Embed Size (px)
Citation preview

THE ROLE OF BMP-6 IN THE
PATHOGENESIS OF COPD
Wannes Van Hooste Stamnummer: 01207894
Promotor: prof. dr. Ken Bracke
Copromotor: dr. Fien Verhamme
Masterproef voorgelegd in het kader tot het behalen van de graad Master of Medicine in de Geneeskunde
Academiejaar: 2017-2018

Deze pagina is niet beschikbaar omdat ze persoonsgegevens bevat.Universiteitsbibliotheek Gent, 2021.
This page is not available because it contains personal information.Ghent University, Library, 2021.

I
Table of Contents
Samenvatting ............................................................................................................................. 1
Abstract ...................................................................................................................................... 1
1. Introduction ......................................................................................................................... 2
1.1 COPD........................................................................................................................... 2
1.1.1 Definition and burden of disease ........................................................................... 2
1.1.2 Risk Factors .......................................................................................................... 2
1.1.3 Pathophysiology .................................................................................................... 3
1.1.4 Signs, symptoms ................................................................................................... 5
1.1.5 Diagnosis .............................................................................................................. 6
1.1.6 Treatment .............................................................................................................. 7
1.1.7 Comorbidities ...................................................................................................... 10
1.1.8 Mechanism of disease ......................................................................................... 10
1.2 BMP-6 ........................................................................................................................ 12
1.2.1 The TGF-β superfamily ....................................................................................... 12
1.2.2 BMP-6 ................................................................................................................. 13
1.2.3 BAMBI ................................................................................................................. 15
2. Objectives ......................................................................................................................... 15
3. Methods ............................................................................................................................ 16
3.1 Human lung tissue...................................................................................................... 16
3.2 Emphysema quantification ......................................................................................... 17
3.3 Murine model of COPD .............................................................................................. 18
3.4 Preparation of lung homogenate ................................................................................ 19
3.5 RNA extraction and RT-qPCR .................................................................................... 20
3.6 Immunohistochemistry ............................................................................................... 21
3.7 Western Blot............................................................................................................... 21
3.8 Statistical analysis ...................................................................................................... 22
4 Results .............................................................................................................................. 23
4.1 Patients with COPD .................................................................................................... 23
4.1.1 Emphysema quantification .................................................................................. 23
4.1.2 RT-qPCR for BAMBI ........................................................................................... 23
4.1.3 RT-qPCR for BMP-6 ............................................................................................ 24
4.1.4 Correlations ......................................................................................................... 25
4.2 Murine model of COPD .............................................................................................. 26

II
4.2.1 RT-qPCR for BAMBI ........................................................................................... 26
4.2.2 RT-qPCR for BMP-6 ............................................................................................ 27
4.2.3 IHC-P .................................................................................................................. 28
4.2.4 Western Blot ....................................................................................................... 32
5 Discussion ......................................................................................................................... 35
5.1 COPD patients ........................................................................................................... 35
5.1.1 BAMBI ................................................................................................................. 35
5.1.2 BMP-6 ................................................................................................................. 35
5.2 Murine model of COPD .............................................................................................. 36
5.2.1 BAMBI ................................................................................................................. 36
5.2.2 BMP-6 ................................................................................................................. 36
5.3 Conclusion ................................................................................................................. 38
6 Acknowledgements ........................................................................................................... 39
7 Bibliography ...................................................................................................................... 40

1
Samenvatting
Chronic Obstructive Pulmonary Disease (COPD) is wereldwijd één van de belangrijkste oorzaken
van mortaliteit en morbiditeit en was in 2010 de op drie na belangrijkste doodsoorzaak wereldwijd.
Het wordt gekenmerkt door een niet reversibele obstructie van de luchtwegen, alsook door
luchtweginflammatie en emfyseem of destructie van het longparenchym. Eén van de belangrijkste
oorzaken van COPD is roken. Totnutoe is er geen curatieve therapie voor COPD voorhanden.
Daarnaast is de pathogenese van COPD onvolledig opgehelderd en is er nood aan beter begrip
van de betrokken mechanismen, hetgeen op zich aanleiding kan geven tot de ontwikkeling van
curatieve therapieën. In deze thesis onderzochten we twee leden van de TGF-β superfamilie,
namelijk BMP-6 en BAMBI omdat een Genome-Wide Association Study BMP-6 correleerde aan
Forced Vital Capacity (FVC). BMP-6 is vooral gekend als regulator van hepcidine.
Gebruikmakende van een muismodel voor COPD alsook van humane longweefselstalen
onderzochten we de mRNA expressie en lokalisatie en proteïne expressie van BMP-6 door middel
van respectivelijk RT-qPCR, immunohistochemie en western blot. We vonden een verlaagde
expressie van BMP-6 mRNA in longweefsel van humane COPD-patiënten en aan rook
blootgestelde muizen. De BMP-6 mRNA expressie correleerde significant met de Forced
Expiratory Volume in 1 second (FEV1), de Tiffeneau index (FEV1/FVC) en het aantal pakjaren.
BMP and activin membrane-bound inhibitor (BAMBI) is een pseudoreceptor die leden van de TGF-
β superfamily uit de circulatie haalt door ze als receptor te binden zonder een signaalfunctie te
hebben. De mRNA expressie van BAMBI werd onderzocht in longweefsel van aan rook
blootgestelde muizen en patiënten met COPD. Terwijl de mRNA expressie van BAMBI niet
verschilde in patiënten met en zonder COPD, was de BAMBI mRNA expressie in aan rook
blootgestelde muizen significant lager in vergelijking met gezonde muizen.
Abstract
Chronic Obstructive Pulmonary Disease is characterized by a not fully reversible airflow
limitation caused by obstruction of the small airways and emphysema. The main cause for
COPD is the smoking of tobacco. It is one of the most prominent worldwide causes of morbidity
and mortality. Up to today, there is no cure and the only action one can to take that influences
the long-term lung function decay is smoking cessation. The pathogenesis of COPD hasn’t been
entirely elucidated and only partial explanations and mechanisms have been proposed so far.
BMP-6 is a member of the TGF-β superfamily and it is best known as an inducer of hepcidin. A
Genome-Wide Association Study (GWAS) found BMP-6 to correlate to Forced Vital Capacity
(FVC) and as such became worthwhile to investigate as possible partaker in the pathogenesis of
COPD. Using both a mouse model of COPD and human lung tissue of COPD patients, we found
BMP-6 mRNA levels to be decreased in the lungs of COPD patients and smoke-exposed mice.
The BMP-6 mRNA levels correlated significantly with packyears, FEV1 and FVC. We also
investigated the location using immunohistochemistry and protein levels using western blot.
BMP and activin membrane-bound inhibitor (BAMBI) is a pseudoreceptor that binds members of
the TGF-β superfamily and thus takes it out of circulation. We investigated the BAMBI mRNA
levels in both human lung tissue and a mouse model of COPD and saw BAMBI to be
significantly decreased in the murine COPD model.

2
1. Introduction
1.1 COPD
1.1.1 Definition and burden of disease
Chronic Obstructive Pulmonary Disease (COPD) is a heterogeneous(1, 2) disease characterized
by a not fully reversible obstruction of the small airways and by emphysema which is destruction
of the lung parenchyma(3). In 2010, COPD was the fourth leading global cause of death(4) and is
estimated to rise to the third place by 2030(5). It is estimated that 384 million people had COPD
in 2010, with a global prevalence of 11,7%(6). These numbers may underestimate the burden of
COPD, as COPD often is not a primary cause of death but rather a strong contributing factor and
as such it is often excluded from the death certificate. Altogether, COPD amounts to a significant
mortality, morbidity and economic burden.
1.1.2 Risk Factors
There are several known risk factors for COPD, smoking tobacco – passive smoking included -
being the most important one(7). Other risk factors include indoor burning of biomass fuels, air
pollution, age and multiple childhood airway infections(8). Because of augmenting numbers of
daily smokers as well as the occurrence of indoor pollution due to burning biomass fuels in badly
ventilated houses, low-and middle-income countries are at risk of seeing their COPD patients
increased should this trend persist. In most high-income countries however, the prevalence of
daily cigarette smoking is estimated to decline by 2025(9).
It has been estimated that about 15-25% percent of the smokers develop COPD(10, 11). Although
there is a consensus about this percentage, some propose that most smokers will exhibit
phenomenon attributable to COPD if they only have smoked enough and long enough. While
smoking is the most important reason for an accelerated annual decline in lung function, other
factors also play a role(12). A genetic factor is thought to play a key role in individual susceptibility
since it has been observed that people suffering from severe α1-antitrypsin deficiency (AAT)
develop early-onset emphysema, especially in synergy with smoking. AAT however only counts
for 2-3% of the total COPD cases. Other genes associated with a faster decline of FEV1 (Forced

3
Expiratory Volume 1 second; this is the maximum amount of air a subject can breathe out in 1
second after maximum inhalation in a forced attempt. This is an important diagnostic parameter
for COPD; see 1.1.5) have also been proposed, as well as other host factors such as bronchial
hyperreactivity. Smokers exhibiting these traits will have a higher tendency towards developing
COPD as these traits work synergistic with the smoking(12, 13).
1.1.3 Pathophysiology
There is a decent understanding of the disease process that explains the clinical features of
COPD. All smokers have some level of airway inflammation, but COPD patients have an
exaggerated inflammatory response to toxic particles resulting in some of the key findings in
COPD. Those are emphysema, airflow limitation, gas trapping, mucus hypersecretion, pulmonary
hypertension and exacerbations.
An important feature is emphysema. Emphysema is the destruction of the lung parenchyma,
where alveolar walls and capillaries are destroyed. Destruction of alveolar walls makes the volume
of alveolar spaces larger compared to the area of the walls where gas exchange can take place.
As such it was found that the extent of emphysema correlates with diffusion capacity of the lungs
for CO (DLCO), a marker of gas exchange(14). Emphysematous destruction of lung parenchyma
is seen as a part of lung remodelling, a process in COPD where the overall architecture of the
lungs is altered as the disease progresses.
Chronic bronchitis is defined as having a chronic productive cough for at least 3 months per year
for two consecutive years. In the Rotterdam study - a population-based cohort study of subjects
aged 45 or more - it was found that COPD patients with chronic bronchitis have an increased risk
of exacerbations and respiratory mortality compared to subjects without chronic productive
cough(15). A result of chronic bronchitis is mucous overproduction due to goblet cell hyperplasia,
squamous metaplasia and an increased size of bronchial submucosal glands. In normal lungs,
ciliary movement functions as a transporter belt to clear mucous out of the lungs, which is then
coughed up and subsequently swallowed to be degraded in the stomach. This constant upwards
stream functions as a cleansing wave through the airway, expectorating pathogens and toxic
particles. Due to the overproduction of mucous in COPD as well as due to ciliary dysfunction, this
mechanism is compromised. This results in the occurrence of a chronic productive cough(16).
All these factors contribute to one of the key findings in COPD: airflow limitation due to small
airway remodelling. Airway remodelling occurs when architectural changes because of

4
proliferation of certain cells, destruction of tissue and deposition of for instance collagen or
fibronectin. The lumen of the small conducting airways is obstructed so patients experience
dyspnoea and have to put more labour into breathing. Because of the difficult exhaling, residual
air tends to stay behind in the lungs distal of the obstruction so that when exhaling, the airway
proximal of the residue collapses. This mechanism leads to hyperinflation or air trapping. Having
troubles breathing due to airway obstruction also explains why the FEV1 is used as a diagnostic
marker. Decreased exercise capacity and shortness of breath in COPD patients are correlated to
the amount of residual air in the lungs(17). Figure 1 portrays a schematic comparison between
healthy airways (left) and COPD airways (right). As can be seen, the COPD airway is clogged due
to mucous, oedema and smooth muscle cell contraction.
Fig. 1: Schematic comparison between COPD airway (left) and healthy airway (right).
On the left, features of airflow limitation are seen such as excessive mucus, contracted
smooth muscle cells and goblet cell hyperplasia. This attributes to airway and airflow
obstruction.
Another aspect of the clinical features of COPD are exacerbations. Exacerbations are defined as
an acute worsening of respiratory symptoms, resulting in the need for additional therapy or
hospitalisation(6). Frequent exacerbations are related to a decline in quality of life(18), less time
spent outdoors resulting in growing sedentarism(19) as well as being a contributing factor to the
economic burden of COPD(20, 21). Exacerbations can be caused by a variety of triggers such as
infection or change in temperature and air quality (22).

5
To summarise COPD is a chronic disease where the lungs undergo a series of changes resulting
in chronic airway inflammation, excessive mucous production, architectural changes around the
small airways and parenchymateous destruction leading to a troubled gas exchange and the
trapping of air in the distal lungs, as well as dyspnoea and coughing(6).
1.1.4 Signs, symptoms
Early symptoms of COPD include chronic cough and progressive dyspnoea. Together with
excessive mucoid production, these symptoms may vary from day to day. They can precede the
airflow limitation by years, explaining why COPD can be easily overlooked(23). The chronic cough
is often regarded by the smoking patient as an expected consequence of smoking and is as such
only a reason to seek out a physician when it affects the quality of life(24). As a result of mucous
overproduction, the cough can be productive. A large Danish longitudinal study that followed a
random population sample (n=876) found respiratory symptoms (including cough and mucus
production) to be a predictor for later hospital admission for COPD. It also predicted treatment for
obstructive lung disease(25).
Dyspnoea or shortness of breath becomes a more important problem when the disease worsens.
In early disease stages, coughing is responsible for the most loss in quality of life. In the later
stages, cough becomes less important while shortness of breath causes the most lost in quality
of life(24). The impact varies from impeding the ability to perform physical exercise to shortness
of breath when at rest.
The feeling of chest tightness has been accredited to the unpleasant awareness of the breathing
muscles contracting. Due to hyperinflation and obstructed airways, COPD patients have to put
more labour into breathing efficiently. The feeling of chest tightness and dyspnoea can induce
anxiety(26).
In the later stages of the disease, pulmonary hypertension can arise. This is a heightened pressure
in the lung circulatory system, mainly due to hypoxic vasoconstriction. In lungs, well ventilated
areas are well supplied of arterial blood. This is in contrast to other tissue in the body, where areas
low on oxygen will receive better perfusion. Due to an overall bad ventilation, the majority of lung
vessels will become constricted, an increased resistance for blood flow is formed and this
increases the afterload for the right ventricle. When this phenomenon persists, the right ventricle
will become hypertrophic(27). The presence of cor pulmonale had a negative influence on overall

6
survival(28, 29). These studies however were conducted before oxygen therapy was used. Severe
cases of cor pulmonale are less common nowadays and are mostly because of factors not related
to hypoxia(27, 30).
1.1.5 Diagnosis
In the majority of cases, a primary care physician will think about the diagnosis when seeing a
patient at risk who has a chronic cough, which can be a first sign. Physicians who are attentive for
patients at risk can rely on case-finding and will reduce the underdiagnosis of early COPD, as well
as providing an extra incentive to talk about smoking cessation with the patient(31). Another sign
that may be occasionally observed is the flattening of the diaphragm on a regular Rx thorax that
may have been taken for another reason such as pneumonia.
Spirometry is necessary for the diagnosis of COPD. Two spirometry variables are required: FEV1
and FVC. FVC or Forced Vital Capacity is the maximum amount of air a person can force himself
to blow out after maximum inhalation. For both FEV1 and FVC the reference values are adjusted
to gender, weight and height of a person. An FEV1/FVC ratio (sometimes called Tiffeneau index)
less than 70% that doesn’t entirely reverse after the use of a bronchodilator is diagnostic for
COPD(3). The inhalation of a fast-acting sympathicomimeticum is important to differentiate
between asthma, where contrarily to COPD the airflow obstruction is reversible. After diagnosis,
patients can be further stratified into 4 COPD GOLD categories based upon the value of FEV1.
This is shown in figure 2.

7
GOLD classification FEV1/FVC FEV1 (% predicted)
GOLD I <70% ≥80
GOLD II <70% 50-80
GOLD III <70% 30-50
GOLD IV <70% <30
Fig. 2: GOLD classification of COPD stages: An FEV1/FVC (Tiffeneau) index less than
70% that doesn’t reverse after bronchodilator use is diagnostic for COPD. Further
classification is possible according to the value of FEV1 compared to the predicted reference
value(6).
It should be noted that COPD is often considered a heterogeneous disease. On CT scan, different
types of findings are reported such as predominantly emphysematous patients, dominant airway
wall thickening and air trapping dominance(2). The same goes for clinical symptoms, where
different patients may display different sets of symptoms and where the subjective loss of function
does not always correlate with the objective lung function as assessed with spirometry(32). As
such, it is important to perform both the diagnostic tests as well as to assess the severity of one’s
symptoms, as these do not always correlate with GOLD stage(33). There are several validated
questionnaires available to assess symptoms. For dyspnoea, the Medical Research Council
(MRC) dyspnoea scale exists(34). As the symptomatology goes beyond only dyspnoea, other
questionnaires such as the St. George’s Respiratory Questionnaire(35) and the COPD
Assessment Test (CAT)(36) were developed. As the St. George’s counts 76 questions, the CAT
proves more practical for use in daily practice.
1.1.6 Treatment
The most important measure in the treatment of COPD is smoking cessation. To date, it is the
only measure that influences the long-term decline in lung function(37, 38). Pharmacological
interventions can reduce the severity, frequency and impact of exacerbations as well as improve
exercise tolerance and quality of life. Figure 3 shows the treatment guidelines as recommended
by the GOLD initiative(6). In this figure, patients are stratified into 4 categories based upon COPD
GOLD stage, history of exacerbations and symptom assessment. The most basic pharmaceutical
intervention is by using a fast-acting bronchodilator. Fast is the keyword rather than short-or long-

8
acting. This reliever medication is used when the patient experiences difficulty with breathing. Note
that this fast-acting β-mimetic should be available for all groups as reliever medication meaning
the patient can use it when needed. As symptoms worsen or the reliever medication proves to be
insufficient, a Long-Acting Muscarinic Antagonist (LAMA) or Long-Acting β-Agonist (LABA) can
be used. These products have to be taken once or twice daily. Recently, products with a 24-hour
duration of action have been developed. These ultra-LABA’s are more user-friendly and improve
compliance(39).
Patients stratified in group C may need a LABA and a LAMA, or a combination of LABA and ICS.
Combination of drugs proved to have better result on lung function and number of
exacerbations(39-41). For group D triple therapy (LAMA + LABA + ICS) could be an option, as
well as oxygen therapy in severe cases(42). Continuous macrolide antibiotic use has been proven
effective in reducing the number of exacerbations as well as improving the quality of life due to
immunomodulatory and anti-inflammatory effects(43). As mentioned, COPD can present itself in
different ways and the attending physician should always treat the patient and not the disease.
For group D, palliative measures should be considered.
All mentioned products but macrolides are preferably administered by inhalation. Inhalation
compared to systemic administration results in less systemic side-effects. Especially systemic
corticoids should be avoided or be kept short in time(44).

9
Treatment options in COPD C
OP
D G
OL
D s
tag
e
1
2
3
4
Group C
LAMA and LABA OR
LABA and ICS
Group D
LAMA and LABA OR
LABA and LAMA and ICS
Consider oxygen Consider macrolide
≥2 o
r ho
sp
italis
atio
n 1
0
Ex
acerb
atio
n H
isto
ry
Group A
Reliever bronchodilator (SABA)
Group B
LAMA or LABA
mMRC 0-1 CAT <10
mMRC 2+ CAT ≥10
Fig. 3 Treatment options in COPD. Based upon the GOLD 2017 report(6). Different
diagnostic and assessment tools can contribute towards making a choice in treatment
rather than only based upon the COPD GOLD stage. Left, there is the COPD GOLD
stage as determined by spirometry. Below is the patient’s score on the mMRC or CAT
scale, assessing subjective symptoms and dyspnoea. On the right is the exacerbation
history in the last year. One exacerbation leading to hospital admission counts as ≥2
exacerbations. It should be noted that while this figure acknowledges the limitations of
FEV1 as a parameter, this figure is merely a guideline and one should always treat the
patient, not the disease. Abbreviations: Long-Acting Muscarinic Antagonist (LAMA),
Long-Acting β agonist (LABA), Inhalation Corticosteroids (ICS), Short-Acting β agonist
(SABA).
Apart from the smoking cessation and pharmaceutic intervention, patient education is important,
resulting in more compliance. Crucial is the inhalation technique. A lot of mistakes are made and
face-to-face teaching proved to result in the best patient inhalator technique(45). Preventive
measures include pneumococcal and influenza vaccinations to reduce the number or severity of
exacerbations. For influenza vaccination there is evidence that it decreases the frequency of

10
exacerbations(46), for the pneumococcal vaccine there is less proof(47). Both vaccines are
recommended for all COPD patients(6).
In selected cases, surgical intervention by the means of a bullectomy may be opportune. A bulla
is a confluence of emphysematous lesions, forming a large air bulb in the lungs.
1.1.7 Comorbidities
Even when correcting for the smoking history, COPD patients are still at risk for a number of
diseases compared to healthy smokers. These include cardiovascular diseases(48, 49),
depression and anxiety(50), diabetes mellitus(51), osteoporosis (52) and possibly stroke(53). The
physician suspecting COPD in a patient should also assess the risk factors for both COPD and
comorbidities such as smoking status, inactivity and cardiovascular risk factors (54). As most
COPD patients have comorbidities and different clinical presentations, it is important to address
these and evolve to a personalised therapy(54, 55). This includes finding the therapy where both
objective lung function tests and patient satisfaction are maximised as well as working on
prevention of comorbidities.
1.1.8 Mechanism of disease
The classic triad explaining the pathogenesis of COPD consists of inflammation,
protease/antiprotease imbalance and oxidative stress(56). Genetics, autoimmunity(57) and
bacterial colonization(58) could also be implied.
Inflammation is triggered by noxious gas particles such as those in cigarette smoke. The
epithelium fulfils an important defensive role as it also secrets defensins to counter pathogens as
well as functioning as a physical barrier(59). Damage by cigarette smoke leads to necrosis and
the release of damage-associated molecular patterns (DAMPs) such as High Mobility Group Box
1 (HMGB1) (60, 61). These DAMPs bind to Pattern Recognition Receptors (PRRs), leading to the
activation and chemotaxis of the innate immune system by releasing pro-inflammatory
chemokines and cytokines such as IL-1β, IL-6 and IL-8(62). HMBG1 for example binds to TLR2,
TLR4 and RAGE(63) inducing IL-1β and the maturation of dendritic cells(64).
Cells belonging to the innate immune system are attracted to the site of injury, most importantly
neutrophils and macrophages. Main neutrophilic attractants are IL-8 as well as C5a, a member of

11
the complement system(65, 66). Monocytes and macrophages are stimulated by TNF-α, IL-1α
and IL-1β to produce IL-8(67). This leads to an accumulation of neutrophils, being a hallmark in
the genesis of COPD. Neutrophils aid in the clearance of infections, but are also potential harmful
to the lung. By releasing neutrophil proteases into the extracellular environment, neutrophils
degrade extracellular matrix components such as collagen and proteoglycans(68) and as such
attribute to emphysema(69). Neutrophil elastase is known to cleave epithelial cadherin,
compromising the epithelial monolayer integrity and cell-cell junctions(70). Collagenase-2 and
gelatinase B are also involved in breaking down the extracellular matrix(71). Neutrophilic
breakdown of collagen results in collagen fragments that act as a neutrophilic chemotaxant,
leading to chronicity and a self-sustaining cycle of neutrophilic inflammation(72).
Furthermore, the adaptive immune system is essential in the pathogenesis of COPD as well. While
asthma is regarded as a predominantly CD4+ T-cell driven disease, CD8+ T-cells are the main
lymphocyte population in COPD, as seen in CD8+ T-cell-deficient mice that did not develop
emphysema after long-term cigarette smoke exposure(73). In contrast, our research group found
that severe combined immunodeficient (SCID) mice did develop emphysema in response to
chronic cigarette smoke exposure, despite the lack of functional B-and T-cells(75). Systemic CD8+
T-cell count was found to correlate with GOLD disease stage(74). Antigen Presenting Cells
(APCs) such as Dendritic Cells (DCs) activate cells belonging to the adaptive immune system by
presenting them with antigens. CD4+ T-cells are also increased in the airways of COPD patients,
but not as much as CD8+ T-cells. Th17 cells attribute to inflammation by stimulating CD8 cells
and Th1 and Th2 cells, as well as providing a feedback loop upregulating itself via IL21(76).
A protease-antiprotease imbalance also fulfils a key role in COPD. This is nicely illustrated in AAT,
where neutrophil elastase that is unopposed by α1-antitrypsin leads to a net proteolytic activity
resulting in emphysematous lesions in the lung parenchyma(77). However, it has become clear
that the development of emphysema can’t be attributed to a single protease. While Matrix
Metalloproteinases (MMPs) and neutrophils take a prominent place, several proteases are at work
and seem to influence each other(78, 79).
Oxidative stress occurs when the antioxidant defence mechanisms fail or become overwhelmed
by oxidative agents (reactive oxygen species, ROS) (80). There are endogenous sources of ROS
such as ROS generated by mitochondrial respiration or ROS used in antimicrobial defence(81).
ROS are scavenged by antioxidants, neutralizing their potential harmful effect(82). Cigarette
smoke however is a source of ROS and smoking may tip the oxidative/antioxidative balance
favouring oxidative stress, thus leading to cell damage.

12
Inflammation, oxidative stress and protease/antiprotease imbalance are intertwined and influence
each other in their contribution to the genesis of COPD. Oxidative stress for example leads to cell
death thus leading to an increased inflammatory response. Proteolytic cleavage of collagen can
lead to molecules promoting inflammation. This may offer an explanation to why inflammation
persists even after smoking cessation.
Autoimmunity is also thought to be a contributing factor. The CD8+ T-cell repertoire in the lungs
of mice continued to expand oligoclonal, even after smoking cessation(83). Smoke components
may alter autologous proteins, creating neo-antigens. Another explanation might be the loss of
self-tolerance due to repetitive stimulation of the immune system by DAMPs. Elastin-specific
CD4+ T-cells were found, correlating with the total amount IFN-γ(84). Another study found that as
the disease worsens, natural killer cells (NK cells) are more drawn to exhibiting toxicity to
autologous structural lungs cells, suggesting autoimmunity is a late effect of COPD(85).
Tertiary lymphoid follicles arise in tissue that harbours a lot of antigens the body wants to clear.
This can be the case in sites of chronic infection or inflammation. Immunocompetent cells infiltrate
the tissue and organize themselves into a functional entity called a tertiary lymphoid follicle (TLO).
These TLOs have been found in lung tissue of severe COPD patients. The number and size of B-
cell follicles increase with CS exposure time and correlate with the enlargement of the alveolar air
space(86). Both protective as well as harmful properties have been attributed to these TLOs.
Protective because they help clearing infections, harmful because autoreactive immunoglobulins
were found, suggesting a role for autoimmunity and tissue destruction. As autoantibodies were
found to be absent in some studies, it was hypothesized they are only implicated in emphysema
predominant types(87). B-cell activating factor (BAFF) expression was found to be significantly
increased in COPD lungs. BAFF is a major activator of B-cells and plays a role in adaptive humoral
responses. Antagonising BAFF in long-term smoke-exposed mice resulted in an attenuated
inflammatory response and a lesser degree of alveolar destruction(88).
1.2 BMP-6
1.2.1 The TGF-β superfamily
The TGF-β superfamily consists of several highly conserved proteins including bone
morphogenetic proteins (BMPs), growth differentiation factors (GDFs), activins and transforming
growth factor β (TGF-β) itself. Even though the superfamily counts over 30 members, there are a
limited number of receptors. Each receptor complex consists of two type I and two type II elements,

13
with both components being Serine/Threonine kinases. Seven type I and five type II receptors are
known to exist in humans(89). While the role of TGF-β1 itself has been studied well in the context
of COPD, little is known about the other members of the TGF-β superfamily.
Each member of the TGF-β superfamily requires a set of type I and type II serine/threonine kinase
receptors. The receptor complex phosphorylates itself. Upon binding, the small mother against
decapentaplegic (Smad) is phosphorylated by the intracellular part of the receptor complex and
will oligomerise with the costimulatory Smad. The complex then translocates to the nucleus. An
inhibitory Smad works as inhibitor of the phosphorylated complex. There is an overlap between
the receptor complexes and Smads the different members of the TGF-β superfamily use.
1.2.2 BMP-6
The bone morphogenetic proteins are well-known for their role in the bone morphogenesis, having
osteoinductive properties. However, they fulfil regulatory roles in the development and
homeostasis of nearly every tissue(90).
As demonstrated in figure 4, upon binding of BMP-6 on the receptor complex consisting of two
type 1 and two type 2 receptors, Small mother against decapentaplegic 1/5/8 (Smad1/5/8) is
phosphorylated. These Smads 1, 5 or 8 are the R-Smads or receptor Smads and will be
phosphorylated by the type I receptor part. Smad4 is needed as a costimulatory molecule and will
oligomerise with the activated R-Smad. The phosphorylated Smad4-Smad1/5/8 complex then
translocates to the nucleus to regulate gene expression. There is an overlap between the different
type-1 and type-2 receptors BMP-6 can bind upon(91). Smad 6/7 functions as the inhibitory Smad.
They block the phosphorylation of the R-Smad by the receptor complex and promote ubiquitination
and degradation of the receptor complex and in doing so inhibit signalling(92).

14
BMP-6 plays a role in iron homeostasis, as BMP-6 levels were found to positively correlate with
hepcidin levels(93, 94). Increased hepcidin and lower levels of free iron are a defence mechanism
against bacterial infection. Bacterial infection is also thought to contribute to the pathogenesis of
COPD. Higher levels of iron were found in alveolar macrophages of COPD patients, correlating
with emphysema severity(95). This increased iron sequestration in macrophages could occur to
protect the lungs from iron-induced oxidative stress or as result from the bacterial ‘war on iron’. In
the same study, airflow limitation correlated with ferritin, ferritin receptor and transferrin in the
broncheoalveolar lavage fluid of COPD patients as compared to non-smokers and healthy
smokers. This suggests that iron dysregulation plays a role in the pathogenesis of COPD,
reserving a possible role for BMP-6.
There are other diseases where BMP-6 was found to play a role. Enhanced expression of BMP-6
was found to exert an antifibrotic effect, inhibiting hepatic fibrosis in non-alcoholic fatty liver
disease(96). Another display of the antifibrotic effect of BMP-6 is that BMP-6 knockout mice
displayed enhanced fibrosis after kidney injury(97). BMP-6 levels were also found to be
augmented in patients with chronic heart failure(98).
Fig. 4: BMP-6 signalling. Upon BMP-6
binding its receptor, Small Mother Against
Decapentaplegic 1, 5 or 8 (Smad 1/5/8)
will become phosphorylated with the
phosphor from the receptor complex.
Smad 4 functions as common mediator.
After complexing with Smad 4, the
phosphorylated Smad 4 – Smad 1/5/8
complex translocates to the nucleus.

15
As of late, BMP-6 was found in a genome-wide association study (GWAS) to be associated with
forced vital capacity (FVC)(99). Little is known about BMP-6 in relation to COPD. There is some
evidence suggesting BMP’s are involved in airway inflammation in asthma, where BMP-2, -4 and
-6 were found to be inducted and BMP-5 and -7 where downregulated in bronchial epithelial cells
during airway inflammation(100).
1.2.3 BAMBI
BMP and activin membrane-bound inhibitor (BAMBI) is a pseudoreceptor able to inhibit signalling
of TGF-β members. It is a highly reserved transmembrane glycoprotein that resembles type I TGF-
β receptors. However, it lacks a functional intracellular domain, taking TGF-β members out of
circulation by binding them(101). It was found that after infection with non-typeable Haemophilus
influenza, BAMBI was upregulated in COPD patients(102). It was also found that in in vitro
experiments on lung tissue, BAMBI protein levels on CD4+ T-cells and plasma mRNA levels were
elevated in COPD patients compared to healthy smokers and healthy non-smokers. The
enhanced BAMBI levels correlated positively with increased TGF-β1 levels and with an increased
Th17/Treg ratio(103). The hypothesis is that by binding TGF-β and thus taking it out of circulation,
BAMBI drives the lung tissue towards inflammation and compromises tissue repair. This again
supports the theory that BAMBI has a role in the inflammatory status in COPD patients, as Th17
secretes pro-inflammatory cytokines and Treg CD4+ cells secrete TGF-β.
2. Objectives
This study will focus on both BMP-6 and BAMBI. First, we will study the expression and location
of BMP-6 in healthy murine lung tissue and in patients with COPD. We will correlate the BMP-6
levels with lung function parameters. Secondly, we aim to confirm the findings in a cigarette
smoke-induced mouse model of COPD. A similar approach has been used for BAMBI

16
3. Methods
3.1 Human lung tissue
The lung tissue originates from patients who underwent a (partial) lung resection. In the majority
of the cases this is following the diagnosis of a solid lung tumour. In the more severe COPD cases
(COPD GOLD IV), tissue was obtained after lung transplantation. All patients were categorized
using the criteria supplied by the Global Initiative for Chronic Obstructive Lung Disease (GOLD).
All subjects provided their written informed consent according to protocols approved by the Ghent
University Hospital Ethical Committee. The pathologist harvested tissue as far as possible from
the tumoral lesion and without signs of pneumonia. Table 1 and table 2 show the characteristics
of the patient populations used for respectively the RT-qPCR for BAMBI and the RT-qPCR for
BMP-6.
Table 1: Characteristics of the human study population used for the RT-qPCR for BAMBI (n=92)
never-smokers healthy smokers COPD GOLD
II COPD GOLD IV
Number 18 26 34 14
Gender ratio (male/female) 6/12 # 19/7 # 31/3 # 8/6 #
Age (years) 65 (56-70) 63 (55-70) 66 (58-69) ç 56 (54-60)* ç ²
Current- / ex-smoker - 16/10 22/12 0/14
Smoking history (PY) 0 (0-0) 28 (15-45)* 45 (40-60)* ç 30 (25-30)*²
FEV1 post (L) 2,7 (2,3-3,2) 2,7 (2,3-3,3) 2,0 (1,8-2,4)* ç 0,7 (0,7-0,9)* ç ²
FEV1 post (% predicted) 102 (92-116) 95 (93-112) 68 (61-75)* ç 26 (20-32)* ç ²
FEV1 / FVC post (%) 78 (75-83) 75 (71-79)* 56 (53-60)* ç 32 (27-35)* ç ²
DLCO (% predicted) 90 (80-105) 80 (61-102) 67 (51-87)* 35 (33-41)* ç ²
KCO (% predicted) 103 (88-123) 91 (68-107)* 87 (62-108)* 59 (50-65)* ç ²
ICS (yes/no) 0/18 # 1/25 # 15/19 # 13/1 #

17
Table 2: Characteristics of the human study population used for the RT-qPCR for BMP-6 (n=84)
never-smokers healthy mokers
COPD II COPD IV
Number 16 24 30 14
Age (years) 64 (51-71) 65 (55-71) 65 (59-69) 56 (54-60) * ç ¶
Gender (m/f) 3/13# 19/5# 29/1# 8/6#
Current-smoker/Ex-smoker NA 12/12# 17/13# 0/14#
Pack-years NA 33 (15-50) † 45 (40-60) † ç 30 (25-30) †¶
FEV1 (% predicted) 110 (92-118) 96 (92-113) 69 (64-74) †§ 23 (20-33) †§¶
FEV1/FVC (%) 78 (75-83) 76 (73-78) 56 (53-60) †§ 32 (27-35) †§¶
DLCO (% predicted) 88 (107-82) 85 (105-74) 73 (87-54) 35 (41-33) ²†§
KCO (% predicted) 95 (116-86) 95 (104-81) 94 (108-69) 56 (65-50) ²† ç
ICS (yes/no) 0/16# 1/23# 13/17# 13/1#
Table legend:
Smoking history expressed as Packyears (PY). This is the equivalent of smoking 1 pack of cigarettes a day
during a year. FEV1 post (L) is the Forced Expiratory Volume in 1 second post bronchodilator, expressed in
liter. FEV1 post (%predicted) is the FEV1 post bronchodilator expressed as percentage reached as
compared to the age-, weight- and gender adjusted reference value. FEV1/FVC post % or Tiffeneau. FVC is
the Forced Vital Capacity or maximum volume one can exhale after bronchodilation and maximum
inhalation. Dlco (% predicted) or Diffusing Lung Capacity for CO is a measure for gas exchange between
lung and blood, expressed as percentage reached of one’s reference value. Kco (% predicted) or CO
transfer coefficient is a measure of the efficiency of alveolar transfer, expressed as percentage reached of
one’s reference value. ICS (yes/no) refers to whether a patient takes Inhalation Corticosteroids.
Indices:
#: Fisher’s exact test, P < 0.001
*: Mann-Whitney-U test, P < 0.05 versus never smokers ç: Mann-Whitney-U test, P < 0.05 versus smokers without COPD
²: Mann-Whitney-U test, P< 0.05 versus COPD GOLD II
†: Mann-Whitney-U test, P<0.001 versus never smokers
§: Mann-Whitney-U test, P<0.001 versus smokers without COPD ¶: Mann-Whitney-U test, P<0.001 versus COPD GOLD II
Data are presented as median (IQR)
3.2 Emphysema quantification
Preoperative CT-scans of patients were scored for their degree of emphysema. The extent of
emphysema was graded into five categories: 0%, 1-5%, 6-25%, 26-50% and >75%. The main
reason for this stratification is because of the heterogeneity of COPD patients. Apart from the

18
Tiffeneau index, different CT phenotypes have been described, all presenting with different
subsets of symptoms(2). Different components of COPD influence different parameters.
Emphysematous prominent patients may mostly have a low DLCO, while those with a lot of air
trapping may have the worst FEV1(104). For the scoring of the preoperative CT scans, a workshop
on visual evaluation of COPD was used as guidelines(105). They compared the visual assessment
with the objective quantitative CT scoring. Figure 5 shows two of the reference images they used.
It was found that the visual grading of the extent of emphysema correlates well to the software-
driven quantitative CT measurements(106).
1-5% emphysema 6-25% emphysema
Figure 5: two of the reference images used to visually grade the extent of emphysema. Note
the emphysematous lesions seen as black attenuated zones on both pictures. Taken from
Lynch et al.(105). The red arrows indicate two of the many emphysematous lesions.
3.3 Murine model of COPD
Animal models function as a link between in vitro studies and studies in humans. They allow the
research of in vivo mechanisms in an ethically acceptable way. The murine genome has been
entirely sequenced and offers the possibility of altering their genetic constitution, allowing for a
thorough research of gene and gene function. While being good as an animal model and having

19
many commercial scientific supplies available, it should be remarked that every model has its
drawbacks and is never totally representative for real-life human systems.
Male BMP-6 knockout mice (C57BL/6Jx129Sv) were mated with C57BL/6J female mice. The
offspring was genotyped(107) and male KO and WT offspring were used. Jackson Laboratory
(Bar Harbor, ME, USA) provided the C57BL/6J mice. All mice had access to ad libitum food and
chlorinated tap water. They were housed in sterilised cages in an artificial 12 hour diurnal cycle.
All in vivo manipulations where approved by the local Ethics Committee for animal experimentation
of the Faculty of Medicine and Health Sciences of Ghent University.
The mice are placed in a smoking chamber connected to a smoking apparatus where they are
exposed to mainstream CS of 5 simultaneously lit cigarettes at a rate of 4 times per day for 5 days
per week with 30 minute smoke-free intervals between the smoking sessions(108). The used
cigarettes are standardised (Kentucky Reference Cigarette 3R4F without filter; University of
Kentucky, Lexington, KY, USA). An optimal smoke/air ratio of 1/6 is used. Control mice were
subjected only to air. The duration of exposure was 4 weeks (subacute) or 24 weeks (chronic).
Mice exposed for 4 weeks showed pulmonary inflammation, whereas chronic exposure resulted
in mice exhibiting pulmonary inflammation and hallmarks of COPD such as emphysema,
peribronchial lymphoid follicles and airway remodelling(75, 108).
After sacrificing the mice by means of an intraperitoneal overdose pentobarbital, lung tissue was
collected for studying. For the lung homogenate, the middle lobe of the right lung is used for
homogenisation. The tissue is snap-frozen using liquid nitrogen to prevent water from forming
crystals when freezing and damaging the sample. It was then stored at -80°C until further analysis.
The small lobe of the right lung is frozen in RNA-later (Qiagen). The left lobe is used for
immunohistochemistry.
3.4 Preparation of lung homogenate
For homogenisation of lungs, the tissue samples were transferred to tubes containing 1 mL RIPA
buffer (Cell Signalling Technology, Danvers, USA) which also contained Halt™ Protease Inhibitor
Cocktail Kit (Thermo Scientific, Waltham, MA, USA) to inhibit endogenous protease activity.
TissueRuptor (Qiagen, Hilden, Germany) was used for tissue disruption and homogenisation.
Samples were next sonicated for enhanced detection of nuclear proteins (4 times for 5 seconds)
and centrifuged (14000 rpm for 10 minutes at 4°C). Subsequently the middle layer was transferred

20
to microcentrifuge tubes. All steps were performed on ice. Total protein concentration was
measured using the Pierce™ BCA Protein Assay Kit (Thermo Scientific). This is an assay to
measure the protein concentration compared to a standard.
3.5 RNA extraction and RT-qPCR
The miRNeasy Mini kit from Qiagen was used to extract RNA from both human and mice lung.
First, tissue stored in RNA-later was added to QIAzol Lysis Reagent (Qiagen, Hilden, Germmany).
This is a phenol/guanidine thiocyanate used to facilitate lysis of tissue, to inhibit RNase and to
extract most of the DNA and proteins. Next, the TissueRuptor (Qiagen, Hilden, Germany) was
used to homogenise the samples for 30 seconds each. After 5 minutes incubation to dissociate
nucleoprotein complexes, chloroform was added. The samples underwent centrifugation at
12000g for 15 minutes at 4°C. This separates the homogenate into aqueous and organic phases.
The upper aqueous phase holds the RNA while DNA partitions to the middle phase and proteins
are in the organic lower phase or interphase. The upper phase is extracted and brought on the
RNeasy Mini spin column along with 100% ethanol. Here, RNA binds to the column. After 15s
centrifugation at 8000g, the contaminants are washed away and thrown away. RWT and RPE
buffers are used to further wash the column, with 15s centrifugation between each step. In the
end, RNA is eluted with RNase-free water. cDNA is obtained using the Transcriptor Universal
cDNA Master Kit (Roche, Basel, Switzerland). Reverse transcriptase is used to create
complemental DNA from the isolated mRNA. The solutions include the necessary nucleotides to
build in, a buffer and random hexamer primers.
To investigate the RNA levels, RT-PCR for BAMBI and BMP-6 on human and murine samples
was performed. Samples from different subjects were used including never smokers, healthy (ex-
)smokers, COPD GOLD II patient and COPD GOLD IV patient (see table 1 and table 2).
Housekeeping genes GAPDH, HPRT1 and SDH were used as loading control on human samples,
GAPHD, HRPRT1 and TFRC were used on the murine samples.
The expression of BAMBI and BMP-6 relative to the three mentioned household genes is
measured using TaqMan Gene Expression assays which includes specific primers and
fluoregenic mix (Applied Biosystems, Halle, Belgium). First there is a 10 minutes denaturation
phase at 95°C, followed by 50 cycles of 10 seconds at 95°C and 15 seconds at 60°C. Serial
dilutions of a mixture of all samples are used to create a standard curve. The cycles are performed
by a Lightcycler 96 detection system (Roche).
21
3.6 Immunohistochemistry
The localisation of BMP-6 in lung tissue was studied using immunohistochemistry on paraffin (IHC-
P). First, paraffin-embedded sections mouse lungs were dewaxed using xylene followed by
rehydrations. Next, antigen retrieval was performed using a citrate buffer. After blocking for
endogenous peroxidase activity because HRP conjugated antibodies were used, an additional
block (1109616001 Roche Blocking Agent, Sigma-Aldrich) was applied. Next, the specimens were
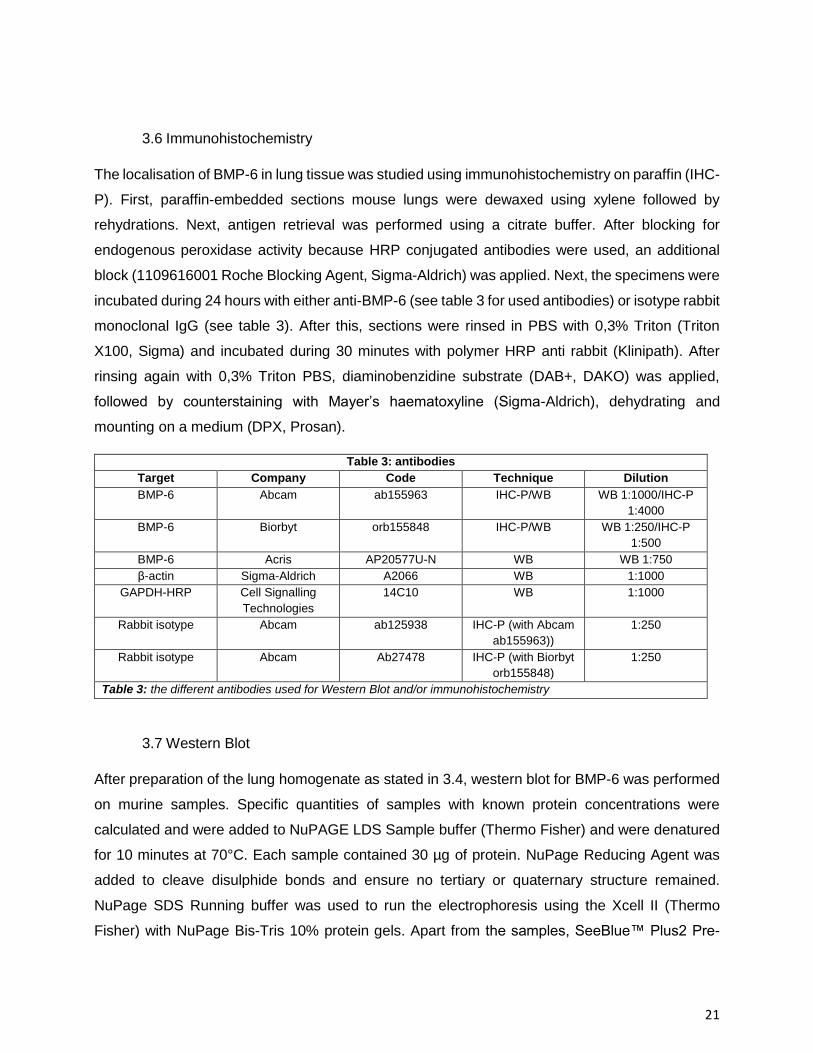
incubated during 24 hours with either anti-BMP-6 (see table 3 for used antibodies) or isotype rabbit
monoclonal IgG (see table 3). After this, sections were rinsed in PBS with 0,3% Triton (Triton
X100, Sigma) and incubated during 30 minutes with polymer HRP anti rabbit (Klinipath). After
rinsing again with 0,3% Triton PBS, diaminobenzidine substrate (DAB+, DAKO) was applied,
followed by counterstaining with Mayer’s haematoxyline (Sigma-Aldrich), dehydrating and
mounting on a medium (DPX, Prosan).
Table 3: antibodies
Target Company Code Technique Dilution
BMP-6 Abcam ab155963 IHC-P/WB WB 1:1000/IHC-P
1:4000
BMP-6 Biorbyt orb155848 IHC-P/WB WB 1:250/IHC-P
1:500
BMP-6 Acris AP20577U-N WB WB 1:750
β-actin Sigma-Aldrich A2066 WB 1:1000
GAPDH-HRP Cell Signalling
Technologies
14C10 WB 1:1000
Rabbit isotype Abcam ab125938 IHC-P (with Abcam
ab155963))
1:250
Rabbit isotype Abcam Ab27478 IHC-P (with Biorbyt
orb155848)
1:250
Table 3: the different antibodies used for Western Blot and/or immunohistochemistry
3.7 Western Blot
After preparation of the lung homogenate as stated in 3.4, western blot for BMP-6 was performed
on murine samples. Specific quantities of samples with known protein concentrations were
calculated and were added to NuPAGE LDS Sample buffer (Thermo Fisher) and were denatured
for 10 minutes at 70°C. Each sample contained 30 µg of protein. NuPage Reducing Agent was
added to cleave disulphide bonds and ensure no tertiary or quaternary structure remained.
NuPage SDS Running buffer was used to run the electrophoresis using the Xcell II (Thermo
Fisher) with NuPage Bis-Tris 10% protein gels. Apart from the samples, SeeBlue™ Plus2 Pre-

22
Stained Protein Standard was loaded into a lane as molecular weight reference. During blotting,
proteins were transferred to PVDF membrane (Novex PVDF Membrane Filter Paper Sandwich
0,2 µm pore size) using the same Xcell II. NuPage Transfer Buffer with NuPage Antioxidant was
used to run the blotting and keep proteins in a reduced state.
For immunodetection, the WesternBreeze™ Chemiluminescent kit (Thermo Fisher) was used. A
blocking solution was used to reduce nonspecific binding and background noise
(Westernbreeze™ Blocker/Diluent A and B). Membranes were incubated with the primary
antibody overnight (see table 3 for used antibodies). After primary antibody incubation, wash steps
(Westernbreeze™ Wash Solution) were performed. Next, the membranes were incubated with
the secondary anti-rabbit antibody for half an hour followed by wash steps. After being treated with
a chemiluminescent substrate (alkaline phosphatase substrate) or HRP, results were visualised
using the Chemidoc system (Bio-Rad, Hercules, USA). Image Lab software was used to analyse
the blot pictures (Bio-Rad, Hercules, USA). The quantitative value of the lanes was normalised
against β-actin or GAPDH (see table 3) in the same blot, resulting in relative quantities and
allowing statistical analysis.
3.8 Statistical analysis
Statistical analysis was performed with SPSS (IBM Corporation) using Mann-Whitney-U test,
Spearman correlation and Kruskal-Wallis test. P-values <0.05 were considered significant (*).

23
4 Results
4.1 Patients with COPD
4.1.1 Emphysema quantification
Figure 6.A: correlation of COPD classification
with the emphysema scoring. Each dot
represents several patients. n=28. P=0.004.
Figure 6.B: correlation of DLCO with
emphysema scoring. n=26. P>0.05.
Figures 6.A and 6.B represent correlations for the emphysema scoring with respectively COPD
classification and DLCO. The correlation with COPD classification was significant. Sadly, no
emphysema scoring of severe COPD (GOLD III-IV) was could be obtained.
4.1.2 RT-qPCR for BAMBI
The mRNA expression of BAMBI in human lung tissue was analysed by RT-qPCR in 92 subjects
(patient characteristics in table 1). The mRNA levels of patients stratified into four groups were
compared. No statistical significant differences between the groups were found (P>0.05). The
results are displayed in figure 7.
24
Figure 7: RT-qPCR for BAMBI on human
lung. RT-qPCR results of patients stratified
into four categories (never smokers, healthy
(ex-)smokers, COPD GOLD II patients and
COPD GOLD IV patients). No significant
difference between the different groups was
found (P=0.961; Kruskal-Wallis).
4.1.3 RT-qPCR for BMP-6
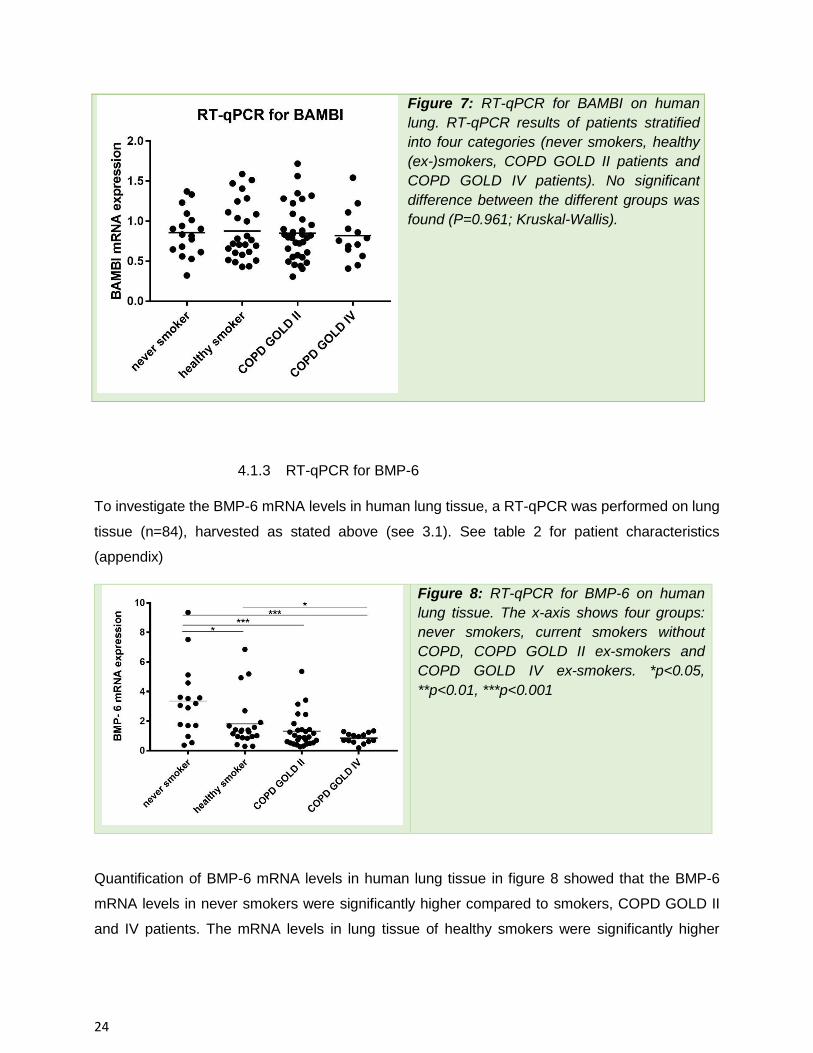
To investigate the BMP-6 mRNA levels in human lung tissue, a RT-qPCR was performed on lung
tissue (n=84), harvested as stated above (see 3.1). See table 2 for patient characteristics
(appendix)
Figure 8: RT-qPCR for BMP-6 on human
lung tissue. The x-axis shows four groups:
never smokers, current smokers without
COPD, COPD GOLD II ex-smokers and
COPD GOLD IV ex-smokers. *p<0.05,
**p<0.01, ***p<0.001
Quantification of BMP-6 mRNA levels in human lung tissue in figure 8 showed that the BMP-6
mRNA levels in never smokers were significantly higher compared to smokers, COPD GOLD II
and IV patients. The mRNA levels in lung tissue of healthy smokers were significantly higher

25
compared to COPD GOLD IV patients. Importantly, patients with very severe COPD have the
lowest levels of BMP-6.
4.1.4 Correlations
Using the BMP-6 mRNA levels we found, we performed some correlations to investigate the
relation of BMP-6 levels to lung function parameters and packyears (PY). Figures 9.A-9.F show
the results. Significant results were found for the correlation of BMP-6 mRNA levels with FEV1,
FEV1/FVC and PY.
Figure 9.A: r= 0,4099 (P=0,0002). n=78 Figure 9.B: r= 0,438 (P<0,0001). n=77
Figure 9.C: r= 0,01945 (P=0,8879). n=55 Figure 9.D: r= 0,2177 (P=0,1069). n=56

26
Figure 9.E: r= -0,1648 (P=0,4020). n=28
emphysema score: 0=0%, 1=1-5%, 2=6-25%,
3=26-50%, 4=>75% emphysema
Figure 9.F: r= -0,4403 (P<0,0001). n=76
4.2 Murine model of COPD
4.2.1 RT-qPCR for BAMBI
Figure 10: RT-qPCR for BAMBI on cigarette
smoke exposed mice for 1 month exposure
and 6 months exposure. There was a
significant (p<0,05) decrease of BAMBI
mRNA levels from both smoke exposure
mice groups compared to their air exposed
equivalents.
Figure 10 shows the RT-qPCR for BAMBI in mice. Two pairs of categories were compared: 1
month air exposure and 1 month smoke exposure, 6 months air exposure and 6 month smoke

27
exposure. In both pairs a significant difference was found, with BAMBI being decreased in smoke-
exposed mice.
4.2.2 RT-qPCR for BMP-6
Figure 11: RT-qPCR for BMP-6 on murine
lungs. Two sets were compared: one
where the mice were exposed to air or
smoke for 1 month and one where they
were exposed for 6 months. **p<0.01
Figure 11 shows the RT-qPCR for BAMBI on air-exposed mice versus smoke-exposed mice in
two groups. In the group where the mice were exposed for 1 months, no significant difference in
BMP-6 mRNA levels was found between air-exposed and smoke-exposed mice. In mice that were
exposed for 6 months, a significant decrease (p=0.0047) in BMP-6 mRNA levels was found in
smoke-exposed mice compared to their air-exposed counterparts.

28
Figure 12: RT-qPCR for BMP-6 on BMP-
6 knockout mice. ****p<0.0001
To determine the BMP-6 deficiency of BMP-6 knockout mice, we also performed a RT-qPCR on
the knockout mice. The results are displayed in figure 12. There was nearly no expression
measured of BMP-6 mRNA in the knockout mice (p<0.0001).
4.2.3 IHC-P
BMP-6 expression and localisation was studied in mouse lungs using immunohistochemistry
staining. In order to validate the antibody’s specificity for BMP-6, we performed knockout validation
for three antibodies (see table 3). Each antibody was tested on wild type mouse and on BMP-6
knockouts. For each there also is an isotype control. BMP-6 was mainly detected in the smooth
muscles around bronchioles and blood vessels. Figure 13 and figure 14 show the results for
respectively the Abcam anti-BMP-6 antibody and the Biorbyt anti-BMP-6 antibody (see table 3 for
antibodies). As seen in figure 13.B and 14.B, the BMP-6 knockout mice show a colouring pattern
similar to the wild types in 13.A and 14.A.

29
Figure 13: BMP-6 immunohistochemistry with antibody from Abcam
13.A: wild type with detail
13.B: BMP-6 knockout with detail
A
B

30
13.C: wild type isotype control 13.D: knockout mouse isotype control
Representative photomicrographs of immunohistochemical stainings for BMP-6 on lung tissue
with close-up of a wild type (fig. 13.A) and BMP-6 knockout mouse (fig. 13.B) with their
corresponding isotype controls. In 13.C and 13.D, isotype controls are displayed.
C D

31
Figure 14: BMP-6 immunohistochemistry with antibody from Biorbyt
14.A: wild type mouse with detail
14.B: BMP-6 knockout with detail
A
B

32
14.C: wild type mouse with isotype 14.D: knockout mouse with isotype
Representative photomicrographs of immunohistochemical stainings for BMP-6 on lung tissue
of a wild type (fig. 14.A) and BMP-6 knockout mouse (fig. 14.B) with their corresponding close-
ups. In 14.C and 14.D, isotype controls are displayed.
4.2.4 Western Blot
To investigate the BMP-6 protein expression, western blots for BMP-6 were performed on mouse
lung tissue samples. We assessed the specificity of 3 anti-BMP-6 antibodies (see table 3) by using
lung homogenates of wild type and BMP-6 knockout mice. Figures 15 and 16 also show a
household protein blot (β-actin and GADPH respectively) for loading control. This allows for a
quantitative analysis of the BMP-6 expression.
Figure 15: Western Blot for BMP-6 using
the Abcam ab155963 monoclonal anti-
BMP-6 antibody. The first 4 lanes were
loaded with wild type mice samples while
the last 4 lanes were loaded with samples
from BMP-6 knockout mice. The bands are
at the predicted 42 kDa.

33
Figure 15 shows the Western Blot for the Abcam ab155963 antibody. As can be seen on the blot,
both the wild type and knockout mouse show intense bands. No loading control household protein
was determined.
Figure 16.A: Western Blot for BMP-6 using
the Biorbyt orb155848 polyclonal anti-BMP-6
antibody. The first 4 lanes were loaded with
wild type mice samples while the last 4 lanes
were loaded with samples from BMP-6
knockout mice. The arrow indicates BMP-6 at
56 kDa.
Figure 16.B: Loading control on the same
western blot, using Sigma-Aldrich A2066 anti-
actin antibody as housekeeping protein. The
β-actin is at 42 kDa.
Figure 16.C: Quantification of the Biorbyt
orb155848 analysis. P<0.05.
Figure 16 shows the result for the Biorbyt orb155848 antibody. The differences in protein
expression for BMP-6 between wild type and knockout mice were found to be statistically
significant (P<0.05), with the expression of BMP-6 in knockout mice being less than in in wild type
mice. The P-value stayed <0.05 when the outlier in the wild type section was taken out of the
calculation (not shown).

34
Figure 17.A: Western Blot for BMP-6 using
the Acris AP20577PU-N polyclonal anti-
BMP-6 antibody. The first 4 lanes were
loaded with wild type mice samples while the
last 4 lanes were loaded with samples from
BMP-6 knockout mice. The bands are at the
predicted 57 kDa.
Figure 17.B: Loading control on the same
western blot, using Cell Signalling
Technology 14C10 anti-GADPH HRP-
conjugated antibody as housekeeping
protein. The bands are at the predicted 37
kDa.
Figure 17.C: Quantification of the Acris
AP20577PU-N analysis. P=0.2.
Figure 17 shows the results for the Acris AP20577PU-N antibody. The protein levels for BMP-6,
as measured by western blot analysis, were not statistically different between wild type and BMP-
6 knockout mice (P=0.2).
In conclusion, all the tested antibodies reacted on both the wild type and knockout mice.

35
5 Discussion
5.1 COPD patients
5.1.1 BAMBI
We attempted to investigate the potential role of BMP and activin membrane-bound inhibitor
(BAMBI) in the pathogenesis of COPD. The hypothesis is that the pseudoreceptor BAMBI takes
TGF-β out of the circulation and drives the system towards inflammation(103). Zhang et al. found
BAMBI to be upregulated in relation to the COPD severity and to correlate with the T17/Treg cell
ratio(76, 103). Drömann et al. found BAMBI to be strongly expressed in COPD lungs and to be
influenced by infection(102). In human lung tissue, we did not find a significant difference between
the BAMBI mRNA levels of never smokers, current healthy (ex-)smokers, COPD GOLD II patients
and COPD GOLD IV patients.
5.1.2 BMP-6
BMP-6 became a suspect because of a GWAS study where it was found to correlate to FVC(99).
However, FVC is not a disease marker of obstructive lung disease but the FEV1/FVC ratio or
Tiffeneau index is. While the FVC can be altered in COPD(109), it is also a marker for restrictive
lung diseases such as lung fibrosis. Little is known about BMP-6 in COPD. It is best described as
a major regulator of hepcidin(94). Iron dysregulation is suspected to play a role in COPD and an
increased iron sequestration in alveolar macrophages of COPD patients has been found(95). We
found BMP-6 mRNA levels to be significantly less in COPD patients compared to never smokers,
reserving a potential role for BMP-6 in the pathogenesis of COPD. It has to be determined if BMP-
6 is an innocent bystander or whether it is a main driver for iron-induced damage in COPD patients.
Our hypothesis is that because of decreased BMP-6 levels in correlation with COPD severity,
more iron-induced reactive oxygen species form. It has already been shown that BMP-6 deficient
mice develop massive iron overload in the liver(110).
Using the results from the RT-qPCR for BMP-6 on human tissue, we performed correlations for
FEV1, FEV1/FVC, emphysema score, packyears, KCO and DLCO. We found a significant result
(P<0,05) for FEV1, FEV1/FVC and packyears. This suggests a correlation to lung function tests
as well as to the main risk factor of COPD being cigarette smoking. However, it is again not clear
whether this is a causal relation or coincidence. A possible explanation why no significant results

36
were found for KCO and DLCO could be because these lung function parameters rely on the gas
exchange capacity. As already stated, different COPD patients will exhibit different symptoms as
well as different CT phenotypes(2). KCO and DLCO are not diagnostic parameters but FEV1/FVC
is, so every diagnosed COPD patient will have an obligate FEV1/FVC ratio lower than 0,7. This
could also explain why the BMP-6 mRNA levels did not correlate with the emphysema score, as
not all COPD patients are dominant emphysematous(2, 104). Another reason may be that the
emphysema scoring only succeeded in 28 patients, resulting in a rather small group that was
available for analysis, lacking sufficient power.
5.2 Murine model of COPD
5.2.1 BAMBI
In contrast to the study by Zhang et al. (103), our results show BAMBI mRNA levels to be
significantly lower in smoke-exposed mice compared to their air exposed counterparts. There
are several reasons as to why our results differ from those in the literature. First, there may be
different conditions involved in the study setup that influences the outcomes. Second, as a
pseudoreceptor, BAMBI may have an ambivalent role, enhancing or attenuating inflammation
depending on the trigger or environment. More research on the role of BAMBI is needed to
assess its role.
5.2.2 BMP-6
We attempted to study the localisation of BMP-6 on the protein level. Here we encountered a
problem as both the wild type and the BMP-6 knockout mice showed a strong signal in the smooth
muscle cells around bronchioles and a weaker sign in the vascular walls and macrophages. Three
different antibodies were used, but the BMP-6 knockout mice never failed to show an equal
reaction as the wild types.
When investigating the protein levels of BMP-6 using western blot, we encountered the same
problem. Only for the Biorbyt orb155848 anti-BMP-6 antibody, a statistical significant difference
between the wild type and knockout mice on western blot was found but this needs to be confirmed
in an independent experiment.

37
There are several possible reasons as to why the knockout mice showed reaction. First of all,
there may have been technical flaws. However, this is unlikely as the procedures are all
standardised, have been previously validated and have been done more than once. This leaves
the mice or the antibodies as potential suspects.
The mice were designed by Solloway et al.(107) by replacing the second exon of the BMP-6 gene
by a Acc1-gene targeting vector carrying a neomycin resistance gene. The offspring mice were
controlled using RNA probes for the mature section of the BMP-6 gene, downstream of the
replaced section. The homozygote knockout mice did not show any reaction to the probes and as
such were deemed full BMP-6 knockout mice. It is however never specified how this results in a
loss-of-function of BMP-6 and one should note that knockout of a gene isn’t the same as deletion.
This may be due to the degrading of the altered mRNA, altering a splice site or by causing a
premature stopcodon (the one in the used Acc1 gene for example). While this results in a
phenotypic knockout, it is possible that the part between the BMP-6 promotor region and the
beginning of the altered exon 2 escapes nonsense mediated decay and gets translated. For the
Abcam anti-BMP-6 antibody, it is known from the datasheet that this antibody has specificity for a
region in the pro-peptide that is later processed. If the other used antibodies (their immunogen
was not included in the datasheet) also have their specificity for a sequence that is included in
exon 1, this may be the reason why the western blot showed equal signal in the wild type and
knockout lanes. For this theory to be true, the BMP-6 exon 1 of the knockout gene should still be
transcribed and translated, providing the epitopes for the antibodies to bind upon. The mice are
still true knockouts, as they don’t have BMP-6 function and exhibit the BMP-6 knockout phenotype.
While western blot uses denatured proteins, immunohistochemistry does not. For the antibodies
to bind the secreted part of the BMP-6 exon 1, it should also have a 3D structure that enables the
antibody to recognise its epitope. The fact that the bands in the knockout lanes on WB were at the
same height as the wild type bands does not agree with our theory that a partial protein is
synthesized. Additionally, we determined by RT-qPCR that the BMP-6 mRNA expression in the
BMP-6 knockout mice is almost absent using a probe against sequences downstream from exon
3. For further analysis, it would be recommended to check the knockout mouse with an antibody
against sequences coded for by the deleted part of exon 2 or further downstream as well a different
antibody towards sequences coded for by exon 1. A more sensitive technique would be the use
of a one-step RT-qPCR with probes against exon 1 compared to probes against exon 3-7. This
would allow to confirm whether exon 1 gets transcribed and translated. It would also explain why
there was a significant difference measured using the Biorbyt antibody. If the used immunogen for
this polyclonal antibody contains both sequences derived from exon 1 and other sequences

38
derived from other exons of BMP-6, the signal from the knockout mice we found could represent
those antibodies reacting to epitopes coded for by exon 1.
Another option is that the antibodies are not specific enough. BMP-6 belongs to the TGF-β
superfamily and shares a strong similarity to other members of its superfamily. Especially BMP-5
and BMP-7 are strongly similar with BMP-6. It is possible that the antibodies aren’t specific enough
and react with other TGF-β members. Studies have found that a lot of commercial antibodies lack
sufficient specificity(111). The percentage of aspecific or polluted antibodies was found to be up
to 30%(112, 113). The lack of sufficient specificity of some commercial antibodies may offer an
explanation as to why the BMP-6 knockout mice showed reaction to anti-BMP-6 antibodies despite
not having BMP-6 protein.
We also studied the mRNA expression in smoke-exposed mice. We found no significant difference
between 1 month smoke-and air-exposed mice. For the 6 months exposed mice, we found that
the BMP-6 mRNA levels in smoke-exposed mice were significantly lower compared to their air-
exposed counterparts. The negative result from the 1 month exposure test can be explained by
the fact that 1 month smoke-exposure only yield airway inflammation but not COPD, while 6
months smoke-exposed mice exhibit hallmarks of COPD.
5.3 Conclusion
We demonstrated a potential role for BMP-6 in the pathogenesis of COPD. An RT-qPCR
performed on human lung tissue from never smokers, smokers, COPD GOLD II patients and
COPD GOLD IV patients showed a statistical significant difference between never smokers and
ex-smokers, GOLD II and GOLD IV. A difference was also found between smokers and GOLD IV
patients. This suggests that BMP-6 is decreased in COPD. In mice, we found a similar result where
smoke-exposed mice showed significantly lower BMP-6 mRNA levels. Correlations between
human BMP-6 mRNA levels and FEV1, FEV1/FVC and packyears were significant. Further
studies should elucidate whether BMP-6 is an active driver of COPD, or only plays a passive role.
While BAMBI has been a suspect after it was found elevated in COPD patients(102, 103), we did
not found significant different BAMBI mRNA levels in humane COPD lung tissue. We did find
significant decreased BAMBI mRNA levels in smoke-exposed mice. Further research should
elucidate the exact role of BAMBI and BMP-6 in the pathogenesis of COPD.

39
6 Acknowledgements
I would like to thank prof. dr. Ken Bracke for his excellent guidance, feedback and for sharing his
ideas. Furthermore, I would like to thank everyone of the laboratory for Translational Research on
Obstructive Pulmonary Diseases for their advice, technical assistance (and availability of coffee).
I would also like to acknowledge prof. dr. Guy Brusselle for his advice on COPD and the pulmonary
CT scans.
In particular I would like to thank dr. Fien Verhamme for her guidance, most helpful feedback, help
with interpretation and patience.
The University of Ghent and UZ Ghent are acknowledged for offering the use of their facilities.

40
7 Bibliography
1. Izquierdo-Alonso JL, Rodriguez-GonzálezMoro JM, de Lucas-Ramos P, Unzueta I, Ribera X, Antón E, et al. Prevalence and characteristics of three clinical phenotypes of chronic obstructive pulmonary disease (COPD). Respiratory Medicine. 2013;107(5):724-31. 2. Mohamed Hoesein FAA, Schmidt M, Mets OM, Gietema HA, Lammers J-WJ, Zanen P, et al. Discriminating dominant computed tomography phenotypes in smokers without or with mild COPD. Respiratory Medicine. 2014;108(1):136-43. 3. Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. American Journal of Respiratory and Critical Care Medicine. 2013;187(4):347-65. 4. Lozano Rea. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. The Lancet. 2012;380(9859):2095-128. 5. CD M, D L. Projections of Global Mortality and Burden
of Disease from 2002 to 2030. PLoS Medicine. 2006;3(11):e442. 6. From the Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2017. Available from: http://goldcopd.org. 7. Eisner MD, Anthonisen N, Coultas D, Kuenzli N, Perez-Padilla R, Postma D, et al. An Official American Thoracic Society Public Policy Statement: Novel Risk Factors and the Global Burden of Chronic Obstructive Pulmonary Disease. American Journal of Respiratory and Critical Care Medicine. 2010;182(5):693-718. 8. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. The Lancet.370(9589):765-73. 9. Bilano V, Gilmour S, Moffiet T, d'Espaignet ET, Stevens GA, Commar A, et al. Global trends and projections for tobacco use, 1990–2025: an analysis of smoking indicators from the WHO Comprehensive Information Systems for Tobacco Control. The Lancet.385(9972):966-76. 10. Løkke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61(11):935-9. 11. Rennard SI, Vestbo J. COPD: the dangerous underestimate of 15%. The Lancet. 2006;367(9518):1216-9. 12. Kerstjens HA, Rijcken B, Schouten JP, Postma DS. Decline of FEV1 by age and smoking status: facts, figures, and fallacies. Thorax. 1997;52(9):820-7. 13. Wain LV, Shrine N, Miller S, Jackson VE, Ntalla I, Artigas MS, et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. The Lancet Respiratory Medicine. 2015;3(10):769-81. 14. D’Anna SE, Asnaghi R, Caramori G, Appendini L, Rizzo M, Cavallaro C, et al. High-Resolution Computed Tomography Quantitation of Emphysema Is Correlated with Selected Lung Function Values in Stable COPD. Respiration. 2012;83(5):383-90. 15. Lahousse L, Seys LJM, Joos GF, Franco OH, Stricker Bruno H, Brusselle GG. Epidemiology and impact of chronic bronchitis in chronic obstructive pulmonary disease. European Respiratory Journal. 2017;50(2). 16. MacNee W. Pathology, pathogenesis, and pathophysiology. BMJ : British Medical Journal. 2006;332(7551):1202-4. 17. O'Donnell DE. Hyperinflation, Dyspnea, and Exercise Intolerance in Chronic Obstructive Pulmonary Disease. Proceedings of the American Thoracic Society. 2006;3(2):180-4.

41
18. Seemungal TAR, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of Exacerbation on Quality of Life in Patients with Chronic Obstructive Pulmonary Disease. American Journal of Respiratory and Critical Care Medicine. 1998;157(5):1418-22. 19. Donaldson GC, Wilkinson TMA, Hurst JR, Perera WR, Wedzicha JA. Exacerbations and Time Spent Outdoors in Chronic Obstructive Pulmonary Disease. American Journal of Respiratory and Critical Care Medicine. 2005;171(5):446-52. 20. Andersson F, Borg S, Jansson S-A, Jonsson A-C, Ericsson Å, PrÜTz C, et al. The costs of exacerbations in chronic obstructive pulmonary disease (COPD). Respiratory Medicine. 2002;96(9):700-8. 21. Punekar YS, Shukla A, Müllerova H. COPD management costs according to the frequency of COPD exacerbations in UK primary care. International Journal of Chronic Obstructive Pulmonary Disease. 2014;9:65-73. 22. Wedzicha JA, Donaldson GC. Exacerbations of Chronic Obstructive Pulmonary Disease. Respiratory Care. 2003;48(12):1204. 23. Miravitlles M, de la Roza C, Morera J, Montemayor T, Gobartt E, Martín A, et al. Chronic respiratory symptoms, spirometry and knowledge of COPD among general population. Respiratory Medicine. 2006;100(11):1973-80. 24. Smith J, Woodcock A. Cough and its importance in COPD. International Journal of Chronic Obstructive Pulmonary Disease. 2006;1(3):305-14. 25. Vestbo J, Rasmussen FV. Respiratory symptoms and FEV1 as predictors of hospitalization and medication in the following 12 years due to respiratory disease. European Respiratory Journal. 1989;2(8):710. 26. Puente-Maestu L, Stringer WW. Hyperinflation and its management in COPD. International Journal of Chronic Obstructive Pulmonary Disease. 2006;1(4):381-400. 27. Sakao S, Voelkel NF, Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. European Respiratory Review. 2014;23(133):350. 28. Burrows B, Kettel LJ, Niden AH, Rabinowitz M, Diener CF. Patterns of Cardiovascular Dysfunction in Chronic Obstructive Lung Disease. New England Journal of Medicine. 1972;286(17):912-8. 29. Traver GA, Cline MG, Burrows B. Predictors of Mortality in Chronic Obstructive Pulmonary Disease. American Review of Respiratory Disease. 1979;119(6):895-902. 30. Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. European Respiratory Journal. 2003;21(5):892. 31. van Schayck CP, Loozen JMC, Wagena E, Akkermans RP, Wesseling GJ. Detecting patients at a high risk of developing chronic obstructive pulmonary disease in general practice: cross sectional case finding study. BMJ : British Medical Journal. 2002;324(7350):1370-. 32. Agusti A, Calverley PMA, Celli B, Coxson HO, Edwards LD, Lomas DA, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respiratory Research. 2010;11(1):122-. 33. Han MK, Muellerova H, Curran-Everett D, Dransfield MT, Washko GR, Regan EA, et al. GOLD 2011 disease severity classification in COPDGene: a prospective cohort study. The Lancet Respiratory Medicine. 2013;1(1):43-50. 34. Bestall JC, Paul EA, Garrod R, Garnham R, Jones PW, Wedzicha JA. Usefulness of the Medical Research Council (MRC) dyspnoea scale as a measure of disability in patients with chronic obstructive pulmonary disease. Thorax. 1999;54(7):581. 35. Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A Self-complete Measure of Health Status for Chronic Airflow Limitation: The St. George's Respiratory Questionnaire. American Review of Respiratory Disease. 1992;145(6):1321-7. 36. Jones PW, Harding G, Berry P, Wiklund I, Chen WH, Kline Leidy N. Development and first validation of the COPD Assessment Test. European Respiratory Journal. 2009;34(3):648.

42
37. Anthonisen NR, Connett JE, Kiley JP, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of fev1: The lung health study. JAMA. 1994;272(19):1497-505. 38. Pauwels RA, Löfdahl C-G, Laitinen LA, Schouten JP, Postma DS, Pride NB, et al. Long-Term Treatment with Inhaled Budesonide in Persons with Mild Chronic Obstructive Pulmonary Disease Who Continue Smoking. New England Journal of Medicine. 1999;340(25):1948-53. 39. Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respiratory Research. 2010;11(1):149-. 40. Rabe KF, Timmer W, Sagkriotis A, Viel K. Comparison of a Combination of Tiotropium Plus Formoterol to Salmeterol Plus Fluticasone in Moderate COPD. Chest. 2008;134(2):255-62. 41. Aaron SD, Vandemheen KL, Fergusson D, et al. Tiotropium in combination with placebo, salmeterol, or fluticasone–salmeterol for treatment of chronic obstructive pulmonary disease: A randomized trial. Annals of Internal Medicine. 2007;146(8):545-55. 42. Bradley JM, O'Neill BM. Short-term ambulatory oxygen for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews. 2005(4). 43. Albert RK, Connett J, Bailey WC, Casaburi R, Cooper JAD, Criner GJ, et al. Azithromycin for Prevention of Exacerbations of COPD. New England Journal of Medicine. 2011;365(8):689-98. 44. Falk JA, Minai OA, Mosenifar Z. Inhaled and Systemic Corticosteroids in Chronic Obstructive Pulmonary Disease. Proceedings of the American Thoracic Society. 2008;5(4):506-12. 45. Pothirat C, Chaiwong W, Phetsuk N, Pisalthanapuna S, Chetsadaphan N, Choomuang W. Evaluating inhaler use technique in COPD patients. International Journal of Chronic Obstructive Pulmonary Disease. 2015;10:1291-8. 46. Poole P, Chacko EE, Wood-Baker R, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews. 2006(1). 47. Walters JAE, Smith S, Poole P, Granger RH, Wood-Baker R. Injectable vaccines for preventing pneumococcal infection in patients with chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews. 2010(11). 48. Williams MC, Murchison JT, Edwards LD, Agustí A, Bakke P, Calverley PMA, et al. Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax. 2014;69(8):718. 49. Takagi H, Umemoto T. A Meta-Analysis of the Association of Chronic Obstructive Pulmonary Disease with Abdominal Aortic Aneurysm Presence. Annals of Vascular Surgery. 2016;34:84-94. 50. Light RW, Merrill EJ, Despars JA, Gordon GH, Mutalipassi LR. Prevalence of Depression and Anxiety in Patients with COPD: Relationship to Functional Capacity. Chest. 1985;87(1):35-8. 51. Meteran H, Backer V, Kyvik KO, Skytthe A, Thomsen SF. Comorbidity between chronic obstructive pulmonary disease and type 2 diabetes: A nation-wide cohort twin study. Respiratory Medicine. 2015;109(8):1026-30. 52. Ferguson GT, Calverley PMA, Anderson JA, Jenkins CR, Jones PW, Willits LR, et al. Prevalence and Progression of Osteoporosis in Patients With COPD: Results From the Towards a Revolution in COPD Health Study. Chest. 2009;136(6):1456-65. 53. Portegies MLP, Lahousse L, Joos GF, Hofman A, Koudstaal PJ, Stricker BH, et al. Chronic Obstructive Pulmonary Disease and the Risk of Stroke. The Rotterdam Study. American Journal of Respiratory and Critical Care Medicine. 2015;193(3):251-8. 54. Van Remoortel H, Hornikx M, Langer D, Burtin C, Everaerts S, Verhamme P, et al. Risk Factors and Comorbidities in the Preclinical Stages of Chronic Obstructive Pulmonary Disease. American Journal of Respiratory and Critical Care Medicine. 2013;189(1):30-8. 55. Agusti A. The path to personalised medicine in COPD. Thorax. 2014;69(9):857.

43
56. Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease–antiprotease imbalance, and inflammation. International Journal of Chronic Obstructive Pulmonary Disease. 2011;6:413-21. 57. Bhavani S, Yuan X, You R, Shan M, Corry D, Kheradmand F. Loss of Peripheral Tolerance in Emphysema. Phenotypes, Exacerbations, and Disease Progression. Annals of the American Thoracic Society. 2015;12(Suppl 2):S164-S8. 58. Yadava K, Pattaroni C, Sichelstiel AK, Trompette A, Gollwitzer ES, Salami O, et al. Microbiota Promotes Chronic Pulmonary Inflammation by Enhancing IL-17A and Autoantibodies. American Journal of Respiratory and Critical Care Medicine. 2016;193(9):975-87. 59. Cole AM, Liao H-I, Stuchlik O, Tilan J, Pohl J, Ganz T. Cationic Polypeptides Are Required for Antibacterial Activity of Human Airway Fluid. The Journal of Immunology. 2002;169(12):6985. 60. Heijink IH, Pouwels SD, Leijendekker C, Bruin HGd, Zijlstra GJ, Vaart Hvd, et al. Cigarette Smoke–Induced Damage-Associated Molecular Pattern Release from Necrotic Neutrophils Triggers Proinflammatory Mediator Release. American Journal of Respiratory Cell and Molecular Biology. 2015;52(5):554-62. 61. Pouwels SD, Heijink IH, ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014;7(2):215-26. 62. Botelho FM, Bauer CMT, Finch D, Nikota JK, Zavitz CCJ, Kelly A, et al. IL-1α/IL-1R1 Expression in Chronic Obstructive Pulmonary Disease and Mechanistic Relevance to Smoke-Induced Neutrophilia in Mice. PLoS ONE. 2011;6(12):e28457. 63. Park JS, Svetkauskaite D, He Q, Kim J-Y, Strassheim D, Ishizaka A, et al. Involvement of Toll-like Receptors 2 and 4 in Cellular Activation by High Mobility Group Box 1 Protein. Journal of Biological Chemistry. 2004;279(9):7370-7. 64. Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. European Journal of Immunology. 2005;35(7):2184-90. 65. Guo R-F, Ward PA. ROLE OF C5A IN INFLAMMATORY RESPONSES. Annual Review of Immunology. 2004;23(1):821-52. 66. Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. Journal of Leukocyte Biology. 1994;56(5):559-64. 67. Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Letters. 1992;307(1):97-101. 68. Hiemstra PS, van Wetering S, Stolk J. Neutrophil serine proteinases and defensins in chronic obstructive pulmonary disease: effects on pulmonary epithelium. European Respiratory Journal. 1998;12(5):1200. 69. Janoff A, Sloan B, Weinbaum G, Damiano V, Sandhaus RA, Elias J, et al. Experimental Emphysema Induced with Purified Human Neutrophil Elastase: Tissue Localization of the Instilled Protease. American Review of Respiratory Disease. 1977;115(3):461-78. 70. Boxio R, Wartelle J, Nawrocki-Raby B, Lagrange B, Malleret L, Hirche T, et al. Neutrophil elastase cleaves epithelial cadherin in acutely injured lung epithelium. Respiratory Research. 2016;17:129. 71. Demedts IK, Brusselle GG, Bracke KR, Vermaelen KY, Pauwels RA. Matrix metalloproteinases in asthma and COPD. Current Opinion in Pharmacology. 2005;5(3):257-63. 72. Overbeek SA, Braber S, Koelink PJ, Henricks PAJ, Mortaz E, LoTam Loi AT, et al. Cigarette Smoke-Induced Collagen Destruction; Key to Chronic Neutrophilic Airway Inflammation? PLoS ONE. 2013;8(1):e55612.

44
73. Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8<sup>+</sup> T Cells Are Required for Inflammation and Destruction in Cigarette Smoke-Induced Emphysema in Mice. The Journal of Immunology. 2007;178(12):8090. 74. Paats MS, Bergen IM, Hoogsteden HC, van der Eerden MM, Hendriks RW. Systemic CD4+ and CD8+ T-cell cytokine profiles correlate with GOLD stage in stable COPD. European Respiratory Journal. 2012;40(2):330. 75. D'Hulst AI, Maes T, Bracke KR, Demedts IK, Tournoy KG, Joos GF, et al. Cigarette smoke-induced pulmonary emphysema in scid-mice. Is the acquired immune system required? Respiratory Research. 2005;6(1):147-. 76. Zhang J, Chu S, Zhong X, Lao Q, He Z, Liang Y. Increased expression of CD4+IL-17+ cells in the lung tissue of patients with stable chronic obstructive pulmonary disease (COPD) and smokers. International Immunopharmacology. 2013;15(1):58-66. 77. Stoller JK, Aboussouan LS. α1-antitrypsin deficiency. The Lancet.365(9478):2225-36. 78. Churg A, Wright JL. Proteases and emphysema. Current Opinion in Pulmonary Medicine. 2005;11(2):153-9. 79. Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4(8):617-29. 80. Kirkham PA, Barnes PJ. Oxidative Stress in COPD. Chest. 2013;144(1):266-73. 81. Turrens JF. Mitochondrial formation of reactive oxygen species. The Journal of Physiology. 2003;552(Pt 2):335-44. 82. Blokhina O, Virolainen E, Fagerstedt KV. Antioxidants, Oxidative Damage and Oxygen Deprivation Stress: a Review. Annals of Botany. 2003;91(2):179-94. 83. Motz GT, Eppert BL, Sun G, Wesselkamper SC, Linke MJ, Deka R, et al. Persistence of Lung CD8 T Cell Oligoclonal Expansions upon Smoking Cessation in a Mouse Model of Cigarette Smoke-Induced Emphysema. The Journal of Immunology. 2008;181(11):8036. 84. Xu C, Hesselbacher S, Tsai C-L, Shan M, Spitz M, Scheurer M, et al. Autoreactive T Cells in Human Smokers is Predictive of Clinical Outcome. Frontiers in Immunology. 2012;3:267. 85. Freeman CM, Stolberg VR, Crudgington S, Martinez FJ, Han MK, Chensue SW, et al. Human CD56+ Cytotoxic Lung Lymphocytes Kill Autologous Lung Cells in Chronic Obstructive Pulmonary Disease. PLoS ONE. 2014;9(7):e103840. 86. Brusselle GG, Demoor T, Bracke KR, Brandsma CA, Timens W. Lymphoid follicles in (very) severe COPD: beneficial or harmful? European Respiratory Journal. 2009;34(1):219. 87. Packard TA, Li QZ, Cosgrove GP, Bowler RP, Cambier JC. COPD is associated with production of autoantibodies to a broad spectrum of self-antigens, correlative with disease phenotype. Immunologic research. 2013;55(0):48-57. 88. Seys LJM, Verhamme FM, Schinwald A, Hammad H, Cunoosamy DM, Bantsimba-Malanda C, et al. Role of B Cell–Activating Factor in Chronic Obstructive Pulmonary Disease. American Journal of Respiratory and Critical Care Medicine. 2015;192(6):706-18. 89. Massagué J. TGFβ signalling in context. Nature reviews Molecular cell biology. 2012;13(10):616-30. 90. Reddi AH. BMPs: From bone morphogenetic proteins to body morphogenetic proteins. Cytokine & Growth Factor Reviews. 2005;16(3):249-50. 91. Verhamme FM, Bracke KR, Joos GF, Brusselle GG. Transforming Growth Factor-β Superfamily in Obstructive Lung Diseases. More Suspects Than TGF-β Alone. American Journal of Respiratory Cell and Molecular Biology. 2014;52(6):653-62. 92. Moustakas A, Souchelnytskyi S, Heldin C-H. Smad regulation in TGF-β signal transduction. Journal of Cell Science. 2001;114(24):4359. 93. Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang R-H, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of <em>Bmp6, Smad7, Id1</em>, and <em>Atoh8</em> in the mouse liver. Blood. 2008;112(4):1503-9.

45
94. Andriopoulos B, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP-6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nature genetics. 2009;41(4):482-7. 95. Philippot Q, Deslée G, Adair-Kirk TL, Woods JC, Byers D, Conradi S, et al. Increased Iron Sequestration in Alveolar Macrophages in Chronic Obtructive Pulmonary Disease. PLoS ONE. 2014;9(5):e96285. 96. Arndt S, Wacker E, Dorn C, Koch A, Saugspier M, Thasler WE, et al. Enhanced expression of BMP6 inhibits hepatic fibrosis in non-alcoholic fatty liver disease. Gut. 2015;64(6):973. 97. Dendooven A, van Oostrom O, van der Giezen DM, Willem Leeuwis J, Snijckers C, Joles JA, et al. Loss of Endogenous Bone Morphogenetic Protein-6 Aggravates Renal Fibrosis. The American Journal of Pathology. 2011;178(3):1069-79. 98. Banach J, Gilewski W, Słomka A, Buszko K, Błażejewski J, Karasek D, et al. Bone morphogenetic protein 6—a possible new player in pathophysiology of heart failure. Clinical and Experimental Pharmacology and Physiology. 2016;43(12):1247-50. 99. Loth DW, Artigas MS, Gharib SA, Wain LV, Franceschini N, Koch B, et al. Genome-wide association analysis identifies six new loci associated with forced vital capacity. Nature genetics. 2014;46(7):669-77. 100. Rosendahl A, Pardali E, Speletas M, ten Dijke P, Heldin C-H, Sideras P. Activation of Bone Morphogenetic Protein/Smad Signaling in Bronchial Epithelial Cells during Airway Inflammation. American Journal of Respiratory Cell and Molecular Biology. 2002;27(2):160-9. 101. Onichtchouk D, Chen Y-G, Dosch R, Gawantka V, Delius H, Massague J, et al. Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature. 1999;401(6752):480-5. 102. Drömann D, Rupp J, Rohmann K, Osbahr S, Ulmer AJ, Marwitz S, et al. The TGF-beta-Pseudoreceptor BAMBI is strongly expressed in COPD lungs and regulated by nontypeable Haemophilus influenzae. Respiratory Research. 2010;11(1):67-. 103. Zhang J-C, Chen G, Chen L, Meng Z-J, Xiong X-Z, Liu H-J, et al. TGF-β/BAMBI pathway dysfunction contributes to peripheral Th17/Treg imbalance in chronic obstructive pulmonary disease. Scientific Reports. 2016;6:31911. 104. Bastos HNe, Neves I, Redondo M, Cunha R, Pereira JM, Magalhães A, et al. Influence of emphysema distribution on pulmonary function parameters in COPD patients. Jornal Brasileiro de Pneumologia. 2015;41(6):489-95. 105. Lynch DA, Murphy JR, Crapo JD, Criner GJ, Galperin-Aizenberg M, Jacobson FL, et al. A combined pulmonary -radiology workshop for visual evaluation of COPD: study design, chest CT findings and concordance with quantitative evaluation. COPD. 2012;9(2):151-9. 106. Kim SS, Seo JB, Lee HY, Nevrekar DV, Forssen AV, Crapo JD, et al. Chronic Obstructive Pulmonary Disease: Lobe-based Visual Assessment of Volumetric CT by Using Standard Images—Comparison with Quantitative CT and Pulmonary Function Test in the COPDGene Study. Radiology. 2013;266(2):626-35. 107. Solloway MJ, Dudley AT, Bikoff EK, Lyons KM, Hogan BLM, Robertson EJ. Mice lacking Bmp6 function. Developmental Genetics. 1998;22(4):321-39. 108. hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. European Respiratory Journal. 2005;26(2):204. 109. Macklem PT. Therapeutic implications of the pathophysiology of COPD. European Respiratory Journal. 2010;35(3):676. 110. Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth M-P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nature Genetics. 2009;41:478. 111. Bordeaux J, Welsh AW, Agarwal S, Killiam E, Baquero MT, Hanna JA, et al. Antibody validation. BioTechniques. 2010;48(3):197-209.

46
112. Spicer SS, Spivey MA, Ito M, Schulte BA. Some ascites monoclonal antibody preparations contain contaminants that bind to selected Golgi zones or mast cells. Journal of Histochemistry & Cytochemistry. 1994;42(2):213-21. 113. Grimsey NL, Goodfellow CE, Scotter EL, Dowie MJ, Glass M, Graham ES. Specific detection of CB1 receptors; cannabinoid CB1 receptor antibodies are not all created equal! Journal of Neuroscience Methods. 2008;171(1):78-86.





























