Embed Size (px)
Citation preview

The Nature of the MineralThe Nature of the Mineral--Water Interface: Water Interface: A Molecular SimulationA Molecular SimulationA Molecular SimulationA Molecular Simulation
and Spectroscopic Investigationand Spectroscopic InvestigationRandall T. Cygan
Geochemistry DepartmentSandia National Laboratories
Albuquerque New Mexico USAAlbuquerque, New Mexico, USACollaborations withJeffery A. Greathouse (Sandia)Michigan State UniversityMichigan State UniversityNorthwestern UniversityPurdue University
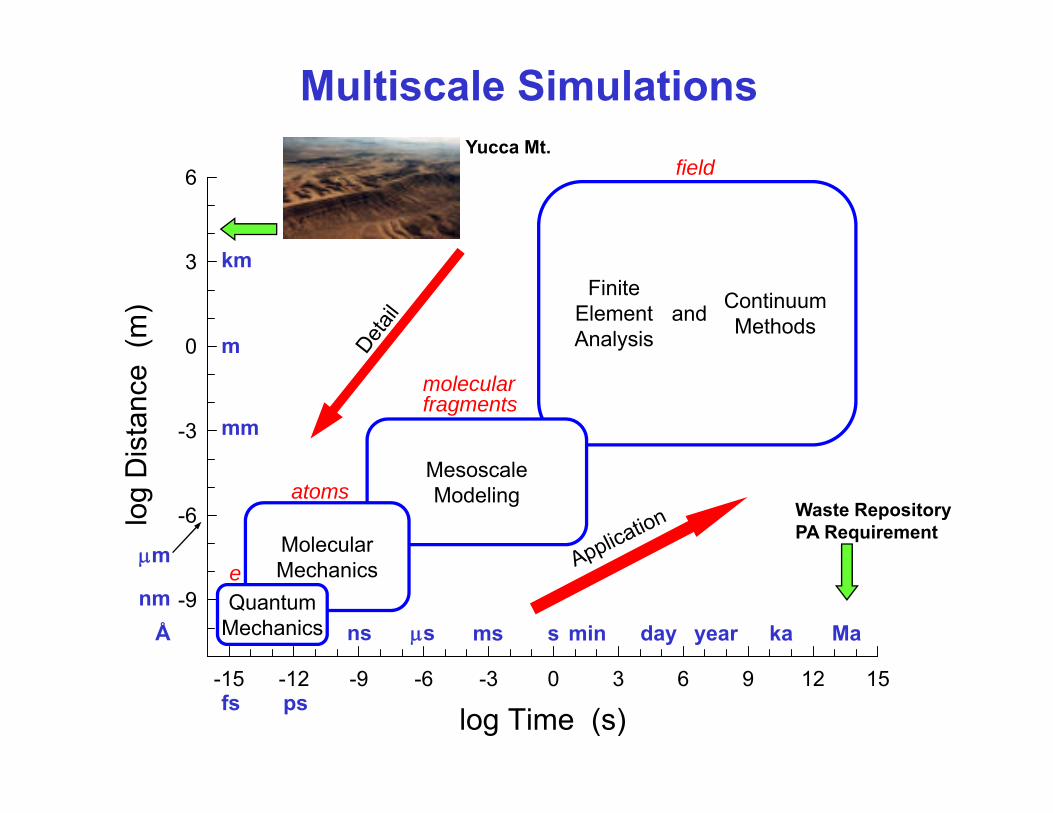
Multiscale SimulationsYucca Mt
6
k
fieldYucca Mt.(m
)
0
3Finite
ElementAnalysis
ContinuumMethodsand
m
km
ista
nce
-3 mm
molecularfragments
log
D
-6
MesoscaleModeling
Molecularμm
atomsWaste RepositoryPA Requirement
-9Mechanics
QuantumMechanicsÅ
μm
min yearday Mamsμsns
enm
s ka
log Time (s)-15 -12 -9 -6 -3 0 3 6 9 12 15
psfs
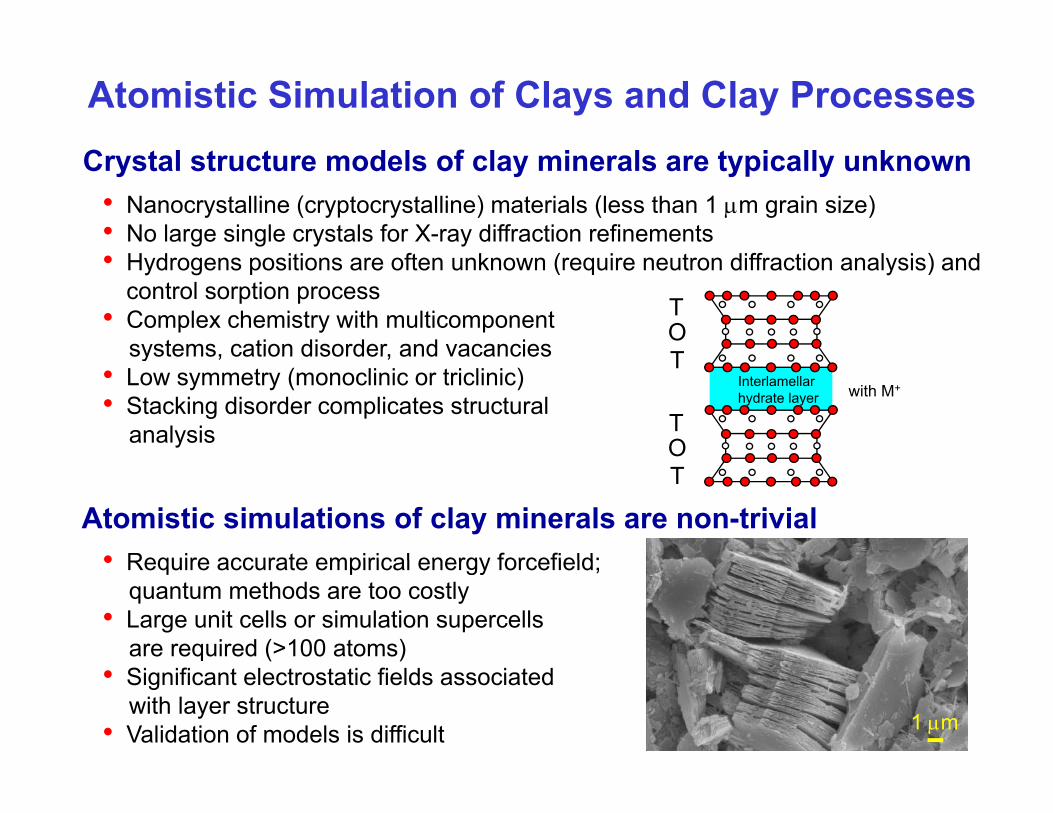
C t l t t d l f l i l t i ll k
Atomistic Simulation of Clays and Clay ProcessesCrystal structure models of clay minerals are typically unknown
• Nanocrystalline (cryptocrystalline) materials (less than 1 μm grain size)• No large single crystals for X-ray diffraction refinements
f ( ff )• Hydrogens positions are often unknown (require neutron diffraction analysis) and control sorption process
• Complex chemistry with multicomponentsystems, cation disorder, and vacancies
TOTsystems, cation disorder, and vacancies
• Low symmetry (monoclinic or triclinic)• Stacking disorder complicates structural
analysis
Interlamellarhydrate layer with M+
T
TO
Atomistic simulations of clay minerals are non-trivial• Require accurate empirical energy forcefield;
T
• Require accurate empirical energy forcefield;quantum methods are too costly
• Large unit cells or simulation supercellsare required (>100 atoms)
• Significant electrostatic fields associatedwith layer structure
• Validation of models is difficult 1 μm
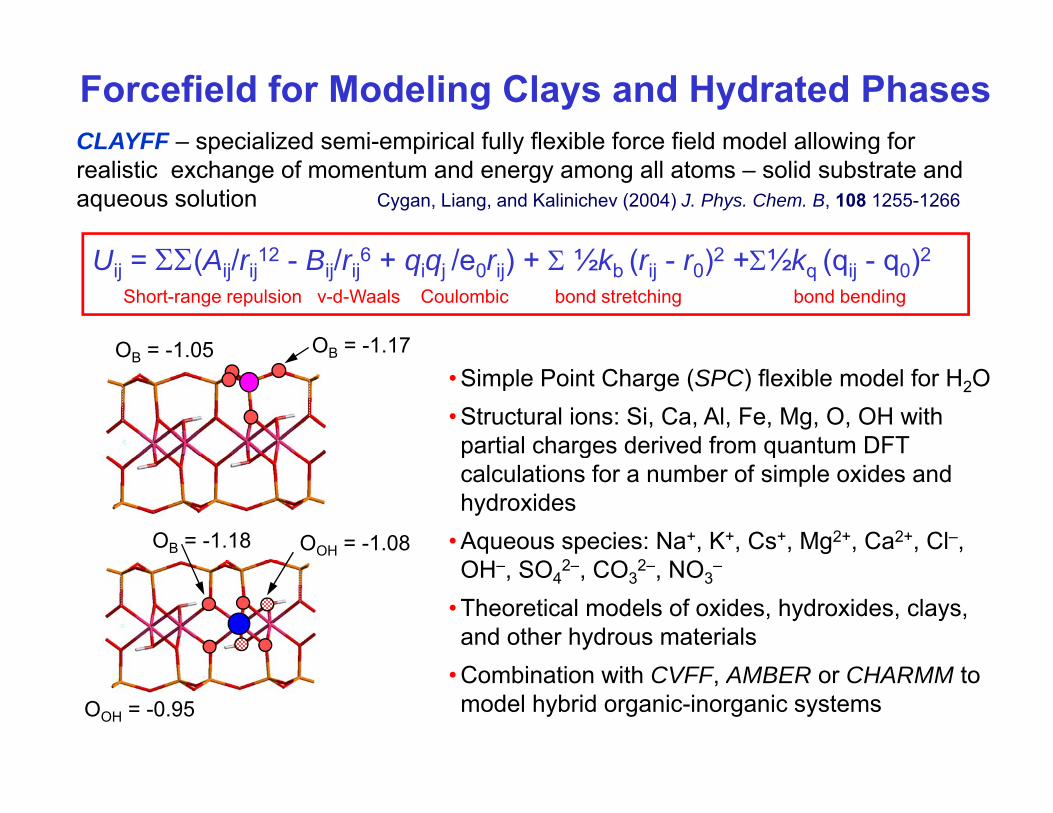
CLAYFF – specialized semi-empirical fully flexible force field model allowing for
Forcefield for Modeling Clays and Hydrated Phases
U ΣΣ(A / 12 B / 6 + / ) + Σ ½k ( )2 +Σ½k ( )2
p p y grealistic exchange of momentum and energy among all atoms – solid substrate and aqueous solution Cygan, Liang, and Kalinichev (2004) J. Phys. Chem. B, 108 1255-1266
Uij = ΣΣ(Aij/rij12 - Bij/rij
6 + qiqj /e0rij) + Σ ½kb (rij - r0)2 +Σ½kq (qij - q0)2
Short-range repulsion v-d-Waals Coulombic bond stretching bond bending
OB = -1.05 OB = -1.17OB 1.05 B
• Simple Point Charge (SPC) flexible model for H2O• Structural ions: Si, Ca, Al, Fe, Mg, O, OH with partial charges derived from quantum DFT
OOH = -1.08OB = -1.18
gcalculations for a number of simple oxides and hydroxides
• Aqueous species: Na+, K+, Cs+, Mg2+, Ca2+, Cl–, O SO 2 CO 2 OOH–, SO4
2–, CO32–, NO3
–
• Theoretical models of oxides, hydroxides, clays, and other hydrous materials
OOH = -0.95• Combination with CVFF, AMBER or CHARMM to model hybrid organic-inorganic systems
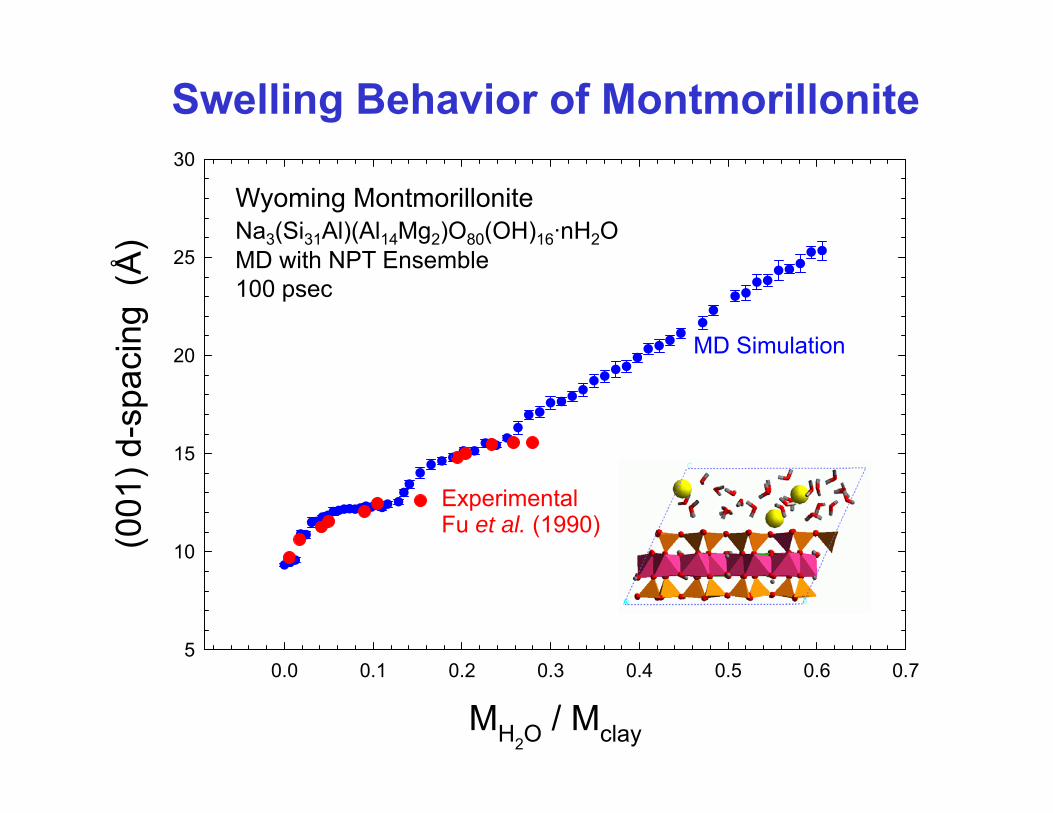
Swelling Behavior of Montmorillonite
25
30Å
)
Na3(Si31Al)(Al14Mg2)O80(OH)16nH2O4 unit cell basis
Wyoming MontmorilloniteNa3(Si31Al)(Al14Mg2)O80(OH)16·nH2OMD ith NPT E bl
20
25
cing
(Å
MD Simulation
4 unit cell basisMD with NPT Ensemble100 psec
15
20
d-sp
ac
10
15
(001
)
ExperimentalFu et al. (1990)
0 0 0 1 0 2 0 3 0 0 0 6 05
MH2O / Mclay
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7
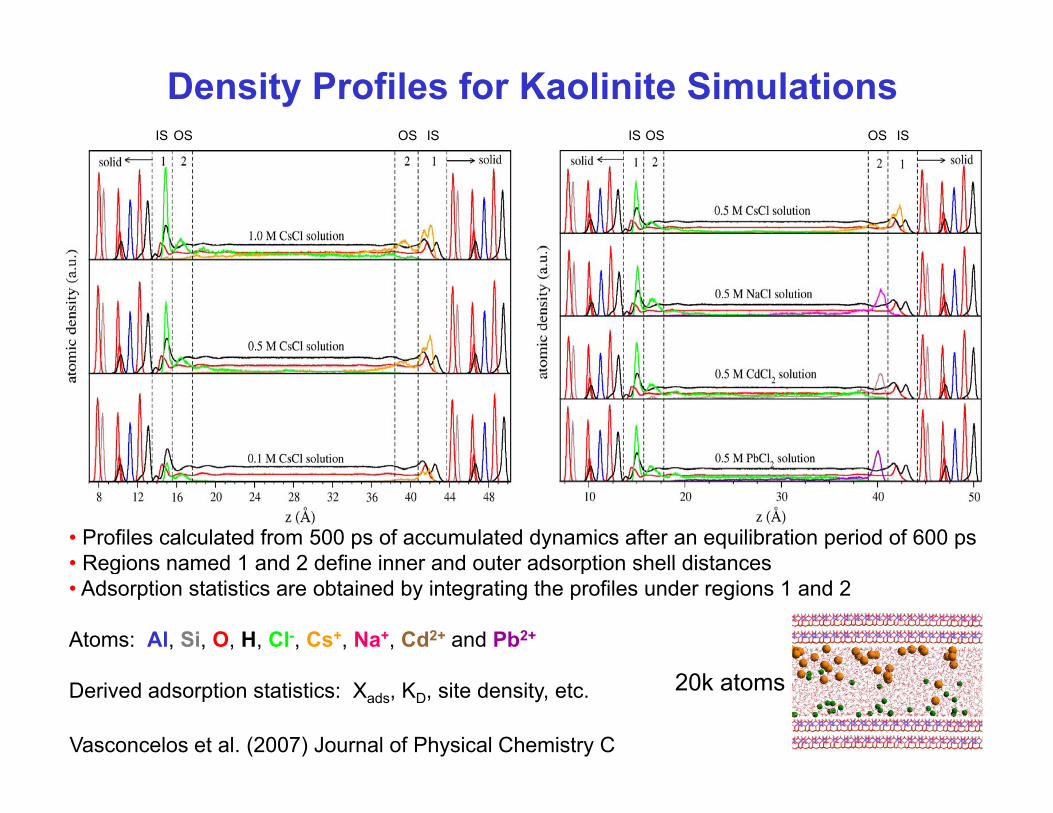
Density Profiles for Kaolinite SimulationsIS IS IS ISOS OS OS OS
• Profiles calculated from 500 ps of accumulated dynamics after an equilibration period of 600 ps• Regions named 1 and 2 define inner and outer adsorption shell distances• Regions named 1 and 2 define inner and outer adsorption shell distances• Adsorption statistics are obtained by integrating the profiles under regions 1 and 2
Atoms: Al, Si, O, H, Cl-, Cs+, Na+, Cd2+ and Pb2+
Derived adsorption statistics: Xads, KD, site density, etc.
Vasconcelos et al. (2007) Journal of Physical Chemistry C
20k atoms
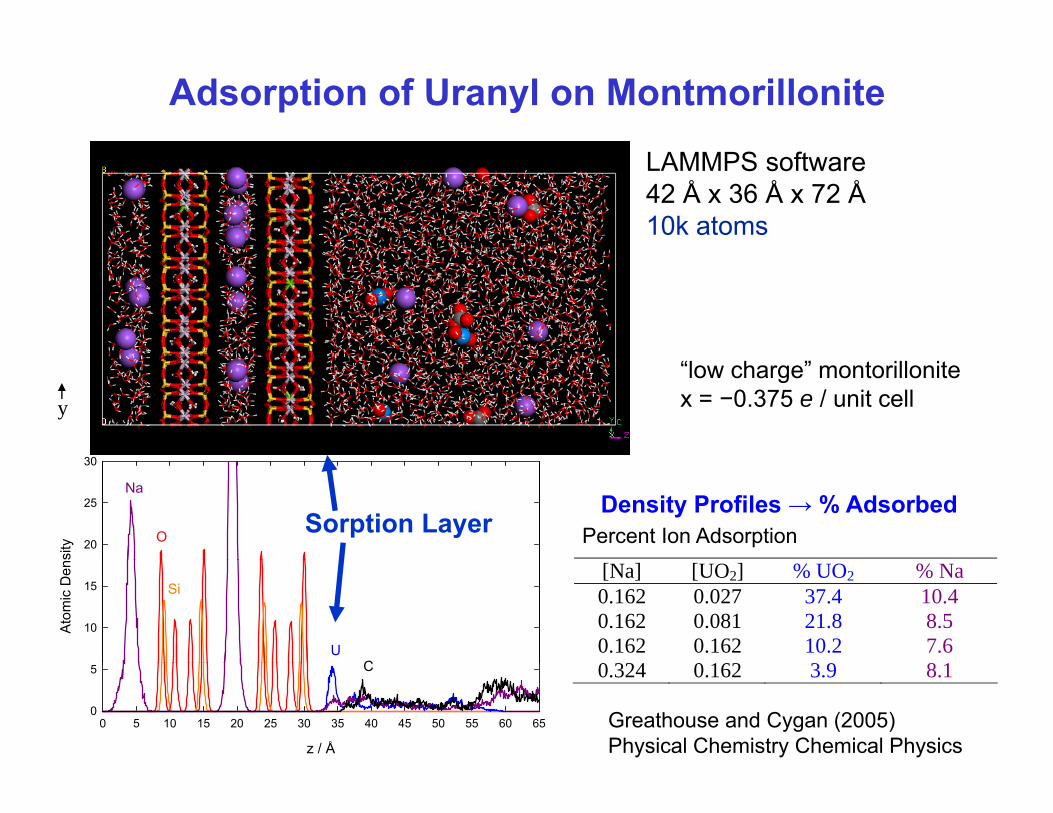
Adsorption of Uranyl on Montmorillonite
LAMMPS software42 Å x 36 Å x 72 Å10k atoms
y
30
“low charge” montorillonitex = −0.375 e / unit cell
sity 20
25
30
O
Na
Sorption Layer Percent Ion AdsorptionDensity Profiles → % Adsorbed
Ato
mic
Den
10
15
UC
Si[Na] [UO2] % UO2 % Na0.162 0.027 37.4 10.4 0.162 0.081 21.8 8.5 0.162 0.162 10.2 7.6 0 324 0 162 3 9 8 1
z / Å
0 5 10 15 20 25 30 35 40 45 50 55 60 650
5 C 0.324 0.162 3.9 8.1
Greathouse and Cygan (2005)Physical Chemistry Chemical Physics
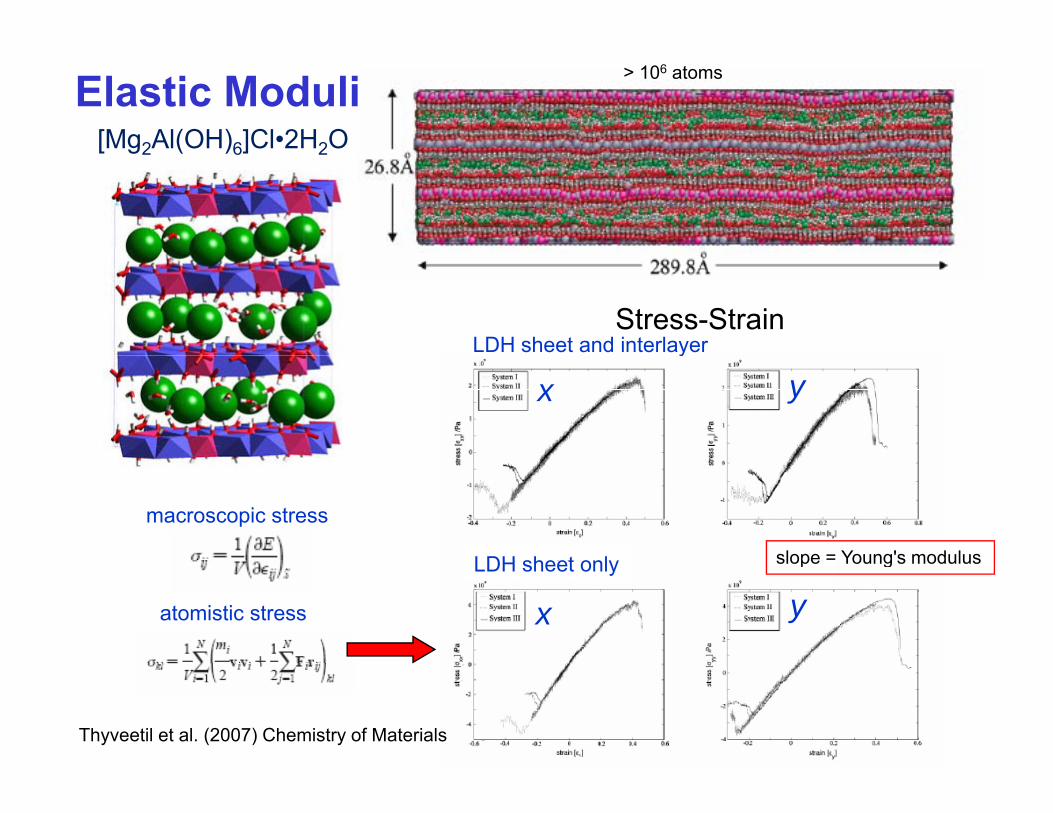
Elastic Moduli > 106 atoms
[Mg2Al(OH)6]Cl•2H2O[ g2 ( )6] 2
Stress-StrainLDH sheet and interlayer
x yy
LDH sheet only
macroscopic stress
slope = Young's modulus
x yLDH sheet only
atomistic stress
slope Young s modulus
Thyveetil et al. (2007) Chemistry of Materials

MD Models of DNA-IntercalatedClay MineralsClay Minerals
Layered Double Hydroxide
TotalAtoms
WaterMolecules
DNA Base Pairs
Thyveetil et al. (2008) Journal of the American Chemical Society
AtomEye visualization(a) 10,142 1,406 12
(b) 256,608 23,328 108
(c) 1,026,432 93,312 480
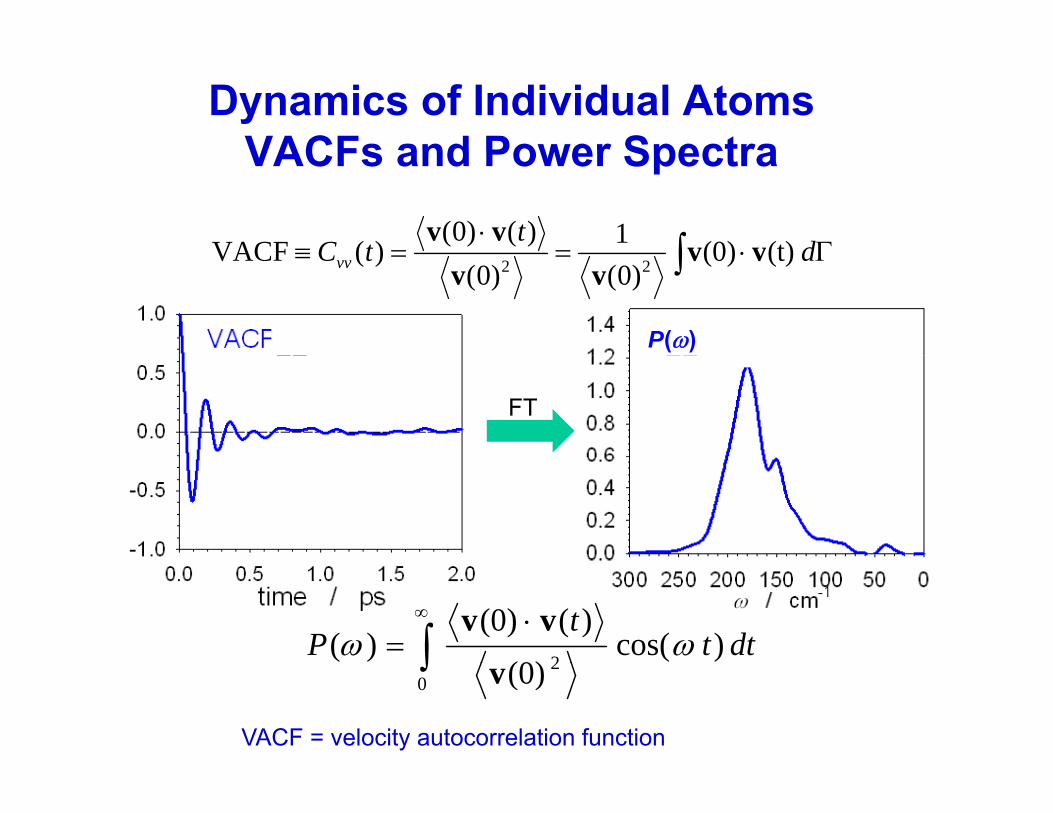
Dynamics of Individual AtomsVACFs and Power Spectra
Γ⋅=⋅
=≡ ∫ dt
tC (t)(0)1)()0()(VACF vv
vv
VACFs and Power Spectra
P(ω)
Γ⋅==≡ ∫ dtCvv (t)(0))0()0(
)(VACF 22 vvvv
FT
dttt
P )cos((0)
)((0))(
2ωω ∫
∞ ⋅=
vv(0)0
2∫ v
VACF = velocity autocorrelation function
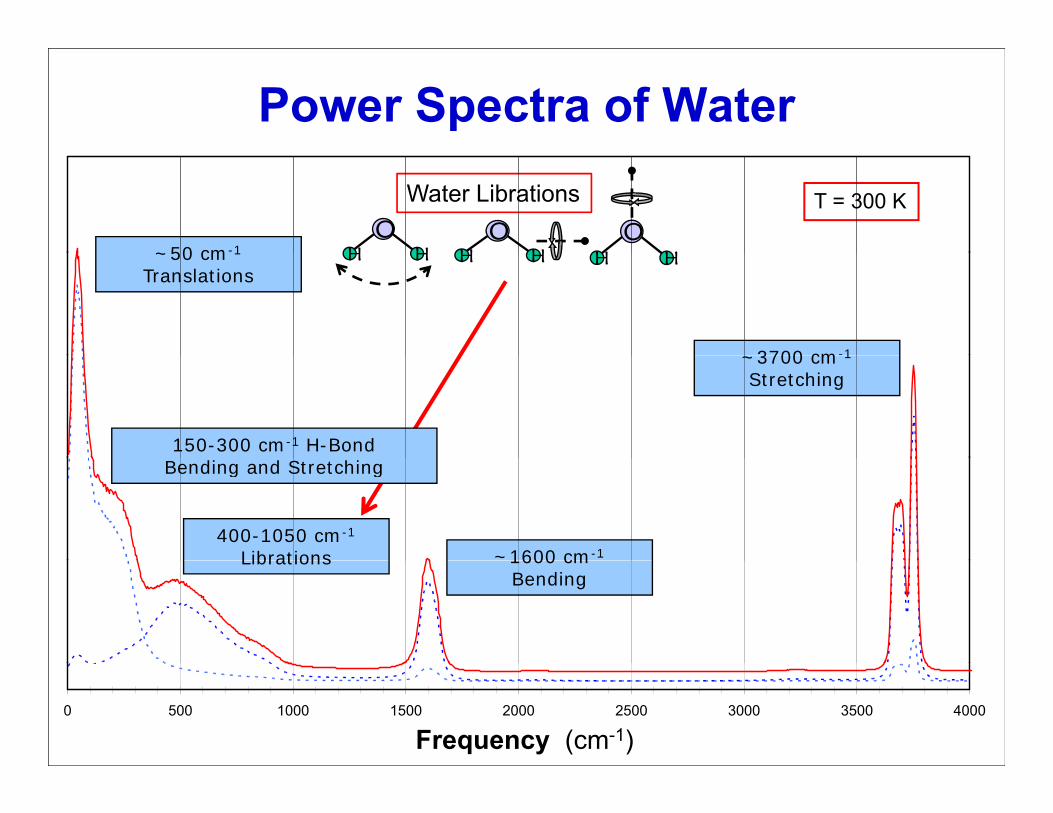
Power Spectra of Water
~50 cm-1
T = 300 KO
H HO
H HO
H H
Water Librations
~50 cm 1
Translations
~3700 cm-1
H H H H H H
~3700 cm 1
Stretching
150-300 cm-1 H-Bond
400-1050 cm-1
Librations ~1600 cm-1
Bending and Stretching
Librations 1600 cmBending
0 500 1000 1500 2000 2500 3000 3500 4000
cm -1Frequency (cm-1)
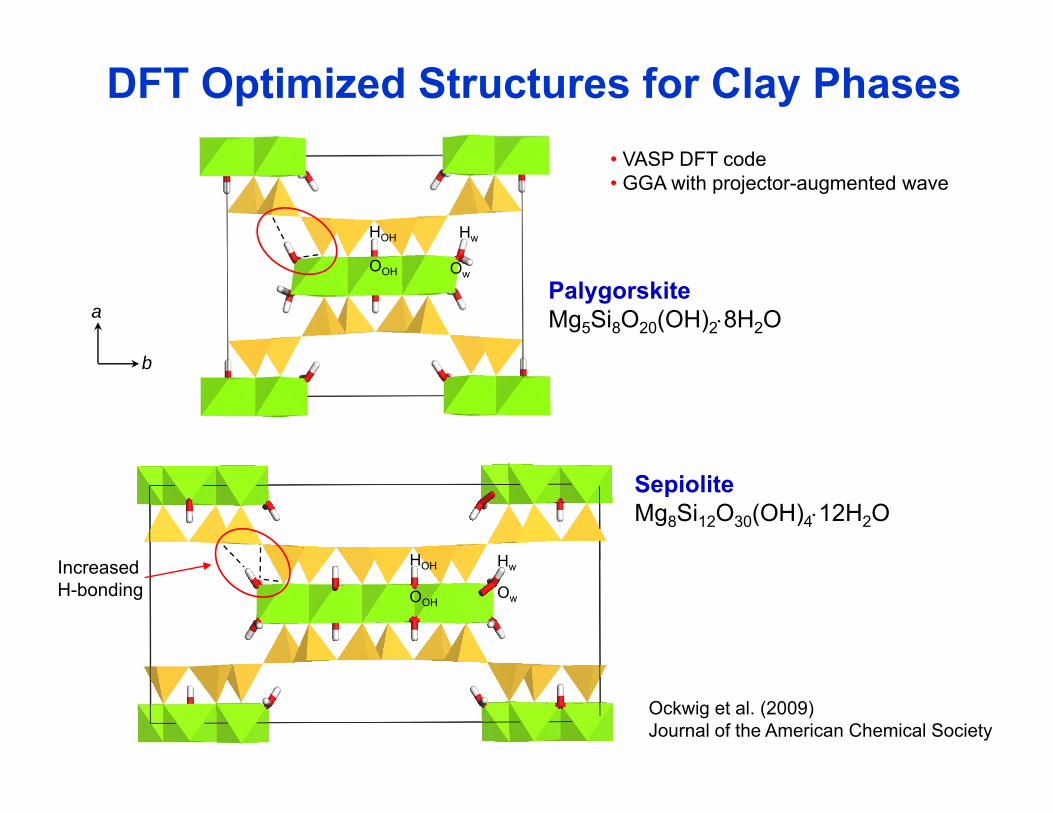
DFT Optimized Structures for Clay Phases
HwHOH
• VASP DFT code • GGA with projector-augmented wave
a
OwOOH
PalygorskiteMg5Si8O20(OH)2⋅8H2O
b
SepioliteMg8Si12O30(OH)4⋅12H2O
HHI d Hw
Ow
HOH
OOH
IncreasedH-bonding
Ockwig et al. (2009)Journal of the American Chemical Society
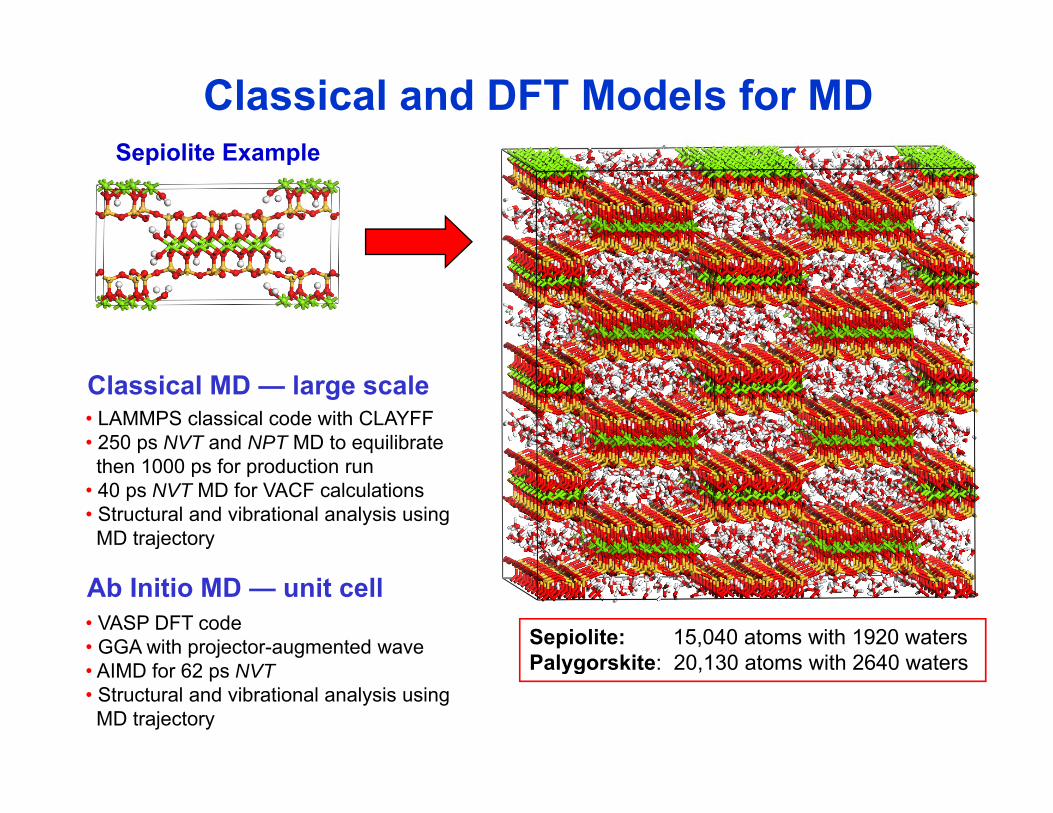
Classical and DFT Models for MDSepiolite ExampleSepiolite Example
• LAMMPS classical code with CLAYFF • 250 ps NVT and NPT MD to equilibrate
Classical MD — large scale
then 1000 ps for production run• 40 ps NVT MD for VACF calculations• Structural and vibrational analysis usingMD trajectory
Sepiolite: 15,040 atoms with 1920 watersPalygorskite: 20 130 atoms with 2640 waters
• VASP DFT code • GGA with projector-augmented wave
f
Ab Initio MD — unit cell
Palygorskite: 20,130 atoms with 2640 waters • AIMD for 62 ps NVT• Structural and vibrational analysis usingMD trajectory
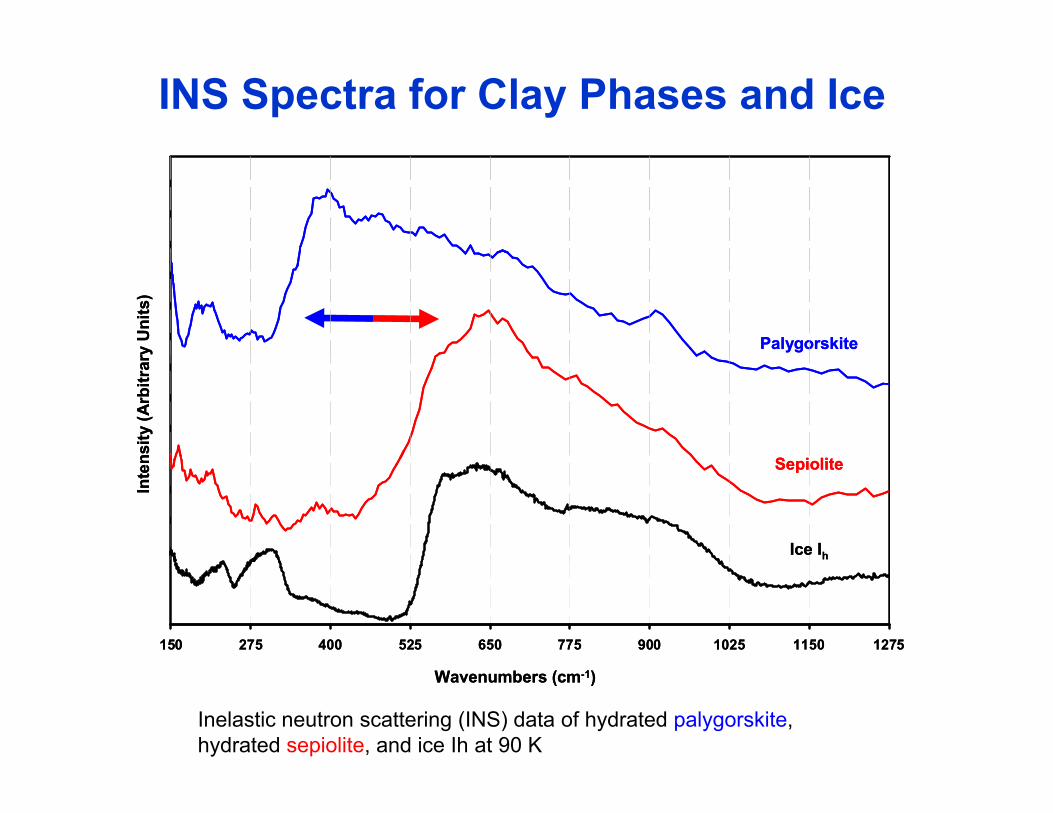
INS Spectra for Clay Phases and Icery
Uni
ts)
Palygorskite
ry U
nits
)
Palygorskite
nsity
(Arb
itrar
S i litnsity
(Arb
itrar
S i lit
Inte
n Sepiolite
Ice Ih
Inte
n Sepiolite
Ice Ih
150 275 400 525 650 775 900 1025 1150 1275150 275 400 525 650 775 900 1025 1150 1275
Wavenumbers (cm-1)Wavenumbers (cm-1)
Inelastic neutron scattering (INS) data of hydrated palygorskite, hydrated sepiolite, and ice Ih at 90 K

LAMMPS SoftwareLarge-scale Atomic/Molecular Massively Parallel Simulator
LAMMPS is a classical molecular dynamics code that models an ensemble of particles in a liquid, solid, or gaseous state. It can model atomic, polymeric, biological, metallic, granular, and coarse-grained systems using a variety of force fields and boundary conditions. LAMMPS can efficiently model up to millions or billions of particles (atoms).
• Runs on a single processor or in parallel• Runs on a single processor or in parallel • Distributed-memory message-passing parallelism (MPI) • Spatial-decomposition of simulation domain for parallelism • Open-source distribution • Highly portable C++ • Optional libraries used: MPI and single-processor FFT • Easy to extend with new features and functionality • Runs from an input scriptRuns from an input script • Syntax for defining and using variables and formulas • Syntax for looping over runs and breaking out of loops • Run one or multiple simulations simultaneously (in parallel) from one script
Steve Plimpton, Sandia National Laboratorieslammps.sandia.gov

Computational Resources
Sandia Geochemistry Facility Sandia ThunderbirdSandia Geochemistry Facility• 42-node AMD and Apple clusters with 100 processors
• More than 53 Teraflops• 4,480 compute nodes • Soon to be decommissioned
Sandia Red Sky• 169 Teraflops• 960 node with 7 680 cores NM Encanto960 node with 7,680 cores• Scalable and extensible design• On-line later this year
• 172 Teraflops• 1,792 nodes with 14,336 processors• New Mexico Computing Alliance Center
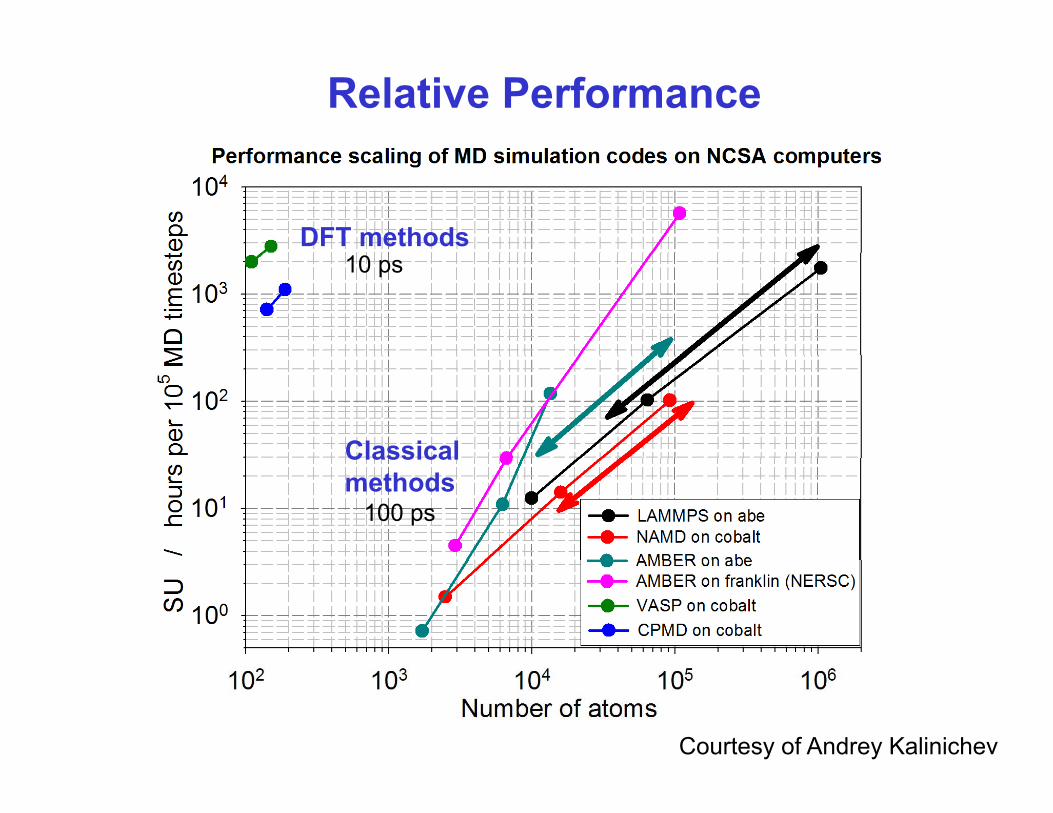
Relative Performance
DFT methods10 ps
ClassicalClassicalmethods
100 ps
Courtesy of Andrey Kalinichev

What’s Needed for Chemical Accuracy?GOAL: Develop computational approaches that are highly accurate
Predict equilibrium chemistry: SelectivityChange in Keq @ 298 K
p p pp g yfor the right system. Get the right answer for the right reason.
Change in Keq @ 298 KKeq = 1 50:50 ∆G = 0 kcal/molKeq = 10 90:10 ∆G = 1.4 kcal/molKeq = 100 99:1 ∆G = 2.8 kcal/mol
Predict accurate rates: ReactivityAbsolute rates @ 298 KFactor of 10 in rate @ 25oC is a change in Ea of 1.4 kcal/mol@ g a
Do this in a complex system where the model represents the system accurately under the relevant conditions
Complexity examples for system size:• 100x100x100 nm box of water molecules would have 4 x 105 H2O molecules• Neutral pH requires 107 H2O molecules per H+/OH- pair• Minimum number of atoms in a molecular dynamics trajectory study will be 105 to 106 atoms for microseconds (10-6 s) with femtosecond (10-15 s) time steps.
Courtesy of David Dixon
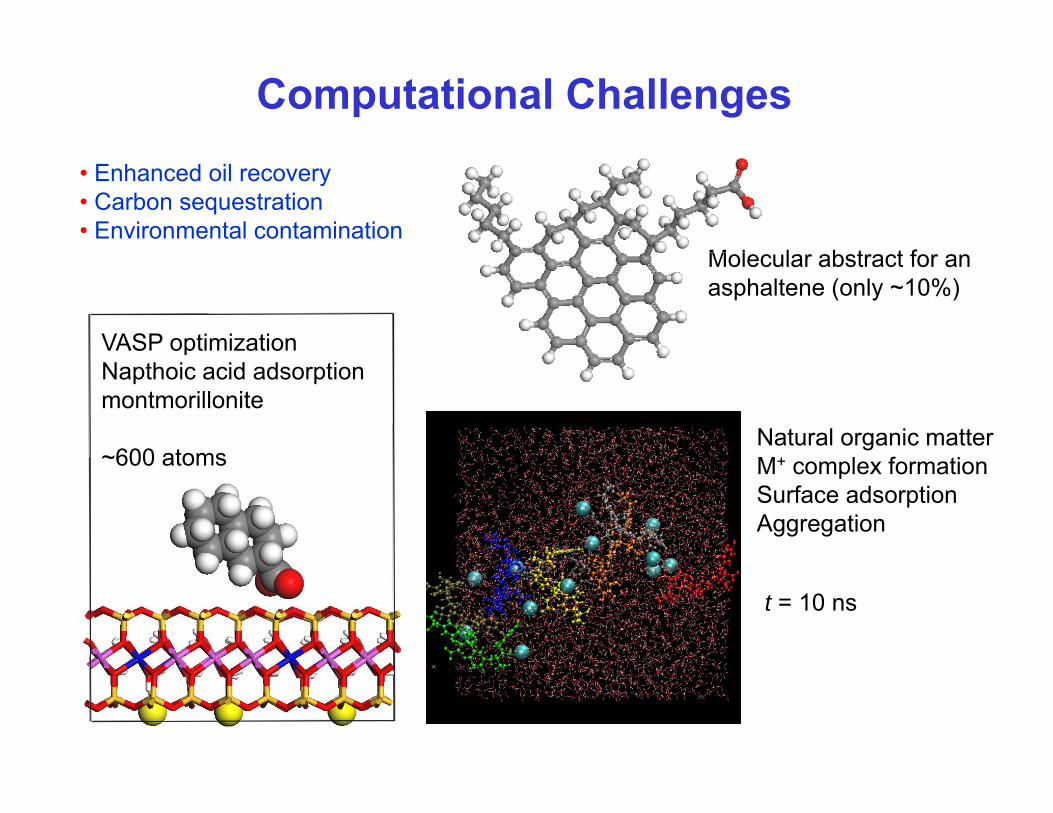
Computational Challenges• Enhanced oil recovery• Carbon sequestration• Environmental contamination
M l l b t t f
VASP optimization
Molecular abstract for anasphaltene (only ~10%)
Napthoic acid adsorptionmontmorillonite
~600 atomsNatural organic matterM+ l f ti~600 atoms M+ complex formationSurface adsorptionAggregation
t = 10 ns
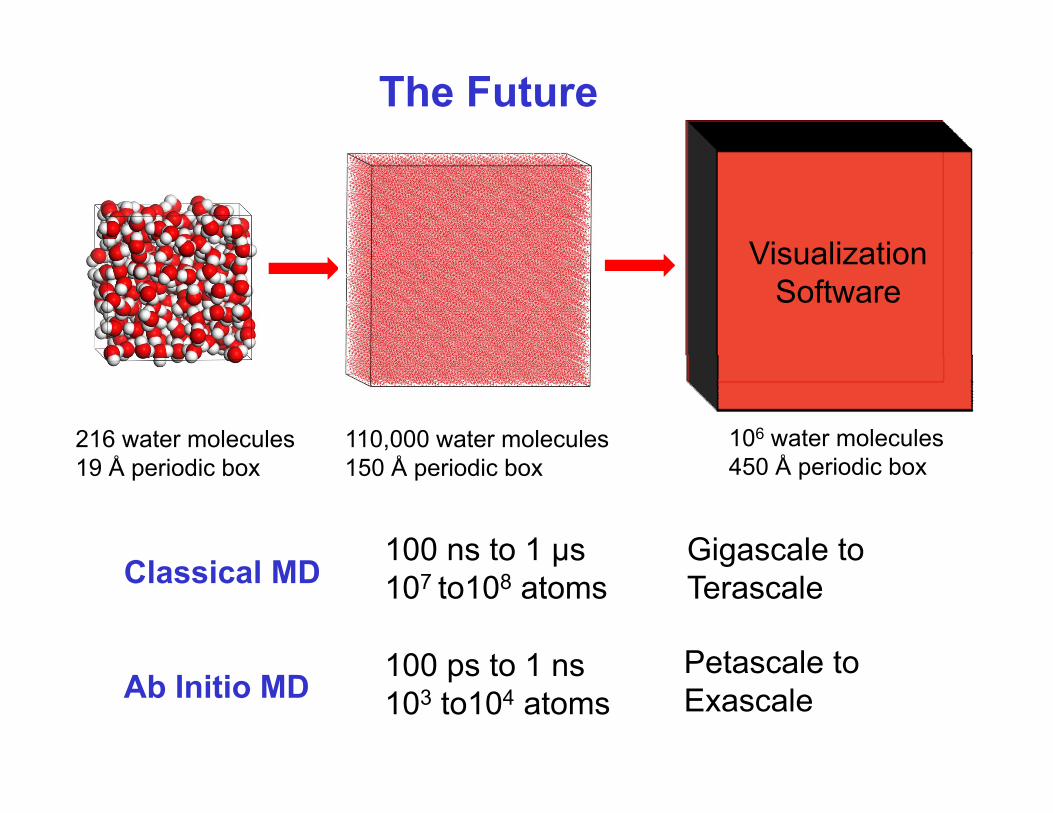
The Future
VisualizationVisualizationSoftware
216 water moleculesÅ
110,000 water moleculesÅ
106 water molecules450 Å i di b
100 ns to 1 µs
19 Å periodic box 150 Å periodic box 450 Å periodic box
Cl i l MDGigascale to 00 s o µs
107 to108 atomsClassical MD
100 ps to 1 ns
G gasca e oTerascale
Petascale toAb Initio MD 100 ps to 1 ns
103 to104 atomsPetascale to Exascale









![Mineral and Petroleum Resources Development Bill [B15B-2002] · and future generations, to ensure ecologically sustainable development of mineral and ... Legal nature of prospecting](https://img.pdfslide.net/doc/110x75/5fcfb5253a172f2534356ae2/mineral-and-petroleum-resources-development-bill-b15b-2002-and-future-generations.jpg)



















