Embed Size (px)
Citation preview

The LDA-1/2 method in excitingRonaldo Rodrigues Pelá
Humboldt Universität zu BerlinInstituto Tecnológico de Aeronáutica
http://exciting-code.org

Outline
DFT-1/2
Exchange-correlation functionals
Exact exchange calculations
Hybrid-functional calculations
Van-der-Waals corrections

DFT-1/2Accuracy: comparable to hybrid functionals
Computational cost: semilocal functionals
Basic idea: introduce a “half-hole”
Approximate QP corrections: band edges

DFT-1/2
AIP Advances 1, 032119 (2011)
Band gaps: binary compounds

DFT-1/2
APL 98, 151907 (2011)J. Phys. Cond. Matt. 27, 505502 (2015)
AlGaN
InGaN
Band gaps: alloys

DFT-1/2
Phys. Rev. B 90, 224102 (2014)Phys. Rev. B 88, 224102 (2013)
EF(Si-insterst.)
Exp = 4.1-5.1 eVLDA = 3.7-3.8 eVLDA-1/2 = 4.6-4.9 eV
Defects and impurities

DFT-1/2
APL 100, 202408 (2012)APL 101, 112403 (2012)
PRB 79, 241312–R (2009)JAP 114, 033709 (2013)
Magnetic semiconductors Band offsets

DFT-1/2
Slater's half occupation scheme
Assuming linearity
0 1/2 1
Janak's theorem

DFT-1/2
0 1/2 1
A calculation for N electrons and with a KS potential given by will work as desired

DFT-1/2
It is easy to calculate Vs for atoms
For more complex systems, it is more difficult
Approximation: Vs for a “complex system” can be obtained from atomic calculations

DFT-1/2
Atomic calculations
Recalling:

DFT-1/2Trimming function
determined by means of a variational process
PRB 78, 125116 (2008)

DFT-1/2Exciting…
<species speciesfile="Si.xml" rmt="2.1"><atom coord="0.00 0.00 0.00"></atom><atom coord="0.25 0.25 0.25"></atom><dfthalfparam exponent="8"
cut="3.90"ampl="1">
<shell number="0" ionization="0.25" /></dfthalfparam>
</species>…

DFT-1/2Exciting…
<species speciesfile="Si.xml" rmt="2.1"><atom coord="0.00 0.00 0.00"></atom><atom coord="0.25 0.25 0.25"></atom><dfthalfparam exponent="8"
cut="3.90"ampl="1">
<shell number="0" ionization="0.25" /></dfthalfparam>
</species>…

DFT-1/2Exciting…
<species speciesfile="Si.xml" rmt="2.1"><atom coord="0.00 0.00 0.00"></atom><atom coord="0.25 0.25 0.25"></atom><dfthalfparam exponent="8"
cut="3.90"ampl="1">
<shell number="0" ionization="0.25" /></dfthalfparam>
</species>…

DFT-1/2Exciting…
<species speciesfile="Si.xml" rmt="2.1"><atom coord="0.00 0.00 0.00"></atom><atom coord="0.25 0.25 0.25"></atom><dfthalfparam exponent="8"
cut="3.90"ampl="1">
<shell number="0" ionization="0.25" /></dfthalfparam>
</species>…

DFT-1/2Exciting…
<species speciesfile="Si.xml" rmt="2.1"><atom coord="0.00 0.00 0.00"></atom><atom coord="0.25 0.25 0.25"></atom><dfthalfparam exponent="8"
cut="3.90"ampl="1">
<shell number="0" ionization="0.25" /></dfthalfparam>
</species>…
Which atomic shell needs to be ionized (the order is the same as in the species file)

DFT-1/2Exciting…
<species speciesfile="Si.xml" rmt="2.1"><atom coord="0.00 0.00 0.00"></atom><atom coord="0.25 0.25 0.25"></atom><dfthalfparam exponent="8"
cut="3.90"ampl="1">
<shell number="0" ionization="0.25" /></dfthalfparam>
</species>…
How much charge should be removed from the atomic shell to obtain Vs

DFT-1/2
Exciting
…<groundstate ngridk="8 8 8"
xctype="GGA_PBE"><dfthalf printVSfile="false"/>
</groundstate>…
Very important: this triggers a DFT-1/2 calculation

DFT-1/2 Tutorial
1) Obtain the gap of Si (given the rcut
of 3.9 Bohr)2) Obtain the r
cut of Si
Egap = 1.2 eV (exp: 1.17 eV)3) Obtain the r
cut of GaN
Egap = 3.4 eV (exp: 3.3 eV)

DFT-1/2 Tutorial
E.g.: SiRefining, to find the maximum

Exchange-correlation functionals
KS equation:

Exchange-correlation functionals
LDA:
GGA:
meta-GGA:
EXX:
Hybrids:

Exchange-correlation functionals
…<groundstate ngridk="8 8 8"
xctype="GGA_PBE"></groundstate>…
exciting input
Specification of the XC functional
“class_name”
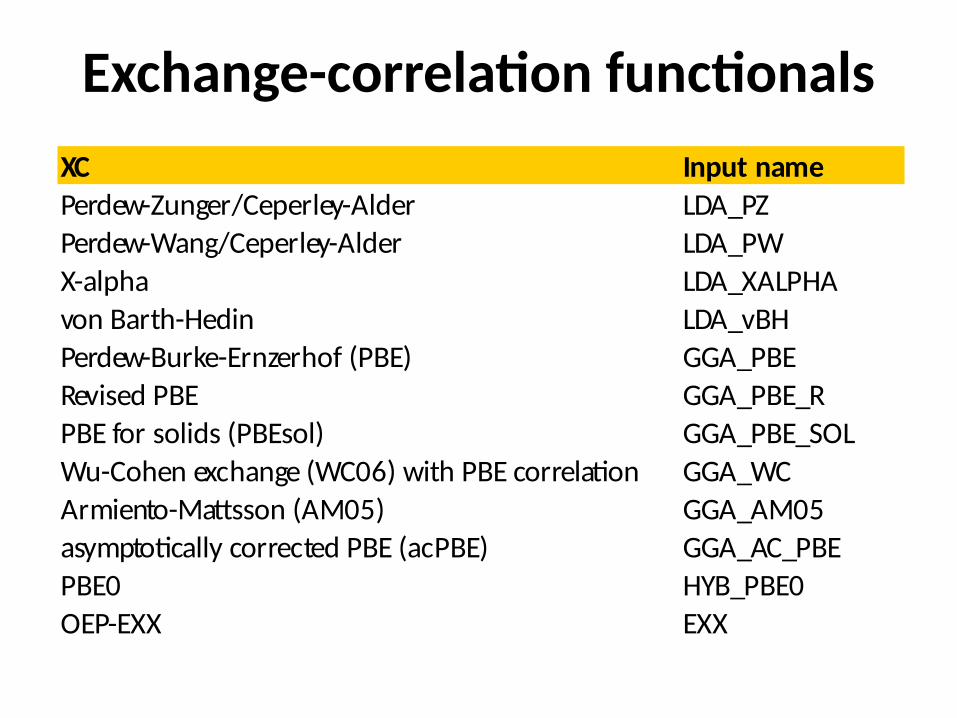
Exchange-correlation functionalsXC Input namePerdew-Zunger/Ceperley-Alder LDA_PZPerdew-Wang/Ceperley-Alder LDA_PWX-alpha LDA_XALPHAvon Barth-Hedin LDA_vBHPerdew-Burke-Ernzerhof (PBE) GGA_PBERevised PBE GGA_PBE_RPBE for solids (PBEsol) GGA_PBE_SOLWu-Cohen exchange (WC06) with PBE correlation GGA_WCArmiento-Mattsson (AM05) GGA_AM05asymptotically corrected PBE (acPBE) GGA_AC_PBEPBE0 HYB_PBE0OEP-EXX EXX

Exchange-correlation functionals
The LIBXC library
www.tddft.org/programs/octopus/wiki/index.php/Libxc [By Miguel A. L. Marques]
Library of XC functionals
Interfaces to various programs
LIBXC

Exchange-correlation functionals
The LIBXC library
…<groundstate ngridk="8 8 8">
<libxc xc = "XC_GGA_XC_LYP" /></groundstate>…

Exchange-correlation functionals
The LIBXC library
…<groundstate ngridk="8 8 8">
<libxc correlation = "XC_GGA_C_PBE"exchange = "XC_GGA_X_PBE"
/></groundstate>…
Combination of different X and C functionalspossible (use with care!)
Available in exciting: L(S)DAs and GGAs

Exchange-correlation functionalsTutorial: Si

Exact exchange calculationsExchange energy
KS potential needed
EXX energy is not an explicit functional of the density. How to get the derivative?

Exact exchange calculationsThe chain rule
PRB 53, 7024 (1996).
Nonlocal matrix elements

Exact exchange calculationsThe chain rule
PRB 53, 7024 (1996).
Density response function
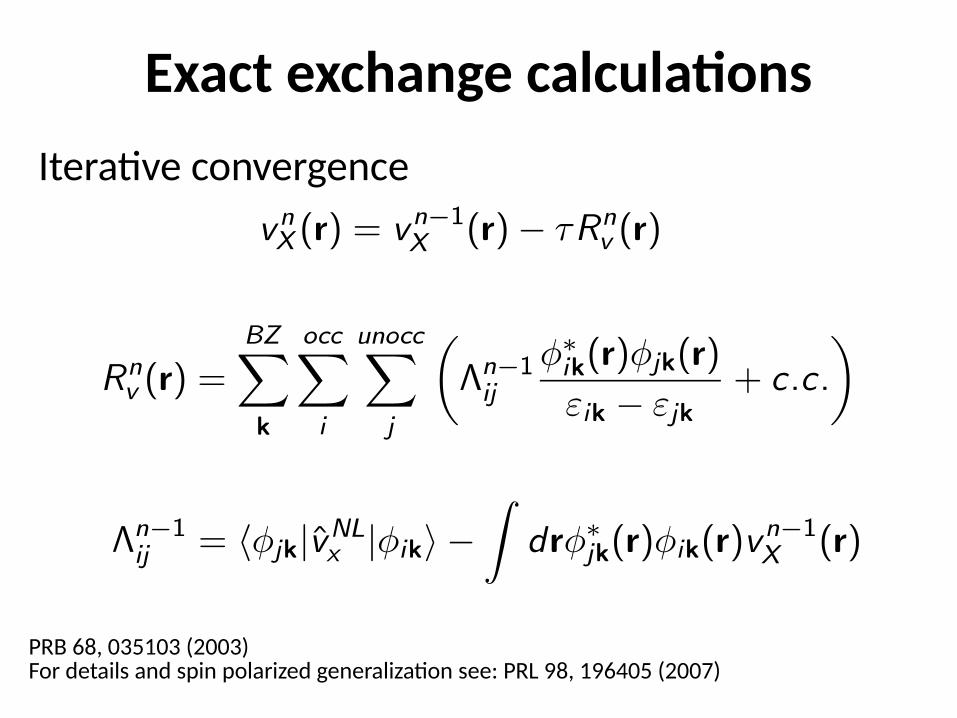
Exact exchange calculationsIterative convergence
PRB 68, 035103 (2003)For details and spin polarized generalization see: PRL 98, 196405 (2007)

Exact exchange calculationsInput parameters
PRB 68, 035103 (2003)For details and spin polarized generalization see: PRL 98, 196405 (2007)
tauoep
maxitoep
ngridk nempty

Exact exchange calculationsInput
…<groundstate
ngridk="3 3 3"xctype="EXX" nempty="30" >
<OEP maxitoep = "XC_GGA_C_PBE"tauoep = "1.0 0.2 1.5"
/></groundstate>…
Initial value Values for subsequent iterations

Exact exchange calculationsTutorial: C

Hybrid functional calculationsInput
…<groundstate
ngridk="3 3 3"xctype="HYB_PBE0" nempty="50" >
<Hybridexchangetype = "HF"excoeff = "0.25"
/></groundstate>…

Hybrid functional calculationsTutorial: C

Hybrid functional calculationsTutorial: C. 8x8x8 k-grid, 100 empty states

Van-der-Waals correctionsInput
Specification of the VdW correction“none”“DFTD2”: J. Comput. Chem. 27, 1787 (2006).“TSvdW”: PRL 102, 073005 (2009)
…<groundstate
ngridk="10 10 4"gmaxvr="20"rgkmax="6.5"xctype="GGA_PBE"vdWcorrection="DFTD2">
</groundstate>…

Van-der-Waals correctionsThe van-der-Waals correction is added to total energy after the last SCF iteration
If forces are calculated, an appropriate dispersion correction is applied Parameters corresponding to each method can be specified using the subelements “DFTD2parameters” and “TSvdWparameters” inside the element “groundstate”
It is also possible to decouple these van-der-Waals corrections from a complete ground-state calculation, using the subelements “DFTD2” and “TSvdW” inside the element properties

Van-der-Waals correctionsTutorial: Graphite

![Generalized Correspondence-LDA Models (GC-LDA) for ... · The GC-LDA and Correspondence-LDA models are extensions of Latent Dirichlet Allocation (LDA) [3]. Several Bayesian methods](https://img.pdfslide.net/doc/110x75/6011a7de37d63b741248406f/generalized-correspondence-lda-models-gc-lda-for-the-gc-lda-and-correspondence-lda.jpg)






















