome (16); and GATA4 hemizy-gosity has been observed in some patients with congenitalheart disease (17). More than 90% of HDR syndrome patientshave hypoparathyroidism and deafness, and more than 80%have renal tract abnormalities (16, 18, 19). The hypoparathy-roidism is characterized by symptomatic or asymptomatic hy-pocalcemia with undetectable or inappropriately normal serumconcentrations of parathyroid hormone (PTH), and normalbrisk increases in plasma cAMP in response to PTH infusion,which indicates normal sensitivity of the PTH receptor (18).The sensorineural deafness is usually bilateral, although thehearing loss may vary in its severity (18, 20–22). The renaltract abnormalities, which may be uni- or bi-lateral, consist of:renal cysts that may cause pelvicalyceal deformities and/orcompression of the glomeruli and tubules that may lead tokidney failure; renal aplasia or hypoplasia; and vesicoureteralreflux (16, 18–22). The precise manner in which GATA3 mu-tations cause these congenital abnormalities of the parathy-roids, inner ear, and kidneys remains to be elucidated. To gainfurther insights into the structure-function relationships ofGATA3, we have studied additional HDR patients for GATA3abnormalities and have investigated the effects of GATA3 mu-tations on DNA binding and protein interactions.
EXPERIMENTAL PROCEDURES
Patients—Ten patients with HDR from seven unrelated familieswere ascertained (Table I). Four families ((identified by anonymouscode designations, in accordance with local ethical research committeeguidelines) 19/1992, 8/2000, 2/2001, and 13/2001) were from NorthernEurope, two families (19/2000 and 16/2001) were from North America,and one family (16/1998) was from Samoa. All 10 patients had hypo-parathyroidism with serum calcium ranging from 1.01 to 2.00 mM, andthis was associated with tetany or seizures in four patients, but wasasymptomatic in six patients (Table I). Bilateral sensorineural deafnesswas found in all 10 patients with the age at diagnosis ranging from �1to �30 years. Renal abnormalities were found in seven patients, ofwhich two patients had developed end-stage renal failure, two hadhypoplastic kidneys, and another two had agenesis of the right kidney.
DNA Sequence Analysis of the GATA3 Gene—Venous blood was ob-
tained after informed consent, as approved by the local ethical commit-tee, and used to extract leukocyte DNA (23). Nine pairs of GATA3-specific primers were used for the PCR amplification of the six exonsand ten intron-exon boundaries (Fig. 1) utilizing 150 ng of genomic DNAas described (24). The DNA sequences of both strands were determinedby Taq polymerase cycle sequencing (24) and resolved on a semi-auto-mated detection system (373 sequencer, Applied Biosystems, FosterCity, CA). DNA sequence abnormalities in the probands, which wereconfirmed either by restriction endonuclease analysis (24), by allele-specific oligonucleotide hybridization (25), or by a modified version ofthe amplification refraction mutation system (26), were demonstratedto co-segregate with the disorder and to be absent in the DNA obtainedfrom 55 unrelated individuals.
Electrophoretic Mobility Shift Assays—COS-1 cells, which do not endo-genously express GATA3, were transfected using LipofectAMINE Plus(Invitrogen, Carlsbad, CA) with either a wild type GATA3 constructprepared in pcDNA 3.1 (GATA3-pcDNA3) (Invitrogen) or a constructharboring the mutation that was introduced by the use of site-directedmutagenesis (QuikChange, Stratagene, La Jolla, CA) (16). Forty-eighthours post-transfection, the cells were harvested, and nuclear extractswere prepared for use in binding reactions that utilized a 32P-labeleddouble-stranded oligonucleotide containing the GATA3 consensus as de-scribed (16). The binding reactions were resolved by non-denaturing 6%PAGE. Western blot analysis using HG3–31 monoclonal antibody againstGATA3 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used todetect the presence of GATA3 protein in the nuclear extracts (16). Fordissociation shift assays (14, 27), unlabeled competitor DNA was added toan 100-fold excess to the binding reactions, and aliquots were removedafter 0, 10, 30, and 60 min for non-denaturing PAGE.
Nuclear Localization Studies Using GATA3-Green Fluorescent Pro-tein Fusion Constructs—The wild type and mutant GATA3 constructswere subcloned in-frame into the mammalian expression vectorpEGFP-C1 (BD Biosciences Clontech, Palo Alto, CA) as previouslydescribed (28). COS-1 cells were transfected with the GATA3-GFPconstructs, using LipofectAMINE Plus (Invitrogen), and after 24 h thecells were replated at lower density onto 70% ethanol-treated coverslipsand cultured for a further 24 h. The cells were then washed withphosphate-buffered saline (PBS), fixed with freshly prepared 4%paraformaldehyde/PBS for 30 min, washed with PBS, and mountedwith 4�,6-diamidino-2-phenylindole (DAPI)-containing Vectashield(Vector Laboratories, Burlingame, CA), as described (28). The DAPI/GFP images were visualized using a Nikon Eclipse E400 microscopewith a Y-FL Epi-fluorescence attachment and a triband DAPI-fluores-cein isothiocyanate-Rhodamine filter (28).
TABLE IClinical and biochemical findings in 10 HDR patients
Family/patient (sex)Hypoparathyroidism Deafness
(sensorineural) Renal abnormalitiese C, H, A, R,S, ERF, (agec)Serum
Ca2� Serum PTHa (assay range) Presentationb Agec Symmetryd Agec
mM
1 8/2000Proband (F) 1.01 13 pg/ml (10–64) Se 20 B 4 H�ERFMother (F) 1.97 28 pg/ml (10–64) As 45 B �25 None
2 13/2001 (M) 1.05 6 pg/ml (10–64) Se/Te 13 B, R�L �1 None
3 2/2001Proband (F) 1.42 1.1 pM (1.0–7.0) As 3 B 3 S�R�C (3)Father (M) 2.00 1.1 pM (1.0–7.0) As �40 B, L�R �30 R
4 16/1998 (F) 1.25 UD Se 4 B �8 A
5 19/1992 (M) 1.91 UD As 11 B 8 S�R�H (0.2), �ERF (9)
6 19/2000Proband (F) 1.60 8 pg/ml (10–64) As 27 B 4 C (23)Mother (F) 1.97 15 pg/ml (10–64) As 51 B 24 None
7 16/2001 (F) 1.72 13 pg/ml (10–64) Se 1 B, R�L 5 A
Normal range 2.15–2.65a PTH normal range given in parenthesis; UD, undetectable.b As, asymptomatic; Se, seizures; Te, tetany.c Age (years) at diagnosis.d B, bilateral; R, right; L, left.e Renal abnormalities: C, cysts; H, hypoplasia; A, agenesis; R, vesicoureteric reflux; S, sepsis; ERF, end stage renal failure.
GATA3 Mutations in HDR Syndrome 22625
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

GATA3 Minigene Construct for mRNA Splicing Studies—A minigenecontaining GATA3 exons 4, 5, and 6 was constructed. Each exon wasPCR-amplified from genomic DNA using exon-specific primers andconditions that were utilized for DNA sequence analysis (16). The PCRproducts were cloned directly into pGEM-T (Promega, Madison, WI)and sequenced to determine orientation and absence of Taq-introducedsecondary mutations. Each exon was excised from pGEM-T using ap-propriate restriction endonucleases and directionally subcloned in afour-way ligation reaction into pcDNA 3.1. COS-1 cells were transfectedwith the plasmid, as described above, and after 48 h the cells wereharvested and RNA prepared (24) for use in reverse-transcription PCR(RT-PCR) that utilized avian myeloblastosis virus reverse-tran-scriptase (Life Sciences Inc., St. Petersburg, FL) and a reversepcDNA3.1 primer to synthesize the first-strand cDNA. A control reac-tion without reverse transcriptase was also performed. The PCR reac-tion contained 1.5 mM MgCl2, 250 �M dNTP (Invitrogen), 0.3 �M of eachprimer (Forward exon 5 primer, 5�-TCTGCAATGCCTGTGGGCTC-TAC-3�, and reverse exon 6 primer, 5�-CTAACCCATGGCGGTGAC-CATGC-3�), and 1 unit of Taq DNA polymerase (Invitrogen) in 50 �l ofstandard PCR buffer (16). Amplification conditions were, denaturationat 95 °C for 5 min, followed by 30 cycles of 94 °C for 15 s, 65 °C for 15 s,and 72 °C for 1 min, followed by final extension at 72 °C for 5 min andrapid cooling to 20 °C.
Yeast Two-hybrid Assays—In vivo interactions between the GATA3N-terminal zinc finger (ZnF1) (Fig. 1) and FOG2 ZnF1, -5, -6, and -8were studied using a yeast two-hybrid system (BD Biosciences Clon-tech) (29). GATA3 ZnF1 (amino acids 261–293) was generated by clon-ing a PCR product, amplified from the wild type GATA3 expression
construct (16), in-frame, into the Gal4 DNA-binding domain (BD)-en-coding plasmid, pGBKT7 (30). Mutations were introduced into thisconstruct by site-directed mutagenesis (QuikChange, Stratagene).FOG2 ZnFs were generated by RT-PCR using human embryonic kidney(HEK) 293 cell RNA as template. Each FOG2 ZnF (ZnF1 amino acids236–290; ZnF5 amino acids 531–617; ZnF6 amino acids 661–747; andZnF8 amino acids 1100–1151) was cloned in-frame into the Gal4 acti-vation domain (AD)-encoding plasmid, pGADT7. The p53-pGBKT7 andLarge T antigen-pGADT7 plasmids (BD Biosciences Clontech) wereused as controls (31, 32). Competent AH109 yeast cells were trans-formed sequentially with the appropriate GATA3 and FOG2 ZnF plas-mid constructs using the LiAc/single-stranded DNA/polyethylene glycolprocedure (33). The transformants were selected on Leu�Trp� (doubledrop-out, DDO) minimal media plates by growth at 30 °C for 3 days.Transformants were then patched onto His�Ade�Leu�Trp� (quater-nary drop-out, QDO) media plates and monitored for growth for up to 3days. Expression of GATA3 and FOG2 Gal4 fusion proteins was con-firmed by preparing protein extracts from each clone according to themanufacturer’s instructions (BD Biosciences Clontech) and analyzingthem by SDS-PAGE in Tris-glycine-SDS buffer (Bio-Rad, Hercules, CA)and electro-transference onto PolyScreen polyvinylidene difluoridetransfer membrane (PerkinElmer Life Sciences, Boston, MA) in CAPSbuffer (10 mM, pH 11; Sigma Chemical Co., St. Louis, MO). Western blotanalysis was performed with antibodies to either the Gal4-AD (FOG2-pGADT7 constructs) or the Gal4 DNA-BD (GATA3-pGBKT7 con-structs), according to the manufacturer’s instructions (BD BiosciencesClontech) except that Gal4 DNA-BD antibody was used at 50 ng/ml andGal4-AD antibody at 100 ng/ml (29). A secondary antibody, goat anti-
FIG. 1. Schematic representation of the genomic structure of the GATA3 gene illustrating the locations of mutations identified inHDR patients. The human GATA3 gene consists of 6 exons that span 20 kb of genomic DNA and encode a 444-amino acid transcriptional factorthat contains two transactivating domains (TA1 and TA2) and two zinc fingers (ZnF1 and ZnF2). The sizes of exons 1, 2, 3, 4, 5, and 6 are 188,610, 537, 146, 126, and 806 bp, respectively. The ATG (translation start) site is in exon 2 and the TAG (stop) site is in exon 6. The locations of theseven HDR mutations identified by the present study are shown (numbers 1–7, which corresponds to mutations detailed in Table II) together withthe six previously reported mutations (labeled a–f: a, R277X; b, R367X; c, deletion frameshift (del, fs) from codon 156; d, insertion frameshift (ins,fs) from codon 301; e, in-frame deletion (del, inf) 316–319; f, W275R (16, 22). In addition, six whole gene deletions (del) have been previouslyreported (16, 22), yielding a total of 19 GATA3 abnormalities identified in HDR patients. Nine of the 10 HDR mutations, which affect the regionencompassing the two zinc fingers and the adjacent C-terminal region, are further detailed above in the amino acid sequence, in which every tenthamino acid is numbered. The amino acids altered by the nine HDR mutations are highlighted in black, and the seven mutations (E263V, C264R,GA268/269QT, P273T, R276Q, D278G, and D278Y) of ZnF1 generated for additional functional studies (Figs. 4 and 5) are highlighted in gray.
GATA3 Mutations in HDR Syndrome22626
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from
mouse horseradish peroxidase (Bio-Rad) was used at 1/5000 and de-tected by using an enhanced chemiluminescence (ECL) kit (AmershamBiosciences, Piscataway, NJ).
GST Fusion Proteins and Pull-down Assays—The glutathione S-transferase (GST) fusion proteins contained FOG2 ZnF1, ZnF5, ZnF6,and ZnF8 fused downstream of the GST protein in the vector pGEX-4T-1 (Amersham Biosciences). The expression of GST fusion proteinswas carried out in Escherichia coli BL21 (34). The 35S-labeled wild-typeor mutant GATA3 proteins were prepared by in vitro transcription/translation (TNT system, Promega, Madison WI) using GATA3-pcDNA3or constructs harboring selected mutations, and aliquots were utilizedto monitor [35S]methionine incorporation by SDS-PAGE (16). In vitrobinding assays using 1 �g of the fusion protein attached to glutathione-Sepharose 4B (Amersham Biosciences), and 1 �l of the radiolabeledGATA3 protein were performed in 300 �l of binding buffer (150 mM
NaCl, 20 mM Tris-HCl, pH 7.5, 0.1% Igepal CA-630, 20 �M ZnSO4,0.25% bovine serum albumin, 1 mM �-mercaptoethanol, 1.5 mM phen-ylmethylsulfonyl fluoride) and incubated with mixing for 1 h at 4 °C(35). The glutathione-Sepharose 4B-FOG2 fusion protein-GATA3 com-plexes were recovered by centrifugation (20,000 � g, 2 min) and washedfour times with 450 �l of cold binding buffer. The proteins were releasedby boiling in 15 �l of Laemmli sample buffer (Bio-Rad) and analyzed bySDS-PAGE (12% polyacrylamide resolving gel in Tris/glycine/SDS run-ning buffer (Bio-Rad). The gel was fixed and then soaked in Amplify(Amersham Biosciences) prior to autoradiography (36).
Computer Modeling of GATA3 ZnF1 Structure—The three-dimen-sional structure of the murine GATA1 N-terminal zinc finger has beenreported (37), and because the N-terminal zinc fingers of GATA1 andGATA3 are over 90% identical, we modeled the position of the GATA3mutants on this framework. The GATA1 ZnF1 three-dimensional struc-ture is archived in the Protein Data Bank (PDB) at the EuropeanBioinformatics Institute (EBI) with the accession number 1GNF (avail-able at oca.ebi.ac.uk/oca-bin/ccpeek?id � 1GNF) and was visualizedusing the MDL Chime program (MDL Information Systems, Inc., SanLeandro, CA)
RESULTS
Mutations in HDR Families—DNA sequence analysis of theentire 1332-bp coding region together with the associated splicesites and 5� and 3� untranslated regions of the GATA3 genefrom each of the seven probands with HDR revealed the pres-
ence of seven heterozygous mutations (Fig. 1 and Table II), sixof which were novel and one of which had previously beenreported in an unrelated Japanese family (22). Thus, two of themutations were nonsense mutations (Fig. 2), two were frame-shifting deletions, two were missense mutations, and one wasan acceptor splice site mutation (Fig. 3). The occurrence of thenonsense, frameshifting deletions and missense mutations inthe probands was confirmed either by restriction enzyme anal-ysis (Fig. 2), or by allele-specific oligonucleotide hybridizationanalysis, or by amplification refraction mutation system (TableII). The acceptor splice site mutation, which involved a g to ttransversion of the invariant ag (Fig. 3) was confirmed byrepeat DNA sequence analysis on independently obtained PCRproducts. This predicted a loss of this acceptor splice site andthe possible use of another naturally occurring, but normallyunused acceptor splice site at codons 351–353 (Fig. 3). Thesepredicted effects on mRNA splicing were assessed by express-ing wild-type and mutant GATA3 minigene constructs thatencompassed exons 4–6 in COS-1 cells. This revealed utiliza-tion of the alternative acceptor splice site that would lead to aloss of 8 nucleotides from the mRNA. This resulted in a frame-shift that, if translated, would produce a missense peptide witha premature termination at codon 367. Co-segregation of theGATA3 mutations and HDR was demonstrated in the availablemembers from families 8/2000 (Fig. 2), 9/2000, and 2/2001,whereas in the probands from families 13/2001 and 16/2001,the mutations were demonstrated to be absent in the parentsand hence were arising de novo (Table II). In addition, theabsence of these DNA sequence abnormalities in 110 allelesfrom 55 unrelated normal individuals indicated that these ab-normalities were mutations and not functionally neutral poly-morphisms that would be expected to occur in �1% of thepopulation. All of the seven mutations, which occurred in exons3–6 (Fig. 1), predict structurally significant changes (Table II).Thus, the E228X (Glu-228 3 Stop) and frameshift deletion
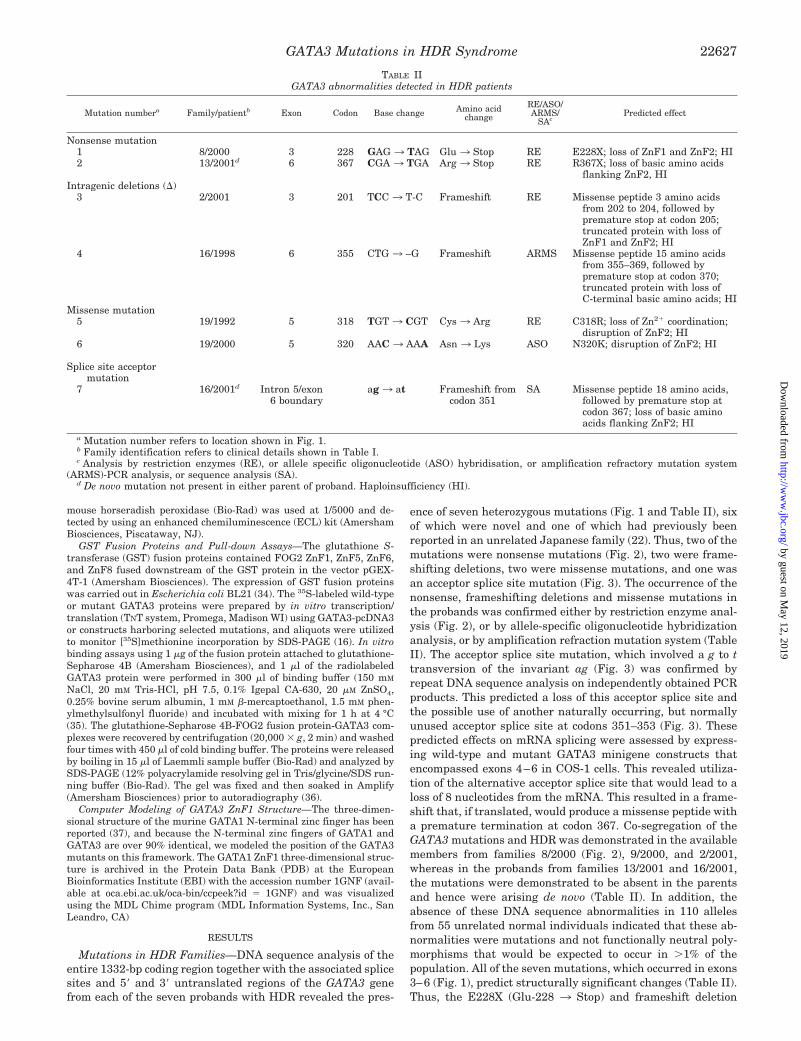
TABLE IIGATA3 abnormalities detected in HDR patients
Mutation numbera Family/patientb Exon Codon Base change Amino acidchange
RE/ASO/ARMS/
SAcPredicted effect
Nonsense mutation1 8/2000 3 228 GAG 3 TAG Glu 3 Stop RE E228X; loss of ZnF1 and ZnF2; HI2 13/2001d 6 367 CGA 3 TGA Arg 3 Stop RE R367X; loss of basic amino acids
flanking ZnF2, HIIntragenic deletions (�)
3 2/2001 3 201 TCC 3 T-C Frameshift RE Missense peptide 3 amino acidsfrom 202 to 204, followed bypremature stop at codon 205;truncated protein with loss ofZnF1 and ZnF2; HI
4 16/1998 6 355 CTG 3 –G Frameshift ARMS Missense peptide 15 amino acidsfrom 355–369, followed bypremature stop at codon 370;truncated protein with loss ofC-terminal basic amino acids; HI
Missense mutation5 19/1992 5 318 TGT 3 CGT Cys 3 Arg RE C318R; loss of Zn2� coordination;
disruption of ZnF2; HI6 19/2000 5 320 AAC 3 AAA Asn 3 Lys ASO N320K; disruption of ZnF2; HI
Splice site acceptormutation
7 16/2001d Intron 5/exon6 boundary
ag 3 at Frameshift fromcodon 351
SA Missense peptide 18 amino acids,followed by premature stop atcodon 367; loss of basic aminoacids flanking ZnF2; HI
a Mutation number refers to location shown in Fig. 1.b Family identification refers to clinical details shown in Table I.c Analysis by restriction enzymes (RE), or allele specific oligonucleotide (ASO) hybridisation, or amplification refractory mutation system
(ARMS)-PCR analysis, or sequence analysis (SA).d De novo mutation not present in either parent of proband. Haploinsufficiency (HI).
GATA3 Mutations in HDR Syndrome 22627
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

occurring in codon 201 are predicted, if translated, to lead totruncated GATA3 proteins that lack both ZnFs; the R367X(Arg-367 3 Stop), the frameshift deletions occurring in codon355, and the acceptor splice site mutation at the intron 5/exon6 boundary are predicted to lead to truncated GATA3 proteinsthat lack the C-terminal region adjacent to ZnF2, and themissense mutations C318R (Cys-3183 Arg) and N320K (Asn-320 3 Lys) are predicted to disrupt ZnF2 of GATA3 (Fig. 1).The effects of these mutations together with the W275R (Trp-275 3 Arg) that was reported in a Japanese HDR patient (22)were further assessed in DNA binding studies. The effects ofthe acceptor splice site mutation found in family 16/2001 (Fig.3) were not assessed separately, because the predicted proteinis almost identical to that resulting from the frameshift dele-tion found in family 16/1998 (Fig. 1 and Table II).
DNA Binding and Subcellular Localization Studies—All of theHDR-associated GATA3 mutations, with the exception of one,W275R, are predicted to disrupt ZnF2 or its adjacent C-terminalregion (Fig. 1), and the results of Western blot analysis areconsistent with this (Fig. 4). ZnF2, which is the C-terminal zincfinger, is essential for DNA binding, and thus all of these HDR-associated GATA3 mutations would predict a disruption of DNAbinding (Table II). However, the W275R mutation lies withinZnF1, and its effects are more difficult to predict, although some
naturally occurring and some engineered GATA1 mutants of theN-terminal zinc finger, ZnF1, have been shown to destabilizeDNA binding or protein-protein interactions (14, 35, 38–40). Wetherefore engineered the equivalent seven GATA3 mutants,E263V (Glu-2633 Val), C264R (Cys-2643 Arg), GA268/269QT(GlyAla-268/269 3 GlnThr), P273T (Pro-273 3 Thr), R276Q(Arg-2763 Gln), D278G (Asp-2783 Gly), and D278Y (Asp-2783 Tyr), so as to facilitate a more comprehensive study of the 25residues forming the GATA3 ZnF1 (Fig. 1). These residues wereselected for engineering mutants, because they either are thenon-conserved residues of ZnF1 when compared with their re-spective ZnF2 counterpart, or they are the equivalent counter-parts to GATA1 disease-causing mutations (38–40). We assessedthese GATA3 mutants (i.e. the ones associated with HDR (Fig. 1)and the seven engineered GATA3 ZnF1 mutations) initially foraltered DNA binding by EMSAs (Fig. 4), using nuclear extractsfrom COS-1 cells transfected with either the wild-type or mutantGATA3 constructs. In addition, an assessment of the subcellularlocalization of the GATA3 mutants, using GATA3-GFP con-structs, was also undertaken, and this revealed that 12 of themutants, which retained ZnF1 (Fig.1), accumulated in the nu-cleus and were indistinguishable from the WT-GATA3 (Fig. 4).However, the two mutants (deletion of C in codon 201 andE228X) that lacked ZnF1 did not accumulate in the nucleus.
FIG. 2. Detection of GATA3 muta-tion in exon 3 in family 8/2000 withHDR by restriction enzyme analysis.DNA sequence analysis of individual III,1 revealed a G to T transversion at codon228, thus altering the wild-type (WT) se-quence GAG, encoding a glutamine to themutant (m) sequence TAG, which is a ter-mination (Stop) codon. This nonsensemutation also resulted in the loss of thewild-type BsoBI restriction enzyme (C/CCGAG), and this facilitated the confir-mation of the mutation (b). PCR amplifi-cation and BsoBI digestion would resultin two products of 167 and 100 bp fromthe normal, i.e. wild type (WT) sequence,but an additional band of 267 bp would beexpected from the mutant (m) sequenceas is illustrated in the restriction map inc. Co-segregation of this E228X mutationand its heterozygosity in the affectedmembers (II, 2 and III, 1) was demon-strated (b), and the absence of this E228Xmutation in 110 alleles from 55 unrelatednormal individuals (N1 and N2 shown) in-dicates that it is not a common DNA se-quence polymorphism. Similar restrictionenzyme analysis was used to confirm anddemonstrate co-segregation of the codon201 deletion, and the C318R and theR367X mutations (Table II). Individualsare represented as: male (squares), fe-male (circles), unaffected (open symbols),affected with hypoparathyroidism (filledupper-left quadrant), affected with deaf-ness (filled lower-left quadrant), affectedwith renal anomalies (filled lower-rightquadrant), deceased (slash through sym-bol) and not available (NA).
GATA3 Mutations in HDR Syndrome22628
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

These findings are consistent with the nuclear localization signalfor GATA3 being contained within residues 249–311 that encom-pass ZnF1 (41). The EMSA studies revealed that the GATA3mutants, which disrupted or lead to a loss of ZnF2 (Fig. 1 andTable II), all resulted in a loss of DNA binding. Furthermore,addition of a 2-fold excess of these mutant GATA3 nuclear ex-tracts to the wild type, did not significantly alter binding byWT-GATA3 (data not shown), thereby suggesting an absence of adominant-negative effect due to heteroduplex formation. This isconsistent with the development of an HDR phenotype in pa-tients who have haploinsufficiency due to a deletion of theGATA3 gene (16). The GATA3 mutants involving ZnF1, all re-tained DNA binding (Fig. 4). However, these ZnF1 mutants dif-fered in the stability of their binding to DNA, which resulted inaltered rates of dissociation. Thus, the HDR-associated mutantW275R and the engineered mutants GA268/269QT, D278G, andD278Y had dissociation rates similar to that of the wild-typeGATA3 (Fig. 4), whereas the engineered mutants E263V, C264R,P273T, and R276Q had a more rapid rate of dissociation (Fig. 4).These results indicate that the ZnF1 GATA3 residues Glu-263,Cys-264, Pro-273, and Arg-276 are critical for stabilizing theDNA binding by ZnF2 and that this is likely to involve interac-tions with other multitype zinc finger proteins, in a mannersimilar to that reported for GATA1 ZnF1 (14, 35, 38–40). Forexample, the engineered GATA1 mutant C204R, which is equiv-alent to the GATA3 C264R, has been reported to destabilize DNAbinding (14) and to abolish the interaction with FOG ZnF6 (35).However, the HDR GATA3 mutant W275R and the engineered
mutants GA268/269QT, D278G, and D278Y did not alter thestability of the DNA binding (Fig. 4), and, to further elucidate therole of these residues and their mutations, we utilized a yeasttwo-hybrid assay.
Yeast Two-hybrid Assay—GATA1 ZnF1 and GATA4 ZnF1interact with the zinc finger proteins FOG1 and FOG2, respec-tively (5–7). We investigated FOG2 for interactions withGATA3, because of their similar temporo-spatial expressionpatterns (6, 12). Thus, in mouse embryos older than 11.5 days,both GATA3 and FOG2 are expressed in the same tissues thatinclude the otic vesicle and the developing kidney (6, 12). Inaddition FOG2 has been shown to interact with GATA3 inmouse embryos (7). These interactions between GATA factorsand the FOG proteins involve the GATA ZnF1 and several ofthe zinc fingers of the FOG protein. For example, the GATA1ZnF1 interacts with four of the nine zinc fingers (ZnF1, -5, -6,and -9) of FOG, and four of the eight zinc fingers (ZnF1, -5, -6,and -8) of FOG2 (42). We selected to investigate the four in-volved zinc fingers (ZnF1, -5, -6, and -8) of FOG2 for interac-tions with wild-type and mutant GATA3 ZnF1 in a yeast two-hybrid assay. One GATA3 construct and one FOG2 constructwere sequentially transformed into the yeast reporter strainAH109, and yeast containing both plasmids were selected onminimal DDO medium that lacked leucine and tryptophan(Fig. 5a). Co-expression of the GATA3 and FOG2 Gal4 fusionproteins was confirmed by Western blotting of yeast proteinextracts, prepared from each clone, and detected using antibod-ies against either the Gal4 DNA-BD or the Gal4-AD (data not
FIG. 3. Detection of acceptor splice site mutation at the intron 5/exon 6 boundary in family 16/2001 (Table II). DNA sequence analysisof the affected proband (Table I) revealed a g to t transversion at the �1 position, which resulted in an alteration of the invariant ag acceptor splicesite (a). Analysis of 110 alleles from 55 unrelated normals revealed the presence of the normal ag acceptor splice site and an absence of the atsequence, thereby indicating that the g to t transversion at position �1 was not a common sequence polymorphism but a likely mutation that wouldalter mRNA splicing (data not shown). In addition, an examination of the DNA sequences of codons 351–353 revealed another naturally occurring,but normally unused, acceptor splice site sequence (ncag) (61, 62). The effects of the likely mutation were therefore investigated by using wild-type(WT) and mutant (m) minigene constructs containing exons 4, 5, and 6 in the mammalian expression vector pcDNA 3.1, and transfecting these intoCOS-1 cells. Total RNA was extracted from the cells and utilized with exon 5- and exon 6-specific primers in RT-PCRs. The mutant RT-PCRproducts are smaller and DNA sequence analysis of these revealed splicing of exon 5 to an internal site in exon 6 that resulted in a new sequence,which encoded a missense peptide with a premature termination at codon 367. Thus, the mutation had resulted in utilization of an alternative,naturally occurring, but normally non-utilized, acceptor splice sequence. Exon sequence (uppercase), intron sequence (lowercase); �, with reversetranscriptase; �, without reverse transcriptase; size markers are in base pairs.
GATA3 Mutations in HDR Syndrome 22629
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

FIG. 4. Analysis of DNA-binding properties and subcellular localization of GATA3 mutants associated with HDR. a, Western blotanalysis of in vitro translated wild-type (WT) and GATA3 mutants revealed the expected 50-kDa WT product. The missense mutations C318R andN320K also yield a 50-kDa product, whereas the nonsense (E228X and R367X) and frameshift deletions (201�C and 355�CT) yield the predicted
GATA3 Mutations in HDR Syndrome22630
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

shown). These yeast colonies were then patched onto minimalQDO medium that lacked leucine, tryptophan, histidine, andadenine to select for those yeast in which a protein-proteininteraction had occurred (Fig. 5b). Interaction between theGATA3 and FOG2 zinc fingers would bring the Gal4 DNA-BDinto close juxtaposition with the AD at the reporter gene pro-moter, thereby enabling transcription of the reporter gene.Disruption of this interaction by the GATA3 mutant would leadto a loss of expression of the reporter genes. The results re-vealed interactions between the wild-type GATA3 ZnF1 andeach of the four FOG2 zinc fingers (ZnF1, -5, -6 and -8) (Fig. 5).However, the GATA3 mutants C264R, E263V, and GA268/269QT did not interact with any of the FOG2 zinc fingers asevidenced by an absence of yeast growth. The GATA3 mutantW275R similarly abolished interaction with FOG2 zinc fingers1, 5, and 8, but retained interaction with ZnF6, whereas theD278G and D278Y mutants retained interaction with all FOG2zinc fingers with the exception of ZnF8. However, the P273Tand R276Q mutants retained interaction with all four FOG2zinc fingers, thereby suggesting that they exert their effectsolely by loss of DNA-binding stabilization. These results of theyeast two-hybrid assay were confirmed by GST pull-downassays.
GST Pull-down Assays—GST pull-down assays were per-formed using full-length GATA3 expressed in a rabbit reticu-locyte system and FOG2 ZnF-GST fusion proteins. The wild-type GATA3, and the P273T and R276Q mutants, wereretained by FOG2 ZnF1, -5, -6, and -8, whereas the W275Rmutant was retained only with FOG2 ZnF6 (Fig. 5c, datashown for wild-type and W275R). In contrast, the E263V,C264R, and GA268/269QT mutants were not retained by any ofthe four FOG2 ZnFs (Fig. 5c, data shown for C264R), whereasthe D278G and D278Y mutants were retained by FOG2 ZnF1,-5, and -6 but not FOG2 ZnF8. These GST pull-down results,which confirm the results of the yeast two-hybrid assay, are inagreement with those previously reported for interactions be-tween GATA1 and FOG2 ZnFs (42).
DISCUSSION
Our results, which have identified seven mutations of theGATA3 gene in seven HDR probands and their families (TableII), expand the spectrum of mutations, report the first acceptorsplice site mutation (Fig. 3), and further establish the role ofGATA3 haploinsufficiency in the etiology of this developmentaldisorder. In addition, our studies of these GATA3 mutationshelp to increase our understanding of the underlying DNAbinding and protein interactions that are involved for the func-tion of this zinc finger transcription factor. Thus, all the mu-tations that disrupt either ZnF2 or the basic amino acids lo-cated C-terminal to it, lead to a loss of DNA binding (Fig. 4),whereas those that disrupt ZnF1 do not lead to a loss of DNAbinding but instead alter interactions with FOG2 (Fig. 5)
and/or change DNA binding affinity (Fig. 4). For example, thetwo missense mutations, C318R and N320K (Fig. 1 and TableII), which result in alterations of evolutionarily conserved res-idues in ZnF2 of the GATA family members, are predicted todisrupt the tertiary structure either directly or via a loss ofco-ordination of the zinc ion. This in turn results in a loss ofDNA binding and hence a likely alteration in the transcriptionof target genes. Similarly, the three mutations (two frameshiftsstarting at codons 351 and 355 and the nonsense mutationR367X) involving the residues on the C-terminal side of ZnF2(Fig. 1 and Table II) also result in a loss of DNA binding (Fig.4). These three mutations involve codons 364–369, whoseequivalents in GATA1 have been shown to be essential for DNAbinding, either by direct contact with DNA or by stabilization ofnearby residues that contact DNA (43).
In contrast to these GATA3 ZnF2 mutants, the 8 ZnF1 mu-tants (the HDR-associated W275R and the seven engineeredmutants) all retained DNA-binding activity (Fig. 4). Thesefindings for human GATA3 ZnF1 are consistent with thosereported for the chicken GATA3 ZnF1 (44), which has beenshown to bind GATA or GATC motifs even in the absence ofZnF2. Such studies (27, 44, 45) have indicated that GATA ZnF1may serve to stabilize the binding of ZnF2 to gene promoters orenhancers that contain double or palindromic GATA sites, andthereby help in distinguishing between genes that are regu-lated by different GATA members. However, the GATA3 ZnF1mutants in our study did show differences in both their DNAbinding affinities (Fig. 4) and in interactions with the four ofthe eight FOG2 ZnFs that were studied (Fig. 5). Thus, themutants E263V and C264R had low DNA binding affinities anda lack of interactions with FOG2 ZnF1, -5, -6, and -8; the P273Tand R276Q mutants had low DNA binding affinities but re-tained interactions with the four FOG2 ZnFs; the W275R,D278G, and D278Y had a normal DNA binding affinity andinteracted with some FOG2 ZnFs, e.g. W275R interacted withFOG2 ZnF6, and D278G and D278Y interacted with FOG2ZnF1, -5, and -6; whereas GA268/269QT had a normal DNAbinding affinity but a lack of interactions with any of the fourFOG2 ZnFs. The altered DNA binding affinities observed withE263V and C264R, but not GA268/269QT, may be attributed tothe disruption of the ZnF1 structure and a lack of zinc ioncoordination that is likely to result with the E263V and C264Rmutants, but not the GA268/269QT mutant that involves sub-stitutions for residues that are present in equivalent positionsin ZnF2 (Fig. 1). However, any further explanation for theseresults is difficult to provide on the basis of the primary struc-ture of GATA3 ZnF1, but an analysis of the predicted three-dimensional structure of ZnF1 (Fig. 6) may be useful, becauseit indicates that there may be specific DNA and protein bindingsurfaces. Thus, Glu-263, Cys-264, Gly-268, and Ala-269 areclustered to form a surface that is important for protein bind-
truncated products (Table II). b, electrophoretic mobility shift assays, EMSAs. COS-1 cells were transfected with either the WT or mutant GATA3constructs, and nuclear extracts were prepared for binding reactions, which used a radiolabeled (32P) double-stranded oligonucleotide containingthe GATA consensus DNA sequence (16). Control binding reactions using untransfected (UT) cells and the oligonucleotide alone (OA), i.e. withoutnuclear extract, were performed. The WT GATA3 bound to double-stranded (ds) DNA, and the method was sensitive enough to detect 10% of theWT GATA3 binding reaction. GATA3 mutants, which disrupted or lead to a loss of ZnF2 (Fig. 1), all resulted in a loss of DNA binding. However,EMSAs revealed normal DNA binding by all the GATA3 ZnF1 missense mutants, whether they were associated with HDR (W275R) or had beenengineered (E263V, C264R, GA268/269QT, P273T, R276Q, D278G, and D278Y), (panel c, 0 min). The stability of the DNA binding of all these eightGATA3 mutants that occur in ZnF1 were further studied using dissociation gel shift assays (c) in which unlabeled dsDNA was added, and theeffects on the binding of GATA3 to the radiolabeled dsDNA measured over a time course of 60 min by autoradiography. The wild-type (WT) GATA3and mutants GA268/269QT, W275R, D278G, and D278Y dissociated from the radiolabeled DNA at similar rates, whereas the E263V, C264R,P273T, and R276Q mutations dissociated more rapidly such that the 100-fold excess of unlabeled DNA had replaced all, or a substantial amount,of the radiolabeled DNA by 30 min. Subcellular localization studies (d) revealed that WT-GATA3 and the 12 mutants (E263V, C264R,GA268/269QT, P273T, W275R, R276Q, D278G, D278Y, C318R, N320K, 355�CT, and R367X) that contained ZnF1 (Fig. 1) accumulated in thenucleus, whereas the two mutants (�201C and E228X) that lacked ZnF1 did not accumulate in the nucleus but instead had a pattern similar tothat observed in the cells transfected with GFP alone (GFP). Green and blue labeling represents GFP and nuclear DAPI staining, respectively.Nuclear GFP staining masks DAPI staining, and hence the presence of blue nuclei represents untransfected cells. The scale bar represents 10 �m.
GATA3 Mutations in HDR Syndrome 22631
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

FIG. 5. Interactions between GATA3 ZnF1 and FOG2 ZnFs using a yeast two-hybrid assay. The interaction between wild-type (WT) ormutant GATA3 N-terminal ZnF1, and FOG2 ZnF1, -5, -6, and -8 was studied in the yeast reporter strain AH109 following transformation with thevectors containing GATA3 ZnF1 (pGBKT7) and each FOG2 ZnF (pGADT7) in turn. Yeast growth was monitored 48 h after streaking andincubation at 30 °C using either double dropout, DDO (Leu�Trp�), media (a) as a control, or quaternary drop out, QDO (Leu�Trp�Ade�His�)media (b) in which growth is dependent on the physical interaction between the GATA3-Gal4 DNA-BD and FOG2-Gal4-AD fusion proteins (29, 63).The SV40 large T antigen and p53 proteins, which are known to interact (32), were used as positive controls. Co-expression of the GATA3 and FOG2Gal4 fusion proteins in the yeast colonies was confirmed in each case by Western blot analysis. The WT GATA3 fusion protein interacted withFOG2 ZnF1, -5, -6, and -8 fusion proteins, whereas the engineered mutant E263V, C264R, and GA268/269QT GATA3 proteins showed an absenceof interaction with these FOG2 ZnFs. However, the W275R mutant, which was reported in an HDR patient (22), significantly interacted with FOG2
GATA3 Mutations in HDR Syndrome22632
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

ing, e.g. with FOG2 ZnFs; whereas Trp-275 and Asp-278 resideon another surface that may be important for interactions withZnF1, -5, and -8, and ZnF8, respectively, and whereas Pro-273and Arg-276 reside on a different surface that is involved inbinding DNA but not FOG2 ZnFs.
The role of the HDR-associated W275R mutation is of furtherinterest in this model. The W275R mutation is located amongresidues (Pro-273 and Arg-276) that form a DNA binding sur-face (Fig. 6), and yet it leads to a loss of protein interactionswith FOG2 ZnF1, -5, and -8 and not an alteration in DNAbinding affinity. This suggests a dual role for the WRR peptide(codons 275–277), which is conserved in both ZnF1 and ZnF2(Fig. 1), in binding to DNA as well as FOG2. These results areconsistent with those reported from studies of GATA4 ZnF2, inwhich the equivalent conserved residues were mutated andshown to be critical for DNA binding and for interactions be-tween GATA4 ZnF2 and the protein p300/CBP (46, 47). Fur-thermore, a GATA1 ZnF1 mutant, which involved the equiva-lent GATA3 residue Arg-276, failed to bind GATA motifs butinteracted normally with FOG (40). All these observations in-dicate that the WRR peptide is involved in separate FOG-GATA-interacting and DNA-binding functions, and a three-dimensional model of ZnF1 (Fig. 6) is consistent with this if thearomatic side chain of the Trp-275 residue projects away fromthe DNA binding surface formed by Pro-273, Arg-276, and
Arg-277, and is thereby available to interact with FOG2 ZnFs.We have concentrated on studying the effects of GATA3 mu-tants on the interactions with FOG2 because of their similartemporo-spatial expression patterns (6, 7). However, GATA3also interacts with other transcription factors that includeGATA1, GATA2 (48, 49), Sma- and Mad-related protein 3(smad3) (50), specificity protein 1 (51), erythroid Kruppel-likefactor (51), and rhombotin 2 (52). GATA2 (53), smad3 (54), andrhombotin 2 (55) are expressed in kidney, whereas specificityprotein 1 is expressed in both kidney (56) and parathyroids(57), and thus, it may be possible for HDR-associated GATA3mutations to disrupt interactions with these proteins, providedthat they were expressed contemporaneously.
An examination of the HDR-associated GATA3 mutationstogether with the observed phenotypes does not establish acorrelation (Tables I and II), and this is well illustrated by thetwo unrelated families from Britain and Japan (22) who had anidentical R367X mutation but different phenotypes. Thus, theBritish patient 13/2001 (Tables I and Table I) had hypopara-thyroidism and deafness but no renal abnormalities, whereasboth Japanese patients had hypoparathyroidism and renal ab-normalities but no deafness (22). Furthermore, even withinfamilies with patients harboring identical GATA3 mutations,there appears to be a variable expression of renal abnormalitiesas illustrated by family 8/2000 (Fig. 2 and Table I). The basis of
ZnF6 but not with FOG2 ZnF1, -5, and -8. The engineered mutants P273T and R276Q interacted with FOG2 ZnF1, -5, -6, and -8, whereas D278Gand D278Y interacted with FOG2 ZnF1, -5, and -6. These results were confirmed by GST pull-down assays (c) that utilized in vitro translated35S-labeled GATA3 and GST-FOG2 ZnF fusion proteins (data shown for wild-type, C264R, and W275R). The input row demonstrates that equalamounts of the wild-type and mutant GATA3 protein were loaded, and the Coomassie-stained gel (d) shows that approximately equal amounts ofGST-FOG2 fusion proteins were used in the GST pull-down assay. These results of GATA3-ZnF1 interactions with FOG2, are consistent with thefindings of the proposed GATA1-ZnF1 three-dimensional model (Fig. 6).
FIG. 6. Three-dimensional structure of the human GATA3-ZnF1 based on the model of murine GATA1 ZnF1 (37). Human GATA1,which consists of 413 amino acids, and human GATA3, which consists of 444 amino acids, belong to the same subfamily (10) and share structuralsimilarities that include two ZnFs (Fig. 1). The three-dimensional structure of the murine GATA1-ZnF1(residues 201–243) has been characterized,and this has 91% identity to the human GATA3-ZnF1 (a), thereby enabling us to use this to construct a three-dimensional model of humanGATA3-ZnF1 (residues 261–303). The residues shown in the ribbon (b) and space-filing (c) models refer to those of the equivalent human (h) GATA3ZnF1, and the corresponding murine (m) GATA1 ZnF1 residues are as follows: hE263 � mE203, hC264 � mC204, hG268 � mG208, hA269 �mA209, hW275 � mW215, hR276 � mR216, and hD278 � mD218. Residues participating in the interaction between mGATA1 and FOG ZnFs,which include the human equivalents of Glu-263, Cys-264, Gly-268, and Ala-269, are shown. They are seen to form a binding surface distinct fromthat containing Trp-275 and Asp-278, which have been shown to interact with different FOG zinc fingers, whereas Arg-276 lies at the DNA bindingsurface and does not participate in binding to FOG2 ZnFs. Pro-273, which also resides at a DNA binding surface, is not visible in the projectionshown. The backbone is shown as dark magenta; hydrophobic side chains as gray; polar side chains as magenta; acidic side chains as red; and basicside chains as blue. This color scheme derives from the Corey, Pauling, Koltun (CPK) color scheme as follows. hydrophobic � carbon; acidic �oxygen; basic � nitrogen; polar but uncharged � a mixture of oxygen (red) and nitrogen (blue), namely magenta. The backbone is polar but lesslikely (dark magenta) than side chains to hydrogen bond to non-backbone moieties, because most backbone hydrogen bonding occurs within thebackbone.
GATA3 Mutations in HDR Syndrome 22633
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

these phenotypic differences in patients with the same muta-tion remains to be elucidated. One possibility is that there maybe different levels of compensation by other GATA family mem-bers in different patients. This hypothesis seems attractive,particularly because GATA2 and GATA3 have been shown tobe able to partially compensate for the loss of GATA1 in thedifferentiation of hematopoietic lineages, when placed underthe control of the GATA1 locus in transgenic mice (58, 59).However, it is important to note that studies of mice lackingGATA1 have demonstrated that GATA2 does not compensatefor the loss of GATA1 function in vivo (60), thereby indicatingthat extrapolation of compensatory mechanisms to the nativesituation requires cautious interpretation. Additional studiesinvestigating for genotype-phenotype correlations in HDR pa-tients and for alterations in the expression of GATA familymembers that may compensate for reduced GATA3 expressionare required. In summary, our studies have shown that HDR-associated GATA3 mutations may either disrupt DNA bindingor protein interactions with FOG2 and that these are consist-ent with the roles of the zinc finger domains and with theproposed three-dimensional model. However, the manner inwhich these GATA3 mutations lead to parathyroid, otic vesicle,and renal anomalies remains to be elucidated.
Acknowledgment—We (M. A. N., M. R. B., B. H., A. A., and R. V. T.)are grateful to the Medical Research Council (UK).
REFERENCES
1. Pandolfi, P. P., Roth, M. E., Karis, A., Leonard, M. W., Dzierzak, E., Grosveld,F. G., Engel, J. D., and Lindenbaum, M. H. (1995) Nat. Genet. 11, 40–44
2. Kuo, C. T., Morrisey, E. E., Anandappa, R., Sigrist, K., Lu, M. M., Parmacek,M. S., Soudais, C., and Leiden, J. M. (1997) Genes Dev. 11, 1048–1060
3. Simon, M. C. (1995) Nat. Genet. 11, 9–114. Orkin, S. H. (1992) Blood 80, 575–5815. Tsang, A. P., Visvader, J. E., Turner, C. A., Fujiwara, Y., Yu, C., Weiss, M. J.,
Crossley, M., and Orkin, S. H. (1997) Cell 90, 109–1196. Tevosian, S. G., Deconinck, A. E., Cantor, A. B., Rieff, H. I., Fujiwara, Y.,
Corfas, G., and Orkin, S. H. (1999) Proc. Natl. Acad. Sci. U. S. A. 96,950–955
7. Svensson, E. C., Tufts, R. L., Polk, C. E., and Leiden, J. M. (1999) Proc. Natl.Acad. Sci. U. S. A. 96, 956–961
8. Haenlin, M., Cubadda, Y., Blondeau, F., Heitzler, P., Lutz, Y., Simpson, P., andRamain, P. (1997) Genes Dev. 11, 3096–3108
9. Fossett, N., Zhang, Q., Gajewski, K., Choi, C. Y., Kim, Y., and Schulz, R. A.(2000) Proc. Natl. Acad. Sci. U. S. A. 97, 7348–7353
10. Weiss, M. J., and Orkin, S. H. (1995) Exp. Hematol. 23, 99–10711. Molkentin, J. D. (2000) J. Biol. Chem. 275, 38949–3895212. George, K. M., Leonard, M. W., Roth, M. E., Lieuw, K. H., Kioussis, D.,
Grosveld, F., and Engel, J. D. (1994) Development 120, 2673–268613. Debacker, C., Catala, M., and Labastie, M. C. (1999) Mech. Dev. 85, 183–18714. Nichols, K. E., Crispino, J. D., Poncz, M., White, J. G., Orkin, S. H., Maris,
J. M., and Weiss, M. J. (2000) Nat. Genet. 24, 266–27015. Wechsler, J., Greene, M., McDevitt, M. A., Anastasi, J., Karp, J. E., Le Beau,
M. M., and Crispino, J. D. (2002) Nat. Genet. 32, 148–15216. Van Esch, H., Groenen, P., Nesbit, M. A., Schuffenhauer, S., Lichtner, P.,
Vanderlinden, G., Harding, B., Beetz, R., Bilous, R. W., Holdaway, I., Shaw,N. J., Fryns, J. P., Van de Ven, W., Thakker, R. V., and Devriendt, K. (2000)Nature 406, 419–422
17. Pehlivan, T., Pober, B. R., Brueckner, M., Garrett, S., Slaugh, R., VanRheeden, R., Wilson, D. B., Watson, M. S., and Hing, A. V. (1999) Am. J.Med. Genet. 83, 201–206
18. Bilous, R. W., Murty, G., Parkinson, D. B., Thakker, R. V., Coulthard, M. G.,Burn, J., Mathias, D., and Kendall-Taylor, P. (1992) N. Engl. J. Med. 327,1069–1074
19. Van Esch, H., Groenen, P., Daw, S., Poffyn, A., Holvoet, M., Scambler, P.,Fryns, J. P., Van de Ven, W., and Devriendt, K. (1999) Clin. Genet. 55,269–276
20. Hasegawa, T., Hasegawa, Y., Aso, T., Koto, S., Nagai, T., Tsuchiya, Y., Kim,K. C., Ohashi, H., Wakui, K., and Fukushima, Y. (1997) Am. J. Med. Genet.73, 416–418
21. Fujimoto, S., Yokochi, K., Morikawa, H., Nakano, M., Shibata, H., Togari, H.,and Wada, Y. (1999) Am. J. Med. Genet. 86, 427–429
22. Muroya, K., Hasegawa, T., Ito, Y., Nagai, T., Isotani, H., Iwata, Y., Yamamoto,K., Fujimoto, S., Seishu, S., Fukushima, Y., Hasegawa, Y., and Ogata, T.(2001) J. Med. Genet. 38, 374–380
23. Thakker, R. V., Bouloux, P., Wooding, C., Chotai, K., Broad, P. M., Spurr,N. K., Besser, G. M., and O’Riordan, J. L. (1989) N. Engl. J. Med. 321,218–224
24. Lloyd, S. E., Pearce, S. H., Fisher, S. E., Steinmeyer, K., Schwappach, B.,Scheinman, S. J., Harding, B., Bolino, A., Devoto, M., Goodyer, P., Rigden,S. P., Wrong, O., Jentsch, T. J., Craig, I. W., and Thakker, R. V. (1996)Nature 379, 445–449
25. Pearce, S. H., Trump, D., Wooding, C., Besser, G. M., Chew, S. L., Grant, D. B.,Heath, D. A., Hughes, I. A., Paterson, C. R., Whyte, M. P., and Thakker,R. V. (1995) J. Clin. Invest. 96, 2683–2692
26. Newton, C. R., Heptinstall, L. E., Summers, C., Super, M., Schwarz, M.,Anwar, R., Graham, A., Smith, J. C., and Markham, A. F. (1989) Lancet 2,1481–1483
27. Trainor, C. D., Ghirlando, R., and Simpson, M. A. (2000) J. Biol. Chem. 275,28157–28166
28. Quadrini, K. J., and Bieker, J. J. (2002) J. Biol. Chem. 277, 32243–3225229. Chien, C. T., Bartel, P. L., Sternglanz, R., and Fields, S. (1991) Proc. Natl.
Acad. Sci. U. S. A. 88, 9578–958230. Louvet, O., Doignon, F., and Crouzet, M. (1997) BioTechniques 23, 816–818,
82031. Li, B., and Fields, S. (1993) FASEB J. 7, 957–96332. Iwabuchi, K., Li, B., Bartel, P., and Fields, S. (1993) Oncogene 8, 1693–169633. Gietz, R. D., Schiestl, R. H., Willems, A. R., and Woods, R. A. (1995) Yeast 11,
355–36034. Smith, D. B., and Johnson, K. S. (1988) Gene (Amst.) 67, 31–4035. Fox, A. H., Kowalski, K., King, G. F., Mackay, J. P., and Crossley, M. (1998)
J. Biol. Chem. 273, 33595–3360336. Perng, G. G., Rulli, R. D., Wilson, D. L., and Perry, G. W. (1988) Anal. Biochem.
173, 387–39237. Kowalski, K., Czolij, R., King, G. F., Crossley, M., and Mackay, J. P. (1999)
J. Biomol. NMR 13, 249–26238. Mehaffey, M. G., Newton, A. L., Gandhi, M. J., Crossley, M., and Drachman,
J. G. (2001) Blood 98, 2681–268839. Freson, K., Matthijs, G., Thys, C., Marien, P., Hoylaerts, M. F., Vermylen, J.,
and Van Geet, C. (2002) Hum. Mol. Genet. 11, 147–15240. Yu, C., Niakan, K. K., Matsushita, M., Stamatoyannopoulos, G., Orkin, S. H.,
and Raskind, W. H. (2002) Blood 100, 2040–204541. Yang, Z., Gu, L., Romeo, P. H., Bories, D., Motohashi, H., Yamamoto, M., and
Engel, J. D. (1994) Mol. Cell. Biol. 14, 2201–221242. Fox, A. H., Liew, C., Holmes, M., Kowalski, K., Mackay, J., and Crossley, M.
(1999) EMBO J. 18, 2812–282243. Omichinski, J. G., Trainor, C., Evans, T., Gronenborn, A. M., Clore, G. M., and
Felsenfeld, G. (1993) Proc. Natl. Acad. Sci. U. S. A. 90, 1676–168044. Pedone, P. V., Omichinski, J. G., Nony, P., Trainor, C., Gronenborn, A. M.,
Clore, G. M., and Felsenfeld, G. (1997) EMBO J. 16, 2874–288245. Trainor, C. D., Omichinski, J. G., Vandergon, T. L., Gronenborn, A. M., Clore,
G. M., and Felsenfeld, G. (1996) Mol. Cell. Biol. 16, 2238–224746. Dai, Y. S., and Markham, B. E. (2001) J. Biol. Chem. 276, 37178–3718547. Lee, Y., Shioi, T., Kasahara, H., Jobe, S. M., Wiese, R. J., Markham, B. E., and
Izumo, S. (1998) Mol. Cell. Biol. 18, 3120–312948. Crossley, M., Merika, M., and Orkin, S. H. (1995) Mol. Cell. Biol. 15,
2448–245649. Yang, H. Y., and Evans, T. (1995) Mol. Cell. Biol. 15, 1353–136350. Blokzijl, A., ten Dijke, P., and Ibanez, C. F. (2002) Curr. Biol. 12, 35–4551. Merika, M., and Orkin, S. H. (1995) Mol. Cell. Biol. 15, 2437–244752. Osada, H., Grutz, G., Axelson, H., Forster, A., and Rabbitts, T. H. (1995) Proc.
Natl. Acad. Sci. U. S. A. 92, 9585–958953. Uchida, S., Matsumura, Y., Rai, T., Sasaki, S., and Marumo, F. (1997) Bio-
chem. Biophys. Res. Commun. 232, 65–6854. Oxburgh, L., and Robertson, E. J. (2002) Mech. Dev. 112, 207–21155. Royer-Pokora, B., Rogers, M., Zhu, T. H., Schneider, S., Loos, U., and Bolitz, U.
(1995) Oncogene 10, 1353–136056. Cohen, H. T., Bossone, S. A., Zhu, G., McDonald, G. A., and Sukhatme, V. P.
(1997) J. Biol. Chem. 272, 2901–291357. Alimov, A. P., Langub, M. C., Malluche, H. H., and Koszewski, N. J. (2003)
Endocrinology 144, 3138–314758. Tsai, F. Y., Browne, C. P., and Orkin, S. H. (1998) Dev. Biol. 196, 218–22759. Takahashi, S., Shimizu, R., Suwabe, N., Kuroha, T., Yoh, K., Ohta, J., Nish-
imura, S., Lim, K. C., Engel, J. D., and Yamamoto, M. (2000) Blood 96,910–916
60. Fujiwara, Y., Browne, C. P., Cunniff, K., Goff, S. C., and Orkin, S. H. (1996)Proc. Natl. Acad. Sci. U. S. A. 93, 12355–12358
61. Mount, S. M. (1982) Nucleic Acids Res. 10, 459–47262. Burset, M., Seledtsov, I. A., and Solovyev, V. V. (2001) Nucleic Acids Res. 29,
255–25963. Silver, P. A., Keegan, L. P., and Ptashne, M. (1984) Proc. Natl. Acad. Sci.
U. S. A. 81, 5951–5955
GATA3 Mutations in HDR Syndrome22634
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from

McWilliams, Susan Rigden, Julian Sampson, Andrew J. Williams and Rajesh V. ThakkerCrowe, Angus Dobbie, Geeta Hampson, Ian Holdaway, Michael A. Levine, Robert
M. Andrew Nesbit, Michael R. Bowl, Brian Harding, Asif Ali, Alejandro Ayala, CarolRenal Dysplasia (HDR) Syndrome
Mutations in the Hypoparathyroidism, Deafness, andGATA3Characterization of
doi: 10.1074/jbc.M401797200 originally published online February 24, 20042004, 279:22624-22634.J. Biol. Chem.
10.1074/jbc.M401797200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/279/21/22624.full.html#ref-list-1
This article cites 63 references, 30 of which can be accessed free at
by guest on May 12, 2019
http://ww
w.jbc.org/
Dow
nloaded from





























