Embed Size (px)
Citation preview

© 2000 Oxford University Press Human Molecular Genetics, 2000, Vol. 9, No. 10 1525–1532
The most frequent constitutional translocation inhumans, the t(11;22)(q23;q11) is due to a highlyspecific Alu-mediated recombinationAlexander S. Hill, Nicola J. Foot, Tracy L. Chaplin and Bryan D. Young+
Imperial Cancer Research Fund, Department of Medical Oncology, St Bartholomew’s and the Royal London Schoolof Medicine and Dentistry, Charterhouse Square, London ECIM 6BQ, UK
Received 22 February 2000; Revised and Accepted 4 April 2000
The t(11;22) is the most common recurrent non-Robertsonian constitutional translocation in humans,having been reported in more than 160 unrelated fami-lies. Balanced carriers are at risk of having offspringwith the derivative 22 syndrome owing to 3:1 meioticnon-disjunction event. Clinical features of the der(22)syndrome include mental retardation, craniofacialabnormalities and congenital heart defects. Thebreakpoints for the t(11;22) translocation have beenmapped to specific Alu repeats on chromosomes 11and 22, indicating that this event is due to an Alu–Alurecombination. Remarkably, in five samples derivedfrom individuals with no apparent common ancestrythe der(11) and der(22) breakpoints appear to bealmost identical at the genomic sequence level. Thesmall number of base differences between thesamples indicates some variation in the position ofthe breakpoints, although this appears to be quitelimited. Indeed, the der(11) breakpoints are all locatedwithin a region of just 32 bp and the der(22) break-points within 21 bp. If, as suggested by current data,the widespread occurrence of this translocation is dueto multiple independent events, our results suggestthat this particular Alu–Alu recombination is subjectto an unprecedented degree of selection.
INTRODUCTION
The t(11;22) is the most common constitutional translocationin humans. In contrast to most constitutional translocations,which appear as single sporadic events, the t(11;22) transloca-tion has been reported in more than 160 unrelated families (1–3).Individuals who carry the balanced form of the translocationhave a risk that their offspring may have the derivative 22[der(22)] syndrome owing to a 3:1 meiotic non-disjunctionevent. Affected offspring usually carry a supernumerary deriv-ative 22 chromosome and are thus effectively trisomic for partof chromosome 22 and part of chromosome 11. The clinicalfeatures of patients with the der(22) syndrome include mentalretardation, cranio-facial abnormalities and congenital heartdefects (4). Segregation analysis has shown that the risk of
unbalanced offspring born to female heterozygotes may be ashigh as 10%, with a significant risk to offspring of male heter-ozygotes (3). The frequency of abortions among offspring ofbalanced carriers is also raised. Although balanced carriers forthis translocation are normal it has been suggested that theymay have a 10-fold increased risk of breast cancer (5).
A variety of evidence suggests the occurrence of multipleoriginal t(11;22) translocation events. A family has beendescribed in which a child carried the translocation, which waslacking in either parent. Non-paternity was excluded throughthe analysis of 10 microsatellite markers distributed on 10different chromosomes and three VNTRs on three differentchromosomes (6). The translocation appears to occur in fami-lies with no common ancestry and a variety of racial and ethnicbackgrounds and, additionally, familial heteromorphic variantsof the derivative 22 have been described.
The trisomic region on chromosome 22 overlaps the regionhemizygously deleted in another congenital anomaly disorder,velo-cardio-facial syndrome/DiGeorge syndrome (VCFS/DGS). The breakpoint on chromosome 22 for the t(11;22)translocation has been mapped to the same interval as a 1.5 Mbdistal deletion breakpoint for VCFS (7). The identification ofthe breakpoints on chromosomes 11 and 22 is therefore animportant aid to our understanding of the nature of these inher-ited defects.
RESULTS
The breakpoint for the t(11;22) translocation, previouslymapped between the markers D11S29 and D11S144 (8,9) wasfurther localized using flow-sorted aliquots of der(11) andder(22) chromosomes. PCR analysis (Fig. 1a) showed thatD11S4516 (one end of YAC 785E12) was distal and thatD11S1340 and D11S1169 were proximal to the breakpoint(10). These data placed the breakpoint within the region ofoverlap between YACs 785E12 and 911F02 (10), consistentwith recent mapping data (11,12).
The apolipoprotein gene cluster (APOA1, APOC3, APOA4)was also found to lie in this region of overlap (data not shown).A series of PACs and a BAC was isolated using primer combi-nations for the APO gene cluster and D11S1169 (Table 1).Markers within this region were PCR mapped against theseclones (Fig. 1b). Fluorescence in situ hybridization (FISH)
+To whom correspondence should be addressed. Tel: +44 20 7882 6002; Fax: +44 20 7882 6004; Email: [email protected]

1526 Human Molecular Genetics, 2000, Vol. 9, No. 10
experiments against the cell line GM06229 determined that theclone 1062M21 (Fig. 1c) was split by the translocation. Thesedata implied that the APO gene cluster was distal to the break-point and effectively narrowed the breakpoint intervaldescribed recently (11).
Pulsed field gel analysis (data not shown) suggested that thebreakpoint lay within 50 kb of the APO gene cluster. ConventionalSouthern analysis was performed on genomic DNAs from the celllines GM06229 and GM06275 derived from balanced carriers andplacenta (Fig. 2a and b). The probe APOP3-P4 (Fig. 3) hybridizedto a germline 21 kb EcoRI fragment in all samples, a 50 and 26 kbnovel fragment in GM06229 and GM06275 DNA, respectively(Fig. 2a). The 21 kb EcoRI fragment was identified within a BACclone 227C10 and subcloned. The probe 1122A1-B1 (Fig. 3) fromthe other end of the 21 kb EcoRI fragment hybridized to the same 21kb germline fragment (Fig. 2b) and a novel 13.5 kb fragment in thecell lines GM06229 and GM06275. These data suggested that thebreakpoint in both cell lines lay within this 21 kb EcoRI fragment.In order to compare the breakpoints from a greater range of sources,DNA was extracted from lymphoblastoid and fibroblast cell linesderived from balanced (GM06229, GM06275 and GM04403) and
unbalanced (GM06228, GM03371 and GM00084A) carriers and ablood sample from a patient A with a balanced translocation. Theprobe APOP3-P4 detected a DraIII germline band of 17.5 kb in allsamples and a rearranged fragment of 8.5 kb in all the samplesexcept the placental control. These data, together with the break-point analysis (below), indicate that the size difference seen for therearranged fragments (Fig. 2A) is due to an EcoRI polymorphism.The 8.5 kb DraIII fragment was expected to contain the der(22)chromosome junction, consistent with its presence in both balancedand unbalanced individuals. The probe ET82-83 (Fig. 3) detects a21 kb EcoRI germline fragment and a 13.5 kb rearranged fragmentin all the samples from balanced carriers and, unexpectedly, in theapparently unbalanced cell line GM03371. The 13.5 kb EcoRI frag-ment was expected to contain the der(11) chromosome junction andtherefore should be present only in balanced carriers. This discrep-ancy was resolved by reanalysing the karyotype of GM03371. Itwas found to have the karotype: 47,XX,inv(9)(p13q21)pat,t(11;22)(q23;q11)mat,+der(22)t(11;22)(q23;q11)mat and thereforecontained a der(11) and two der(22) chromosomes. Such akaryotype has been observed previously and is thought to arise fromalternate segregation at meiosis I followed by either meiosis II or
Figure 1. (a) PCR amplification of flow-sorted der(22) and der(11) chromosomes from the cell line GM06229, placental DNA and a water blank with primers forthe loci; D11S4516 (785E12L1 and 785E12L2), D11S1340 (D11S1340A and D11S1340B) and ZPR1-4 (ZPR1 and ZPR4). (b) A diagrammatic representation ofthe region of chromosome 11q23, which contains the t(11;22)(q23;q11) breakpoint. The relative order of the loci D11S4516, APOP3-P4, ET82-83, ZPR1-4(D11S1169 ) and D11S1340 is shown and the position of the breakpoint is indicated by the dotted line. The solid horizontal bars represent two YAC clones (911F02and 785E12), five PACs (676 L8, 1062 M21, 1068 M21, 1124 I19 and 764 F24) and a single BAC (227 C10), the presence or absence of a particular locus in theseclones is represent by a solid vertical bar. (c and d) Metaphase cells from the cell line GM06229 showing FISH of the PAC clones; 1062 M21 (c) and 3090 O16(d). The chromosomes are counter-stained blue with DAPI. (c) The probe 1062 M21 localization seen as red signals (arrowed) on chromosomes 11q23, der(11)and der(22). (d) The probe 3090 O16 localization seen as red signals (arrowed) on chromosome 22q11, der(11) and der(22).

Human Molecular Genetics, 2000, Vol. 9, No. 10 1527
post-zygotic non-disjunction of the der(22) (11,13–15). TheSouthern data indicated that all the breakpoints clustered in a 15 kbregion of chromosome 11 between APOP3-P4 and ET82-83, thesize similarity of the rearrangements suggesting a highly specificform of recombination.
In order to isolate the breakpoint, long-range inverse PCR(LR-IPCR) (16) was performed on DNA from GM06229digested with EcoRI. A 1.8 kb product was amplified,subcloned and sequenced (data not shown) (GenBank acces-sion no. AF226670). Comparison with chromosome 11 germ-line sequence from the BAC clone 442e11 (GenBankaccession no. AC007707) indicated that novel sequence waspresent following an Alu repeat. Database searches revealedthat this novel sequence was present in a previously sequencedBAC b563b9 (GenBank accession no. AC007708) attributedto chromosome 22. Significantly, this chromosome 22 germ-line sequence also contained a truncated (100 bp) Alu elementat the fusion point. It was noted that non-identical sequencesrelated to the chromosome 22-specific sequence were presentin other BACs consistent with the mapping of the chromosome22 breakpoint to a low copy repeat (LCR22) sequence (7,17).Another BAC clone, 3090 O16, was identified in the genomesurvey sequence (GSS) database and was used in FISH exper-
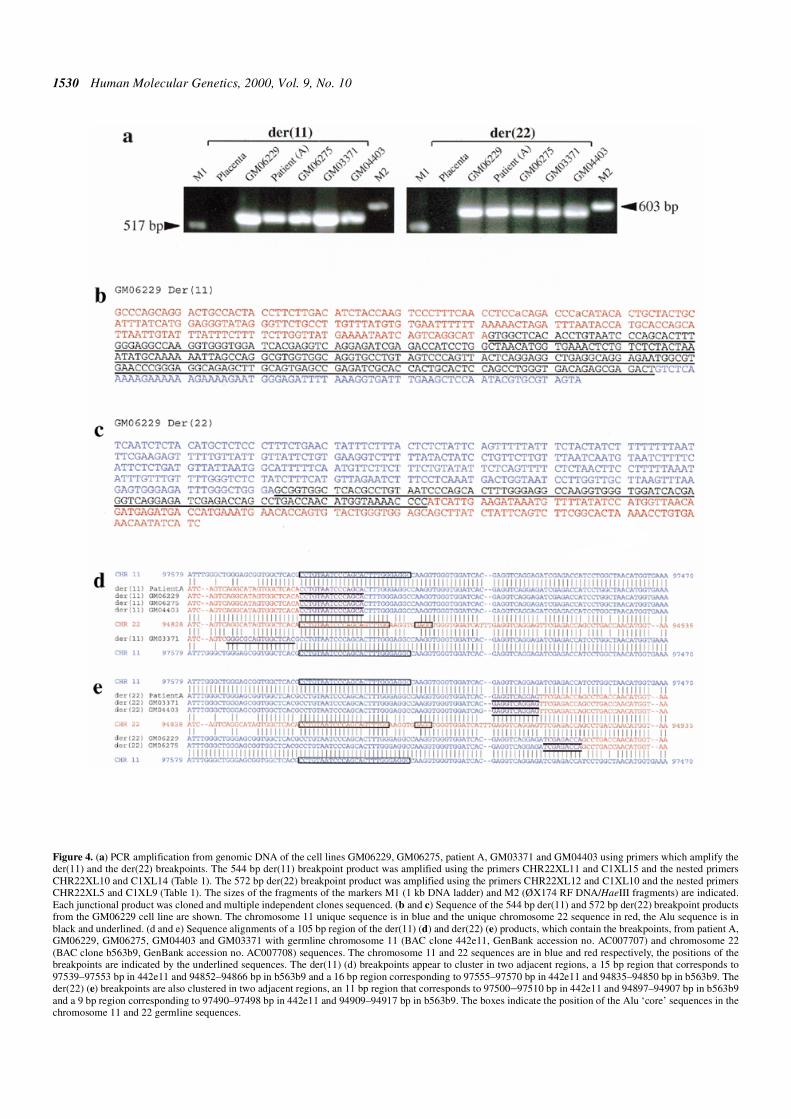
iments against metaphase cells from the cell line GM06229(Fig. 1d) demonstrating signals on the normal chromosome 22,the der(11) and the der(22). These data indicated that theGM06229 der(11) breakpoint was contained within the 1.8 kbIPCR product and was located within an Alu repeat. Primersplaced on chromosome 11 and 22 sequence in the 1.8 kbproduct, external to the Alu repeat, amplified a junctional540 bp product from GM06229, GM06275, GM03371,GM04403 and patient A DNA but not from normal placentalDNA (Fig. 4a). The sequences of all these products (Fig. 4b)show a high level of similarity i.e. >98%. The sequence of theder(22) breakpoint (the inverse of the 1.8 kb LR-IPCR and 540bp PCR product) could be predicted using sequences from thetwo genomic clones 442e11 and b563b9. The predicted break-point sequence was confirmed by PCR using DNA from thecell lines GM06229, GM06275, GM03371, GM04403 andpatient A (Fig. 4a). With the exception of the placental controla reciprocal junctional 570 bp product was amplified andsequenced (Fig. 4c). These products also show a high level ofsimilarity (>98%).
Given the high degree of similarity between the breakpointsobserved by PCR and Southern blotting, it is important toexclude the possibility of contamination during either cell
Table 1. Chromosome 11 and 22 primers, sequence and where applicable the position within the clones 442e11(GenBank accession no. AC007707) (a) or b563b9 (GenBank accession no. AC007708) (b)
Primer Sequence 5′–3′ Position
1122A1 GCTACATTTATTATCCGATTTC 86241–86262 (a)
C30XL1R CCTTCTTACTACACATCCAGAATGAGTCAG 86450–86488 (a)
C30XL1REXT GAATAAGGTACCTGCTAACCTTTACCAATC 86502–86531 (a)
1122B1 GAAAGACTCTAGAAGATCTTC 86622–86642 (a)
ET82 CAATAACGTGGCATTGAAGTGCAC 89653–89676 (a)
ET83 CTCCAGGCCGGAGTCCCACTC 90032–90052 (a)
C1XL1 CCTTTCTCAAGACACTCTACAGCCGTCTGC 96673–96702 (a)
C1XL2 CAAATCTGTCATGTCTACCCTGTGAGGGAG 96727–96756 (a)
C1XL15 GTGGATCACCCTGCAGTGACATTCATAGTC 97193–97222 (a)
C1XL14 TACTACGCACGTATTGGAGCTTCAAATCAC 97229–97258 (a)
C1XL9 TCAATCTCTACATGCTCTCCCTTTCTGAAC 97879–97908 (a)
C1XL10 TGAGCCACTGCGCCCGGCGTGGATCCTCTC 97932–97961 (a)
APOP4 CTAAGCTCTGCCCAGTCATGG 106348–106368 (a)
APOP3 AGCACCTTCAAGGAGAAAGAG 106689–106709 (a)
APOP2 CTGAGACCTCAGCTTCTGCAG 125489–125509 (a)
APOP1 CATGTGCTGACTGGGACAAAC 125782–125802 (a)
ZPR1 GATGAGTTCATTGTCAAACTG N/A
ZPR4 GATAACCTCCTTAAAGTGAGG N/A
785E12L1 GGGGATGTATGTTTGGAATC N/A
785E12L2 CTTACCTCACCAGCAACTCT N/A
D11S1340A GCTGAATGAGTCCTGAGTAATAA N/A
D11S1340B GGCCTAGACGTTCTTTTGTG N/A
CHR22XL11 CATGCTAGGATTCAACCACAGCATCCTGTC 94580–94609 (b)
CHR22XL10 GCCCAGCAGGACTGCCACTACCTTCTTGAC 94631–94660 (b)
CHR22XL5 GATGATATTGTTTCACAGGTTTTAGTGCCG 95041–95070 (b)
CHR22XL6 GCTCATCAGTTCCATGACTTGGAAACTGTC 95111–95140 (b)
CHR22XL12 GCTTTGAAAACCTGTTTACTTGTTAATGCC 95205–95234 (b)

1528 Human Molecular Genetics, 2000, Vol. 9, No. 10
culture or DNA extraction. It is unlikely that these junctionalproducts amplified by PCR arise from contamination due to anumber of consistent base differences and the lack of productsfrom normal genomic DNA. Another possible reason for thesimilarity between the breakpoints is that GM06229,GM06275, GM03371, GM04403 and patient A are not trulyindependent. However, the individuals from whom the celllines were derived and who provided the blood sample are notdirectly related. Contamination of the cell lines during culturealso seems unlikely as two are lymphoid and the other two arederived from fibroblasts and the sample from patient A was notcultured. Furthermore, there are distinct karyotypic differencesbetween the cell lines (Table 2). GM03371 is of particularinterest as this cell line is derived from a black individual andhas a highly distinctive karyotype. The cell type, age, ethnicorigin and karyotpye for all the samples used are summarizedin Table 2.
Since the t(11;22) is due to an Alu–Alu recombination it isnot possible to identify the precise fusion points. However,
alignment (Fig. 4d and e) of the junctional sequences with thegermline sequences of the clones 442e11 and b563b9 limits theder(11) breakpoints of GM06229, GM06275, patient A andGM04403 to a 15 bp region (Fig. 4d) that is identical within theAlu repeats on chromosomes 11 and 22. The GM03371der(11) breakpoint appears to be located in an adjacent 16 bp.The der(22) breakpoints (Fig. 4e) of patient A, GM03371 andGM04403 are located in an 11 bp region, whereas the break-points in GM06229 and GM06275 are located in an adjacent 9bp region.
DISCUSSION
The location of the breakpoint on chromosome 11 had previ-ously been predicted from haplotype analysis to be in an AT-rich repeat (12). However, our analysis has placed the break-point within a particular Alu repeat 350 bp proximal to the AT-rich sequence. Although the breakpoint region on chromosome11 is rich in Alu repeats (18 Alus in the 21 kb EcoRI fragment)the recombination occurs consistently in a particular Alu of theSb subtype (18), 9 kb proximal to the APOA4 gene and 20 kbdistal to a putative APOA4-like gene (Fig. 3). Similarly, thebreakpoint on chromosome 22 is located in a truncated Aluelement (Alu–Sq subtype) located in one of the LCR22 repeatregions between COMT (distal) and ZNF74 (proximal) (7) andwithin a gap in the recently defined chromosome 22 sequences(19). A probe from the chromosome 22 sequence close to thebreakpoint hybridized to approximately seven differently sizedbands in EcoRI-digested genomic DNA (data not shown). Thisnot only supported the idea that the breakpoint lies within anLCR22 but, when compared with the highly consistent rear-ranged fragment sizes (Fig. 2), strongly suggested that thebreakpoint is consistently within this LCR22 and does notinvolve other LCR22 members on chromosome 22. The PCRanalysis of the breakpoint sequences (Fig. 4) is consistent withthis interpretation. It has been proposed that the LCR22s couldmediate duplications and deletions of the 22q11 region byhomologous recombination mechanisms (17). The distalbreakpoint for a 1.5 Mb deletion associated with DGS and withVCFS has also been mapped to the same interval (7) and couldinvolve breakage of the same region. The study of other consti-tutional translocations supports the concept that the LCR22smay be preferentially involved. The breakpoints for a t(17;22)translocation associated with neurofibromatosis type I havebeen shown to map to the inverted sub-repeats within the prox-imal and distal LCR22s (20,21). Also, a constitutional t(1;22)translocation associated with an ependymoma has been shownto involve breakage in the same LCR22 as that for the 1.5 MbVFCS/DGS deletion breakpoint (22).
An examination of the sequences on chromosomes 11 and 22surrounding the t(11;22) breakpoints does not reveal anyparticular homologies other than the presence of Alu repeats.Alu-mediated recombination is known to be responsible for avariety of genetic disorders. These events include alterations tothe LDL receptors (23), α-globin (24) and β-globin genes (25),the c-sis oncogene (26) and apolipoprotein B (27). Both Alusegments involved in the t(11;22) translocation contain a‘core’ Alu sequence (5′-CCTGTAATCCCAGCACTTTGG-GAGGC-3′) (Fig. 4d and e) which has been associated withother Alu-mediated recombinations (28). This ‘core’ sequencecontains a pentanuceotide motif CCAGC, which is also part of
Figure 2. (a and b) Southern blot of DNA from GM06229, GM06275 and pla-centa digested with EcoRI and probed with APOP3-P4 (a) and 1122A1-B1 (b).(c and d) Southern blot of DNA from placenta, GM06229, GM06228,GM06275, patient A, GM03371, GM00084A and GM04403 digested withDraIII (c) and EcoRI (d) and probed with APOP3-P4 (c) and ET82-83 (d),respectively. The DNA samples were derived from both balanced (B) andunbalanced (U) carriers as indicated below the Southern blots. *GM03371 isderived from an individual with one der(11) and two der(22) chromosomes.

Human Molecular Genetics, 2000, Vol. 9, No. 10 1529
chi, an 8 bp sequence known to stimulate recBC-mediatedrecombination in Escherichia coli (29). It is not entirely clearwhether this ‘core’ sequence is recombinogenic or if the asso-ciation with Alu repeats involved in recombination is circum-stantial due to it being highly conserved between differentclasses of Alu
One possible explanation for the remarkable similaritybetween the breakpoints in unrelated individuals is that thet(11;22) translocation is a single ancestral event and the differ-ences we have observed represent subsequent mutations.However, the available evidence suggests that the relativelyhigh frequency of the t(11;22) in the human population may bedue to multiple independent events (3), with a case of a de novopaternal translocation having been described (6). If the t(11;22)breakpoints we have characterized are due to independentevents it would appear to involve a form of Alu-mediatedrecombination with a remarkable degree of specificity notobserved with other recurrent translocations such as thoseassociated with cancer. Although cancer-associated transloca-tions are not usually thought to be directly mediated by Alurecombination, there is some evidence that the t(9;22) translo-cation associated with chronic myeloid leukemia and thet(11;22) translocation associated with Ewing sarcoma may beassociated with repeated elements including Alu repeats(30,31). In these examples, however, there is variation in thegenomic breakpoint positions although it is clear that there is aselection for fusions that result in the expression of in-framechimeric mRNAs. It therefore may be speculated that the
constitutional t(11;22) translocation event is selected for by asimilar mechanism. It is conceivable that the high degree ofspecificity observed here is due to the requirement to express aparticular fused mRNA transcript. The effect of the transloca-tion on the expression of the genes surrounding the breakpointis currently unknown and, in particular, the possibility that afused transcript may be expressed remains to be determined.
Although individuals with a balanced t(11;22) translocationdo not appear to be affected adversely, a 10-fold increased riskof breast cancer was reported (5) in a small study of eight fami-lies. The molecular characterization of the t(11;22) will facili-tate the analysis of a cohort of breast cancer patients toinvestigate whether there is an association between this trans-location and sporadic breast cancer. In this context it is inter-esting to note that this region of chromosome 11 has beenshown to undergo loss of heterozygosity in this region in awide variety of cancers (32–35).
MATERIALS AND METHODS
Chromosome flow sorting
Metaphase chromosome suspensions were prepared from thelymphoblastoid cell line GM06229 as described previously(36) and stained with chromomycin A3 and Hoechst 33258.Flow analysis identified the peaks due to the der(11) andder(22) and these were sorted into 500 chromosome aliquots inpreparation for PCR analysis.
Table 2. The translocation-bearing cells used in this study
L, lymphoblastoid cells; F, fibroblast cells; BS, blood sample. The ages and ethnic origin are as listed in the Coriell catalogue (web site:http://locus.umdnj.edu/nigmsl/ ).
Source Cell type Age Ethnic origin Karyotype
GM06229 L 23 years WH 46,XX,t(11;22)(q23;q11)
GM06228 L 9 months – 47,XX,+der(22)t(11;22)(q23;q11)
GM06275 L 29 years WH 46,XX,t(11;22)(q23;q11)
GM03371 F 1 month BL 47,XX,inv(9)(p13q21)pat,t(11;22)(q23;q11)mat,+der(22)t(11;22)(q23;q11)mat
GM00084A F 1 month WH 47,XY,+der(22)t(11;22)(q23;q11)
GM04403 F 30 years WH 46,XX,t(11;22)(q23;q11)
Patient (A) BS WH 46,XX,t(11;22)(q23;q11)
Figure 3. A diagrammatic representation of the regions of chromosomes 11 and 22 which contain the t(11;22) breakpoints represented by the vertical dashed line.This is based on our sequence analysis of the 21 kb EcoRI fragment together with genomic sequence data available for BACs 442e11 (GenBank accession no.AC007707) and b563b9 (GenBank accession no. AC007708). Chromosome 11, the position and structure of the three genes APOA1, APOC3, APOA4, a putativegene APOA4L and the marker D11S1169 is shown. The location of three probes: APOP3-P4, ET82-83 and 1122A1-B1 is also shown. The restriction sites forEcoRI (E) and NotI (N) are indicated. The positions of 18 Alu repeats are represented by black boxes with arrows showing their orientation in the 21 kb EcoRIfragment. Alu repeats outside this region are not shown. Chromosome 22, the position and orientation of eight Alu repeats in a 21 kb region around the breakpointis shown.
1530 Human Molecular Genetics, 2000, Vol. 9, No. 10
Figure 4. (a) PCR amplification from genomic DNA of the cell lines GM06229, GM06275, patient A, GM03371 and GM04403 using primers which amplify theder(11) and the der(22) breakpoints. The 544 bp der(11) breakpoint product was amplified using the primers CHR22XL11 and C1XL15 and the nested primersCHR22XL10 and C1XL14 (Table 1). The 572 bp der(22) breakpoint product was amplified using the primers CHR22XL12 and C1XL10 and the nested primersCHR22XL5 and C1XL9 (Table 1). The sizes of the fragments of the markers M1 (1 kb DNA ladder) and M2 (ØX174 RF DNA/HaeIII fragments) are indicated.Each junctional product was cloned and multiple independent clones sequenced. (b and c) Sequence of the 544 bp der(11) and 572 bp der(22) breakpoint productsfrom the GM06229 cell line are shown. The chromosome 11 unique sequence is in blue and the unique chromosome 22 sequence in red, the Alu sequence is inblack and underlined. (d and e) Sequence alignments of a 105 bp region of the der(11) (d) and der(22) (e) products, which contain the breakpoints, from patient A,GM06229, GM06275, GM04403 and GM03371 with germline chromosome 11 (BAC clone 442e11, GenBank accession no. AC007707) and chromosome 22(BAC clone b563b9, GenBank accession no. AC007708) sequences. The chromosome 11 and 22 sequences are in blue and red respectively, the positions of thebreakpoints are indicated by the underlined sequences. The der(11) (d) breakpoints appear to cluster in two adjacent regions, a 15 bp region that corresponds to97539–97553 bp in 442e11 and 94852–94866 bp in b563b9 and a 16 bp region corresponding to 97555–97570 bp in 442e11 and 94835–94850 bp in b563b9. Theder(22) (e) breakpoints are also clustered in two adjacent regions, an 11 bp region that corresponds to 97500–97510 bp in 442e11 and 94897–94907 bp in b563b9and a 9 bp region corresponding to 97490–97498 bp in 442e11 and 94909–94917 bp in b563b9. The boxes indicate the position of the Alu ‘core’ sequences in thechromosome 11 and 22 germline sequences.

Human Molecular Genetics, 2000, Vol. 9, No. 10 1531
Cell lines and DNA isolation
The cell lines GM06229, GM06228, GM0275, GM03371,GM00084A and GM04403 were obtained from the Coriell CellRepository (Camden, NJ). These cell lines were cultured inRPMI 1640 with Glutamax (Gibco BRL, Paisley, UK), 20%fetal calf serum, penicillin and streptomycin. Genomic DNAwas isolated from blood and cell lines using the Pure-geneDNA isolation kit (Gentra Systems, Ashby de la Zouch, UK).
Isolation of genomic clones and DNA purification
The YACs 911 F02 and 785 E12 were obtained from the CEPHmega YAC libray held at the MRC UK Human GenomeMapping Project Resource Centre (Hinxton, Cambridge, UK).The screening of the BAC and PAC libraries was carried out byResearch Genetics (Huntsville, AL) using the probes ZPR1-4and APOP3-P4. The clone 3090 O16 was also obtained fromResearch Genetics.
YAC DNA was isolated using the Pure-gene DNA isolationkit (Gentra Systems) from 2 day cultures in AHC medium at30°C. Plasmid DNA was isolated using the Wizard DNA Puri-fication System (Promega, Southampton, UK) from overnightcultures in Luria–Bertani broth with the appropriate antibioticat 37°C.
Polymerase chain reaction (PCR)
PCR was performed using Taq polymerase supplied by theICRF (London, UK). A 50 µl reaction comprised 1× buffer(Promega, containing MgCl), 200 µM dNTPs, 50 µmol oligo-nucleotide primers and 1 U Taq polymerase. For PCR fromgenomic DNA ~500 ng of DNA was added and for clonedDNA ~1 ng was added to each reaction. Samples weresubjected to 94°C for 1 min followed by 30 cycles of 94°C for20 s, 56°C for 20 s, 1 min at 72°C, followed by a final incuba-tion at 72°C for 10 min. This was repeated for nested reactions.LR-IPCR was performed as described by Willis et al. (16)using the XL-PCR kit (PE Applied Biosystems, Warrington,UK) on genomic DNA from GM06229 digested with EcoRIwith the primers C30XL1REXT, C1XL1, C30XL1R andC1XL2 (Table 1) in nested LR-IPCRs. The primers used formapping with the flow-sorted chromosomes, YAC, PAC, BACclones and generating probes for Southern analysis were785E12L1 and 785E12L2 (D11S4615), APOP3 and APOP4(APOP3-P4), ET82 and ET83 (ET82-83), ZPR1 and ZPR4(ZPR1-4, D11S1169), D11S1340A and D11S1340B(D11S1340) and 1122A1 and 1122B1 (1122A1-B1) (Table 1).The der(11) breakpoints were amplified using the primersCHR22XL11, C1XL15, CHR22XL10 and C1XL14 in nestedPCRs. The der(22) primers were amplified using the primersCHR22XL12, C1XL10, CHR22XL5 and C1XL9 (Table 1) innested PCRs. PCR products were then either cloned using theTOPO or TOPO-XL TA cloning kits (Invitrogen, Groningen,The Netherlands) or used directly.
FISH analysis
FISH was performed as described by Pinkel et al. (37) usingfluorescently labelled PAC and BAC clones (1062 M21, 676L8, 227 C10 and 3090 O16) hybridized to metaphase chromo-some spreads from GM06229 counter-stained with DAPI.
DNA sequence analysis
Sequencing reactions were performed using the BigDyeTerminator Cycle Sequencing Ready Reaction DNAsequencing kit (PE Applied Biosystems) according to themanufacturer’s instructions using specific and M13 forwardand reverse primers. Samples were either plasmid preparationspurified using the Wizard DNA purification system (Promega)or PCR products purified using centricon 100 concentrators(Amicon, Beverley, MA). The reaction products were resolvedon a 4.8% polyacrylamide gel run using the ABI PRISM 377DNA sequencer (PE Applied Biosystems).
Gel electrophoresis and Southern blot analysis
Genomic DNA was digested with the enzymes EcoRI andDraIII (Promega) according to the manufacturer’s instructions.Standard 20 × 25 cm agarose gels (0.4–0.7%) were run in1× TBE for between 24 and 48 h depending on the size of frag-ments to be resolved. Gels were blotted on to Hybond N+(Amersham, Little Chalfont, UK). The probes were labelledwith [32P]dCTP using the multi-prime labelling kit(Amersham) and hybridized overnight at 65°C in a solutioncomprising 6× SSC, 0.5% SDS and 5× Denhardt’s. The filterswere then washed twice in 2× SSC at 65°C for 15 min followedby 2× SSC, 0.1% SDS at 65°C for 30 min and finally 0.1× SSCat 65°C for 10 min. The pattern of hybridization was visualizedusing phosphor screens (Kodak, Hemel Hempstead, UK)exposed to the washed filters and scanned with a Storm 840(Molecular Dynamics, Sunnyvale, CA) PhosphoImager.
ACKNOWLEDGEMENTS
We thank John Sgouros, ICRF for valuable discussions in thecourse of this work, Professor Joy Delhanty for provision ofblood from t(11;22) patients and Dr Louise Jones for invalu-able technical assistance.
REFERENCES
1. Fraccaro, M., Lindsten, J., Ford, C.E. and Iselius, L. (1980) The 11q;22qtranslocation: a European collaborative analysis of 43 cases. Hum. Genet.,56, 21–51.
2. Iselius, L., Lindsten, J., Aurias, A., Fraccaro, M., Bastard, C., Bottelli,A.M., Bui, T.H., Caufin, D., Dalprà, L., Delendi, N. et al. (1983) The11q;22q translocation: a collaborative study of 20 new cases and analysisof 110 families. Hum. Genet., 64, 343–355.
3. Zackai, E.H. and Emanuel, B.S. (1980) Site-specific reciprocaltranslocation, t(11;22) (q23;q11), in several unrelated families with 3:1meiotic disjunction. Am. J. Med. Genet., 7, 507–521.
4. Lin, A.E., Bernar, J., Chin, A.J., Sparkes, R.S., Emanuel, B.S. and Zackai,E.H. (1986) Congenital heart disease in supernumerary der(22), t(11;22)syndrome. Clin. Genet., 29, 269–275.
5. Lindblom, A., Sandelin, K., Iselius, L., Dumanski, J., White, I.,Nordenskjöld, M. and Larsson, C. (1994) Predisposition for breast cancerin carriers of constitutional translocation 11q;22q. Am. J. Hum. Genet., 54,871–876.
6. Dawson, A.J., Mears, A.J., Chudley, A.E., Bech-Hansen, T. andMcDermid, H. (1996) Der (22)t (11;22) resulting from a paternal de novotranslocation, adjacent 1 segregation, and maternal heterodisomy ofchromosome 22. J. Med. Genet., 33, 952–956.
7. Funke, B., Edelmann, L., McCain, N., Pandita, R.K., Ferreira, J.,Merscher, S., Zohouri, M., Cannizzaro, L., Shanske, A. and Morrow, B.E.(1999) Der (22) syndrome and velo-cardio-facial syndrome/DiGeorgesyndrome share a 1.5-Mb region of overlap on chromosome 22q11. Am. J.Hum. Genet., 64, 747–758.

1532 Human Molecular Genetics, 2000, Vol. 9, No. 10
8. Budarf, M., Sellinger, B., Griffin, C. and Emanuel, B.S. (1989)Comparative mapping of the constitutional and tumor-associated 11;22translocations. Am. J. Hum. Genet., 45, 128–139.
9. Junien, C., van Heyningen, V., Evans, G., Little, P. and Mannens, M.(1992) Report of the second chromosome 11 workshop. Genomics, 12,620–625.
10. Arai, Y., Hosoda, F., Nakayama, K. and Ohki, M. (1996) A yeast artificialchromosome contig and NotI restriction map that spans the tumorsuppressor gene (s) locus, 11q22.2–q23.3. Genomics, 35, 196–206.
11. Shaikh, T.H., Budarf, M.L., Celle, L., Zackai, E.H. and Emanuel, B.S.(1999) Clustered 11q23 and 22q11 breakpoints and 3:1 meioticmalsegregation in multiple unrelated t (11;22) families. Am. J. Hum.Genet., 65, 1595–1607.
12. Edelmann, L., Spiteri, E., McCain, N., Goldberg, R., Pandita, R.K.,Duong, S., Fox, J., Blumenthal, D., Lalani, S.R., Shaffer, L.G. et al.(1999) A common breakpoint on 11q23 in carriers of the constitutionalt(11;22) translocation. Am. J. Hum. Genet., 65, 1608–1616.
13. Lockwood, D.H., Farrier, A., Hecht, F. and Allanson, J. (1989) Not allchromosome imbalance resulting from the 11q;22q translocation is due to3:1 segregation in first meiosis. Hum. Genet., 83, 287–288.
14. Simi, P., Ceccarelli, M., Barachini, A., Floridia, G. and Zuffardi, O.(1992) The unbalanced offspring of the male carriers of the 11q;22qtranslocation: nondisjunction at meiosis II in a balanced spermatocyte.Hum. Genet., 88, 482–483.
15. Petkovic, I., de Capoa, A., Giancotti, P. and Barisic, I. (1996) Unusualsegregation of t(11;22) resulting from crossing-over followed by 3:1disjunction at meiosis I. Clin. Genet., 50, 515–519.
16. Willis, T.G., Jadayel, D.M., Coignet, L.J., Abdul-Rauf, M., Treleaven,J.G., Catovsky, D. and Dyer, M.J. (1997) Rapid molecular cloning ofrearrangements of the IGHJ locus using long-distance inverse polymerasechain reaction. Blood, 90, 2456–2464.
17. Edelmann, L., Pandita, R.K., Spiteri, E., Funke, B., Goldberg, R.,Palanisamy, N., Chaganti, R.S., Magenis, E., Shprintzen, R.J. andMorrow, B.E. (1999) A common molecular basis for rearrangementdisorders on chromosome 22q11. Hum. Mol. Genet., 8, 1157–1167.
18. Jurka, J. and Smith, T.A. (1988) A fundamental division in the Alu familyof repeated sequences. Proc. Natl Acad. Sci. USA, 85, 4775–4779.
19. Dunham, I., Shimizu, N., Roe, B.A., Chissoe, S., Hunt, A.R., Collins, J.E.,Bruskiewich, R., Beare, D.M., Clamp, M., Smink, L.J. et al. (1999) TheDNA sequence of human chromosome 22. Nature, 402, 489–495.
20. Kehrer-Sawatzki, H., Häussler, J., Krone, W., Bode, H., Jenne, D.E.,Mehnert, K.U., Tümmers, U. and Assum, G. (1997) The second case of at(17;22) in a family with neurofibromatosis type 1: sequence analysis ofthe breakpoint regions. Hum. Genet., 99, 237–247.
21. Edelmann, L., Pandita, R.K. and Morrow, B.E. (1999) Low copy repeatsmediate the common 3 Mb deletion in velo-cardio-facial syndromep[atients on 22q11. Am. J. Hum. Genet., 64, 1076–1086.
22. Rhodes, C.H., Call, K.M., Budarf, M.L., Barnoski, B.L., Bell, C.J.,Emanuel, B.S., Bigner, S.H., Park, J.P. and Mohandas, T.K. (1997)Molecular studies of an ependymoma-associated constitutional t(1;22)(p22;q11.2). Cytogenet. Cell Genet., 78, 247–252.
23. Hobbs, H.H., Brown, M.S., Goldstein, J.L. and Russell, D.W. (1986)Deletion of exon encoding cysteine-rich repeat of low density lipoproteinreceptor alters its binding specificity in a subject with familialhypercholesterolemia. J. Biol. Chem., 261, 13114–13120.
24. Nicholls, R.D., Fischel-Ghodsian, N. and Higgs, D.R. (1987)Recombination at the human alpha-globin gene cluster: sequence featuresand topological constraints. Cell, 49, 369–378.
25. Henthorn, P.S., Mager, D.L., Huisman, T.H. and Smithies, O. (1986) Agene deletion ending within a complex array of repeated sequences 3′ tothe human beta-globin gene cluster. Proc. Natl Acad. Sci. USA, 83, 5194–5198.
26. Smidt, M., Kirsch, I. and Ratner, L. (1990) Deletion of Alu sequences inthe fifth c-sis intron in individuals with meningiomas. J. Clin. Invest., 86,1151–1157.
27. Huang, L.S., Ripps, M.E., Korman, S.H., Deckelbaum, R.J. and Breslow,J.L. (1989) Hypobetalipoproteinemia due to an apolipoprotein B geneexon 21 deletion derived by Alu–Alu recombination. J. Biol. Chem., 264,11394–11400.
28. Rudiger, N.S., Gregersen, N. and Kielland-Brandt, M.C. (1995) One shortwell conserved region of Alu-sequences is involved in human generearrangements and has homology with prokaryotic chi. Nucleic AcidsRes., 23, 256–260.
29. Stahl, F.W. (1979) Special sites in generalized recombination. Annu. Rev.Genet., 13, 7–24.
30. Jeffs, A.R., Benjes, S.M., Smith, T.L., Sowerby, S.J. and Morris, C.M.(1998) The BCR gene recombines preferentially with Alu elements incomplex BCR-ABL translocations of chronic myeloid leukaemia. Hum.Mol. Genet., 7, 767–776.
31. Obata, K., Hiraga, H., Nojima, T., Yoshida, M.C. and Abe, S. (1999)Molecular characterization of the genomic breakpoint junction in at(11;22) translocation in Ewing sarcoma. Genes Chromosomes Cancer,25, 6–15.
32. Negrini, M., Rasio, D., Hampton, G.M., Sabbioni, S., Rattan, S., Carter,S.L., Rosenberg, A.L., Schwartz, G.F., Shiloh, Y., Cavenee, W.K. et al.(1995) Definition and refinement of chromosome 11 regions of loss ofheterozygosity in breast cancer: identification of a new region at 11q23.3.Cancer Res., 55, 3003–3007.
33. Keldysh, P.L., Dragani, T.A., Fleischman, E.W., Konstantinova, L.N.,Perevoschikov, A.G., Pierotti, M.A., Della Porta, G. and Kopnin, B.P.(1993) 11q deletions in human colorectal carcinomas: cytogenetics andrestriction fragment length polymorphism analysis. Genes ChromosomesCancer, 6, 45–50.
34. Hampton, G.M., Mannermaa, A., Winquist, R., Alavaikko, M., Blanco,G., Taskinen, P.J., Kiviniemi, H., Newsham, I., Cavenee, W.K. andEvans, G.A. (1994) Loss of heterozygosity in sporadic human breastcarcinoma: a common region between 11q22 and 11q23.3. Cancer Res.,54, 4586–4589.
35. Carter, S.L., Negrini, M., Baffa, R., Gillum, D.R., Rosenberg, A.L.,Schwartz, G.F. and Croce, C.M. (1994) Loss of heterozygosity at 11q22-q23 in breast cancer. Cancer Res., 54, 6270–6274.
36. Young, B.D. (1990) Chromosome analysis and sorting. In Ormerod, M.G.(ed.), Flow Cytometry: A Practical Approach. IRL Press, Oxford, UK, pp.145–159.
37. Pinkel, D., Gray, J.W., Trask, B., van den Engh, G., Fuscoe, J. andvan Dekken, H. (1986) Cytogenetic analysis by in situ hybridization withfluorescently labeled nucleic acid probes. Cold Spring Harbor Symp.Quant. Biol., 51, 151–157.
![Translocation (11;14)(q13;q32) and preferential ...with MCL is t(11;14)(q13;q32), but other variants such as t(11;22) have also been described [23]. Deletion 6q15 and monosomy 13 are](https://img.pdfslide.net/doc/110x75/6050220ab1a6ff33e8485b4d/translocation-1114q13q32-and-preferential-with-mcl-is-t1114q13q32.jpg)




























