Embed Size (px)
Citation preview

The Low Density Lipoprotein Receptor-Related Protein/alpha 2-Macroglobulin
Receptor is a Receptor for Connective Tissue Growth Factor (CTGF)
Patricia R. Segarini +,%, James E. Nesbitt#,+ , Dongxia Li+, Lara G. Hayes*,@, John R. Yates
III *,$ and David F. Carmichael+
#To whom correspondence should be addressed.([email protected])
+FibroGen, Inc.225 Gateway Blvd.
South San Francisco, CA 94080phone: 650-866-7200FAX: 650-866-7205
%current address :Aspira Biosystems571 Eccles Ave.
South San Francisco, CA 94080
*University of WashingtonDepartment of Molecular Biotechnology
Box 357730Seattle, Washington 98195
@current address: Zymogenetics
1201 Eastlake Ave East Seattle, WA 98102
$current address:The Scripps Research Institute
Department of Cell Biology, SR1110550 North Torrey Pines Road
1
Copyright 2001 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on August 22, 2001 as Manuscript M105180200 by guest on M
arch 19, 2018http://w
ww
.jbc.org/D
ownloaded from

La Jolla, CA 92037
2
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

RUNNING TITLE
LRP is a CTGF Receptor
3
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

SUMMARY
Connective tissue growth factor (CTGF) expression is regulated by transforming growth factor-ß
(TGF-ß) and strong upregulation occurs during wound healing; in situ hybridization data
indicate that there are high levels of CTGF expression in fibrotic lesions. Recently the binding
parameters of CTGF to both high and lower affinity cell surface binding components have been
characterized1. Affinity crosslinking and SDS-PAGE analysis demonstrated the binding of
CTGF to a cell surface protein with a mass of ~620 kDa. We report here the purification of this
protein by affinity chromatography on CTGF coupled to Sepharose and sequence information
obtained by mass spectroscopy. The binding protein was identified as the multi-ligand receptor,
low density lipoprotein receptor-related protein / alpha-2-macroglobulin receptor (LRP). The
identification of LRP as a receptor for CTGF was validated by several studies: (1) binding
competition with many ligands that bind to LRP, including Receptor-Associated Protein (RAP);
(2) immunoprecipitation of CTGF-receptor complex with LRP antibodies; and (3) cells that are
genetically deficient for LRP were unable to bind CTGF. Lastly, CTGF is rapidly internalized
and degraded and this process is LRP-dependent. In summary, our data indicate that LRP is a
receptor for CTGF, and may play an important role in mediating CTGF biology.
4
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

INTRODUCTION
Connective tissue growth factor2 (CTGF; Mr approximately 38,000 Da), is a member of
the CCN family of growth factors, which is characterized by the presence and conserved spacing
of some 38 cysteine residues. CTGF was initially identified in and subsequently purified from
HUVEC conditioned media (1). Early studies demonstrated the strong induction of CTGF
expression by transforming growth factor beta (TGF-ß), and the promoter region of the CTGF
gene contains a unique TGF-ß response element not shared by other members of the CCN
family (2). In an anchorage-independent growth assay of TGF-ß, neutralization of CTGF
activity with antibodies or inhibition of CTGF expression with antisense oligonucleotides
reduced the ability of the cells to form colonies (3), suggesting that CTGF is a necessary part of
the cascade for induction of anchorage-independent growth. More recently, CTGF was shown
to be positively regulated by vascular endothelial growth factor (VEGF) (4,5), epidermal growth
factor, fibroblast growth factor (4), plasma clotting factor VIIa (6,7), thrombin (7,8), and by
lysophosphatidic acid and serotinin activation of heptahelical receptors (9), but negatively
regulated by tumor necrosis factor alpha (10) and the Wilms tumor suppressor WT1 (11).
The initial discovery of CTGF in vascular endothelium (1), and the subsequent
demonstration that CTGF is involved in the proliferation and migration of vascular endothelial
cells (12), suggests that CTGF is also an angiogenic factor. The isolation of CTGF from uterine
fluid and localization in embryonic and placental tissues suggests of role for CTGF in embryo
implantation (13,14).
CTGF is expressed at high levels during granulation tissue deposition in normal healing
5
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

wounds (15,16). Expression of the extracellular matrix proteins fibronectin, α5 integrin, and
type I collagen is regulated by CTGF (16-18). Co-administration of CTGF with TGF-ß
produced a persistent fibrotic reaction that lasted for 14 days, but had resolved and was absent by
7 days in animals treated with TGF-ß-alone (19). The overexpression of TGF-ß in fibrotic
lesions is well documented (reviewed in (20,21)), and now many reports indicate that CTGF, too,
is overexpressed in many fibrotic lesions ((22-28) and reviewed in (29)). The emerging
understanding that CTGF is actively involved in the induction and/or maintenance of persistent
fibrosis has provided a target for the modulation of matrix overproduction in fibrotic disease.
The low density lipoprotein receptor-related protein (LRP)/alpha-2-macroglobulin
receptor (α2mR) (hereto referred to as LRP), is a member of the family of low density
lipoprotein (LDL) receptors (30). The LDL receptor family includes two subfamilies: one
containing “small” receptor members of approximately 120 kDa, including the LDL receptor
(LDLR), apo E receptor-2 (apoER2), and very low density lipoprotein receptor (VLDLR); and
one containing “large” receptor members of approximately 600 kDa, including LRP, epithelial
glycoprotein 330/megalin, and a related protein, sorLA. LDL family receptors bind multiple
ligands. At least two of these protein ligands, apolipoprotein E (apoE) and a 39 kDa Receptor-
Associated Protein (RAP), bind to all LDL receptor family members (30). A number of
functionally and structurally distinct ligands bind LRP and the diversity of these ligands suggests
that it may function in a variety of distinct physiological processes, such as lipoprotein
metabolism, protease regulation, tissue repair and remodeling, and embryonic development
(reviewed in (30)).
Here we have further characterized the major CTGF binding protein1. This protein was
6
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

affinity purified and identified as LRP. Competitive inhibition of CTGF binding using other
LRP ligands and immunoprecipitation of CTGF receptor complexes with LRP antibodies has
confirmed that LRP is a receptor for CTGF. Additionally, this report shows that CTGF is rapidly
internalized and degraded by cells through an LRP-dependent pathway. This is the first
identification of direct interaction of a growth factor with LRP. Our findings suggest that LRP
has a regulatory role in the biology of CTGF.
7
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

EXPERIMENTAL PROCEDURES
Materials
A baculovirus expression system was used for the preparation of recombinant human
CTGF (rhCTGF) and the protein was purified as described1. Apolipoprotein E (apoE), low
density lipoprotein (LDL), lactoferrin (LF), alpha 2-macroglobulin (α2m), and chloroquine were
purchased from Sigma Chemical Co. (St. Louis, MO). Membrane grade Triton® X-100 was
purchased from Boehringer Mannheim Corp. (Indianapolis, IN). Heparin was purchased from
Gibco/BRL (Bethesda, MD). A panel of receptor grade detergents, including Nonidet® P-40
(NP40), Triton® X-100, Tween® 20, digitonin, dodecyl-ß-D-maltoside, octylglucoside,
deoxycholic acid, and CHAPS, was purchased from Boehringer Mannheim Corp.
Cell lines
The cell lines MG63, MEF, PEA10, and PEA13 were purchased from the American Type
Culture Collection (ATCC). Cells were maintained as recommended by the ATCC. The murine
bone marrow stromal cell line BMS-2 was graciously provided by Dr. Jeff Gimble (formerly of
the Oklahoma Medical Center, currently at Artecel Sciences, Inc., Durham, NC). BMS-2 cells
were maintained in DMEM, containing pyruvate, 55 µM ß-mercaptoethanol, and 10% fetal
bovine serum.
Binding of CTGF to monolayer cultures
rhCTGF was iodinated with chloramine-T (Sigma Chemical Co.) by the procedures
described1. For binding analysis, cells were plated at 2 5 x 104 cells/cm2 in 24 well dishes, 16 –
24 hours prior to a binding experiment. The cells were washed twice with binding buffer
(Dulbecco’s phosphate buffered saline (PBS) (Life Technologies, Inc.) containing 0.2% bovine
8
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

serum albumin and 0.2% sodium azide) at 4oC. Binding experiments were performed by
incubating monolayers of cells with various concentrations of 125I-rhCTGF in binding buffer
for 4 hours at 4oC with gentle rocking. Duplicate wells were incubated with at least 100-fold
excess of unlabeled rhCTGF for the determination of nonspecific binding.
We determined the kinetics of binding of 125I-rhCTGF to cells as follows. Supernatants
were collected from the cells and directly counted on a Beckman Gamma 5500B counter.
Radioactivity measured from this was called the “free” fraction. The cells were then washed
four times with cold binding buffer, and lysed with 1% Triton® X-100. The lysis fraction was
counted as above, and contained the “total bound” fraction. The nonspecific bound fraction was
also determined and subtracted from each point to yield specific bound. The specific bound and
free fractions were analyzed for one or two site binding using non-linear regression (GraphPad
Prism, version 3.0, GraphPad Software, Inc., San Diego, CA). The same protocol and analysis
was used for competitive binding experiments.
Affinity labeling and crosslinking of monolayer cultures
Cells to be affinity labeled were washed twice with cold binding buffer, then incubated
with 50 - 100 pM 125I-rhCTGF in binding buffer at 4oC for 3 – 4 hours. The binding medium
was removed and replaced with 0.5 mM Bis-[succinimidyl suberate] (BS3, Pierce Chemical Co.,
Rockville, IL). Alternatively, 0.1 mM ethylene glycobis [sulfo-succinimidyl succinate] (Sulfo-
EGS, Pierce Chemical Co.) was also used for crosslinking. Crosslinking proceeded at room
temperature for 15 minutes (BS3) or at 4oC for 30 min (Sulfo-EGS) and was terminated by
removing the medium and washing the cells several times with buffer containing 250 mM
9
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

sucrose, 10 mM Tris pH 7.4, and 10 mM EDTA at 4oC. The complexes were collected in the
soluble fraction following cell lysis with 1% Triton® X-100 (unless indicated otherwise) in PBS
with a cocktail of protease inhibitors (Calbiochem®, San Diego, CA). In some experiments,
0.5% NP40 was used for cell lysis.
Preparation of crude cell membranes
Membranes were prepared from BMS2 cells by a modification of the procedure described
by Atkinson (31). The cells were grown to confluence in roller bottles, and then dissociated with
5 mM EDTA in Dulbecco’s PBS lacking calcium and magnesium. Cell pellets were then
collected by centrifugation, and washed. The cells were then suspended in hypotonic phosphate
buffer (7.5 mM NaPO4, pH 7.2) and incubated 10 min on ice. The membranes were disrupted
by sonication, and the nuclei were stabilized in buffer containing10 mM NaPO4, pH 7.2,
10 mM NaCl, and 3 mM MgCl2. The nuclei and whole cells were removed by centrifugation for
5 min at 800 x g, and the supernatant was collected. The supernatant was centrifuged over a
cushion of 45% sucrose in Dulbecco’s PBS, pH 7.2, for one hour at 24,000 x g. The membrane
fraction located at the sucrose/PBS interface was carefully collected, diluted, and concentrated
by centrifugation at 100,000 x g for 15 min. The membrane pellet was resuspended in
Dulbecco’s PBS, and protein content estimated with the Pierce BCA reagent against an albumin
standard (Pierce Chemical Co.). From a preparation of an estimated 109 cells, 10 – 20 mg of
crude membrane protein was frequently obtained.
Affinity chromatography
One milligram of rhCTGF prepared from baculovirus was immobilized on Reacti-Gel®
10
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

GF-2000 (Pierce Chemical Co.) according to the manufacturer’s specifications. The membranes
(10 – 15 mg) were solubilized in 0.2% Triton® X-100, 20% glycerol, in Dulbecco’s PBS
(Buffer A) containing a cocktail of protease inhibitors, centrifuged (14,000 x g) for 10 min at
4oC to remove insoluble material, and applied to the CTGF affinity matrix. The sample was re-
circulated over the column 5 – 10 times. The flowthrough was collected and the column was
washed with 20 column volumes of Buffer A. The bound sample was eluted with a gradient of
0.135 – 2M NaCl in buffer containing 0.2% Triton X-100, 20% glycerol, and PBS. The CTGF
receptor-containing fractions were identified using a solution binding assay (described below).
Further purification was achieved by electrophoresis of the fractions in 5% SDS-PAGE under
non-reducing conditions.
Solution binding assay
Preparations of membrane proteins (5 µg each) were solubilized in 0.2% Triton® X-100,
20% glycerol in Dulbeccos PBS (Buffer A) and the insoluble material was removed by
centrifugation (14,000 x g) for 10 min at 4oC. Fractions from the affinity column were used
directly in the solution binding assay (10 µl/fraction). The samples were incubated with 0.2 nM
125I-rhCTGF for 3 4 hours. The crosslinker, BS3, was added to a final concentration of
0.5 mM, and the reaction proceeded at room temperature for 15 min. Gel sample buffer was
added to each sample, the samples were then heated for 2 min at 100oC, and applied to 5%
SDS-PAGE. Following electrophoresis, the gels were dried and analyzed by autoradiography.
Mass spectroscopy
The gel band of interest (migrating above the 220 kDa marker) was excised with a fresh
11
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

razor band, destained, and subjected to trypsin digestion (32). The recovered peptide fragments
were analyzed by LC/MS/MS. Microelectrospray columns of 360 µm o.d. x 100 µm i.d. fused
silica capillary were packed with 10 12 cm POROS 10R2, a reversed phase packing material
(PerSeptive Biosystems, Framingham, MA) (33). The flow from the HPLC pump (typically
150 µl/min) was split pre-column to achieve a flow rate of 500 nl/min. The mobile phase for the
gradient elution consisted of (A) 0.5% acetic acid and (B) acetonitrile/water 80:20 (v/v)
containing 0.5% acetic acid. The gradient was linear from 0 – 60% B in 30 min. Mass spectra
were recorded on an LCQ ion trap mass spectrometer (Finnigan MAT, San Jose, CA) equipped
with a microelectrospray ionization source (33). Tandem mass spectra were acquired during the
entire gradient automatically as previously described (34). Protein sequence databases were
searched with the tandem mass spectra using the computer program SEQUEST (35). SEQUEST
correlates tandem mass spectra of peptides with amino acid sequences from protein and
nucleotide databases (15 public databases available). The FBSC Non-Redundant Protein
Database (NRP) database was obtained as an ASCII file in the FASTA format from Frederick
Biomedical Supercomputing Center (ncbi.nlm.nih.gov in /pub/nrdb) by anonymous ftp. Each
sequence produced by SEQUEST was verified by manually inspecting the fit of the amino acid
sequence to the corresponding tandem mass spectrum.
Purification of receptor-associated protein (RAP)
Plasmid DNA containing the human RAP protein sequence was purchased from the
ATCC as the I.M.A.G.E. Consortium CloneID 511113 (36). The RAP cDNA sequence was
excised from the vector with the restriction enzymes BamHI and XhoI, and the 1 kb fragment
was gel purified. The pGEX-4T-1 vector (Pharmacia) was cut with BamHI and XhoI, into
12
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

which the RAP cDNA was ligated. Expression of RAP from this expression construct results in
the formation a fusion protein of RAP and glutathione S-transferase. Correct RAP cDNA
insertion was verified by sequence analysis. The RAP-GST fusion protein and the GST protein
alone (control vector) were expressed in E. coli and purified as described (37).
Antibodies and immunoprecipitation
A mouse monoclonal antibody reactive to the C-terminus of LRP was prepared from
IgG-11H4 hybridoma supernatant (ATCC) and purified by Protein G-Sepharose (Pharmacia).
Monoclonal antibodies reactive with the extracellular domains of the α chain and ß chain of LRP
were purchased from American Diagnostica (catalog #3402 and #3501 respectively).
Affinity labeled cells were extracted with 1% Triton® X-100 or 0.5% NP40 in PBS
containing a cocktail of protease inhibitors. The extracts were incubated for 2.5 hours at 4oC
with 1 µg of antibody to LRP. Protein G-Sepharose was added to the sample and the incubation
continued at 4oC for 1 hour. The sample that bound to the suspension was collected by
centrifugation, washed four times with PBS, then eluted by boiling with Laemmli gel buffer.
The eluted proteins were applied to 5% SDS-PAGE, and analyzed by autoradiography.
Internalization experiments
To determine the kinetics of internalization of CTGF following binding to the cell
surface, MG63 cells grown in 6-well dishes were affinity labeled at 4oC with 0.2 nM 125I-
rhCTGF for 3 hours in binding buffer prepared with Minimal Essential Medium containing
0.2% bovine serum albumin (MEM/BSA). Nonspecific binding, internalization, and degradation
were determined by the addition of 50 nM unlabeled CTGF and subtracted from experimental
13
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

values. In some samples, 3 µg/ml heparin or 250 nM lactoferrin was added to the cells during
binding. After the binding period, the binding medium was removed and the cells were carefully
rinsed twice with fresh, cold MEM/BSA. Pre-warmed MEM/BSA was then added to the cells
and the cells were incubated at 37oC in a CO2 humidified incubator. At 0, 15, 30, 60, 120, and
180 min after transfer to 37oC, the cells were harvested. The medium was collected, treated with
10% trichloroacetic acid (TCA), and the precipitable fraction was separated by centrifugation. In
these experiments, the supernatant resulting from TCA precipitation contained the degradation
products of 125I-rhCTGF that had been internalized, degraded, and returned to the extracellular
space (i.e., to the conditioned media). The internalized fraction was determined following
trypsinization of the cell layer for 10 minutes. The resulting cell pellet was collected by
centrifugation. The radioactivity associated with the trypsin-resistant pellet represented the
internalized fraction of 125I-rhCTGF. To demonstrate LRP-mediated endocytosis of CTGF,
MG63 cells were affinity labeled with 0.2 nM 125I-rhCTGF in MEM/BSA in the presence or
absence of 50 nM RAP-GST. Nonspecific binding, internalization, and degradation of 125I-
rhCTGF were determined by the addition of 50 nM unlabeled CTGF and subtracted from
experimental values. To inhibit lysosomal activity, 200 µM of chloroquine was added to some
of the cultures. After a 2 hour incubation period at 37oC, the degraded and internalized fractions
were determined as described above.
14
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

RESULTS
Solubilized Receptor Assay.
The bone marrow stromal cell (BMS2) line was chosen as a source for purification of the
receptor due to their high level of CTGF-binding to a low affinity receptor1. The ease of growth
and subculture combined with the high level of CTGF binding were factors involved in choosing
these cells for receptor purification. Prior to affinity purification, we optimized the binding and
solubilization conditions that would allow for solubilization of cell membrane proteins as well as
retain the capacity to bind CTGF. Crude membranes were prepared from BMS2 cells by
homogenization and differential centrifugation. A panel of receptor-grade detergents was
examined individually in order to solubilize the membrane proteins. Solubilized membranes
were incubated with 0.2 nM 125I-rhCTGF in the presence or absence of 200-fold excess
unlabeled CTGF. The samples were crosslinked with BS3 and separated by 5% SDS-PAGE.
Competitive crosslinking and binding was achieved using 1% Triton® X-100 containing
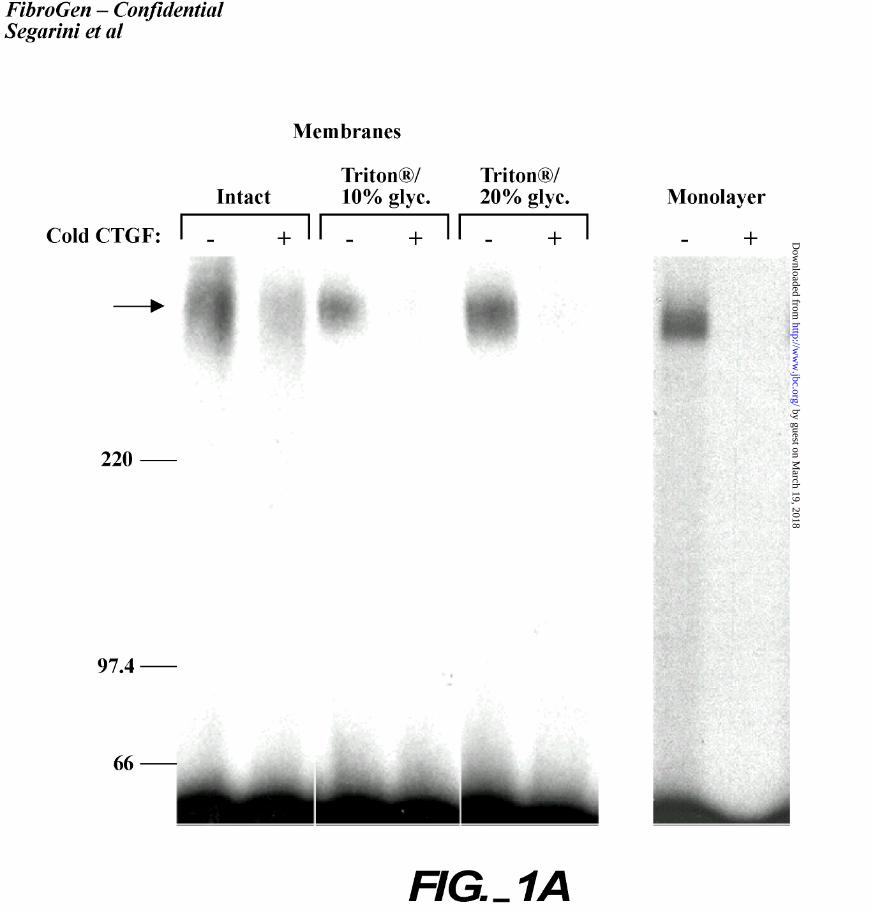
glycerol to help stabilize the membrane proteins (Figure 1A). The left panel demonstrates CTGF
binding and crosslinking to intact membrane fragments; the addition of 10% and 20% glycerol to
the solubilized membranes gave similar binding results, with the inclusion of 20% glycerol
providing more favorable results (middle and right panels, Figure 1A). These conditions were
used for the affinity purification of the receptor.
Affinity Purification.
Membranes prepared from BMS2 cells were solubilized and applied to a column of
CTGF coupled to Sepharose, washed with the same solvent, and bound proteins were eluted with
15
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

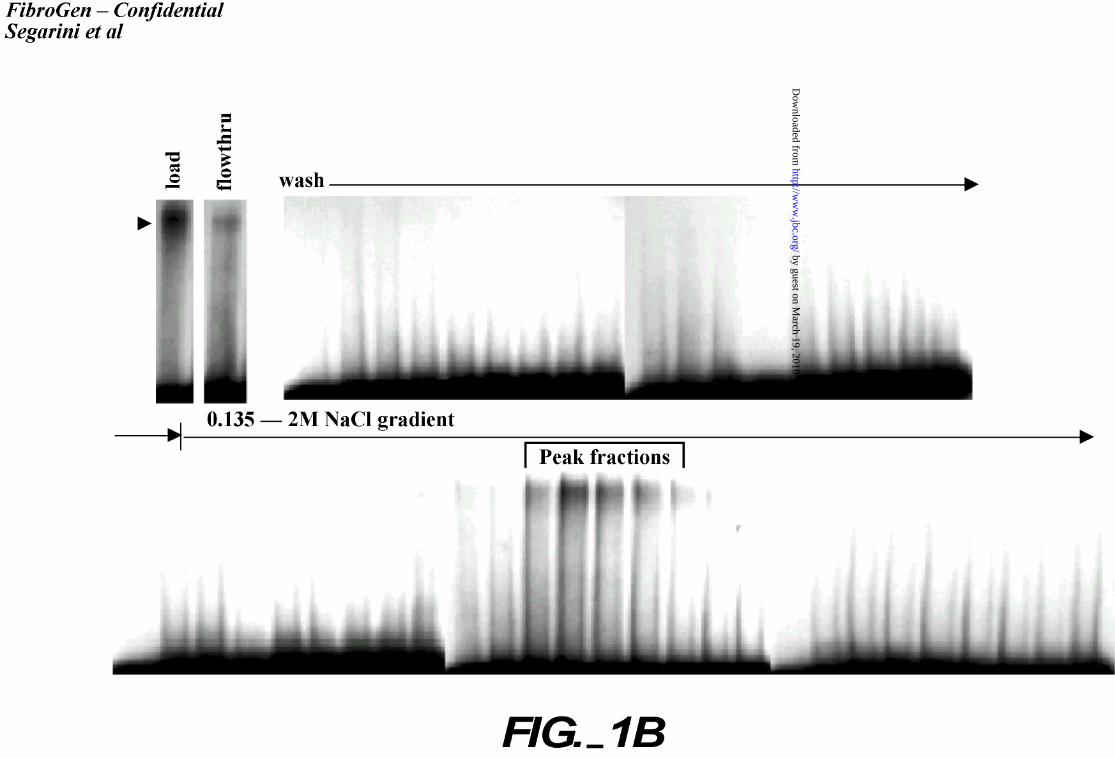
a salt gradient as described above. Fractions were collected and incubated with 125I-rhCTGF to
assay for CTGF binding activity (Figure 1B). The peak CTGF binding fractions eluted between
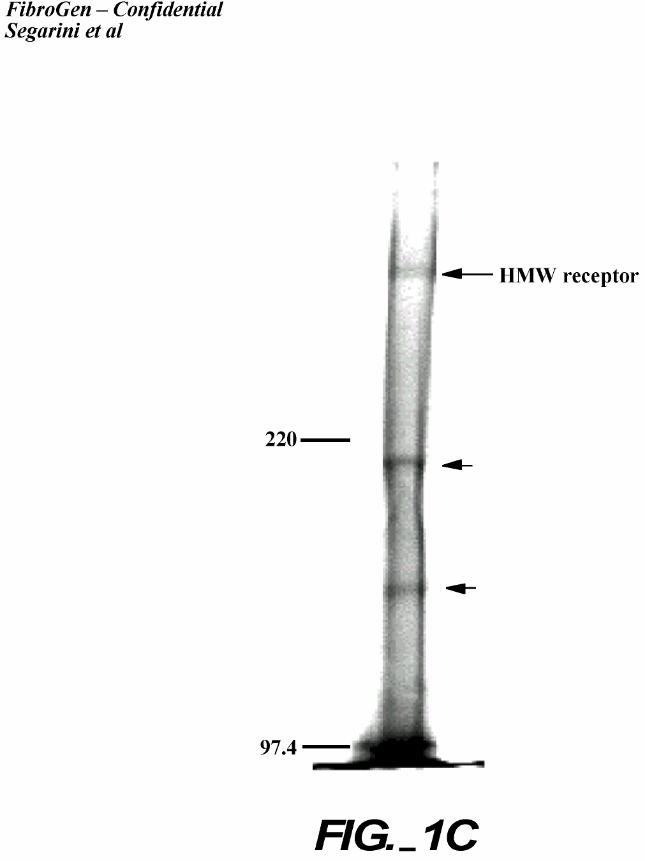
50 – 75% buffer B (1.15M – 1.65M NaCl). Peak fractions were pooled and separated by SDS-
PAGE (Figure 1C). A band migrating above the highest Mr standard (220 kDa) and estimated as
>400 kDa was observed in the Coomassie stained gel. We occasionally observed additional
proteins migrating at Mr = 200 kDa, and Mr = 150 kDa (as shown in Figure 1C). These bands
were excised and analyzed by mass spectroscopy. The >400 kDa band contained the peptides
listed in Table I and database analysis identified the protein as LRP. Repeat purification and
mass spectroscopy analysis confirmed the result. Identification of the Mr 200 kDa and 150 kDa
proteins was not achieved.
LRP Ligands Compete for CTGF binding.
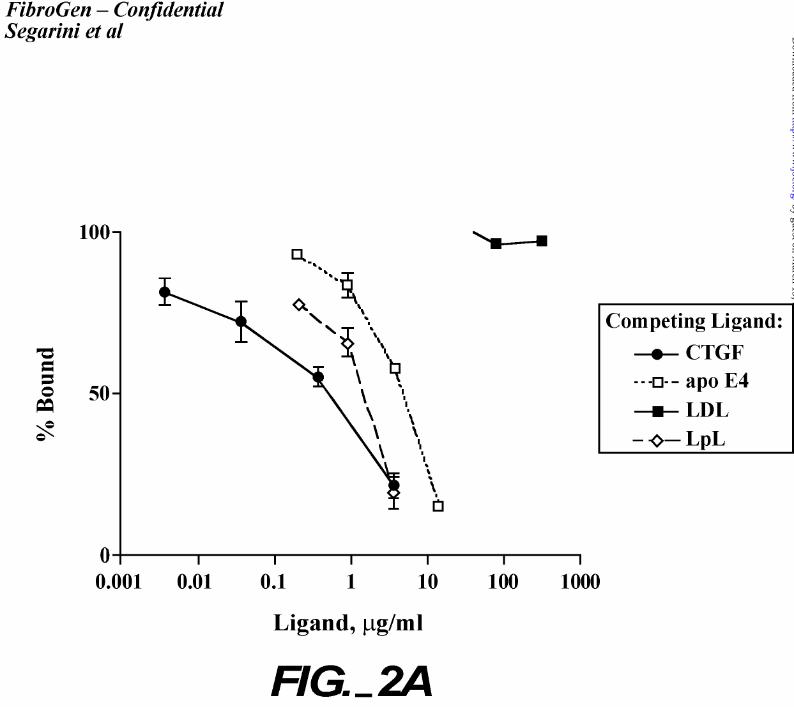
That LRP was a binding protein for CTGF was further confirmed. A number of
commercially available LRP ligands were tested for their ability to compete with CTGF for
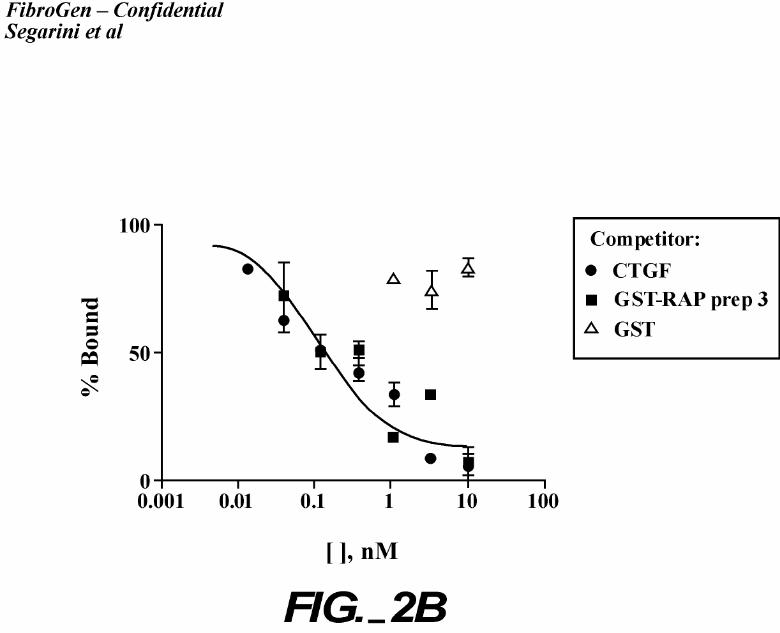
binding to cells (Figure 2). All ligands tested inhibited 125I-rhCTGF binding to cells (Figure
2A) albeit with 5 – 10 fold lower affinity than that of CTGF. Additionally, receptor associated
protein, RAP, which is able to displace all LRP ligands, prevented CTGF binding (Figure 2B).
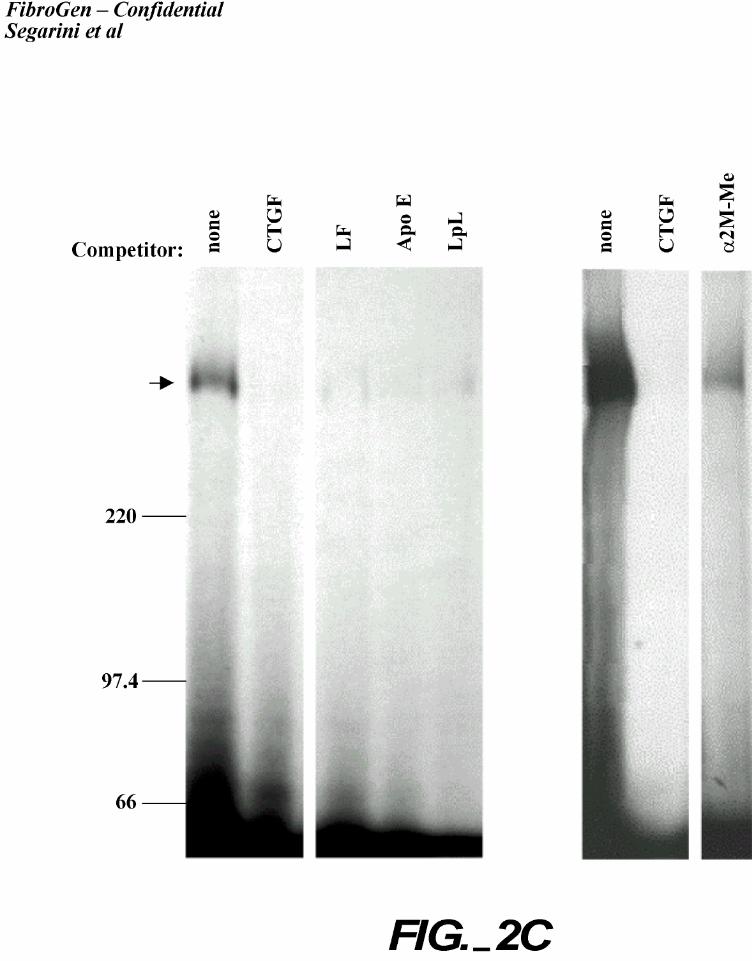
Crosslinking analyses confirmed that the inhibition was due to lack of binding to the high Mr
receptor protein (Figure 2C). These data support the identity of LRP as a CTGF binding protein.
Binding of CTGF to LRP Deficient Cells.
The availability of cells isolated from LRP gene deletion mouse embryos provided an
opportunity to study CTGF binding on cells genetically lacking LRP. The cell lines examined
16
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

include a homozygous LRP-deficient mouse embryo fibroblast cell line, PEA 13 (-/-), a
heterozygous LRP-deficient cell line, PEA 10 (+/-), and a wildtype mouse embryo fibroblast,
MEF1, (+/+).
Each of these cell lines was examined for CTGF binding in a CTGF-binding assay. The
binding parameters obtained (determined by nonlinear regression analysis) are summarized in
Table II. MEF1 (+/+) and PEA10 (+/-) cells bound 125I-rhCTGF with single site binding
kinetics, while the LRP-deficient cells, PEA13, did not bind 125I-rhCTGF in this assay. The
heterozygous cell line, PEA10, appeared to have approximately one-fifth the number of CTGF
binding sites as observed in wild-type MEF1 cells, yet had the same Kd for binding. These data
are suggestive of a gene dosage affect on LRP protein expression levels, but not on the kinetics
of ligand association/dissociation.
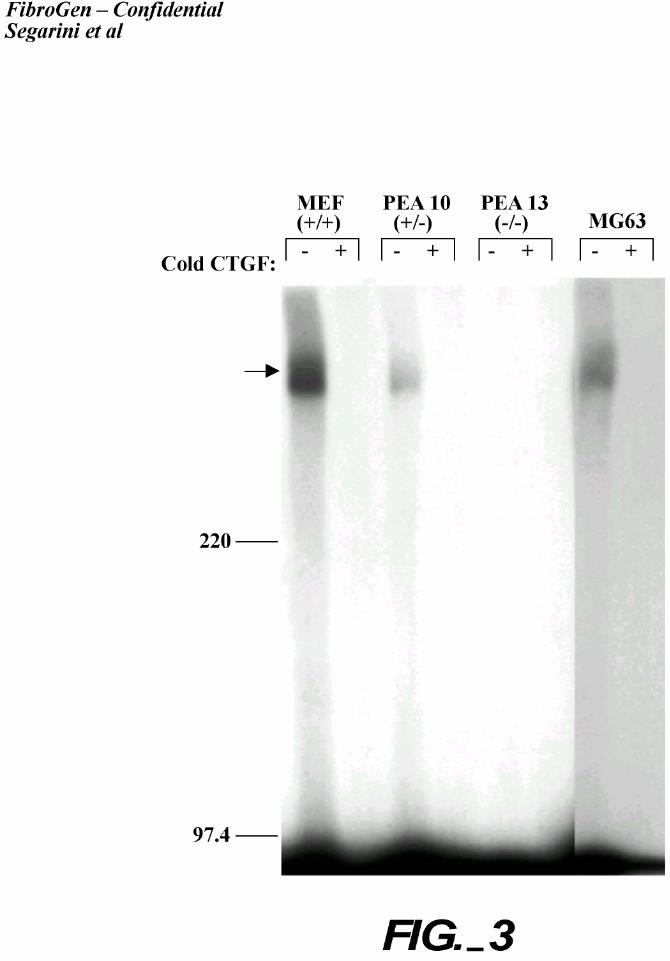
Crosslinking analysis was performed using these LRP-deficient cells to confirm that the
observed binding of CTGF to these cells involved the high Mr protein (Figure 3). Both MEF
(+/+) and PEA10 (+/-) cells, but not PEA 13 (-/-) cells, bound and could be cross-linked to
CTGF to the high Mr protein. The binding of CTGF to the LRP-expressing cells, but not to the
LRP-deficient cells, supports the identification that LRP is a CTGF binding protein.
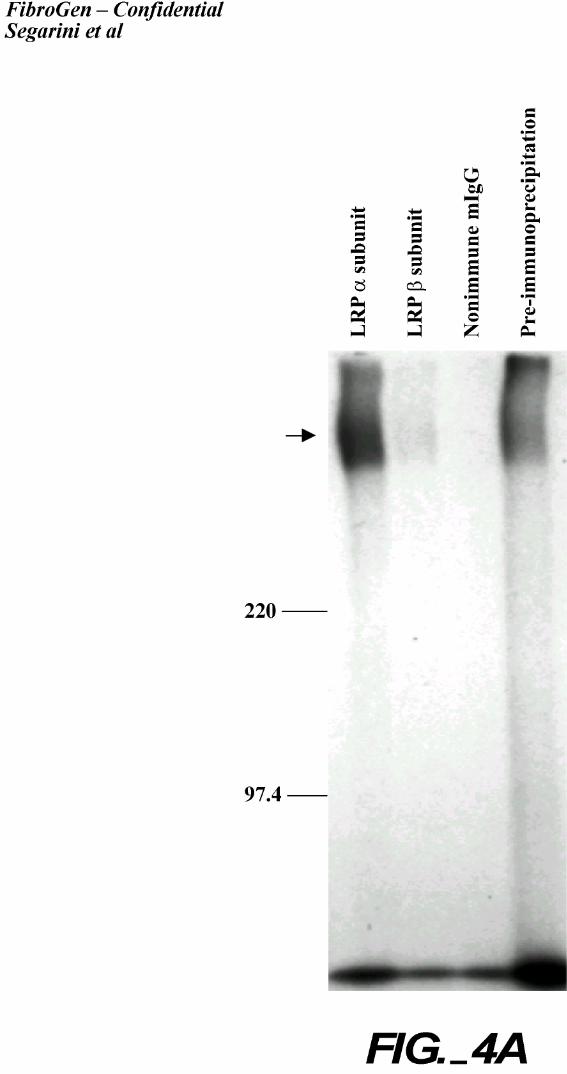
Immunoprecipitation of CTGF/High Mr Complexes.
Further confirmation that LRP is the high Mr CTGF binding protein was demonstrated
using LRP antibodies. Detergent lysates were prepared from 2 cell types, BMS2 and MG63, that
had been affinity labeled with 125I-rhCTGF, cross-linked, and immunoprecipitated with
17
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

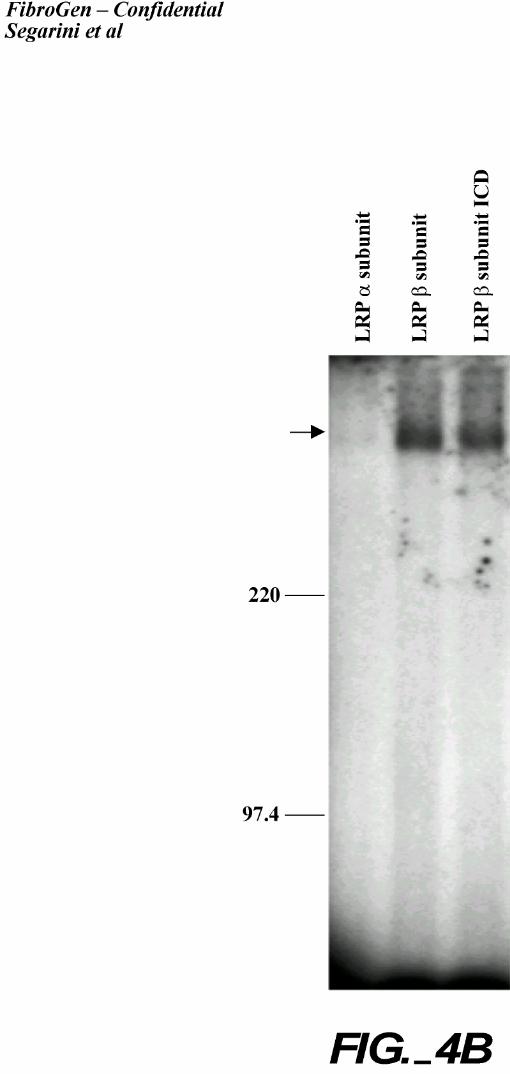
antibodies directed to LRP (Figure 4). Antibodies specific to the extracellular epitopes in both
the α and β subunit chains of LRP, as well as an antibody that recognizes a cytoplasmic domain
of LRP, immunoprecipitated the CTGF-containing complex, while control normal murine IgG
did not. CTGF, migrating at the front of the gel, was poorly immunoprecipitated with these LRP
antibodies. Notably, the antibody recognizing the LRP α chain was most active for MG63
(human origin) cells while the antibody recognizing the LRP β chain was most active for BMS2
(murine origin) cells. The LRP α chain antibody is immunoreactive with the human protein
only. While the β chain antibody is reactive with both human and rodent protein, the difference
in recognition could be due to different detergent extraction conditions.
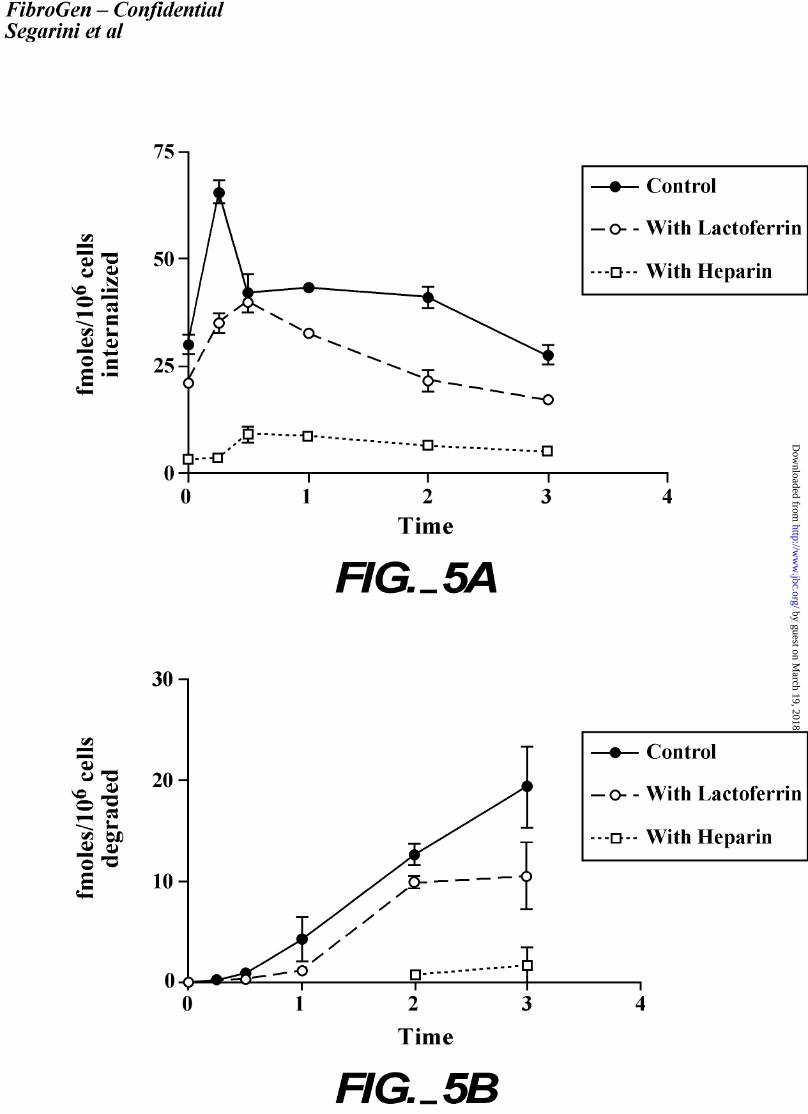
Internalization of CTGF.
The internalization kinetics of CTGF following binding was examined. For these
experiments, the MG63 cell line was utilized because the conditions often promoted lifting of
BMS2 cells from monolayers whereas the MG63 cultures remained intact. Monolayer cultures
of MG63 cells were incubated at 4oC with 0.2 nM 125I-rhCTGF in medium. The cells were
then transferred to 37oC and internalization and degradation of 125I-rhCTGF were followed at
specific time points (Figure 5). In one third of the cultures, an LRP ligand, lactoferrin, was
included to compete for LRP binding. To the other third of the cultures, heparin was added to
inhibit binding of CTGF to the cell surface. The results demonstrated that CTGF is rapidly
internalized by cells, occurring within 30 minutes following the temperature shift to 37°C.
Degradation of CTGF, determined by the release of label into the medium, was detected after 30
minutes, and continued throughout the time course of the experiment. Lactoferrin moderately
reduced internalization and degradation, suggesting LRP-mediated internalization.
18
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

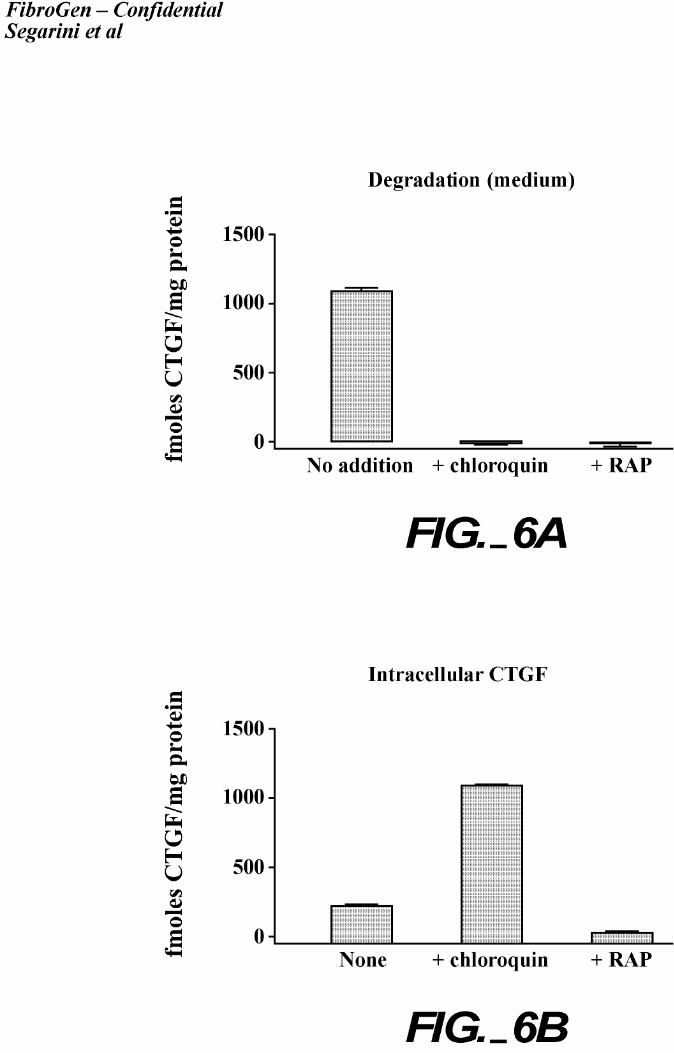
An LRP internalization pathway transports ligands through an endosomal compartment,
wherein the acidic lumen of the endosome promotes ligand dissociation, and the ligand is
subsequently degraded (38). This process is sensitive to the drug, chloroquine, which raises the
pH of the endosome and thus inhibiting the pH-dependant ligand dissociation. We examined
whether CTGF is internalized by LRP and degraded through the same pathway (Figure 6). In
these experiments, cultures of MG63 cells were incubated at 37oC with 125I-rhCTGF in the
presence or absence of the LRP antagonist, RAP, or with chloroquine. MG63 cells treated with
chloroquine showed in an increased level of intracellular 125I-rhCTGF. Degraded 125I-
rhCTGF was barely detectable in these chloroquine-treated cell cultures, whereas the control
cultures had high levels of degraded CTGF. These results support an endosomal-dependent
pathway for the LRP-mediated internalization of CTGF, with ligand dissociation required for its
degradation. RAP addition to the cultures eliminated CTGF internalization and degradation.
These results provide evidence that CTGF uptake and degradation by cells are LRP dependent.
19
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

DISCUSSION
In this study, we have presented evidence that the major cell membrane protein to which
CTGF binds is a high Mr protein, identified previously as LRP. This study provides the first
report of a growth factor that binds directly to LRP, although recently the Wnt family of secreted
molecules has been demonstrated to bind to other members of the LRP family (39). Previous
suggestion of growth factor interaction with LRP has been through complexes with α2m as a
clearance molecule (40).
We were unable to obtain sequence information for two lower Mr proteins that were
occasionally affinity co-purified with LRP (estimated Mr of 150 and 200 kDa). These proteins
were not detected in the crosslinking/binding assay. CTGF, and the structurally related protein,
Cyr61, bind to a number of integrins, including αvß3, αIIbß3 and α6ß1 (41-44). Recently it has
been demonstrated that CTGF-dependent cell adhesion induces cell signaling through integrin-
mediated pathways (42). While the identity of the co-purifying bands is unclear, the Mr
estimates are suggestive for some of the integrin subunits. The possibility exists that these bands
represented breakdown products of the large LRP protein. Further purification and analysis will
be necessary for unambiguous sequence identity of these proteins.
Our data indicate that at least part of the CTGF-binding site on LRP is similar or
common to the binding site utilized for many of the LRP ligands, as these ligands competed for
CTGF binding, although with lower affinity (Figure 2). LRP contains multiple copies of
cysteine-rich repeats known as LDLR class A repeats (complement-type repeats) arranged in 4
clusters, forming ligand binding sites (45-49). The binding site for many LRP ligands has been
20
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

sub-localized (50), and NMR solution structure of the complement-like repeat CR3 from LRP
has recently been determined (51). It will be of interest to sub-localize the binding site for
CTGF within the extracellular domain of LRP.
With the exception of RAP, which binds to LRP with very high affinity (Kd = 3 nM
(52)), most LRP ligands bind LRP with a moderate affinity. For example, hepatic lyase binds
LRP with a Kd of 52 nM (53); PEA binds LRP with a Kd of 14 nM (54); and, coagulation factor
VIII binds LRP with a Kd of approximately 50 nM to LRP (55). Urokinase-type plasminogen
activator (uPA) is synthesized as a single chain zymogen, pro-urokinase, and binds LRP with a
Kd of 45 nM; the active two-chain enzyme (tc-uPA) binds LRP with a Kd of 60 nM. Complex
formation with plasminogen activator inhibitor type I (PAI-1) increases the affinity of uPA to
LRP, such that the uPA/PAI-1 complex binds LRP with a Kd of 1-3 nM (56,57).
Many LRP ligands are polyanionic proteins that bind heparin, suggesting that their
binding to cell surface HSPG may be required for interaction with LRP (58). HSPG may serve
to concentrate the ligands at the cell surface, promoting binding with the relatively low affinity
interaction with LRP. The observed Kd for CTGF binding to LRP is 0.5 -1 nM (the present
study, for MEF1 and PEA10 cells; and in Nesbitt et al.1), which is a much higher binding
affinity than reported for other LRP ligands. In Nesbitt et al.1, we showed that, while CTGF is a
heparin binding protein, CTGF is capable of binding to the surface of cells depleted of GAGs.
Perhaps the higher affinity of CTGF for LRP obviates concentration of CTGF on the cell surface
by HSPGs. Nevertheless, the significance of the high affinity binding of CTGF to LRP is
21
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

unclear at this time, but suggests an important role for LRP in CTGF biology.
Experiments aimed at creating mice lacking LRP by genetic deletion of the LRP allele
failed to produce viable embryos (59), suggesting an important role for LRP during early
development. CTGF is highly expressed in uterine epithelium and in decidualizing endometrial
stromal cells during early pregnancy, as well as in embryonic ectoderm, endoderm, and at the
ectoplacental cone after implantation (60). LRP is localized to invading trophoblastic cells, in
decidualizing tissue, and at the ectoplacental cone after implantation (61,62). The similar
developmental localization patterns support possible interaction between CTGF and LRP during
embryonic development.
We have demonstrated that CTGF is rapidly internalized and degraded by cells, and that
this represents an LRP-mediated process. We have experienced a rapid disappearance of CTGF
from culture medium after addition of CTGF to cell cultures. A rapid internalization and
degradation pathway would account for the inability to detect CTGF in conditioned medium.
We suggest that one function of LRP in CTGF biology is to modulate the concentration of free
CTGF in the extracellular space or at the cell surface.
Whether or not LRP serves as a signaling receptor for CTGF bioactivity remains to be
determined. LRP binds and internalizes numerous ligands that would necessitate separate
ligand-specific signaling mechanisms; yet, signaling involving LRP has been suggested in at
least some systems. Most recently, signaling by the Wnt family of proteins requires the LRP-5
or LRP-6 members of the LRP family to function as co-receptors with the Frizzled family of
receptors (39,63,64). In a different system, LRP mediates long-term potentiation in
hippocampal neurons (65) and activation with activated α2M promotes calcium influx in
22
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

neuronal cells (66). Other members of the LDLR family have recently been demonstrated to
transduce extracellular signals. For instance, VLDLR and apoER2 function as receptors in the
Reelin/disabled-mediated neuronal migration pathway (67-70). The short cytoplasmic domain
of LRP contains multiple potential endocytosis motifs including 2 NPXY motifs; but, a recent
study demonstrated that a YXXL motif serves as the dominant signal for LRP endocytosis (71),
leaving the NPXY motifs available for interaction with other signaling or adaptor proteins.
Disabled-1 (dab-1) interacts with the NPXY motifs of LRP in neuronal cells (69,70,72,73).
When tyrosine phosphorylated, mDab1 binds nonreceptor tyrosine kinases, such as src, fyn and
abl (74). Another member of this family, mDab2/p96/DOC-2, has approximately 50% sequence
conservation with the amino terminal sequence of mDab1, but is expressed in a wider variety of
cells (75). Dab2/p96/DOC-2 has been shown to compete with SOS to bind Grb2, suggesting a
negative regulatory role in the Ras signaling cascade (76) and may negatively regulate
mitogenesis (77). Whether Dab2/p96/DOC-2 or an unidentified member of the Dab family
binds to LRP remains unknown and is of much interest to investigate.
We have been unable to detect tyrosine phosphorylation of LRP in cells treated with
CTGF (data not shown). In addition, we have examined LRP purified by CTGF affinity
chromatography in kinase assays. Although an associated kinase activity towards casein co-
purified with LRP, this kinase activity was found not from LRP itself, but rather from an
associating protein. Most importantly, the kinase activity did not respond to CTGF stimulation3.
Therefore it remains unclear whether LRP phosphorylation plays any role in mediating
biological functions of CTGF.
LRP and many of its ligands have been localized to the senile plaques of Alzheimer’s
23
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

disease (78-80), in plaque-associated activated astrocytes, but not in resting astrocytes (80).
Recent biochemical data support a role for LRP in the pathogenesis of Alzheimer’s disease
(81,82). There is an increase in LRP expression in monocytes from patients with coronary heart
disease (83), and LRP is abundantly expressed by smooth muscles cells and macrophages in
human atherosclerotic lesions (84). In the renal ablation model of experimental kidney fibrosis,
LRP expression is significantly increased in the glomeruli and interstitium with preferential
localization to fibrotic lesions (85). While to date CTGF has not been documented in
Alzheimer’s lesions, it is highly expressed in fibrotic kidney disease (22) and atherosclerotic
plaques (24). With co-localization of CTGF and LRP in these diseases, it will be important to
understand the functional role of the high affinity interaction of these proteins.
It will be of interest to examine whether altered LRP expression is a general marker of
disease characterized by impaired wound healing or in fibroproliferative disorders. Future work
examining the role of LRP in CTGF signaling by itself or as a co-receptor to a yet unidentified
CTGF receptor, and the effect of the modulation of LRP on CTGF activity will be important
studies to probe the biology of CTGF and for the design of therapeutics for intervention in
fibrotic disease.
24
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

ACKNOWLEDGMENTS
We acknowledge and thank Miss Cathyrn Baker for preparation of cell membranes and Dr. Leah
Allen for subcloning the RAP gene into the GST fusion vector. We thank Dr. George R. Martin
for valuable advice and critical review of this work.
25
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

REFERENCES
1. Bradham, D. M., Igarashi, A., Potter, R. L., and Grotendorst, G. R. (1991) J. Cell Biol.
114, 1285 - 1294
2. Grotendorst, G. R., Okochi, H., and Hayashi, N. (1996) Cell Growth And Differentiation
7, 469-480
3. Kothapalli, D., Frazier, K. S., Welply, A., Segarini, P. R., and Grotendorst, G. R. (1997)
Cell Growth Diff. 8, 61 - 68
4. Wunderlich, K., Senn, B. C., Todesco, L., Flammer, J., and Meyer, P. (2000) Graefes
Arch Clin Exp Ophthalmol 238, 910-915.
5. Suzuma, K., Naruse, K., Suzuma, I., Takahara, N., Ueki, K., Aiello, L. P., and King, G.
L. (2000) J. Biol. Chem. 275, 40725-40731
6. Camerer, E., Gjernes, E., Wiiger, M., Pringle, S., and Prydz, H. (2000) J. Biol. Chem.
275, 6580 - 6585
7. Pendurthi, U. r., Allen, K. E., Ezban, M., and Rao, L. V. M. (2000) J. Biol. Chem. 275,
14632 - 14641
8. Chambers, R. C., Leoni, P., Blanc-Brude, O. P., Wembridge, D. E., and Laurent, G. J.
(2000) J. Biol. Chem. 275, 35584-35591
9. Hahn, A., Heusinger-Ribeiro, J., Lanz, T., Zenkel, S., and Goppelt-Struebe, M. (2000) J.
Biol. Chem. 275, 37429-37435
10. Abraham, D. J., Shiwen, X., Black, C. M., Sa, S., Xu, Y., and Leask, A. (2000) J Biol
Chem 275, 15220-15225.
11. Stanhope-Baker, P., and Williams, B. R. G. (2000) J. Biol. Chem. 275, 38139-38150
26
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12. Shimo, T., Nakanishi, T., Kimura, Y., Nishida, T., Ishizeki, K., Matsumura, T., and
Takigawa, M. (1998) Journal Of Biochemistry 124, 130-140
13. Surveyor, G. A., Wilson, A. K., and Brigstock, D. R. (1998) Biology Of Reproduction
59, 1207-1213
14. Brigstock, D. R., Steffen, C. L., Kim, G. Y., Vegunta, R. K., Diehl, J. R., and Harding, P.
A. (1997) J. Biol. Chem. 272, 20275-20282
15. Igarashi, A., Okochi, H., Bradham, D. M., and Grotendorst, G. R. (1993) Molecular
Biology Of The Cell 4, 637-645
16. Frazier, K., Williams, S., Kothapalli, D., Klapper, H., and Grotendorst, G. R. (1996) J.
Invest. Dermatol. 107, 404 - 411
17. Riser, B. L., Denichilo, M., Cortes, P., Baker, C., Grondin, J. M., Yee, J., and Narins, R.
G. (2000) J Am Soc Nephrol 11, 25-38.
18. Fan, W. H., Pech, M., and Karnovsky, M. J. (2000) Eur J Cell Biol 79, 915-923.
19. Mori, T., Kawara, S., Shinozaki, M., Hayashi, N., Kakinuma, T., Igarashi, A., Takigawa,
M., Nakanishi, T., and Takehara, K. (1999) J. Cell Physiol. 181, 153-159
20. Wahl, S. M. (1994) J. Exp. Med. 180, 1587 - 1590
21. Border, W. A., and Ruoslahti, E. (1992) J. Clin. Invest. 90, 1-7
22. Ito, Y., Aten, J., Bende, R. J., Oemar, B. S., Rabelink, T. J., Weening, J. J., and
Goldschmeding, R. (1998) Kidney Int. 53, 853-861
23. Lasky, J. A., Ortiz, L. A., Tonthat, B., Hoyle, G. W., Corti, M., Athas, G., Lungarella, G.,
Brody, A., and Friedman, M. (1998) Am. J. Physiol. 275, L365 - L371
24. Oemar, B. S., Werner, A., Garnier, J. M., Do, D. D., Godoy, N., Nauck, M., Marz, W.,
27
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Rupp, J., Pech, M., and Luscher, T. F. (1997) Circulation 95, 831-839
25. Kikuchi, K., Kadono, T., Ihn, H., Sato, S., Igarashi, A., Nakagawa, H., Tamaki, K., and
Takehara, K. (1995) J. Invest. Derm. 105, 128 - 132
26. Igarashi, A., Nashiro, K., Kikuchi, K., Sato, S., Ihn, H., Grotendorst, G. R., and Takehara,
K. (1995) J. Invest. Dermatol. 105, 280 - 284
27. Schwab, J. M., Postler, E., Nguyen, T. D., Mittelbronn, M., Meyermann, R., and
Schluesener, H. J. (2000) Neuropathol Appl Neurobiol 26, 434-440.
28. Wunderlich, K., Pech, M., Eberle, A. N., Mihatsch, M., Flammer, J., and Meyer, P.
(2000) Curr Eye Res 21, 627-636.
29. Brigstock, D. R. (1999) Endocrine Reviews. 20, 189 - 206
30. Gliemann, J. (1998) Biol. Chem. 379, 951 - 964
31. Atkinson, P. H. (1973) in Methods in Cell Biology (Prescott, D. M., ed) Vol. VII, pp. 157
- 188, Academic Press, New York
32. Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996) Anal. Chem. 68, 850 - 858
33. Gatlin, T., Kleeman, G., Hays, L. G., Link, A. J., and Yates, J. R., III. (1998) Anal.
Biochem. 262, 93 - 101
34. Link, A. J., Hays, L. G., Carmack, E. B., and Yates, J. R., III. (1997) Electrophoresis 18,
1314 - 1334
35. Eng, J., McCormack, A. L., and Yates, J. R., III. (1994) J. Am. Soc. Mass Spectrom. 5,
976 - 989
36. Lennon, G., Auffray, C., Polymeropoulos, M., and Soares, M. B. (1996) Genomics 33,
151-152
28
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

37. Warshawsky, I., Bu, G., and Schwartz, A. L. (1993) J. Biol. Chem. 268, 22046 - 22054
38. Herz, J., Kowal, R. C., Ho, Y. K., Brown, M. S., and Goldstein, J. L. (1990) J. Biol.
Chem. 265, 21355 - 21362
39. Tamai, K., Semenov, M., Kato, Y., Spokony, R., Liu, C., Katsuyama, Y., Hess, F., Saint-
Jeannet, J. P., and He, X. (2000) Nature 407, 530-535.
40. LaMarre, J., Wollenberg, G. K., Gonias, S. L., and Hayes, M. A. (1991) Laboratory
Investigation 65, 3-14
41. Chen, N., Chen, C. C., and Lau, L. F. (2000) J Biol Chem 275, 24953-24961
42. Chen, C.-C., Chen, N., and Lau, L. F. (2001) J. Biol. Chem. 276, 10443-10452
43. Jedsadayanmata, A., Chen, C. C., Kireeva, M. L., Lau, L. F., and Lam, S. C. (1999) J
Biol Chem 274, 24321-24327.
44. Kireeva, M. L., Lam, S. C.-T., and Lau, L. F. (1998) J. Biol. Chem. 273, 3090 - 3096
45. Moestrup, S. K., Holtet, T. L., Etzerodt, M., Thogersen, H. C., Nykjaer, A., Andreasen, P.
A., Rasmussen, H. H., Sottrup-Jensen, L., and Gleimann, J. (1993) J. Biol. Chem. 268,
13691 - 13696
46. Bu, G., and Rennke, S. (1996) J. Biol. Chem. 271, 22218 - 22224
47. Willnow, T. E., Orth, K., and Herz, J. (1994) J. Biol. Chem. 269, 15827 - 15832
48. Horn, I. R., van den Berg, B. M., van der Meijden, P. Z., Pannekoek, H., and van
Zonneveld, A. J. (1997) J. Biol. Chem. 272, 13608 - 13613
49. Vash, B., Phung, N., Zein, S., and Decamp, D. (1998) Blood 92, 3277 - 3285
50. Neels, J. G., van den Berg, M. M., A., L., Olivecrona, G., Pannekoek, H., and van
Zonneveld, A.-J. (1999) J. Biol. Chem. 274, 31305 - 31311
29
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

51. Dolmer, K., Huang, W., and Gettins, P. G. (2000) J. Biol. Chem. 275, 3264-3269
52. Iadonato, S. P., Bu, G., Maksymovitch, E. A., and Schwartz, A. L. (1993) Biochem. J.
296, 867 - 875
53. Kounnas, M. Z., Chappell, D. A., Wong, H., Argraves, W. S., and Strickland, D. K.
(1995) J. Biol. Chem. 270, 9307 - 9312
54. Kounnas, M. Z., Morris, R. E., Thompson, M. R., Fitzgerald, D. J., Strickland, D. K., and
Saelinger, C. B. (1992) J. Biol. Chem. 267, 12420 - 12423
55. Lenting, P. J., Neels, J. G., van den Berg, B. M. M., Clijsters, P. P. F. M., Meijerman, D.
W. E., Pannekoek, H., van Mourik, J. A., Mertens, K., and van Zonneveld, A.-J. (1999)
J. Biol. Chem. 274, 23734 - 23739
56. Kounnas, M. Z., Henkin, J., Argraves, W. S., and Strickland, D. K. (1993) J. Biol. Chem.
268, 21862 - 21867
57. Kasza, A., Petersen, H. H., Heegaard, C. W., Oka, K., Christensen, A., Dubin, A., Chan,
L., and Andreasen, P. A. (1997) Eur. J. Biochem. 248, 270 - 281
58. Crisp, R. J., Knauer, D. J., and Knauer, M. F. (2000) J Biol Chem 275, 19628-19637
59. Herz, J., Clouthier, D. E., and Hammer, R. E. (1992) Cell 71, 411 - 421
60. Surveyor, G. A., Wilson, A. K., and Brigstock, D. R. (1998) Biol. Reprod. 59, 1207 -
1213
61. Teesalu, T., Blasi, F., and Talarico, D. (1996) Mechanisms Of Development 56, 103-116
62. Coukos, G., Gåfvels, M. E., Wisel, S., Ruelaz, E. A., Strickland, D. K., Strauss, J. F. r.,
and Coutifaris, C. (1994) American Journal Of Pathology 144, 383-392
63. Wehrli, M., Dougan, S. T., Caldwell, K., O’Keefe, L., Schwartz, S., Vaizel-Ohayon, D.,
30
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Schejter, E., Tomlinson, A., and DiNardo, S. (2000) Nature 407, 527-530.
64. Pinson, K. I., Brennan, J., Monkley, S., Avery, B. J., and Skarnes, W. C. (2000) Nature
407, 535-538.
65. Zhuo, M., Holtzman, D. M., Li, Y., Osaka, H., DeMaro, J., Jacquin, M., and Bu, G.
(2000) J. Neurosci. 20, 542 - 549
66. Bacskai, B. J., Xia, M. Q., Strickland, D. K., Rebeck, G. W., and Hyman, B. T. (2000)
Proc Natl Acad Sci U S A 97, 11551-11556.
67. Hiesberger, T., Trommsdorff, M., Howell, B. W., Goffinet, A., Mumby, M. C., Cooper, J.
A., and Herz, J. (1999) Neuron 24, 481-489.
68. D’Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D. S., Sheldon, M., and Curran, T.
(1999) Neuron 24, 471-479.
69. Trommsdorff, M., Borg, J. P., Margolis, B., and Herz, J. (1998) J. Biol.Chem. 273,
33556-33560
70. Trommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger, W., Nimpf, J.,
Hammer, R. E., Richardson, J. A., and Herz, J. (1999) Cell 97, 689-701.
71. Li, Y., Marzolo, M. P., van Kerkhof, P., Strous, G. J., and Bu, G. (2000) J. Biol. Chem.
275, 17187 - 17194
72. Le, N., and Simon, M. A. (1998) Molecular And Cellular Biology 18, 4844-4854
73. Howell, B. W., Lanier, L. M., Frank, R., Gertler, F. B., and Cooper, J. A. (1999) Mol Cell
Biol 19, 5179-5188.
74. Howell, B. W., Gertler, F. B., and Cooper, J. A. (1997) EMBO J 16, 121-132
75. Xu, X. X., Yang, W., Jackowski, S., and Rock, C. O. (1995) Journal Of Biological
31
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Chemistry 270, 14184-14191
76. Xu, X. X., Yi, T., Tang, B., and Lambeth, J. D. (1998) Oncogene 16, 1561-1569
77. Fulop, V., Colitti, C. V., Genest, D., Berkowitz, R. S., Yiu, G. K., Ng, S. W., Szepesi, J.,
and Mok, S. C. (1998) Oncogene 17, 419-424
78. Rebeck, G. W., Harr, S. D., Strickland, D. K., and Hyman, B. T. (1995) Annals Of
Neurology 37, 211-217
79. Rebeck, G. W., Reiter, J. S., Strickland, D. K., and Hyman, B. T. (1993) Neuron 11, 575
- 580
80. Thal, D. R., Schober, R., and Birkenmeier, G. (1997) Brain Res 777, 223-227
81. Ulery, P. G., Beers, J., Mikhailenko, I., Tanzi, R. E., Rebeck, G. W., Hyman, B. T., and
Strickland, D. K. (2000) J. Biol. Chem. 275, 7410-7415
82. LaDu, M. J., Shah, J. A., Reardon, C. A., Getz, G. S., Bu, G., Hu, J., Guo, L., and van
Eldik, L. J. (2000) J Biol Chem 275, 33974-33980.
83. Handschug, K., Schulz, S., Schnurer, C., Köhler, S., Wenzel, K., Teichmann, W., and
Gläser, C. (1998) Journal Of Molecular Medicine 76, 596-600
84. Luoma, J., Hiltunen, T., Särkioja, T., Moestrup, S. K., Glieman, J., Kodama, T., Nikkari,
T., and Ylä-Herttuala, S. (1994) J. Clin. Invest. 93, 2014 - 2021
85. van Goor, H., Diamond, J. R., Ding, G., and Kaysen, G. (1999) Exp. Nephrol. 7, 35 - 43
32
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

FOOTNOTES
1. Abbreviations: LRP, Low density lipoprotein- receptor related protein/α2-
macroglobulin receptor; CTGF, Connective Tissue Growth Factor; rhCTGF, recombinant
human CTGF; TGF-ß, Transforming Growth Factor Beta; PEA Pseudomonas Exotoxin
A; IgG, Immunoglobulin gamma; RAP,Receptor associated protein; RAP-GST,RAP
fusion protein wit2. h glutathione S-transferase; α2m, alpha 2-
macroglobulin; α2m-Me, methylamine activated α2M; α2mR, α2m receptor; LF,
lactoferrin; ApoE; apolipoprotein E; LDL, low density lipoprotein; VLDLR, very low
density lipoprotein receptor; TCA, trichloroacetic acid; MEM/BSA, minimal essential
medium with 0.2% bovine serum albumin
3. Nesbitt, J. E., Segarini, P. R., Carmichael, D. F. Manuscript submitted.
4. Li, D. Unpublished observations.
33
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE LEGENDS
Table I. The gel band of interest (migrating above the highest Mr standard of 220 kDa) was
excised with a clean razor blade, destained and subjected to trypsinization. The recovered
peptide fragments were analyzed by LC/MS/MS and mass measurements were analyzed using
the SEQUEST analysis program.
Table II. Binding parameters of CTGF with LRP wildtype and deficient cells. Equilibrium
binding was performed on monolayer cultures of cells and analyzed as described in Materials
and Methods.
Figure 1. Purification of a CTGF receptor.
A. The solubilized receptor binds CTGF. Crude membranes were prepared from BMS2 cells
and solubilized in 1% Triton in PBS with 10% glycerol, or 20% glycerol, and a cocktail of
protease inhibitors. Intact membranes were used as a binding control (far left panel). The
membranes were affinity labeled with 0.2 nM 125I-rhCTGF and crosslinked. Half the
samples also contained a 200 fold excess of unlabeled CTGF. The crosslinked samples were
separated on 5% SDS-PAGE under non-reducing conditions. The gels were dried and
visualized by autoradiography. Molecular size markers are indicated at the left in kDa, and an
arrow indicates the CTGF-receptor complex. The far right panel demonstrates 125I-
rhCTGF crosslinked to BMS2 cells in monolayer culture.
B. Binding assay of solubilized membrane proteins separated by CTGF affinity
chromatography. Crude membrane preparations were solubilized in 1% Triton® X-100 in
34
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

PBS with 20% glycerol and protease inhibitors. The sample was applied to a CTGF affinity
matrix and washed through with Buffer A. The bound material was eluted with a NaCl
gradient. An aliquot of the loaded sample, the flowthrough, and each fraction collected
during wash and elution were analyzed by affinity labeling, crosslinking, and separation on
non-reducing 5% SDS-PAGE. Peak fractions are indicated with a bracket. An arrowhead
indicates the CTGF-receptor complex.
C. Coomassie staining of affinity purified receptor. The peak fractions were pooled and
separated on non-reducing 5% SDS-PAGE. The upper band (long arrow) migrates at the
expected position for the previously identified complex. Short arrows indicate 2 other
proteins co-purifying on the column. The three indicated bands were excised for further
analysis.
Figure 2. Competition of LRP/α2mR ligands with CTGF.
A. Monolayers of BMS2 cells were incubated with 100 pM 125I-rhCTGF in the presence of
indicated concentrations of unlabeled CTGF, apo E4, LpL or LDL. The bound material was
collected and determined by counting. Bound is represented as a fraction of total (non-
competing) CTGF bound. Each point is the average of duplicate samples and error bars
indicate the standard deviation.
B. Monolayers of BMS2 cells were incubated with 50 pM 125I-rhCTGF in the presence of
indicated concentrations of unlabeled CTGF, RAP or RAP-GST. The bound material was
collected and determined by counting. Bound is represented as a fraction of total (non-
competing) CTGF bound. Each point is the average of duplicate samples and error bars
indicate the standard deviation.
35
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

C. Monolayers of BMS2 cells were incubated with 50 pM 125I-rhCTGF in the presence of
unlabeled competitor: 21.5 nM CTGF, 250 nM LF, 574 nM apo E4, 400 nM LpL or 100 nM
α2M-Me. The bound material was crosslinked to the cells. Cell lysates were separated by
SDS-PAGE, dried and visualized by autoradiography. An arrow indicates the position of the
CTGF-receptor complex.
Figure 3. Affinity labeling and crosslinking of 125I-rhCTGF to LRP/α2mR -deficient cells.
MEF1 (+/+), PEA 10 (+/-) and PEA 13 (-/-) cells were grown in monolayer culture; affinity
labeling with 125I-rhCTGF and crosslinking were performed. The Triton® X-100 soluble
fraction was run on SDS-PAGE; the gel was dried and visualized by autoradiography. An arrow
indicates the position of the CTGF-receptor complex.
Figure 4. Immunoprecipitation of CTGF complexes with anti- LRP/α2mR antibodies.
A. Immunoprecipitation from MG63 cells. MG63 cells were affinity labeled and crosslinked
with 125I-rhCTGF. The complexes were extracted with Triton® X-100 and
immunoprecipitated with anti LRP-α, antiLRP-β or nonimmune murine IgG. A portion of
the extract was saved and applied to the gel without immunoprecipitation. An arrow
indicates the CTGF-receptor complex.
B. Immunoprecipitation from BMS2 cells. BMS2 cells were affinity labeled and crosslinked
with 125I-rhCTGF. The complexes were extracted with NP40 and immunoprecipitated with
anti LRP-α, antiLRP-β or mAb11H4. An arrow indicates the CTGF-receptor complex.
Figure 5. Kinetics of the internalization and degradation of CTGF. MG63 cells were affinity-
labeled with 125I-rhCTGF alone (solid circles), with 3 µg/ml heparin (open squares), or with
36
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

250 nM lactoferrin (open circles), at 4oC. After 3 hours, the cells were shifted to 37oC and at
the indicated time points, the internalized (A) or degraded (B) fractions were determined.
Internalized CTGF was measure as the trypsin-resistant cell pellet. Degraded CTGF was
measure in the supernatant of TCA-treated cell medium. Each point is the average of
triplicate samples and error bars indicate the standard deviation.
Figure 6. Internalization and degradation of CTGF is LRP/α2mR dependent. MG63 cells were
incubated with 125I-rhCTGF alone, with RAP-GST, or with chloroquine for 2 hours at
37oC. After 2 hours, the internalized (A) and degraded (B) fractions were measured as described
in the legend to Figure 5 and as detailed in the methods section. Each point is the average of
triplicate samples and error bars indicate the standard deviation.
37
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

TABLE I
First round of mass spectroscopy yielded following peptides:(R)AALSGANVLTLIEKDIR(K)NAVVQGLEQPHGLVVHPLR(R)SERPPIFEIR(K)TVLWPNGLSLDIPAGR(R)TTLLAGDIEHPR(R)YVVISQGLDKPR
Second round of mass spectroscopy yielded following peptides:(R)DGILFWTDWDASLPR(R)GWDTLYWTSYTTSTITR(R)IFFSDIHFGNIQQINDDGSGR(R)ILWIDAR(K)ITWPNGLTVDYVTER(K)NAVVQGLEQPHGLVVHPLR(R)SERPPIFEIR(R)TTLLAGDIEHPR(K)TVLWPNGLSLDIPAGR
38
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

TABLE II.
Cell line Phenotype Kd (pM) Bmax (fmol/106cells) #receptors/cell
MEF1 +/+ 489 40 24,008PEA10 +/- 456 9 5227PEA13 -/- -- <0 --
39
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from

David F. CarmichaelPatricia R. Segarini, James E. Nesbitt, Dongxia Li, Lara G. Hayes, John R. Yates III and
is a receptor for connective tissue growth factor (CTGF)The low density lipoprotein receptor-related protein/alpha 2-Macroglobulin receptor
published online August 22, 2001J. Biol. Chem.
10.1074/jbc.M105180200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 19, 2018
http://ww
w.jbc.org/
Dow
nloaded from